

Abstract
Microfluidic techniques are widely used for high-throughput quantification and discrete analysis of micron-scale objects but are difficult to apply to molecular-scale targets. Instead, single-molecule methods primarily rely on low-throughput microscopic imaging of immobilized molecules. Here we report that commercial-grade flow cytometers can detect single nucleic acid targets following enzymatic extension and dense labeling with multiple distinct fluorophores. We focus on microRNAs, short nucleic acids that can be extended by rolling circle amplification (RCA). We labeled RCA-extended microRNAs with multicolor fluorophores to generate repetitive nucleic acid products with submicron sizes and tunable multispectral profiles. By cross-correlating the multiparametric optical features, signal-to-background ratios were amplified 1600-fold to allow single-molecule detection across 4 orders of magnitude of concentration. The limit of detection was measured to be 47 fM, which is 100-fold better than gold-standard methods based on polymerase chain reaction. Furthermore, multiparametric analysis allowed discrimination of different microRNA sequences in the same solution using distinguishable optical barcodes. Barcodes can apply both ratiometric and colorimetric signatures, which could facilitate high-dimensional multiplexing. Because of the wide availability of flow cytometers, we anticipate that this technology can provide immediate access to high-throughput multiparametric single-molecule measurements and can further be adapted to the diverse range of molecular amplification methods that are continually emerging.
Keywords: miRNA, microRNA, biomarker, multiplexing, RCA, amplification, diagnostics
Graphical Abstract
Single-molecule analytical techniques have had a transformative impact on analytical chemistry,1–5 biophysics,6–11 enzymology,12–16 and cellular biology,17–22 garnering numerous Nobel Prizes.23–27 For the most common experimental approaches, individual molecules are immobilized through surface adsorption, cooling, or optical trapping and are optically probed or imaged using high-resolution microscopy.28–30 Using these techniques, diverse molecular processes have been revealed in inhomogeneous media and complex systems, and the resulting ultrahigh analytical sensitivity is beginning to benefit clinical molecular diagnostics.31–35 However, a fundamental bottleneck for the application of these methods is the low-throughput nature of microscopy-based single-molecule analyses, which typically require long data acquisition times and computationally expensive analysis pipelines.36–38 Experimental results can also be limited by subjective image processing steps that bias sampling39,40 and insufficient statistical power to analyze rare events or to characterize heterogeneous molecular populations.29,36,41 These technical challenges limit broad adoption of single-molecule techniques and limit the rigorous interpretation of reported outcomes.
For objects with micron-scale dimensions, microfluidic instruments have solved the problem of achieving both high accuracy and high-throughput analysis at the resolution of individual objects.42,43 Fluorescence-based flow cytometers are microfluidic instruments containing a flow cell in which dispersed objects in a microscale laminar flow stream are rapidly passed across high-speed, high-sensitivity optical detectors aligned with laser excitation sources. These instruments measure fluorescence and scattering of individual events in microseconds, allowing counting of 105 to 107 cells in just seconds to minutes to generate large statistical power from small volume samples. These instruments are the standard platform for quantitative single-cell analysis in biology and are used routinely in clinical diagnostic laboratories for characterization of blood cells. For basic research studies, these flow-based methods have been extended from individual mammalian cells44 to colloids with submicron dimensions, including chromosomes,45 virions,46 extracellular vesicles,47 and synthetic nanoparticles.48 Individual molecules have also been analyzed in fluidic systems for nearly 30 years,49,50 however customized micro- and nanofluidic designs and optics that are not available in commercial instruments are needed to achieve ultrasensitive detection at the level of single molecules,50–52 preventing adoption by applied scientists. Meanwhile, the past two decades have seen a tremendous advancement of commercial flow cytometry instruments that allow high-dimensional multiparametric event analyses, including optical detection instruments measuring 19 coincident optical channels using numerous lasers and detectors precisely aligned in series along the flow path.43,53 This results in an extraordinary combination of rapid data collection in conjunction with high-dimensional characterization. Furthermore, advances in software for both data collection and statistical analysis have made these techniques easily adoptable for new users in the biomedical sciences.54 However, small molecules and biomacromolecules remain below the detection limit of commercial-grade flow cytometers and have not benefitted from the advances in throughput, multiparametric analysis, and simplicity.
Here we describe the quantification of soluble macro-molecules at the single-molecule level using commercial-grade flow cytometers. The key development is a method for molecular amplification and multispectral fluorescent labeling to generate densely labeled and fluorescently barcoded products which can be detected with high fidelity. Optical barcodes are the critical advance that enables cross-correlation analysis to eliminate background signals intrinsic to individual optical channels. We demonstrate the technique for detection of microRNA (miR), which are short ~22-mer single-stranded RNAs. We focus on miR-375, an exosomal blood biomarker that is a prognostic indicator for survival in castrate resistant prostate cancer.55,56 As depicted in Figure 1, each miR is extended from its 3′ end as single-stranded DNA (ssDNA) by rolling circle amplification (RCA) using a circular ssDNA template and DNA polymerase. This reaction appends long sequential repeats of a designed nucleic acid sequence to each miR, resulting in products that we call “miR amplicons.”57–59 In a second step, miR amplicons are fluorescently labeled through sequence-specific binding of dye-conjugated ssDNA probes (dye-DNA) through hybridization, in addition to an intercalating dye (SYBR Green) that exhibits a 1000-fold fluorescence enhancement when bound to double-stranded DNA (dsDNA).60,61 This results in amplicon products labeled with precise ratios of spectrally distinct fluorophores. Finally, amplicons are injected into the flowstream of a benchtop fluorescence-based flow cytometer to measure optical scattering and multichannel fluorescence intensity for each detected event. Notably, RCA has previously been used to quantify miRs at the ensemble level by measuring intensities from radioisotopic or fluorescent nucleic acids probes.62,63 By instead measuring fluorescence intensity spikes in a flow cytometer, individual molecules derived from RCA and labeled with multispectral probes can be discretely measured and classified by their multispectral profiles. These profiles can be precisely controlled in both color and ratiometric intensity to generate diverse barcodes to differentiate and quantify distinct molecules in a mixture. This methodology could, therefore, serve as a framework for multiparametric single-molecule detection that is not possible at the ensemble level and could be applied to the rapidly diversifying techniques that use high density fluorescence labeling of molecular targets, especially those focused on RNA.64,65
Figure 1.
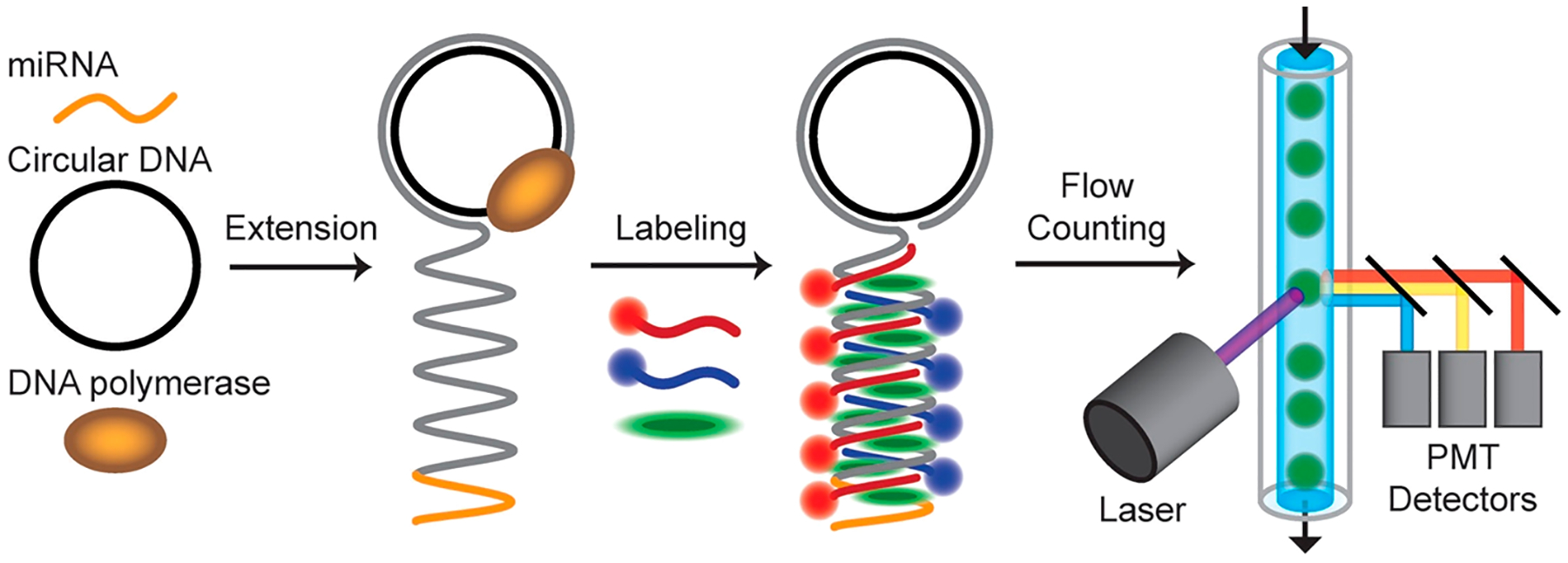
Schematic diagram depicting fluorescent labeling and fluorescence detection of single nucleic acid molecules using a flow cytometer. In this example, ~22-mer microRNA is extended by rolling circle amplification using a circular DNA template and DNA polymerase to generate large molecules with repeating sequence motifs. The products are then labeled with a mixture of multicolor intercalating dyes and dye-DNA molecules that bind through hybridization. The products are then analyzed as individual events by multispectral fluorescence and scattering in a flow cytometer. The schematics illustrate molecular mechanisms but are not drawn to scale. The specific dye-DNA probes applied determine the density of labeling and the fraction of amplicon that is double-stranded.
RESULTS AND DISCUSSION
Molecular Extension and Fluorescent Labeling.
After RCA amplification and dye-DNA probe hybridization, miR amplicons exhibit intense fluorescence that can be distinguished amid a large excess of dye-DNA probes. Figure 2a shows a comparison between the fluorescently labeled amplicons and probes alone after immobilization on a glass coverslip, showing much higher fluorescence spot intensities of the amplicons. As a result of the RCA reaction, 22 base miRs are extended to an average length near 48 kilobases based on gel electrophoresis (SI Figure S1), which is orders of magnitude longer than their original size. The amplicon products have a polydisperse length distribution, which is discussed further below. Using a 70 base circular template, miR amplicons are labeled with an average of 691 concatamer repeats which allow binding of as many as 691 dyes through dye-DNA probe hybridization. When characterized using fluorescence microscopy, a portion of amplicons are even larger than the ~200 nm optical diffraction limit (SI Figure S2). Confocal imaging of a diffraction-limited spot in the reaction solution also reveals fluorescent molecules with intensities much brighter than the dye-DNA background signals (Figure 2b) with diffusion coefficients that are nearly 2 orders of magnitude smaller than those of the dye-DNA probes alone (SI Figure S3).
Figure 2.
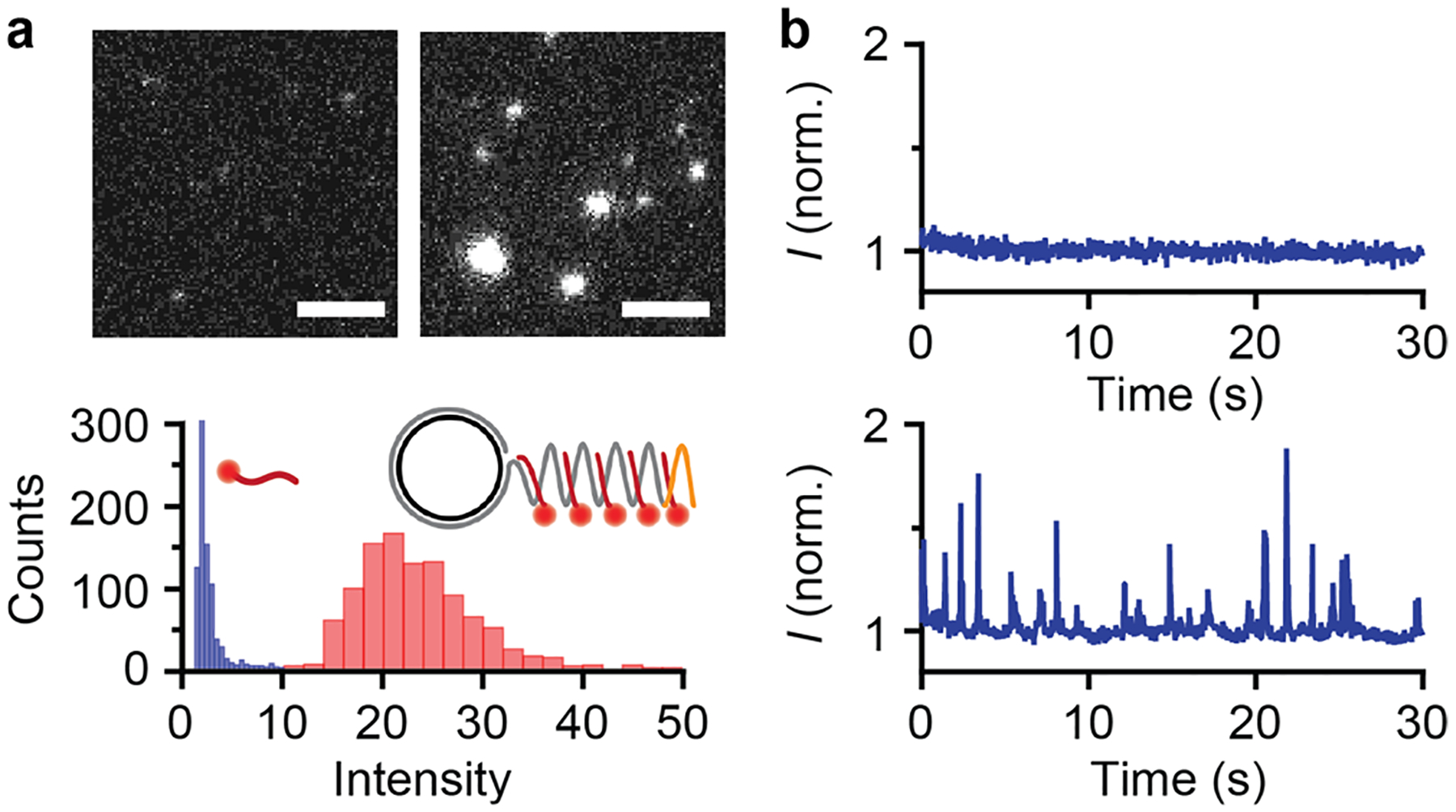
Characterization of extended and fluorescently labeled microRNA at the single molecule level. (a) Fluorescence micrographs of Cy3-conjugated dye-DNA probes (left) and miR-375 amplicons labeled with the same dye-DNA probes (right). Scale bars represent 5 μm. Intensity histograms are shown below for probes alone (blue) and labeled microRNA products (red). (b) Temporal fluorescence intensity in a diffraction limited confocal spot for dye-DNA probes in solution (top) and labeled microRNA products (bottom).
Molecular Detection Using a Flow Cytometer.
Following extension and labeling, we injected the solution of miR amplicons into the inlet flow valve of a commercial-grade fluorescence-based flow cytometer with photomultiplier tube detectors and laser illumination sources, instrumentation typically used to detect, count, and characterize large scattering particles with micron dimensions. Figure 3a,b shows scatter plots of optically detected events in the solution, showing single events exhibiting high-intensity side-scattering of light (SSC) as well as high-intensity fluorescence deriving from the SYBR Green intercalating dye and two dye-DNA probes (Cy3-DNA and Cy5-DNA) bound through sequence-specific hybridization. With this labeling scheme, less than 20% of the amplicon is labeled with dye-DNA probes and therefore 80% remains single-stranded, providing molecular regions for multispectral color-coding (described below). These results show that this extension and labeling method generates individual miR amplicons that are sufficiently intense to register as distinguishable optical events. However, high levels of background counts were detected in each independent optical channel (SI Figure S4). In conventional flow cytometry, true events for individual cells are isolated from events corresponding to noncellular matter through an initial analysis of forward and side scattering, as individual cells exhibit distinctive scattering signatures. This metric was insufficiently specific for the recognition of noncolloidal molecules due to the absence of a distinguishable forward scattering signal from the labeled miR amplicons (SI Figure S5), so an alternative methodology was needed.
Figure 3.
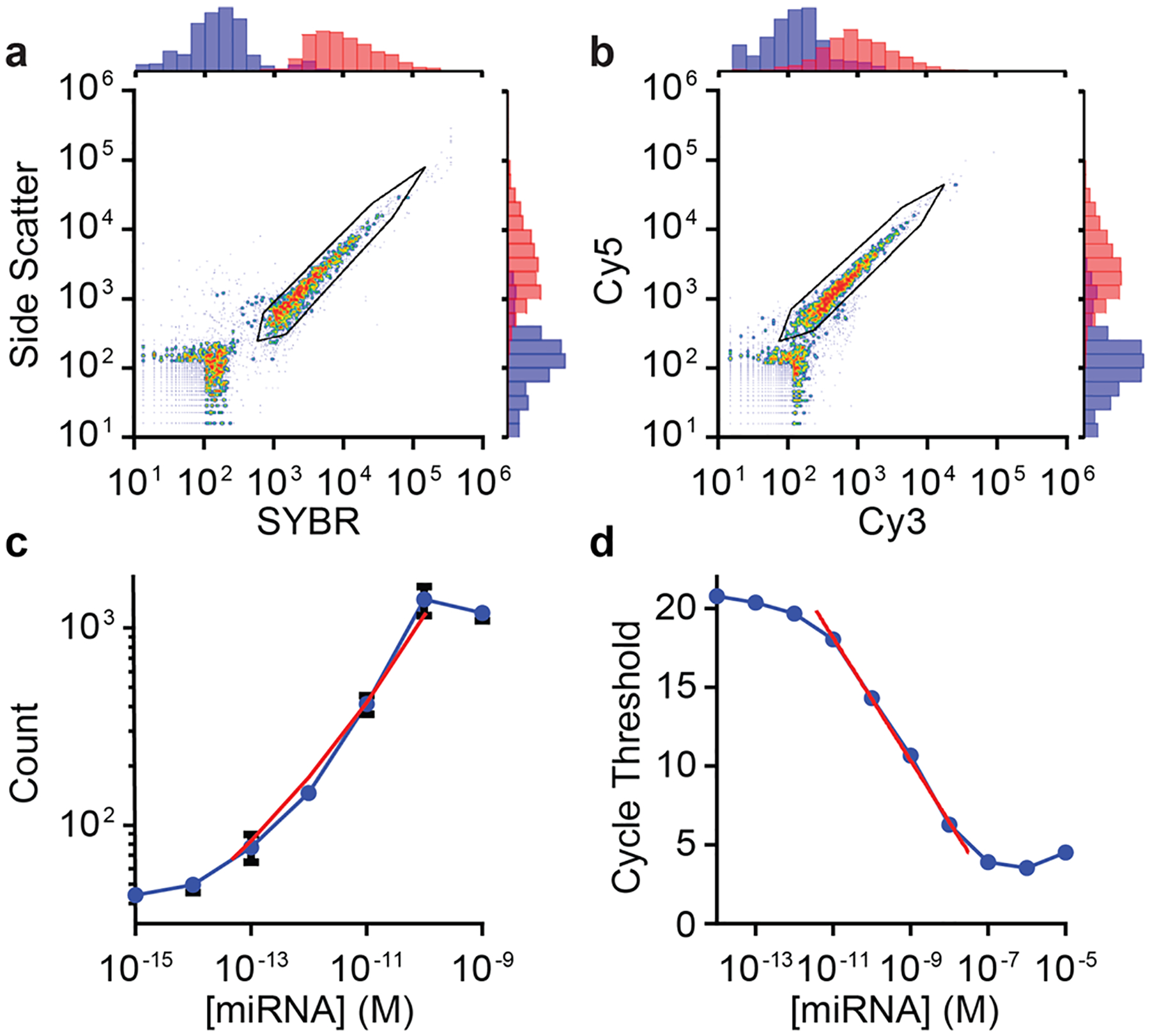
Detection of fluorescently labeled and extended microRNA in a flow cytometer. (a,b) Scatter plots show events detected for microRNA amplicons labeled with both dye-DNA probes and SYBR Green. (a) Correlation between side scattering and SYBR Green fluorescence intensity. Black lines indicate gates. (b) Same measurement as (a) showing Cy5 and Cy3 intensity channels for each event. (c) Correlation between molecular counts and microRNA concentration with all other conditions held constant. Red line indicates the linear dynamic range. (d) microRNA quantification through reverse transcription polymerase chain reaction. Plot shows the measured cycle threshold for each microRNA concentration using TaqMan qRT-PCR reagents and a real-time PCR instrument. All data points and error bars represent the mean and standard deviation, respectively, of three technical replicates.
In order to differentiate true miR amplicon signals from aberrant events measured for any individual optical parameter, we developed a method to cross-correlate the signals from multiple fluorescent labels and optical scattering for each event. Specifically, we used two-step biparametric gating of the four-dimensional (4D) data sets for each event composed of SSC intensities and fluorescence intensities from SYBR Green, Cy3-DNA, and Cy5-DNA. These gates are depicted as black polygonal boundaries in event scatter plots (see Figure 3a,b). Figure 3a shows a first level of gating based on SSC signal and the fluorescence intensity of SYBR Green. Data points within the gate show a band of events with nearly linear proportionality (R2 = 0.959), a relationship resulting from the dependence of SSC on the size of dielectric matter48 and the linearity of SYBR Green fluorescence enhancement when bound to dsDNA.60,61 Likewise, Figure 3b shows that the vast majority of gated counts correspond to molecules simultaneously bound by sequence specific dye-DNA probes labeled with Cy5 and Cy3, which exhibit a similar linear proportionality (R2 = 0.954).
The vast majority of events binned within the gated regions in Figure 3a,b were correlated across all four optical parameters (SI Figure S4), as each parameter pair in the 4D data set exhibited a Pearson correlation coefficient between 0.884 and 0.976. These correlated events corresponded to a population only present in RCA reactions containing miR targets (SI Figure S4). For control RCA reactions without miR-375 added, 5456 events were detected within an individual channel, of which 5453 were outside of the multiparametric gates, allowing exclusion of 99.95% of the nonspecific background counts. Simultaneously, for RCA reactions containing miR-375, 1732 out of 1973 total detected events were within all gates, allowing retention of 87.79% of the miR-specific events. Therefore, compared to samples analyzed without gating, this 4D gating strategy resulted in a 1576-fold improvement in signal-to-background ratio. Notably, it is optimal to tune gates for specific sample matrices, as fluorescence and scattering intensities of the background solution can differ; however, once optimized, gates can be consistently applied across different samples in the same matrix for miR quantification (see below). Importantly, this analytical methodology for multispectral fluorescence analysis of individual molecules is enabled by the standard features of commercial grade flow cytometers, whereas other single molecule techniques for nucleic acid quantification typically only allow single- or two-color detection.
We further measured the miR count as a function of RCA reaction time, which showed an abrupt rise and plateau (SI Figure S6). This is unlike the trend observed when amplicon growth is measured by real time enhancement of SYBR fluorescence, which exhibits a progressively increasing signal, indicating that further amplicon extension occurs after reaching a detectable size in the flow cytometer (SI Figure S6). This finding indicates that, despite significant heterogeneity in amplicon length in the population (SI Figure S1), miRs undergoing labeling reactions were successfully extended to a size that is stably detectable after a 1 h reaction. We note that it is unlikely that the entirety of miRs in solution are detected as counts across all concentrations, as many of the amplicons may not pass directly through the focal volume in microfluidic flow streams optimized for micron-scale objects. In addition, gating strategies were driven primarily to maximize fidelity of miR-specific signals, so many true target signals were discarded.
miR Quantification.
Gated counts of miRs from flow cytometry were stoichiometrically proportional to the number of miR targets present. Figure 3c shows counts from solutions containing seven different concentrations of miR-375 with all other conditions held constant (see also SI Figure S7). Using this readout as an assay, miR-375 could be quantified across a dynamic range spanning more than 4 orders of magnitude, from an upper limit of 100 pM to a lower limit of 47.0 fM, defined as the concentration for which counts were equal to three standard deviations above background counts. The sensitivity and wide dynamic range observed for this test are important for miRs due to the wide range of concentration of distinct sequences in biological fluids and cells.66 Further, the specificity for miR-375 was sufficient to quantitatively measure targets in RNA extracts from human plasma (SI Figure S8). A higher number of background counts was detected in these samples when using the same gates, likely due to interactions between dye-DNA probes and long RNA sequences present in the matrix. This could be eliminated by either using miR purification kits or choosing more stringent gating for the extract matrix. However, neither were necessary for the application of the assay methodology for miR-375 quantification, as the dynamic range and limit of detection were similar to those performed in pure solutions. This demonstrates the compatibility of this platform with complex sample media, although we recommend analyzing the contribution of matrix to background signals for different sample classes. In addition, a standard curve using spiked samples may be needed for new matrices to calibrate absolute native concentrations of the target miR sequence. We also determined that this same methodology and platform could be used for the detection and quantification of short DNA, resulting in a similar limit of detection (136 fM) and nearly 5 orders of magnitude of dynamic range (SI Figure S9).
In comparison with a flow cytometry-based readout, miR quantification based on net SYBR Gold intensity from RCA reactions measured using a quantitative polymerase chain reaction (qPCR) instrument gave a limit of detection of 36.5 pM (SI Figure S10), nearly a thousand times higher than flow cytometry-based detection. Likewise, the detection limit for gold-standard quantitative reverse transcriptase PCR (qRT-PCR) was 3.7 pM (Figure 3d), nearly 100 times higher. For this assay, we used a commercial grade Applied Biosystems TaqMan qPCR kit for miR-375, which gave similar results as the QIAGEN miScript PCR System (data not shown).67 Notably, some qRT-PCR assays for miR sequences have been reported to have higher sensitivity, however this is sequence-specific and lower sensitivity can result from nonoptimal primer design, differing efficiencies of multiple enzymatic steps, and interference from secondary structures, particularly for GC-rich miR sequences like miR-375.68,69 Notably, it is likely that sensitivity in flow cytometry-based detection could be further improved, as the high target specificity deriving from 4D gating in flow cytometry results in the exclusion of some labeled miRs which do not achieve a minimum intensity threshold in all four of the optical channels (SI Figure S4). This challenge could be addressed through the use of more sensitive optical instruments or dyes with higher fluorescence intensity. Sensitivity could also be boosted by using longer data acquisition periods, as the overall count number in flow cytometry increases linearly with acquisition time (SI Figure S11).
In addition to quantifying miR targets with perfect sequence complementarity to the full target region on the circular DNA template, we analyzed quantitative differences across the most common miR-375 isoforms (isomiRs miR-375.1, miR-375.2, miR-375.3, miR-375.4; sequences in SI Table S1). The isomiR sequences were derived from next-generation sequencing of exosomal RNA extracts from plasma, revealing polymorphisms primarily at the 3′ end.70 Both gold-standard qRT-PCR and RCA assays were performed using synthetic isomiRs, revealing differing degrees of selectivity for the isomiRs across the two analytical platforms (SI Figure S12). All four were detectable to differing degrees by RCA, whereas qRT-PCR was highly selective for isomiRs 1 and 3. Unlike sequencing-based quantification methods, hybridization-based nucleic acid assays exhibit lower degrees of sequence specificity and can be challenging to predict and interpret. In the case of miR-375, a broader degree of detectability across the isomiRs is desirable for our purposes, as the clinical correlation for prostate cancer prognostics was developed by summing the concentrations of all isomiRs.55 However, for some applications it is desirable to measure a single isoform alone with high specificity. This can be achieved with RCA amplification by using linear padlock probes in the place of circular templates with sequence-specific circularization mediated by SplintR Ligase.71–73 We determined that RCA amplicons deriving from padlock probes with SplintR Ligase can be labeled and detected in the same way as RCA alone, which can enable higher sequence specificity (SI Figure S13).
Molecular Barcoding.
In addition to increasing amplicon detection specificity, multiparametric analysis can be used to independently identify and quantify amplicons with spectrally distinct fluorescent profiles.74–78 To this end, circular RCA templates were designed to contain 5 distinct and tunable probe sites that were each 12−13 bases in length. The amplicons generated from RCA could then be labeled by hybridization with different combinations of up to five dye-DNA probes to generate distinguishable multispectral fingerprints (Figure 4a).74 The probe sites were designed to be as short as possible to maximize the number of color-coding regions for a fixed RCA template length, which will maximize multiplexing capacity. Shorter dye-DNA probes also increase the total density of fluorescent labeling, which can improve sensitivity, but also result in decreased melting temperatures, which can reduce probe binding affinity. We therefore balanced these effects to ensure that lengths were sufficient for efficient hybridization at room temperature, finding that 12-to 13-mer dye-DNA probes worked well. Probe sequences and binding mechanisms can likely be further optimized to tune multiplexing capacity and detection sensitivity.
Figure 4.
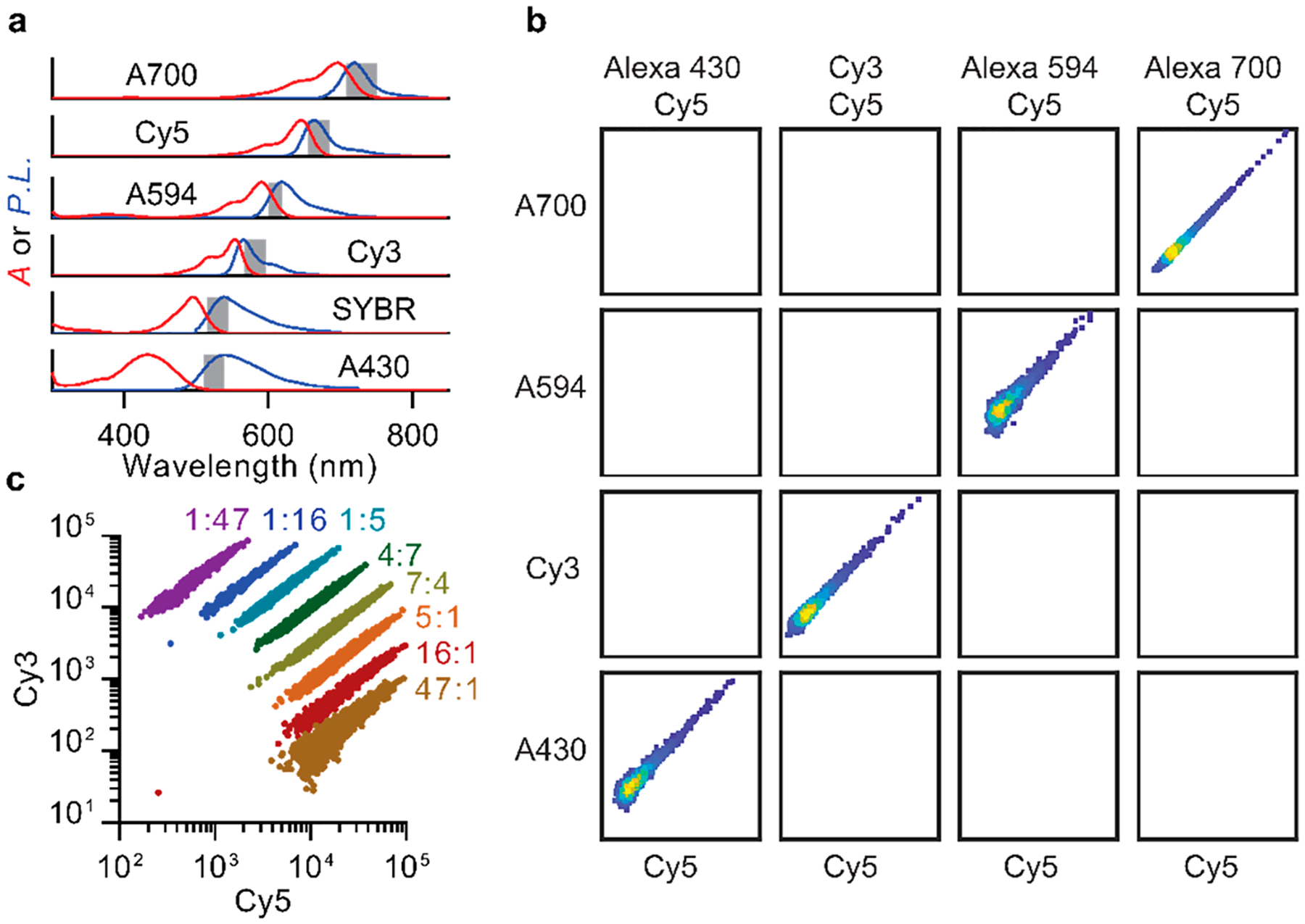
Multispectral labeling of extended microRNA through hybridization with multicolor dye-DNA. (a) Fluorophore absorption (A, red) and photoluminescence (P.L., blue) spectra for tested dyes are shown with normalized intensities. Gray highlight indicates emission bandpass filters. (b) Representative data for event counts in biparametric color channels using the dye combinations indicated at top. Axes show logarithmically scaled fluorescence intensity for indicated fluorophores. (c) Ratiometric labeling of miR amplicons with Cy3/Cy5 dye pairs shows discrete bands with narrow intensity profiles. Data were collected for 10 pM microRNA concentrations and >79 000 counts.
To characterize the capacity to differentiate amplicons using biparametric colorimetric gating, 10 pairs of 5 distinct fluorophores were analyzed. Figure 4b shows representative examples, revealing densely concentrated events within target color channels and nearly zero events in off-target channels. We also characterized the capacity to ratiometrically label single miR amplicons to generate distinguishable optical signatures within a single biparametric channel based on relative intensities. Figure 4c shows ratiometrically differentiated signatures for targets generated from eight different mixtures of Cy3 and Cy5 dye-DNA probes that bind to the same region of an miR amplicon. The resulting optical signatures are widely tunable and comprise narrow and minimally overlapping bands. These results indicate that single molecule flow cytometry measurements are compatible with a variety of fluorophores and that both colorimetric and ratiometric barcodes can be generated and distinguished.
To investigate the capacity of flow cytometry to independently detect and quantify multiple miR sequences in the same mixture, a second RCA assay was developed to target miR-30a-5p. miR-30a-5p is an important target for comparison with miR-375, as it serves as an endogenous internal reference for normalizing miR levels in serum exosome extract from prostate cancer patients.55 The circular template for miR-30a-5p was designed to generate amplicons that bind to two dye-DNA probes labeled with Alexa-430 and Alexa-594 and to exhibit minimal binding to miR-375 and its distinct dye-DNA probes labeled with Cy3 and Cy5. The templates and probes are thereby orthogonally reactive and distinguishable in separate spectral channels, as depicted in Figure 5a,b (sequences are provided in SI Table S1). Figure 5c,d shows that when all of the templates and probes are mixed together, amplicons generated in the presence of miR-375 result in events detected in the Cy3/Cy5 channels, with minimal events in the Alexa-430/Alexa-594 channels. Likewise, amplicons generated in the presence of miR-30a-5p result in events detected in the Alexa-430/Alexa-594 channels with minimal events in the Cy3/Cy5 channels (Figure 5e,f). The labeled amplicons are therefore amplified independently and can be detected independently in their distinguishable fluorescence channels without contributing to background counts in off-target channels. Figure 5g shows miR-375 channel counts from solutions containing seven different concentrations of miR-375 in the presence and absence of a high concentration of miR-30a-5p, showing a wide linear range of detection with only a small impact from the presence of the second target sequence. The lower detection limits were similar, 99.2 fM and 86.5 fM, in the presence and absence of miR-30a-5p, respectively. A similar result was observed for the detection of miR-30a-5p (Figure 5h), yielding comparable 772 fM and 835 fM detection limits in the presence and absence of miR-375, respectively. Therefore, both miR-375 and miR-30a-5p could be independently quantified from reactions containing both miR targets. Small differences in absolute counts due to the presence of multiple targets likely arise from differing reaction rates between the two sequences in the competing RCA reactions, leading to differing rates of reagent consumption.
Figure 5.
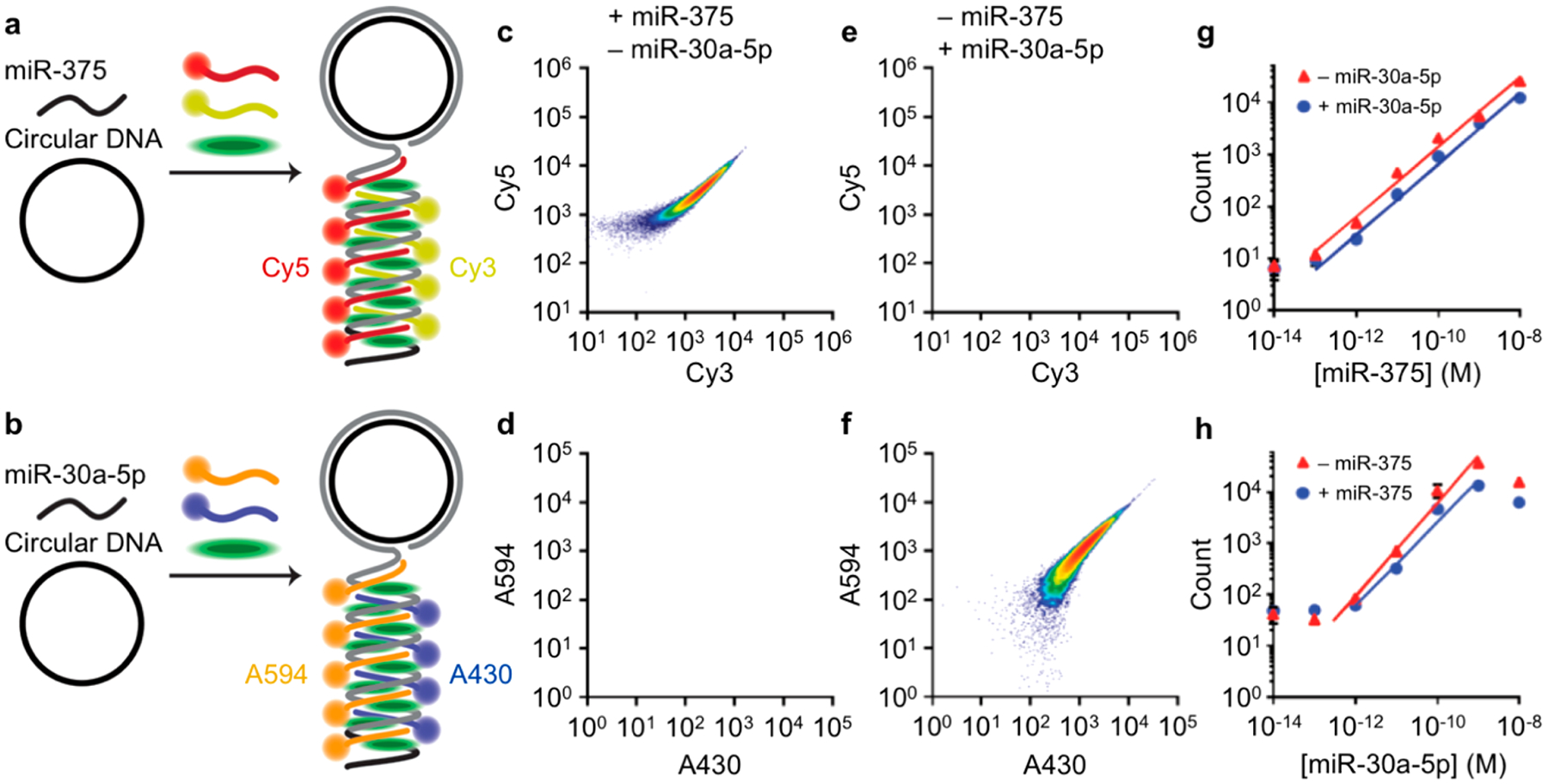
Multiplexed detection and quantification of microRNA in a flow cytometer. (a,b) Schematic diagrams depict fluorescent labeling and detection of (a) miR-375 using Cy3-DNA and Cy5-DNA probes or (b) miR-30a-5p using A430-DNA and A594-DNA probes with each amplicon labeled with SYBR Green. All templates and hybridization probes are mixed together to perform the multiplexed assay. The schematics illustrate molecular mechanisms but are not drawn to scale. (c,e) Scatter plots show events detected in the miR-375-specific Cy3/Cy5 biparametric color channel or the (d,f) Alexa-430/Alexa-594 color channel for amplicons generated through RCA reactions containing (c,d) 10 pM miR-375 alone or (e,f) 10 pM miR-30a-5p alone. Events were filtered by SYBR Green and side-scatter prior to gating in dye-DNA channels. (g) Correlation between Cy3/Cy5 counts and miR-375 concentration in the presence and absence of 10 pM miR-30a-5p. Trend lines represent the linear dynamic range. (h) Correlation between Alexa-430/Alexa-594 counts and miR-30a-5p concentration in the presence and absence of 10 pM miR-375. Trend lines represent the linear dynamic range.
Based on these experiments validating the capacity for multispectral optical barcoding, we calculated the theoretical capacity for sequence multiplexing. A primary success metric for multispectral barcoding is the capacity to minimize signal crosstalk between distinguishable channels. In the case of fluorescent dye-DNA signatures, optical codes are derived from the sequence-specific binding sites designed into the RCA template (Figure 6a). For colorimetric barcoding using biparametric gates with 10 pairs of 2 spectrally distinct fluorophores from Figure 4, we determined that 87 of 90 off-target channels analyzed yielded no detectable off-target events with an overall average occurrence of fewer than one in one million false positive counts (Figure 6b). Likewise, off-target events were nearly absent when using two fluorophores for ratiometric labeling, and false positive counts were only observed between a single pair of adjacent gates (Figure 6c). Using these data, we calculated the absolute number of potential molecular barcodes that could be independently distinguished at different degrees of specificity. On the basis of the observation that ratiometric amplicon populations fit well to elliptical Gaussian functions (R2 > 0.95; SI Figure S14), we calculated the overlap integrals for all pairwise ratios and summed these values to calculate the total false positive rate for each barcode. This process was repeated iteratively for all distinct ratiometric and colorimetric combinations, and parameters were optimized to maximize the number of codes resulting in a maximum false positive rate of either 10−3 or 10−6. For the Cy5/Cy3 pair analyzed with a ratio range between 1:47 and 47:1 (Figure 4c), as many as 16 distinguishable codes could be generated with fewer than one in one million false positive counts in any single channel. Figure 6d shows the calculated total number of distinguishable optical barcodes that could be generated by combining both color-coding and ratiometric labeling schemes (solid lines), showing the effects of signal crosstalk between channels resulting in false positive rates of either 10−3 (dotted lines) or 10−6 (dashed lines). When the color channel number is five or higher, hundreds to thousands of barcodes could be theoretically discriminated with minimal crosstalk.
Figure 6.
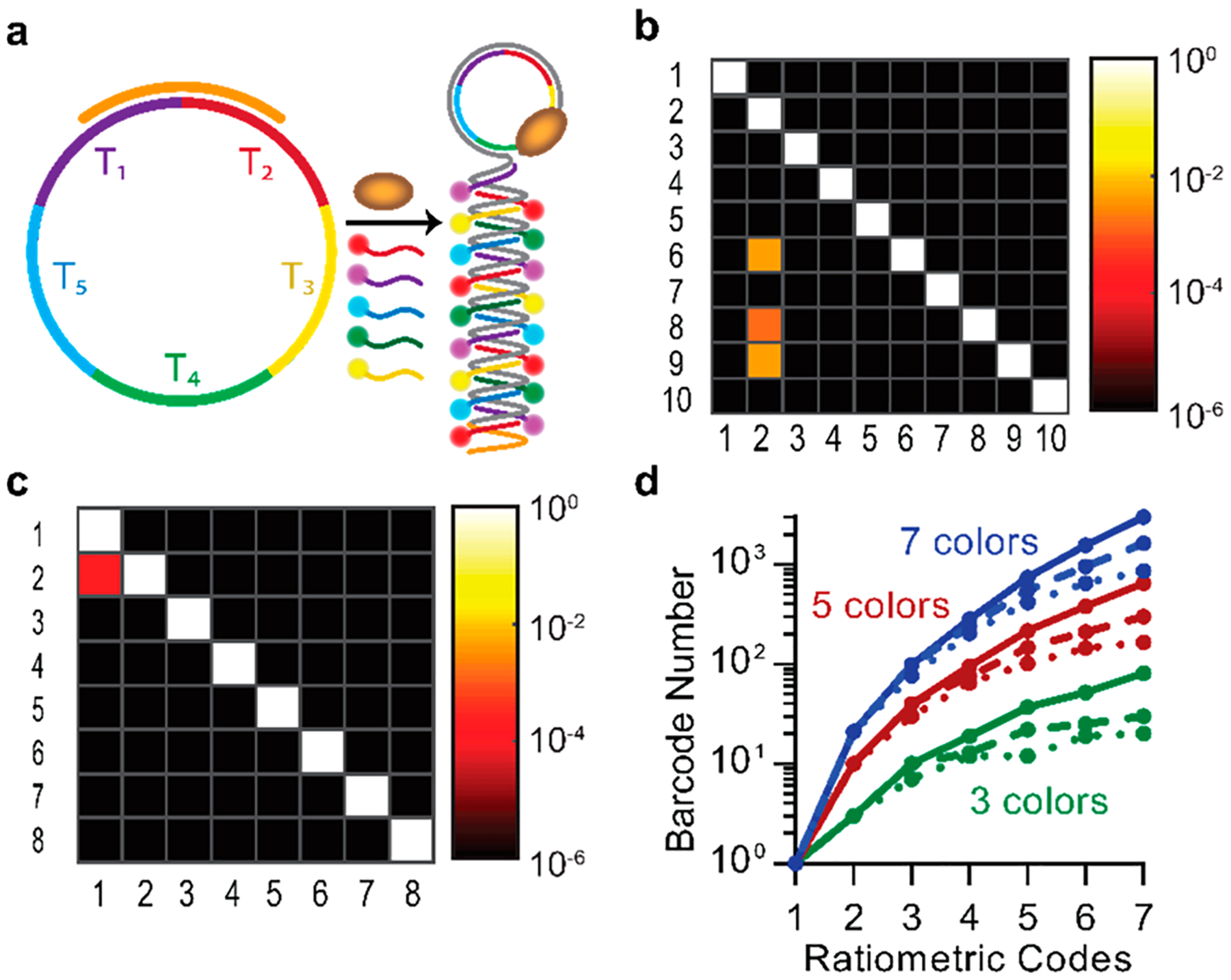
Optical differentiation and barcoding of microRNAs. (a) Schematic depiction of multicolor labeling based on sequence-specific dye-DNA hybridization. (b) Heat map shows false positive count rates for the biparametric color channels from Figure 4b. Fluorophore pairs are plotted on the y-axis, and optical channels are plotted on the x-axis. Channels and fluorophores corresponding to numerical labels are provided in SI Table S2. (c) Heat map of empirical false positive count rates between ratiometric channels from Figure 4c. Fluorophore pairs are plotted on the y-axis, and optical channels are plotted on the x-axis. Channels and fluorophores corresponding to numerical labels are provided in SI Table S3. (d) Calculated optical barcode number for indicated color code number and ratiometric code number. Colors indicate three (green), five (red), or seven (blue), color combinations. Solid curves show the total code numbers, whereas dashed and dotted lines indicate code numbers with maximum false positive count rates of 10−3 and 10−6, respectively.
While we demonstrated the effectiveness of this multiplexed detection and quantification approach for two sequences using colorimetric codes (Figure 5), further empirical optimization of orthogonally reactive template sequences and dye-DNA probe sequences will be needed to approach the theoretical limits of multiplexing using ratiometric codes. Most importantly, amplification reactions will need to be finely tuned to allow detection of numerous sequences that may be present at widely differing concentrations. For RCA reactions in particular, multiple competing reactions could lead to disproportionate consumption of free dNTPs in solution depending on target concentrations, resulting in amplicons with different size distributions, which could skew the measured results. This could be avoided by ensuring that reactions take place independently, such as with the use of emulsion-based reaction compartmentalization in droplets that has now become common for PCR.79
CONCLUSIONS
Flow-based measurements of single molecules have previously required intricately designed optical and fluidic setups that are not widely available.50–52 As a result, single-molecule quantification methods primarily rely on low-throughput microscopy-based analyses of immobilized targets. As shown here, benchtop flow cytometers are sufficiently sensitive to robustly detect individual molecules after dense labeling with multiple emitters that provide distinct optical signatures. Sensitive miR quantification can be achieved even in complex media with a detection limit that is better than that of conventional qRT-PCR by 2 orders of magnitude. Multiple sequences can also be independently quantified in a mixture with the potential to generate hundreds to thousands of optical barcodes. This provides an exciting opportunity to multiplex the detection of nucleic acids in solution with an optical capacity approaching the number of endogenous human miRs, currently estimated at 4076.80 While we demonstrated the applicability of flow cytometer-based detection for miRs and short DNA, we have not yet explored applicability for longer sequences of mRNA or genomic DNA. However, we expect that similar assays can be developed based on RCA using priming methods that have already been successfully applied to these nucleic acid classes.81–83 As a result of the high sensitivity and throughput of flow cytometers, their capacity for extensive barcoding, and their wide availability, we anticipate that multiparametric flow cytometer-based molecular analysis can be broadly adopted in research as well as clinical applications.
METHODS
Chemicals and Reagents.
DNA and RNA oligonucleotides with sequences shown in SI Table S1 were purchased from Integrated DNA Technologies. The oligos were resuspended in molecular biology grade water (Corning), centrifuged (5 min, 5000×g), analyzed for concentration and purity by absorption at 260 and 280 nm, and stored at −20 °C prior to use. CircLigase II was purchased from Lucigen. Deoxynucleotide Solution Mix (dNTP), Φ29 DNA polymerase, Murine RNase Inhibitor, SplintR Ligase, and E. coli exonuclease I were purchased from New England Biolabs. SUPERase-In RNase Inhibitor, SYBR Green I Nucleic Acid Gel Stain (SYBR Green), SYBR Gold Nucleic Acid Gel Stain (SYBR Gold), sodium hydroxide (>97%), glacial acetic acid (>99.7%), sodium bicarbonate (>99.7%) tris(hydroxymethyl)-aminomethane (Tris), Tween 20, ethylenediaminetetraacetic acid (EDTA), 6× DNA Gel Loading Dye, and TRIzol LS Reagent were purchased from ThermoFisher Scientific. 10× TBE was purchased from Bio-Rad Laboratories. Phosphate buffered saline (PBS) was purchased from Corning. Monomethoxy monosuccinimidyl ester poly(ethylene glycol) (mPEG5000-NHS, 5000 Da) and monoazido monosuccinimidyl ester poly(ethylene glycol) (azide-PEG5000-NHS, 5000 Da) were purchased from Nanocs, Inc. In-house purified Milli-Q water was used throughout. Unless specified, all other chemicals and solvents were purchased from Sigma-Aldrich and used without further purification. No. 1.5 coverglass was purchased as 50-well chambers from Electron Microscopy Sciences. Pooled human plasma was purchased from Innovative Research. Flow-Check Fluorospheres were purchased from Beckman Coulter.
DNA Template Circularization.
miR-375 or miR-30a-5p RCA Template (SI Table S1) with 5′-phosphoryl modification was circularized at 2 μM using CircLigase II (2.5 U μL−1) in 0.33 M Tris-acetate, 0.66 M potassium acetate, 2.5 mM MnCl2, and 5 mM dithiothreitol at pH 7.5 for 1 h at 60 °C. Unreacted linear DNA was removed by reaction with exonuclease I for 1 h at 37 °C followed by a 10 min incubation at 80 °C.
Rolling Circle Amplification.
miR or DNA was hybridized with corresponding circular RCA templates (100 nM) in polymerase buffer (50 mM Tris-HCl, 10 mM MgCl2, 10 mM (NH4)2SO4, 4 mM dithiothreitol, 0.5 U μL−1 SUPERase-In RNase Inhibitor, and 0.5 U μL−1 Murine RNase Inhibitor at pH 7.5) for 4 h at room temperature. RCA was then initiated through the addition of a 2× reaction mixture (0.2 U μL−1 Φ29 DNA polymerase, 2 mM dNTPs, 0.2 mg mL−1 BSA, and 0.002% SYBR Gold). Reactions were performed using an Eppendorf Realplex 4S Real-time PCR system with SYBR Gold fluorescence monitored using LED excitation at 470 nm and emission at 520 ± 10 nm at 30 s intervals. Reactions were allowed to proceed for 1 h at 37 °C prior to polymerase heat inactivation at 95 °C for 5 min. For time course studies, reaction mixtures were stored on ice until reaction initiation at 37 °C, and all reactions were heat inactivated simultaneously at 95 °C for 5 min. All samples were then stored at −20 °C prior to analysis.
Padlock Probe Mediated Rolling Circle Amplification.
A linear miR-375 Padlock Probe sequence with 5′-phosphoryl modification (SI Table S1) was mixed with miR-375 at different concentrations for 2 h at room temperature in a solution containing 0.625 U uL−1 SplintR Ligase prepared using SplintR Ligase reaction buffer (50 mM Tris-HCl, 10 mM MgCl2, 1 mM ATP, and 10 mM dithiothreitol, pH 7.5). RCA was then performed as described above using the 2× reaction mixture.
Amplicon Labeling.
RCA reaction solutions were diluted 20-fold in a solution containing dye-DNA probes at a final concentration of 3 nM in phosphate buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 2% BSA, pH 7.4) containing SYBR Green at a final concentration of 0.0316%. Samples were mixed thoroughly and denatured by incubation at 50 °C for 10 min, followed by 4 h of annealing at room temperature. Unless otherwise indicated, a combination of Cy3-DNA and Cy5-DNA were used to detect all miR-375 amplicons.
Data Collection by Flow Cytometry.
Labeled miR amplicons were transferred to round-bottom polystyrene tubes for loading into a BD LSR Fortessa Flow Cytometry Analyzer. All acquisition parameters were tested prior to each experiment using Flow-Check Fluorospheres to confirm that emission parameters were within expected intensity ranges. All events were recorded for which intensities were greater than the minimum detection threshold of 200 r.f.u. for side scattering, SYBR Green, and optical channels corresponding to all the dye-DNA probes used. Photomultiplier voltage of 450 V was used for all samples. Data were collected with a 15 s acquisition time and a total of 8.75 μL. Lasers and filters used for each probe are provided in SI Table S4. Fluidics were cleaned with desalinated water to remove residual sample between experiments.
Data Analysis.
Data from the flow cytometer were analyzed using FCS Express 6 software (De Novo Software). Events were first gated based on the biparametric intensities of side scattering and SYBR Green fluorescence and then based on the biparametric intensities of dye-DNA fluorescence unless otherwise noted. When analyzing two or more fluorophore combinations, intensity values were compensated to account for overlap between fluorophore channels. Compensation values were generated based on miR amplicons labeled with only one fluorophore, and automated compensation levels were manually optimized to maximize discrimination between different populations.
Multiplexed Detection of miR-375 and miR-30a-5p.
Reactions mixtures were prepared with miR-375, miR-30a-5p, or both miRs, together with miR-375 RCA Template and miR-30a-5p RCA Template, each at a 100 nM concentration in polymerase buffer (SI Table S1). When evaluating the effect of off-target miRs on reaction performance, off-target miRs were added at 10 pM concentration. Templates and miRs were hybridized for 4 h at room temperature and RCA was initiated with the addition of the 2× reaction mixture; reactions were performed for 1 h at 37 °C prior to polymerase heat inactivation at 95 °C for 5 min. All samples were then stored at −20 °C until analysis. RCA reaction solutions were then diluted 20-fold in a solution containing Cy3-DNA, Cy5-DNA T2, A430-DNA T3, and A594-DNA T4 dye-DNA probes, each at 3 nM concentration in phosphate buffer containing 0.0316% SYBR Green. Data were then collected as described above with events gated using SSC, SYBR, Cy3, Cy5, Alexa 430, and Alexa 594 channels.
Imaging MicroRNA Amplicons on Glass Coverslips.
Glass coverslips were cleaned by sonication in 1 M NaOH for 10 min, washed with water 10 times, and dried by nitrogen flush or centrifugation. The surfaces were further cleaned with plasma and then incubated in a solution of methanol, glacial acetic acid, and 3-aminopropyltriethoxysilane (93.46:4.67:1.87 by volume, respectively) for 10 min at room temperature, for 1 min with sonication, and for an additional 10 min at room temperature. The coverslips were then washed with water 10 times, dried, and treated with a 100 mM sodium bicarbonate solution containing mPEG5000-NHS (2.375% w/v) and azide-PEG5000-NHS (0.125% w/v) for 3 h. The surfaces were then washed 10 times with water, dried, and stored at −20 °C until use. Dibenzocyclooctyneconjugated Amplicon Capture DNA (40 nM) was then added to the azide-functionalized coverslips in a high salt Tris buffer (900 mM sodium chloride, 20 mM Tris Base, 1 mM EDTA, 0.1% Tween 20, pH 7.4) for 4 h at room temperature. The surfaces were washed four times with PBS before miR amplicon labeling reaction solutions were applied for 1 h at room temperature. Surfaces were then washed twice with PBS and imaged immediately.
Single Molecule Fluorescence Imaging.
Imaging was performed with wide-field illumination on a Zeiss Axio Observer.Z1 inverted microscope with a 100× 1.45 NA alpha PlanFluar oil immersion microscope objective using a 120 W X-Cite 120Q (Excelitas Technologies) excitation light source with a 550/25 nm bandpass excitation filter and a 605/70 nm bandpass emission filter. Images were collected on a Photometrics eXcelon Evolve 512 electron-multiplying charge-coupled device with 200 ms integration time and an electron multiplying gain of 500, controlled with Zeiss Zen software. Images were exported as 8-bit 512×512 TIFF files.
Single Molecule Fluorescence Image Analysis.
Single-molecule images were analyzed using an automated MATLAB script to identify fluorescent spots. Spot intensities were calculated from 33 pixel squares centered about the centroid of each detected spot, using 1000 individual spots from 10 different images for each sample.
Confocal Fluorescence Measurements in Solution.
Fluorescence intensities of diffraction limited spots were collected from RCA reaction samples with 0 or 1 nM miR concentration, labeled with Alexa-430-DNA. Measurements were performed using an Alba FCS single-molecule fluorescence confocal microscope (ISS) with 470 nm diode laser excitation. All intensity data were normalized so that the mean background intensity was 1.
Agarose Gel Electrophoresis.
Agarose gels (1.5% w/v) were prepared by dissolving 0.75 g of agarose in 1× TBE buffer at 100 °C. The solution was transferred to a horizontal gel electrophoresis apparatus and allowed to solidify for 30 min at room temperature. All samples were prepared using 6× DNA Loading Dye prior to loading in gel lanes. Electrophoresis was performed at 25 V for 1 h, followed by 50 V for 5 h. Gels were then removed and incubated in 1× TBE buffer containing a 1:10 000 dilution of SYBR Gold with shaking for 1 h at room temperature. Gels were then washed with deionized water and imaged using a Bio-Rad Gel Doc XR+ System with ultraviolet illumination.
Quantitative Reverse Transcription Polymerase Chain Reaction.
Reverse transcription of miR-375 was performed using a TaqMan MicroRNA Reverse Transcription Kit, and PCR was performed using a TaqMan Universal PCR Master Mix, following instructions provided by Thermo Fisher Scientific. The reverse transcription master mix was mixed with miR samples before reaction in an Eppendorf Realplex 4S Real-time PCR system with incubation at 16 °C for 30 min, 42 °C for 30 min, and 85 °C for 5 min. A qPCR reaction mixture was then prepared and added to each sample before running on an Eppendorf qRT-PCR machine at 95 °C for 10 min for enzyme activation followed by 40 PCR cycles consisting of a 15 s denaturation step at 95 °C and a 1 min annealing step at 60 °C. Results were analyzed using DataAssist software (Thermo Fisher Scientific) with custom threshold values set to optimize the assay limit of detection.
IsomiR Quantification.
For RCA, reactions were prepared as described above using miR-375 isomiRs (SI Table S1) diluted to 10 pM in PBS, except for miR-375.1, which was prepared across a log(10)-spaced concentration range. RCA was then performed as described above with SYBR Gold fluorescence signal measured using an Eppendorf Realplex 4S Real-time PCR system. The signal measured for each miR-375.1 concentration was then used to prepare a standard curve to calculate a linear fit for the dependence of the fluorescence signal on concentration. The effective concentration of each of the isomiRs was then calculated using the measured fluorescence signal and the linear fit. For qRT-PCR, reactions were performed as described above using isomiRs diluted to 10 pM in PBS, except for miR-375.1, which was prepared across a log(10)-spaced concentration range. After reverse transcription and PCR reactions, the effective concentration of each isomiR was calculated based on a standard curve for miR-375.1, using the same analysis methodology used for RCA.
RNA Extraction from Human Plasma.
RNA present in pooled healthy human plasma (Innovative Research) was extracted with the addition of 0.75 mL of TRIzol LS Reagent (Invitrogen) to 0.25 mL of plasma. Samples were then homogenized through pipetting and allowed to incubate for 5 min at room temperature. Chloroform (0.2 mL) was then added to each tube and allowed to incubate for 3 min. Samples were then centrifuged for 15 min at 12 000×g. RNA in the aqueous phase was then transferred to a new tube, precipitated with 0.5 mL of isopropanol, and incubated for 10 min before centrifugation at 12 000×g for 15 min at 4 °C. The supernatant was then removed, and RNA was resuspended in 75% ethanol prior to centrifugation at 7500×g for 5 min at 4 °C. The supernatant was then removed, and the RNA was allowed to air-dry for 10 min before resuspension in RNase free water. Samples were stored at −20 °C until use.
Measuring Crosstalk Between Color Channels.
RCA amplicons from 1 nM miR-375 were labeled as described above with the following modifications. All binary combinations of 5 dye-DNA probes were used to dilute the amplicon solution (2.5 μL) 50-fold. Automatic compensation was then performed using samples containing individual dye-DNA probes, followed by manual optimization to minimize signal overlap. Side scattering and SYBR Green gates were then applied to isolate amplicon signals, and independent gates were generated for each of the 2-fluorophore combinations. Counts corresponding to the number of amplicons present in each of the gates were then recorded for each of the 2-fluorophore combinations, and data were normalized based on the total number of counts in each gate. Any events within 2-fluorophore gates other than the channels corresponding to the used dyes were counted as false positives.
Measuring Crosstalk Between Ratiometic Labels.
RCA amplicons from 1 nM miR-375 were labeled as described above with the following modifications. Mixtures of Cy3-DNA and Cy5-DNA probes at a fixed concentration but varying Cy3:Cy5 ratios were used to dilute RCA reactions 50-fold by volume. After color channel compensation, side scattering and SYBR Green gates were applied to isolate amplicon signals from background fluorescence. Finally, independent gates were generated for each of the samples containing different ratios of Cy3-DNA and Cy5-DNA probes, and event counts were recorded. Any events within gates other than the gates corresponding to the applied dye ratios were counted as false positives.
Estimating Optical Barcode Numbers.
A Matlab code was written to generate all possible barcodes for a given combination of fluorescent label numbers and hybridization probe binding sites on the circular DNA template. Codes were removed if they yielded equivalent multicolor intensity ratios due to the broad linear range of intensities observed for the miR amplicon bands. Crosstalk between different ratiometric intensities was estimated based on data in Figure 4c and Figure 6b for Cy3 and Cy5 labels, and crosstalk between color channels was assumed to be zero, consistent with the vast majority of data in Figure 4b and Figure 6c. Each plot was fit to a two-dimensional elliptical Gaussian function to calculate overlap integrals between pairs of ratiometric barcodes with equal enclosed volumes. Gaussian function symmetry was assumed to extend in all color dimensions. The overlap integrals for all pairwise ratios were then summed to calculate the total false positive rate for each code, and codes with the highest false positive count were then individually eliminated. This process was repeated iteratively until the maximum number of codes was generated for which all of the false positive rates were a maximum of either 10−3 or 10−6.
Data Availability.
All data sets generated or analyzed during this study are included in this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (R00CA153914 to A.M.S.; R01CA227699 to A.M.S., B.T.C., M.K., and L.W.; R01CA212097 to M.K. and L.W.), the Department of Defense (W81XWH-15-1-0634), and the Mayo-Illinois Alliance.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b09498.
Agarose gel electrophoresis of RCA products (Figure S1), single molecule fluorescence imaging (Figure S2), fluorescence correlation spectroscopy (Figure S3), flow cytometry gating examples (Figure S4), flow cytometry side and forward scattering data (Figure S5), reaction time dependence of ensemble fluorescence and flow cytometry count (Figure S6), representative miR quantification data (Figure S7), miR quantification in RNA extracts (Figure S8), DNA detection in a flow cytometer (Figure S9), real-time RCA fluorescence analysis (Figure S10), acquisition time dependence of flow cytometry count (Figure S11), isomiR specificity by qRT-PCR and RCA (Figure S12), flow cytometer-based detection of miRs using padlock probes (Figure S13), example of flow cytometry histogram fitting (Figure S14), oligonucleotide sequences (Table S1), multiplexing fluorophore pairs (Table S2), multiplexing fluorophore ratios (Table S3), and flow cytometer lasers and optical filters (Table S4) (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.9b09498
The authors declare no competing financial interest.
Contributor Information
Lucas D. Smith, Department of Bioengineering and Holonyak Micro and Nanotechnology Laboratory, University of Illinois at Urbana−Champaign, Urbana, Illinois 61801, United States;.
Yang Liu, Department of Bioengineering and Holonyak Micro and Nanotechnology Laboratory, University of Illinois at Urbana−Champaign, Urbana, Illinois 61801, United States;.
Mohammad U. Zahid, Department of Bioengineering and Holonyak Micro and Nanotechnology Laboratory, University of Illinois at Urbana−Champaign, Urbana, Illinois 61801, United States
Taylor D. Canady, Holonyak Micro and Nanotechnology Laboratory and Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana−Champaign, Urbana, Illinois 61801, United States
Liang Wang, Department of Tumor Biology, H. Lee Moffitt Cancer Center, Tampa, Florida 33612, United States.
Manish Kohli, Department of Genitourinary Oncology, H. Lee Moffitt Cancer Center, Tampa, Florida 33612, United States.
Brian T. Cunningham, Holonyak Micro and Nanotechnology Laboratory, Carl R. Woese Institute for Genomic Biology, Department of Electrical and Computer Engineering, and Cancer Center at Illinois, University of Illinois at Urbana−Champaign, Urbana, Illinois 61801, United States
Andrew M. Smith, Department of Bioengineering, Holonyak Micro and Nanotechnology Laboratory, Carl R. Woese Institute for Genomic Biology, Cancer Center at Illinois, and Department of Materials Science and Engineering, University of Illinois at Urbana−Champaign, Urbana, Illinois 61801, United States; Carle Illinois College of Medicine, Urbana, Illinois 61801, United States;.
REFERENCES
- (1).Eid J; Fehr A; Gray J; Luong K; Lyle J; Otto G; Peluso P; Rank D; Baybayan P; Bettman B; Bibillo A; Bjornson K; Chaudhuri B; Christians F; Cicero R; Clark S; Dalal R; Dewinter A; Dixon J; Foquet M; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [DOI] [PubMed] [Google Scholar]
- (2).Xie XS; Trautman JK Optical Studies of Single Molecules at Room Temperature. Annu. Rev. Phys. Chem 1998, 49, 441–480. [DOI] [PubMed] [Google Scholar]
- (3).Geiss GK; Bumgarner RE; Birditt B; Dahl T; Dowidar N; Dunaway DL; Fell HP; Ferree S; George RD; Grogan T; James JJ; Maysuria M; Mitton JD; Oliveri P; Osborn JL; Peng T; Ratcliffe AL; Webster PJ; Davidson EH; Hood L; et al. Direct Multiplexed Measurement of Gene Expression with Color-Coded Probe Pairs. Nat. Biotechnol 2008, 26, 317–325. [DOI] [PubMed] [Google Scholar]
- (4).Clarke J; Wu H-C; Jayasinghe L; Patel A; Reid S; Bayley H Continuous Base Identification for Single-Molecule Nanopore DNA Sequencing. Nat. Nanotechnol 2009, 4, 265–270. [DOI] [PubMed] [Google Scholar]
- (5).Jain A; Liu R; Ramani B; Arauz E; Ishitsuka Y; Ragunathan K; Park J; Chen J; Xiang YK; Ha T Probing Cellular Protein Complexes Using Single-Molecule Pull-Down. Nature 2011, 473, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Smith SB; Finzi L; Bustamante C Direct Mechanical Measurements of the Elasticity of Single DNA Molecules by Using Magnetic Beads. Science 1992, 258, 1122–1126. [DOI] [PubMed] [Google Scholar]
- (7).Finer JT; Simmons RM; Spudich JA Single Myosin Molecule Mechanics - Piconewton Forces and Nanometer Steps. Nature 1994, 368, 113–119. [DOI] [PubMed] [Google Scholar]
- (8).Yang H; Luo GB; Karnchanaphanurach P; Louie TM; Rech I; Cova S; Xun LY; Xie XS Protein Conformational Dynamics Probed by Single-Molecule Electron Transfer. Science 2003, 302, 262–266. [DOI] [PubMed] [Google Scholar]
- (9).Cecconi C; Shank EA; Bustamante C; Marqusee S Direct Observation of the Three-State Folding of a Single Protein Molecule. Science 2005, 309, 2057–2060. [DOI] [PubMed] [Google Scholar]
- (10).Neuman KC; Nagy A Single-Molecule Force Spectroscopy: Optical Tweezers, Magnetic Tweezers and Atomic Force Microscopy. Nat. Methods 2008, 5, 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Roy R; Hohng S; Ha T A Practical Guide to Single-Molecule FRET. Nat. Methods 2008, 5, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Funatsu T; Harada Y; Tokunaga M; Saito K; Yanagida T Imaging of Single Fluorescent Molecules and Individual ATP Turnovers by Single Myosin Molecules in Aqueous Solution. Nature 1995, 374, 555–559. [DOI] [PubMed] [Google Scholar]
- (13).Lu HP; Xun LY; Xie XS Single-Molecule Enzymatic Dynamics. Science 1998, 282, 1877–1882. [DOI] [PubMed] [Google Scholar]
- (14).Xie XS; Lu HP Single-Molecule Enzymology. J. Biol. Chem 1999, 274, 15967–15970. [DOI] [PubMed] [Google Scholar]
- (15).Min W; English BP; Luo GB; Cherayil BJ; Kou SC; Xie XS Fluctuating Enzymes: Lessons from Single-Molecule Studies. Acc. Chem. Res 2005, 38, 923–931. [DOI] [PubMed] [Google Scholar]
- (16).English BP; Min W; van Oijen AM; Lee KT; Luo GB; Sun HY; Cherayil BJ; Kou SC; Xie XS Ever-Fluctuating Single Enzyme Molecules: Michaelis-Menten Equation Revisited. Nat. Chem. Biol 2006, 2, 87–94. [DOI] [PubMed] [Google Scholar]
- (17).Sako Y; Minoguchi S; Yanagida T Single-Molecule Imaging of EGFR Signalling on the Surface of Living Cells. Nat. Cell Biol 2000, 2, 168–172. [DOI] [PubMed] [Google Scholar]
- (18).Douglass AD; Vale RD Single-Molecule Microscopy Reveals Plasma Membrane Microdomains Created by Protein-Protein Networks That Exclude or Trap Signaling Molecules in T Cells. Cell 2005, 121, 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cai L; Friedman N; Xie XS Stochastic Protein Expression in Individual Cells at the Single Molecule Level. Nature 2006, 440, 358–362. [DOI] [PubMed] [Google Scholar]
- (20).Elf J; Li GW; Xie XS Probing Transcription Factor Dynamics at the Single-Molecule Level in a Living Cell. Science 2007, 316, 1191–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li G-W; Xie XS Central Dogma at the Single-Molecule Level in Living Cells. Nature 2011, 475, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Coulon A; Chow CC; Singer RH; Larson DR Eukaryotic Transcriptional Dynamics: From Single Molecules to Cell Populations. Nat. Rev. Genet 2013, 14, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Srinivasan R 1997 Nobel Prize for Physics: Laser Cooling and Trapping of Atoms. Curr. Sci 1998, 74, 106–110. [Google Scholar]
- (24).Moeckl L; Lamb DC; Braeuchle C Super-Resolved Fluorescence Microscopy: Nobel Prize in Chemistry 2014 for Eric Betzig, Stefan Hell, and William E. Moerner. Angew. Chem., Int. Ed 2014, 53, 13972–13977. [DOI] [PubMed] [Google Scholar]
- (25).Shen PS The 2017 Nobel Prize in Chemistry: Cryo-EM Comes of Age. Anal. Bioanal. Chem 2018, 410, 2053–2057. [DOI] [PubMed] [Google Scholar]
- (26).Killian JL; Ye F; Wang MD Optical Tweezers: A Force to Be Reckoned With. Cell 2018, 175, 1445–1448. [DOI] [PubMed] [Google Scholar]
- (27).Colquhoun D Neher and Sakmann Win Nobel Prize for Patch-Clamp Work. Trends Pharmacol. Sci 1991, 12, 449. [DOI] [PubMed] [Google Scholar]
- (28).Walter NG; Huang CY; Manzo AJ; Sobhy MA Do-It-Yourself Guide: How to Use the Modern Single-Molecule Toolkit. Nat. Methods 2008, 5, 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Efcavitch JW; Thompson JF Single-Molecule DNA Analysis. Annu. Rev. Anal. Chem 2010, 3, 109–128. [DOI] [PubMed] [Google Scholar]
- (30).Cordes T; Blum SA Opportunities and Challenges in Single-Molecule and Single-Particle Fluorescence Microscopy for Mechanistic Studies of Chemical Reactions. Nat. Chem 2013, 5, 993–999. [DOI] [PubMed] [Google Scholar]
- (31).Ma F; Li Y; Tang B; Zhang C -y. Fluorescent Biosensors Based on Single-Molecule Counting. Acc. Chem. Res 2016, 49, 1722–1730. [DOI] [PubMed] [Google Scholar]
- (32).Walt DR Optical Methods for Single Molecule Detection and Analysis. Anal. Chem 2013, 85, 1258–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gooding JJ; Gaus K Single-Molecule Sensors: Challenges and Opportunities for Quantitative Analysis. Angew. Chem., Int. Ed 2016, 55, 11354–11366. [DOI] [PubMed] [Google Scholar]
- (34).Shivanandan A; Deschout H; Scarselli M; Radenovic A Challenges in Quantitative Single Molecule Localization Microscopy. FEBS Lett. 2014, 588, 3595–3602. [DOI] [PubMed] [Google Scholar]
- (35).Mir KU Ultrasensitive RNA Profiling: Counting Single Molecules on Microarrays. Genome Res. 2006, 16, 1195–1197. [DOI] [PubMed] [Google Scholar]
- (36).Holzmeister P; Acuna GP; Grohmann D; Tinnefeld P Breaking the Concentration Limit of Optical Single-Molecule Detection. Chem. Soc. Rev 2014, 43, 1014–1028. [DOI] [PubMed] [Google Scholar]
- (37).Chenouard N; Smal I; de Chaumont F; Maska M; Sbalzarini IF; Gong YH; Cardinale J; Carthel C; Coraluppi S; Winter M; Cohen AR; Godinez WJ; Rohr K; Kalaidzidis Y; Liang L; Duncan J; Shen HY; Xu YK; Magnusson KEG; Jalden J; et al. Objective Comparison of Particle Tracking Methods. Nat. Methods 2014, 11, 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shen H; Tauzin LJ; Baiyasi R; Wang WX; Moringo N; Shuang B; Landes CF Single Particle Tracking: From Theory to Biophysical Applications. Chem. Rev 2017, 117, 7331–7376. [DOI] [PubMed] [Google Scholar]
- (39).Smith L; Kohli M; Smith AM Expanding the Dynamic Range of Fluorescence Assays through Single-Molecule Counting and Intensity Calibration. J. Am. Chem. Soc 2018, 140, 13904–13912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Colabrese S; Castello M; Vicidomini G; Del Bue A Machine Learning Approach for Single Molecule Localisation Microscopy. Biomed. Opt. Express 2018, 9, 1680–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hill FR; Monachino E; van Oijen AM The More the Merrier: High-Throughput Single-Molecule Techniques. Biochem. Soc. Trans 2017, 45, 759–769. [DOI] [PubMed] [Google Scholar]
- (42).Brown M; Wittwer C Flow Cytometry: Principles and Clinical Applications in Hematology. Clin. Chem 2000, 46, 1221–1229. [PubMed] [Google Scholar]
- (43).Fan HC; Fu GK; Fodor SPA Combinatorial Labeling of Single Cells for Gene Expression Cytometry. Science 2015, 347, 1258367. [DOI] [PubMed] [Google Scholar]
- (44).Krutzik PO; Nolan GP Fluorescent Cell Barcoding in Flow Cytometry Allows High-Throughput Drug Screening and Signaling Profiling. Nat. Methods 2006, 3, 361–368. [DOI] [PubMed] [Google Scholar]
- (45).Brind’Amour J; Lansdorp PM Analysis of Repetitive DNA in Chromosomes by Flow Cytometry. Nat. Methods 2011, 8, 484–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lippe R Flow Virometry: A Powerful Tool to Functionally Characterize Viruses. J. Virol 2018, 92, No. e01765–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).van der Pol E; Coumans FA; Grootemaat AE; Gardiner C; Sargent IL; Harrison P; Sturk A; van Leeuwen TG; Nieuwland R Particle Size Distribution of Exosomes and Microvesicles Determined by Transmission Electron Microscopy, Flow Cytometry, Nanoparticle Tracking Analysis, and Resistive Pulse Sensing. J. Thromb. Haemostasis 2014, 12, 1182–1192. [DOI] [PubMed] [Google Scholar]
- (48).Mage PL; Csordas AT; Brown T; Klinger D; Eisenstein M; Mitragotri S; Hawker C; Soh HT Shape-Based Separation of Synthetic Microparticles. Nat. Mater 2019, 18, 82–89. [DOI] [PubMed] [Google Scholar]
- (49).Brooks Shera E; Seitzinger NK; Davis LM; Keller RA; Soper SA Detection of Single Fluorescent Molecules. Chem. Phys. Lett 1990, 174, 553–557. [Google Scholar]
- (50).Horrocks MH; Li H; Shim J.-u.; Ranasinghe RT; Clarke RW; Huck WTS; Abell C; Klenerman D Single Molecule Fluorescence under Conditions of Fast Flow. Anal. Chem 2012, 84, 179–185. [DOI] [PubMed] [Google Scholar]
- (51).Huang B; Wu H; Bhaya D; Grossman A; Granier S; Kobilka BK; Zare RN Counting Low-Copy Number Proteins in a Single Cell. Science 2007, 315, 81–84. [DOI] [PubMed] [Google Scholar]
- (52).Neely LA; Patel S; Garver J; Gallo M; Hackett M; McLaughlin S; Nadel M; Harris J; Gullans S; Rooke J A Single-Molecule Method for the Quantitation of microRNA Gene Expression. Nat. Methods 2006, 3, 41–46. [DOI] [PubMed] [Google Scholar]
- (53).Perfetto SP; Chattopadhyay PK; Roederer M Seventeen-Colour Flow Cytometry: Unravelling the Immune System. Nat. Rev. Immunol 2004, 4, 648–655. [DOI] [PubMed] [Google Scholar]
- (54).Saeys Y; Van Gassen S; Lambrecht BN Computational Flow Cytometry: Helping to Make Sense of High-Dimensional Immunology Data. Nat. Rev. Immunol 2016, 16, 449–462. [DOI] [PubMed] [Google Scholar]
- (55).Huang X; Yuan T; Liang M; Du M; Xia S; Dittmar R; Wang D; See W; Costello BA; Quevedo F; Tan W; Nandy D; Bevan GH; Longenbach S; Sun Z; Lu Y; Wang T; Thibodeau SN; Boardman L; Kohli M; et al. Exosomal miR-1290 and miR-375 as Prognostic Markers in Castration-Resistant Prostate Cancer. Eur. Urol 2015, 67, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wang Y; Lieberman R; Pan J; Zhang Q; Du M; Zhang P; Nevalainen M; Kohli M; Shenoy NK; Meng H; You M; Wang L miR-375 Induces Docetaxel Resistance in Prostate Cancer by Targeting SEC23A and YAP1. Mol. Cancer 2016, 15, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Blanco L; Bernad A; Lazaro JM; Martin G; Garmendia C; Salas M Highly Efficient DNA Synthesis by the Phage Phi 29 DNA Polymerase. Symmetrical Mode of DNA Replication. J. Biol. Chem 1989, 264, 8935–8940. [PubMed] [Google Scholar]
- (58).Lizardi PM; Huang XH; Zhu ZR; Bray-Ward P; Thomas DC; Ward DC Mutation Detection and Single-Molecule Counting Using Isothermal Rolling-Circle Amplification. Nat. Genet 1998, 19, 225–232. [DOI] [PubMed] [Google Scholar]
- (59).Ali MM; Li F; Zhang Z; Zhang K; Kang DK; Ankrum JA; Le XC; Zhao W Rolling Circle Amplification: A Versatile Tool for Chemical Biology, Materials Science and Medicine. Chem. Soc. Rev 2014, 43, 3324–3341. [DOI] [PubMed] [Google Scholar]
- (60).Tuma RS; Beaudet MP; Jin X; Jones LJ; Cheung CY; Yue S; Singer VL Characterization of SYBR Gold Nucleic Acid Gel Stain: A Dye Optimized for Use with 300-nm Ultraviolet Transilluminators. Anal. Biochem 1999, 268, 278–288. [DOI] [PubMed] [Google Scholar]
- (61).Vitzthum F; Geiger G; Bisswanger H; Brunner H; Bernhagen J A Quantitative Fluorescence-Based Microplate Assay for the Determination of Double-Stranded DNA Using SYBR Green I and a Standard Ultraviolet Transilluminator Gel Imaging System. Anal. Biochem 1999, 276, 59–64. [DOI] [PubMed] [Google Scholar]
- (62).Cheng Y; Zhang X; Li Z; Jiao X; Wang Y; Zhang Y Highly Sensitive Determination of microRNA Using Target-Primed and Branched Rolling-Circle Amplification. Angew. Chem., Int. Ed 2009, 48, 3268–3272. [DOI] [PubMed] [Google Scholar]
- (63).Jonstrup SP; Koch J; Kjems J A microRNA Detection System Based on Padlock Probes and Rolling Circle Amplification. RNA 2006, 12, 1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Larsson C; Grundberg I; Soderberg O; Nilsson M In Situ Detection and Genotyping of Individual mRNA Molecules. Nat. Methods 2010, 7, 395–397. [DOI] [PubMed] [Google Scholar]
- (65).Shah S; Lubeck E; Schwarzkopf M; He TF; Greenbaum A; Sohn CH; Lignell A; Choi HM; Gradinaru V; Pierce NA; Cai L Single-Molecule RNA Detection at Depth by Hybridization Chain Reaction and Tissue Hydrogel Embedding and Clearing. Development 2016, 143, 2862–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Liang Y; Ridzon D; Wong L; Chen CJBG Characterization of microRNA Expression Profiles in Normal Human Tissues. BMC Genomics 2007, 8, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chen C; Ridzon DA; Broomer AJ; Zhou Z; Lee DH; Nguyen JT; Barbisin M; Xu NL; Mahuvakar VR; Andersen MR; Lao KQ; Livak KJ; Guegler KJ Real-Time Quantification of microRNAs by Stem-Loop RT-PCR. Nucleic Acids Res. 2005, 33, No. e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Udvardi MK; Czechowski T; Scheible W-R Eleven Golden Rules of Quantitative RT-PCR. Plant Cell 2008, 20, 1736–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Chugh P; Dittmer DP Potential Pitfalls in microRNA Profiling. WIRES RNA 2012, 3, 601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Yuan T; Huang X; Woodcock M; Du M; Dittmar R; Wang Y; Tsai S; Kohli M; Boardman L; Patel T; Wang L Plasma Extracellular RNA Profiles in Healthy and Cancer Patients. Sci. Rep 2016, 6, 19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Li R; Wang Y; Wang P; Lu J A Dual Discrimination Mode for Improved Specificity Towards let-7a Detection Via a Single-Base Mutated Padlock Probe-Based Exponential Rolling Circle Amplification. Luminescence 2017, 32, 1574–1581. [DOI] [PubMed] [Google Scholar]
- (72).Liu H; Li L; Duan L; Wang X; Xie Y; Tong L; Wang Q; Tang B High Specific and Ultrasensitive Isothermal Detection of microRNA by Padlock Probe-Based Exponential Rolling Circle Amplification. Anal. Chem 2013, 85, 7941–7947. [DOI] [PubMed] [Google Scholar]
- (73).Jin J; Vaud S; Zhelkovsky AM; Posfai J; McReynolds LA Sensitive and Specific miRNA Detection Method Using SplintR Ligase. Nucleic Acids Res. 2016, 44, No. e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Ullal AV; Peterson V; Agasti SS; Tuang S; Juric D; Castro CM; Weissleder R Cancer Cell Profiling by Barcoding Allows Multiplexed Protein Analysis in Fine-Needle Aspirates. Sci. Transl. Med 2014, 6, 219ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Frei AP; Bava FA; Zunder ER; Hsieh EW; Chen SY; Nolan GP; Gherardini PF Highly Multiplexed Simultaneous Detection of RNAs and Proteins in Single Cells. Nat. Methods 2016, 13, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Maetzig T; Ruschmann J; Lai CK; Ngom M; Imren S; Rosten P; Norddahl GL; von Krosigk N; Sanchez Milde L; May C; Selich A; Rothe M; Dhillon I; Schambach A; Humphries RK A Lentiviral Fluorescent Genetic Barcoding System for Flow Cytometry-Based Multiplex Tracking. Mol. Ther 2017, 25, 606–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Akkaya B; Miozzo P; Holstein AH; Shevach EM; Pierce SK; Akkaya M A Simple, Versatile Antibody-Based Barcoding Method for Flow Cytometry. J. Immunol 2016, 197, 2027–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Krutzik PO; Clutter MR; Trejo A; Nolan GP Fluorescent Cell Barcoding for Multiplex Flow Cytometry. Curr. Protoc. Cytom 2011, 55, 6.31.1–6.31.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Choi K; Ng AHC; Fobel R; Wheeler AR Digital Microfluidics. Annu. Rev. Anal. Chem 2012, 5, 413–440. [DOI] [PubMed] [Google Scholar]
- (80).Chou CH; Shrestha S; Yang CD; Chang NW; Lin YL; Liao KW; Huang WC; Sun TH; Tu SJ; Lee WH; Chiew MY; Tai CS; Wei TY; Tsai TR; Huang HT; Wang CY; Wu HY; Ho SY; Chen PR; Chuang CH; et al. Mirtarbase Update 2018: A Resource for Experimentally Validated microRNA-Target Interactions. Nucleic Acids Res. 2018, 46, D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Gu L; Yan W; Liu L; Wang S; Zhang X; Lyu M Research Progress on Rolling Circle Amplification (RCA)-Based Biomedical Sensing. Pharmaceuticals 2018, 11, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Deng R; Zhang K; Sun Y; Ren X; Li J Highly Specific Imaging of mRNA in Single Cells by Target RNA-Initiated Rolling Circle Amplification. Chem. Sci 2017, 8, 3668–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Christian AT; Pattee MS; Attix CM; Reed BE; Sorensen KJ; Tucker JD Detection of DNA Point Mutations and mRNA Expression Levels by Rolling Circle Amplification in Individual Cells. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 14238–14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data sets generated or analyzed during this study are included in this article.