

Abstract
Endogenous biomarkers remain at the forefront of early disease detection efforts, but many lack the sensitivities and specificities necessary to influence disease management. Inspired by emerging adoptive cell transfer immunotherapies and the natural migration of immune cells to pathology, here we describe a new class of cell-based in vivo sensors for ultrasensitive disease detection. In our proof of concept, we perform adoptive transfer of syngeneic macrophages which were engineered to produce a synthetic biomarker upon adopting a ‘tumor-associated’ metabolic profile. Notably, the macrophage sensor detected tumors as small as 25-50 mm3, effectively tracked the immunological response in two models of acute inflammation, and was more sensitive than both protein and nucleic acid cancer biomarkers. This technology establishes a clinically translatable approach to early cancer detection and provides a conceptual framework for the use of engineered immune cells for the monitoring of many disease states in addition to cancer.
Keywords: Cancer diagnostics, Synthetic biology, Adoptive cell transfer, Macrophages
Introduction
Early detection of primary disease and recurrence are promising avenues towards reducing cancer mortality1. To date, efforts that rely on detection of endogenous biomarkers such as circulating tumor cells2, circulating tumor DNA3, and cancer exosomes4 are still unable to reliably detect early stage disease, at least in part due to rapid biomarker clearance, high background signal, and low concentrations in circulation5.
An alternative diagnostic strategy is the systemic delivery of probes which are selectively activated and generate signal in the presence of disease6–9. By no longer relying on natural biomarker shedding, these approaches can likely achieve sensitivities and signal-to-noise ratios greater than what is possible with endogenous biomarkers10. While promising, these strategies are limited by either (i) biocompatibility of the probe, (ii) efficient delivery of the probe to sites of pathology, and (iii) the ability of the probe to assay multiple features of a disease environment.
To overcome these challenges, here we leverage metabolic alterations occurring in tumor-infiltrating immune cells11 and describe a new class of cellular sensors for highly sensitive cancer detection. Specifically, we exploit the M1/M2 metabolic dichotomy of macrophages12 as the basis of a cell sensor. While most pathologies polarize macrophages to a pro-inflammatory M1 phenotype, a smaller subset, including solid tumors, reprogram macrophages into the anti-inflammatory M2 state12. We thus aimed to use M2 polarization as a diagnostic surrogate for detection of small tumors.
Leveraging advances in adoptive cell transfer, here we engineer macrophages to express a dual secreted and intracellular protein biomarker upon taking on a “tumor associated” metabolic profile, inject the cells intravenously to allow homing to sites of disease13, and subsequently image the host and monitor the blood for presence of the biomarker (Figure 1). In doing so, we demonstrate the first example of a biocompatible, personalized, and generalizable immune cell sensor for early disease detection.
Figure 1. Schematic of diagnostic adoptive cell transfer.
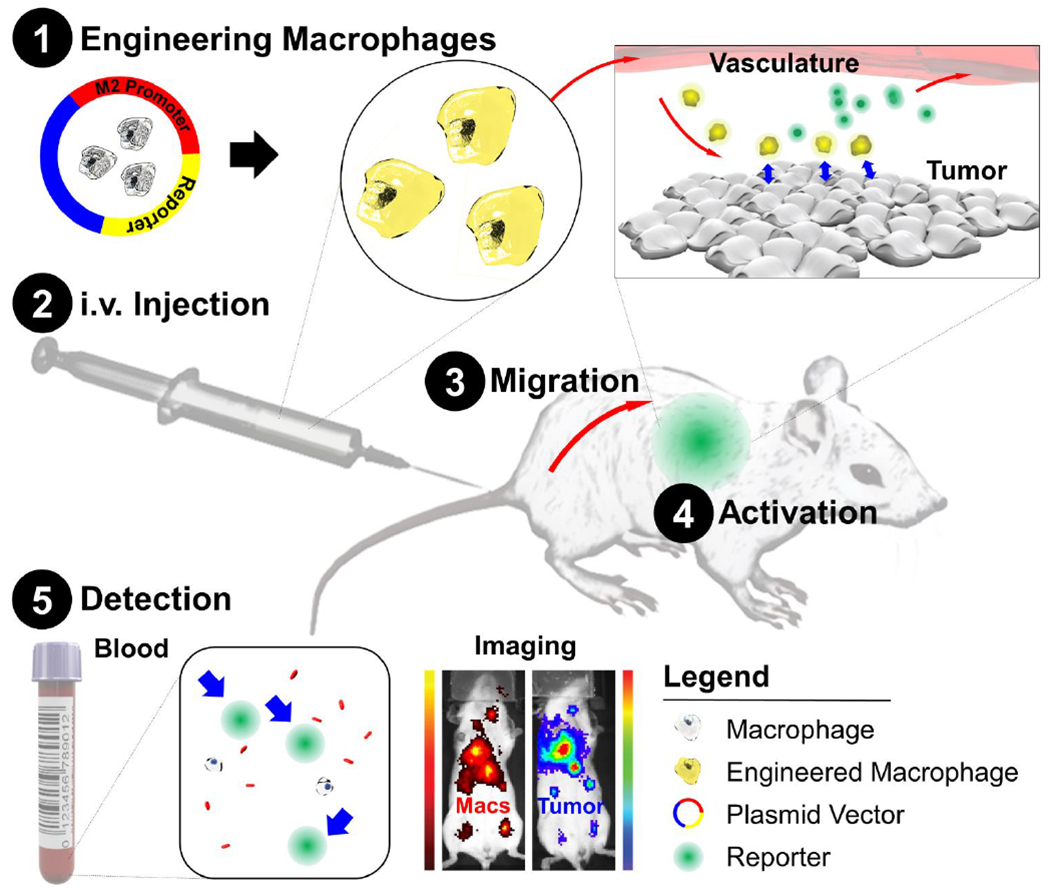
Macrophages are genetically engineered to secrete a synthetic biomarker upon adopting a “tumor associated” metabolic profile. The engineered macrophages are injected intravenously in syngeneic hosts and allowed to home to existing sites of pathology. A blood test can then be used to monitor for secretion of the biomarker which would indicate the presence of disease. This system can also provide spatial information of immune cell activation with use of an imageable synthetic biomarker. Macs, Macrophages.
Methods
Tumor infiltrating leukocyte profiting
The fractional immune cell makeup from transcriptomic profiling of various cancers was obtained from the immune prediction of clinical outcomes from genomic profiles (iPRECOG) database14 (https://precog.stanford.edu/iPRECOG.php). As multiple cohorts of tumor samples are listed under each cancer type, we calculated a weighted average of the immune cell fractions for each cancer based on the number of tumor samples analyzed in each cohort.
Bone marrow-derived macrophage/monocyte (BMDM) preparation and electroporation
Femurs and tibias from 6-8 week old female BALB/c mice were isolated and bone marrow flushed with 5 mL cold PBS. Marrow was re-suspended into a homogenous solution by repeated pipetting and passed through a 40 μm filter to eliminate debris. After centrifugation for 5 min at 300 × g, marrow was re-suspended in ACK lysis buffer (Invitrogen, Waltham, MA) for 5 min on ice. ACK was diluted 10-fold in PBS, and the solution centrifuged again for 5 min at 300 × g. To make BMD macrophages for in vitro activation assays, cells were plated at a density of 4 × 106 cells/10 cm petri dish in 10 mL of IMDM (ThermoFisher, Waltham, MA) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% antibacterial/antimycotic (A/A) solution (ThermoFisher), and 10 ng/mL murine macrophage colony stimulating factor (M-CSF, Peprotech, Rocky Hill, NJ). Cells were maintained in a humidified, 5% CO2 incubator at 37 °C for 5 days prior to harvesting with a cell lifter for downstream use. To make BMD monocytes15 for all in vivo studies, cells were instead plated at a density of 6 × 106 cells/well in 6-well Corning® Costar® Ultra-Low Attachment Plates (Corning, Corning, NY) in 6 mL of media supplemented with 20 ng/mL M-CSF. After 5 days, adherent cells (macrophages) were discarded and the non-adhered cell were collected. Purity was determined to >96% by flow cytometry via staining for F4/80 (Biolegend, San Diego, CA) compared to an isotype control (Supplementary Figure S1a).
Transient transfection was performed by electroporation using a Nucleofector kit for mouse macrophages (Lonza, Basel, Switzerland) and protocol Y-001 on the associated Nucleofector 2b Device. Each reaction contained 1 × 107 BMDMs and 100 μg plasmid.
Cell Lines
RAW264.7 murine macrophage, CT26 murine colon carcinoma, 4T1 murine breast cancer, and LS174T human colorectal adenocarcinoma were obtained from ATCC (Manassas, VA) and cultured in either DMEM (RAW264.7, LS174T) or RPMI (CT26, 4T1) supplemented with 10% FBS and 1% antibacterial/antimycotic solution (ThermoFisher). Cells were maintained in a humidified, 5% CO2 incubator at 37 °C. CT26 eGFP-firefly luciferase (Fluc) and 4T1 eGFP-Fluc cell lines were generated by lentiviral transduction followed by three rounds of sorting for the highest 2.5% of eGFP expressers. The RAW264.7 arginase-1 promoter driving Gaussia luciferase (pARG1-Gluc) cell line was generated by transfection with Lipofectamine 3000 (ThermoFisher) and three rounds of sorting for the highest 2.5% of eGFP expressers.
in vitro macrophage activation
Macrophages (RAW264.7 or BMDM) were plated at a density of 1 × 106 cells/well in 6-well plates in 2.5 mL of medium. After 24 hours, media was either replaced with tumor conditioned media (TCM), or was supplemented with IL-4, IL-13, lactic acid, tumor necrosis factor alpha (TNFα), interferon gamma (IFNγ), or lipopolysaccharide (LPS). Unless otherwise noted, activation was performed with 25 ng/mL IL-4 or IL-13, 100 ng/mL TNFα or IFNγ, or 200 ng/mL LPS from Escherichia coli serotype O55:B5 (Sigma-Aldrich, St. Louis, MO). “High” and “low” TCM were generated by culturing 2 × 106 or 3 × 106 CT26 cells respectively in 2.5 mL media per well in a 6-well plate for 24 hours. For Gluc activation experiments with BMDMs, “high” and “low” TCM were generated with 2.5 × 105 or 1 × 105 CT26 cells respectively due to BMDM toxicity at the higher cell numbers. Conditioned media was centrifuged for 10 min at 300 × g to eliminate debris prior to use. After 3, 6, 12, or 24 hours, macrophages were either harvested for RNA extraction or 20 μL of culture media was collected to assay for Glue using a BioLux Gaussia Luciferase Assay Kit (New England BioLabs, Ipswich, MA) according to manufacturer’s instructions. Luminescence measurements were performed on a TD 20/20 luminometer (Turner Designs, San Jose, CA) with 10 seconds of integration and luminescence expressed in relative light units (RLU).
Arginase gene expression assays
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) following manufacturer’s instructions. Extraction of RNA from macrophages in cell culture was performed by direct lysis within the well, while extraction from tumor- and spleen-infiltrating macrophages was performed by direct sorting into RNeasy lysis buffer during flow cytometry. cDNA synthesis was performed using the iScript cDNA synthesis kit (Bio-rad, Hercules, CA) following manufacturer’s instructions. Quantitative PCR (qPCR) reactions were performed in 20 μL volumes containing 1× SsoAdvanced Universal Probes Supermix (Bio-Rad), 1 μL of gene-specific hydrolysis probe, 2 μL of cDNA, and nuclease-free water (Bio-rad). FAM fluorophore-conjugated hydrolysis probes for Ym1, FIZZ1, HIF1α, MRC1, ARG1, and GAPDH were commercially obtained (Bio-Rad). Thermal cycling for both cDNA synthesis and qPCR were performed using a CFX96 Real-Time System C1000 Touch Thermal Cycler (Bio-Rad) using the following protocols: 25°C for 5 min, 46°C for 20 min, 95°C for 1 min (cDNA synthesis) and 95°C for 3 min, followed by 60 cycles of: 95°C for 15 seconds and 59°C for 30 seconds (qPCR). Technical replicates for all samples were performed in duplicate. Negative controls were performed with nuclease free water instead of cDNA. The cycle threshold was a single threshold determined automatically (using the CFX Manager Software Version 3.1) with all Cq values falling within the linear quantifiable range of the assay.
Arginase activity assay
Macrophages were washed once with PBS, harvested, and lysed in 100 μL Pierce IP Lysis Buffer (ThermFisher) containing 1× Halt Protease Inhibitor Cocktail (ThermoFisher) for 10 min on ice. Lysate was centrifuged at 4°C for 10 min at 14,000 × g and supernatant arginase activity was measured using the colorimetric QuantiChrom Arginase Assay Kit (BioAssay Systems, Hayward, CA) following manufacturer instructions. Optical density at 430 nm was measured on a Synergy 4 microplate reader (BioTek, Winooski, VT).
In vitro migration assay
In vitro migration assays were performed using 6.5 mm Transwell tissue culture-treated inserts with 8.0 μM pore size polyester membranes (Corning, Corning, NY). 1 × 105 RAW264.7 macrophages were seeded in 100 μL of DMEM on the top of the membrane chamber and allowed to adhere for 10 min prior to submerging of the chamber into wells containing either “high” tumor conditioned or unconditioned media. After 24 hours, the insert was removed, non-migrated cells on the top of the membrane were removed with a cotton swab, and the insert was fixed in 600 μL of 70% ethanol for 10 min. Membrane was allowed to dry for 15 min and then submerged into 600 μL of 0.2% crystal violet (CV) solution for 10 min for cell staining. Finally, the membrane was washed with PBS, removed from the insert, and number of migrated cells were counted in brightfield in 10 random 10× fields of view on an EVOS imaging system (ThermoFisher).
In vivo migration and blocking assay
All animal experiments were performed in accordance with a protocol approved by the Stanford University Administrative Panels on Laboratory Animal Care (APLAC). 6-8 week old female BALB/c mice (Charles River, Wilmington, MA) were subcutaneously implanted with 1 × 106CT26 eGFP-Fluc cells in 100 μL PBS on the right shoulder. After seven days, tumors were imaged by bioluminescence imaging (BLI) on an IVIS Spectrum (PerkinElmer, Waltham, MA) device after intraperitoneal injection of 30 mg/kg D-Luciferin in 100 μL PBS to confirm tumor intake. Ten days after tumor implantation, 1 × 107 syngeneic RAW264.7 macrophages were labeled with a near infrared fluorescent membrane dye (VivoTrack 680, PerkinElmer) and injected intravenously in 100 μL PBS. In vivo fluorescence imaging with the 640 nm excitation and 700 nm emission filter set was performed using the IVIS Spectrum on days one, three, and five after macrophage injection to visualize migration to the tumor. In cytokine blocking studies, 150 pg of neutralizing antibody (BioXCell, West Lebanon, NH) against CCL2 or CSF1 or their isotype control (Rat IgG and Armenian Hamster IgG respectively) were administered intraperitoneally daily starting two days prior to RAW264.7 macrophage injection and continuing until 4 days after macrophage injection. Regions of interest (ROIs) and line traces were drawn using Living Image 4.5.2 software to quantitate extent of macrophage migration and co-localization with tumor respectively based on average radiance in photons-s−1.cm−2steradian−1.
Immunofluorescence
6-8 week old female BALB/c mice bearing 10-day old CT26 tumors were injected intravenously with 1 × 107 BMDMs labeled with CellBrite™ Fix 640 dye (Biotium, Fremont, CA) according to manufacturer’s instructions. After four days, tumors were harvested 90 minutes following intraperitoneal injection of 60 mg/kg pimonidazole hydrochloride (Hypoxyprobe, Burlington, MA) for detection of hypoxia. Tumors were frozen in optimal cutting temperature compound (Sakura Finetek, Torrance, CA) immediately following excision, cut into 10 μm-thick sections using a microtome, and mounted onto frosted microscope slides. Tissue slides were then blocked for 30 minutes with immunofluorescence blocking buffer (Cell Signaling Technology, Danvers, MA) prior to staining with 1:50 (1.2 μg/mL) FITC anti-pimonidazole (Hypoxyprobe) overnight at 4°C. Slides were washed three times with PBS and coverslips mounted using ProLong® Gold Antifade Reagent with DAP I (ThermoFisher) prior to sealing with clear nail polish. Images were acquired using a NanoZoomer 2.0-RS whole slide imager (Hamamatsu, Hamamatsu City, Japan).
Macrophage staining and cell sorting
Resected tumors were mechanically dissociated with scissors and digested in 5 mL Flank’s Balanced Salt Solution (FIBSS) containing 10 μg/mL DNase I (Sigma-Aldrich) and 25 μg/mL Liberase (Roche, Basel, Switzerland) for 45 min at 37 °C. The solution was then diluted with cold PBS, filtered through a 70 μm filter, and centrifuged for 5 min at 300 × g prior to resuspension in FACS buffer (PBS + 2% FBS + 1% A/A). Harvested spleens were pressed through 40 μm filters using the back of a 1 mL syringe plunger and washed through with PBS. After spinning for 5 min at 300 × g, splenocytes were re-suspended in 5 mL of ACK lysis buffer and put on ice for 5 min. The red blood cell free fraction was then centrifuged and re-suspended in FACS buffer. Harvested lungs and livers were perfused with the described digestion buffer, mechanically dissociated, and stirred for 30 min at 37 °C. Digested tissues were then filtered through 70 μm filters. Cell suspensions were pelleted, resuspended in 33% Percoll (GE Healthcare, Chicago, IL), and spun at 2,200 × rpm for 30 min. Isolated cells were washed twice and resuspended in FACS buffer.
For macrophage sorting experiments, single cell suspensions were stained in 100 μL HBSS containing 0.2 μg of each antibody against F4/80 and CD11 b (Biolegend) as well as a live/dead stain (propidium iodide) and sorted on a FACSAria II benchtop cell sorter (Becton Dickinson, Franklin Lakes, NJ). For receptor staining experiments, single cell suspensions were stained in 100 μL HBSS containing 0.2 μg of each antibody against F4/80, CD11b, CSF1R, CCR2, and CCR5 (Biolegend) as well as a live/dead stain (propidium iodide) and analyzed on a LSRII analyzer (Becton Dickinson). In receptor staining experiments, fluorescence from RAW264.7 macrophages stably expressing eGFP was used to gate adoptively transferred macrophages. Compensation was performed using AbC Total Antibody Compensation Beads (ThermoFisher) and single-stained cells for VivoTrack 680. In all experiments positive and negative cells were gated using fluorescence minus one controls.
Reporter plasmid construction
Plasmids were constructed using standard PCR-based cloning techniques and sequenced by Sequetech (Mountain View, CA). The pARG1-Gluc reporter plasmid (Supplementary Figure S2) was formed by cloning the −31/−3810 ARG1 promoter/enhancer (Addgene, Cambridge, MA) sequence described previously16 upstream of the sequence for Gaussia Dura Luciferase (Genecopoeia, Rockville, MD).
Mouse tumor models and blood collection
A syngeneic model of metastatic breast cancer was established by intravenous injection of 2.5 × 105 4T1 eGFP-Fluc cells in 150 μL PBS into 6-8 week old female BALB/c mice. In the localized model of disease, macrophage sensor was injected after seven days when disease was still likely restricted to the lungs as visualized by BLI. A dose of 3 × 106 cells was used for both RAW264.7 and BMDM sensors with the injected BMDM dose accounting for electroporation efficiency and viability measured by flow cytometry. In the metastatic model, sensor was injected after 14 days once tumor burden had spread beyond the lung. Mice were bled from the submandibular vein 24 hours after RAW264.7 sensor injection or 72 hours after BMDM sensor injection (due to transient non-specific activation of the sensor upon electroporation), blood was collected in K3-EDTA tubes (Greiner, Baden-Württemberg, Germany), and then centrifuged at 4 °C for 10 min at 1,000 × g. Gluc was assayed from 20 μL plasma as described previously. Activated macrophages were imaged by BLI 48 hours after RAW264.7 injection and 72 hours after BMDM injection by intravenous administration of 35 μg coelenterazine (CTZ) substrate (Promega, Madison, WI) diluted in 150 μL of PBS.
A syngeneic subcutaneous colorectal cancer model was established as described in the migration studies with tumor volumes ranging from 0-250 mm3 prior to engineered macrophage injection. Tumor volumes were approximated by the equation V = 0.5 × L × W2 with L and W representing the longer and shorter immediately perpendicular diameters of the tumor spheroid respectively. Dimensions were measured with a digital caliper. Mice were bled (50 μL) from the submandibular vein at 24-hour intervals for up to four days following RAW264.7 sensor injection or 72 hours after BMDM sensor injection, and BLI of activated macrophages was performed 48 hours after injection of RAW264.7 sensor. When assessing the effect of sensor on tumor progression, mice with subcutaneous colorectal tumors were injected intravenously with either 3 × 106 BMDM sensor in 100 μL PBS or vehicle once tumor volumes exceeded 15 mm3 Tumors were measured by caliper every third day after macrophage injection. Mice were sacrificed if any tumor dimension exceeded 15 mm.
Murine inflammation models
In the model of muscle inflammation, 6-8 week old female BALB/c mice were injected intramuscularly with 30 μL turpentine oil (Sigma-Aldrich) in the right hind limb. Healthy contralateral muscle injected with PBS and inflamed muscle from mice sacrificed on days one, three, seven, and ten after injection were collected and fixed in 10% formalin for 48 hours, embedded in paraffin, and processed for Hematoxylin & Eosin (H&E) staining following standard protocols (Histo-Tec Laboratory, Hayward, CA). RAW264.7 macrophage sensor (3 × 106 cells) was injected intravenously in 100 μL PBS either the same day as or seven days after turpentine oil injection. Blood collection, plasma Glue measurement, and BLI of activated macrophages occurred 24 hours after cell injection.
In the LPS-induced model of acute lung inflammation, 6-8 week old female BALB/c mice were inoculated intranasally with 50 μg of LPS resuspended in 20 μL PBS. Control mice received no vehicle as intranasal administration of saline can induce lung inflammation. Healthy lungs or lungs at 7, 24, 48, 72 hours after LPS administration were fixed and processed for H&E staining as previously described. In the monitoring of wound healing, BMDM sensor (3 × 106 live, transfected cells) was injected intravenously in 100 μL PBS and LPS was administered either 0, 24, 48, or 65 hours after injection. Since plasma Gluc from BMDM sensor is assayed 72 hours after injection, this schedule allowed for interrogation of sensor activity at either 72, 48, 24, or 7 hours of inflammation respectively. In the model of co-occurring tumors and acute inflammation, Balb/c mice bearing metastatic 4T1 tumors were injected with BMDM sensor and inoculated intranasally with LPS 65 hours afterwards (7 hours prior to assaying plasma Gluc). BLI of activated BMDM sensor was performed immediately following blood collection.
Carcinoembryonic antigen (CEA) release model
LS174T cells (2 × 106 in 50 uL PBS) were implanted subcutaneously on the right shoulder of female immunodeficient BALB/c NU/NU mice (Charles River). Tumor volumes were approximated by caliper measurements and blood (50 uL) was collected every three days starting on the tenth day post-implantation. RAW264.7 macrophage sensor (3 × 106 cells in 100 μL PBS) was injected on day 1 (10 days after implantation) with 50 uL blood collected 24 hours afterwards for Gluc and CEA detection. Plasma CEA concentration was measured with a commercial enzyme linked immunosorbent assay (ELISA) kit (ThermoFisher) with detection limit of 200 pg/mL.
Cell-free DNA model
Subcutaneous CT26 tumors were grown to volumes between 0-2000 mm3 and mice were terminally bled from the submandibular vein. Cell-free DNA (cfDNA) was extracted from the plasma using the NextPrep-Mag™ cfDNA Isolation Kit (Bioo Scientific, Austin, TX) and quantitated using the Quant-iT™ High-Sensitivity dsDNA Assay Kit (ThermoFisher). We confirmed that the cfDNA exhibited a primarily mononucleosome size profile (140-180 base pairs) using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA) and excluded samples with contamination of large fragment genomic DNA. Primer and locked nucleic acid (LNA) probes were obtained from IDT (San Jose, CA) with sequences shown in Supplementary Table 1. Thermal cycling for qPCR were performed using a CFX96 Real-Time System C1000 Touch Thermal Cycler (Bio-Rad) using the following protocol: 95°C for 3 min, followed by 50 cycles of 95°C for 15 seconds and 69.4°C (chr7_13872039_del) or67.9°C (chr19_39237841_del) for 15 seconds. Allele frequency limit of detection experiments were performed with genomic DNA isolated from the CT26 cells using the PureLink™ Genomic DNA Mini Kit (ThermoFisher) that was diluted with healthy Balb/c genomic DNA (Sigma-Aldrich) to obtain allele frequencies of 5%, 1%, and 0.1% based on initial allele frequencies of 100% (chr7_13872039_del) and 9% (chr19_39237841_del) previously reported17 Reactions contained 5 ng of cfDNA, forward and reverse primer concentrations of 500 nM, and probe concentration of 200 nM in 20μL volume total.
Statistical Analysis
Statistical analysis was performed using parametric unpaired t tests. The ROUT method was used to identify and remove outliers (only a single point was removed). All statistical analysis was performed in GraphPad Prism version 7.03.
Results
Arginase-1 expression identifies tumor-infiltrating macrophages
In designing a broadly applicable sensor, we wanted to select an immune subset present across a range of human cancers. Analysis of the fractional prevalence of leukocytes across 5,782 tumors from the iPRECOG dataset14 showed M2 macrophages are the predominant immune cells in the majority of solid tumors (Figure 2a). We thus selected macrophages as our candidate for a pan-cancer diagnostic sensor.
Figure 2. M2 macrophages are highly represented across a range of human cancers and arginase-1 identifies these macrophages in vitro and in vivo.
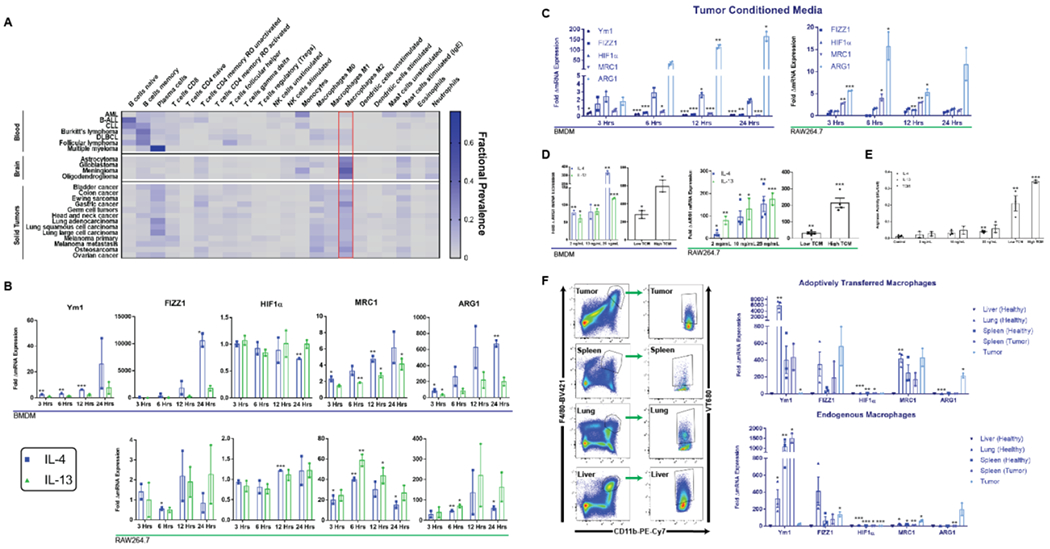
(A) Heat map depicting relative fractions of various immune cells across a range of human cancers. (B) Murine BMDMs and RAW264.7 murine macrophages exhibit time dependent increases in expression of M2-associated genes in response to IL-4, IL-13, and (C) TCM as measured with qPCR. RAW264.7 macrophages did not express Ym1. (D) ARG1 exhibits a dose-dependent response to IL-4, IL-13, and TCM on both the mRNA (BMDM and RAW264.7) and (E) protein level (RAW264.7) as measured by arginase activity assays. (F) Endogenous macrophages and adoptively transferred (ACT) RAW264.7 macrophages were isolated by flow cytometry from tumors, spleens, lungs, and livers of subcutaneous tumor bearing or healthy mice (left) and fold elevations of M2 gene expression in different tissues relative to liver-resident (for endogenous) or liver-homing (for ACT) macrophages are shown (right). * indicates statistical significance at p < 0.05, ** indicates statistical significance at p < 0.01, and *** indicates statistical significance at p < 0.001. Error bars depict standard error of the mean (s.e.m). BMDM, bone marrow-derived macrophage; TCM, tumor conditioned media.
In mice, tumor-associated M2-polarization is characterized by upregulation of genes involved in fostering an immunosuppressive microenvironment18. The tumor microenvironment (TME) is characterized by acidosis, hypoxia, elevated concentrations of IL-4 and IL-13, and tumor-derived cytokines19. Both BMDMs and RAW264.7 macrophages exhibited a time dependent increase in expression of established18 M2 markers Ym1, FIZZ1, MRC1, and ARG1 upon stimulation with IL-4 and IL-13 as measured with qPCR. BMDMs and RAW264.7 macrophages also upregulated ARG1 up to 600-fold upon exposure to tumor-conditioned media (TCM) from a CT26 colon carcinoma cell line (Figure 2c). This effect may be mediated by tumor derived cytokines and metabolic intermediates including lactic acid (Supplementary Figure S3)18. ARG1 also exhibited dose-dependent induction upon stimulation with IL-4, IL-13, and TCM (Figure 2d–e), which is important for the dynamic range of an eventual diagnostic.
To assess whether exogenous macrophages adopt a tumor associated phenotype in tumor bearing mice, we labeled RAW264.7 cells with VivoTrack 680 (VT680) dye (Supplementary Figure S4) prior to intravenous injection into healthy BALB/c mice or mice harboring syngeneic 25-50 mm3 subcutaneous colorectal tumors. Five days post injection of labeled macrophages, tumors, spleens, livers, and lungs were harvested, and endogenous (CD11b+, F4/80+, VT680-) and adoptively transferred (CD11b+, F4/80+, VT680+) macrophages were isolated by flow cytometry (Figure 2f, Supplementary Figure S5). As predicted in vitro, ARG1 exhibited the most compelling differential gene expression profile with both endogenous and adoptively transferred tumor-infiltrating macrophages exhibiting a ~200-fold increase in ARG1 levels compared to liver-homing and resident macrophages respectively.
Adoptively transferred macrophages migrate to and accumulate in tumor microenvironments
One barrier to translation of probe-based diagnostics is inefficient delivery to sites of disease10. Since immune cell recruitment is a feature of many diseases, we hypothesized that a macrophage sensor could naturally migrate to sites of malignancy. Macrophages exhibited concentration dependent migration towards TCM up to 4-fold greater than toward unconditioned media in transwell assays (Figure 3a). In vivo, VT680 labeled adoptively transferred macrophages migrated to 25-50 mm3 tumors as visualized by fluorescence imaging (Figure 3b). Fluorescence signal co-localized with bioluminescence signal from Fluc transfected CT26 tumors (Figure 3c). Consistent with previous reports20, immunofluorescence of resected tumors revealed co-localization of macrophages with regions of hypoxia (Supplementary Figure S6).
Figure 3. Macrophages exhibit chemokine mediated migration towards tumors in vitro and in vivo.
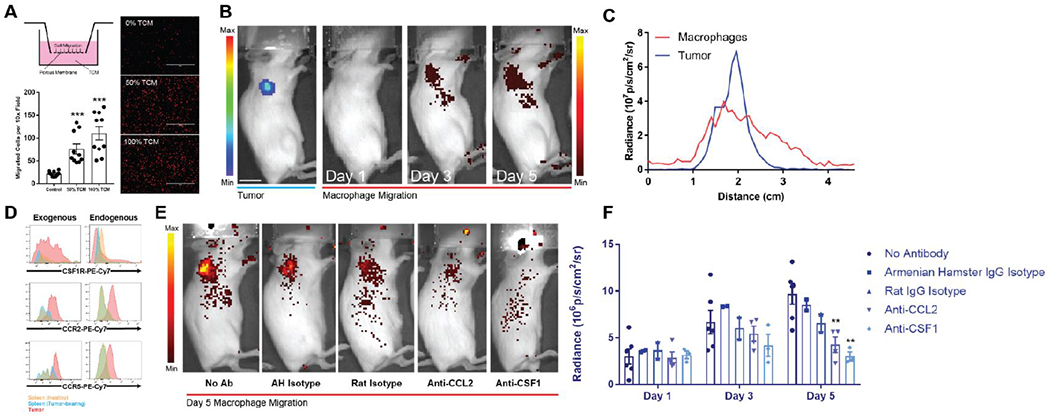
(A) Transwell assays performed with tumor conditioned media as the chemoattractant revealed a concentration dependent increase in RAW264.7 macrophage migration by optical microscopy (right) with quantification of migrated cells per 10× field revealing greater than four-fold increases in migration (bottom). Macrophages are shown in red false-color. Scale bars measure 400 μm. (B) VivoTrack 680 labeled macrophages demonstrate a time-dependent accumulation in a subcutaneous Fluc-expressing tumor from days 1 to 5 after intravenous injection as visualized with in vivo fluorescence microscopy. Scale bar measures 1 cm. Left and right radiance scales apply to the tumor and macrophage signals respectively. (C) Tumor and macrophage signal strongly co-localize on radiance line traces as a function of distance from scruff across the right shoulder. (D) Flow cytometry of tumor-resident macrophages (endogenous) and adoptively transferred tumor-infiltrating macrophages (exogenous) shows increased expression of CSF1R, CCR2, and CCR5 compared to splenic macrophages in both healthy and tumor bearing mice. (E-F) Neutralizing doses of anti-CCL2 (n = 4, p = 0.0077) and anti-CSF1 (n = 3, p = 0.0049) antibody interferes with macrophage migration to subcutaneous tumors more so than their respective isotype control antibodies. Radiance values are background subtracted. * indicates statistical significance at p < 0.05, ** indicates statistical significance at p < 0.01, and *** indicates statistical significance at p < 0.001. Error bars depict s.e.m. TCM, tumor conditioned media; Ab, antibody; AH, Armenian hamster.
To investigate the mechanism of recruitment, we analyzed surface expression of chemokine receptors known20 to play a role in migration: CSF1, CCL2, and CCL5. Both endogenous and adoptively transferred tumor-infiltrating macrophages exhibited increased expression of the cognate receptors CSF1R, CCR2, and CCR5 relative to splenic macrophages (Figure 3d). To delineate the role of chemokines in mediating either recruitment to or maintenance in the tumor, we administered neutralizing doses21, 22 of antibody against CCL2 and CSF1 and again monitored the migration of VT680+ macrophages. Neutralization of either chemokine significantly diminished migration compared and confirmed the mechanism of recruitment (Figure 3e–f).
Secreted biomarkers enable non-invasive monitoring of macrophage activation
In order to non-invasively assay macrophage polarization in vivo, we engineered macrophages such that activation of ARG1 would be coupled to production of a secreted biomarker. Glue is a primarily (95%) secreted luciferase which also allows for BLI of intracellularly trapped Glue23. We thus cloned the ARG1 promoter and enhancer region upstream of Glue and engineered a RAW264.7 cell line with stable expression of the pARG1-Gluc reporter. In activation experiments, sampling of culture media revealed a concentration and time dependent increase in Glue signal of up to 2-fold for IL-4/IL-13 and 60-fold for “high” TCM over 24 hours (Figure 4a).
Figure 4. Macrophage sensors enable detection and visualization of small tumors in vivo.
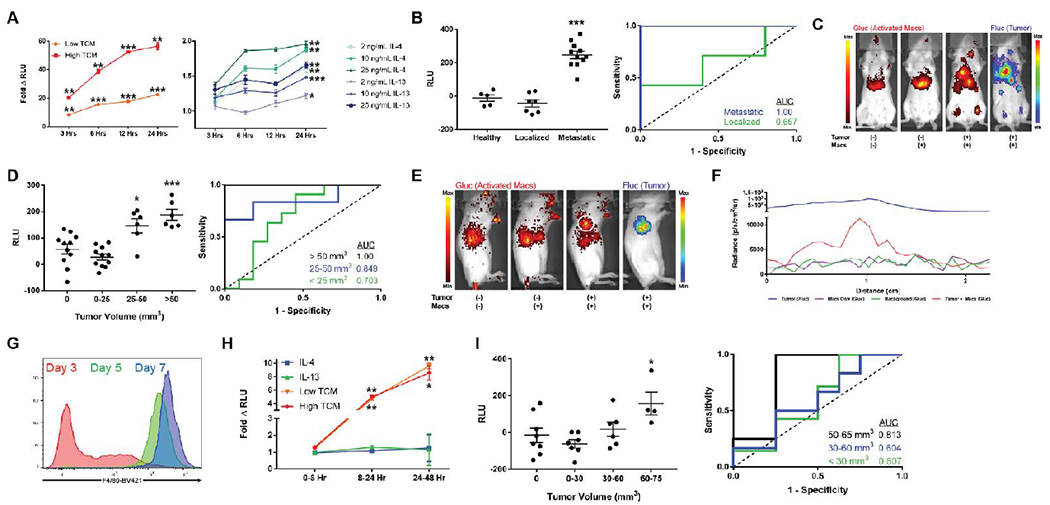
(A) RAW264.7 macrophages engineered to express the pARGI-Gluc reporter exhibit a time and concentration dependent secretion of Gluc when stimulated with tumor conditioned media (left) and IL-4/IL-13 (right) as assayed from culture media. Statistical significance in the right plot only shown for the 24 hour time point. Fold change is calculated relative to non-activated macrophage sensor. (B) RLU values assayed from plasma of 4T1 tumor bearing mice injected with sensor showed no elevations above healthy controls (n = 7, AUC = 0.657, 95% CI 0.335-0.979, p = 0.372) in localized lung microtumors but significant elevations in disseminated disease (n = 11, AUC = 1.00, p = 0.0018). RLU values are background subtracted to eliminate non-specific signal from healthy plasma. (C) BLI of activated macrophages and tumor dissemination reveals marked co-localization of macrophage sensor activation with sites of disease. (D) In a localized subcutaneous CT26 model, background subtracted plasma Gluc from activated macrophage sensor could reliably detect > 50 mm3 tumors with an AUC = 1.00 (n = 6, p = 0.0009) and 25-50 mm3 tumors with an AUC = 0.849 (n = 6, 95% CI 0.620-1.00, p = 0.021). (E) BLI and (F) right shoulder radiance line traces show visible spatial overlap of activated macrophages (white circle) with right shoulder tumors. (G) Bone marrow derived cells cultured in M-CSF exhibit increasing levels of monocyte/macrophage maturity marker F4/80 with a monocyte phenotype present at day 5 of culture. (H) BMDMs electroporated with the pARG1-Gluc reporter exhibit time dependent secretion of Gluc with tumor conditioned media as assayed from culture media. (I) Background subtracted plasma Gluc from activated BMDM sensors shows significant elevation (n = 4, p = 0.0342) when localized subcutaneous CT26 tumors reach volumes of 60-75 mm3 (AUC = 0.813, 95% CI 0.555-1.00, p = 0.0894). Scale bars measure 1 cm. Left and right radiance scales in (C) and (E) apply to the activated macrophages and tumor signals respectively. * indicates statistical significance at p < 0.05, ** indicates statistical significance at p < 0.01, and *** indicates statistical significance at p < 0.001. Error bars depict s.e.m. TCM, tumor conditioned media; RLU, relative luminescence units; AUC, area under the curve.
An engineered macrophage sensor can detect sub 50 mm3 tumors
To determine whether our sensor could detect tumors in vivo, we introduced it intravenously into tumor bearing mice and subsequently assayed the plasma for Glue. We first employed a syngeneic model of metastatic breast cancer24, wherein intravenously injected 4T1 cells form localized microtumors in the lung followed by emergence of diffuse metastatic disease. Our sensor discriminated metastatic disease from healthy controls with 100% sensitivity and specificity (area under the curve (AUC) = 1.00, n = 11, p = 0.0018) by plasma Gluc measurements (Figure 4b). Furthermore, imaging of both activated macrophages (intracellular Gluc) and metastases (Fluc) revealed co-localization of activated macrophages and sites of metastasis (Figure 4c). Notably, when tumor burden was localized to lung microtumors early after 4T1 injection (Figure 4b, Supplementary Figure S7), our sensor was unable to detect disease possibly due to a poorly developed TME in the highly oxygenated lung.
To determine the smallest tumor volumes that could be detected with the sensor, we next applied the macrophage sensor to a subcutaneous model of CT26 with tumor volumes of 0-250 mm3. Tumor size was measured with a caliper (Supplementary Figure S8a) on the day of sensor injection and plasma was assayed on subsequent days. At 24 hours post-sensor injection, we could detect tumors with volumes 50-250 mm3 (average 117.19 +/− 74.87 mm3) with 100% sensitivity and specificity (AUC = 1.00, n =6, p = 0.0009) (Figure 4d). Notably, tumor volumes of 25-50 mm3 (average 39.43 +/− 7.90) were also discriminated from healthy controls with an AUC = 0.849 (95% CI 0.620-1.00, n = 6, p = 0.021). Visibly necrotic tumors with volumes > ~1500 mm3 were not detected (Supplementary Figure S8b), possibly due to limited immune infiltration into poorly vascularized cores. Co-localization of activated macrophages and tumors was again observed with BLI (Figure 4e–f).
This localized model also allowed us to interrogate features of the sensor that are difficult to study in a metastatic model. For example, assaying of plasma Glue up to four days following sensor injection revealed a consistent decline after 24 hours (Supplementary Figure S8c), potentially due to the immunogenicity of the biomarker and/or the RAW264.7 cells. Additionally, the minimum required dose of macrophages was determined empirically to be ~3 million cells (Supplementary Figure S8d). These studies support early plasma sampling and can help guide further dose optimization.
As further evidence for the translatability of the approach, we also demonstrated tumor detection with primary BMDMs. We first generated monocytes from bone marrow and confirmed their phenotype by intermediate expression of the maturation marker F4/8015 (Figure 4g). The pARG1-Gluc construct was introduced by electroporation with efficiencies greater than 80% (Supplementary Figure S1b) and the resulting sensor was activated ~10-fold by both “low” and “high” TCM (Figure 4h). The BMDM sensor also detected CT26 tumor volumes as low as 60-75 mm3 in vivo (n = 4, p = 0.0342) with an AUC of 0.813 (95% Cl 0.555-1.00, n = 4, p = 0.0894) (Figure 4i). Tumors from mice injected with BMDMs exhibited an initial regression (Day 4, p = 0.058) but did not exhibit altered growth kinetics thereafter (Supplementary Figure S9) suggesting that sensor M2 polarization likely does not accelerate tumor progression25.
Macrophage sensor specificity in models of inflammation and wound healing
Inflammation is a significant confounding disease state for cancer diagnostics26. We thus investigated the degree of sensor activation upon exposure to inflammatory conditions. BMDMs did not exhibit an elevation in ARG1 expression by qPCR upon stimulation with inflammatory cytokines such as IFNγ/LPS and TNFα (Figure 5a). RAW264.7 ARG1 expression was similarly unaffected by TNFα, but was surprisingly induced by IFNγ/LPS. This induction is mediated largely by LPS as high doses of IFNγ or TNFα did not significantly affect ARG1 expression. Stimulation of the pARG1-Gluc engineered BMDMs and RAW264.7 macrophages with these same inflammatory mediators led to minimal increases in Glue secretion (Figure 5b).
Figure 5. Macrophage sensors reflect immunological timeframe in two models of acute inflammation and wound healing.
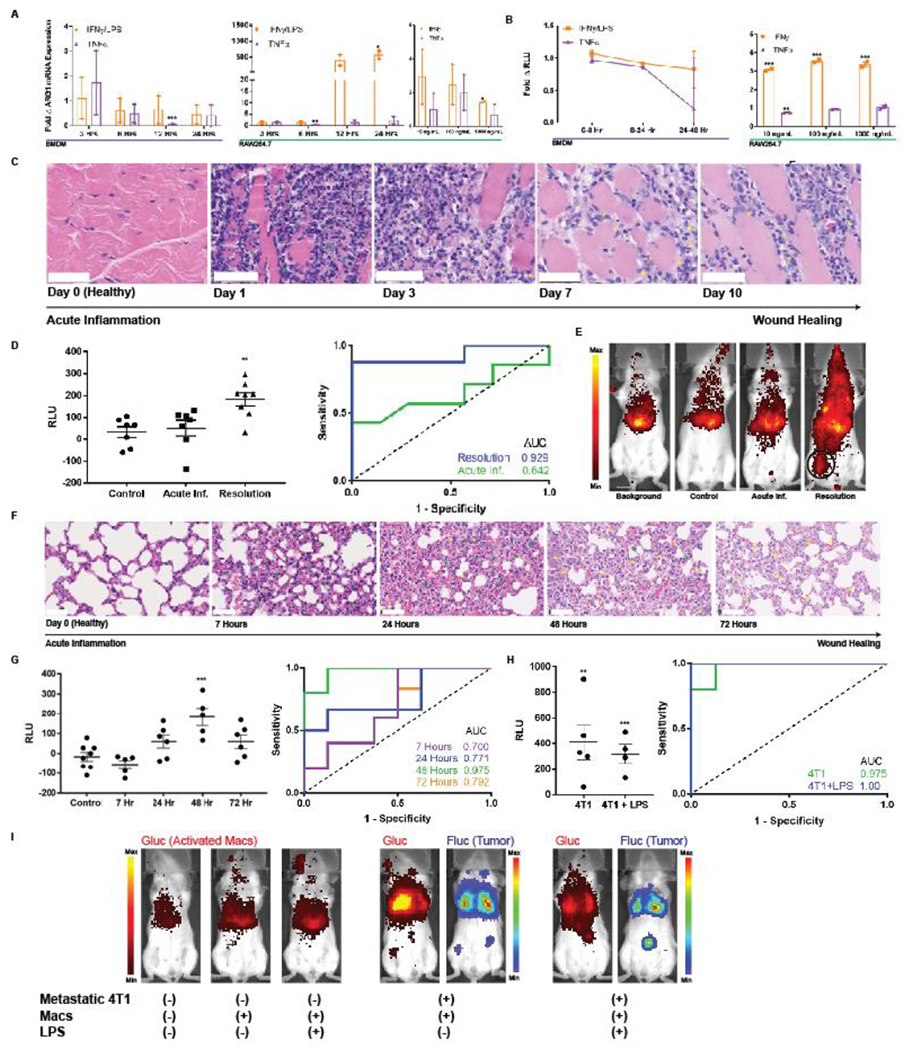
(A) BMDMs and RAW264.7 macrophages exhibit minimal elevations in ARG1 mRNA upon exposure to classic pro-inflammatory cytokines IFNγ and TNFα as quantified by qPCR. BMDM ARG1 levels are similarly not affected by LPS. (B) The engineered pARG1-Gluc expressing macrophage sensors do not result in notably increased Glue levels in culture media upon stimulation with the same pro-inflammatory cytokines. (C) H&E stains of hind leg muscle from days 0-10 post intramuscular injection of turpentine oil exhibit a classical timeline of acute inflammation with a primary neutrophilic (green arrows) response followed by infiltration of macrophages (yellow arrows) in the later stages of inflammation resolution. Scale bars measure 50 pm. (D) Background subtracted plasma Glue levels from the macrophage sensor either on day 1 (n = 6) or day 7 (n = 8) of inflammation (left) revealed no elevation in the acute inflammatory phase (day 1) but significant elevation and macrophage activation in the resolution phase (day 7). This is also reflected by an undiscriminating AUC = 0.643 (95% CI 0.332-0.953, p = 0.371) during acute inflammation but a robust AUC = 0.929 (95% CI 0.783-1.00, p = 0.006) during the wound healing phase. (E) BLI of intracellular Glue from activated macrophages reveals comparable signal from background, non-inflamed mice injected with sensor (control), and acutely-inflamed mice injected with sensor (Acute Inf.). This contrasts with BLI images when macrophage sensor is injected during the resolution phase on day 7 wherein localized activation of macrophages at the site of wound healing (black circle) is clearly visible. (F) H&E stains of lungs following intranasal inoculation with LPS exhibit a similar timeline of acute inflammation with a neutrophilic infiltrate (green arrows) present at 7 hours followed by gradual replacement with macrophages (yellow arrows) as the wound healing process progresses. Wound healing peaks at 48 hours after LPS inoculation and by 72 hours there is some restoration of healthy lung architecture. Scale bars measure 50 μm. (G) Plasma Glue measurements of mice injected with BMDM sensor reflect the acute inflammation and wound healing kinetics peaking at 48 hours with an AUC = 0.975 (95% CI 0.900-1.00, n = 5, p = 0.0054). (H) The BMDM sensor can robustly discriminate metastatic 4T1 tumors both in the absence (AUC = 0.975, 95% CI 0.900–1.00, n = 5, p = 0.0054) and presence (AUC = 1.00, 95% CI 1.00-1.00, n = 4, p = 0.0066) of LPS-induced acute inflammation via plasma Glue measurements as well as (I) via BLI of activated macrophages * indicates statistical significance at p < 0.05, ** indicates statistical significance at p < 0.01, and *** indicates statistical significance at p < 0.001. Error bars depict s.e.m. LPS, lipopolysaccharide; RLU, relative luminescence units; AUC, area under the curve.
Sensor specificity in inflammation was evaluated in vivo first in a model of hind leg inflammation27. Histology of muscle following injection of turpentine oil revealed a stereotypical timeline of inflammation starting with a neutrophil influx (day 1-3) that was replaced by macrophages (day 7-10)28 (Figure 5c). Administration of RAW264.7 sensor on the same day as turpentine oil did not yield significantly elevated plasma Glue, corroborating the sensor’s specificity (Figure 5d). Further, since M2 macrophages are involved in wound healing29, we hypothesized that injection of the sensor during the resolution phase would lead to sensor activation. Consistent with the known biology of M2 polarization during wound healing, plasma Glue was elevated in this case (AUC = 0.929, 95% CI 0.783-1.00, n = 8, p = 0.006) and activated macrophages were visible in the hind leg (Figure 5e).
Similar trends were observed using the BMDM sensor in an LPS-induced model of lung inflammation. Histology confirmed the previously described30, 31 kinetics of this model, revealing acute inflammation and an influx of neutrophils at 7 hours, wound healing and an influx of macrophages peaking at 48 hours, and a gradual restoration of healthy alveolar morphology at 72 hours (Figure 5f). Intranasal administration of LPS either 7, 24, 48, or 72 hours prior to plasma sampling recapitulated these kinetics. Plasma Glue was not elevated during acute inflammation at the 7 hour time point, providing further evidence for sensor specificity. As expected, plasma Glue exhibited a gradual elevation during wound healing at 24 hours (AUC = 0.771, 95% CI 0.501-1.00, n = 6, p = 0.093) and eventually peaked at 48 hours (AUC = 0.975, 95% CI 0.900-1.00, n = 5, p = 0.0054) (Figure 5g). Levels began to decrease towards baseline by 72 hours (AUC = 0.792, 95% CI 0.540-1.00, n = 6, p = 0.071) as normal lung architecture was restored.
We also investigated whether co-occurring inflammation affected the sensor’s ability to detect tumors in vivo. Employing the model of metastatic 4T1, we observed no significant differences in plasma Glue either in the absence (AUC = 0.975, 95% CI 0.900-1.00, n = 5, p = 0.0054) or presence (AUC = 1.00, 95% CI 1.00-1.00, n = 4, p = 0.0066) of LPS-induced acute lung inflammation when using BMDM sensors (Figure 5h). BLI of activated BMDM sensor was similarly unaffected (Figure 5i).
Macrophage sensor outperforms clinically used cancer biomarkers
Finally, we evaluated the sensitivity of the macrophage sensor against that of existing cancer biomarkers. First, we developed a subcutaneous model of colorectal adenocarcinoma using the LS174T cell line, which aggressively6 sheds CEA. Once tumors reached an average volume of > 25 mm3, we measured plasma CEA by ELISA every third day. The macrophage sensor was injected 24 hours prior to first CEA measurement. CEA was detectable no earlier than the second day of plasma sampling when tumors were of average volume 136.62 +/− 110.71 mm3 (Figure 6a–b). Of note, during the first plasma sampling, tumors of average volume 44.82 +/− 40.12 mm3 (n = 7) were not discriminated using CEA but could be on the basis of Gluc measurements (Figure 6c). This is reflected in the improved AUC from 0.829 (95% CI 0.590-1, p =0. 062) with CEA to 0.914 (95% CI 0.738-1.00, p = 0.019) with the macrophage sensor (Figure 6d).
Figure 6. Macrophage sensors outperform clinically used cancer biomarkers.
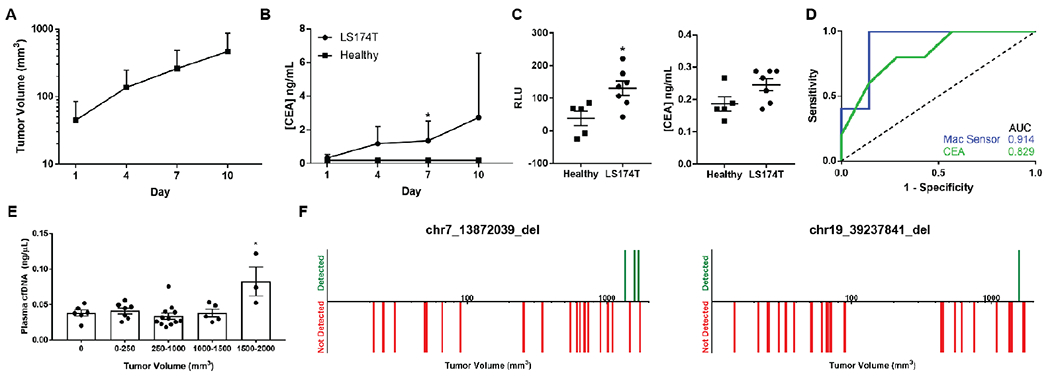
(A) Subcutaneously implanted LS174T tumors exhibit exponential growth in nu/nu mice (n = 12) with (B) increasing levels of plasma CEA detected by ELISA. (C) On day one of plasma sampling, the background subtracted plasma Gluc measurements from a macrophage sensor (left) were better able to discriminate tumor bearing (n = 7) and healthy mice (n = 5) compared to CEA measurements (right). (D) This improved sensitivity and specificity is reflected in improved AUC values on the receiver operator curve with the macrophage sensor (0.914, 95% CI 0.738-1.00, p = 0.019) compared to CEA (0.829, 95% CI 0.590-1, p = 0.062). (E) Plasma concentration of cfDNA was not significantly increased above healthy levels until subcutaneous CT26 tumor volumes reached 1500-2000 mm3. (F) Neither assayed mutation was detectable by qPCR in mouse plasma cfDNA (n = 23, left; n = 28, right) until tumors reached volumes of > ~1300 mm3. Downward red bars indicate a tumor-bearing mouse wherein the mutation was not detected in plasma cfDNA and vertical green bars indicate that the mutation was detected. * indicates statistical significance at p < 0.05. Error bars depict standard deviation (A, B) and s.e.m. (C, E). AUC, area under the curve.
We further characterized how the sensitivity of our sensor compared to that of a second diagnostic modality, cfDNA. Since we had previously shown that our macrophage sensor could detect 25-50 mm3 CT26 tumors (Figure 4d), we determined the smallest size tumors that could be detected by either quantitation of cfDNA concentration32 or detection of mutations in the plasma. Using a database of mutations in the CT26 cell line17, we identified two deletions and designed qPCR assays with allele frequency limits of detection of 0.1% and 1% respectively, which are similar to sensitivities of existing sequencing methods3 (Supplementary Figure S10). CfDNA concentration could not discriminate healthy from tumor bearing mice until tumors reached volumes of 1500-2000 mm3 (Figure 6e). Similarly, neither deletion mutation was detectable in the plasma until tumors had reached a minimum volume of 1300 mm3 (Figure 6f). While the generalizability of our model is limited by variables such as tumor vascularization, rate of cell death, and kinetics of tumor DNA release, the data suggests that the macrophage sensor can potentially detect tumors an order of magnitude smaller than possible with cfDNA even given a priori knowledge of the mutations.
Discussion
Encouraged by emerging cellular immunotherapies33, here we describe a novel platform technology for early disease detection using engineered immune cells as diagnostic sensors. By leveraging disease-specific metabolic alterations in macrophages, we show that an immune cell sensor can detect tumors as small as 4 mm in diameter and exhibit greater sensitivity than existing cancer biomarkers. The modest tumor volumes (25-50 mm3) and diameters detected are below the limit of detection of current clinical PET molecular imaging (~200 mm3 and 7 mm respectively34) and the estimated >10,000 mm3 tumor volumes needed for cfDNA detection35. We further show through models of inflammation that such a sensor can potentially achieve high specificity and be applied to monitoring of disease states other than cancer.
The case for disease-activatable probes stems from biological and mathematical limitations faced by endogenous biomarkers. In cfDNA, for example, mutation allele frequency decreases with disease burden, leading to an increasing probability that there will not exist a single copy of a mutation in a 10 mL blood draw36. It is estimated35, 37 that tumors must reach volumes greater than 1,000 mm3 (corresponding to allele frequencies of 0.01 %) for there to exist even one genome equivalent of tumor DNA in 4 mL of plasma. Our studies mirror these findings as the macrophage sensor could detect tumors up to 50-fold smaller in volume than possible with cfDNA mutation detection.
Our approach has the additional advantage of modularity in choice of disease, immune cell, and reporter. While our proof-of-concept uses macrophages, other immune subtypes including T-cells38, B-cells39, and natural killer cells40 all modulate metabolic genes in tumors and could be engineered similarly. Further, while we use Gluc given its ease of measurement, other non-immunogenic synthetic biomarkers such as secreted placental alkaline phosphatase (SEAP)41 could be used.
Our approach does have limitations which must be addressed prior to clinical translation. For one, M2 macrophages could potentially negatively impact tumor progression, though we note that the quantity of injected cells is relatively small (<3% of the total macrophage content of a mouse42) and the doses used in our study did not accelerate tumor growth. Future iterations can include genetically encoded suicide genes43 for sensor elimination. Second, our sensor did not detect visibly necrotic tumors which suggests that metabolic sensors could underestimate the extent of disease. As such, we envision our sensors to be complementary to other biomarkers which are effective in high disease burden settings.
Another limitation is the sub-optimal disease specificity of a single metabolic marker. Exclusion criteria will be needed for patients with confounding comorbidities, including those having recently undergone surgery44, asthmatics45, or those with recent myocardial infarction46 In addition, multiplexing metabolic gene reporters could improve specificity. In practice, this would involve using multiple promoters driving different secreted proteins, artificial microRNAs, or halves of a split reporter molecule47. Finally, spatial information would also be a valuable tool to improve clinical decision making. Our lab recently reported on the tracking of adoptively transferred chimeric antigen receptor (CAR) T-cells containing the herpes simplex virus type 1 (HSV1-tk) reporter gene using PET48. A similar technique could be adapted here by replacing Glue with HSV1-tk and performing PET to visualize regions of M2 polarization.
Lastly, while adoptive cell transfer is too costly as a routine screening tool, techniques such as in vivo gene delivery could generate sensors in situ without requiring cell isolation49. Alternatively, one could design low-cost allogeneic “off-the-shelf” sensors that perform a diagnostic function and are thereafter recognized and eliminated by the host immune system as foreign. In any case, we envision this technology first being utilized in populations at high risk of developing primary disease or recurrence, or in patients already underdoing therapeutic adoptive cellular therapy. As adoptive cell transfer becomes streamlined, there will be a strong case for use of such sensors as frontline diagnostics. In this way, we are optimistic that immune cell sensors can become prominent members in the fight for early disease detection and ultimately improve patient outcomes across a wide range of pathologies.
Supplementary Material
Acknowledgements:
Funding for this work was provided by the Canary Foundation (S.S.G.). A.A. has received support from the National Institutes of General Medical Sciences Medical Scientist Training Program T32 training grant GM007365, the Paul and Daisy Soros Fellowship, and the Bio-X Graduate Student Fellowship. We would like to thank Ms. Pauline Chu for help with tissue processing.
Footnotes
Author Conflict of Interest: A.A. and S.S.G. are co-inventors of a patent filed on the subject of this work which has been licensed by Earli, Inc. S.S.G. is a co-founder of Earli, Inc.
The authors declare that the data supporting the findings of this study are available within the paper [and its supplementary information files].
References
- 1.Etzioni R et al. The case for early detection. Nature reviews. Cancer 3, 243–252 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Park S.-m. et al. Molecular profiling of single circulating tumor cells from lung cancer patients. Proceedings of the National Academy of Sciences 113, E8379–E8386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phallen J et al. Direct detection of early-stage cancers using circulating tumor DNA. Science Translational Medicine 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheridan C Exosome cancer diagnostic reaches market. Nat Biotech 34, 359–360 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Hori SS & Gambhir SS Mathematical Model Identifies Blood Biomarker-Based Early Cancer Detection Strategies and Limitations. Science Translational Medicine 3, 109ra116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwong GA et al. Mass-encoded synthetic biomarkers for multiplexed urinary monitoring of disease. Nat Biotech 31, 63–70 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin KY, Kwong GA, Warren AD, Wood DK & Bhatia SN Nanoparticles That Sense Thrombin Activity As Synthetic Urinary Biomarkers of Thrombosis. ACS Nano 7, 9001–9009 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riglar DT et al. Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nature biotechnology 35, 653–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronald JA, Chuang H-Y, Dragulescu-Andrasi A, Hori SS & Gambhir SS Detecting cancers through tumor-activatable minicircles that lead to a detectable blood biomarker. Proceedings of the National Academy of Sciences 112, 3068–3073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slomovic S, Pardee K & Collins JJ Synthetic biology devices for in vitro and in vivo diagnostics. Proceedings of the National Academy of Sciences of the United States of America 112, 14429–14435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biswas Subhra K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 43, 435–449 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Murray Peter J. et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Headley MB et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531, 513–517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nature medicine 21, 938–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner M et al. Isolation and Intravenous Injection of Murine Bone Marrow Derived Monocytes. JoVE, e52347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pauleau AL et al. Enhancer-Mediated Control of Macrophage-Specific Arginase I Expression. The Journal of Immunology 172, 7565–7573 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Castle JC et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics 15, 190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colegio OR et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balkwill FR, Capasso M & Hagemann T The tumor microenvironment at a glance. Journal of Cell Science 125, 5591 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Murdoch C, Giannoudis A & Lewis CE Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 104, 2224 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Conde P et al. DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance. Immunity 42, 1143–1158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X et al. CD47 Blockade Triggers T cell-mediated Destruction of Immunogenic Tumors. Nature medicine 21, 1209–1215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tannous BA Gaussia luciferase reporter assay for monitoring of biological processes in culture and in vivo. Nature protocols 4, 582–591 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rashid OM et al. Is tail vein injection a relevant breast cancer lung metastasis model? Journal of Thoracic Disease 5, 385–392 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quail DF & Joyce JA Microenvironmental regulation of tumor progression and metastasis. Nature medicine 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chechlinska M, Kowalewska M & Nowak R Systemic inflammation as a confounding factor in cancer biomarker discovery and validation. Nature reviews. Cancer 10, 2–3 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Rivera S & Ganz T Animal Models of Anemia of Inflammation. Seminars in hematology 46, 351–357 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh TJ & DiPietro LA Inflammation and wound healing: The role of the macrophage. Expert reviews in molecular medicine 13, e23–e23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wynn TA & Vannella KM Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox G IL-10 enhances resolution of pulmonary inflammation in vivo by promoting apoptosis of neutrophils. American Journal of Physiology-Lung Cellular and Molecular Physiology 271, L566–L571 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Ulich TR et al. The intratracheal administration of endotoxin and cytokines. I. Characterization of LPS-induced IL-1 and TNF mRNA expression and the LPS-, IL-1-, and TNF-induced inflammatory infiltrate. The American Journal of Pathology 138, 1485–1496 (1991). [PMC free article] [PubMed] [Google Scholar]
- 32.Szpechcinski A et al. Cell-free DNA levels in plasma of patients with non-small-cell lung cancer and inflammatory lung disease. British Journal Of Cancer 113, 476 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadelain M, Riviere I & Riddell S Therapeutic T cell engineering. Nature 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erdi YE Limits of Tumor Detectability in Nuclear Medicine and PET. Molecular Imaging and Radionuclide Therapy 21, 23–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diamandis E & Fiala C Can circulating tumor DNA be used for direct and early stage cancer detection? [version 1; referees: 2 approved]. F1000Research 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haque IS & Elemento O Challenges in Using ctDNA to Achieve Early Detection of Cancer. bioRxiv (2017). [Google Scholar]
- 37.Fiala C, Kulasingam V & Diamandis EP Circulating Tumor DNA for Early Cancer Detection. The Journal of Applied Laboratory Medicine 3, 300 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Kouidhi S, Noman MZ, Kieda C, Elgaaied AB & Chouaib S Intrinsic and Tumor Microenvironment-Induced Metabolism Adaptations of T Cells and Impact on Their Differentiation and Function. Frontiers in Immunology 7, 114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somasundaram R et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nature Communications 8, 607 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vitale M, Cantoni C, Pietra G, Mingari MC & Moretta L Effect of tumor cells and tumor microenvironment on NK-cell function. European Journal of Immunology 44, 1582–1592 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Berger J, Hauber J, Hauber R, Geiger R & Cullen BR Secreted placental alkaline phosphatase: a powerful new quantitative indicator of gene expression in eukaryotic cells. Gene 66, 1–10 (1988). [DOI] [PubMed] [Google Scholar]
- 42.Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. The Journal of Experimental Medicine 161, 475–489 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonifant CL, Jackson HJ, Brentjens RJ & Curran KJ Toxicity and management in CAR T-cell therapy. Molecular Therapy Oncolytics 3, 16011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klinkert K et al. Selective M2 Macrophage Depletion Leads to Prolonged Inflammation in Surgical Wounds. European Surgical Research 58, 109–120 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Girodet P-O et al. Alternative Macrophage Activation Is Increased in Asthma. American Journal of Respiratory Cell and Molecular Biology 55, 467–475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Troidl C et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. Journal of Cellular and Molecular Medicine 13, 3485–3496 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim SB, Sato M & Tao H Split Gaussia Luciferase-Based Bioluminescence Template for Tracing Protein Dynamics in Living Cells. Analytical Chemistry 81, 67–74 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Keu KV et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Science Translational Medicine 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith TT et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nano 12, 813–820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.