

Summary
Immunological homeostasis in T cells is maintained by a tightly regulated signaling and transcriptional network. Full engagement of effector T cells occurs only when signaling exceeds a critical threshold that enables induction of immune response genes carrying an epigenetic memory of prior activation. Here we investigate the underlying mechanisms causing the suppression of normal immune responses when T cells are rendered anergic by tolerance induction. By performing an integrated analysis of signaling, epigenetic modifications, and gene expression, we demonstrate that immunological tolerance is established when both signaling to and chromatin priming of immune response genes are weakened. In parallel, chromatin priming of immune-repressive genes becomes boosted, rendering them sensitive to low levels of signaling below the threshold needed to activate immune response genes. Our study reveals how repeated exposure to antigens causes an altered epigenetic state leading to T cell anergy and tolerance, representing a basis for treating auto-immune diseases.
Keywords: T cell, tolerance, chromatin, epigenetic, gene regulation, signaling, transcription factor, CTLA4, IL-10, Cbl-b
Graphical Abstract
Highlights
-
•
Activation of immune response genes is suppressed in tolerant T cells
-
•
Epigenetic priming of repressive genes is boosted when tolerance is established
-
•
Inhibitory receptor genes have a lower threshold of activation in tolerant cells
-
•
Induction of tolerance by peptides points toward a therapy for multiple sclerosis
Autoimmune diseases such as multiple sclerosis (MS) are caused by T cells reacting to host proteins. Bevington et al. use escalating doses of a MS auto-antigen to induce tolerance in T cells in mice, and define a mechanism whereby antigen activates repressive genes while silencing immune response genes.
Introduction
The maintenance of immunological homeostasis requires T cell responses to antigen (Ag) to be both tightly regulated and receptive to negative feedback. The engagement of effector T cells is dependent on the activation of multiple parallel signaling pathways, thereby ensuring appropriate immune responses. The magnitude of the response to T cell receptor (TCR) signaling can also be suppressed by inducing an anergic state in response to: (1) repeated exposure to Ag, resulting in tolerance (Sabatos-Peyton et al., 2010); (2) lack of co-receptor stimulation (Schwartz, 2003); or (3) negative feedback from inhibitory receptors, such as PD-1, CTLA4, LAG3, TIM3, and TIGIT, as seen in tumor-infiltrating lymphocytes (TILs) and exhausted T cells facing chronic exposure to viral Ags (Anderson et al., 2016, Wherry et al., 2007, Wherry and Kurachi, 2015). PD-1 reduces the strength of TCR signaling below the threshold for many immune response genes, but not for a subset of other inducible genes that have a lower activation threshold (Shimizu et al., 2020). However, the molecular basis for this phenomenon is unknown.
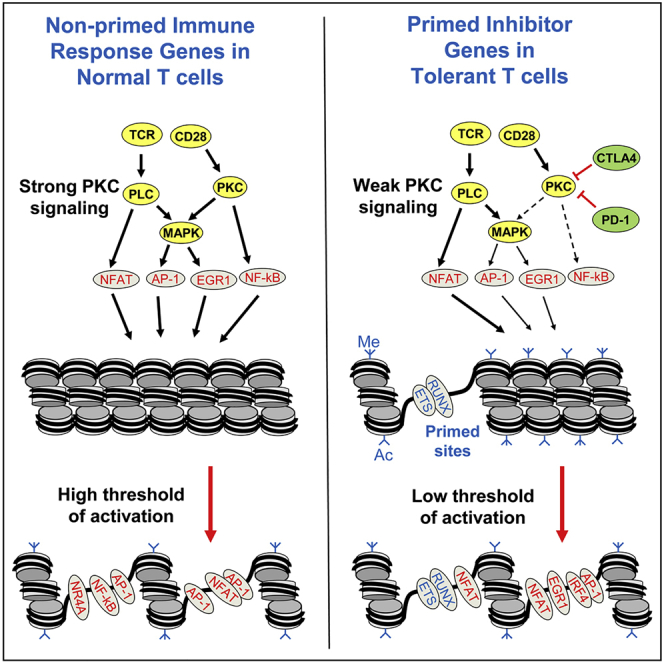
During a normal immune response, the co-activation of TCR and CD28 signaling in effector T cells leads to the transcriptional activation of hundreds of genes via inducible transcription factors (TFs), such as NFAT, AP-1, nuclear factor κB (NF-κB), and EGR1 (Yukawa et al., 2020; Figure 1A). These same TFs are also strongly induced by the phorbol ester phorbol myristate acetate (PMA) and the calcium ionophore A23187 (PI), which cooperate by activating diverging signaling pathways downstream of phospholipase C (PLC) and protein kinase C (PKC) (Figure 1A; Brignall et al., 2017). The pathways then re-converge in the nucleus, where NFAT and AP-1 bind cooperatively to activate gene expression (Chen et al., 1998, Johnson et al., 2004). Little is known about how inhibitory receptors suppress these signaling pathways and alter the T cell response. However, numerous studies point to a role for the ubiquitin ligase Cbl-b, which is activated by CTLA4 and PD-1 (Fujiwara et al., 2017, Li et al., 2004, Li et al., 2019). Cbl-b functions as a gatekeeper of T cell activation by antagonizing PLC, phosphatidylinositol 3-kinase (PI3K), and PKC signaling (Fang and Liu, 2001, Heissmeyer et al., 2004, Jeon et al., 2004). Furthermore, Cbl-b expression is raised in T cells when anergy is induced by calcineurin signaling to NFAT in the absence of PKC activation (Heissmeyer et al., 2004) or by tolerizing signals (Jeon et al., 2004).
Figure 1.

Gene Expression Analyses Comparing Tolerized T Cells with Naive T Cells
(A) Model of the activation of immune response genes by TCR and CD28 signaling to inducible TFs. Calcium ionophore A23187 and PMA act on the same signaling pathways downstream of PLC.
(B) The transcription factors activated by signaling (NFAT, AP-1) induce chromatin remodeling at immune response genes at both priming elements and inducible enhancers. Enhancer activation initiates transcription of their target genes. When signaling ceases, primed DHSs are stably maintained as regions of methylated (me) and acetylated (ac) chromatin by the retention of constitutively expressed transcription factors (ETS, RUNX).
(C) Schematic showing the dose escalation protocol used in tolerizing Tg4 mice by subcutaneous injections with MBP Ac1-9-specific TCR.
(D) Principle-component analysis of the RNA-seq datasets for the top 500 most variable genes for N0, T0, NAg, TAg, NPI, and TPI. RNA-seq data were taken from three biological replicates.
(E and F) Normalized average RNA-seq counts for classical immune response (E) and immune-modulatory genes (F). Error bars represent the standard deviation from three biological replicates.
We previously showed that a single cycle of activation of TCR/CD28 signaling pathways was sufficient to epigenetically reprogram chromatin domains encompassing immune response genes in naive T cells, resulting in the stable maintenance of altered chromatin states regulating gene accessibility and expression (Bevington et al., 2016, Bevington et al., 2017). When activated by Ag for the first time, quiescent naive T cells undergo a slow transformation into rapidly dividing T blast (TB) cells, during which they remodel broad chromatin domains carrying activating histone modifications and encompassing inducible genes and their regulatory elements (Figure 1B). In response to TCR signaling, these regulatory elements form open chromatin regions detectable as DNase I hypersensitive sites (DHSs). Although inducible DHSs (iDHSs) are opened up transiently by factors such as AP-1 and NFAT, and typically function as inducible transcriptional enhancers or promoters, ∼3,000 of the DHSs formed during blast cell transformation are stably maintained as primed DHSs (pDHSs) in TB and in memory T cells without influencing steady-state transcription. Although these pDHSs are initially opened in response to transient activation of AP-1, they are then maintained by stable binding of constitutively expressed TFs such as ETS-1 and RUNX1, which introduce activating histone modifications into surrounding regions, thereby maintaining an epigenetic memory of TCR activation (Bevington et al., 2016, Bevington et al., 2017).
Several previous studies of anergy in the context of tolerance have investigated the roles and regulation of production of Tr1 cells, which are a Foxp3−ve subset of regulatory T cells, which limit damage resulting from responses to infection and suppress auto-immunity by secreting factors such as interleukin (IL)-10 (O’Garra and Vieira, 2007, Roncarolo et al., 2014, Trinchieri, 2007). Intra-nasal or subcutaneous administration of peptides can induce an anergic IL-10-secreting Tr1-like population in vivo (Burton et al., 2014, Gabrysová et al., 2009, Sundstedt et al., 2003), and this treatment is effective in establishing tolerance (Burton et al., 2014) and protecting against autoimmunity (Burkhart et al., 1999, Clemente-Casares et al., 2016, Gabrysová et al., 2009, O’Neill et al., 2006). Tr1-like cells can also be generated in vitro by culturing T cells in IL-27 (Pot et al., 2009). The Tr1 gene expression signature resembles that of both exhausted TILs and exhausted T cells associated with chronic viral infections (Chihara et al., 2018). Tr1-like cells can also be induced by repeated anti-CD3ε antibody exposure (Mayo et al., 2016) or by nano-particles coated with peptide-bound major histocompatibility complex (MHC) class II (Clemente-Casares et al., 2016). These studies show that TCR signaling is important in generating tolerance (Wraith, 2016). However, although there is a consensus surrounding the importance of Tr1-like cells in a variety of immunological contexts, the molecular mechanisms that lead to the generation of Tr1-like tolerant cells and their altered response to Ag remain obscure.
To investigate the underlying basis of T cell tolerance, we performed genome-wide profiling of gene-regulatory networks in T cells before and after induction of tolerance, and after reactivation of TCR signaling. For this, we employed a transgenic TCR model (Tg4) (Liu et al., 1995) based on desensitization of mice in response to escalating doses of a tolerizing peptide Ag (Figure 1C; Burton et al., 2014). Tg4 mouse T cells recognize the Ac1-9 N-terminal peptide AcASQKRPSQR from myelin basic protein (MBP), an encephalitogenic auto-Ag associated with multiple sclerosis, and can be rendered tolerant by repeated exposure to the higher affinity, MHC-binding MBP Ac1-9[4Y] analog AcASQYRPSQR (4Y) (Burton et al., 2014). This approach may form the basis of future therapies in auto-immune disease because we have established that it alleviates symptoms of multiple sclerosis in patients (Chataway et al., 2018). To define epigenetic mechanisms maintaining an anergic tolerant state in Tg4 T cells, we identified DHSs on a genome-wide scale, together with genome-wide RNA-seq. These integrated studies demonstrated that the tolerized state is associated with two distinct mechanisms. First, tolerized T cells specifically maintain chromatin priming at a subset of pDHSs within archetypal T cell tolerance signature genes, allowing them to be activated at a signaling threshold below that of immune response genes. Second, receptor signaling to AP-1 is suppressed, and tolerized T cells fail to activate classic immune response genes in vivo in response to TCR stimulation by peptide. Hence it is altered epigenetic states that shift the balance between immune response and immune repression following tolerization.
Results
T Cell Tolerization Reprograms Inducible Gene Expression Potential
In the Tg4 mouse model, tolerance induction correlates with the induction of anergic CD4+ T cells with a Tr1 phenotype (Gabrysová et al., 2009). To address the molecular basis of this phenomenon, we first undertook genome-wide analyses of changes in gene expression (RNA sequencing [RNA-seq]) associated with tolerance in both the steady state and following challenge with a specific Ag. Tg4 transgenic mice were tolerized by repeated subcutaneous injection of the 4Y MBP peptide according to the schedule outlined in Figure 1C. To examine TCR responses in tolerized T cells (T) and in non-tolerant predominantly naive T cells (N) from control mice, we harvested cells 2 h after a final injection with 80 μg 4Y peptide (TAg and NAg) or PBS vehicle (T0 and N0). We also examined in vitro responses of T cells after a 2-h stimulation with 20 ng/mL PMA and 2 μM calcium ionophore A23187 (TPI and NPI) so as to distinguish between mechanisms acting at the level of TCR/CD28 signaling and transcriptional mechanisms involving inducible TFs downstream of PLC (Figure 1A). After normalization of the data, we defined 17,179 loci where the average RNA-seq value was at least 3 for at least one set of conditions (Table S1). Principle-component analysis of the 500 most variable genes using three biological replicates revealed substantial differences between naive and tolerized cells under all conditions (PC1 versus PC3; Figure 1D) and robust responses to Ag in both naive and tolerant cells (PC1 versus PC2; Figure S1A). Analysis of RNA-seq data for genes where values changed by at least 2-fold after tolerization (Figure S1B), and at least one value was above 50, revealed 475 loci higher and 107 loci lower in T0 than N0, (Table S2) and 568 loci higher and 381 loci lower in TAg than NAg (Table S3). Hierarchical clustering was performed for 637 of these genes where at least one of the T0:N0, TAg:NAg, TAg:T0, or NAg:N0 ratios showed at least a 10-fold difference (Figure S1C). The combined analyses revealed immune response genes, such as Il2, Il3, Tnf, Csf2, Ccl1, Cd40lg, Nr4a3, and Nfkb1, which were less inducible in tolerized cells treated with Ag (Figures 1E, S1C, and S1D), and anergy or tolerance-associated genes, which were more inducible, including the inhibitory receptors Ctla4, Tigit, Lag3, Havcr2 (TIM3), and Pdcd1 (PD1), the phosphatase Dusp6, the TFs Nfil3 and Maf, and the immune-suppressive cytokines Il10 and Il21 (Figures 1F, S1C, and S1E). For many immune response genes, this loss of responsiveness to TCR signaling could be bypassed by direct stimulation with PI (Figures 1E and S1D), suggesting that tolerance involves a membrane-proximal block in TCR signaling to inducible TFs. In contrast, the weak response to TCR signaling seen for many immuno-suppressive genes in naive cells could not be boosted by PI. Similar responses were seen over an extended time course of stimulation, demonstrating that it is not just the kinetics but the magnitude of activation that varies (Figures S1F and S1G). Some genes were also expressed at higher levels in tolerized cells even prior to stimulation (Figures 1F, S1C, and S1E; Table S2). Many of the genes upregulated in T0 had also been previously shown to be upregulated in Tr1-like cells generated in vitro with IL-27, suggesting similarities between these two model systems (Chihara et al., 2018; Table S4). These include Maf, Il10, Il21, Havcr2, Lag3, Nfil3, and Prdm1. These data suggest a potential model whereby tolerized T cells exhibit both: (1) suppression of TCR/CD28 signaling to a level below the threshold needed for activation of many immune response genes; and (2) a heightened sensitivity of immuno-regulatory genes that still allows activation at reduced levels of TCR signaling, consistent with the actions of inhibitory receptors such as PD-1 (Shimizu et al., 2020). Significantly, TCR-inducible activation of Il2, Csf2, and Tnf is dependent on CD28 signaling (Yukawa et al., 2020).
Modification of the Epigenetic Landscape in Tolerant Cells
We next investigated whether the more efficient activation of a subset of genes in tolerant cells was due to epigenetic reprogramming at the level of chromatin modifications, as observed in TB cells (Bevington et al., 2016). To this end, we globally identified DHSs in N0 and T0 cells using DNase sequencing (DNase-seq). We ranked the DHSs based on the fold change of DNase-seq signal between N0 and T0 for two independent replicates (Figure S2A). We selected high-confidence subsets of DHSs that were at least 2-fold different in both experiments. These analyses identified 1,033 T0-specific DHSs (tDHSs) and 635 N0-specific DHSs (Figures 2A, 2B, and S2A; Data S1). We also used DESeq2 as an alternative method of statistical analysis to screen for 2-fold changes and p < 0.05, and again identified the vast majority of the 1,033 tDHSs defined above (84%; Figure S2B).
Figure 2.

DNase-Seq Analyses of Open Chromatin in Naive and Tolerized T Cells
(A and B) Average DHS signal at the 1,033 T0-specific DHSs (A) and at the 635 N0-enriched DHSs (B) in the replicate N0, T0, and T0M samples.
(C) DNase-seq sequence tag density plots showing all 26,227 peaks detected in replicate 1 of T0 and N0 ordered by increasing fold change (FC) of sequence tag count for T0 compared with N0. Alongside is the FC in mRNA level for the closest gene to the DHSs as ranked in the density plots. The FC is shown between T0 and N0 (left), and TAg is compared with NAg (right). The locations of TF consensus binding motifs within the same DHSs are shown alongside for AP-1, NFAT, MAF, and LEF family proteins. Plotted alongside are published TF binding data from ChIP-seq analyses for TCF1 in thymocytes (Dose et al., 2014), c-MAF in Th17 cells (Ciofani et al., 2012), and ectopic CA-RIT-NFAT1 in CD8 T cells (Martinez et al., 2015).
(D and E) HOMER de novo identification of TF motifs enriched in the 1,033 T0-specific DHSs (D) and the 635 N0-specific DHSs (E).
(F) Average ChIP signal at the 1,033 T0-specific DHSs and 635 N0-specific DHSs for TCF1, c-MAF, and CA-RIT-NFAT1.
(G) UCSC Genome Browser tracks showing DNase-seq data from replicate samples of non-stimulated naive cells (N0), non-stimulated tolerant cells (T0), and tolerant cells 3 weeks after the final dose of peptide (T0M), plus published DNase-seq data for CD4 T blast (TB) cells (Bevington et al., 2016). Published ChIP-seq data are shown for the TFs TCF1, c-MAF, and CA-RIT-NFAT1 and the histone modifications H3K27ac and H3K4me2 in TB cells. Representative RNA-seq data from one of three replicates are also shown for resting and in vivo peptide-stimulated naive and tolerant cells (0 and Ag).
(H and I) Bar graphs showing the percentage and proximity of genes that are preferentially induced in T0 or N0 (H) and TAg or NAg (I), which are found within 100 kb of the 1,033 T0-specific DHSs.
We also investigated the stability of the tDHSs after a time interval sufficient for peak effector T cell responses to have subsided and for memory T cells to form (T0M, Figure 1C). We performed DNase-seq on tolerized cells that had received the final dose of Ag 3 weeks prior to harvest, and we still reproducibly detected over half of the 1,033 tDHSs (Figure S2C) that maintained an elevated average DHS signal (Figures 2A and 2C). These data suggest that tolerization involves long-term maintenance of epigenetic reprograming of a specific subset of DHSs in the absence of sustained TCR signaling. To ascertain whether the changes in chromatin structure had any impact on the nearby genes, we measured the difference in mRNA expression for the genes closest to the ranked DHSs for replicate 1 (Figure 2C). We observed an increase in both steady-state (T0 compared with N0) and inducible (TAg compared with NAg) gene expression in parallel with the increase in DHS strength. This is consistent with a role for epigenetic priming in the imprinting of a transcriptional memory of TCR activation, thereby enabling both higher steady-state expression in T0 and more efficient re-activation in TAg compared with NAg.
To gain insight into transcriptional mechanisms underlying tolerization, we used HOMER to perform de novo identification of TF motifs in the different specific subsets of DHSs (Figures 2D, 2E, and S2B). The tDHSs were more highly enriched in AP-1 and NFAT motifs, whereas the pre-existing N0-specific DHSs contained a higher proportion of LEF/TCF motifs. Both subsets were enriched in RUNX, ETS, and CREB/ATF motifs. The distribution of AP-1, NFAT, MAF, and LEF/TCF motifs is depicted in Figure 2C, where their direct correlation with the coordinates of the DHSs is shown. Published chromatin immunoprecipitation sequencing (ChIP-seq) data of TCF1 binding in thymocytes was consistent with these in silico observations (Dose et al., 2014), with TCF1 binding concentrated in both the N0-specific and shared peaks but depleted in the tDHSs (Figures 2C and 2F). Conversely, MAF binding was more enriched in the tDHSs than the N0-specific DHSs (Ciofani et al., 2012; Figures 2C and 2F). MAF has been shown to be important in regulating Il10 expression (Xu et al., 2009) and is implicated in repressing Il2 (Gabryšová et al., 2018). Maf is also upregulated in T0 and TAg cells (Figure 1F), and MAF can dimerize with other CREB/AP-1 family proteins to bind to AP-1 sites (Kataoka et al., 1994), a motif that is enriched in the tDHSs (Figures 2C and 2D). The enrichment of NFAT motifs in tDHSs may also be functionally relevant because NFAT plays a role in T cell anergy (Macián et al., 2002) and T cell exhaustion (Martinez et al., 2015). Indeed, when we aligned our DNase-seq data with ChIP-seq data generated using a constitutively active modified form of NFAT1/NFATC2 (CA-RIT-NFAT), which is known to promote exhaustion (Martinez et al., 2015), we observed that NFAT binding was more strongly enriched in the tDHSs (Figures 2C and 2F). Although it is unlikely/unknown whether NFAT is binding to these sites in T0 cells before stimulation, the presence of the motif at an open DHS should enable rapid binding when the cells are re-exposed to Ag.
Examples of tDHSs with binding sites for MAF and NFAT were found at the tolerance-associated Il10, Ctla4, Tigit, and Nrp1 loci (Figure 2G), where there are higher levels of either constitutive or inducible mRNA expression in tolerized cells (Figures 1F and 2G). At these loci, tDHSs were present in T0, but not in N0, and these sites were largely still maintained 3 weeks later in T0M. The tDHSs were more likely to bind CA-RIT-NFAT and/or c-MAF, but not TCF1. Furthermore, recently activated CD4 TB cells generated by in vitro activation exhibit many of the same DHSs as tolerant cells, and these regions are flanked by the active chromatin modifications H3K27ac and H3K4me2 in TB cells (Figure 2G). It is likely that the tDHSs in tolerant cells will be similarly embedded within extensive active chromatin domains that have been formed during the tolerization process.
T0-Specific DHSs Are Proximal to T0- and TAg-Specific Genes
Previously activated T cells maintain transcriptional memory by employing pDHSs to establish active chromatin domains encompassing adjacent inducible regulatory elements (Figure 1B). To assess the roles of the equivalent tDHSs in the regulation of inducible gene expression in tolerant cells, we integrated the mRNA data for differentially expressed genes with the differential DHS peak data. Starting with the gene sets for mRNA values greater than 50 defined in Figure S1B, we selected the annotated genes that have been assigned to a specific genomic locus, and further selected inducible genes on the basis that the TAg- and NAg-specific genes are also 2-fold inducible compared with T0 and N0, respectively. This allowed us to define genes expressed higher in T0 compared with N0 (460), N0 compared with T0 (80), and inducible genes expressed higher in TAg compared with NAg (138) and in NAg compared with TAg (226) (Table S5). We then measured the distances from the transcription start sites (TSSs) of differentially expressed genes to the closest of the 1,033 tDHSs (Figures 2H and 2I) and the closest of the 635 N0-specific DHSs (Figures S2D and S2E). These analyses revealed that the 1,033 tDHSs were preferentially located closer to the TSSs of T0- and TAg-specific genes, with approximately 25% of the TAg-specific genes located within 50 kb of a tDHS (Figure 2I). Hence these data suggest that tDHSs do indeed prime closely linked inducible loci for rapid re-activation in cells re-exposed to Ag. For example, tDHSs exist at the Il10, Ctla4, and Tigit genes, which are more strongly induced in TAg than in NAg (Figure 2G).
In contrast, priming seems to play little role in defining inducible responses in naive cells, with only 10% of the 226 NAg-specific gene TSSs within 50 kb of N0-specific DHSs (Figure S2D). Instead, the loss of DHSs in N0 correlates more closely with a loss of expression in N0, whereby 25% of the 80 N0-specific genes existed within 50 kb of N0-specific DHSs (Figure S2E). An example of this is shown at the naive T cell-specific gene Satb1 (Figure S2F), which has been implicated in repressing Pdcd1 (PD-1) expression in T cells (Stephen et al., 2017). Notably, the DHSs at Satb1 can bind TCF1, consistent with the enrichment of LEF/TCF motifs at the 635 N0-specific sites.
Activation of Signaling Pathways Induces a Different DHS Profile in Tolerant Cells
To investigate whether epigenetic priming at tDHSs supports inducible chromatin remodeling in tolerant T cells, we performed DNase-seq on NAg and TAg, and identified the differential DHSs by ranking the regions based on the fold change of signal (Figures 3A and S3A). We confirmed that the formation of TAg-specific DHSs was generally correlated with increased gene expression by measuring the difference in mRNA expression in TAg compared with NAg for the genes that are adjacent to the corresponding DHSs (Figure 3A). To define high-confidence subsets of specific DHSs, we intersected the peaks that were at least 2-fold enriched in each sample for two independent pairs of replicates. This gave 2,680 NAg-specific peaks and 1,959 TAg-specific peaks (Figure S3A). As we were specifically interested in iDHSs, we filtered them further to include only peaks that were at least 3-fold higher in NAg compared with N0, or in TAg compared with T0 (Figures S3B and S3C). This revealed 1,824 NAg-specific iDHSs and 682 TAg-specific iDHSs (Data S1; Figure 3B), which were highly specific for just naive or tolerant cells (Figure 3C). The same sets of specific DHSs were also defined by a parallel analysis using DESeq2 and the same fold change for each subset (p < 0.05), which identified 2,161 NAg-specific iDHSs and 923 TAg-specific iDHSs, including 88% of the 1,824 NAg sites and 90% of the 682 sites TAg (Figure S3D). Examples of TAg-specific iDHSs can be observed at the tolerance-associated Il10, Tigit, and Ctla4 loci (Figure 3D), where iDHS induction correlated with increased mRNA gene expression in TAg cells. Furthermore, 19% of the TAg-specific iDHSs were located within 50 kb of a tDHS (Figure 3E), consistent with locus priming enabling more efficient activation of closely linked enhancers residing in the same active chromatin domain (Figure 1B). Many of the same iDHSs that are induced in TAg are also induced in TB cells stimulated with PI (Figure 3D). In Figures 2G and 3D, it is also evident that regions associated with iDHSs induced by PI in TB cells were already marked with H3K4me2 and H3K27ac modifications before Ag stimulation. In contrast with tolerant cells, naive T cells are not preferentially primed by pDHSs at the NAg-specific genes in N0 cells, but nevertheless still induce NAg-specific iDHSs (Figure 3F).
Figure 3.

DNase-Seq Analyses of In Vivo-Activated Naive and Tolerized T Cells
(A) DNase-seq tag density plots showing all peaks detected in replicate 1 of NAg and TAg ranked according to FC of sequence tag count for TAg compared with NAg. Alongside is the FC in mRNA level for the closest gene to the DHSs for TAg compared with NAg.
(B and C) Average DHS signal at the 682 TAg-specific iDHSs (left) and at the 1,824 NAg-specific iDHSs (right) for 0 compared with Ag (B) and NAg compared with TAg (C).
(D) UCSC genome browser tracks showing DNase-seq and RNA-seq data from resting cells (N0 and T0), antigen-stimulated cells (NAg and TAg), and PI-stimulated cells (NPI and TPI). DNase-seq, H3K27ac ChIP, and H3K4me2 ChIP tracks are shown for CD4 TB PI.
(E) Bar graph showing the percentage of iDHSs that are preferentially induced in TAg or NAg that are found within 50 kb of the 1,033 tDHSs.
(F) UCSC genome browser tracks as for (D).
(G) Average DNase-seq signal for the 1,824 NAg-specific iDHSs and the 682 TAg-specific iDHSs in non-stimulated (N0 and T0) and PI-treated (NPI and TPI) cells.
NAg-Specific iDHSs Can Be Induced in Tolerant Cells by Bypassing TCR/CD28 Signaling
The treatment of tolerant cells with PI enabled the induction of many NAg-specific genes in tolerant cells, whereas the TAg-specific genes could not be induced by PI in naive cells (Figures 1E and 1F). To determine whether this same trend was observed at the chromatin level, we measured DNase I accessibility in naive and tolerant cells treated with PI. The mRNA induction correlated closely with the changes in chromatin structure whereby TAg-specific iDHSs could not be induced in naive cells, most likely due to the lack of epigenetic priming, whereas NAg-specific iDHSs were induced in tolerant cells treated with PI. This was observed at both the local level (Figures 3D and 3F) and globally at the 1,824 NAg and 682 TAg iDHSs (Figure 3G). The ability of PI to overcome the block in tolerant cells is consistent with suppression of TCR signaling upstream of PLC and PKC to inducible TFs, a block that could be bypassed by direct activation of PKC and RAS signaling by PI (Figure 1A).
NAg- and TAg-Specific iDHSs Are Governed by Distinct Gene-Regulatory Networks
Tolerance involves the expression and activation of inhibitory receptors that antagonize TCR/CD28 signaling to the inducible TFs, which create iDHSs. To identify gene-regulatory signatures within the specific subsets of NAg and TAg DHSs, we performed HOMER de novo motif analyses of enriched TF motifs (Figures 4A and 4B) and plotted the positions of the identified motifs over the DHS coordinates, together with published ChIP-seq data for these same TFs (Figure 4C). The NAg-specific iDHSs had a complex TF motif signature reflecting the TCR/CD28-inducible TFs AP-1, NFAT, NF-κB, and NR4A, but not EGR or IRF family TFs (Figures 4A and 4C). The TAg-specific DHSs were enriched for EGR and IRF motifs, but not motifs for the TCR/CD28-induced TFs NF-κB and NR4A. The latter could be explained in part by reduced expression for Nr4a3 and Nfkb1 in TAg compared with NAg (Figure 1E) and the absence of NAg-specific iDHSs induced at these loci (Figure 3F). These analyses were also confirmed using DESeq2, which identified essentially the same subsets of NAg and TAg DHSs with the same motif composition as those identified by the above pairwise analysis (Figures S4A and S4B).
Figure 4.

Analyses of TF Interactions with DHSs in Naive and Tolerized T Cells
(A and B) Homer de novo identification of enriched TF motifs in the 1,824 NAg-specific iDHSs (A) and the 682 TAg-specific iDHSs (B).
(C) Locations of transcription factor binding motifs (middle) at all DNase I peaks in the Ag-stimulated samples ordered according to the FC in sequence tag density for TAg compared with NAg (left). Aligned on the same axis are published ChIP-seq data for JunB in TB cells (Bevington et al., 2016), IRF4, BATF in CD4 T cells (Li et al., 2012) and constitutively active CA-RIT-NFAT and endogenous PI-induced NFAT in CD8 T cells (Martinez et al., 2015) (right).
(D) Venn diagrams depicting the overlaps between the published data for 4,4441 peaks detected in ChIP assays for CA-RIT-NFAT1 and/or endogenous (WT) NFAT1 in PI-stimulated T cells (Martinez et al., 2015) with the 682 TAg-specific iDHSs (upper) or 1,824 NAg-specific iDHSs (lower).
(E) Average ChIP-seq signal for WT NFAT1 and CA-RIT-NFAT in PI-stimulated T cells at the 377 NFAT TAg-specific iDHSs (upper) and 666 NFAT-bound NAg-specific iDHSs (lower).
(F) HOMER de novo identification of inducible TF motifs within the 377 and 666 sites.
(G) Representative example of western blot assays of Fos, FosB, JunB, Jun, JunD, and B2M proteins in T cells.
Evidence for Distinct AP-1, NFAT, and IRF Complexes in TAg and NAg Cells
Although the TAg and NAg iDHSs both contained NFAT and AP-1 motifs, the TAg-specific iDHSs were more enriched for NFAT motifs, whereas the NAg iDHSs had a higher proportion of AP-1 binding motifs (Figures 4A–4C). Consistent with this, published ChIP-seq data reveal higher levels of binding of the constitutively active CA-RIT-NFAT (Martinez et al., 2015) at the TAg-specific iDHSs and higher levels of JunB AP-1 binding at the NAg-specific iDHSs in TB cells stimulated with PI (Bevington et al., 2016; Figure 4C). Because CA-RIT-NFAT has modifications that prevent cooperative interactions with AP-1, these data may indicate that much of the NFAT binding seen at TAg-specific DHSs is independent of AP-1, as seen in other models of exhaustion and in TILs (Macián et al., 2002, Martinez et al., 2015, Mognol et al., 2017). In contrast, published ChIP-seq data for endogenous NFAT1 (NFATc2) in CD8 T cells stimulated with PI, where AP-1 will be efficiently induced (Martinez et al., 2015), revealed NFAT binding to many NAg-specific iDHSs that are unable to bind CA-RIT-NFAT (Figure 4C), suggesting direct cooperation between NFAT and AP-1.
To investigate NFAT further, we identified all 44,441 ChIP peaks that could bind either CA-RIT-NFAT1 or wild-type (WT) NFAT1, and then intersected these with the 1,824 NAg and 682 TAg iDHSs (Figure 4D). This analysis revealed that 55% of the 682 TAg iDHSs (377 sites) were capable of binding at least one form of NFAT, whereas just 36% of the NAg-specific iDHSs could bind NFAT. The average level of binding of CA-RIT-NFAT1 detected at the NAg iDHSs was also weaker than endogenous NFAT1 binding (Figure 4E), again suggesting that cooperative binding with AP-1 is more important at NAg iDHSs. This concept was further supported by an enrichment of the composite NFAT/AP-1 element detected by HOMER in the NAg, but not the TAg, NFAT binding sites (Figure 4F). Examples of these patterns of binding were observed at the TAg-specific genes Tigit and Ctla4, where the TAg-specific iDHSs bind NFAT independently of AP-1 (CA-RIT NFAT) (Figure S4C). In contrast, the NAg-specific genes Fosl2 and Nfkb1 bind WT NFAT only when the cells are stimulated with PI and AP-1 is present (WT NFAT PI) (Figure S4D). Furthermore, consensus motifs for cooperative NFAT/AP-1 binding were found at the NAg-specific genes, whereas the TAg-specific iDHSs lacked the true composite element with correctly spaced NFAT and AP-1 motifs (Chen et al., 1998). The above observations and conclusions were supported by a reduction in the total amount of the activating AP-1 proteins c-Fos, Fosb, c-Jun, and JunB in TAg compared with NAg, whereas levels of JunD were not reduced (Figure 4G). JunD has immuno-suppressive functions in lymphocytes (Meixner et al., 2004), suggesting that the ratio between JunD and other activating AP-1 proteins may influence the activity of AP-1 target genes in tolerized cells.
JunD has previously been detected in AP-1/IRF complexes in T cells (Li et al., 2012). This is significant here because IRF family proteins also play a role in anergy, in association with BATF/Jun AP-1-like complexes, whereby these factors can bind to composite IRF1/AP-1 motifs, including at the Il10 locus (Glasmacher et al., 2012, Li et al., 2012). Although the composite IRF/AP-1 motif was not initially detected in the 682 TAg iDHSs (Figure 4B), a direct search detected it in 12% of these sites, compared with less than 3% of the 1,824 NAg iDHSs, and the IRF/AP-1 motif was identified by HOMER within the specific subset of 377 NFAT-bound NAg iDHSs (Figure 4F). Furthermore, BATF has been identified in IRF/AP-1 complexes in anergic Tr1 cells (Karwacz et al., 2017), and here we observed that published ChIP peaks for both BATF and IRF4 are enriched in TAg, but not NAg, DHSs (Figure 4C).
TF Occupancy Differs in Tolerant and Naive Cells
To look for further evidence of differential TF occupancy, we generated higher read depth DNase-seq data for one sample from each group and performed digital DNase I footprinting of protected TF motifs. Using the Wellington footprinting algorithm (Piper et al., 2013), we identified 1,490 protected sites in NAg-specific iDHSs and 1,038 protected sites in TAg-specific iDHSs (Figures 5A and 5B). Consistent with the results depicted in Figure 4, more AP-1 sites were occupied in NAg compared with TAg, and more NFAT and EGR sites were protected in TAg compared with NAg, while NF-κB and IRF sites were protected only in NAg and TAg, respectively (Figures 5C and 5D). To further validate these results, we plotted the forward and reverse DNase I cuts surrounding all the NF-κB motifs that were present in the 1,824 NAg-iDHSs and 682 TAg-iDHSs (Figure 5E). NF-κB sites in the 1,824 iDHSs were more protected in the NAg sample, indicating that this TF is most likely bound here. Examples are shown in Figures 5F and S5A, where publically available ChIP-seq data (Oh et al., 2017) show NF-κB binding correlating with these footprints. Furthermore, in some cases where an NF-κB motif was present within an iDHS that was induced in both NAg and TAg, more efficient protection was observed in the NAg cells (Figures 5G and S5B). This further suggests a reduction in signaling to NF-κB in tolerized cells.
Figure 5.

Footprinting Analyses of TF Occupation in Naive and Tolerized T Cells
(A and B) Footprints within (A) NAg- and (B) TAg-specific iDHSs ordered according to the Wellington footprinting occupancy score.
(C and D) HOMER de novo identification of TF motifs enriched within footprints in the 1,824 NAg-specific iDHSs (C) and the 682 TAg-specific iDHSs (D).
(E) Average profile showing the DNase I cuts around all NF-κB sites in the 1,824 NAg-specific iDHSs in NAg (upper) and 682 TAg-specific iDHSs in TAg (lower).
(F and G) Examples of Wellington digital footprinting of DNase-seq data showing protection of an NF-κB site at a NAg-specific iDHS (F) and at a shared iDHS (G). DNase-seq data are shown for NAg and TAg, and ChIP-seq data for NF-κB (Oh et al., 2017).
(H) Average profile showing the DNase I cuts around all AP-1/IRF sites in the 1,824 NAg-specific iDHSs in NAg (upper) and 682 TAg-specific iDHSs in TAg (lower).
(I) Examples of Wellington digital footprinting of DNase-seq data showing protection of NFAT and composite AP-1/IRF motifs. DNase-seq data are shown for NAg and TAg, and ChIP-seq data for NFAT1 (Martinez et al., 2015), IRF4, and BATF (Li et al., 2012).
We similarly investigated the occupancy of AP-1/IRF motifs and determined that these composite motifs had a greater level of protection in the 682 TAg-specific iDHSs than in the 1,824 NAg iDHSs (Figure 5H). The TAg-specific genes Il10, Ikzf2, and Nrp1 loci each encompass strongly protected AP-1/IRF motifs (Figures 5I and S5C). Furthermore, ChIP-seq data from previously published studies (Li et al., 2012) showed that these sites could bind both IRF4 and BATF (Figures 5I and S5C). At the Il10 −9 kb and Nrp1 +177 kb DHSs, in addition to IRF4 and BATF binding, NFAT peaks were identified in published ChIP data for CA-RIT-NFAT, at sites where NFAT motifs were footprinted in TAg cells, reinforcing the role of these three factors in gene-regulatory networks in tolerized T cells as has been shown in exhausted T cells (Man et al., 2017).
TCR Signaling Complexes Are Disrupted in Tolerized T Cells
To look for direct evidence of defective signaling in tolerant cells, we used microscopy to investigate TCR signaling molecules at the immune synapse formed after engagement of antigen-presenting cells (APCs) with either in vitro-generated T helper 1 (Th1) cells or T cells tolerized by repeated intra-nasal delivery of the 4Y peptide (Figure 6). We investigated the localization of Zap70 and PKCθ just downstream of TCR and PKC signaling (Figure 1A). These analyses revealed that although CD28 clustering at the TCR synapse occurs in both Th1 and tolerized T cells, Zap70 and PKCθ can no longer be efficiently recruited to the synapse in tolerized T cells (Figures 6A–6C). Quantitation of the proportion of signal at the synapse confirmed that there was a global decrease in the amount of Zap70 and PKCθ that migrates to the synapse upon engaging APCs in tolerized T cells compared with Th1 cells (Figure 6D). This breakdown of signaling from the synapse correlated with a reduction of PKCθ T538 phosphorylation in tolerized cells (Figures 6E). We also assessed the opposing roles played by PKCθ and Cbl-b, which functions as the gatekeeper of TCR/CD28 signaling (Li et al., 2004, Qiao et al., 2013, Zhang et al., 2002; Figure 6F). Total internal reflection fluorescence (TIRF) microscopy revealed lower levels of Zap70 and PKCθ in the same plane as the TCR in tolerized T cells engaged with surface-bound CD3 Abs and higher levels of Cbl-b, compared with Th1 cells (Figures S6A–S6D). There was a negative correlation between PKCθ and Cbl-b co-localizing in activated T cells for Th1 and tolerized T cells (Figure 6G). Significantly, PKCθ is just downstream of signaling from CD28 and PI3K, and both PI3K and PKC are repressed by Cbl-b (Figure 6F). PKCθ is also a direct target of PMA, and Ras proteins are indirect targets of PMA, thereby accounting for the ability of PMA to overcome the block in signaling imposed by CTLA4, PD-1, and Cbl-b (Figure 6F). Furthermore, a previous study found that ubiquitination of the p85 subunit of PI3K by Cbl-b blocks co-association of CD28 and the TCR, and thereby suppresses TCR signaling (Fang and Liu, 2001).
Figure 6.

Altered Immunological Synapse Morphology and Reduced TCR-Proximal Signaling in Tolerant T Cells
T cells were tolerized by either intra-nasal (IN) delivery of peptides (A–E and G) or sub-cutaneous injection of peptides (F).
(A–C) Confocal microscopy of Th1-like cells or IN-tolerized T cells coupled with bone marrow APCs presenting cognate peptide for 10 min and imaged by immunofluorescence confocal microscopy following immune-labeling for (A) CD28, (B) PKCθ, or (C) Zap70.
(D) Quantification of confocal microscopy images showing mean enrichment of PKCθ and Zap70 at the T cell-APC interface as in (B) and (C). Values were derived from at least three biological replicates. p values were calculated by Mann-Whitney test, error bars are SEM.
(E) Western blot analyses of PKCθ T538 phosphorylation in Tg4 Th1 or IN-tolerant T cells following activation with cross-linked anti-CD3/CD28 for the indicated amounts of time.
(F) Model depicting TCR and CD28 signaling pathways that can be antagonized when Cbl-b is activated by CTLA4 or PD-1, or bypassed by PMA.
(G) TIRF microscopy of Cbl-b and PKCθ enrichment at the T cell immunological synapse in Th1-like cells or tolerized T cells.
Scale bars: 10 μM.
Discussion
A Two-Step Model Accounting for T Cell Tolerance
Increasing evidence shows that overactive immune responses in allergy and autoimmunity can be corrected by Ag-specific immunotherapy. We established that cells exposed to soluble peptide epitopes become anergic and switch from a pro-inflammatory phenotype to an anti-inflammatory Tr1-like phenotype (Burkhart et al., 1999, Sundstedt et al., 2003). These Foxp3−ve T cells are anergic and capable of suppressing other immune cells through secretion of IL-10, which inhibits the Ag-presenting machinery of APCs such as dendritic cells (Gabrysová et al., 2009). In the current study, our genomic analyses utilized an in vivo model to answer many of the outstanding questions about the epigenetic mechanisms underlying and maintaining tolerance. Building upon published data, we propose a comprehensive inter-connected model explaining T cell tolerance: (1) the increased activity of inducible inhibitory receptors lowers TCR/CD28 signaling strength below the threshold required for induction of many immune response genes; and (2) epigenetic reprograming of inhibitory receptor genes allows them to be induced at a lower threshold of TCR/CD28 signaling than that required for immune response genes (Figure 7). Therefore, immune response genes, such as Il2, Csf2, Tnf, Cd40lg, Nr4a3, and Ccl1, can be rapidly induced in naive T cells and in effector T cells, but not in tolerized T cells. The activation of TCR signaling in the absence of CD28 co-stimulation also leads to loss of activation of Il2, Csf2, and Tnf (Yukawa et al., 2020), consistent with suppression of CD28 signaling in tolerized cells. Some of the immune response genes induced in naive T cells, such as Nr4a3 (Figure 3F), are not epigenetically primed, and thus require an elevated threshold of signaling for their induction. Conversely, epigenetic priming creates an accessible chromatin environment at immuno-suppressive genes such as Ctla4 and Il10, meaning that they can still be induced in tolerized cells in the presence of reduced levels of TCR/CD28 signaling (Figure 7). However, these genes are not yet primed in naive T cells, meaning that their original epigenetic state was altered after the first round of activation by epigenetic priming at DHSs in close proximity to inducible enhancers and promoters, allowing more efficient induction as described by us previously for memory T cells and TB cells (Bevington et al., 2016).
Figure 7.

A Two-Step Model Accounting for T Cell Tolerance
In a normal T cell immune response, efficient co-activation of TCR and CD28 signaling leads to strong activation of inducible TFs and immune response genes. In tolerized T cells, inhibitory receptors activate repressive factors, such as the ubiquitin ligase Cbl-b, which weaken but do not eliminate TCR/CD28 signaling. This reduced level of signaling is sufficient to epigenetically prime and activate inhibitory receptor genes, but not most immune response genes.
Inhibitory Receptors Suppress TCR/CD28 Signaling in Tolerized Cells
We demonstrated that tolerization leads to both high steady-state levels and higher inducible levels of inhibitory receptors. The nature of the suppressive pathways downstream of most inhibitory receptors, including TIM3, LAG3, and TIGIT, still remains poorly understood. However, it is known that both CTLA4 (Li et al., 2004, Li et al., 2019) and PD-1 (Fujiwara et al., 2017) mediate activation of the repressive ubiquitin ligase Cbl-b, and our data suggest a role for Cbl-b in limiting genomic responses in the tolerant state. Cbl-b functions in setting the threshold of T cell activation and is essential for both anergy (Tang et al., 2019, Zhang et al., 2002) and the development of inducible regulatory T cells (Qiao et al., 2013). In its absence, the response to TCR signaling is uncoupled from co-receptor signaling, and mice develop spontaneous auto-immunity (Bachmaier et al., 2000). In the anergic state associated with tolerance and exhaustion, Cbl-b represses TCR/CD28 signaling by: (1) targeting the p85 subunit of PI3K for ubiquitination and blocking its association with CD28 (Fang and Liu, 2001); (2) directing ubiquitination of PKCθ and targeting it for degradation (Heissmeyer et al., 2004); and (3) directing the ubiquitination of PLCγ1, blocking its activation (Jeon et al., 2004; Figure 7). Conversely, CD28 signaling promotes ubiquitination and degradation of Cbl-b in effector T cells (Zhang et al., 2002). Furthermore, effector T cells normally express Satb1, which represses Pdcd1 (PD-1) expression (Stephen et al., 2017). Our data show that Pdcd1 activation in tolerized T cells occurs in parallel with repression of Satb1, and we confirmed that suppressive pathways involving Cbl-b have been triggered in tolerized cells. Others have also recently demonstrated that PD-1 engagement in T cells leads to loss of expression of genes that rely on a high threshold of signaling, such as Il2, Il3, Csf2, Ccl1, and Cd40lg (Shimizu et al., 2020). Consistent with these findings, we demonstrate here that anergy in tolerized T cells involves a membrane-proximal block in cell signaling because Zap70 and PKCθ fail to localize efficiently at the immune synapse, thus failing to induce AP-1 proteins efficiently. Furthermore, we show that the block can be largely bypassed using PMA to directly activate DAG-inducible pathways downstream of PKCθ and Ras (Figure 2F).
The Inducible Gene-Regulatory Network Is Rewired in Tolerized T Cells
Our data establish that activation of repressive pathways during tolerization leads to loss of both epigenetic priming and inducible activation of many immune response genes. Our genome-wide analyses demonstrate that the TCR/CD28-inducible gene-regulatory network is rewired compared with both naive T cells and normal effector T cells after tolerization. It is likely that much of the epigenetic reprogramming described for TB and memory T cells is either aborted or erased as part of the tolerization process. Indeed, tolerized cells successfully prime only 3% of the pDHSs defined by us previously in memory T cells.
The altered signaling network present in tolerized T cells activates different TF networks compared with the ones detected in naive T cells in both resting and activated cells. Maf and Prdm1 levels were elevated in resting tolerant cells, and we observed much higher inducible activation of Nfil3, Maf, and Prdm1 in tolerant cells than in naive T cells. Maf expression correlates directly with Il10 expression in tolerized cells (Burton et al., 2014) and consistent with these findings, Gabryšová et al. (2018) have shown that c-Maf controls IL-10 production in Th1, Th2, and Th17 subsets. Here we show that induction of tolerance involves the opening of chromatin at known c-Maf binding sites (Figure 2), and that the tDHSs are enriched for AP-1 motifs where c-Maf is able to bind with other AP-1 proteins (Kataoka et al., 1994). Furthermore, NFIL3, c-Maf, and Blimp1 are all likely to be important drivers of anergy and immunosuppression because they were previously identified as regulators of Il10, Havcr2, Tigit, and/or Pdcd1 expression in either T cells suppressed by IL-27 (Chihara et al., 2018, Zhu et al., 2015) or TILs derived from acute myeloid leukemia patients (Zhu et al., 2017). These observations were confirmed using regulatory T cells lacking Blimp1 (Cretney et al., 2018). Overexpression of c-Maf is partially sufficient to induce the exhausted T cell program, and Maf-deficient T cells are more efficient at targeting tumors in vivo (Giordano et al., 2015).
The gene-regulatory network that is activated in Ag-stimulated tolerant cells has diverged from that detected in naive T cells or TB cells. By integrating data identifying the TF motifs present within the NAg- or TAg-specific iDHSs with protein expression levels and foot-printing analyses, we have been able to infer a model of how signaling to TFs is de-regulated during tolerization. This includes: (1) a suppression of signaling to AP-1 and NF-κB, (2) an increased utilization of NFAT in the absence of AP-1, and (3) an increased occupancy of composite AP-1/IRF elements. Indeed, increased binding of AP-1/IRF may occur through the suppression of certain AP-1 family members. The AP-1/IRF complex has been shown to contain BATF, JunD, and/or JunB, but not c-Fos (Glasmacher et al., 2012, Li et al., 2012). Although we observed a reduction in c-Fos, FosB, and JunB protein levels in TAg cells, the level of JunD remained stable, which is of interest here because JunD can function to suppress T cell responses (Meixner et al., 2004). The data also imply redistribution of AP-1 proteins away from conventional NFAT/AP-1 sites, which are typically bound by Jun/Fos heterodimers (Chen et al., 1998) and regulate immune response genes such as CSF2 (Johnson et al., 2004), to AP-1/IRF sites found at immune-modulatory genes, such as Il10 and Nrp1. By comparing our data with published data, we found that TAg-specific DHSs were associated with a gain of BATF and IRF4 binding and a loss of JunB, which is consistent with our model. Furthermore, these same sets of TFs have been implicated in rewiring gene-regulatory networks in exhausted CD8 T cells ,where BATF, IRF4, and NFAT bound at the same sites at the immuno-regulatory genes Lag3, Havcr2, Tigit, Ctla4, and Pdcd1 (Man et al., 2017), and in Tr1 cells, where IRF1 and BATF are key for preparing the chromatin landscape for induction of the Tr1 gene program (Kröger, 2017).
The Tolerized State Shares Features with Anergic and Exhausted T Cells
Our previous genome-wide mRNA analyses (Burton et al., 2014) and the data described here demonstrate that tolerized T cells have moved away from the normal effector T cell program and closer to that of the anergic state. For example, 43% of the 1,033 tDHSs identified here in tolerized CD4 cells were also reprogrammed in a chronic LCMV (lymphocytic choriomeningitis virus) infection model of CD8 T cell exhaustion, including the Ctla4 and Il10 loci, whereas most of the epigenetic priming seen in memory T cells is absent (Sen et al., 2016). Tolerized cells resemble, but are not identical to, other anergic states, including exhausted T cells produced during chronic infections, exhausted TILs, T cells rendered anergic by unbalanced TCR/CD28 signaling, and in vitro-derived Tr1 cells (Figure S7). In each case a concerted program of inhibitory receptor gene expression is activated, which typically includes Lag3, Havcr2 (TIM3), Tigit, Ctla4, and Prdm1 (Blimp1) (Mognol et al., 2017, Singer et al., 2016, Wherry and Kurachi, 2015), which oppose the functions of co-activators such as CD28 (Chen and Flies, 2013). These anergic states also involve activation of a gene regulation network driven by NFAT binding independently of AP-1 (Martinez et al., 2015), and we find that certain AP-1 family proteins are downregulated in tolerized cells.
We also observed some differences between our in vivo-generated tolerant T cells and some other models of anergy. Studies of exhausted T cells from TILs (Mognol et al., 2017) and in vitro-derived tolerant T cells (Liu et al., 2019) found evidence that TCR signaling to NR4A family TFs is maintained, whereas we found that this pathway was suppressed in tolerized T cells. We also observed a loss of LEF/TCF consensus binding motifs in TAg DHSs, whereas TCF-1 plays an important role in controlling the differentiation of precursors that give rise to exhausted T cells during chronic infections (Chen et al., 2019). A direct comparison of mRNA data described above with two independent studies of anergic T cells suggests that tolerized cells, Tr1 cells, and CD8+ve TILs all upregulate expression of TIM3, LAG3, Blimp1, and NFIL3. However, tolerant and Tr1 cells preferentially express Maf, IL-10, and IL-21, whereas tolerant T cells and TILs preferentially express PD-1 (Figure S7).
The sum of these studies reveals that different anergic and regulatory T cell states do indeed share the common utilization of immuno-suppressive factors by virtue of preferential epigenetic priming of their genes, thereby rendering them more responsive. The deficiency of the immune response is a direct consequence of inhibitory receptors blocking induction of most inducible genes. These studies also give encouragement for the expanded use of Ag-inducible tolerization as a strategy for combatting auto-immune disorders, and give clues as to how immune responses might be boosted in cancer and chronic infections. Significantly, this is no longer a distant goal, because peptide therapy is already being tested in auto-immune diseases. Our recent phase 1 and 2 clinical trials show that the MBP peptide tolerization strategy described above is giving positive results as a therapy for multiple sclerosis (Chataway et al., 2018), and a similar protocol using thyrotropin receptor peptides has delivered improvements in patients with Graves’ hyperthyroidism (Pearce et al., 2019). The current study has now provided a comprehensive understanding of the molecular basis of this therapeutic approach, which may one day become common practice.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-c-Jun Rabbit monoclonal | Cell Signaling Technologies | Cat# 9165; RRID: AB_2130165 |
| anti-c-Fos Rabbit monoclonal | Cell Signaling Technologies | Cat# 2250; RRID: AB_2247211 |
| anti-FosB Rabbit Monoclonal | Cell Signaling Technologies | Cat# 2251; RRID: AB_2106903 |
| anti-JunB Rabbit Polyclonal | Santa Cruz Biotechnology | Cat# sc-46X; RRID: AB_2130022 |
| anti-JunD Rabbit Polyclonal | Santa Cruz Biotechnology | Cat# sc-74X; RRID: AB_2130711 |
| anti-Beta 2 Microglobulin. | Abcam | Cat# ab75853; RRID: AB_1523204 |
| anti-PKCθ | BD BioScience | Cat# 610089; RRID:AB_397496 |
| anti-PKCθ pT538 polyclonal | Cell Signaling Technologies | Cat# 9377; RRID:AB_2172071 |
| biotinylated anti-CD3ε | BD BioScience | Cat# 553059; RRID:AB_394592 |
| biotinylated anti-CD28 | BD BioScience | Cat# 553296; RRID:AB_394765 |
| anti-Zap70 | Cell Signaling Technologies | Cat# 3165; RRID:AB_2218656 |
| anti-CD28 | Abcam | Cat# ab25234; RRID:AB_470416 |
| anti-Cbl-b polyclonal H121 | Santa Cruz | Cat# sc-8006; RRID: AB_2070711 |
| goat anti-rabbit IgG-DyLight488 | Jackson Immunoresearch | Cat# 305-486-006; RRID:AB_2339508 |
| donkey anti-mouse IgG-Cy3 | Jackson Immunoresearch | Cat# 715-165-150; RRID:AB_2340813 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MBP Ac1-9[4Y] 90% Purity (AcASQYRPSQR) | GL Biochem Shanghai | Custom product |
| Streptavidin | Jackson Immunoresearch | Cat# 016-000-084; RRID:AB_2337233 |
| LPS | Sigma Aldrich | Cat# L8274 |
| PFA | Electron Microscopy Sciences | Cat# 15710. |
| Murine rIL-12 | Peprotech | Cat# 210-12P80H |
| Human rIL-2 | R&D Systems | Cat# 202-IL |
| Murine rGM-CSF | Mitenyi Biotech | Cat# 130-094-043 |
| DNase I | Worthington | DPPF Grade |
| Calcium Ionophore A23187 | Sigma Aldrich | Cat# C7522 |
| Phorbol 12-myristate 13-acetate | Sigma Aldrich | Cat# P8139 |
| Critical Commercial Assays | ||
| MagniSort Mouse CD4 T cell Enrichment Kit | Thermofisher | Cat# 8804-6821-74 |
| CD4+ T cell Isolation Kit II | Miltenyi Biotech | Cat# 130-095-248 |
| PicoPure RNA Isolation Kit | Thermofisher | Cat# KIT0204 |
| Superscript IV reverse transcriptase | Thermofisher | Cat# 18090010 |
| Applied Biosystems SYBR green master mix | Thermofisher | Cat# 4309155 |
| NEBNext rRNA depletion kit | NEB | Cat# E6310L |
| NEBNext Ultra II kit | NEB | Cat# E7760S |
| NEBNext oligonucleotides | NEB | Cat# E7335S |
| NextSeq® High Output kit v2.5 150 cycles | Illumina | Cat# 20024907 |
| NextSeq® 500/550 High Output kit v2 75 cycles | llumina | Cat# FC 404-2005 |
| Kapa Hyper-Prep kit | Kapa Biosystems | Cat# KK8500 |
| Kapa Library Quantification Kit | Kapa Biosystems | Cat# KK4824 |
| MinElute Gel Extraction Kit | QIAGEN | Cat# 2860 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE147268 |
| TCF1 ChIP - thymocytes | (Dose et al., 2014) | GEO: GSE46662 |
| c-MAF ChIP-seq - Th17 cells | (Ciofani et al., 2012) | GEO: GSM1004799 |
| NFAT-CA-RIT-NFAT1, mock NFAT1, NFAT-CA-RIT-NFAT1 PI, mock NFAT1 PI in CD8 T cells | (Martinez et al., 2015) | GEO: GSM1570758 |
| IRF4 ChIP-seq in CD4 T cells and BATF in CD4 +IL-21 T cells | (Li et al., 2012) | GEO: GSE39756 |
| JUNB ChIP-seq in CD4 TB PI cells, H3K4me2 and H3K27ac ChIP-seq in CD4 TB and TB PI cells, and DNase I in CD4 TB and CD4 TB PI | (Bevington et al., 2016) | GEO: GSE67443 |
| p65 ChIP-seq in Tconv cells stimulated with CD3/CD28 | (Oh et al., 2017) | GEO: GSE99319 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tg4-H2u | (Liu et al., 1995) | https://pubmed.ncbi.nlm.nih.gov/7584132 |
| Mouse: B10.PL mice (B10.PL-H2uH2-T18a/(73NS) SnJ | Jackson Laboratory | |
| Oligonucleotides | ||
| Oligonucletide primers are listed in Table S6 | N/A | |
| Software and Algorithms | ||
| TopHat (version 2.1.1) | (Kim et al., 2013) | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTSeq-count (version 0.9.1) | (Anders et al., 2015) | https://pypi.org/project/HTSeq/ |
| DESeq2 (version 1.18) | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| EdgeR (version 3.24) | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| R (version 3.4.3) | The R project for Statistical Computing | https://www.r-project.org/ |
| Bowtie2 v2.2.3 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACs version 1.4.2 | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| MACS2 callpeak (Galaxy Version 2.1.1) | (Zhang et al., 2008) | https://usegalaxy.org/ |
| HOMER v4.9.1 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/index.html |
| Java Treeview v1.1 | (Saldanha, 2004) | https://sourceforge.net/projects/jtreeview/ |
| Bedtools (Galaxy Version 2.26.0.0) | (Quinlan and Hall, 2010) . | https://bedtools.readthedocs.io/en/latest/ |
| Trimmomatic v0.32 | (Bolger et al., 2014) | http://www.usadellab.org/cms/index.php?page=trimmomatic |
| Wellington Method - pyDNase 0.2.4 | (Piper et al., 2013) | https://pythonhosted.org/pyDNase/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Peter Cockerill (p.n.cockerill@bham.ac.uk).
Material Availability
This study did not generate any unique reagents.
Data and Code Availability
The accession number for the data reported in this paper is GEO: GSE147268. The article also includes previously published datasets: TCF1 ChIP-Seq in thymocytes – GSE46662 (Dose et al., 2014), c-MAF ChIP-seq in Th17 cells - GEO: GSM1004799 (Ciofani et al., 2012), NFAT-CA-RIT-NFAT1, WT NFAT1, NFAT-CA-RIT-NFAT1 PI, WT NFAT1 PI in CD8 T cells - GEO: GSM1570758 (Martinez et al., 2015). IRF4 ChIP-seq in CD4 T cells and BATF in CD4 +IL-21 T cells GEO: GSE39756 (Li et al., 2012), JUNB ChIP-seq in CD4 TB PI cells, H3K4me2 and H3K27ac ChIP-seq in CD4 TB and TB PI cells, and DNase I in CD4 TB and CD4 TB PI, GEO: GSE67443 (Bevington et al., 2016) and p65 ChIP-seq in Tconv cells stimulated with CD3/CD28 - GEO: GSE99319 (Oh et al., 2017).
Experimental Model and Subject Details
Tg4 transgenic Mice
Tg4-H2u mice expressing the αβ TCR (Vα4, Vβ8.2) of the MBP Ac1-9-specific hybridoma 1934.4, derived from an encephalitogenic T cell clone have been described previously (Liu et al., 1995). Tg4 mice have a skewed CD4+ T cell repertoire where over 90% of the CD4+ T cells are Vβ8+. Male and female mice aged between 6-12 weeks were used. Animals were housed under specific pathogen-free conditions, and experiments were performed in accordance with the UK Home Office Project License held by D.C.W. and approved by the University of Birmingham ethical review committee. T cells from the peripheral blood of Tg4 mice were phenotyped for CD4 and Vβ8 by flow cytometry.
Cells
Th1 effector cells were generated in vitro by culturing splenocytes, from which red blood cells were depleted, from Tg4 mice with 10 μg/ml MBP Ac1-9 [4K] and 5 ng/ml rmIL-12 (Peprotech) for 72 hours in complete RPMI. Cells were further expanded for 6-8 days in complete RPMI supplemented with 20 U/ml rhIL-2 (R&D) (Hill et al., 2013). Splenocytes from tolerized mice were also expanded in vitro before analysis by culturing for 5 days in complete RPMI with 10 μg/ml MBP Ac1-9 [4K] and 20 U/ml rhIL-2 (Anderson et al., 2005). Bone marrow-derived dendritic cells were generated from B10.PL mice (B10.PL-H2u H2-T18a/(73NS) SnJ originally from the Jackson Laboratory) by culturing the non-adherent fraction of disaggregated bone marrow with 10 ng/ml rmGM-CSF (Miltenyi Biotec) in complete RPMI for 10-12 days (Inaba et al., 1992). BM-DC were routinely > 85% MHC-II+ as measured by flow cytometry.
Method Details
Induction of tolerance by sub-cutaneous injection of peptides
For most of the tolerized T cell experiments described here (T0 and TAg), tolerance was induced by administering 200 μL doses of the MBP Ac1-9[4Y] subcutaneously in the flank of un-anaesthetized mice every 3-4 days for 7 doses. The amount of peptide was increased until the maximum dose was reached and maintained, as defined in Figure 1C (0.08 μg, 0.8 μg, 8 μg, 80 μg peptide/0.2ml PBS). For N0, 200 μL of PBS was administered in the same dosing strategy. 2 hours prior to sacrifice a final dose of 200 μL of PBS (N0 and T0) or 200 μL of 400 μg/ml MBP Ac1-9 [4Y] (NAg and TAg) was given. For T0M, mice were sacrificed 3 weeks after the final dose.
Induction of tolerance by intra-nasal peptides
In Figures 6A–6G and S6, Tg4 mice were tolerized by intranasal administration of 10 doses of 80 μg MBP Ac1-9 [4Y]) at 3-4 day intervals (Gabrysová et al., 2009, Sundstedt et al., 2003).
CD4+ T cell Selection
For most experiments CD4+ T cells were purified using the MagniSort Mouse CD4 T cell Enrichment Kit (8804-6821-74) according to the manufacturer’s instructions. For the western blotting in Figure 6E and microscopy experiments Figures 6 and S6, CD4+ T cells were purified by magnetic separation (Miltenyi Isolation Kit II) according to the manufacturer’s instruction.
In vitro activation of T cells
For western blot analyses, CD4+ T cells were activated by incubating with 10 μg/ml biotinylated anti-CD3ε (clone 145-2C11, BD) and 10 μg/ml biotinylated anti-CD28 (clone 37.51, BD) for 30 minutes followed by crosslinking with 10 μg/ml streptavidin (Jackson Immunoresearch) at 37°C for the indicated times. For mRNA and DNase-Seq analyses, CD4+ T cells were re-suspended in complete RPMI with 20 ng/ml phorbol myristate acetate (PMA) and 2 μM Calcium Ionophore A23187 (CaI) (NPI and TPI). For the mRNA time course cells were harvested at 30’, 60’, 120’, 240’ and 480’. For DNase-Seq and RNA-seq experiments cell were harvested at 120’.
Western blot analyses of protein expression
Proteins were extracted in 50 mM Tris, 120 mM NaCl, 1 mM EDTA with 1% IGEPAL CA-630 and protease and phosphatase inhibitor cocktails (Thermo) before SDS-PAGE and western blotting. The antibodies used in this study were anti-PKCθ clone 27/PKCθ (BD Bioscience), anti-PKCθ pT538 ployclonal (Cell Signaling Technologies #9377) , anti-c-Jun Rabbit monoclonal (Cell Signaling Technologies #9165), anti-c-fos Rabbit monoclonal (Cell Signaling Technologies #2250), anti-fosB Rabbit monoclonal (Cell Signaling Technologies #2251), anti-JunB Rabbit polyclonal (Santa Cruz Biotehnology #sc-46X) anti-JunD Rabbit polyclonal (Santa Cruz Biotehnology #sc-74X) and anti-beta-2-microglobulin (Abcam #AB75853).
Microscopy sample preparation
Before imaging, BM-DC were incubated with 1 μg/ml LPS (Sigma) for 16-18 hours, washed and then incubated for 2 hours with 1 μg/ml MBP Ac1-9[4Y]. DC-T cell couples for confocal microscopy were prepared by combining activated and peptide-loaded BM-DC and magnetically enriched CD4+ T cell at a ratio of 1:2 at 3x106 cells/ml. Coupling was synchronized by brief centrifugation (75 g) in a round bottom plate and the cells were allowed to interact for 5 minutes at 37°C before the cells were gently transferred to pre-warmed glass slides and incubated for a further 5 minutes. Cells were fixed in 4% buffered PFA before immune-labeling. For TIRF-M imaging, glass coverslips (Mat-Tek II) were pre-coated with 2 μg/ml anti-CD3ε clone 145-2C11, BD) and 1 μg/ml anti-CD28 (clone 37.51, BD). CD4+ T cells were allowed to interact with the antibody coated surface for 8 minutes at 37°C before fixation and immune-labeling. Primary antibodies used were anti-PKCθ clone 27/PKCθ (BD Bioscience) at 2.5 μg/ml, anti-Zap70 clone D1C10E (Cell Signaling Technologies) at 2.5 μg/ml, anti-CD28 clone PV.1 (Abcam) at 1 μg/ml and anti-Cbl-b polyclonal H121 (Santa Cruz - discontinued) at 5 μg/ml. Secondary antibodies, goat anti-rabbit IgG-DyLight488, donkey anti-mouse IgG-Cy3 and goat anti-Armenian hamster-Cy2. (Jackson Immunoresearch) were used at 2.5 μg/ml.
Microscopy
Confocal laser scanning microscopy was performed on a DMI 6000 inverted epifluorescence microscope equipped with an SP5-AOBS confocal scanning system (Leica Microsystems). Total internal reflection fluorescence microscopy (TIRF-M) was performed on a DMI 6000 inverted epifluorescence microscope attached to an AM TIRF MC system (Leica). An estimated penetration depth of 100 nm was used. Multi-color imaging was performed using sequential laser scanning. Conjugates between APC and T cells were identified using the bright field channel without reference to fluorescence channels and laser power, gain, offset and averaging/additive functions were kept constant between samples which were to be compared. Image analysis was performed with Volocity 5 (Perkin Elmer). Analysis of synaptic protein enrichment in DC-T cell couples were performed by measuring the fluorescence intensity of the T cell membrane-proximal region at areas of the interface and distal from the interface. The enrichment of a protein at the interface was represented as the ratio if these values. In TIRF-M experiments, protein accumulation at the interface was assessed by measuring the relative fluorescence intensity within the cell footprint, defined by bright field images. Where indicated, the ratio of two fluorescence channels is presented in pseudo color using the ‘Ratio’ tool in Volocity 5.
mRNA isolation and quantitative PCR
Total RNA was isolated using PicoPure RNA Isolation Kit (KIT0204) according to the manufacturer’s instructions. RNA was reverse transcribed using Superscript IV reverse transcriptase (Thermo Fisher) according to the manufacturer’s protocol. qPCR was carried out using Applied Biosystems SYBR green master mix (Thermo Fisher). Primers used for qPCR are listed in Table S6.
RNA-seq library preparation
Ribosomal RNA was depleted using NEBNext rRNA depletion kit (E6310L) according to the manufacturer’s instructions. RNA-seq libraries were prepared from 30 ng RNA using the NEBNext Ultra II kit (E7760S) using the NEBNext oligonucleotides (E7335S) according to the manufacturer’s instructions. Samples were sequenced using the NextSeq® High Output kit v2.5 150 cycles (Illumina, 20024907).
DNase I hypersensitive site analysis
DNase I digestions were carried out as described previously (Bevington et al., 2016). In brief, cells were re-suspended at 1x107 cells/ml in DNase I buffer (60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 100 mM Tris-HCl pH 7.4, 1 mM EGTA pH 7.4, 0.3 M sucrose, 0.2% NP40, 1 mM CaCl2 and DNase I). 1x106 cells were digested for 3′ at 22°C before the reaction was terminated by the addition of SDS (final concentration 0.5%). Samples were treated with 0.5 mg/ml Proteinase K at 37°C overnight followed by 0.2 mg/ml RNase A for 1 hour. DNA was purified by phenol/chloroform extraction and fragments were separated by agarose gel electrophoresis. Small DNA fragments (50-250 bp) were extracted and purified (QIAGEN Minelute Gel extraction Kit) before library preparation.
Library preparation
DNase I libraries were prepared using the Kapa Hyper-Prep kit (KAPA Biosystems) according the manufacturer’s instructions. Libraries were amplified by PCR and fragments of 200-300bp were gel purified (QIAGEN Minelute Gel extraction Kit) before qPCR validation and quantification (KAPA Library Quantification Complete kit -ABI Prism®). Samples were pooled and sequenced using the NextSeq® 500/550 High Output kit v2 75 cycles (Illumina, FC 404-2005).
Quantification and Statistical Analysis
RNA-seq Analysis
Paired-end reads were aligned to the mouse genome (version mm9, build 37) using TopHat (version 2.1.1) (Kim et al., 2013, Kim and Salzberg, 2011, Langmead et al., 2009, Trapnell et al., 2009). Transcript counts were calculated with HTSeq-count (version 0.9.1) (Anders et al., 2015) using gene models from Ensembl as reference. Differential gene expression analysis was carried out using the DESeq2 (version 1.18) (Love et al., 2014) and EdgeR (version 3.24) (McCarthy et al., 2012, Robinson et al., 2010) packages in R (version 3.4.3). Only genes that could be reproducibly identified as statistically significant by both DESeq2 (FDR < 0.05) and EdgeR (p < 0.05) were retained for further analysis. This was done to reduce the number of false positive results and ensure that the gene sets used in downstream analyses were robust. Genes that had a fold-change of at least 2 (as calculated by DESeq2) were deemed to be differentially expressed. PCA and heatmap plots were generated using ggplot in R.
Definition of gene expression groups
The 460 T0 and 80 N0 specific gene mRNAs were defined as 2-fold upregulated between T0 and N0 and N0 and T0 respectively with a minimum read count of 50. The 138 TAg and 226 NAg specific genes were defined as 2 fold upregulated between TAg and NAg and NAg and TAg respectively with a minimum read count and 50. In addition these genes were filtered to only include genes which were 2 fold upregulated when T0 or N0 were stimulated with Ag.
DNase-Seq - alignment, coverage and peak detection
Raw sequencing reads were aligned to mm9 using bowtie2 (Galaxy Version 2.3.2.2) (Langmead and Salzberg, 2012) with the preset –very-sensitive-local. Coverage files were generated using MACs version 1.4.2 using –g mm –keep-dup auto –w -S as parameters (Zhang et al., 2008).
Normalization of DNase-Seq datasets
The BAM files from the duplicate samples of N0, T0, NAg and TAg were merged using BamTools (Galaxy Version 0.0.2). A master set of peaks were determined using MACS2 callpeak (Galaxy Version 2.1.1). 69743 summits were identified across all samples. The sequence tags ± 200bp from the peak summit were counted for the 69743 peaks for each individual sample using the annotatePeaks function of the HOMER package (Heinz et al., 2010). The samples were normalized using a correction factor based on the median of the top 25,000 peaks for each sample. Correction factors were then used to normalize genome browser scales, average sequence tag density plots and contrast levels in heatmaps.
Unions of DNase-Seq data
For each sample the sequence tags were counted ± 200bp for the 69743 peaks and the significant peaks were determined using a cut off which excluded background and insignificant peaks. Two samples were compared by merging the peaks using the sort and merge function of the bedtools package (Quinlan and Hall, 2010)to give a union which included common and unique peaks. For the unions between T0 and N0 peaks were further filtered to eliminate sine elements. The sequence tags were counted ± 200bp from the summits of the peaks in the union, normalized using the correction factor and the fold change difference was calculated between the 2 samples. The data were visualized as sequence tag density profiles ordered according to the fold change difference in sequence tag density of one sample compared to the other.
Sequence tag density profiles
Sequence tag density profiles were generated using the annotatePeaks function of the HOMER package using -hist 10 -ghist -size 2000 as parameters. Images were generated using Java Treeview v1.1 (Saldanha, 2004).
Average sequence tag density plots
Average sequence tag density plots ± 1 kb around the DHS summit were generated using the annotatePeaks function of the HOMER package using -hist 10 -size 2000 as parameters.
Differential peak analysis using DESeq2
Only peaks which were present in both samples for each condition were used in analyses. Reads were counted using featurecounts and DESeq2 (Galaxy version 2.11.40.6) was used to determine differential peaks between 2 conditions. Only regions with p < 0.05 were included.
RNA fold change heatmaps
Peaks were assigned to the closest gene within 100 kb of the transcription start site (TSS) using the annotatePeaks.pl function in Homer v4.9.1 (Heinz et al., 2010). The fold-change values for these genes were then plotted as a heatmap using Java TreeView v1.1 (Saldanha, 2004).
Distance analyses
The distance between the DHSs and the TSSs of upregulated genes was calculated using the BEDTools closestBed function (Galaxy Version 2.26.0.0). Genes were grouped according to distance from the DHSs to the TSSs of the genes in intervals.
Motif discovery
De novo motif analysis was performed using the findMotifsGenome.pl function of the HOMER package. Motifs were identified ± 100 bp from the peak summit.
Footprinting analysis
Raw sequencing data from high-depth DNase-Seq experiments were processed to remove low-quality reads and sequencing adaptors with Trimmomatic v0.32 (Bolger et al., 2014). The processed reads were then aligned to the mouse genome (version mm9) with Bowtie2 v2.2.3 (Langmead and Salzberg, 2012) using the option–very-sensitive-local. Footprints were identified using the Wellington method implemented in pyDNase 0.2.4 (Piper et al., 2013). DNase I cut profiles were calculated using the in-built functions in pyDNase.
Acknowledgments
This study was supported by the Medical Research Council (UK). We thank David Bending and Constanze Bonifer for their input into the manuscript. We thank Genomics Birmingham at the University of Birmingham for help with DNA sequencing.
Author Contributions
S.L.B., S.T.H.N., and G.J.B. performed the experiments; S.L.B., S.T.H.N., and P.K. analyzed the data; and S.L.B., D.C.W., and P.N.C. conceived the study and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: June 9, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107748.
Contributor Information
David C. Wraith, Email: d.wraith@bham.ac.uk.
Peter N. Cockerill, Email: p.n.cockerill@bham.ac.uk.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P.O., Sundstedt A., Yazici Z., Minaee S., O’Neill E.J., Woolf R., Nicolson K., Whitley N., Li L., Li S. IL-2 overcomes the unresponsiveness but fails to reverse the regulatory function of antigen-induced T regulatory cells. J. Immunol. 2005;174:310–319. doi: 10.4049/jimmunol.174.1.310. [DOI] [PubMed] [Google Scholar]
- Anderson A.C., Joller N., Kuchroo V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmaier K., Krawczyk C., Kozieradzki I., Kong Y.Y., Sasaki T., Oliveira-dos-Santos A., Mariathasan S., Bouchard D., Wakeham A., Itie A. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- Bevington S.L., Cauchy P., Piper J., Bertrand E., Lalli N., Jarvis R.C., Gilding L.N., Ott S., Bonifer C., Cockerill P.N. Inducible chromatin priming is associated with the establishment of immunological memory in T cells. EMBO J. 2016;35:515–535. doi: 10.15252/embj.201592534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevington S.L., Cauchy P., Cockerill P.N. Chromatin priming elements establish immunological memory in T cells without activating transcription: T cell memory is maintained by DNA elements which stably prime inducible genes without activating steady state transcription. BioEssays. 2017;39:1600184. doi: 10.1002/bies.201600184. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignall R., Cauchy P., Bevington S.L., Gorman B., Pisco A.O., Bagnall J., Boddington C., Rowe W., England H., Rich K. Integration of Kinase and Calcium Signaling at the Level of Chromatin Underlies Inducible Gene Activation in T Cells. J. Immunol. 2017;199:2652–2667. doi: 10.4049/jimmunol.1602033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart C., Liu G.Y., Anderton S.M., Metzler B., Wraith D.C. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: a role for IL-10. Int. Immunol. 1999;11:1625–1634. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- Burton B.R., Britton G.J., Fang H., Verhagen J., Smithers B., Sabatos-Peyton C.A., Carney L.J., Gough J., Strobel S., Wraith D.C. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nat. Commun. 2014;5:4741. doi: 10.1038/ncomms5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chataway J., Martin K., Barrell K., Sharrack B., Stolt P., Wraith D.C., ATX-MS1467 Study Group Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology. 2018;90:e955–e962. doi: 10.1212/WNL.0000000000005118. [DOI] [PubMed] [Google Scholar]
- Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Glover J.N., Hogan P.G., Rao A., Harrison S.C. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–48. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- Chen Z., Ji Z., Ngiow S.F., Manne S., Cai Z., Huang A.C., Johnson J., Staupe R.P., Bengsch B., Xu C. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity. 2019;51:850–855.e5. doi: 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara N., Madi A., Kondo T., Zhang H., Acharya N., Singer M., Nyman J., Marjanovic N.D., Kowalczyk M.S., Wang C. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. 2018;558:454–459. doi: 10.1038/s41586-018-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M., Madar A., Galan C., Sellars M., Mace K., Pauli F., Agarwal A., Huang W., Parkhurst C.N., Muratet M. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente-Casares X., Blanco J., Ambalavanan P., Yamanouchi J., Singha S., Fandos C., Tsai S., Wang J., Garabatos N., Izquierdo C. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016;530:434–440. doi: 10.1038/nature16962. [DOI] [PubMed] [Google Scholar]
- Cretney E., Leung P.S., Trezise S., Newman D.M., Rankin L.C., Teh C.E., Putoczki T.L., Gray D.H., Belz G.T., Mielke L.A. Characterization of Blimp-1 function in effector regulatory T cells. J. Autoimmun. 2018;91:73–82. doi: 10.1016/j.jaut.2018.04.003. [DOI] [PubMed] [Google Scholar]
- Dose M., Emmanuel A.O., Chaumeil J., Zhang J., Sun T., Germar K., Aghajani K., Davis E.M., Keerthivasan S., Bredemeyer A.L. β-Catenin induces T-cell transformation by promoting genomic instability. Proc. Natl. Acad. Sci. USA. 2014;111:391–396. doi: 10.1073/pnas.1315752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D., Liu Y.C. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat. Immunol. 2001;2:870–875. doi: 10.1038/ni0901-870. [DOI] [PubMed] [Google Scholar]
- Fujiwara M., Anstadt E.J., Clark R.B. Cbl-b Deficiency Mediates Resistance to Programmed Death-Ligand 1/Programmed Death-1 Regulation. Front. Immunol. 2017;8:42. doi: 10.3389/fimmu.2017.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrysová L., Nicolson K.S., Streeter H.B., Verhagen J., Sabatos-Peyton C.A., Morgan D.J., Wraith D.C. Negative feedback control of the autoimmune response through antigen-induced differentiation of IL-10-secreting Th1 cells. J. Exp. Med. 2009;206:1755–1767. doi: 10.1084/jem.20082118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabryšová L., Alvarez-Martinez M., Luisier R., Cox L.S., Sodenkamp J., Hosking C., Pérez-Mazliah D., Whicher C., Kannan Y., Potempa K. c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells. Nat. Immunol. 2018;19:497–507. doi: 10.1038/s41590-018-0083-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano M., Henin C., Maurizio J., Imbratta C., Bourdely P., Buferne M., Baitsch L., Vanhille L., Sieweke M.H., Speiser D.E. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J. 2015;34:2042–2058. doi: 10.15252/embj.201490786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E., Agrawal S., Chang A.B., Murphy T.L., Zeng W., Vander Lugt B., Khan A.A., Ciofani M., Spooner C.J., Rutz S. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012;338:975–980. doi: 10.1126/science.1228309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissmeyer V., Macián F., Im S.H., Varma R., Feske S., Venuprasad K., Gu H., Liu Y.C., Dustin M.L., Rao A. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- Hill E.V., Oakley C.M., Ng T.H.S., Burton B.R., Wraith D.C. Glycogen synthase kinase-3 regulates IL-10 production in Th1 cells. Immunology. 2013;140:144. [Google Scholar]
- Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M.S., Atfield A., Venuprasad K., Krawczyk C., Sarao R., Elly C., Yang C., Arya S., Bachmaier K., Su L. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–177. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Johnson B.V., Bert A.G., Ryan G.R., Condina A., Cockerill P.N. Granulocyte-macrophage colony-stimulating factor enhancer activation requires cooperation between NFAT and AP-1 elements and is associated with extensive nucleosome reorganization. Mol. Cell. Biol. 2004;24:7914–7930. doi: 10.1128/MCB.24.18.7914-7930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwacz K., Miraldi E.R., Pokrovskii M., Madi A., Yosef N., Wortman I., Chen X., Watters A., Carriero N., Awasthi A. Critical role of IRF1 and BATF in forming chromatin landscape during type 1 regulatory cell differentiation. Nat. Immunol. 2017;18:412–421. doi: 10.1038/ni.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K., Noda M., Nishizawa M. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol. Cell. Biol. 1994;14:700–712. doi: 10.1128/mcb.14.1.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Salzberg S.L. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröger A. IRFs as competing pioneers in T-cell differentiation. Cell. Mol. Immunol. 2017;14:649–651. doi: 10.1038/cmi.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Gál I., Vermes C., Alegre M.L., Chong A.S., Chen L., Shao Q., Adarichev V., Xu X., Koreny T. Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J. Immunol. 2004;173:7135–7139. doi: 10.4049/jimmunol.173.12.7135. [DOI] [PubMed] [Google Scholar]
- Li P., Spolski R., Liao W., Wang L., Murphy T.L., Murphy K.M., Leonard W.J. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. 2012;490:543–546. doi: 10.1038/nature11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Gong L., Gu H. Regulation of immune system development and function by Cbl-mediated ubiquitination. Immunol. Rev. 2019;291:123–133. doi: 10.1111/imr.12789. [DOI] [PubMed] [Google Scholar]
- Liu G.Y., Fairchild P.J., Smith R.M., Prowle J.R., Kioussis D., Wraith D.C. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- Liu X., Wang Y., Lu H., Li J., Yan X., Xiao M., Hao J., Alekseev A., Khong H., Chen T. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567:525–529. doi: 10.1038/s41586-019-0979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macián F., García-Cózar F., Im S.H., Horton H.F., Byrne M.C., Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- Man K., Gabriel S.S., Liao Y., Gloury R., Preston S., Henstridge D.C., Pellegrini M., Zehn D., Berberich-Siebelt F., Febbraio M.A. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity. 2017;47:1129–1141.e5. doi: 10.1016/j.immuni.2017.11.021. [DOI] [PubMed] [Google Scholar]
- Martinez G.J., Pereira R.M., Äijö T., Kim E.Y., Marangoni F., Pipkin M.E., Togher S., Heissmeyer V., Zhang Y.C., Crotty S. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity. 2015;42:265–278. doi: 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo L., Cunha A.P., Madi A., Beynon V., Yang Z., Alvarez J.I., Prat A., Sobel R.A., Kobzik L., Lassmann H. IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain. 2016;139:1939–1957. doi: 10.1093/brain/aww113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meixner A., Karreth F., Kenner L., Wagner E.F. JunD regulates lymphocyte proliferation and T helper cell cytokine expression. EMBO J. 2004;23:1325–1335. doi: 10.1038/sj.emboj.7600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mognol G.P., Spreafico R., Wong V., Scott-Browne J.P., Togher S., Hoffmann A., Hogan P.G., Rao A., Trifari S. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl. Acad. Sci. USA. 2017;114:E2776–E2785. doi: 10.1073/pnas.1620498114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Garra A., Vieira P. T(H)1 cells control themselves by producing interleukin-10. Nat. Rev. Immunol. 2007;7:425–428. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- Oh H., Grinberg-Bleyer Y., Liao W., Maloney D., Wang P., Wu Z., Wang J., Bhatt D.M., Heise N., Schmid R.M. An NF-κB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity. 2017;47:450–465 e5. doi: 10.1016/j.immuni.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill E.J., Day M.J., Wraith D.C. IL-10 is essential for disease protection following intranasal peptide administration in the C57BL/6 model of EAE. J. Neuroimmunol. 2006;178:1–8. doi: 10.1016/j.jneuroim.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce S.H.S., Dayan C., Wraith D.C., Barrell K., Olive N., Jansson L., Walker-Smith T., Carnegie C., Martin K.F., Boelaert K. Antigen-Specific Immunotherapy with Thyrotropin Receptor Peptides in Graves’ Hyperthyroidism: A Phase I Study. Thyroid. 2019;29:1003–1011. doi: 10.1089/thy.2019.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper J., Elze M.C., Cauchy P., Cockerill P.N., Bonifer C., Ott S. Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res. 2013;41:e201. doi: 10.1093/nar/gkt850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pot C., Jin H., Awasthi A., Liu S.M., Lai C.Y., Madan R., Sharpe A.H., Karp C.L., Miaw S.C., Ho I.C., Kuchroo V.K. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 2009;183:797–801. doi: 10.4049/jimmunol.0901233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao G., Zhao Y., Li Z., Tang P.Q., Langdon W.Y., Yang T., Zhang J. T cell activation threshold regulated by E3 ubiquitin ligase Cbl-b determines fate of inducible regulatory T cells. J. Immunol. 2013;191:632–639. doi: 10.4049/jimmunol.1202068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncarolo M.G., Gregori S., Bacchetta R., Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr. Top. Microbiol. Immunol. 2014;380:39–68. doi: 10.1007/978-3-662-43492-5_3. [DOI] [PubMed] [Google Scholar]
- Sabatos-Peyton C.A., Verhagen J., Wraith D.C. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr. Opin. Immunol. 2010;22:609–615. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha A.J. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Schwartz R.H. T cell anergy. Annu. Rev. Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- Sen D.R., Kaminski J., Barnitz R.A., Kurachi M., Gerdemann U., Yates K.B., Tsao H.W., Godec J., LaFleur M.W., Brown F.D. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K., Sugiura D., Okazaki I.-M., Maruhashi T., Takegami Y., Cheng C., Ozaki S., Okazaki T. PD-1 Imposes Qualitative Control of Cellular Transcriptomes in Response to T Cell Activation. Mol. Cell. 2020;77:937–950.e6. doi: 10.1016/j.molcel.2019.12.012. [DOI] [PubMed] [Google Scholar]
- Singer M., Wang C., Cong L., Marjanovic N.D., Kowalczyk M.S., Zhang H., Nyman J., Sakuishi K., Kurtulus S., Gennert D. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell. 2016;166:1500–1511.e9. doi: 10.1016/j.cell.2016.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen T.L., Payne K.K., Chaurio R.A., Allegrezza M.J., Zhu H., Perez-Sanz J., Perales-Puchalt A., Nguyen J.M., Vara-Ailor A.E., Eruslanov E.B. SATB1 Expression Governs Epigenetic Repression of PD-1 in Tumor-Reactive T Cells. Immunity. 2017;46:51–64. doi: 10.1016/j.immuni.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundstedt A., O’Neill E.J., Nicolson K.S., Wraith D.C. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J. Immunol. 2003;170:1240–1248. doi: 10.4049/jimmunol.170.3.1240. [DOI] [PubMed] [Google Scholar]
- Tang R., Langdon W.Y., Zhang J. Regulation of immune responses by E3 ubiquitin ligase Cbl-b. Cell. Immunol. 2019;340:103878. doi: 10.1016/j.cellimm.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-10 production by effector T cells: Th1 cells show self control. J. Exp. Med. 2007;204:239–243. doi: 10.1084/jem.20070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E.J., Ha S.J., Kaech S.M., Haining W.N., Sarkar S., Kalia V., Subramaniam S., Blattman J.N., Barber D.L., Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Wraith D. Autoimmunity: Antigen-specific immunotherapy. Nature. 2016;530:422–423. doi: 10.1038/nature17300. [DOI] [PubMed] [Google Scholar]
- Xu J., Yang Y., Qiu G., Lal G., Wu Z., Levy D.E., Ochando J.C., Bromberg J.S., Ding Y. c-Maf regulates IL-10 expression during Th17 polarization. J. Immunol. 2009;182:6226–6236. doi: 10.4049/jimmunol.0900123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukawa M., Jagannathan S., Vallabh S., Kartashov A.V., Chen X., Weirauch M.T., Barski A. AP-1 activity induced by co-stimulation is required for chromatin opening during T cell activation. J. Exp. Med. 2020;217:e20182009. doi: 10.1084/jem.20182009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Bárdos T., Li D., Gál I., Vermes C., Xu J., Mikecz K., Finnegan A., Lipkowitz S., Glant T.T. Cutting edge: regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J. Immunol. 2002;169:2236–2240. doi: 10.4049/jimmunol.169.5.2236. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C., Sakuishi K., Xiao S., Sun Z., Zaghouani S., Gu G., Wang C., Tan D.J., Wu C., Rangachari M. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 2015;6:6072. doi: 10.1038/ncomms7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Kong Y., Zhang J., Claxton D.F., Ehmann W.C., Rybka W.B., Palmisiano N.D., Wang M., Jia B., Bayerl M. Blimp-1 impairs T cell function via upregulation of TIGIT and PD-1 in patients with acute myeloid leukemia. J. Hematol. Oncol. 2017;10:124. doi: 10.1186/s13045-017-0486-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the data reported in this paper is GEO: GSE147268. The article also includes previously published datasets: TCF1 ChIP-Seq in thymocytes – GSE46662 (Dose et al., 2014), c-MAF ChIP-seq in Th17 cells - GEO: GSM1004799 (Ciofani et al., 2012), NFAT-CA-RIT-NFAT1, WT NFAT1, NFAT-CA-RIT-NFAT1 PI, WT NFAT1 PI in CD8 T cells - GEO: GSM1570758 (Martinez et al., 2015). IRF4 ChIP-seq in CD4 T cells and BATF in CD4 +IL-21 T cells GEO: GSE39756 (Li et al., 2012), JUNB ChIP-seq in CD4 TB PI cells, H3K4me2 and H3K27ac ChIP-seq in CD4 TB and TB PI cells, and DNase I in CD4 TB and CD4 TB PI, GEO: GSE67443 (Bevington et al., 2016) and p65 ChIP-seq in Tconv cells stimulated with CD3/CD28 - GEO: GSE99319 (Oh et al., 2017).