

Abstract
Alzheimer’s disease (AD) is a debilitating disorder characterized by age-related dementia, which has no effective treatment to date. β-Amyloid depositions and hyperphosphorylated tau proteins are the main pathological hallmarks, along with oxidative stress, N-methyl-d-aspartate (NMDA) receptor-mediated excitotoxicity, and low levels of acetylcholine. Current pharmacotherapy for AD only provides symptomatic relief and limited improvement in cognitive functions. Many molecules have been explored that show promising outcomes in AD therapy and can regulate cellular survival through different pathways. To have a vivid approach to strategize the treatment regimen, AD physiopathology should be better explained considering diverse etiological factors in conjunction with biochemical disturbances. This Review attempts to discuss different disease modification approaches and address the novel therapeutic targets of AD that might pave the way for new drug discovery using the well-defined targets for therapy of the disease.
Keywords: proteoglycans, sirtuins, apolipoprotein, immune system, RanBP9, PrPC
The neurodegenerative disorder Alzheimer’s disease (AD) accounts for almost 70% of the cases of dementia. It is primarily diagnosed in older people with symptoms of memory loss and behavioral and cognitive abnormalities.1 Over 46 million people worldwide have dementia.2 Likely, the number will almost double every 20 years, to 81.1 million in 2040. The current total estimated worldwide healthcare cost related to dementia is US$818 billion, and it is projected that by 2030 it will become a trillion-dollar disease,2,3 having an enormous economic impact.
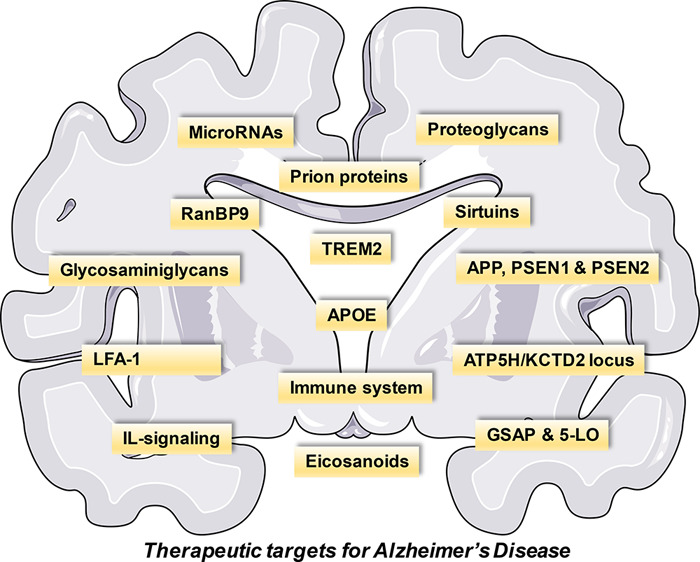
AD is classified on the basis of the age of onset of the condition and whether it is a result of genetic mutation or is developed spontaneously. Early-onset AD is diagnosed before the age of 65 and is mainly caused by genetic mutations that are passed on to a child from a parent.4 Fewer than 10% of all AD patients have a familial type of AD.5 Mutations may arise in one of three genes—amyloid precursor protein (APP), Presenilin 1 (PSEN1), or PSEN2—which over time leads to amyloid plaque formation.4 Several genetic, lifestyle, and environmental factors are said to contribute toward late-onset or sporadic AD.4 This accounts for about 90% of the cases and usually occurs after 65 years of age.5 A variant of the gene ApoE called E4 allele is reported to increase the risk of developing late-onset AD.4 Late-onset AD affects almost half of all people over the age of 85.5 AD is an outcome of multiple pathogenic conditions with inherently complex biology and manifesting mainly as a cognitive deficit. While treating the symptoms, it is of utmost importance to address the cause and find its unknown. Exploring new targets for treating the disease may result in the development of drugs that may impede neurodegeneration. Table 1 summarizes the various disease-modifying strategies along with genetic and immune targets for AD.
Table 1. Overview of Novel Targets for AD.
| target | subcategories | mechanism |
|---|---|---|
| disease modifying strategies | proteoglycans (PGs) and glycosaminoglycans (GAGs) | The 13–16 amino acid region of beta-amyloid (Aβ) is the binding site of PGs and GAGs. PGs and GAGs promote aggregation of Aβ into insoluble amyloid fibrils. |
| sirtuins | Sirtuins act via regulation of serine/threonine Rho kinase ROCK1, known for its role in the inhibition of the non-amyloidogenic α-secretase processing of amyloid precursor protein (APP). Sirtuins are potential modulators of cholesterol biosynthesis. | |
| gamma-secretase activating protein (GSAP) and 5-lipoxygenase (5-LOX) | 5-LOX directly and specifically activates caspase-3, which in turn leads to formation of the GSAP-active fragment, potentiating γ-secretase activity. | |
| C5 bonding in the beta sheets | C5 hydrogen bonds especially stabilize the flat β-sheets of the amyloid state. | |
| genetic targets | APOE | Apo-E (encoded as APOE) brings about binding and internalization of soluble Aβ by glial cells and disruption of Aβ clearance at the blood–brain barrier (BBB) in an isoform-dependent manner (Apo-E4 > Apo-E3 > Apo-E2). |
| APP, PSEN1, and PSEN2 | Mutations on | |
| • Chromosome 21: formation of abnormal APP. | ||
| • Chromosome 14: abnormal presenilin 1 (PSEN1) formation | ||
| • Chromosome 1: abnormal presenilin 2 (PSEN1) formation. | ||
| PSEN1 and PSEN2 are vital components of the γ-secretase complexes responsible for cleavage and release of Aβ. APP is broken down into smaller molecules of Aβ. | ||
| ATP5H/KCTD2 | Potassium conductance alteration and abolition of ATP to maintain neuronal survival during oxygen deprivation. | |
| TREM2 | TREM2 modulates microglial functions, as it facilitates Aβ(1–42) phagocytosis and inhibits Aβ(1–42)-triggered proinflammatory responses. This modulation is dependent on DAP12. TREM2 risk variants impair TREM2 function by decreasing affinity of TREM2 for its natural ligands and affecting its downstream products. Decreased function of TREM2 causes a decrease in phagocytic clearance of amyloid proteins or cellular debris and impairs the survival mechanism, resulting in systemic inflammatory response and neuronal death. | |
| immune system | IL-12/IL-23 signaling | The mechanism is unknown, but IL-12 and IL-23 act via a common subunit p40, which can be targeted using small-molecule therapy. |
| inflammatory eicosanoids | Thromboxane A2 (TXA2)-prostanoid (TP) receptor: increase of APP mRNA stability and enhanced APP expression and Aβ production. | |
| EP1, EP3, and CysLT1 receptors: additional Gα q-linked G protein-coupled receptors, which can modulate APP and Aβ expression. | ||
| LFA-1 integrin | Aβ initiates the transition of LFA-1 from its low-affinity state to its higher-affinity state, thereby enhancing neutrophil adhesion. High-affinity LFA-1 may be critical for neutrophil accumulation and neutrophil-dependent damage during AD. | |
| The engagement of LFA-1 triggers neutrophil extracellular trap (NET) formation, potentially damaging the BBB and neural cells. | ||
| Migrating neutrophils produce IL-17. IL-17 is directly toxic to neurons and the BBB and may recruit more neutrophils. | ||
| RanBP9 | RanBP9 functions as a scaffold upon which APP, BACE1, and LRP are brought together. | |
| It inhibits cell adhesion by accelerating the endocytosis of β1-integrin complexes. It also promotes apoptosis by activating the actin and mitochondria-associated protein cofilin. RanBP9 exerts pro-apoptotic activity by regulating Bcl-2 and Bax protein levels in mitochondria. | ||
| prion proteins | Cellular prion protein (PrPC) is a mediator of Aβ oligomer-induced synaptic dysfunction. | |
Pathophysiology of AD
Several pathogenic conditions are believed to accelerate the progression of the disease, and in the early stages of the disease these factors cause significant destruction of brain areas like the cortex and hippocampus.1 Several hypotheses have been put forward for the pathophysiology of AD, including cholinergic hypothesis, amyloid hypothesis, tau hypothesis, and NMDA excitotoxicity theory.
The degeneration of cholinergic pathways and deposition of beta-amyloid (Aβ) are hallmarks of the disease. In the cholinergic hypothesis, the acetylcholine (ACh) neurotransmitter level is found to be lower, which is mainly due to increased activity of acetylcholinesterase (AchE) enzyme and cholinergic neurodegeneration.6 The levels of ACh are reduced in the cortical and hippocampal regions, which are involved in memory function.6 The pharmacotherapy targeting this hypothesis is the administration of anti-cholinesterase drugs, like Donepezil, Rivastigmine, and Galantamine. Apart from AChE, butyrylcholinesterase (BuChE) is also targeted to improve cognitive dysfunction. Rivastigmine is an example of a drug that inhibits both AChE and BuChE enzymes.
In the amyloid hypothesis, aggregates of Aβ(40–42) peptide lead to further activation of inflammatory mediators like TLR4 and JNK, leading to the death of neurons.6 The enzymes responsible for the breakdown of APP and β- and γ-secretases lead to the formation of insoluble toxic aggregates called Aβ fragments.7 These fragments are considered to be the primary cause of neurodegeneration in AD.6 The enzyme α-secretase acts upon APP under physiological conditions to produce soluble APPα (sAPPα) fragments. These fragments remain within the extracellular space. Other fragments that are formed are the carboxy-terminal 83-amino-acid (C83) fragments, which anchor the plasma membrane.8−10 sAPPα is responsible for the modulation of neuronal excitability, increase in neuronal resistance to metabolic and oxidative stresses, and improvement in synaptic plasticity, learning, and memory.8 During a neuropathological situation, β-secretase-1 (BACE1) cleaves APP into sAPPβ within the extracellular space and a membrane-bound fraction having 99 amino acids (C99). Processing by γ-secretase of the C99 fragment further leads to the formation of either Aβ(1–40) or Aβ(1–42) peptides. These peptides are said to be responsible for the generation of senile plaques8−12 (Figure 1). While sAPPα is beneficial, Aβ peptides may cause alteration of energy metabolism, synaptic loss, decreased neuronal plasticity, induction of oxidative stress, and mitochondrial dysfunction and may trigger disturbances in the cellular calcium homeostasis.8,9 One of the major goals of AD treatment is to bring about clearance or inhibit the formation of these Aβ fragments, along with improving patient’s quality of life, survival, and function. Numerous Aβ-targeted therapeutic strategies are being pursued, including inhibition of Aβ aggregation, modulation of Aβ production, immunotherapy targeted at Aβ, and enhancement of Aβ degradation.13 Many drugs targeting Aβ peptides have failed in clinical trials, and ongoing attempts are being made to address the issue. Many of the genes known to be risk factors of the disease act by increasing the levels of Aβ peptides and bring about neurodegeneration.
Figure 1.
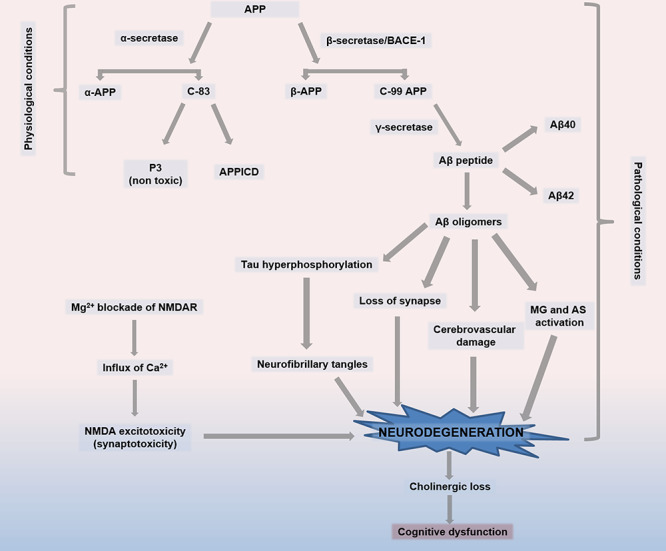
Pathophysiology of Alzheimer’s disease. (1) Formation of beta-amyloid (Aβ) peptides through BACE. Under physiological conditions, APP is acted upon by the enzyme α-secretase to produce sAPPα fragments, which remain in the extracellular space, and carboxy-terminal 83-amino-acid (C83) fragments, which anchor in the plasma membrane. During a neuropathological situation, APP is first preferentially cleaved by BACE, which breaks down APP into sAPPβ in the extracellular space and a 99-amino-acid membrane-bound fraction (C99). Further processing of the C99 fragment by γ-secretase results in the formation of either Aβ(1–40) or Aβ(1–42) peptides, which are thought to be responsible for senile plaque formation. (2) Tau hyperphosphorylation resulting in neurofibrillary tangle formation, loss of synapse, cerebrovascular damage, and MG-AS activation due to Aβ oligomers. (3) NMDA excitotoxicity is caused by Mg2+ blockade of NMDAR, leading to calcium influx and neuronal death. (4) Cholinergic neuronal loss and cognitive dysfunction. Abbreviations: APP, amyloid precursor protein; sAPP-α, soluble fragment formed from APP after cleavage by α-secreatase; sAPP-β, soluble fragment formed from APP after cleavage by β-secreatase; BACE, β-secretase 1; C83, carboxy-terminal 83-amino-acid fragment; C99, carboxy-terminal 99-amino-acid fragment; MG, microglia; AS, astrocytes; NMDAR, N-methyl-d-aspartate receptor.
Tau is a protein that stabilizes the microtubule, particularly in the axons.14 Tau hyperphosphorylation causes neurodegeneration via the formation of neurofibrillary tangles (NFTs), which are insoluble paired helical filaments.6 Hyperphosphorylated tau protein and Aβ plaques are the known hallmarks of AD. Curative therapy of the disease will be called so only if it halts or reverses the disease symptoms in the patient.
Chronic “excitotoxicity” leads to neurodegeneration, which is a result of mild and chronic activation of NMDA receptors. The blockade of the NMDA receptor channel by Mg2+ can be removed even by modest depolarization of the plasma membrane,6,15 triggering a pathological inflow of Ca2+ within the postsynaptic neurons. This Ca2+ overload over a prolonged period leads to the loss of synaptic functioning, followed by synaptotoxicity and cell death.6 Drugs like Memantine, which are administered at a later stage of AD to antagonize NMDA excitotoxicity, provide only symptomatic relief.16
By targeting Aβ plaques and the intracellular tau-containing neurofibrillary tangles, most therapeutic interventions have been focused on the inhibition of the aggregation of these proteins in the brain. Several other targets have also been dissected to explain AD etiology and physiopathology, but none of the attempts brought a convincing outcome. The present review attempts to address the novel therapeutic targets of AD that might pave the way for drug discovery using a known target and provide a successful pharmacotherapy for the complete treatment of the disease.
Disease Modifying Strategies
Proteoglycans and Glycosaminoglycans
Proteoglycans (PGs) play an integral role in the formation of amyloid proteins,17 which includes promoting the formation of insoluble amyloid fibrils due to the accumulation of Aβ.18 These fibrils contribute to the increased neurotoxicity of Aβ.18 Self-aggregating Aβ peptides in vitro freely form amyloid fibrils. But amyloid aggregation and fibril formation are dependent on their interaction with heparin and PGs.18−21 Carbohydrate portions of PGs, along with sulfate moiety in glycosaminoglycans (GAGs), are essential for forming amyloid fibrils.17,18,22 Colocalization of PGs and Aβ within the AD brain has been reported in NFTs and senile plaques.23 The Aβ region of 13–16 amino acids (His-His-Gln-Lys) has been identified as a binding site of PGs and Aβ, representing a distinctive site of a target for inhibiting the formation of the amyloid fibril. Mainly the His13 region is critical for interacting with GAGs.18,24 Formation of β-plated structures can be inhibited by low-molecular-weight heparins (LMWHs), e.g., enoxaparin and dalteparin, and also reverse amyloidosis.25,26 At the same time, it also decreases amyloidosis associated with inflammation.25 Anionic disulfides are said to inhibit Aβ fibril aggregation. These compounds also cross the blood–brain barrier (BBB) and have been shown to have a protective effect against Aβ associated cytotoxicity.27 Heparin oligosaccharides (e.g., neuroparin, CSPG-DS) have inhibitory effects on the assembly of proteoglycans and demonstrate anti-inflammatory activity.28,29 Other drugs interfering with the interaction of GAGs with amyloidogenic proteins include eprodisate and its structural analog, tramiprosate.30 The drugs prevent the polymerization of amyloid fibrils and their deposition in the tissues.30
Sirtuins
The sirtuins are a group of enzymes controlling a range of diverse and fundamental cellular functionings.31 Modulation of sirtuin activities results in the activation of many cellular processes, namely, anti-inflammatory, anti-apoptotic, and anti-stress responses. Sirtuins, at the same time regulate the aggregation of proteins, which are intricately involved in the progression of neurodegenerative disorders.32 Recently, in various models of neurodegeneration, sirtuins were seen to act as disease modifiers.33 SIRT1 overexpression or its pharmacological activation using NAD+ promotes the activity of α-secretase and mitigates Aβ peptide generation, as seen in vitro in Tg2576 embryonic mouse neurons.34 This process takes place via the regulation of serine/threonine Rho-kinase ROCK1, a protein that is well known for its role in inhibiting the non-amyloidogenic α-secretase processing of APP.34
SIRT1 is recognized to regulate the process of cellular cholesterol biosynthesis, demonstrating its role in neuroprotection.34 Culture-based models of AD tauopathy and ALS have shown that activation of SIRT1 by resveratrol, a well-proven antioxidant, promoted survival of neurons.35 Resveratrol has been shown to reduce hippocampal neurodegeneration and prevented learning impairments in an inducible transgenic mouse model of AD tauopathy.36 This process correlates with the decreased acetylation of PGC-1α and p53, which are the known SIRT1 substrates.33 Injecting lentivirus expressing SIRT1 within the hippocampal region of transgenic mice provided substantial protection against neurodegeneration.33 Such studies suggest the positive therapeutic benefits of SIRT1 activators for tauopathies. To date, resveratrol is the only SIRT1 activator for which a double-blind, randomized, and placebo control trial has been carried out.37 Although resveratrol did not provide any significant beneficial outcome as compared to the placebo, it was, however, found to be well tolerated even at very high doses, cross the BBB, and alter the pattern of AD biomarkers.38
Gamma-Secretase Activating Protein and 5-Lipoxygenase
Aβ formation is catalyzed by γ-secretase.39 A novel γ-secretase activating protein (GSAP) was recently discovered, which has been shown to significantly and selectively increase the synthesis of Aβ through mechanisms involving interactions between APP carboxy-terminal fragment (APP-CTF) and γ-secretase.40 Recombinant GSAP stimulates Aβ production in vitro.41 In AD mouse model, knockdown of GSAP reduced the levels of Aβ and plaque development.41 Thus, GSAP can serve as a therapeutic agent with an Aβ-lowering potential to inhibit γ-secretase.39 Imatinib, an anti-cancer agent, is a direct target of GSAP, prevents activation of γ-secretase activity, and was found to reduce Aβ production.42 However, a recent study has shown uncertainty in the role of GSAP and imatinib in regulating Aβ generation and γ-secretase activity.43
The role of the enzyme 5-lipoxygenase (5LO) in the pathogenesis of AD was recently highlighted by demonstrating its association in Aβ formation and its deposition.44−46 Within the GSAP precursor protein sequence, a caspase-3 processing domain was identified, and a piece of experimental evidence for its involvement in the formation of active fragments 16 kDa GSAP along with Aβ peptides biogenesis was shown.47 Further, 5LO was discovered to be an endogenous GSAP formation regulator.45 It is shown to act by specifically and directly activating caspase-3, which was confirmed using transgenic mouse models of AD where 5LO activity was regulated pharmacologically or genetically.46,48 These results provide support toward 5LO as a viable therapeutic target for AD lowering Aβ without having the concomitant toxic effect of the classical γ-secretase inhibitors.41
Approaches to Lower the Impact of Genetic Abnormalities for AD
Majority of the strategies available for AD treatment target either the Aβ pathway or the symptomatic pathology. In the view of rising failure in several clinical trials for drugs targeting Aβ, today, there is a vital necessity to identify new targets and develop other novel therapeutic strategies aimed toward the treatment of AD.49 Another approach seen in the picture is called the genome-wide association study (GWAS).50 This approach is being used to identify regions of interest within the genome, which increases the susceptibility of an individual toward the development of late-onset AD.51 Thirty-three such regions were reported by 2015.50 Recent approaches that include the use of gene and cell therapies in preclinical and clinical studies have shown encouraging results.
APOE
The cause of late-onset AD is not much clear as it does not run in families, although some cases have been reported in several families. It is probably related to variations in one or more genes, as well as lifestyle and environmental factors.4 Apolipoprotein E (Apo-E) is an essential carrier of cholesterol52 and, along with repairing tissue injury, supports the transport of lipids in the brain.49 The gene APOE, which expresses apolipoprotein, has been studied as a risk factor extensively for the disorder.4 An individual’s chances of developing late-onset AD increases with the presence of the apolipoprotein E (APOE) gene on chromosome 19 AD.49
There are several forms of APOE. APOE2 isoform is relatively rare and, in certain instances, provide some sort of defense against the disease.50 The most common allele, APOE3, has a neutral role in the disease progression-neither, reducing, nor elevating the risk. The APOE4 allele raises the risk for AD and is also responsible for the early onset of the condition. An individual can have zero, one, or two APOE4 alleles, amplifying the risk of developing AD with an increasing number of alleles.50
Synthesis of Apo-E primarily takes place in the microglia and astrocytes. It is followed by the lipidation of the protein by ATP-binding cassette A1 (ABCA1) transporter, which helps in the formation of lipoprotein particles.53 Soluble Aβ then binds to the lipidated Apo-E facilitating the uptake of Aβ through several cell-surface receptors, which includes low-density lipoprotein receptors (LDLR), low-density lipoprotein receptor-related protein 1 (LRP1), and the HSPG.53,54 Apo-E brings about binding followed by soluble Aβ internalization by glial cells and disturbance in the clearance of Aβ at the BBB takes place in an isoform-dependent manner (Apo-E4 > Apo-E3 > Apo-E2).55 This impacts cerebral amyloid angiopathy (CAA) pathogenesis, whose risk is elevated by the presence of APOE4 allele.55 This also influences the age-related cognitive decline.56 For delivering lipids, binding of Apo-E lipoproteins takes place with various cell-surface receptors53,54,57 and also to hydrophobic Aβ peptide.58 Aβ peptide aggregation and its clearance in the brain are differentially regulated by the different Apo-E isoforms. Each isoform performs a distinct function for modulating the brain lipid transport, glucose metabolism, neuronal signaling, neuroinflammation, and mitochondrial function.49 AD pathogenesis is known to involve Apo-E4, which takes place via an Aβ-independent mechanism involving cholesterol homeostasis, synaptic plasticity, neuroinflammation, and neurovascular functions49 (Figure 2).
Figure 2.
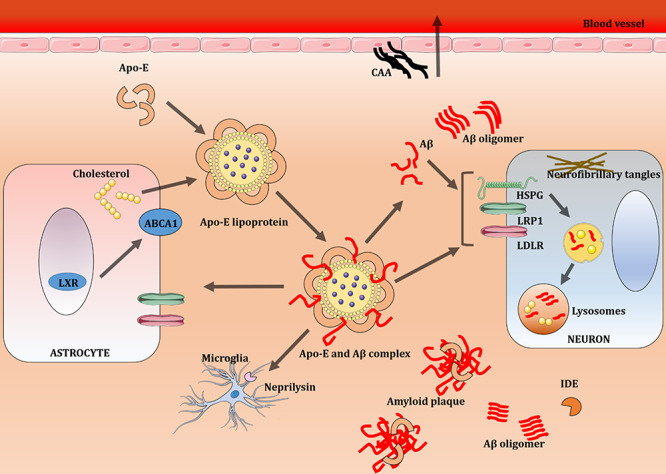
ApoE lipoprotein and Aβ metabolism in the brain. Aβ clearance is brought about by receptor-mediated uptake by glia and neurons, drainage into interstitial fluid, or through the BBB, and also by proteolytic degradation by IDE and neprilysin. Astrocytes and microglia synthesize Apo-E, which is lipidated by the ABCA1 transporter to form lipoprotein particles. This apolipoprotein binds to soluble Aβ and facilitates Aβ uptake through cell-surface receptors like LRP1, LDLR, and HSPG. Apo-E enhances binding and internalization of soluble Aβ by glial cells, disrupts Aβ clearance at the BBB, and influences CAA pathogenesis. Abbreviations: Aβ, beta-amyloid; ABCA1, ATP-binding cassette A1; Apo-E, apolipoprotein E; BBB, blood–brain barrier; CAA, cerebral amyloid angiopathy; HSPG, heparan sulfate proteoglycan; IDE, insulin-degrading enzyme; LDLR, low-density lipoprotein receptor; LRP1, low-density lipoprotein receptor-related protein 1; LXR, liver X receptor.49 Adapted from Servier Medical Art, licensed under a Creative Commons Attribution 3.0 Unported License (https://smart.servier.com/).
Genin et al.59 used the Rochester (USA) incidence data to calculate AD Lifetime Risks (LTR) from 10 132 controls and 7351 cases from Caucasian ancestry. The AD LTR without reference to APOE genotype was 14% in females, while 11% in males at the age of 85. LTR for APOE44 female carriers increased up to 60%, 30% for APOE34 female carriers, 51% for APOE44 male carriers, and 23% for APOE34 male carriers for the same age.59 These results urge a shift of category to “major gene” from the “risk factor” of the APOE gene.59
Numerous approaches have been sought out for targeting APOE. The use of Aβ antibodies that target the Apo-E binding site is one of the approaches.60 Aβ12–28p antibody reduced Aβ accumulation, microgliosis, and tau phosphorylation both in vitro and in vivo.61 CPO_Aβ17–21p, an Aβ12–28p derivative, also decreased Aβ load and its mediated neurotoxicity. This peptoid antibody treatment reduced glial activation and improved both spatial and short-term memory in mice.62 The use of a direct Apo-E antibody has also been suggested. One of the Apo-E antibody, HJ6.3, targets all forms of Apo-E to prevent its binding with Aβ. HJ6.3 is, however, reported to have safety issues that limit its use.63 Recently, anti-human Apo-E4 (HAE4) was developed and found to be more specific than HJ6.3. Both of these antibodies worked by a microglia inflammation pathway to reduce the Aβ plaque load.64
Small molecules that change the structure of Apo-E are also under consideration. Apo-E small-molecule structural correctors (SMSC) changed the structure of Apo-E4 into Apo-E3-like structure, reducing the detrimental effects of Apo-E4 on the pathology.65 PH-002 is one such molecule, which in in vitro studies showed positive results. This molecule reduced Aβ production and decreased tau phosphorylation.65,66 Overexpressing Apo-E2 is another approach, as this allele is said to be the protective allele against AD. The use of gene therapy to express Apo-E2 has shown benefits in preclinical studies.67 Lentiviral mediated Apo-E2 delivery to the hippocampus was able to increase the Apo-E load. However, lentiviral-mediated delivery is less efficient due to its local distribution.68 Adeno-associated virus (AAV) based Apo-E2 delivery was found to be more effective. Hippocampal injection of AAVrh.10hAPOE2 in mice exhibited increased Apo-E2 load along with a reduction in insoluble Aβ42 and Aβ40.69 AAVrh.10hAPOE2 may be closest to being in clinical therapy because of its efficacy, wider distribution, and safety.60
Agonists or antagonists of the nuclear receptors peroxisome proliferator-activated receptor-γ (PPARγ) and liver X receptors (LXRs), which form complexes with Retinoid X receptors (RXRs) and control the expression of Apo-E,70,71 act as potential candidates for modulating Apo-E.49 LXRs, apart from Apo-E, also modulate ABCA1, promoting cholesterol efflux.72 Aβ fibrils deposition requires Apo-E, as demonstrated in mouse amyloid models.73 Interrupting the interaction between Aβ and Apo-E using Aβ-mimicking peptides could decrease Aβ aggregation and deposition, and this approach may be an effective therapy for treating AD.
APP, PSEN1, and PSEN2
Early-onset familial AD accounts for a tiny proportion (<1%) of all AD cases and typically develops before 65 years of age.49 It may be a result of several different single-gene mutations occurring on chromosomes numbers 1, 14, and 21. These mutations lead to the formation of abnormal proteins.50 Mutations on chromosome 21 are responsible for the formation of abnormal APP. Chromosome 14 mutation results in abnormal formation of presenilin 1, and a mutation on chromosome 1 leads to the formation of abnormal presenilin 2.50 PSEN1 and PSEN2 are responsible for the cleavage and release of Aβ.49
APP is broken down into visible clumps of Aβ.6 These clumps or plaques of Aβ are a hallmark of AD.9 Jonsson et al. explored the association between the variants of APP with AD. The most significant association was found with single nucleotide polymorphism (SNP) rs63750847. Allele A of SNP rs63750847-A results in the substitution of alanine by threonine at 673 positions in APP (A673T) and was found to be relatively common in the elderly control group than in the AD group. It is, therefore, protective against AD.74 The amino acid is close to the site where the enzyme BACE1 ordinarily cleaves APP into smaller Aβ moieties—and the alteration is enough to reduce the efficacy of the enzyme.74 Behavioral improvement and reduction in Aβ pathology were observed in the AD mouse model following lentiviral delivery of siRNA, which mediated knockdown of BACE1.75 Pharmaceutical companies have been working on developing “BACE inhibitors” for more than a decade, and several are now under clinical trials. The drug that would mimic the effects of the mutation would have the potential both to prevent AD and to slow down cognitive decline. Downregulating APP using siRNA in APP overexpressing mice by herpes simplex virus (HSV) short hairpin (shRNA) has been shown to reduce Aβ pathology.76 Gene therapy mediated by AAV acts on the proteolytic fragments of APP, which in APP/dE9 mice may have demonstrated neurotrophic effects and improved working memory.77
ATP5H/KCTD2 Locus
Destefano and Gonza78 recognized a novel association within the H+ transporting, ATP synthase, and mitochondrial F0 (ATP5H)/potassium channel tetramerization domain-containing protein 2 (KCTD2) locus in AD.78KCTD2 gene is responsible for functions like DNA transcription,79 voltage-dependent potassium channel function, and GABAB receptor hetero-multimeric composition,80 regulating proteasome physiology81 and degradation of ubiquitinated proteins. This gene is also a member of the KCTD family. The ATP5H gene regulates cell energy production via respiration and is embedded in the third intron of the KCTD2 gene.82 Mitochondrial ATP synthase brings about ATP synthesis and comprises of two multi-subunit complexes, namely, membrane-spanning component, F0, which contains the proton channel and the soluble catalytic core, F1. ATP5H gene encodes the “d” subunit of a total of nine subunits of the Fo complex.83 The oxidative stress hypothesis for AD is well known, and AD cases express a significantly low level of the ATP5H gene encoding subunits.84,85KCTD2, ATP5H, or both are attractive candidates for AD risk86 and therapeutic targets. The ATP5H/KCTD2 expression can be used as a diagnostic tool for late-onset AD. AD is usually diagnosed when the disease progresses to the late stage. Thus, early diagnosis and prophylaxis are of utmost importance. Since mitochondria have a significant role to play in AD, future therapy targeting mitochondrial components like ATP Synthase subunits may have a novel approach in the treatment for AD.87
TREM2
The membrane protein triggering receptor expressed on myeloid cells 2 (TREM2) is expressed in the peripheral tissues by myeloid cells.88 It is encoded by the TREM2 gene and forms a receptor-signaling complex with the tyrosine kinase-binding protein (TYROBP) or DAP12, which triggers in macrophages and dendritic cells the activation of immune responses.89 In microglia, TREM2 brings about signaling in two ways. One of the signaling pathways regulates phagocytosis90 while the other pathway suppresses cytokine production and secretion.91 This may support survival by prompting the secretion of tumor necrosis factor (TNF) through tumor necrosis factor receptor 2 (TNFR2) at levels that potentiate survival and repair pathways.92
Heterozygous rare variants of the TREM2 gene predispose to AD.92TREM2 risk variants impair TREM2 function, which can be by decreasing the affinity of TREM2 to its natural ligands and affecting its downstream products. Reduced function of TREM-2 causes a decrease in phagocytic clearance of amyloid proteins or cellular debris and impairs the survival mechanism, resulting in a systemic inflammatory response and neuronal death.93
Jiang et al.94 demonstrated that during the disease progression, there was an upregulation in the levels of TREM2 in the microglia, which was credited to the increased levels of Aβ(1–42) in the brain. TREM2 was also shown to regulate the functions of microglia by inhibiting Aβ(1–42) triggered pro-inflammatory responses and facilitating Aβ(1–42) phagocytosis in primary cultured microglia where TREM2 was knocked down or overexpressed. Overexpression of TREM2 in brains of APPswe/PS1dE9 mice markedly ameliorated the AD-related neuropathologies, accompanied by an improved spatial cognitive function.94 This suggests that TREM2 upregulation serves as a compensatory response to Aβ(1–42), and by regulating the microglial function, it protects against AD progression.94 TREM2 expression rises in parallel with a rise in Aβ levels in the cortex. Other components of the cascade, such as DAP12, are not dysregulated. Thus, for controlling microglial responses, TREM2 acts as a gateway.92
Antibodies that stimulate the signaling of TREM2 are under development. TREM2 activating antibodies in vitro have been shown to activate ERK, and Ca2+ signaling in human dendritic cells.95 Overexpressing TREM2 has been shown to reduce inflammation and promote phagocytosis.96 Lentiviral mediated increase in TREM2 expression in mice brain attenuated neuropathologic and cognitive alterations.97 However, the use of lentiviral mediated approaches is not favored in humans due to the risk of developing oncogenic transformation.95
Other Possible Targets
Along with the familial AD variants in APP and PSEN1/2, coding variants in BIN1, ABCA7, ADAM10, EPHA1, CD2AP, CLU, CR1, MS4A4A/MS4A6A, SORL1, PICALM, and PLD3 are associated with late-onset AD. They have been identified by targeted sequencing or exome sequencing.98
Targeting Immune System
The immune system plays a major role in the pathogenesis of AD, and this has been supported by findings from studies that reported reduced incidence of the pathology in transgenic AD mice following pharmacological and genetic manipulation of the IL-12/IL-23 signaling pathway.99,100 Modulation of the immune system is widely accepted to influence the progression of the disease, although there is a lack of clarity on whether the process of inflammation is spontaneous or is an associated event of the pathology. Epidemiological studies propose that non-steroidal anti-inflammatory drugs (NSAIDs) can diminish the risk of AD, but numerous prospective placebo-controlled trials evaluating the efficacy of NSAIDs in AD have been futile in demonstrating the same.101,102
Following Aβ release and deposition, certain hallmarks of inflammation have been reported in AD-like proinflammatory cytokines upregulation (tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β)) and generation of reactive oxygen species.103 In AD mice models, it was observed that inhibition of IL-1β or IL-6 signaling did not alter the plaque load while deletion of CD40 ligand (CD40L) or neutralization of prostaglandin E2 resulted in a slight reduction in Aβ plaque load.99 Expression patterns of inflammatory mediators in AD are not well defined104 Microglia, which is closely associated with Aβ plaques, and other glial cells are described to be the primary source of proinflammatory cytokines in many CNS diseases, including AD.103 TNF-α, in particular, has been reported to have a significant influence on neurodegeneration in mice.105,106
Modulating IL-Signaling
AD pathology is marked by an increase of proinflammatory cytokines as a response to Aβ. p40 is an IL-12/IL-23 signaling molecule and a shared subunit of IL-12 and IL-23. Increased production of this subunit p40 by microglia was found in the APPPS1 AD mouse model. The knockout of p40 resulted in decreased cerebral amyloid load. Likewise, a decrease in cerebral amyloid load was observed in APPPS1 mice following the peripheral administration of a neutralizing p40-specific antibody. When p40 antibodies were delivered intracerebroventricularly (icv), there was a significant reduction in the level of soluble Aβ species and a reversal of deficits in cognition in aged APPPS1 mice. In cerebrospinal fluid of AD patients amount of p40 was found to be increased, suggesting that inhibiting the IL-12/IL-23 pathway by targeting the p40 subunit may attenuate AD pathology and its associated cognitive deficits.107 However, the real challenge lies in delivering mAb-based therapy across the BBB. Alternatively, small-molecule inhibitors of IL-12/IL-23 can be easily made to get across the BBB.108
Anti-inflammatory cytokines may have beneficial effects in AD, as demonstrated by various studies. Delivery of IL-2 via AAV viral vector into the brains of APP/PS1dE9 mice reportedly enhanced memory function increased synaptic plasticity and restored spine density.109 AVV-mediated enhanced IL-10 gene expression in APP/PS1 mice hippocampus led to amelioration of cognitive dysfunction, reduced astrogliosis, and promoted neurogenesis.110 However, a similar study carried out in TgCRND8 mice and Tg2576 mice demonstrated opposite effects. The study reported an increase in the plaque load, reduction in synaptic proteins, and memory impairment.111 Delivery of IL-4 AVV viral vector in AAP/PS1 mice hippocampus and frontal cortex at 3 months of age was able to reduce the accumulation of soluble and insoluble Aβ following 3 months of treatment.112 In a different study, IL-4 gene delivery in the hippocampus of AAP/PS1 mice showed increased neurogenesis, improved cognitive function, and suppressed Aβ pathology.75
Inflammatory Eicosanoids
Increased microglial activation observed in areas surrounding the senile plaques in the AD brain results in inflammation.113 The thromboxane A2 (TXA2)-prostanoid (TP) receptor located on the neurons is activated, leading to increased stability of APP mRNA, which increased Aβ production and APP expression.114,115 Activated microglia produce TXA2 leading to an increase in its concentration in the AD brain.116
An important finding is the identification of the CysLT1, EP1, and EP3 receptors as supplementary Gα q-linked G protein-coupled receptors which regulate Aβ and APP expression. The knockout of these receptors leads to a substantial decrease in plaque load in APP transgenic mice.117−119 A similar knockout of PGE2 EP4 receptor or its pharmacological inhibition also reduces plaque burden as observed in an APP transgenic mice model.120 The increase of APP and Aβ in AD due to eicosanoids may be potentially prevented by the inhibiting eicosanoid production. However, the approach has yielded varying results. In culture systems, an approximate 2-fold rise in APP levels was seen following multiple Gα q-linked eicosanoid receptor activations, which is coherent with enhanced levels of APP in AD patient brains, as well as in plaque-bearing 5XFAD mice an increase in APP expression was observed. Limiting the synthesis of eicosanoids in aged 5XFAD mice brings about a reduction in the total levels of APP and decreases the APP fragments with COOH-terminal. This further drives researchers to scrutinize the involvement of these inflammatory mediators along with their receptors toward Aβ plaque pathology.121
Lymphocyte Function-Associated Antigen 1 (LFA-1 Integrin)
Neutrophils are involved in the destruction of tissue during inflammation and also known to be the first in line for defense against inbound pathogens.122−124 Aβ induces adhesion of neutrophils, which is dependent on LFA1, by initiating the shift of LFA-1 to its higher-affinity state from its low-affinity state, thereby increasing adhesion of neutrophil. The high-affinity LFA-1 act as a stop signal for the arrested neutrophils, which are present in the parenchyma, which suggests that high-affinity LFA-1 might be crucial for neutrophil-dependent damage and its accumulation during AD.
LFA-1 integrin blockade or neutrophil depletion at the inception AD had a significant ability to decrease memory deficits and other neuropathological hallmarks in AD-like mouse models.125 This clearly demonstrates the role of neutrophils in the induction of cognitive dysfunction. Also, at the earlier stages of the disease, brief suspension of neutrophils had an effective long-term effect on older animals, which shows that neutrophils are indeed vital for the development of a chronic disorder. Antibodies blocking LFA-1 in mice displayed evidence of memory restoration in behavioral tests as compared to control animals.126 Also, mice that lack LFA-1 integrin showed improved cognition with lesser neuropathological changes as compared to the wild-type mice.126
Silva127 showed that blocking LFA-1 integrin leads to neutrophil depletion and inhibition of neutrophil trafficking, which dramatically reduces the activation of microglial. These two processes may create a series of feedback loops that sustain and amplify their activation.127,128 This hints toward the probable existence of a neutrophil–microglia crosstalk, which formerly has not been hypothesized in AD. The engagement of LFA-1 triggers neutrophil extracellular traps (NET) formation, potentially damaging the neural cells and BBB. Furthermore, confocal microscopy confirmed the presence of NETs within the parenchyma and cortical vessels of patients with AD.129 Evidence from AD patients and AD animal models suggests that targeting NET components, such as DNase I, protein arginine deiminases (PAD), and NADPH oxidase, may help to limit tissue damage.130,131 Migrating neutrophils produce IL-17, which is directly toxic to neurons and BBB and may further recruit more neutrophils.124,132 Therefore, targeting the neutrophil-dependent inflammatory mechanisms may hinder early pathogenesis and, during the disease progression, may protect AD patients from cerebral injury.129
RanBP9
The scaffolding protein RanBP9 interacts with LRP, APP, and BACE1 via its cytoplasmic tails. It acts as a scaffold on top of which APP, BACE1, and LRP are brought together.133 Such interactions of RanBP9 with APP and BACE1 promote APP endocytosis, considerably increases BACE1 cleavage, and generate Aβ fragments in cultured cells and in vivo.134,135 Also, in the brains of AD patients, there is found to be a robust increase of a 60 kDa proteolytic fragment of RanBP9, which strongly accelerates Aβ generation through BACE1 processing of APP.136 Moreover, RanBP9 inhibits adhesion of cells by potentiating the β1-integrin complex endocytosis.137 By activating the actin and mitochondria-associated protein cofilin, it promotes apoptosis.138 A 4-fold increase in RanBP9 proteins was found in mutant APP transgenic mice, and a significant increase in neurodegeneration, gliosis, spatial memory deficits, and increased synapse loss in RanBP9 transgenic mice was observed.138 Woo et al. showed that APP-PS1 mice exhibited a 3.5-fold increase in the levels of RanBP9, and in the same mice, they demonstrated that reduction in RanBP9 protected neuronal cells against synaptic damage, cofilin-actin pathology, Aβ accumulation, and gliosis.139 A study showed that in DNA damage-induced apoptosis, RanBP9 exerted pro-apoptotic activity. It acts by regulating B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X (Bax) protein levels within the mitochondria.140 Physical association with the C-terminal of p73a (a tumor suppressor protein) with RanBP9 has also been reported. This interaction modulates the exogenously expressed p73a levels along with the nuclear translocation of RanBP9.141 Apoptosis can also be induced via nuclear and non-nuclear pathways by p73a.142 Liu et al.143 found that RanBP9 and p73 together induce abnormal changes within the mitochondria and initiate apoptosis that depends on their supportive actions. This demonstrates the critical role of the RanBP9/p73 pathway in maintaining apoptosis in mitochondria mediated manner during neurodegeneration (Figure 3).
Figure 3.
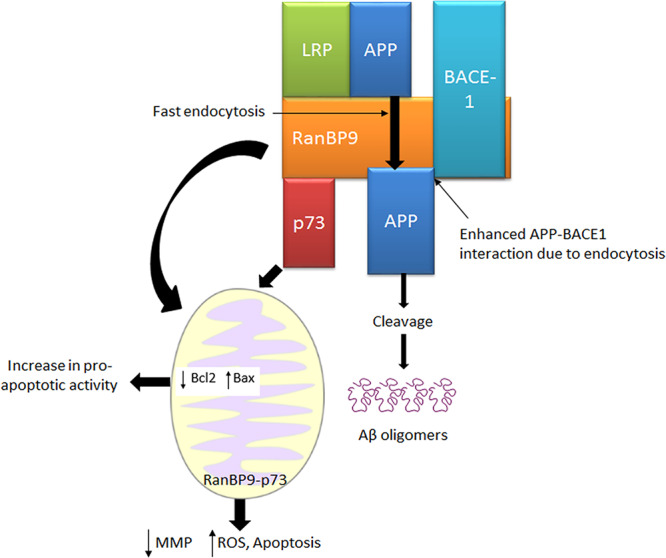
Interactions of RanBP9 with APP, LRP, and BACE1. RanBP9 promotes the endocytosis of APP and considerably increases its BACE1 cleavage to generate Aβ fragments. RanBP9 exerts pro-apoptotic activity by regulating B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X (Bax) protein levels in mitochondria. RanBP9 and p73 together induce abnormal changes in mitochondria (MMP, superoxide levels, apoptotic proteins, and fission) and bring about apoptosis.
Prion Proteins (PrP)
AD progression has been shown to correlate with soluble non-fibrillar forms of Aβ protein.144 These soluble Aβ oligomers (Aβo) block the long-term potentiation (LTP) in the hippocampus at nanomolar concentrations145 and impair rodent spatial memory.146 Antibodies targeting PrP prevent Aβ-oligomer binding to cellular prion proteins (PrPC) and ameliorate synaptic plasticity in hippocampal slices.147 PrPC have demonstrated a high affinity for Aβo. PrPC are highly expressed in the mammalian nervous system and are a glycosyl-phosphatidylinositol-anchored protein located on the plasma membrane.148 Interaction of PrPC with Aβo in AD models has been shown to mediate synaptic loss, aberrant signaling pathway, and decline in cognition.149 This binding has been shown to recruit metabotropic glutamate receptors type 5 (mGluR5) and abnormally activate Fyn kinase to impair synaptic function.150 Salazar et al. studied the effects of PrPC ablation in advanced AD stages in APP/PS1 mice and demonstrated an increase in protein synthesis, which restored neuronal activity. This improvement in neuronal activity was attributed to blockage of increased phosphorylation of eEF2.149 A study by Li and Gotz showed that PrPC deletion had a positive influence on tau hyperphosphorylation as a somatodendritic accumulation of Tau has been linked with Fyn.151 Thus, the Aβo-PrPC-mGluR5 interaction can be a potential therapeutic target for AD as it plays a significant role in Aβo-induced synaptic dysfunction. It was found that the constitutive knockout of PrPC expression in a mouse model of AD upturned several pathological phenotypes152 as did the peripheral treatment with an anti-PrP antibody on the same mouse model.153In vitro data of LTP recordings from PrP null mice and congenic wild-type mice have confirmed the PrPC dependence of the inhibitory effect of Aβ-derived diffusible ligands (ADDLs) on synaptic plasticity.144
However, the ability of a few Aβ preparations to integrate with PrP and form a complex is questioned by one report.154 Different synthetic Aβ preparations lack characterization and have heterogeneity.155 This makes it imperative to state the active PrP-independent, PrP-dependent, and benign species to draw a parallel to their presence with the pathogenesis of AD and its progression. Electron microscopy has revealed that the PrP-binding protofibrils have a triple-helical structure and are called Aβ nanotubes. These are present within the human brain and play a significant role in disease progression. This has been demonstrated by a study that shows that PrP expression is requisite for protofibril synaptotoxicity, and similar results have been demonstrated by water-soluble AD brain extracts blocking LTP in a PrP-dependent manner. Thus, using humanized anti-PrP monoclonal antibodies to target PrP–Aβ nanotube interactions with maybe an essential therapy for AD.156
Multi-Target-Directed Ligand (MTDL)
At present, another area of emerging therapeutics is multi target-directed ligand (MTDL) strategy, which emphasizes targeting multiple enzymes like AChE, BChE, BACE1, and MAO.157 In the future, MTDLs may offer enhanced efficacy against AChE and Aβ plaque formation or offer metal-complexing, neuroprotective, antioxidant, inhibition of glutamate-induced excitotoxicity, voltage-dependent calcium channel antagonistic activity, cannabinoid CB1 receptor antagonism, histamine H3 receptor antagonism, and BACE1 inhibition to nip the evil of disease with multiple arms.158 This will eventually lead to the mitigation of AD symptoms resulting in better therapeutic outcomes and enhanced patient compliance.
MicroRNAs (miRNAs)
miRNAs within the neurons play a vital role in regulating synaptic plasticity, neuronal differentiation, neurite outgrowth, and maintaining dendritic spine morphology.159 Dysfunction of miRNAs is now well recognized in AD, and their use as a therapy for AD is increasing widely. Antagonists or mimics of miRNAs are known to modulate the expression of various genes involved in the pathogenesis of AD.160 miRNAs can be modulated in two ways: first, by inhibiting the functioning of miRNA by using a single-stranded complementary antisense oligonucleotide (ASO), and second, by increasing its expression by administration of compounds that stimulate its secretion or using a double-stranded synthetic oligonucleotide miRNA which mimics the function of the endogenous miRNA.159 Studies from cell cultures and animal models have shown that increased expression of specific miRNAs may inhibit Aβ and tau accumulation by acting at different molecular levels.160
Higaki et al. identified miRNAs from human neuroblastoma cells and murine primary neurons, miR200b and miR200c, to be downregulators of Aβ secretion by regulating the mTOR pathway.161 Overexpression of miR-124 in SHSY5Y cells suppressed the expression of BACE1, while its inhibition increased BACE1 levels.162 Transfecting SHSY5Y cells with miR-15b inhibits BACE1, thereby reducing Aβ.162,163 miR-302/367 has been shown to convert astrocytes into neurons, replacing the dead ones in animal models of AD.164 Neuronal stem cells transfected with miR-9 increased differentiation of stem cells into neurons.165 Improvement in synaptic and cognitive activity was observed in AD mouse models following upregulation of miR-188.166 miR-214 and miR-let-7f-5p have been shown to inhibit autophagy and decrease caspase-mediated apoptosis.167,168 Inhibition of miR-128 in cell cultures decreased Aβ cytotoxicity by inactivating NFκB pathway.169
Several pharmacological compounds can be utilized for modulating miRNA expression. NSAIDs and simvastatin are known to modulate miRNA expression and may prevent the progression of AD.170,171 Natural compounds such as resveratrol and osthole act as potential neuroprotective effects by modulating miRNAs levels.165,172 Recent studies have demonstrated that exosomes may be utilized as therapeutic agents in AD patients to deliver miRNAs or siRNAs.173
The use of miRNAs for therapy of AD patients looks enticing and will be expanded in the coming years with the help of computerized gene analyses. However, limitations in its clinical applicability persist due to difficulty in delivering to the target site and determination of the specificity of miRNA silencing.174 Also, altering the miRNA levels may have an undesirable effect in AD patients as every single miRNA is known to have a plethora of effects on the biological functioning of the body.175
Stem Cells
Stem cell therapy for AD is being explored widely. Both preclinical, as well as clinical trial results, have supported the use of stem cells as a therapy for AD. Stem cells are an attractive means of replenishing neurons that are damaged or lost by means of neurogenesis.176 They provide a ray of hope for developing disease-modifying therapies for AD. The ability of stem cells to proliferate, differentiate, self-renew, and reprogram themselves into different lineages make them excellent candidates for AD therapy.177 Different types of stem cells have been identified, which are useful for AD therapy. These include pluripotent stem cells (induced pluripotent and embryonic stem cells (ESCs)) and adult stem cells (mesenchymal stem cells (MSCs), hematopoietic stem cells (HSCs), neural stem cells (NSCs), and olfactory ensheathing cells (OECs)).176,178Table 2 lists the ongoing clinical trials with stem cells for AD. Although promising, however, scientists are trying to address answers to some technical difficulties that arise with the use of stem cells.176 The time point for stem cell intervention is still under debate as many trials have failed following intervention failure at the appropriate time.176 Gender-based differences exist and have to be taken into account for establishing a proper therapeutic regimen with stem cells.179
Table 2. List of Stem Cells under Clinical Trials for ADa.
| NCT number | type of stem cells | phase | status | place |
|---|---|---|---|---|
| NCT02600130 | HMSCs | 1 | recruiting | United States |
| NCT02833792 | HMSCs | 2 | recruiting | United States |
| NCT02672306 | HUCMSCs | 1 and 2 | active, not recruiting | China |
| NCT01547689 | HUCMSCs | 1 and 2 | unknown | China |
| NCT02054208 | HUCMSCs | 1 and 2 | recruiting | Korea |
| NCT01297218 | HUCMSCs | 1 | completed | Korea |
| NCT00927108 | PSCs | 2 | unknown | Thailand |
| NCT03117738 | HAMSCs | 1 and 2 | active, not recruiting | United States |
| NCT03172117 | HUCMSCs | 1 and 2 | recruiting | Korea |
| NCT02912169 | ADSVFCs | 1 and 2 | withdrawn | United States |
| NCT02899091 | HPMSCs | 1 and 2 | unknown | Korea |
| NCT03724136 | BMSCs | N/A | recruiting | United States/United Arab Emirates |
| NCT01696591 | HUCMSCs | N/A | unknown | Korea |
Abbreviations: HMSCs, human mesenchymal stem cells; HUCMSCs, human umbilical cord mesenchymal stem cells; PSCs, progenitor stem cells; HAMSCs, human adipose mesenchymal stem cells; ADSVFCs, adipose-derived stromal vascular fraction cells; HPMSCs, human placental mesenchymal stem cells; BMSCs, bone marrow stem cells; N/A, not available. Data from clinicaltrials.gov.
Clinical Trials
Results from clinical trials have been disappointing and have pushed scientists toward intervening the disease in its earlier stages. However, this poses a challenge as determining therapeutic benefits in subjects who are healthy in the early stages of the disease becomes difficult.180,181 Keeping in view, a new draft was issued by the U.S. FDA in 2018 for conducting clinical trials in AD. The taxonomy of AD was expanded to contain four stages, namely, Stage 1: Preclinical; Stage 2: Preclinical/Prodromal; Stage 3: Prodromal; Stage 4: Dementia.180 Candidates under clinical trial for AD treatment majorly include therapies that are targeted against Aβ, those which decrease Aβ production, and those which reduce the formation of neurofibrillary tangles.182 Other candidates include 5-HT6 and H3 receptor antagonists, AGE inhibitors, glucagon-like peptide-1 (GLP-1) agonists, calcium channel inhibitors, and TNF-α inhibitors. Table 3 shows a selected list of AD drugs under clinical trials.183 As of 2019, nine trials for eight interventions targeting the amyloid pathway are in phase III, and over 30 interventions targeting the non-amyloid pathway are in phase III.180,184 The number of drugs in clinical trials targeting the amyloid pathway lowered in 2019, as compared to 2018 and 2017. Also, trials now have moved toward the earlier prodromal or preclinical stages of the disease.184
Table 3. List of Selected Anti-AD Drugs in Active Clinical Trialsa.
| NCT number | drug | mechanism | phase |
|---|---|---|---|
| NCT01966666 | TPI-287 | microtubule stabilization | I |
| NCT02991235 | JNJ-63733657 | tau elimination | I |
| NCT03698695 | THN 201 | N/A | I |
| NCT02947893 | Nilotinib | Aβ inhibition and tau stabilization | II |
| NCT03352557 | BIIB092 | tau stabilization | II |
| NCT03790982 | AD-35 | inhibits neuroinflammation | II |
| NCT02292238 | Benfotiamine | slows decline in brain glucose metabolism | II |
| NCT02615002 | Piromelatine | melatonin and serotonin receptor agonist | II |
| NCT01767311 | BAN-2401 | Aβ inhibition | II |
| NCT03417986 | Thiethylperazine (TEP) | Aβ inhibition | II |
| NCT03289143 | RO7105705 | N/A | II |
| NCT02880956 | ABBV-8E12 | tau stabilization | II |
| NCT03367403 | LY3002813 | Aβ inhibition | II |
| NCT01409915 | Sagramostim | immunostimulator | II |
| NCT02756858 | ANAVEX2-73 | muscarinic receptor agonist | II |
| NCT01843075 | Liraglutide | slows decline in brain glucose metabolism and prevents Aβ accumulation | II |
| NCT03518073 | LY3303560 | tau stabilization | II |
| NCT01998841 | Crenezumab | Aβ inhibition | II |
| NCT02788513 | BI 425809 | modulates NMDA receptor function | II |
| NCT03131453 | CNP520 | BACE1 inhibitor | II and III |
| NCT03036280 | Elenbecestat | BACE inhibitor | III |
| NCT02484547 | Aducanumab | Aβ inhibition | III |
| NCT01872598 | Masitinib | reduces neuroinflammation | III |
| NCT02051608 | Gantenerumab | eliminates Aβ | III |
| NCT02008357 | Solanezumab | inhibits Aβ aggregation | III |
As of September 2019. Data from clinicaltrials.gov.
Over the past few years, several promising candidates for AD have failed in clinical trials. Table 4 provides a list of selected drugs that have recently failed in clinical trials. The majority of the candidates who failed were the monoclonal antibodies (MAbs). MAbs such as bapineuzumab, gantenerumab, crenezumab, and solanezumab act by binding to Aβ and neutralizing it, but these agents failed to become a promising therapy due to several reasons.185−189 Bapineuzumab trial was prematurely terminated due to lack of clinical efficacies between ApoE4 carriers and non-carriers. In both the cases, fluctuation of Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-Cog/11) and Disability Assessment for Dementia (DAD) total score at 78 weeks was the primary efficacy endpoint which was not improved when compared with placebo.185 Similarly, gantenerumab clinical trial was stopped earlier due to futility. Groupwise comparison of primary Clinical Dementia Rating-Sum of Boxes (CDR-SB) looked at changes from baseline between placebo and two different doses of drug that were considered as the endpoints. The exploratory analysis for biomarkers suggests a necessary study with higher dose.186 Solanezumab clinical trial failed, as no significant improvement in cognitive outcome was observed with respect to placebo.187 Crenezumab study at phase 2 failed because the primary and secondary endpoints on ADAS-Cog/12 and CDR-SB score were not met.188 Two γ-secretase inhibitors, avagacestat and semagacestat, failed mainly due to lack of any clinical efficacy and reports of worsening of cognitive abilities. The narrow therapeutic window of these agents also deterred their use.190,191 Similar results were reported in the case of BACE inhibitors elenbecestat, atabecestat, verubecestat, and umibecestat.192−195 Neurochemical modulators such as idalopiridine and dimebon, which act by increasing Ach release and inhibiting H1 receptor, respectively, failed due to their lack of efficacy.196,197 Encenicline specifically failed as it caused gastrointestinal toxicity.198 Experts point toward various reasons for the failure of drugs, one of which being targeting the wrong pathological substrates.199 The question still remains whether one should target the plaque or the monomeric, oligomeric or the protofibrillar form of Aβ. Second, the methodology pursued from trial to trial. It is essential to look out for discrepancies that might exist between trials. Third, the timing of the treatment is of importance, and scientists are pushing toward starting treatment at the early stages of the disease. However, the patients selected for clinical trials may sometimes be far too advanced in terms of pathology, and the therapy might be late for such patients.199
Table 4. List of AD Drugs Failed in Clinical Trials.
| treatment strategy | drug | mechanism of action | reason for failure | ref |
|---|---|---|---|---|
| Disease Modifying Treatment | ||||
| Monoclonal Antibodies | gantenerumab | binds to and neutralizes Aβ | lack of clinical efficacy and futility | (186) |
| solanezumab | binds to and neutralizes Aβ | no change found in levels of AD proteins and no significant improvement in cognitive outcome | (187) | |
| crenezumab | converts Aβ from insoluble to a soluble form | no clinical benefits noticed along with primary and secondary end points on ADAS-Cog/12 and CDR-SB score were not improved compared to placebo | (188) | |
| bapineuzumab | binds to and neutralizes Aβ | lack of clinical efficacy as ADAS-Cog/11 and CDR-SB score were not improved in comparison to placebo | (185) | |
| aducanumab | binds to and neutralizes Aβ | inability to meet primary end point | (189) | |
| gamma-secretase inhibitors | semagacestat | inhibits γ-secretase | lack of clinical efficacy and worsening of cognitive abilities | (190) |
| avagacestat | inhibits γ-secretase | narrow therapeutic window | (191) | |
| BACE inhibitors | elenbecestat | inhibits enzyme BACE | unfavorable risk–benefit ratio | (192) |
| verubecestat | inhibits enzyme BACE | lack of clinical efficacy | (193) | |
| atabecestat | inhibits enzyme BACE | elevated liver enzymes in participants | (194) | |
| umibecestat | inhibits enzyme BACE | worsening of cognitive abilities | (195) | |
| Symptomatic Treatment | ||||
| neurochemical modulators | idalopiridine | increases Ach release by inhibiting 5HT6 receptors | lack of clinical efficacy | (196) |
| encenicline | nicotinic Ach receptor agonists | GI toxicity | (198) | |
| dimebon | H1 receptor antagonist | lack of clinical efficacy | (197) | |
Conclusion
Currently, there are no curable treatments available for AD, but several symptomatic treatment regimens like the use of anti-cholinesterase drugs, NMDA antagonists, etc. are available. These treatments available result in a temporary improvement in cognitive functions. Targeting neuronal degeneration directly or its underlying cause would offer a better cure for AD. The targets and intervention strategies discussed above can be explored for the development of new chemical entities using several drug discovery techniques. This may find a lead that may further prove useful in the treatment of the disease and pave the way for novel drug regimens for AD.
Acknowledgments
This work is supported by the Department of Pharmaceuticals, Ministry of Chemical and Fertilizers, Government of India, and National Institute of Pharmaceutical Education and Research (NIPER), Ahmedabad, Gandhinagar, Gujarat, India. The authors also want to express their thanks to the Director, NIPER Ahmedabad, for providing necessary facilities and infrastructure.
Author Contributions
# S.B. and D.S. contributed equally.
The authors declare no competing financial interest.
References
- Begum M.; Biswas K.; Sarker A.; Huq T.; Sarwar A.; et al. (2015) Anticholinesterase and Antioxidant Potentials of a Medicinal Plant Abroma augusta: Implications for the Alternative Treatment Therapy of Cognitive Deficits in Alzheimer’s disease. Clin. Pharmacol. Biopharm. 4, 2. 10.4172/2167-065X.1000148. [DOI] [Google Scholar]
- Prince M., Wilmo A., Guerchet M., Ali G.-C., Wu Y.-T., and Prina M. (2015) World Alzheimer Report 2015, The Global Impact of Dementia: An analysis of prevalence, incidence, cost and trends, https://www.alz.co.uk/research/world-report-2015.
- El-Hayek Y. H.; Wiley R. E.; Khoury C. P.; Daya R. P.; Ballard C.; Evans A. R.; Karran M.; Molinuevo J. L.; Norton M.; Atri A. (2019) Tip of the Iceberg: Assessing the Global Socioeconomic Costs of Alzheimer’s Disease and Related Dementias and Strategic Implications for Stakeholders. J. Alzheimer’s Dis. 70, 323. 10.3233/JAD-190426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan B.; Ezeriņa D.; Amoako T. N.; Riemer J.; Seedorf M.; Dick T. P. (2013) Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat. Chem. Biol. 9, 119. 10.1038/nchembio.1142. [DOI] [PubMed] [Google Scholar]
- The BootsWebMD, In Alzheimer's Disease guide, (2009), U.K.
- Danysz W.; Parsons C. G. (2012) Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. British journal of pharmacology 167, 324–352. 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M.; DeKosky S. T.; Fitzpatrick A. L.; Furtado J. D.; Lopez O. L.; Kuller L. H.; Mackey R. H.; Hughes T. M.; Mukamal K. J.; Jensen M. K. (2018) Apolipoproteins and Alzheimer’s pathophysiology. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 10, 545–553. 10.1016/j.dadm.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C.; Kaether C.; Thinakaran G.; Sisodia S. (2012) Trafficking and proteolytic processing of APP. Cold Spring Harbor Perspect. Med. 2, a006270. 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L.; Selkoe D. J. (2012) Neurotoxicity of Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring Harbor Perspect. Med. 2, a006338. 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello M. A.; Soriano S. (2013) Rational heterodoxy: Cholesterol reformation of the amyloid doctrine. Ageing Res. Rev. 12, 282–288. 10.1016/j.arr.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Castello M. A.; Soriano S. (2014) On the origin of Alzheimer’s disease. Trials and tribulations of the amyloid hypothesis. Ageing Res. Rev. 13, 10–12. 10.1016/j.arr.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Drachman D. A. (2014) The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimer's Dementia 10, 372–380. 10.1016/j.jalz.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Citron M. (2004) Strategies for disease modification in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 677–685. 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- Folch J.; Petrov D.; Ettcheto M.; Abad S.; Sánchez-López E.; García M. L.; Olloquequi J.; Beas-Zarate C.; Auladell C.; Camins A. (2016) Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2016, 8501693. 10.1155/2016/8501693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner N. W.; Dingledine R. (1991) Regulation of hippocampal NMDA receptors by magnesium and glycine during development. Brain Res. Mol. Brain Res. 11, 151–159. [PubMed] [Google Scholar]
- van Marum R. J. (2009) Update on the use of memantine in Alzheimer’s disease. Neuropsychiatric Disease and Treatment 5, 237–247. 10.2147/NDT.S4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow A. D.; Willmer J. P.; Kisilevsky R. (1987) Sulfated glycosaminoglycans in Alzheimer’s disease. Hum. Pathol. 18, 506–510. 10.1016/S0046-8177(87)80036-9. [DOI] [PubMed] [Google Scholar]
- Fraser P. E.; Nguyen J. T.; Chin D. T.; Kirschner D. A. (1992) Effects of sulfate ions on Alzheimer beta/A4 peptide assemblies: implications for amyloid fibril-proteoglycan interactions. J. Neurochem. 59, 1531–1540. 10.1111/j.1471-4159.1992.tb08470.x. [DOI] [PubMed] [Google Scholar]
- Ariga T.; Miyatake T.; Yu R. K. (2010) Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: amyloidogenesis and therapeutic strategies--a review. J. Neurosci. Res. 88, 2303–2315. 10.1002/jnr.22393. [DOI] [PubMed] [Google Scholar]
- Sipe J. D.; Cohen A. S. (2000) Review: history of the amyloid fibril. J. Struct. Biol. 130, 88–98. 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- Cohlberg J. A.; Li J.; Uversky V. N.; Fink A. L. (2002) Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 41, 1502–1511. 10.1021/bi011711s. [DOI] [PubMed] [Google Scholar]
- Castillo G. M.; Lukito W.; Wight T. N.; Snow A. D. (1999) The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J. Neurochem. 72, 1681–1687. 10.1046/j.1471-4159.1999.721681.x. [DOI] [PubMed] [Google Scholar]
- Snow A. D.; Mar H.; Nochlin D.; Kimata K.; Kato M.; Suzuki S.; Hassell J.; Wight T. N. (1988) The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. American journal of pathology 133, 456–463. [PMC free article] [PubMed] [Google Scholar]
- van Horssen J.; Wesseling P.; van den Heuvel L. P.; de Waal R. M.; Verbeek M. M. (2003) Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol. 2, 482–492. 10.1016/S1474-4422(03)00484-8. [DOI] [PubMed] [Google Scholar]
- Kisilevsky R.; Szarek W. A. (2002) Novel glycosaminoglycan precursors as anti-amyloid agents part II. J. Mol. Neurosci. 19, 45–50. 10.1007/s12031-002-0009-3. [DOI] [PubMed] [Google Scholar]
- Kisilevsky R.; Lemieux L. J.; Fraser P. E.; Kong X.; Hultin P. G.; Szarek W. A. (1995) Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer’s disease. Nat. Med. 1, 143–148. 10.1038/nm0295-143. [DOI] [PubMed] [Google Scholar]
- Gervais F.; Chalifour R.; Garceau D.; Kong X.; Laurin J.; Mclaughlin R.; Morissette C.; Paquette J. (2001) Glycosaminoglycan mimetics: a therapeutic approach to cerebral amyloid angiopathy. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis 8, 28–35. [PubMed] [Google Scholar]
- Rose M.; Dudas B.; Cornelli U.; Hanin I. (2003) Protective effect of the heparin-derived oligosaccharide C3, on AF64A-induced cholinergic lesion in rats. Neurobiol. Aging 24, 481–490. 10.1016/S0197-4580(02)00093-3. [DOI] [PubMed] [Google Scholar]
- Borysik A.; Morten I.; Radford S.; Hewitt E. (2007) Specific glycosaminoglycans promote unseeded amyloid formation from β2-microglobulin under physiological conditions. Kidney Int. 72, 174–181. 10.1038/sj.ki.5002270. [DOI] [PubMed] [Google Scholar]
- Gervais F.; Paquette J.; Morissette C.; Krzywkowski P.; Yu M.; Azzi M.; Lacombe D.; Kong X.; Aman A.; Laurin J.; et al. (2007) Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 28, 537–547. 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Herskovits A. Z.; Guarente L. (2013) Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 23, 746–758. 10.1038/cr.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavu S.; Boss O.; Elliott P. J.; Lambert P. D. (2008) Sirtuins [mdash] novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discovery 7, 841–853. 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- Outeiro T. F.; Marques O.; Kazantsev A. (2008) Therapeutic role of sirtuins in neurodegenerative disease. Biochim. Biophys. Acta, Mol. Basis Dis. 1782, 363–369. 10.1016/j.bbadis.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Kim D.; Nguyen M. D.; Dobbin M. M.; Fischer A.; Sananbenesi F.; Rodgers J. T.; Delalle I.; Baur J. A.; Sui G.; Armour S. M.; Puigserver P.; Sinclair D. A.; Tsai L.-H. (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179. 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anekonda T. S. (2006) Resveratrol--a boon for treating Alzheimer’s disease?. Brain Res. Rev. 52, 316–326. 10.1016/j.brainresrev.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Mohan S.; Gobinath T.; Salomy A.; Nisha M.; Kandasamy M.; Essa M. M.; Jayachandran K. S.; Anusuyadevi M. (2018) Biophysical interaction of resveratrol with sirtuin pathway: Significance in Alzheimer’s disease. Front. Biosci., Landmark Ed. 23, 1380–1390. 10.2741/4650. [DOI] [PubMed] [Google Scholar]
- Wong S. Y.; Tang B. L. (2016) SIRT1 as a therapeutic target for Alzheimer’s disease. Rev. Neurosci. 27, 813–825. 10.1515/revneuro-2016-0023. [DOI] [PubMed] [Google Scholar]
- Turner R. S.; Thomas R. G.; Craft S.; Van Dyck C. H.; Mintzer J.; Reynolds B. A.; Brewer J. B.; Rissman R. A.; Raman R.; Aisen P. S. (2015) A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 85, 1383–1391. 10.1212/WNL.0000000000002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J.; Li J.-G.; Hoffman N. E.; Stough A. M.; Madesh M.; Praticò D. (2015) Regulation of gamma-secretase activating protein by the 5Lipoxygenase: in vitro and in vivo evidence. Sci. Rep. 5, 11086. 10.1038/srep11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami M.; Nagashima Y.; Sano Y.; Ishihara S.; Morishima-Kawashima M.; Funamoto S.; Ihara Y. (2009) gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052. 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G.; Luo W.; Li P.; Remmers C.; Netzer W. J.; Hendrick J.; Bettayeb K.; Flajolet M.; Gorelick F.; Wennogle L. P.; Greengard P. (2010) Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98. 10.1038/nature09325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G.; Luo W.; Li P.; Remmers C.; Netzer W. J.; Hendrick J.; Bettayeb K.; Flajolet M.; Gorelick F.; Wennogle L. P.; Greengard P. (2010) Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98. 10.1038/nature09325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain I.; Fabregue J.; Anderes L.; Ousson S.; Borlat F.; Eligert V.; Berger S.; Dimitrov M.; Alattia J.-R.; Fraering P. C.; Beher D. (2013) The role of γ-secretase activating protein (GSAP) and imatinib in the regulation of γ-secretase activity and amyloid-β generation. J. Biol. Chem. 288, 2521–2531. 10.1074/jbc.M112.370924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firuzi O.; Zhuo J.; Chinnici C. M.; Wisniewski T.; Prarico D. (2008) 5-Lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer’s disease. FASEB J. 22, 1169–1178. 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J.; Pratico D. (2011) 5-lipoxygenase as an endogenous modulator of amyloid beta formation in vivo. Ann. Neurol. 69, 34–46. 10.1002/ana.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannopoulos P. F.; Chu J.; Joshi Y. B.; Sperow M.; Li J. L.; Kirby L. G.; Praticò D. (2014) Gene Knockout of 5-Lipoxygenase Rescues Synaptic Dysfunction and Improves Memory in the Triple-Transgenic Model of Alzheimer’s Disease. Mol. Psychiatry 19, 511–518. 10.1038/mp.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chu J.; Li J. G.; Joshi Y. B.; Giannopoulos P. F.; Hoffman N. E.; Madesh M.; Pratico D. (2015) Gamma secretase-activating protein is a substrate for caspase-3: implications for Alzheimer’s disease. Biol. Psychiatry 77, 720–728. 10.1016/j.biopsych.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J.; Lauretti E.; Craige C. P.; Pratico D. (2014) Pharmacological modulation of GSAP reduces amyloid-beta levels and tau phosphorylation in a mouse model of Alzheimer’s disease with plaques and tangles. J. Alzheimer’s Dis. 41, 729–737. 10.3233/JAD-140105. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Kanekiyo T.; Xu H.; Bu G. (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Dept. of Health and Human Services , MedlinePlus, 2017.
- Giri M.; Zhang M.; Lü Y. (2016) Genes associated with Alzheimer’s disease: an overview and current status. Clin. Interventions Aging 11, 665–681. 10.2147/CIA.S105769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoz C.; Schaefer E. J.; Cupples L. A.; Wilson P. W.; Levy D.; Osgood D.; Parpos S.; Pedro-Botet J.; Daly J. A.; Ordovas J. M. (2001) Apolipoprotein E genotype and cardiovascular disease in the Framingham Heart Study. Atherosclerosis 154, 529–537. 10.1016/S0021-9150(00)00570-0. [DOI] [PubMed] [Google Scholar]
- Kim J.; Castellano J. M.; Jiang H.; Basak J. M.; Parsadanian M.; Pham V.; Mason S. M.; Paul S. M.; Holtzman D. M. (2009) Overexpression of Low Density Lipoprotein Receptor in the Brain Markedly Inhibits Amyloid Deposition and Increases Extracellular Aβ Clearance. Neuron 64, 632–644. 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T.; Zhang J.; Liu Q.; Liu C. C.; Zhang L.; Bu G. (2011) Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J. Neurosci. 31, 1644–1651. 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R. J.; Olichney J. M.; Thal L. J.; Mirra S. S.; Morris J. C.; Beekly D.; Heyman A. (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology 46, 1592–1596. 10.1212/WNL.46.6.1592. [DOI] [PubMed] [Google Scholar]
- Bender A. R.; Raz N. (2012) Age-Related Differences in Memory and Executive Functions in Healthy APOE ε4 Carriers: The Contribution of Individual Differences in Prefrontal Volumes and Systolic Blood Pressure. Neuropsychologia 50, 704–714. 10.1016/j.neuropsychologia.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Trotter J.; Zhang J.; Peters M. M.; Cheng H.; Bao J.; Han X.; Weeber E. J.; Bu G. (2010) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 30, 17068–17078. 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J.; Chen Y. (2006) Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 7, 850–859. 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- Genin E.; Hannequin D.; Wallon D.; Sleegers K.; Hiltunen M.; Combarros O.; Bullido M. J.; Engelborghs S.; De Deyn P.; Berr C.; Pasquier F.; Dubois B.; Tognoni G.; Fievet N.; Brouwers N.; Bettens K.; Arosio B.; Coto E.; Del Zompo M.; Mateo I.; Epelbaum J.; Frank-Garcia A.; Helisalmi S.; Porcellini E.; Pilotto A.; Forti P.; Ferri R.; Scarpini E.; Siciliano G.; Solfrizzi V.; Sorbi S.; Spalletta G.; Valdivieso F.; Vepsalainen S.; Alvarez V.; Bosco P.; Mancuso M.; Panza F.; Nacmias B.; Bossu P.; Hanon O.; Piccardi P.; Annoni G.; Seripa D.; Galimberti D.; Licastro F.; Soininen H.; Dartigues J. F.; Kamboh M. I.; Van Broeckhoven C.; Lambert J. C.; Amouyel P.; Campion D. (2011) APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907. 10.1038/mp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. (2019) New APOE-related therapeutic options for Alzheimer’s disease. AIP Conference Proceedings 2058, 020002. 10.1063/1.5085515. [DOI] [Google Scholar]
- Sadowski M.; Pankiewicz J.; Scholtzova H.; Ripellino J. A.; Li Y.; Schmidt S. D.; Mathews P. M.; Fryer J. D.; Holtzman D. M.; Sigurdsson E. M.; Wisniewski T. (2004) A synthetic peptide blocking the apolipoprotein E/β-amyloid binding mitigates β-amyloid toxicity and fibril formation in vitro and reduces β-amyloid plaques in transgenic mice. Am. J. Pathol. 165, 937–948. 10.1016/S0002-9440(10)63355-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Park S.; Allington G.; Prelli F.; Sun Y.; Martá-Ariza M.; Scholtzova H.; Biswas G.; Brown B.; Verghese P. B.; et al. (2017) Targeting apolipoprotein E/amyloid β binding by peptoid CPO_Aβ17–21 P ameliorates Alzheimer’s disease related pathology and cognitive decline. Sci. Rep. 7, 1–12. 10.1038/s41598-017-08604-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Eltorai A. E.; Jiang H.; Liao F.; Verghese P. B.; Kim J.; Stewart F. R.; Basak J. M.; Holtzman D. M. (2012) Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J. Exp. Med. 209, 2149–2156. 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F.; Li A.; Xiong M.; Bien-Ly N.; Jiang H.; Zhang Y.; Finn M. B.; Hoyle R.; Keyser J.; Lefton K. B.; et al. (2018) Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Invest. 128, 2144–2155. 10.1172/JCI96429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Najm R.; Xu Q.; Jeong D.-e.; Walker D.; Balestra M. E.; Yoon S. Y.; Yuan H.; Li G.; Miller Z. A.; et al. (2018) Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. 10.1038/s41591-018-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.-K.; Liu Z.; Meyer-Franke A.; Brodbeck J.; Miranda R. D.; McGuire J. G.; Pleiss M. A.; Ji Z.-S.; Balestra M. E.; Walker D. W.; et al. (2012) Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem. 287, 5253–5266. 10.1074/jbc.M111.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vélez J.; Lopera F.; Sepulveda-Falla D.; Patel H.; Johar A.; Chuah A.; Tobon C.; Rivera D.; Villegas A.; Cai Y.; et al. (2016) APOE* E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol. Psychiatry 21, 916–924. 10.1038/mp.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart J.-C.; Marr R. A.; Koistinaho M.; Gregersen B. M.; Malkani S.; Verma I. M.; Paul S. M. (2005) Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 102, 1211–1216. 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Gottesdiener A. J.; Parmar M.; Li M.; Kaminsky S. M.; Chiuchiolo M. J.; Sondhi D.; Sullivan P. M.; Holtzman D. M.; Crystal R. G.; Paul S. M. (2016) Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 44, 159–172. 10.1016/j.neurobiolaging.2016.04.020. [DOI] [PubMed] [Google Scholar]
- Cramer P. E.; Cirrito J. R.; Wesson D. W.; Lee C. Y.; Karlo J. C.; Zinn A. E.; Casali B. T.; Restivo J. L.; Goebel W. D.; James M. J.; Brunden K. R.; Wilson D. A.; Landreth G. E. (2012) ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science (Washington, DC, U. S.) 335, 1503–1506. 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A.; Boisvert W. A.; Lee C. H.; Laffitte B. A.; Barak Y.; Joseph S. B.; Liao D.; Nagy L.; Edwards P. A.; Curtiss L. K.; Evans R. M.; Tontonoz P. (2001) A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 7, 161–171. 10.1016/S1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- Vaya J.; Schipper H. M. (2007) Oxysterols, cholesterol homeostasis, and Alzheimer disease. J. Neurochem. 102, 1727–1737. 10.1111/j.1471-4159.2007.04689.x. [DOI] [PubMed] [Google Scholar]
- Bales K. R.; Verina T.; Dodel R. C.; Du Y.; Altstiel L.; Bender M.; Hyslop P.; Johnstone E. M.; Little S. P.; Cummins D. J.; Piccardo P.; Ghetti B.; Paul S. M. (1997) Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 17, 263–264. 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Jonsson T.; Atwal J. K.; Steinberg S.; Snaedal J.; Jonsson P. V.; Bjornsson S.; Stefansson H.; Sulem P.; Gudbjartsson D.; Maloney J.; Hoyte K.; Gustafson A.; Liu Y.; Lu Y.; Bhangale T.; Graham R. R.; Huttenlocher J.; Bjornsdottir G.; Andreassen O. A.; Jonsson E. G.; Palotie A.; Behrens T. W.; Magnusson O. T.; Kong A.; Thorsteinsdottir U.; Watts R. J.; Stefansson K. (2012) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Loera-Valencia R.; Piras A.; Ismail M.; Manchanda S.; Eyjolfsdottir H.; Saido T.; Johansson J.; Eriksdotter M.; Winblad B.; Nilsson P. (2018) Targeting Alzheimer’s disease with gene and cell therapies. J. Intern. Med. 284, 2–36. 10.1111/joim.12759. [DOI] [PubMed] [Google Scholar]
- Hong C.; Goins W.; Goss J.; Burton E.; Glorioso J. (2006) Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer’s disease-related amyloid-β peptide in vivo. Gene Ther. 13, 1068. 10.1038/sj.gt.3302719. [DOI] [PubMed] [Google Scholar]
- Fol R.; Braudeau J.; Ludewig S.; Abel T.; Weyer S. W.; Roederer J.-P.; Brod F.; Audrain M.; Bemelmans A.-P.; Buchholz C. J.; et al. (2016) Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol. 131, 247–266. 10.1007/s00401-015-1498-9. [DOI] [PubMed] [Google Scholar]
- Boada M.; Antunez C.; Ramirez-Lorca R.; DeStefano A. L.; Gonzalez-Perez A.; Gayan J.; Lopez-Arrieta J.; Ikram M. A.; Hernandez I.; Marin J.; Galan J. J.; Bis J. C.; Mauleon A.; Rosende-Roca M.; Moreno-Rey C.; Gudnasson V.; Moron F. J.; Velasco J.; Carrasco J. M.; Alegret M.; Espinosa A.; Vinyes G.; Lafuente A.; Vargas L.; Fitzpatrick A. L.; Launer L. J.; Saez M. E.; Vazquez E.; Becker J. T.; Lopez O. L.; Serrano-Rios M.; Tarraga L.; van Duijn C. M.; Real L. M.; Seshadri S.; Ruiz A. (2014) ATP5H/KCTD2 locus is associated with Alzheimer’s disease risk. Mol. Psychiatry 19, 682–687. 10.1038/mp.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X. F.; Luo C.; Ren K. Q.; Zhang J.; Zhou J. L.; Hu X.; Liu R. S.; Wang Y.; Gao X.; Zhang J. (2008) Characterization and expression of a human KCTD1 gene containing the BTB domain, which mediates transcriptional repression and homomeric interactions. DNA Cell Biol. 27, 257–265. 10.1089/dna.2007.0662. [DOI] [PubMed] [Google Scholar]
- Schwenk J.; Metz M.; Zolles G.; Turecek R.; Fritzius T.; Bildl W.; Tarusawa E.; Kulik A.; Unger A.; Ivankova K.; Seddik R.; Tiao J. Y.; Rajalu M.; Trojanova J.; Rohde V.; Gassmann M.; Schulte U.; Fakler B.; Bettler B. (2010) Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature 465, 231–235. 10.1038/nature08964. [DOI] [PubMed] [Google Scholar]
- Bayón Y.; Trinidad A. G.; de la Puerta M. L.; del Carmen Rodríguez M.; Bogetz J.; Rojas A.; De Pereda J. M.; Rahmouni S.; Williams S.; Matsuzawa S.-i.; Reed J. C.; Crespo M. S.; Mustelin T.; Alonso A. (2008) KCTD5, a putative substrate adaptor for cullin3 ubiquitin ligases. FEBS J. 275, 3900–3910. 10.1111/j.1742-4658.2008.06537.x. [DOI] [PubMed] [Google Scholar]
- Hirai K.; Aliev G.; Nunomura A.; Fujioka H.; Russell R. L.; Atwood C. S.; Johnson A. B.; Kress Y.; Vinters H. V.; Tabaton M.; Shimohama S.; Cash A. D.; Siedlak S. L.; Harris P. L.; Jones P. K.; Petersen R. B.; Perry G.; Smith M. A. (2001) Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023. 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. J.; Sugnet C. W.; Furey T. S.; Roskin K. M.; Pringle T. H.; Zahler A. M.; Haussler D. (2002) The human genome browser at UCSC. Genome Res. 12, 996–1006. 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terni B.; Boada J.; Portero-Otin M.; Pamplona R.; Ferrer I. (2010) Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol. 20, 222–233. 10.1111/j.1750-3639.2009.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W. S.; Reiman E. M.; Valla J.; Dunckley T.; Beach T. G.; Grover A.; Niedzielko T. L.; Schneider L. E.; Mastroeni D.; Caselli R.; Kukull W.; Morris J. C.; Hulette C. M.; Schmechel D.; Rogers J.; Stephan D. A. (2008) Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U. S. A. 105, 4441–4446. 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton S. L.; Prentice H. M. (2007) Beyond Anoxia: The Physiology of Metabolic Downregulation and Recovery in the Anoxia-Tolerant Turtle. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 147, 277–290. 10.1016/j.cbpa.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Chen G.-J. (2016) Mitochondria as a therapeutic target in Alzheimer’s disease. Genes & Diseases 3, 220–227. 10.1016/j.gendis.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws M. R.; Lanier L. L.; Seaman W. E.; Ryan J. C. (2001) Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 31, 783–791. . [DOI] [PubMed] [Google Scholar]
- Paloneva J.; Manninen T.; Christman G.; Hovanes K.; Mandelin J.; Adolfsson R.; Bianchin M.; Bird T.; Miranda R.; Salmaggi A.; Tranebjaerg L.; Konttinen Y.; Peltonen L. (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662. 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S.; Burbach G. J.; Bonin M.; Walter M.; Streit W.; Bechmann I.; Deller T. (2008) TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447. 10.1002/glia.20710. [DOI] [PubMed] [Google Scholar]
- Piccio L.; Buonsanti C.; Mariani M.; Cella M.; Gilfillan S.; Cross A. H.; Colonna M.; Panina-Bordignon P. (2007) Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur. J. Immunol. 37, 1290–1301. 10.1002/eji.200636837. [DOI] [PubMed] [Google Scholar]
- Guerreiro R.; Wojtas A.; Bras J.; Carrasquillo M.; Rogaeva E.; Majounie E.; Cruchaga C.; Sassi C.; Kauwe J. S. K.; Younkin S.; Hazrati L.; Collinge J.; Pocock J.; Lashley T.; Williams J.; Lambert J.-C.; Amouyel P.; Goate A.; Rademakers R.; Morgan K.; Powell J.; St. George-Hyslop P.; Singleton A.; Hardy J. (2013) TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 368, 117–127. 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaghmoor F.; Noorsaeed A.; Alsaggaf S.; Aljohani W.; Scholtzova H.; Boutajangout A.; Wisniewski T. (2014) The Role of TREM2 in Alzheimer’s Disease and Other Neurological Disorders. Journal of Alzheimer’s Disease & Parkinsonism 4, 160. 10.4172/2161-0460.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T.; Tan L.; Zhu X. C.; Zhang Q. Q.; Cao L.; Tan M. S.; Gu L. Z.; Wang H. F.; Ding Z. Z.; Zhang Y. D.; Yu J. T. (2014) Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 39, 2949–2962. 10.1038/npp.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratuze M.; Leyns C. E.; Holtzman D. M. (2018) New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13, 66. 10.1186/s13024-018-0298-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K.; Rochford C. D.; Neumann H. (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657. 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T.; Tan L.; Zhu X.-C.; Zhang Q.-Q.; Cao L.; Tan M.-S.; Gu L.-Z.; Wang H.-F.; Ding Z.-Z.; Zhang Y.-D.; Yu J.-T. (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 2949–2962. 10.1038/npp.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiteri C.; Mostafavi S.; Honey C. J.; De Jager P. L.; Bennett D. A. (2016) Genetic variants in Alzheimer disease - molecular and brain network approaches. Nat. Rev. Neurol. 12, 413–427. 10.1038/nrneurol.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. (2006) Inflammation in Alzheimer disease: driving force, bystander or beneficial response?. Nature medicine 12, 1005–1015. 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Town T.; Tan J.; Flavell R. A.; Mullan M. (2005) T-cells in Alzheimer’s disease. NeuroMol. Med. 7, 255–264. 10.1385/NMM:7:3:255. [DOI] [PubMed] [Google Scholar]
- Szekely C. A.; Zandi P. P. (2010) Non-steroidal anti-inflammatory drugs and Alzheimer’s disease: the epidemiological evidence. CNS Neurol. Disord.: Drug Targets 9, 132–139. 10.2174/187152710791012026. [DOI] [PubMed] [Google Scholar]
- Du X.; Wang X.; Geng M. (2018) Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 7, 2. 10.1186/s40035-018-0107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S. E.; Allison E. K.; El Khoury J. (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit W. J.; Mrak R. E.; Griffin W. S. (2004) Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 1, 14. 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A.; Suri-Payer E.; Veltkamp C.; Doerr H.; Sommer C.; Rivest S.; Giese T.; Veltkamp R. (2009) Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 15, 192–199. 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- He P.; Zhong Z.; Lindholm K.; Berning L.; Lee W.; Lemere C.; Staufenbiel M.; Li R.; Shen Y. (2007) Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 178, 829–841. 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Berg J.; Prokop S.; Miller K. R.; Obst J.; Kalin R. E.; Lopategui-Cabezas I.; Wegner A.; Mair F.; Schipke C. G.; Peters O.; Winter Y.; Becher B.; Heppner F. L. (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. 18, 1812–1819. 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- Lou K.-j. (2012) Targeting interleukins in Alzheimer’s disease. SciBX 5, 1276. 10.1038/scibx.2012.1276. [DOI] [Google Scholar]
- Wang X.; Zhu M.; Hjorth E.; Cortés-Toro V.; Eyjolfsdottir H.; Graff C.; Nennesmo I.; Palmblad J.; Eriksdotter M.; Sambamurti K.; et al. (2015) Resolution of inflammation is altered in Alzheimer’s disease. Alzheimer's Dementia 11, 40–50.e2. 10.1016/j.jalz.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Puerta E.; Cedazo-Minguez A.; Hjorth E.; Schultzberg M. (2015) Insufficient resolution response in the hippocampus of a senescence-accelerated mouse model—SAMP8. J. Mol. Neurosci. 55, 396–405. 10.1007/s12031-014-0346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P.; Li A.; Ceballos-Diaz C.; Eddy J. A.; Funk C. C.; Moore B.; DiNunno N.; Rosario A. M.; Cruz P. E.; Verbeeck C.; et al. (2015) IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 85, 519–533. 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirkis D. W.; Bonham L. W.; Aparicio R. E.; Geier E. G.; Ramos E. M.; Wang Q.; Karydas A.; Miller Z. A.; Miller B. L.; Coppola G.; Yokoyama J. S. (2016) Rare TREM2 variants associated with Alzheimer’s disease display reduced cell surface expression. Acta Neuropathologica Communications 4, 98. 10.1186/s40478-016-0367-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga S.; Akai K.; Ishii T. (1989) Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. Acta Neuropathol. 77, 569–575. 10.1007/BF00687883. [DOI] [PubMed] [Google Scholar]
- Shineman D. W.; Zhang B.; Leight S. N.; Pratico D.; Lee V. M.-Y. (2008) Thromboxane Receptor Activation Mediates Isoprostane-Induced Increases in Amyloid Pathology in Tg2576 Mice. J. Neurosci. 28, 4785–4794. 10.1523/JNEUROSCI.0684-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soper J. H.; Sugiyama S.; Herbst-Robinson K.; James M. J.; Wang X.; Trojanowski J. Q.; Smith A. B. 3rd; Lee V. M.; Ballatore C.; Brunden K. R. (2012) Brain-penetrant tetrahydronaphthalene thromboxane A2-prostanoid (TP) receptor antagonists as prototype therapeutics for Alzheimer’s disease. ACS Chem. Neurosci. 3, 928–940. 10.1021/cn3000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D.; Corpuz M.; Richmond B.; Wendt E.; Hall E. R. (1996) Activated microglia are the principal glial source of thromboxane in the central nervous system. Neurochem. Int. 29, 65–76. 10.1016/0197-0186(95)00140-9. [DOI] [PubMed] [Google Scholar]
- Zhen G.; Kim Y. T.; Li R. C.; Yocum J.; Kapoor N.; Langer J.; Dobrowolski P.; Maruyama T.; Narumiya S.; Dore S. (2012) PGE2 EP1 receptor exacerbated neurotoxicity in a mouse model of cerebral ischemia and Alzheimer’s disease. Neurobiol. Aging 33, 2215–2219. 10.1016/j.neurobiolaging.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Wang Q.; Johansson J. U.; Liang X.; Woodling N. S.; Priyam P.; Loui T. M.; Merchant M.; Breyer R. M.; Montine T. J.; Andreasson K. (2012) Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann. Neurol. 72, 788–798. 10.1002/ana.23677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Wang Q.; Hand T.; Wu L.; Breyer R. M.; Montine T. J.; Andreasson K. (2005) Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J. Neurosci. 25, 10180–10187. 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino T.; Namba T.; Takehara M.; Murao N.; Matsushima T.; Sugimoto Y.; Narumiya S.; Suzuki T.; Mizushima T. (2012) Improvement of cognitive function in Alzheimer’s disease model mice by genetic and pharmacological inhibition of the EP(4) receptor. J. Neurochem. 120, 795–805. 10.1111/j.1471-4159.2011.07567.x. [DOI] [PubMed] [Google Scholar]
- Herbst-Robinson K. J.; Liu L.; James M.; Yao Y.; Xie S. X.; Brunden K. R. (2016) Sci. Rep. 5, 18286. 10.1038/srep18286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson M.; Kubes P. (2011) The neutrophil in vascular inflammation. Nat. Med. 17, 1381–1390. 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E.; Kubes P. (2013) Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175. 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- Siffrin V.; Radbruch H.; Glumm R.; Niesner R.; Paterka M.; Herz J.; Leuenberger T.; Lehmann S. M.; Luenstedt S.; Rinnenthal J. L.; Laube G.; Luche H.; Lehnardt S.; Fehling H. J.; Griesbeck O.; Zipp F. (2010) In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity 33, 424–436. 10.1016/j.immuni.2010.08.018. [DOI] [PubMed] [Google Scholar]
- Zenaro E.; Constantin G. (2017) Targeting neuroinflammation in the treatment and prevention of Alzheimer’s disease. Drugs Future 42, 21. 10.1358/dof.2017.042.01.2564104. [DOI] [Google Scholar]
- Zenaro E.; Pietronigro E.; Della Bianca V.; Piacentino G.; Marongiu L.; Budui S.; Turano E.; Rossi B.; Angiari S.; Dusi S.; et al. (2015) Neutrophils promote Alzheimer’s disease–like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880. 10.1038/nm.3913. [DOI] [PubMed] [Google Scholar]
- Silva M. T. (2010) Neutrophils and macrophages work in concert as inducers and effectors of adaptive immunity against extracellular and intracellular microbial pathogens. J. Leukocyte Biol. 87, 805–813. 10.1189/jlb.1109767. [DOI] [PubMed] [Google Scholar]
- Soehnlein O.; Lindbom L. (2010) Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 10, 427–439. 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- Zenaro E.; Pietronigro E.; Della Bianca V.; Piacentino G.; Marongiu L.; Budui S.; Turano E.; Rossi B.; Angiari S.; Dusi S.; Montresor A.; Carlucci T.; Nani S.; Tosadori G.; Calciano L.; Catalucci D.; Berton G.; Bonetti B.; Constantin G. (2015) Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. 10.1038/nm.3913. [DOI] [PubMed] [Google Scholar]
- Barnado A.; Crofford L. J.; Oates J. C. (2016) At the Bedside: Neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J. Leukocyte Biol. 99, 265–278. 10.1189/jlb.5BT0615-234R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietronigro E. C.; Della Bianca V.; Zenaro E.; Constantin G. (2017) NETosis in Alzheimer’s Disease. Front. Immunol. 8, 211. 10.3389/fimmu.2017.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebir H.; Kreymborg K.; Ifergan I.; Dodelet-Devillers A.; Cayrol R.; Bernard M.; Giuliani F.; Arbour N.; Becher B.; Prat A. (2007) Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13, 1173–1175. 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorantla N. V.; Chinnathambi S. (2018) Tau Protein Squired by Molecular Chaperones During Alzheimer’s Disease. J. Mol. Neurosci. 66, 356–368. 10.1007/s12031-018-1174-3. [DOI] [PubMed] [Google Scholar]
- Lakshmana M. K.; Yoon I. S.; Chen E.; Bianchi E.; Koo E. H.; Kang D. E. (2009) Novel role of RanBP9 in BACE1 processing of amyloid precursor protein and amyloid beta peptide generation. J. Biol. Chem. 284, 11863–11872. 10.1074/jbc.M807345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmana M. K.; Hayes C. D.; Bennett S. P.; Bianchi E.; Reddy K. M.; Koo E. H.; Kang D. E. (2012) Role of RanBP9 on amyloidogenic processing of APP and synaptic protein levels in the mouse brain. FASEB J. 26, 2072–2083. 10.1096/fj.11-196709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmana M. K.; Chung J. Y.; Wickramarachchi S.; Tak E.; Bianchi E.; Koo E. H.; Kang D. E. (2010) A fragment of the scaffolding protein RanBP9 is increased in Alzheimer’s disease brains and strongly potentiates amyloid-beta peptide generation. FASEB J. 24, 119–127. 10.1096/fj.09-136457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J. A.; Roh S. E.; Lakshmana M. K.; Kang D. E. (2012) Pivotal role of RanBP9 in integrin-dependent focal adhesion signaling and assembly. FASEB J. 26, 1672–1681. 10.1096/fj.11-194423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J. A.; Jung A. R.; Lakshmana M. K.; Bedrossian A.; Lim Y.; Bu J. H.; Park S. A.; Koo E. H.; Mook-Jung I.; Kang D. E. (2012) Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell Death Differ. 19, 1413–1423. 10.1038/cdd.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J.; Boggess T.; Uhlar C.; Wang X.; Khan H.; Cappos G.; Joly-Amado A.; De Narvaez E.; Majid S.; Minamide L.; et al. (2015) RanBP9 at the intersection between cofilin and Aβ pathologies: rescue of neurodegenerative changes by RanBP9 reduction. Cell Death Dis. 6, e1676. 10.1038/cddis.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atabakhsh E.; Bryce D. M.; Lefebvre K. J.; Schild-Poulter C. (2009) RanBPM has proapoptotic activities that regulate cell death pathways in response to DNA damage. Mol. Cancer Res. 7, 1962–1972. 10.1158/1541-7786.MCR-09-0098. [DOI] [PubMed] [Google Scholar]
- Kramer S.; Ozaki T.; Miyazaki K.; Kato C.; Hanamoto T.; Nakagawara A. (2005) Protein stability and function of p73 are modulated by a physical interaction with RanBPM in mammalian cultured cells. Oncogene 24, 938–944. 10.1038/sj.onc.1208257. [DOI] [PubMed] [Google Scholar]
- John K.; Alla V.; Meier C.; Putzer B. M. (2011) GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria. Cell Death Differ. 18, 874–886. 10.1038/cdd.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Roh S. E.; Woo J. A.; Ryu H.; Kang D. E. (2013) Cooperative role of RanBP9 and P73 in mitochondria-mediated apoptosis. Cell Death Dis. 4, e476. 10.1038/cddis.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freir D. B.; Nicoll A. J.; Klyubin I.; Panico S.; Mc Donald J. M.; Risse E.; Asante E. A.; Farrow M. A.; Sessions R. B.; Saibil H. R.; Clarke A. R.; Rowan M. J.; Walsh D. M.; Collinge J. (2011) Nat. Commun. 2, 336. 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Klyubin I.; Fadeeva J. V.; Cullen W. K.; Anwyl R.; Wolfe M. S.; Rowan M. J.; Selkoe D. J. (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Lesne S.; Koh M. T.; Kotilinek L.; Kayed R.; Glabe C. G.; Yang A.; Gallagher M.; Ashe K. H. (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lima-Filho R. A.; Oliveira M. M. (2018) A Role for Cellular Prion Protein in Late-Onset Alzheimer’s Disease: Evidence from Preclinical Studies. J. Neurosci. 38, 2146. 10.1523/JNEUROSCI.3307-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhao Y.; Zhang L.; Yu W.; Wang Y.; Chang W. (2019) Cellular prion protein as a receptor of toxic amyloid-β42 oligomers is important for Alzheimer’s disease. Front. Cell. Neurosci. 13, 339. 10.3389/fncel.2019.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar S. V.; Strittmatter S. M. (2017) Cellular prion protein as a receptor for amyloid-β oligomers in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 483, 1143–1147. 10.1016/j.bbrc.2016.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas L. T.; Salazar S. V.; Smith L. M.; Zhao H. R.; Cox T. O.; Herber C. S.; Degnan A. P.; Balakrishnan A.; Macor J. E.; Albright C. F.; Strittmatter S. M. (2017) Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing Alzheimer’s mouse phenotypes. Cell Rep. 20, 76–88. 10.1016/j.celrep.2017.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Götz J. (2017) Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J. 36, 3120–3138. 10.15252/embj.201797724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2011) State of aggregation. Nat. Neurosci. 14, 399–399. 10.1038/nn0411-399. [DOI] [PubMed] [Google Scholar]
- Chung E.; Ji Y.; Sun Y.; Kascsak R. J.; Kascsak R. B.; Mehta P. D.; Strittmatter S. M.; Wisniewski T. (2010) Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 11, 130. 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels H. W.; Nguyen L. N.; Nabavi S.; Malinow R. (2010) The prion protein as a receptor for amyloid-beta. Nature 466, E3–4. 10.1038/nature09217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel D. A.; Nygaard H. B.; Coffey E. E.; Gunther E. C.; Lauren J.; Gimbel Z. A.; Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll A. J.; Panico S.; Freir D. B.; Wright D.; Terry C.; Risse E.; Herron C. E.; O’Malley T.; Wadsworth J. D. F.; Farrow M. A.; Walsh D. M.; Saibil H. R.; Collinge J. (2013) Nat. Commun. 4, 2416. 10.1038/ncomms3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knez D.; Coquelle N.; Pislar A.; Zakelj S.; Jukic M.; Sova M.; Mravljak J.; Nachon F.; Brazzolotto X.; Kos J.; Colletier J.-P.; Gobec S. (2018) Multi-target-directed ligands for treating Alzheimer’s disease: Butyrylcholinesterase inhibitors displaying antioxidant and neuroprotective activities. Eur. J. Med. Chem. 156, 598–617. 10.1016/j.ejmech.2018.07.033. [DOI] [PubMed] [Google Scholar]
- Leon R.; Garcia A. G.; Marco-Contelles J. (2013) Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 33, 139–189. 10.1002/med.20248. [DOI] [PubMed] [Google Scholar]
- Angelucci F.; Cechova K.; Valis M.; Kuca K.; Zhang B.; Hort J. (2019) Micro RNAs in Alzheimer’s disease: diagnostic markers or therapeutic agents?. Front. Pharmacol. 10, 665. 10.3389/fphar.2019.00665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos S. L.; Miller R. L.; Schoch K. M.; Holmes B. B.; Kebodeaux C. S.; Wegener A. J.; Chen G.; Shen T.; Tran H.; Nichols B.; et al. (2017) Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 9, eaag0481. 10.1126/scitranslmed.aag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higaki S.; Muramatsu M.; Matsuda A.; Matsumoto K.; Satoh J.-i.; Michikawa M.; Niida S. (2018) Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS One 13, e0196929. 10.1371/journal.pone.0196929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An F.; Gong G.; Wang Y.; Bian M.; Yu L.; Wei C. (2017) MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 8, 114065. 10.18632/oncotarget.23119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Wang H. (2018) miR-15b reduces amyloid-β accumulation in SH-SY5Y cell line through targetting NF-κB signaling and BACE1. Biosci. Rep. 38, 51. 10.1042/BSR20180051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemi-Kasman M.; Shojaei A.; Gol M.; Moghadamnia A. A.; Baharvand H.; Javan M. (2018) miR-302/367-induced neurons reduce behavioral impairment in an experimental model of Alzheimer’s disease. Mol. Cell. Neurosci. 86, 50–57. 10.1016/j.mcn.2017.11.012. [DOI] [PubMed] [Google Scholar]
- Li S.-H.; Gao P.; Wang L.-T.; Yan Y.-H.; Xia Y.; Song J.; Li H.-Y.; Yang J.-X. (2017) Osthole stimulated neural stem cells differentiation into neurons in an Alzheimer’s disease cell model via upregulation of microRNA-9 and rescued the functional impairment of hippocampal neurons in APP/PS1 transgenic mice. Front. Neurosci. 11, 340. 10.3389/fnins.2017.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Hu M.; Teng Z.; Tang Y.-P.; Chen C. (2014) Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188–3p in a mouse model of Alzheimer’s disease. J. Neurosci. 34, 14919–14933. 10.1523/JNEUROSCI.1165-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Li Q.; Liu C.; Gao S.; Ping H.; Wang J.; Wang P. (2016) MiR-214–3p attenuates cognition defects via the inhibition of autophagy in SAMP8 mouse model of sporadic Alzheimer’s disease. NeuroToxicology 56, 139–149. 10.1016/j.neuro.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Han L.; Zhou Y.; Zhang R.; Wu K.; Lu Y.; Li Y.; Duan R.; Yao Y.; Zhu D.; Jia Y. (2018) MicroRNA Let-7f-5p promotes bone marrow mesenchymal stem cells survival by targeting caspase-3 in alzheimer disease model. Front. Neurosci. 12, 333. 10.3389/fnins.2018.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L.; Zhang T.; Liu W.; Chen Y. (2018) Inhibition of miR-128 abates Aβ-mediated cytotoxicity by targeting PPAR-γ via NF-κB inactivation in primary mouse cortical neurons and Neuro2a cells. Yonsei Med. J. 59, 1096–1106. 10.3349/ymj.2018.59.9.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Li Z.; Zhao L.; Zhao W. (2017) Simvastatin ameliorate memory deficits and inflammation in clinical and mouse model of Alzheimer’s disease via modulating the expression of miR-106b. Biomed. Pharmacother. 92, 46–57. 10.1016/j.biopha.2017.05.060. [DOI] [PubMed] [Google Scholar]
- Shadfar S.; Hwang C. J.; Lim M.-S.; Choi D.-Y.; Hong J. T. (2015) Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharmacal Res. 38, 2106–2119. 10.1007/s12272-015-0648-x. [DOI] [PubMed] [Google Scholar]
- Kou X.; Chen N. (2017) Resveratrol as a natural autophagy regulator for prevention and treatment of Alzheimer’s disease. Nutrients 9, 927. 10.3390/nu9090927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.-j.; Zhao B.; Zhao J.; Li S. (2017) Potential roles of exosomal microRNAs as diagnostic biomarkers and therapeutic application in Alzheimer’s disease. Neural Plast. 2017, 7027380. 10.1155/2017/7027380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X.; Zhao Y.; Wang J.; Zhou Q.; Xu L.; Kang D.; Liu A.-L.; Du G.-H. (2017) The bioinformatic analysis of the dysregulated genes and microRNAs in entorhinal cortex, hippocampus, and blood for Alzheimer’s disease. BioMed Res. Int. 2017, 9084507. 10.1155/2017/9084507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E.; Mouradian M. M. (2012) MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 133, 142–150. 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipour M.; Nabavi S. M.; Arab L.; Vosough M.; Pakdaman H.; Ehsani E.; Shahpasand K. (2019) Stem cell therapy in Alzheimer’s disease: possible benefits and limiting drawbacks. Mol. Biol. Rep. 46, 1425–1446. 10.1007/s11033-018-4499-7. [DOI] [PubMed] [Google Scholar]
- Bali P.; Lahiri D.; Banik A.; Nehru B.; Anand A. (2017) Potential for stem cells therapy in Alzheimer’s disease: Do neurotrophic factors play critical role?. Curr. Alzheimer Res. 14, 208–220. 10.2174/1567205013666160314145347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinley L. M.; Kashlan O. N.; Bruno E. S.; Chen K. S.; Hayes J. M.; Kashlan S. R.; Raykin J.; Johe K.; Murphy G. G.; Feldman E. L. (2018) Human neural stem cell transplantation improves cognition in a murine model of Alzheimer’s disease. Sci. Rep. 8, 14776. 10.1038/s41598-018-33017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2015) 2015 Alzheimer's disease facts and figures. Alzheimer’s Dementia 11, 332–384. 10.1016/j.jalz.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Rafii M. S.; Aisen P. S. (2019) Alzheimer’s Disease Clinical Trials: Moving Toward Successful Prevention. CNS Drugs 33, 99–106. 10.1007/s40263-018-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung S.-Y.; Fu W.-M. (2017) Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 24, 47. 10.1186/s12929-017-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmaleh D. R.; Farlow M. R.; Conti P. S.; Tompkins R. G.; Kundakovic L.; Tanzi R. E. (2019) Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions. J. Alzheimer's Dis. 71, 715. 10.3233/JAD-190507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J.; Lee G.; Ritter A.; Sabbagh M.; Zhong K. (2019) Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 5, 272–293. 10.1016/j.trci.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.-K.; Chao S.-P.; Hu C.-J. (2020) Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 27, 18. 10.1186/s12929-019-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R.; Rinne J. O.; Boada M.; Katayama S.; Scheltens P.; Vellas B.; Tuchman M.; Gass A.; Fiebach J. B.; Hill D.; Lobello K.; Li D.; McRae T.; Lucas P.; Evans I.; Booth K.; Luscan G.; Wyman B. T.; Hua L.; Yang L.; Brashear H. R.; Black R. S. (2016) Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimer's Res. Ther. 8, 18. 10.1186/s13195-016-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki S.; Lasser R. A.; Dorflinger E.; Scheltens P.; Barkhof F.; Nikolcheva T.; Ashford E.; Retout S.; Hofmann C.; Delmar P.; Klein G.; Andjelkovic M.; Dubois B.; Boada M.; Blennow K.; Santarelli L.; Fontoura P. (2017) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimer's Res. Ther. 9, 95. 10.1186/s13195-017-0318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig L. S.; Vellas B.; Woodward M.; Boada M.; Bullock R.; Borrie M.; Hager K.; Andreasen N.; Scarpini E.; Liu-Seifert H.; Case M.; Dean R. A.; Hake A.; Sundell K.; Poole Hoffmann V.; Carlson C.; Khanna R.; Mintun M.; DeMattos R.; Selzler K. J.; Siemers E. (2018) Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 378, 321–330. 10.1056/NEJMoa1705971. [DOI] [PubMed] [Google Scholar]
- Cummings J. L.; Cohen S.; van Dyck C. H.; Brody M.; Curtis C.; Cho W.; Ward M.; Friesenhahn M.; Rabe C.; Brunstein F.; Quartino A.; Honigberg L. A.; Fuji R. N.; Clayton D.; Mortensen D.; Ho C.; Paul R. (2018) ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90, e1889–e1897. 10.1212/WNL.0000000000005550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A.; Benet L. Z. (2020) Late-Stage Failures of Monoclonal Antibody Drugs: A Retrospective Case Study Analysis. Pharmacology 105, 145. 10.1159/000505379. [DOI] [PubMed] [Google Scholar]
- Doody R. S.; Raman R.; Farlow M.; Iwatsubo T.; Vellas B.; Joffe S.; Kieburtz K.; He F.; Sun X.; Thomas R. G.; Aisen P. S.; Siemers E.; Sethuraman G.; Mohs R. (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 369, 341–350. 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- Coric V.; Salloway S.; van Dyck C. H.; Dubois B.; Andreasen N.; Brody M.; Curtis C.; Soininen H.; Thein S.; Shiovitz T.; Pilcher G.; Ferris S.; Colby S.; Kerselaers W.; Dockens R.; Soares H.; Kaplita S.; Luo F.; Pachai C.; Bracoud L.; Mintun M.; Grill J. D.; Marek K.; Seibyl J.; Cedarbaum J. M.; Albright C.; Feldman H. H.; Berman R. M. (2015) Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol 72, 1324–1333. 10.1001/jamaneurol.2015.0607. [DOI] [PubMed] [Google Scholar]
- Panza F.; Lozupone M.; Watling M.; Imbimbo B. P. (2019) Do BACE inhibitor failures in Alzheimer patients challenge the amyloid hypothesis of the disease?. Expert Review of Naurotherapeutics 19, 599–602. 10.1080/14737175.2019.1621751. [DOI] [PubMed] [Google Scholar]
- Egan M. F.; Kost J.; Tariot P. N.; Aisen P. S.; Cummings J. L.; Vellas B.; Sur C.; Mukai Y.; Voss T.; Furtek C.; et al. (2018) Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 378, 1691–1703. 10.1056/NEJMoa1706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan M. F.; Kost J.; Voss T.; Mukai Y.; Aisen P. S.; Cummings J. L.; Tariot P. N.; Vellas B.; van Dyck C. H.; Boada M.; Zhang Y.; Li W.; Furtek C.; Mahoney E.; Harper Mozley L.; Mo Y.; Sur C.; Michelson D. (2019) Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 380, 1408–1420. 10.1056/NEJMoa1812840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. (2019) Alzheimer prevention failure rattles field, anew. Nat. Rev. Drug Discovery 18, 656. 10.1038/d41573-019-00139-z. [DOI] [PubMed] [Google Scholar]
- Atri A.; Frolich L.; Ballard C.; Tariot P. N.; Molinuevo J. L.; Boneva N.; Windfeld K.; Raket L. L.; Cummings J. L. (2018) Effect of Idalopirdine as Adjunct to Cholinesterase Inhibitors on Change in Cognition in Patients With Alzheimer Disease: Three Randomized Clinical Trials. Jama 319, 130–142. 10.1001/jama.2017.20373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I. (2010) The rise and fall of Dimebon. Drug News Perspect. 23, 518. 10.1358/dnp.2010.23.8.1500435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S.; Kumar A.; Tripathi T.; Kumar A. (2018) Muscarinic and nicotinic acetylcholine receptor agonists: current scenario in Alzheimer’s disease therapy. J. Pharm. Pharmacol. 70, 985–993. 10.1111/jphp.12919. [DOI] [PubMed] [Google Scholar]
- Mehta D.; Jackson R.; Paul G.; Shi J.; Sabbagh M. (2017) Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Invest. Drugs 26, 735–739. 10.1080/13543784.2017.1323868. [DOI] [PMC free article] [PubMed] [Google Scholar]