

Abstract
We have demonstrated that ivacaftor displays synergistic antibacterial activity in combination with polymyxin B against polymyxin-resistant Pseudomonas aeruginosa that commonly colonizes the lungs of people with cystic fibrosis (CF). However, the underlying mechanism(s) remain unclear. In the present study, we employed untargeted metabolomics to investigate the synergistic killing mechanism of polymyxin B in combination with ivacaftor against a polymyxin-susceptible P. aeruginosa FADDI-PA111 (polymyxin B MIC = 2 mg/L) and a polymyxin-resistant CF P. aeruginosa FADDI-PA006 (polymyxin B MIC = 8 mg/L). Metabolites were extracted at 3 h after treatments with polymyxin B alone (2 μg/mL for FADDI-PA111 and 4 μg/mL FADDI-PA006 P. aeruginosa), ivacaftor alone (8 μg/mL), and in combination. Polymyxin B monotherapy induced significant perturbations in the glycerophospholipid and fatty acid metabolism pathways against FADDI-PA111 and to a lesser extent in FADDI-PA006. In both strains, treatment with ivacaftor alone induced more pronounced perturbations in glycerophospholipid and fatty acid metabolism pathways than that with polymyxin B alone. This highlights the unique antimicrobial mode of action of ivacaftor. Pathway analysis revealed that in combination treatment, polymyxin B mediated killing is elevated by ivacaftor, largely due to the inhibition of cell envelope biogenesis via suppression of key membrane lipid metabolites (e.g., sn-glycerol 3-phosphate and sn-glycero-3-phosphoethanolamine) as well as perturbations in peptidoglycan and lipopolysaccharide biosynthesis. Furthermore, significant perturbations in the levels of amino sugars and nucleotide sugars, glycolysis, the tricarboxylic acid cycle, and pyrimidine ribonucleotide biogenesis were observed with the combination treatment. These findings provide novel mechanistic information on the synergistic antibacterial activity of polymyxin–ivacaftor combination.
Keywords: cystic fibrosis, ivacaftor, CFTR modulator, CFTR potentiators, polymyxin B, polymyxins, metabolomics, synergy, Pseudomonas aeruginosa
Introduction
Cystic fibrosis (CF) is a genetically acquired, life-shortening chronic illness that is caused by deficient or defective cystic fibrosis transmembrane conductance regulator (CFTR) protein.1 The main function of CFTR is the transport of anions including chloride and bicarbonate across the epithelial cell membrane. A deficient or defective CFTR protein leads to ion imbalance and dehydration of epithelial surfaces affecting exocrine mucus glands in multiple organs including the lung, liver, pancreas, and intestines.2,3 Despite CF being a multiorgan disease, the greatest cause of morbidity is rooted in the respiratory tract where mucus obstruction of the airways results in chronic infections and inflammation, ultimately resulting in respiratory failure.4 There is a close link between inflammation and establishment of chronic infections, such as Pseudomonas aeruginosa and/or Staphylococcus aureus, with declining respiratory function in these patients.5 During infancy, lung infections are associated with high abundances of S. aureus and Haemophilus influenzae; however, in older patients, infections caused by P. aeruginosa, Burkholderia spp., and Achromobacter spp. are associated with more severe outcomes of CF.6 In addition to indirectly changing airway microbiota by addressing CFTR dysfunction and its effects on mucus hydration and mucociliary clearance, there is growing evidence that ivacaftor has direct antimicrobial properties.7,8 We have previously shown that ivacaftor (IVA) and polymyxin B (PMB) monotherapy were ineffective when used against multidrug-resistant (MDR) P. aeuruginosa, although when used in combination (COM), there was a synergistic antibacterial effect against these isolates.7 The mechanisms of this synergistic action have not been fully elucidated; however, we have shown that the resulting outer membrane damage to P. aeruginosa cells imparted by the combination treatment was distinct from the effect of each compound per se.(7) Moreover, in our previous studies, ivacaftor was also shown to be a weak inhibitor of the bacterial DNA gyrase and topoisomerase IV with no effect on either human type I or type II topoisomerases, despite its structural similarity to quinolone antibiotics.
The relatively large genome of P. aeruginosa (5.9–6.3 Mb) has noteworthy metabolic fluidity, allowing isolates to rapidly adapt to external influences such as antibiotic treatments.9 Mechanisms of resistance in P. aeruginosa include the target alteration, induction of efflux pumps, and enzymatic inactivation of antibiotics.10,11 Unfortunately, reports of resistance against last-line antibiotics such as polymyxins (polymyxin B and colistin) are becoming more common.12 Colistin is clinically used as its inactive prodrug colistimethate sodium (CMS).13 More recently, CMS dry powder pod-inhaler (Colobreathe) or nebulizer solution (Coly-Mycin) is used for the treatment of respiratory infections caused by P. aeruginosa in CF patients.14 An emerging repurposing approach to combat multidrug resistance is to combine FDA-approved nonantibiotic drugs that show synergistic antibacterial killing when combined with antibiotics such as polymyxins.7,15,17−19 Polymyxin B has previously been shown to display synergistic antibacterial effects when combined with nonantibiotic drugs such as ivacaftor, closantel, tamoxifen, raloxifene, toremifene, mitotane, and zidovudine.7,15,17,18,20−22
Systems pharmacology allows deciphering of the complex interplay between cellular pathways in response to drug treatments.23 Metabolomics provide the opportunity to gain a system-wide picture of cellular biochemical networks under defined conditions to shed light upon the complex modes of action and bacterial cellular processes in response to drug treatment. To the best of our knowledge, we are the first to conduct an untargeted metabolomics analysis of the synergistic killing mechanism of the novel CFTR potentiator ivacaftor with the antibiotic polymyxin B in combination.
Results
Multivariate and Univariate Analyses of the Metabolites Affected by Polymyxin B and Ivacaftor in P. aeruginosa
Multivariate data analysis using one-way analysis of variance (ANOVA) followed by partial least-squares discriminant analysis (PLSDA) was performed to determine the significant metabolites (FDR ≤ 0.05; Fisher’s LSD, p ≤ 0.05) induced by different treatments (i.e., polymyxin B or ivacaftor monotherapy and combination therapy) against polymyxin-susceptible (FADDI-PA111) and polymyxin-resistant (FADDI-PA006) P. aeruginosa (Figure S1).The reproducibility of metabolite semi-quantitation was acceptable in both strains, where the median RSD was (18–22%) for all untreated (control) groups and (15–25%) for all treated samples (Table S1).
Notably, the PLSDA plots showed that the untreated control and combination treated samples were significantly differentiated in the polymyxin-susceptible (FADDI-PA111) and polymyxin-resistant (FADDI-PA006) P. aeruginosa (Figure S1A,1B). According to the PLSDA of FADDI-PA111, ivacaftor treatment samples resembled the combination treatment samples, whereas polymyxin B and the untreated control treatments overlapped, which suggests that ivacaftor elevates the antibacterial killing of polymyxin B (Figure S1A). Samples treated with ivacaftor alone and polymyxin B alone were comparable with the untreated control in FADDI-PA006 (Figure S1B). These differences between the treatment groups is reflected in the heat maps of both strains (P. aeruginosa FADDI-PA111 and FADDI-PA006; Figure S2A,B). The heat maps show opposing effects of PMB and IVA on the PA111 metabolome and an approximately equal influence of both compounds in the combination treatment. In PA0006, however, there is a strong ivacaftor-driven effect on the peptides, nucleotides, cofactors and vitamins, lipids, and glycan biosynthesis metabolites, while the effects carbohydrate, amino acid, and undefined metabolites seem to be more polymyxin B driven.
A total of 926 (FADDI-PA111) and 680 (FADDI-PA006) putatively identified metabolites were detected by using hydrophilic interaction liquid chromatography (HILIC)-based high-resolution accurate mass LC-MS. Among metabolite classes, lipid intermediates constituted the highest proportion, ∼20 and ∼40%, in polymyxin-susceptible (FADDI-PA111) and polymyxin-resistant (FADDI-PA006) P. aeruginosa, respectively. This is followed by amino acids, peptides, and carbohydrates metabolites (Figure S3).
The combination caused significant perturbations in a total of 202 in FADDI-PA111 and 183 in FADDI-PA006. Fewer perturbations were observed following ivacaftor monotherapy with a total of 108 and 53 metabolites in FADDI-PA111 and FADDI-PA006, respectively (Figure S4). Treatment of FADDI-PA111 with polymyxin B alone showed a significant effect in polymyxin-susceptible strain FADDI-PA111 by perturbing 190 metabolites; however, this displayed a trivial effect of significantly perturbing only 5 metabolites in FADDI-PA006 (Figure S4). The Venn diagrams showed that there were 85 uniquely significantly impacted metabolites induced by the combination treatment in FADDI-PA111 and 144 in FADDI-PA006 (Figure 1A,B). Notably, there were 32 significant metabolites in common between polymyxin B, ivacaftor, and their combination in FADDI-PA111 and 3 shared metabolites in FADDI-PA006 (Figure 1A).
Figure 1.
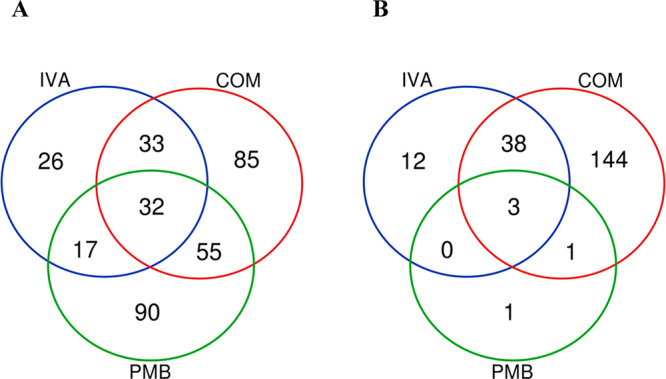
Venn diagrams represent the number of metabolites significantly affected by each treatment for (A) polymyxin-susceptible (FADDI-PA111) and (B) polymyxin-resistant (FADDI-PA006) P. aeruginosa. Significant metabolites were selected (≥0.59-log2-fold, p ≤ 0.05; FDR ≤ 0.05).
The statistically significant metabolites influenced by each treatment (one-way ANOVA, FDR ≤ 0.05; Fisher’s LSD, p ≤ 0.05) were allocated into seven different metabolite classes: carbohydrates, amino acids, lipids, peptides, nucleotides, energy, glycan, and others (the latter includes cofactors and vitamins, secondary metabolites, and metabolites that could not be mapped into pathways). The number of significant metabolites from each class that increased or decreased in abundance compared to the untreated control group is illustrated in Figure 2.
Figure 2.

Summary number of significantly changed metabolites classified according to different metabolite classes after polymyxin B, ivacaftor, and combination treatment of P. aeruginosa FADDI-111 and FADDI-PA006 (changes >0.59-log2-fold, p ≤ 0.05; FDR ≤ 0.05).
Impact of Polymyxin B, Ivacaftor, and Their Combination on Lipid Metabolism
In the polymyxin-susceptible strain FADDI-PA111, polymyxin B treatment alone induced significant perturbations in the levels of a wide range of fatty acids (FAs) such as 3R-hydroxy-tetradecanoic acid (log2 FC = −9.3307), one of the key fatty acids (FA) of the lipid A component of bacterial lipopolysaccharides (LPS),24 and to a lesser extent in glycerophospholipids (GPLs) (Figure 3A). Notably, the abundance of essential bacterial phospholipids and precursors increased following polymyxin B monotherapy, including sn-glycero-3-phosphocholine (log2 FC = 4.4009) and sn-glycero-3-phosphoethanolamine (log2 FC = 2.349). Likewise, ivacaftor monotherapy caused a greater number of significant perturbations in FAs and fewer in GPLs. However, among GPLs, a marked reduction in the levels of essential bacterial phospholipids such as sn-glycero-3-phosphoethanolamine (log2 FC = −1.9094) and sn-glycerol 3-phosphate (log2 FC = −0.63925) was observed (Figure 3A). Compared to each monotherapy, the combination treatment showed a more pronounced impact on FADDI-PA111 lipid metabolism (FAs and GPLs) as manifested by declining levels of 3R-hydroxy-tetradecanoic acid (log2 FC = −9.5105), tetradecanoic acid (myristic acid) (log2 FC = −0.92399), and FA(16:0) (palmitic acid) (log2 FC = −1.3352). Notably, these are the most abundant FAs in the Gram-negative membrane.25 Furthermore, essential bacterial phospholipids (sn-glycerol 3-phosphate, sn-glycero-3-phosphoethanolamine, sn-glycero-3-phosphocholine, and choline) were substantially decreased after combination therapy (≥−2.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 3A).
Figure 3.

Significantly impacted lipids in (A) P. aeruginosa FADDI-PA111 and (B) FADDI-PA006 following treatment with polymyxin B (PMB, green), ivacaftor (IVA, blue), and their combination (COM, red). Lipid names are putatively assigned based on accurate mass, (≥0.59-log2-fold, p ≤ 0.05; FDR ≤ 0.05).
In contrast, against polymyxin-resistant FADDI-PA006, polymyxin B monotherapy did not impact any metabolites involved in FAs and GPLs metabolism; however, a significant effect on three bacterial membrane phospholipid precursors, namely, sn-glycerol 3-phosphate, 2-acyl-sn-glycero-3-phosphoethanolamine, and sn-glycero-3-phosphocholine was seen (≥−1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 3B). The effect of ivacaftor monotherapy was more profound than that of polymyxin B alone as seen by a significant suppression in the levels of FAs and GPLs intermediates, including FA hydroxy(18:1), LysoPC(16:0), and LysoPC(18:0) (≥−1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 3B). The combination treatment induced an even higher level of suppression of a wide range of FAs and GPLs such as FA oxo(16:0) (3-oxopalmitic acid), LysoPC(16:0), (2-palmitoyl-sn-glycero-3-phosphoethanolamine), and 2-acyl-sn-glycero-3-phosphoethanolamine (≥1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05). Additionally, the combination treatment also caused significant perturbation in the main precursors of bacterial phospholipids formation, namely, sn-glycerol 3-phosphate, sn-glycero-3-phosphoethanolamine, and sn-glycero-3-phosphocholine (log2 FC = −1.272, −4.4605 and −2.2024, respectively) (Figure 3B). Our results suggest that the combination of polymyxin B and ivacaftor reduced the main precursors of bacterial membrane lipids which appeared to be driven by ivacaftor elevating polymyxin-mediated bacterial killing. The impact of polymyxin B, ivacaftor alone and the combination treatment on glycerophospholipid metabolism in polymyxin-susceptible FADDI-PA111 as shown in Figure 4A,B. In FADDI-PA111, polymyxin B and combination therapy caused a significant reduction in total of five essential intermediates of glycerophospholipid metabolism, namely, sn-glycero-3-phosphocholine, choline, sn-glycerol 3-phosphate, sn-glycero-3-phosphoethanolamine, and 2-acyl-sn-glycero-3-phosphoethanolamine (≥−0.59-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 4A). Meanwhile, only three metabolites, namely, sn-glycerol 3-phosphate, sn-glycero-3-phosphoethanolamine, and choline, underwent a significant reduction after ivacaftor monotherapy (≥−0.59-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 4A).
Figure 4.

(A) Schematic diagram depicting glycerophospholipids metabolism in P. aeruginosa FADDI-PA111 and (B) bar charts for the significantly impacted metabolites involved in glycerophospholipids metabolism of FADDI-PA111 and FADDI-PA006 following treatment with polymyxin B (PMB, green), ivacaftor (IVA, blue), and their combination (COM, red) (≥1.0-log2-fold, p ≤ 0.05; FDR ≤ 0.05). Data represent geometric means of relative intensity ± standard deviation (n = 4).
In the polymyxin-resistant P. aeruginosa FADDI-PA006, a slight alteration in glycerophospholipid metabolism was observed after polymyxin B and ivacaftor monotherapies compared to that with combination treatment. The abundance of sn-glycero-3-phosphoethanolamine, sn-glycero-3-phosphocholine, and sn-glycerol 3-phosphate were markedly reduced after treatment with polymyxin B alone (log2 FC = −2.61, −1.17, and −1.29, respectively). Ivacaftor was slightly effective in decreasing the levels of sn-glycero-3-phosphoethanolamine (log2 FC = −2.78) and sn-glycero-3-phosphocholine (log2 FC = −1.3618) (Figure 4B). The combination treatment significantly reduced the abundance of five crucial intermediates of glycerophospholipid metabolism, including ethanolamine phosphate (a key intermediate used by the bacteria to modify LPS to attain polymyxin resistance), sn-glycerol 3-phosphate, sn-glycero-3-phosphoethanolamine, 2-acyl-sn-glycero-3-phosphoethanolamine, and sn-glycero-3-phosphocholine (≥−1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 4B).
Significantly Impacted Metabolites in Amino–Sugar and Sugar–Nucleotide Metabolism and Pentose Phosphate Pathway (PPP) and Their Downstream Peptidoglycan and Lipopolysaccharide Biosynthesis
The impact of polymyxin B, ivacaftor, and their combination on amino sugar and sugar nucleotide, PPP, and their interrelated pathways in P. aeruginosa FADDI-PA111 is shown in Figure 5A. In FADDI-PA111, five intermediates of amino sugar and sugar nucleotides were induced after polymyxin B and ivacaftor monotherapy. The combination produced a more pronounced effect in the abundance of nine components of amino sugar and sugar nucleotide metabolism including UDP-N-acetyl-d-glucosamine, N-acetyl-d-glucosamine, N-acetyl-d-glucosamine 6-phosphate, and UDP-N-acetylmuramate (≥1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 5A,B). The abundance of an essential metabolite of PPP (d-sedoheptulose 7-phosphate) and the downstream LPS intermediate [3-deoxy-d-manno-octulosonate (KDO)] were perturbed as a consequence of the aforementioned perturbations; 3-deoxy-d-manno-octulosonate was significantly altered by both polymyxin B and ivacaftor monotherapies (log2 FC = −1.7218 and 1.7542, respectively) (Figure 5A,B). The downstream peptidoglycan pathway was significantly perturbed by polymyxin B monotherapy and combination treatment in FADDI-PA111, with greater levels of perturbation produced by the combination treatment. This is indicated by marked changes in the levels of 3 fundamental components (d-alanyl-d-alanine, UDP-MurNAc-l-Ala-γ-d-Glu-m-DAP, and d-isoglutamine) and four key precursors (d-alanyl-d-alanine, UDP-MurNAc-l-Ala-γ-d-Glu-m-DAP, l-Ala-d-Glu-meso-A2 pm, and d-isoglutamine) following polymyxin B monotherapy and the combination treatment, respectively (≥1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 5A,B).
Figure 5.

(A) Schematic diagram depicted the complex interrelated pathways (amino sugar and sugar nucleotide metabolism, PPP, and peptidoglycan and LPS biosynthesis) in P. aeruginosa FADDI-PA111 and (B) fold changes for the significantly impacted metabolites involved in amino sugar and sugar nucleotide metabolism and PPP and their direct downstream peptidoglycan and LPS biosynthesis in FADDI-PA111 and FADDI-PA006 following treatment with polymyxin B (PMB, green), ivacaftor (IVA, blue), and their combination (COM, red), (≥1.0-log2-fold, p ≤ 0.05; FDR ≤ 0.05).
In polymyxin-resistant P. aeruginosa FADDI-PA006, both monotherapies were ineffectual, and only the combination treatment produced significant perturbations in amino sugar and sugar nucleotide metabolism and its downstream peptidoglycan and LPS biogenesis (Figure 5B). The abundances of four key precursors of amino sugar and sugar nucleotide, namely, d-glucosamine 6-phosphate, UDP-N-acetyl-d-glucosamine, d-glucosamine, and UDP-N-acetylmuramate (≥1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05), were disproportionately increased following combination treatment. Simultaneously, a similar number of metabolites involved in peptidoglycan biogenesis experienced a significant perturbation, including d-alanyl-d-alanine, UDP-MurNAc-l-Ala-γ-d-Glu-m-DAP, UDP-MurNAc-l-Ala-γ-d-Glu-m-DAP-d-Ala-d-Ala, and l-Ala-d-Glu-meso-A2 pm (≥1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 5B). The intracellular concentration of fundamental LPS core metabolite 3-deoxy-d-manno-octulosonate was altered remarkably after combination treatment (≥4.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 5B).
Impact of Polymyxin B, Ivacaftor, and Combination Treatment on Pantothenate and Coenzyme A Pathway Intermediates
Treatments with polymyxin B and ivacaftor alone and in combination caused significant perturbations to several metabolites involved in pantothenate and coenzyme A (CoA) biosynthesis in FADDI-PA111 (Figure 6A). A higher level of alteration in this pathway, seen from more metabolites altered, was caused in polymyxin-resistant FADDI-PA006 after combination treatment only (Figure 6B). Phosphopantetheine and CoA are vital for enzymes involved in the synthesis and degradation of fatty acids, the synthesis of phospholipids, and the tricarboxylic acid (TCA) cycle.26 The levels of seven key precursors of pantothenate and CoA biosynthesis were significantly increased after the combination treatment in FADDI-PA111, including (R)-pantoate, pantotenate, panthetine 4′-phosphate, 3-methyl-2-oxobutanoic acid, panteteine, adenosine 3′,5′-biphosphate, and CoA (≥1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05) (Figure 6A). Notably, in FADDI-PA111, polymyxin B monotherapy produced minimal impact on pantothenate and CoA biosynthesis, wherein four components underwent a significant depletion, namely, pantothenate (log2 FC = −1.34), pantetine 4′-phosphate (log2 FC = −3.3775), 3-methyl-2-oxobutanoic acid (log2 FC = −2.5263), and pantotenate (log2 FC = −1.3397) (Figure 6A), while ivacaftor monotherapy significantly perturbed only two essential intermediates which underwent a dramatic increase in FADDI-PA111, namely, (R)-pantoate (log2 FC = 1.242) and 3-methyl-2-oxobutanoic acid (log2 FC = 1.1558) (Figure 6A).
Figure 6.

Schematic diagrams and whisker plots showing pantothenate and CoA biosynthesis in P. aeruginosa (A) FADDI-PA111 and (B) FADDI-PA006 following treatment with polymyxin B (PMB, green), ivacaftor (IVA, blue), and their combination (COM, red) (≥1.0-log2-fold, p ≤ 0.05; FDR ≤ 0.05). Data represent geometric means of relative intensity ± standard deviation (n = 4).
In polymyxin-resistant P. aeruginosa FADDI-PA006, the highest level of perturbation was observed only with combination treatment. The abundance of CoA, pantetheine, adenosine 3′,5′-bisphosphate, pantothenate, pantetheine 4′-phosphate, (R)-pantoate, and 3-methyl-2-oxobutanoic acid were increased dramatically after combination treatment (≥1.0-log2-fold, p ≤ 0.05, FDR ≤ 0.05), whereas there was no significant impact for polymyxin B and ivacaftor monotherapies on pantothenate and CoA biosynthesis in FADDI-PA006 (Figure 6B).
Discussion
Polymyxin-containing nebulizers and inhalers are commonly used for the treatment of respiratory infections caused by P. aeruginosa in CF patients;14 however, unfortunately reports of resistance are becoming more common.12 Therefore, rational combination therapy is strongly recommended. Reznikov et al. published positive interactions of the CFTR potentiator ivacaftor with other antibiotics suggesting additivity or synergy.8 Previously, our group has reported the synergistic antibacterial activity of ivacaftor with the last-line antibiotic polymyxin B against P. aeruginosa.7 To our knowledge, the present study is the first to demonstrate that the synergistic killing of polymyxin B in combination with the CFTR potentiator ivacaftor is elevated by ivacaftor.
Polymyxin B was examined at 2 μg/mL for FADDI-PA111 and 4 μg/mL for FADDI-PA006 P. aeruginosa to ensure the clinical relevance of our findings.27 In addition, clinically achievable concentrations of ivacaftor (8 μg/mL) were used for the present study.28,29 Neither 4 μg/mL polymyxin B nor 8 μg/mL ivacaftor had a significant killing effect on resistant FADDI-PA006 P. aeruginosa using an inoculum of 108 CFU/mL. Consistent with phenotypic data previously reported by our group, the combination of polymyxin B and ivacaftor exhibited synergistic killing against polymyxin-resistant P. aeruginosa.7 Bacterial metabolic profiles were examined at 3 h following combination treatment in order to understand the molecular basis of the dynamic widespread killing. In line with phenotypic data, including scanning and transmission electron microscopy imaging, our metabolomic results demonstrated that the effect of the combination treatment was clearly separated from each monotherapy per se. Metabolomic analysis highlighted that the combination treatment effect was attributed to key metabolic pathways including bacterial membrane lipids biogenesis, cell wall (LPS and peptidoglycan) formation, and central carbohydrate metabolism.
Due to the similarity of ivacaftor to quinolone antibiotics, it has been hypothesized that ivacaftor employs a mode of action that is similar to quinolones.8 The antibacterial action of quinolone antibiotics involves the inhibition of the related DNA gyrase and topoisomerase IV enzymes which are involved in DNA replication.30 In our previous study, we have reported that antibacterial activity of ivacaftor does not involve a quinolone-like mode of action, as quinolones are strong interfacial poising agents while ivacaftor is not (it is a weak catalytic inhibitory compound).7 Consistent with this previous finding, we report ivacaftor and its combination did strongly affect metabolites from the glycerophospholipids and fatty acids metabolism as well as bacterial membrane remodeling intermediates (e.g., ethanolamine phosphate) at 3 h, highlighting that the metabolic changes induced by ivacaftor or the combination treatment do not resemble the changes observed with quinolone antibiotics.31 Our study highlights that the synergistic effects of the combination treatment is driven by ivacaftor elevating polymyxin B mediated bacterial killing. It is also largely due to the inhibition of cell envelope biogenesis via suppression of key membrane lipid metabolites (e.g., sn-glycerol 3-phosphate and sn-glycero-3-phosphoethanolamine) as well as perturbations in peptidoglycan and lipopolysaccharide biosynthesis. Our metabolomics results highlight the potential of a combination therapy of polymyxin B and ivacaftor in minimizing the development of resistance in P. aeruginosa. The lipopeptide antibiotic polymyxin E (colistin) has been commercially available in the clinic as its inactive prodrug colistimethate sodium (CMS) as both nebulizer solution and dry powder pod-inhaler for the treatment of respiratory infections caused by P. aeruginosa in CF patients. Hence, combining ivacaftor with polymyxin B as an inhaled therapy incorporates several synergistic elements: (1) combination treatment of lung infections via unique mechanisms and (2) ivacaftor correction of the defective CFTR and enhancement of mucociliary clearance.32 The possibility of introducing these drugs together into a nebulizer or inhalation device has potential possibilities and deserves further study.
Conclusions
Given that polymyxin B is an outer-membrane-active antibiotic which exerts a disruptive/permeabilizing effect, the concerted action of ivacaftor against bacterial membrane lipid biogenesis provides an ideal/targeted synergistic effect. Taken together, the metabolomics mechanistic data presented herein support the repurposing of polymyxin B in combination with ivacaftor for the treatment of problematic polymyxin-resistant P. aeruginosa lung infections in CF patients.
Material and Methods
Materials
Ivacaftor was purchased from (SelleckChem, USA); stock solutions were prepared in DMSO. Polymyxin B (catalog number 81334, ≥6500 IU/mg) was purchased from Sigma-Aldrich (Australia) and prepared in Milli-Q water (Millipore, NSW, Australia). Stock solutions of both drugs were freshly prepared prior to each experiment and sterilized by filtration with a 0.22 μM Millex GP filter (Millipore, Bedford, MA). All other reagents were purchased from Sigma-Aldrich (Australia) and are of the highest commercial grade available.
Bacterial Isolates
Polymyxin-susceptible P. aeruginosa FADDI-PA111 (PMB MIC = 2 μg/mL, IVA MIC > 128 μg/mL) and polymyxin-resistant P. aeruginosa isolated from the lungs of a CF patient (FADDI-PA006, (PMB MIC = 8 μg/mL, IVA MIC > 32 μg/mL) were employed for this study. Polymyxin–ivacaftor death curves were obtained for various doses and treatment durations by cell counts of cultures at bacterial colony forming unit (CFU) density that would provide a suitable biomass for metabolomic analysis (8–9 × log10 cells mL–1) (data not shown). We selected drug concentrations of polymyxin B alone (2 μg/mL for FADDI-PA111 and 4 μg/mL FADDI-PA006 P. aeruginosa), ivacaftor alone (8 μg/mL), and for combination therapy that mediated an approximately 20% reduction of bacteria within the treatment window. The isolates were stored in tryptone soy broth (Oxoid) with 20% glycerol (Ajax Finechem, Seven Hills, New South Wales, Australia) in cryovials at −80 °C. Before use, FADDI-PA111 and FADDI-PA006 were grown in cation-adjusted Mueller–Hinton broth (CAMHB; 20–25 mg/L Ca2+ and 10–12.5 mg/L Mg2+) for 24 and 48 h, respectively.
Metabolomics Experiments
A single bacterial colony was used to inoculate 10–15 mL of cation-adjusted Mueller–Hinton broth (CAMHB; Oxoid, England; 20–25 mg/L Ca2+ and 10–12.5 mg/L Mg2+) in 50 mL Falcon tubes (Thermo Fisher, Australia) and incubated in a water bath at 37 °C (shaking speed, 180 rpm) overnight. Following overnight incubation, each culture was transferred to a 500 mL conical flask with 200 mL of fresh CAMHB at ∼50–100-fold dilution. Flasks were further incubated at 37 °C with shaking at 180 rpm for ∼2 h to log phase (OD600 ∼0.5). Polymyxin B and ivacaftor treatments alone and combination treatment were added to three of the four flasks to give a final concentrations as follows: polymyxin B, 2 μg/mL for FADDI-PA111 and 4 μg/mL for FADDI-PA006; ivacaftor, 8 μg/mL for both strains. The remaining flask acted as a drug-free control. The concentrations were chosen based on a serial of optimization and adjustment steps including time kill studies using high starting inoculum size (∼108 CFU/mL) to avoid excessive antibacterial effects (Figure S5A,B). The flasks were further incubated at 37 °C. After 3 h, the OD600 was read for each flask and normalized to ∼0.5 with fresh CAMHB. For quenching and extraction, 15 mL of each flask were transferred to 50 mL Falcon tubes (Thermo Fisher, Australia). Four biological samples per isolate were prepared for each treatment to account for inherent random variation.
Metabolite Extraction for Metabolomic Studies
Following bacterial culture preparation, extraction of metabolites was immediately carried out in order to decrease further drug effects on metabolite levels. Initially, samples were centrifuged at 3220 × g at 4 °C for 20 min. Supernatants were then removed, and bacterial pellets washed twice in 1 mL of cold normal saline followed by centrifugation at 3220 × g at 4 °C for 10 min to remove residual extracellular metabolites and medium components. Then, 300 μL of cold chloroform/methanol/water (CMW; 1:3:1, v/v) extraction solvent containing 1 μM each of the internal standards (CHAPS, CAPS, PIPES, and TRIS) was added to the washed pellets. The used internal standards are physiochemically different small molecules which do not naturally exist in any microorganism. Samples were then thrice immersed in liquid nitrogen, thawed on ice, and vortexed to liberate the intracellular metabolites. The samples were centrifuged for 10 min at 3220 × g at 4 °C after a third freeze–thaw cycle, whereby 300 μL samples of the supernatants were taken to 1.5 mL Eppendorf tubes. Centrifugation at 14 000 × g at 4 °C for 10 min was used to detach any particles from samples, and 200 μL samples were transferred into the injection vials for storage in a −80 °C freezer. For LC-MS analysis (described below), the samples were taken out from the −80 °C freezer to thaw, and 10 μL of each sample was transferred to a vial and used as a pooled quality control sample (QC), namely, a sample that contains all the analytes that will be encountered during the analysis.33
LC-MS Analysis
Metabolites were identified with HILIC high-resolution mass spectrometry (HRMS) using a Dionex high-performance liquid chromatography (HPLC) system (RSLC U3000, Thermo Fisher) with a ZIC-pHILIC column (5 μm, polymeric, 150 × 4.6 mm; SeQuant, Merck). The system was coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher) operated in both positive and negative electro-spray ionization (ESI) mode (rapid switching) at 35 000 resolution with a detection range of 85–1275 m/z. Two LC solvents, (A) 20 mM ammonium carbonate and (B) acetonitrile, were used which operated via a multistep gradient system. The gradient started at 80% B which declined to 50% B over 15 min and then reduced from 50% B to 5% B over 3 min, followed by a wash with 5% B for another 3 min, and finally 8 min of re-equilibration with 80% B at a flow rate of 0.3 mL/min.34 The injection sample volume was 10 μL, and the total run time was 32 min. All samples were analyzed as a single LC-MS batch to avoid batch-to-batch variation. Mixtures of pure standards containing over 300 metabolites were also included in the analysis batch to aid metabolite identification.
Data Processing, Bioinformatics, and Statistical Analyses
Conversion of LC-MS raw data to metabolite levels was conducted using IDEOM software (http://mzmatch.sourceforge.net/ideom.php),35 which initially employed ProteoWizard to convert raw LC-MS data to mzXML format and XCMS to pick peaks with Mzmatch.R to convert to peakML files.36,37 Mzmatch.R was subsequently used for the alignment of samples and the filtering of peaks using a minimum peak intensity threshold of 100 000, relative standard deviation (RSD) < 0.5 (reproducibility), and peak shape (codadw) > 0.8. Mzmatch was also used to retrieve missing peaks and annotate related peaks. Default IDEOM parameters were used to eliminate unwanted noise and artifact peaks. Loss or gain of a proton was corrected in negative and positive ESI mode, respectively, followed by putative identification of metabolites by the exact mass within 2 ppm. Univariate statistical analysis was conducted using one-way ANOVA for multiple group comparisons, and the p-value was corrected using the Benjamini–Hochberg method to ensure false discovery rates (FDR) < 0.05.38 Furthermore, metabolites were identified by a two-level identification process: Retention times of authentic standards and accurate mass time with authentic standards as indicated by IDEOM confidence score of 9 or 10 (corresponding to Metabolomics Standards Initiative Guidelines) were used to confirm the identification of each metabolite (level 1 identification based on MSI standards). Other metabolites were putatively identified (level 2 identification based on MSI standards) using exact mass and predicted retention time based time to achieve an IDEOM confidence score of 6 or greater (metabolomics standards initiative level 2/3); metabolites showing a change of ≥2-fold are further examined and subjected to pathway analysis according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway as well as MetaCyc and LIPIDMAPS databases, using preference to bacterial metabolites annotated in EcoCyc in cases where isomers could not be clearly differentiated by retention time.39−41 Raw peak intensity was used to quantify each metabolite. The free online tool MetaboAnalyst 3.0 was used for the statistical analysis. Briefly, putative metabolites with median RSD ≤ 0.2 (20%) within the QC group and IDEOM confidence level of ≥5 were incorporated into a table and uploaded to MetaboAnalyst. Features with >50% missing values were replaced by half of the minimum positive value in the original data. Interquartile ranges (IQRs) were utilized to filter data, then log2 transformation and autoscaling were used to normalize the data. Partial least-squares discriminant analysis (PLSDA) was performed to identify and remove outliers. PLSDA is normally used to reduce the dimension of variables from a large data set.42 One-way ANOVA was used to identify metabolites with significant level changes between all samples, and Fisher’s least-squares difference (LSD) was used to determine the metabolites with significant level changes between treatment and control groups. Statistically significant metabolites were selected using a false discovery rate of ≤0.1 for one-way ANOVA and p ≤ 0.05 for Fisher’s LSD. KEGG mapper was used to determine the pathway modules by statistically significant metabolites containing the KEGG compound numbers.
Acknowledgments
The authors thank Dr Maytham Hussein (University of Melbourne) for technical assistance and Prof Jian Li (Monash University) and A/Prof Tony Velkov (The University of Melbourne) for technical support. E.K.S.-F. is supported by the Peter Phelan Research Award from The Thoracic Society of Australia and New Zealand and by the Australian National Health and Medical Research Council (NHMRC) as a Biomedical Research Fellow.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00030.
Additional information on data precision of individual samples (Table S1), analysis of global metabolic changes (Figures S1–S4), as well as killing kinetics (Figure S5) (PDF)
Author Contributions
# R.A. and D.P.G. contributed equally to this work
Parts of this work were delivered at the Australian Society of Clinical and Experimental Pharmacology and Toxicology Annual Meeting 2019 within the Women Early Career Achievement Lecture.
The authors declare no competing financial interest.
Supplementary Material
References
- Bobadilla J. L.; Macek M. Jr.; Fine J. P.; Farrell P. M. (2002) Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum. Mutat. 19 (6), 575–606. 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- Schneider E. K.; Huang J. X.; Carbone V.; Baker M.; Azad M. A.; Cooper M. A.; Li J.; Velkov T. (2015) Drug-drug plasma protein binding interactions of ivacaftor. J. Mol. Recognit. 28 (6), 339–48. 10.1002/jmr.2447. [DOI] [PubMed] [Google Scholar]
- O’Sullivan B. P.; Flume P. (2009) The clinical approach to lung disease in patients with cystic fibrosis. Seminars in respiratory and critical care medicine 30 (5), 505–13. 10.1055/s-0029-1238909. [DOI] [PubMed] [Google Scholar]
- Schneider E. K.; Reyes-Ortega F.; Li J.; Velkov T. (2017) Can cystic fibrosis patients finally catch a breath with Orkambi?. Clin. Pharmacol. Ther. 101, 130. 10.1002/cpt.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treggiari M. M.; Rosenfeld M.; Retsch-Bogart G.; Gibson R.; Ramsey B. (2007) Approach to eradication of initial Pseudomonas aeruginosa infection in children with cystic fibrosis. Pediatr Pulmonol 42 (9), 751–6. 10.1002/ppul.20665. [DOI] [PubMed] [Google Scholar]
- Goss C. H.; Burns J. L. (2007) Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax 62 (4), 360–7. 10.1136/thx.2006.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E. K.; Azad A. K.; Han M.-L.; Zhou Q.; Wang J.; Huang J. X.; Cooper M. A.; Doi Y.; Baker M. A.; Bergen P. J.; Muller M.; Li J.; Velkov T. (2016) An “Unlikely” Pair: The Antimicrobial Synergy of Polymyxin B in Combination with the Cystic Fibrosis Transmembrane Conductance Regulator Drugs KALYDECO and ORKAMBI. ACS Infect. Dis. 2, 478. 10.1021/acsinfecdis.6b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikov L. R.; Abou Alaiwa M. H.; Dohrn C. L.; Gansemer N. D.; Diekema D. J.; Stoltz D. A.; Welsh M. J. (2014) Antibacterial properties of the CFTR potentiator ivacaftor. J. Cystic Fibrosis 13 (5), 515–9. 10.1016/j.jcf.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradali M. F.; Ghods S.; Rehm B. H. (2017) Pseudomonas aeruginosa lifestyle: a paradigm for adaptation, survival, and persistence. Front. Cell. Infect. Microbiol. 7, 39. 10.3389/fcimb.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert P. (2002) Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J. R. Soc. Med. 95, 22–26. [PMC free article] [PubMed] [Google Scholar]
- Lister P. D.; Wolter D. J.; Hanson N. D. (2009) Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22 (4), 582–610. 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz S. M.; Brannon M. K.; Dasgupta N.; Pier M.; Sgambati N.; Miller A. K.; Selgrade S. E.; Miller S. I.; Denton M.; Conway S. P.; Johansen H. K.; Hoiby N. (2012) PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 56 (2), 1019–30. 10.1128/AAC.05829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun B.; Azad M. A.; Wang J.; Nation R. L.; Thompson P. E.; Roberts K. D.; Velkov T.; Li J. (2015) Imaging the distribution of polymyxins in the kidney. J. Antimicrob. Chemother. 70 (8), 827–829. 10.1093/jac/dku441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov T.; Abdul Rahim N.; Zhou Q. T.; Chan H. K.; Li J. (2015) Inhaled anti-infective chemotherapy for respiratory tract infections: Successes, challenges and the road ahead. Adv. Drug Delivery Rev. 85, 65–82. 10.1016/j.addr.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein M. H.; Schneider E. K.; Elliott A. G.; Han M.; Reyes-Ortega F.; Morris F.; Blaskovich M. A. T.; Jasim R.; Currie B.; Mayo M.; Baker M.; Cooper M. A.; Li J.; Velkov T. (2017) From Breast Cancer to Antimicrobial: Combating Extremely Resistant Gram-Negative ″Superbugs″ Using Novel Combinations of Polymyxin B with Selective Estrogen Receptor Modulators. Microb. Drug Resist. 23 (5), 640–650. 10.1089/mdr.2016.0196. [DOI] [PubMed] [Google Scholar]
- Tran T. B.; Bergen P. J.; Creek D. J.; Velkov T.; Li J. (2018) Synergistic Killing of Polymyxin B in Combination With the Antineoplastic Drug Mitotane Against Polymyxin-Susceptible and -Resistant Acinetobacter baumannii: A Metabolomic Study. Front. Pharmacol. 9, 359. 10.3389/fphar.2018.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T. B.; Wang J.; Doi Y.; Velkov T.; Bergen P. J.; Li J. (2018) Novel Polymyxin Combination With Antineoplastic Mitotane Improved the Bacterial Killing Against Polymyxin-Resistant Multidrug-Resistant Gram-Negative Pathogens. Front. Microbiol. 9, 721. 10.3389/fmicb.2018.00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E. K.; Reyes-Ortega F.; Velkov T.; Li J. (2017) Antibiotic-non-antibiotic combinations for combating extremely drug-resistant Gram-negative ’superbugs’. Essays Biochem. 61 (1), 115–125. 10.1042/EBC20160058. [DOI] [PubMed] [Google Scholar]
- Lin Y. W.; Abdul Rahim N.; Zhao J.; Han M. L.; Yu H. H.; Wickremasinghe H.; Chen K.; Wang J.; Paterson D. L.; Zhu Y.; Rao G. G.; Zhou Q. T.; Forrest A.; Velkov T.; Li J. (2019) Novel Polymyxin Combination with the Antiretroviral Zidovudine Exerts Synergistic Killing against NDM-Producing Multidrug-Resistant Klebsiella pneumoniae. Antimicrob. Agents Chemother. 63 (4), e02176-18. 10.1128/AAC.02176-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein M.; Han M. L.; Zhu Y.; Schneider-Futschik E. K.; Hu X.; Zhou Q. T.; Lin Y. W.; Anderson D.; Creek D. J.; Hoyer D.; Li J.; Velkov T. (2018) Mechanistic Insights From Global Metabolomics Studies into Synergistic Bactericidal Effect of a Polymyxin B Combination With Tamoxifen Against Cystic Fibrosis MDR Pseudomonas aeruginosa. Comput. Struct. Biotechnol. J. 16, 587–599. 10.1016/j.csbj.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T. B.; Cheah S. E.; Yu H. H.; Bergen P. J.; Nation R. L.; Creek D. J.; Purcell A.; Forrest A.; Doi Y.; Song J.; Velkov T.; Li J. (2016) Anthelmintic closantel enhances bacterial killing of polymyxin B against multidrug-resistant Acinetobacter baumannii. J. Antibiot. 69 (6), 415–21. 10.1038/ja.2015.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R.; Weinshilboum R. M. (2014) Pharmacometabolomics: implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 95 (2), 154–167. 10.1038/clpt.2013.217. [DOI] [PubMed] [Google Scholar]
- TMIC . 3-Hydroxytetradecanoic acid (HMDB0061656). https://hmdb.ca/metabolites/HMDB0061656 (accessed 23/3/2020).
- Rietschel E. T.; Wollenweber H.-W.; Zähringer U.; Lüderitz O. (1982) Lipid A, the lipid component of bacterial lipopolysaccharides: relation of chemical structure to biological activity. Klin. Wochenschr. 60 (14), 705–709. 10.1007/BF01716559. [DOI] [PubMed] [Google Scholar]
- Leonardi R.; Jackowski S. (2007) Biosynthesis of pantothenic acid and coenzyme A. EcoSal Plus 2, 3.6.3.4. 10.1128/ecosalplus.3.6.3.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri A. M.; Landersdorfer C. B.; Jacob J.; Boniatti M. M.; Dalarosa M. G.; Falci D. R.; Behle T. F.; Bordinhao R. C.; Wang J.; Forrest A.; Nation R. L.; Li J.; Zavascki A. P. (2013) Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin. Infect. Dis. 57, 524–531. 10.1093/cid/cit334. [DOI] [PubMed] [Google Scholar]
- Committee for Medicinal Products for Human Use (CHMP) . Assessment report KALYDECO ivacaftor; EMEA/H/C/002494//0000; European Medicines Agency, 2012.
- Masson A.; Schneider-Futschik E. K.; Baatallah N.; Nguyen-Khoa T.; Girodon E.; Hatton A.; Flament T.; Le Bourgeois M.; Chedevergne F.; Bailly C.; Kyrilli S.; Achimastos D.; Hinzpeter A.; Edelman A.; Sermet-Gaudelus I. (2019) Predictive factors for lumacaftor/ivacaftor clinical response. J. Cystic Fibrosis 18 (3), 368–374. 10.1016/j.jcf.2018.12.011. [DOI] [PubMed] [Google Scholar]
- Chu D. T.; Fernandes P. B. (1989) Structure-activity relationships of the fluoroquinolones. Antimicrob. Agents Chemother. 33 (2), 131–5. 10.1128/AAC.33.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. W.; Han M. L.; Zhao J.; Zhu Y.; Rao G.; Forrest A.; Song J.; Kaye K. S.; Hertzog P.; Purcell A.; Creek D.; Zhou Q. T.; Velkov T.; Li J. (2019) Synergistic Combination of Polymyxin B and Enrofloxacin Induced Metabolic Perturbations in Extensive Drug-Resistant Pseudomonas aeruginosa. Front. Pharmacol. 10, 1146. 10.3389/fphar.2019.01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghelani D. P.; Schneider-Futschik E. K. (2020) Emerging Cystic Fibrosis Transmembrane Conductance Regulator Modulators as New Drugs for Cystic Fibrosis: A Portrait of in Vitro Pharmacology and Clinical Translation. ACS Pharmacol. Transl. Sci. 3, 4. 10.1021/acsptsci.9b00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gika H. G.; Theodoridis G. A.; Wingate J. E.; Wilson I. D. (2007) Within-day reproducibility of an HPLC-MS-based method for metabonomic analysis: application to human urine. J. Proteome Res. 6 (8), 3291–303. 10.1021/pr070183p. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Creek D. J.; Barrett M. P.; Blackburn G.; Watson D. G. (2012) Evaluation of coupling reversed phase, aqueous normal phase, and hydrophilic interaction liquid chromatography with Orbitrap mass spectrometry for metabolomic studies of human urine. Anal. Chem. 84 (4), 1994–2001. 10.1021/ac2030738. [DOI] [PubMed] [Google Scholar]
- Creek D. J.; Jankevics A.; Burgess K. E.; Breitling R.; Barrett M. P. (2012) IDEOM: an Excel interface for analysis of LC-MS-based metabolomics data. Bioinformatics 28 (7), 1048–9. 10.1093/bioinformatics/bts069. [DOI] [PubMed] [Google Scholar]
- Scheltema R. A.; Jankevics A.; Jansen R. C.; Swertz M. A.; Breitling R. (2011) PeakML/mzMatch: a file format, Java library, R library, and tool-chain for mass spectrometry data analysis. Anal. Chem. 83 (7), 2786–93. 10.1021/ac2000994. [DOI] [PubMed] [Google Scholar]
- Smith C. A.; Want E. J.; O’Maille G.; Abagyan R.; Siuzdak G. (2006) XCMS: Processing mass spectrometry data for metabolite profiling using Nonlinear peak alignment, matching, and identification. Anal. Chem. 78 (3), 779–787. 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Keseler I. M.; Collado-Vides J.; Gama-Castro S.; Ingraham J.; Paley S.; Paulsen I. T.; Peralta-Gil M.; Karp P. D. (2004) EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res. 33, D334–D337. 10.1093/nar/gki108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salek R. M.; Steinbeck C.; Viant M. R.; Goodacre R.; Dunn W. B. (2013) The role of reporting standards for metabolite annotation and identification in metabolomic studies. GigaScience 2 (1), 13. 10.1186/2047-217X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulesteix A.-L.; Strimmer K. (2006) Partial least squares: a versatile tool for the analysis of high-dimensional genomic data. Briefings Bioinf. 8 (1), 32–44. 10.1093/bib/bbl016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.