

Abstract
The pharmacokinetics of nanomedicines are complicated by the unique dispositional characteristics of the drug carrier. Most simplistically, the carrier could be a solubilizing platform that allows administration of a hydrophobic drug. Alternatively, the carrier could be stable and release the drug in a controlled manner, allowing for distribution of the carrier to influence distribution of the encapsulated drug. A third potential dispositional mechanism is carriers that are not stably complexed to the drug, but rather bind the drug in a dynamic equilibrium, similar to the binding of unbound drug to protein; since the nanocarrier has distributional and binding characteristics unlike plasma proteins, the equilibrium binding of drug to a nanocarrier can affect pharmacokinetics in unexpected ways, diverging from classical protein binding paradigms. The recently developed stable isotope tracer ultrafiltration assay (SITUA) for nanomedicine fractionation is uniquely suited for distinguishing and comparing these carrier/drug interactions. Here we present the the encapsulated, unencapsulated, and unbound drug fraction pharmacokinetic profiles in rats for marketed nanomedicines, representing examples of controlled release (doxorubicin liposomes, Doxil; and doxorubicin HCl liposome generic), equilibrium binding (paclitaxel cremophor micelle solution, Taxol generic), and solubilizing (paclitaxel albumin nanoparticle, Abraxane; and paclitaxel polylactic acid micelle, Genexol-PM) nanomedicine formulations. The utility of the SITUA method in differentiating these unique pharmacokinetic profiles and its potential for use in establishing generic nanomedicine bioequivalence are discussed.
Keywords: nanomedicine, pharmacokinetics, bioequivalence, bioanalytical methods, generic drugs, nonbiological complex drugs
The complex pharmacokinetics of nanomedicine drugs requires the measurement of all drug fractions to accurately define drug disposition, including encapsulated, unencapsulated, unbound, as well as total drug. The importance of measuring these drug fractions is highlighted by recent Food and Drug Administration (FDA) and European Medicines Agency (EMA) regulatory guidances, recommending their measurement in order to establish generic nanomedicine bioequivalence.1−6 Unfortunately, generalized methods to accurately measure these drug fractions without sample processing artifacts are lacking.7 Conventional fractionation methods, primarily solid phase extraction, are chemically harsh techniques that have the potential to disrupt formulations and inflate unencapsulated drug fractions, while simultaneously being incapable of delineating unencapsulated bound and unbound drug fractions. Alternatively, simple ultrafiltration or equilibrium dialysis techniques, while capable of measuring unbound drug, cannot differentiate nanomedicine encapsulated and unencapsulated bound drug fractions.
Optimally, methods to quantify drug fractions in biological matrices should be able to not only distinguish the release characteristics of stable and unstable formulations, but also identify dynamic binding of drug to protein and formulation components that can influence drug disposition in novel ways. The equilibrium binding of unbound drug to formulation components is similar in theory to equilibrium binding of unbound drug to protein. However, the physicochemical characteristics of nanocarriers result in drug binding and tissue distribution characteristics that can influence drug disposition in unique ways that diverge from classic protein binding paradigms.8 It is therefore important that these drug–carrier interactions be identified, so that their significance to overall drug disposition can be determined.
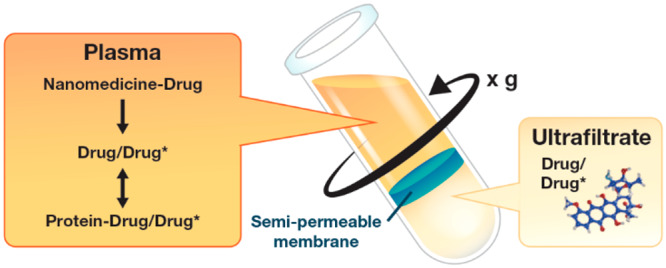
Recently, the Nanotechnology Characterization Laboratory developed a method that accurately and precisely measures all nanomedicine drug fractions by defining the unbound–bound drug relationship using stable isotope tracers of the analyte.9 The stable isotope tracer ultrafiltration assay (SITUA) is based on the concept that a tracer amount of isotopically labeled drug in plasma will behave identically with regard to protein and formulation equilibrium (on–off) binding as drug that is released from the nanomedicine (Figure 1). Therefore, the isotopically labeled tracer added to nanomedicine containing plasma becomes a measure of the free drug fraction in the system, which can then be used to calculate nanomedicine encapsulated, unencapsulated, protein bound, and unbound drug fractions simultaneously (see Materials and Methods section). This use of a tracer to determine drug fractions is novel and is based on the epidemiological and ecological concept of mark and recapture.10 Previously, stable isotopes have been used in drug metabolism-pharmacokinetics as analytical internal standards, to define metabolism mechanisms, differentiate exogenous and endogenous analytes, in controlled release bioavailability studies and to evaluate the pharmacokinetics of high clearance and chronically administered drugs.11
Figure 1.
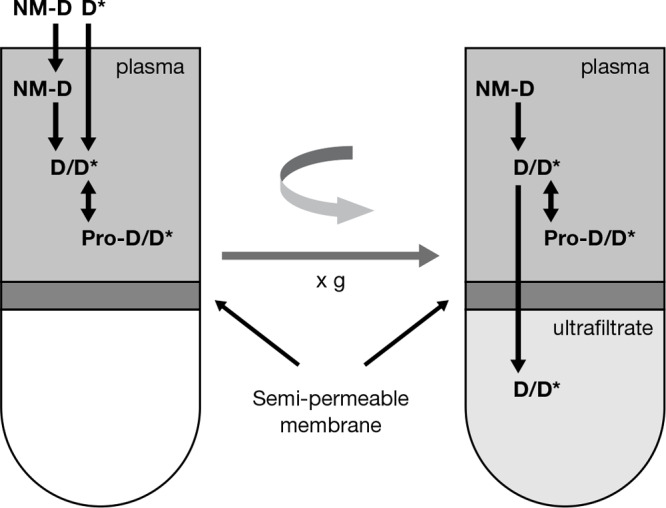
Stable isotope tracer ultrafiltration assay (SITUA). Stable isotopically labeled tracer (D*) is spiked into nanomedicine (NM-D) containing plasma and behaves identically to normoisotopic drug (D) with regard to protein and formulation binding (Pro-D/D*). After reaching binding equilibrium, the sample is transferred to an ultrafiltration device and the filtrate is separated by centrifugation. The stable isotope tracer unbound drug fraction, represented as the ultrafilterable drug fraction, can be used to calculate protein bound, unencapsulated, and encapsulated drug fractions. Reproduced with permission from ref (12). Copyright 2018 Springer.
At this time, the SITUA is the only bioanalytical method that the authors are aware of that has the ability to distinguish all types of drug/nanocarrier interactions in vitro and in vivo for a broad array of drugs and delivery platforms. This method is finding great utility in characterization and optimization of novel nanomedicines, as well as potential for evaluating pharmacokinetic sameness (e.g., bioequivalence) for purposes of regulatory approval. Here, we evaluate the pharmacokinetics of several types of nanomedicines: doxorubicin liposome (Doxil and doxorubicin HCl liposome generic); paclitaxel cremophor micelle solution (Taxol generic); and paclitaxel albumin nanoparticle (Abraxane) and paclitaxel PEG–PLA polymeric nanoparticle micelle (Genexol-PM), representing examples of controlled release, equilibrium binding, and solubilizing nanomedicine formulations, respectively. These also represent products that are approved therapeutic equivalents (i.e., the liposomal doxorubicin products) and products that have the same active ingredient (i.e., paclitaxel), but are not therapeutically equivalent due to differences in formulation/dosage form and lack bioequivalence testing. Using the SITUA method, the unique pharmacokinetic attributes of these formulations were identified and contrasted.
Results
In Vitro Drug Release for Doxorubicin and Paclitaxel Nanomedicines
A series of control studies were performed to evaluate assay accuracy and identify possible processing artifacts. A 5 ng/mL doxorubicin (DXR) spike into 1 μg/mL generic liposomal DXR in plasma was recovered within 20% of the theoretical spike concentration (an encapsulated/unencapsulated ratio of 200). A 200 ng/mL paclitaxel (PTX) spike into 1 μg/mL Abraxane, Genexol-PM, or Taxol generic containing plasma were recovered within 20% of the theoretical spike concentration (an encapsulated/unencapsulated ratio of 5). These spike recoveries were considered sufficient to accurately measure encapsulated and unencapsulated drug based on individual formulation stabilities. Double processing and organic solvent stable isotope spike of generic liposomal DXR, Abraxane, Genexol-PM, or Taxol generic plasma samples did not alter formulation stability (data not shown).
Doxil and generic liposomal DXR drug release was evaluated at 0.5, 1, and 5 μg/mL DXR equivalents in rat plasma at 37 °C over a 6 h period. Solvent-solubilized DXR was included at identical concentrations and time points as a control. The solvent DXR controls averaged between 93 and 114% of theoretical for all concentrations and time points (Figure 2A), and % binding of the DXR and DXR_C13 tracer to protein and formulation components were highly correlated (R2 = 0.94, Figure 2B), supporting similar binding kinetics. The Doxil and generic liposomal DXR drug release was similar, at approximately 2% release over the 6 h period, although there were instances of statistically significant differences owing to the high precision of the assay. The drug release was without a clear temporal trend, although release was lower at higher concentrations for all formulations and all time points (Figure 2C).
Figure 2.

(A) Solvent DXR control data for liposome formulations. Displayed is the calculated percent release for the solvent DXR control data. (mean ± SD, N = 3). (B) DXR vs DXR_C13 tracer percent binding correlation. Displayed is the percent binding correlation for DXR vs DXR_C13 tracer for the solvent DXR control study, with the dotted line showing the linear regression. (C) Doxil and generic liposomal DXR drug release comparison. Displayed are the 10 min–6 h calculated percent release data for the Doxil and generic liposomal DXR (mean ± SD, N = 3), *p ≤ 0.05, Student’s t test.
Drug release for Abraxane, Genexol-PM, and Taxol (generic) were evaluated at 0.5, 5, 25, and 100 μg/mL PTX equivalents in rat plasma at 37 °C over a 2 h period. Solvent PTX at identical concentrations and time points was included as a control. The solvent PTX controls averaged between 95 and 109% of theoretical values for all concentrations and time points (Figure 3A), and the percent binding of the solvent PTX and PTX_C13 tracer were highly correlated (R2 = 0.85, Figure 3B), supporting similar protein binding kinetics. The Abraxane, Genexol-PM, and Taxol generic drug release was similar, with approximately 100% release at the earliest 10 min time point, without a clear concentration-dependent or temporal trend (Figure 3C). However, there were significant differences in PTX_C13 tracer binding between the formulations over the concentration range (Figure 3D). Abraxane and Genexol-PM displayed saturated binding in which the unbound drug fraction increased with increasing drug concentration, similar to the solvent paclitaxel, suggesting that drug binding is exclusively due to protein binding and the Abraxane human albumin nanoparticle; the Genexol PLA micelle formulation components do not contribute. Taxol generic, however, displayed concentration-dependent binding, suggesting the cremophor micelle strongly binds to the unbound drug. Concentration-dependent trends in unbound drug concentration were not observed for the DXR liposome formulations (data not shown).
Figure 3.

(A) Solvent PTX control data for PTX formulations. Displayed is the calculated percent release for the solvent PTX control data. (mean ± SD, N = 3). (B) PTX vs PTX_C13 tracer percent binding correlation. Displayed is the percent binding correlation for PTX vs PTX_C13 tracer for the solvent PTX control study, with the dotted line showing the linear regression. (C) Abraxane, Genexol-PM, and Taxol (generic) drug release comparison. Displayed are the 10 min–2 h calculated percent release data for the Abraxane, Genexol-PM, and Taxol (generic). (mean ± SD, N = 3); *p ≤ 0.05, ANOVA, with Tukey’s posthoc test; 1,2 designates significantly different from Abraxane and Genexol-PM, respectively. (D) Abraxane, Genexol-PM, and Taxol (generic) percent unbound drug fraction vs concentration comparison. Displayed are the calculated percent unbound fraction vs concentration for Abraxane, Genexol-PM, and Taxol (generic), and linear regression fitting to the data points.
Pharmacokinetics of Doxil and Generic Liposomal DXR in Rats
Rats were administered 5 mg/kg DXR equivalents of either Doxil or generic liposomal DXR i.v., and blood was collected at 0.25, 0.5, 1, 4, 8, 12, 24, 48, and 96 h. The Doxil and generic liposomal DXR encapsulated, unencapsulated and unbound drug concentration–time profiles, determined by the SITUA method, were very similar (Figure 4), as were the calculated pharmacokinetic parameters (Table 1). Since the total plasma drug profile was dominated by the encapsulated drug, as is expected for a stable liposome formulation, the total drug profile data was omitted as it overlapped with the encapsulated profile. The encapsulated drug volume of distribution apparent (Vd) and volume of distribution steady state (Vss) values were close to the theoretical plasma volume for rats of 40 mL/kg,13 supporting retention of the liposome encapsulated drug within the plasma space (Table 1). For encapsulated drug, the concentration time zero (C0) and concentration maximum (Cmax) were lower, and Vd was higher, for Doxil in comparison to generic liposomal DXR. It is important to note that this pharmacokinetic (PK) comparison only represents a single lot of each product and is a small sampling number; as such, additional lots and more testing would be warranted to conclude that these statistical differences are biologically significant or rather includes the acceptable lot-to-lot variability in the products. However, it does illustrate the sensitivity of the SITUA method to identify differences. The remaining encapsulated pharmacokinetic parameters, T1/2, AUCall, AUCinf, CL, and MRTinf, were not different between the two treatment groups. The half-life (T1/2) of ∼30 h for both groups is consistent with previous published pharmacokinetic data for Doxil total drug distribution half-life in the rat.14
Figure 4.

Encapsulated, unencapsulated, and unbound DXR comparison. Presented is the encapsulated, unencapsulated, and unbound DXR concentration–time comparison for Doxil and generic liposomal DXR treatment groups (mean ± SD, N = 8).
Table 1. PK Comparison of Doxil vs Generic Liposomal DXRa.
| Co | Cmax | T1/2 | Tmax | AUCall | AUCinf | Vd | CL | MRTinf | Vss | |
|---|---|---|---|---|---|---|---|---|---|---|
| ng/mL | ng/mL | h | h | ng·h/mL | ng·h/mL | mL/kg | mL/(h·kg) | h | mL/kg | |
| Doxil Encapsulated | ||||||||||
| avg | 164338 | 160923 | 29 | - | 5780633 | 6605784 | 31 | 0.77 | 40 | 62 |
| SD | 19799 | 12804 | 7 | 901285 | 1070015 | 4 | 0.13 | 9 | 9 | |
| Generic Liposomal DXR Encapsulated | ||||||||||
| avg | 186841b | 184708b | 34 | - | 6717656 | 7911722 | 26b | 0.67 | 47 | 61 |
| SD | 15480 | 12988 | 7 | 1173239 | 1780233 | 2 | 0.20 | 10 | 3 | |
| Doxil Unencapsulated | ||||||||||
| avg | - | 186 | 88 | 36 | 12515 | - | - | - | - | - |
| SD | 46 | 30 | 31 | 1742 | ||||||
| Generic Liposomal DXR Unencapsulated | ||||||||||
| avg | - | 157 | 86 | 33 | 10129b | - | - | - | - | - |
| SD | 52 | 67 | 12 | 2360 | ||||||
| Doxil Unbound | ||||||||||
| avg | - | 28 | 60 | 24 | 1804 | - | - | - | - | - |
| SD | 7 | 12 | 31 | 516 | ||||||
| Generic Liposomal DXR Unbound | ||||||||||
| avg | - | 24 | 64 | 36 | 1381 | - | - | - | - | - |
| SD | 13 | 16 | 13 | 429 | ||||||
Displayed are the average animal pharmacokinetic parameters for the Doxil and generic liposomal DXR encapsulated, unencapsulated, and unbound drug profiles: concentration time zero (C0); half-life (T1/2); area under the time concentration curve to time infinity (AUCinf); maximum concentration (Cmax); area under the time concentration curve all time points (AUCall); volume of distribution (Vd); clearance (CL); mean residence time (MRTinf); volume of distribution steady state (Vss); time of maximum concentration (Tmax); half-life (T1/2).
(N = 8) p ≤ 0.05, Student’s t test.
The unencapsulated drug AUCall was higher for Doxil in comparison to generic liposomal DXR, and there was a trend toward higher unbound drug AUCall as well, which is consistent with the lower C0 and Cmax for the Doxil encapsulated drug fraction (Table 1). Most interestingly, the terminal half-lives (T1/2) for Doxil and generic liposomal DXR unencapsulated and unbound drug were much longer than for encapsulated drug (60–88 h vs 33–36 h) (Figure 4, Table 1). The ratio of the encapsulated to unencapsulated drug concentrations ranged over 2 orders of magnitude, from ∼10 000 at early time points to ∼100 at later time points, consistent with the in vitro rat plasma drug release study and supporting the stability and slow drug release of the liposomal DXR formulations.
Pharmacokinetics of Taxol (generic), Abraxane, and Genexol-PM in Rats
Rats were administered 6 mg/kg PTX equivalents of either Abraxane, Genexol-PM, or Taxol (generic) i.v., and blood was collected at 0.25, 0.5, 2, 4, 6, 8, 24, 48, and 72 h. Encapsulated drug, as measured by the SITUA method, was only observed at the earliest 15 min time point for the Taxol (generic) and Genexol-PM formulations, but not the Abraxane formulation. This is consistent with stable cremophor and PEG-polylactic acid micelles at this early time point for the Taxol (generic) and Genexol-PM formulations, respectively, corresponding with peak excipient concentrations. However, the shape of the Abraxane, Genexol-PM, and Taxol (generic) concentration–time profiles for total, unencapsulated, and unbound drug were all identical (Figure 5), displaying a triphasic curve.
Figure 5.

Total, unencapsulated, and unbound PTX comparison. Presented is the total, unencapsulated, and unbound PTX concentration–time comparison for Taxol, Abraxane, and Genexol-PM treatment groups (mean ± SD, N = 8).
For the Abraxane and Genexol-PM formulations, the total drug PK parameters were not significantly different. This is consistent with preliminary clinical bioequivalence results (NCT02064829) comparing Abraxane and Genexol-PM, which supports our finding that the total drug PK of the two products are equivalent.15 Notably, the clinical bioequivalence study did not evaluate unencapsulated or unbound drug concentrations. In addition to total drug PK, the unencapsulated and unbound drug PK parameters were very similar for Abraxane and Genexol-PM in our study, although there were some instances of significant differences (Table 2).
Table 2. PK Comparison of Taxol vs Abraxane vs Genexol-PMa.
| Co | Cmax | T1/2 | AUCall | AUCinf | Vd | CL | MRTinf | Vss | |
|---|---|---|---|---|---|---|---|---|---|
| ng/mL | ng/mL | h | ng·h/mL | ng·h/mL | mL/kg | mL/h·kg | h | mL/kg | |
| Taxol Total | |||||||||
| avg | 19294b | 11066b | 11 | 19737b | 19770b | 325b | 324b | 3b | 964b |
| SD | 4890 | 2303 | 2 | 5597 | 5635 | 66 | 85 | 1 | 325 |
| Abraxane Total | |||||||||
| avg | 1726 | 1091 | 17 | 2291 | 2324 | 4115 | 2627 | 7 | 20512 |
| SD | 498 | 298 | 9 | 343 | 332 | 715 | 379 | 2 | 6780 |
| Genexol-PM Total | |||||||||
| avg | 1413 | 853 | 15 | 2176 | 2214 | 4267 | 3060 | 8 | 16649 |
| SD | 481 | 264 | 2 | 896 | 897 | 1861 | 1083 | 3 | 5900 |
| Taxol Unencapsulated | |||||||||
| avg | 12238b | 9283b | 12 | 17699b | 17735b | 469b | 362b | 3b | 980b |
| SD | 1765 | 2041 | 3 | 5014 | 5053 | 111 | 99 | 1 | 189 |
| Abraxane Unencapsulated | |||||||||
| avg | 1772 | 1117 | 22 | 2319 | 2373 | 4063b | 2600 | 7 | 18766 |
| SD | 631 | 347 | 14 | 466 | 450 | 1219 | 457 | 2 | 6697 |
| Genexol-PM Unencapsulated | |||||||||
| avg | 995 | 744 | 18 | 2035 | 2066 | 5976 | 3251 | 8 | 21521 |
| SD | 354 | 237 | 4 | 786 | 797 | 1620 | 1126 | 3 | 4575 |
| Taxol Unbound | |||||||||
| avg | 446b | 239b | 16 | 438b | 442b | 17525b | 14523b | 4b | 52359b |
| SD | 183 | 57 | 6 | 127 | 132 | 4649 | 3706 | 2 | 14110 |
| Abraxane Unbound | |||||||||
| avg | 174 | 117 | 21 | 283 | 320 | 36518 | 20872b | 8 | 303918 |
| SD | 38 | 23 | 9 | 73 | 123 | 11545 | 6582 | 3 | 248205 |
| Genexol-PM Unbound | |||||||||
| avg | 145 | 99 | 19 | 199 | 202 | 38773 | 32603 | 8 | 259769 |
| SD | 34 | 50 | 5 | 77 | 78 | 16073 | 9470 | 2 | 131868 |
Displayed are the average animal pharmacokinetic parameters for the Taxol (generic), Abraxane, and Genexol-PM total, unencapsulated, and unbound drug profiles: concentration time zero (C0); half-life (T1/2); area under the time concentration curve to time infinity (AUCinf); maximum concentration (Cmax); area under the time concentration curve all time points (AUCall); volume of distribution (Vd); clearance (CL); mean residence time (MRTinf); volume of distribution steady state (Vss); half-life (T1/2).
Significantly different than other groups, p ≤ 0.05, ANOVA with Tukey’s posthoc test.
As noted earlier, encapsulated drug was only observed at the earliest time point for Genexol-PM and not at any of the time points for Abraxane, and this may be reflected in the higher unencapsulated Cmax and significantly lower apparent volume of distribution, Vd, for Abraxane in comparison to Genexol-PM. While Abraxane and Genexol have the same active ingredient (i.e., paclitaxel), they are not therapeutically equivalent due to differences in formulation/dosage form and lack of bioequivalence testing. The lack of Abraxane and Genexol-PM stability observed in vivo is supported by our in vitro studies (Figure 3C), in which both formulations released drug immediately, and the unbound drug fraction–concentration profiles were identical to that of solvent solubilized drug (Figure 3D). These data strongly support the idea that both Abraxane and Genexol-PM are solubilizing formulations that do not maintain significant encapsulated drug fractions and do not affect the the unbound drug fraction by binding to unbound drug in equilibrium.
Taxol (generic) demonstrated significantly lower total, unencapsulated, and unbound CL, Vd and Vss than Genexol-PM and Abraxane (Table 2). The lower CL and volume of distribution for Taxol (generic) in comparison to Abraxane has been observed in previous studies in rats evaluating total drug PK and is consistent with strong equilibrium binding to stable cremophor micelles.16 The equilibrium binding of PTX to the cremophor micelle was reflected in the nonlinear/reduced unbound drug fraction at high total drug concentrations for the Taxol (generic) formulation, as determined for the stable isotope tracer (Figure 6). Here nonlinear binding is defined as a decrease or increase in the unbound drug fraction with increasing total drug concentration.17 By comparison, the Abraxane and Genexol-PM unbound drug fraction did not display this concentration-dependency. The same relationship between Taxol (generic) concentration and the PTX unbound fraction was observed in vitro as well (Figure 3D).
Figure 6.

Stable isotope tracer %Fu vs total PTX concentration. Presented are the individual stable isotope tracer % fraction unbound (%Fu) vs total PTX concentration for Abraxane, Genexol-PM, and Taxol (generic) (N = 8).
Discussion
Doxil and generic liposomal DXR released DXR to a similar extent in rat plasma, ∼2% release over the 6 h period. By contrast, Abraxane, Genexol-PM, and Taxol generic showed approximately 100% PTX release at the earliest 10 min time point. Significant differences in PTX_C13 tracer binding were observed between the PTX formulations, with Abraxane and Genexol-PM displaying saturated binding, similar to solvent paclitaxel. Taxol generic, by contrast, displayed concentration-dependent binding suggesting Taxol cremophor micelle contributes to drug binding and the Abraxane human albumin nanoparticle and Genexol PLA micelle formulation components do not.
Although both pegylated liposomal doxorubicin (PLD) formulations studied had similar PK in rats, important differences were observed between the present PK study using the SITUA method to separate encapsulated/unencapsulated/unbound drug fractions and previous PLD PK studies in rats. Previous PLD PK studies in rats have relied on traditional solid phase extraction (SPE) methods which are based on interaction between DXR and a hydrophobic solid phase to separate encapsulated and unencapsulated drug fractions. Unencapsulated drug concentrations measured in our study were much lower than in previous PLD studies, resulting in encapsulated/unencapsulated drug ratios as high as 10 000 compared to a high of ∼100 for the previous SPE method studies.18,19 Further, the terminal slope of the unencapsulated profile in our study did not parallel the encapsulated profile, as it does for rat PLD PK studies using the conventional SPE fractionation method,18,19 with a half-life that was 2–3-fold longer than that of the encapsulated drug profile.
The long terminal plasma half-life for unencapsulated and unbound drug observed in our study is similar to the tumor PLD drug release half-life observed in previous rat studies,20 which is consistent with formation-rate limited PK for controlled release drugs.7 Formation-rate limited kinetics for controlled release drugs, similar to absorption-rate limited kinetics for delayed release oral drugs, results when drug release is slower than drug clearance, and this slower release rate is then reflected in the terminal profile rather than the actual clearance rate (also known as flip-flop kinetics).7 By contrast, the paralleling of the unencapsulated and encapsulated drug profiles observed with the conventional SPE fractionation methods is inconsistent with formation-rate limited PK, as the encapsulated drug half-life is a tissue distribution half-life not a drug release half-life.
Another difference noted between our SITUA PK study and a previous SPE-based study is that unencapsulated Cmax is ∼20-fold lower for our study, 0.2 μg/mL vs 3.4 μg/mL, which is consistent with a slow controlled release formulation.18 In fact, the unencapsulated Cmax of 3.4 μg/mL measured in the previous SPE study for a 6 mg/kg dose of PLD is similar to the Cmax measured for a separate 6 mg/kg bolus DXR HCl dose study in rats,21 which is not possible for a controlled release formulation. These data strongly suggest that the conventional SPE method results in artifactual release of liposome encapsulated DXR during PLD SPE sample processing which confounds accurate measurement of unencapsulated drug, and supports the validity of the SITUA method over the conventional SPE method for fractionation of doxorubicin liposome.
In the PTX formulation rat PK study, marked differences were noted between the Abraxane/Genexol and the Taxol treatment groups, with Taxol having lower CL and Vss for all drug fractions, total, unencapsulated, and unbound. Importantly, the SITUA method explained these findings by allowing discrimination between drug stably encapsulated in cremophor micelles and dynamic drug binding to micelles in equilibrium with unbound drug. Current pharmacological theory states that changes in protein binding do not affect the unbound drug PK for low extraction drugs, such as paclitaxel, administered parenterally.22 Since unbound drug is the active drug fraction, this lack of effect on unbound drug PK implies that alterations in drug binding should be without clinical consequence. Surprisingly, and inconsistent with this theory of drug binding to protein, unbound drug PK in our study was, apparently, significantly altered by equilibrium binding to the cremophor micelles.
Although cremophor has been shown to inhibit paclitaxel metabolism at high concentrations in vitro,23 the differences in unbound drug PK for the Taxol (generic) formulation in comparison to the Abraxane and Genexol-PM formulations do not appear to be the result of changes in drug metabolism, since changes in metabolic clearance alone would have resulted in altered drug half-life (T1/2 = ln 2/(Vss/CL)), which was not the case. By contrast, alterations in unbound drug fraction for a high volume of distribution drug such as paclitaxel result in compensatory changes in both volume of distribution and clearance resulting in no change in drug half-life, as observed.8 This is an important finding, as it would suggest that the binding of paclitaxel to the Taxol cremophor micelle affects the unbound drug PK differently than changes in the binding to protein would, and can influence both hepatic drug extraction and tissue distribution of the unbound drug, even for a drug that is not high extraction and perfusion limited.
This effect of cremophor on unbound PTX tissue distribution is consistent with previous Taxol findings in the isolated liver perfusion and liver slice models,23 in which researchers observed a decrease in tissue uptake of PTX which indirectly resulted in decreased metabolism, as well as studies demonstrating decreased RBC partitioning of PTX in vitro.16In vitro and in vivo studies also support the ability of cremophor to suppress PTX transport and tissue distribution,23 and limit tissue distribution of coadministered drugs.24,25 By decreasing the tissue distribution of active, unbound drug, the Taxol cremophor micelle could potentially influence drug therapy, and this may help explain differences between Abraxane and Taxol pharmacology.8 While Abraxane has been approved for treatment of pancreatic cancer,26 Taxol has not, and the reasons for this lack of Taxol efficacy may be due to the underlying pharmacology of the cremophor formulation, not simply the fact that Abraxane can safely be administered at higher doses. Indeed, higher PTX tissue exposure was observed for Abraxane in comparison to Taxol for pancreas and xenograft tumor in preclinical models at identical doses,16,27 supporting the lower unbound volume of distribution observed in the present study.
It has long been contended that a key aspect of Abraxane’s clinical advantage over Taxol is the preferential tumor distribution of stable PTX-bound albumin nanoparticles mediated by albumin transport proteins.28 On the basis of the rapid and complete solubility of PTX from the Abraxane particulate formulation observed in our in vitro and in vivo studies, the proposed clinical mechanism of Abraxane does not appear to include the sustained systemic circulation and tissue uptake of PTX particles. In support of our current findings of Abraxane nanoparticle instability in vivo, a recent European bioequivalence study comparing Abraxane and a micellar paclitaxel formulation, Paclical, found the pharmacokinetics of unbound drug to be identical and concluded that the Abraxane nanoparticle must dissociate immediately upon administration.29 Further, recent clinical studies have not shown a correlation between albumin transporter expression and clinical efficacy of Abraxane.30,31 These mechanistic and formulation properties are important to consider when developing nanoformulations of PTX. As of 2017, there were 18 companies developing such PTX products.30
Although the SITUA method offers several advantages over conventional drug fractionation techniques, there are still challenges associated with the implementation of this method. The SITUA requires the availability of two stable isotope standards of the drug, one for use as a tracer in the assay and the second as an analytical internal standard. There is also the requirement that the tracer behave identically to the normoisotopic drug with regard to protein binding, and the drug not have high binding to the ultrafiltration apparatus. The SITUA also requires that the plasma sample be processed at the time of collection unless the nanomedicine is stable to freeze–thaw, which is often not the case. While this processing requirement can be an added logistical and financial burden for preclinical and clinical studies, it is also a requirement of all conventional drug fractionation techniques such as solid phase extraction.
Another major limitation of this method is that it can only be used for plasma samples and has not yet been adapted to measurement of drug fractions in tissue. Without an understanding of the amount of active, unbound drug delivered to tissue, we have an incomplete picture of pharmacokinetic–pharmacodynamic relationships. At this time there are very few methods available in the literature for tissue drug fraction measurement, and these methods that do exist have limitations with regard to the drugs and formulations to which they can be applied.32,33 PK modeling has also been used to estimate tissue free drug concentrations, but without actual free drug concentration data with which to compare, the predictability of these models is uncertain.34 Therefore, this lack of unbound tissue concentration data is a problem for the nanomedicine field. A future direction for further development of the SITUA method is application to measurement of tissue drug fractions.
The established SITUA method allows for measurement of all systemic drug fractions simultaneously. In the case of liposomal doxorubicin formulations, Doxil and generic liposomal DXR, the SITUA method demonstrated utility in characterizing the formulations’ in vitro plasma release profiles, as well as the similarity of the encapsulated, unencapsulated, and unbound drug PK profiles in vivo. On the basis of the differential PK of Taxol (generic), and Abraxane and Genexol-PM, unbound drug PK appears to be a discriminating criterion for drug formulations that influence drug pharmacology by equilibrium binding; the SITUA method is the only bioanalytical technique currently capable of identifying changes in unbound drug fraction resulting from nanocarrier equilibrium binding. Overall, the SITUA method is a precise and accurate method for identifying and comparing the unique PK attributes of controlled-release, solubilizing, and equilibrium binding nanomedicine formulations, and is finding ever increasing application in drug optimization and potential in bioequivalence.
Materials and Methods
Chemicals and Rreagents
Paclitaxel (PTX) was purchased from LC Laboratories, Woburn, MA (Cat. No. P-9600). 13C6-Paclitaxel (PTX_C13) (Cat. No. sc-477982) and 2H5-paclitaxel (PTX_d5) (Cat. No. sc-219546) were purchased from Santa Cruz Biotechnology, Inc., Dallas, TX. Doxorubicin hydrochloride (DXR) was purchased from Sigma-Aldrich, St. Louis, MO (Cat. No. D1515). 13C,2H3-Doxorubicin trifluoroacetate salt (DXR_C13) was purchased from Alsachim, Duisburg, Germany (Cat. No. C2451). Aclarubicin hydrochloride was purchased from Alfa Aesar, Haverhill, MA (Cat. No. J66842). Acetonitrile (ACN) was purchased from VWR, Radnor, PA (Cat. No. BJLC015-1). Formic acid was purchased from Thermo Scientific, Waltham, MA (Cat. No. 28905). Zorbax-SB-C18 RRHD 1.8 μm particle, 2.1 × 100 mm (Cat. No. 858700-902) and Zorbax-SB-C18 1.8 μm particle, 2.1 × 5 mm (Cat. No. 821725-902) were purchased from Agilent Technologies, Inc., Santa Clara, CA. Amicon Ultra-4 centrifugal filter unit with Ultracel 30 membrane (Cat. No. UFC80302) and Microcon 10 kDa MWCO centrifugal filter unit with Ultracel 10 membrane (Cat. No. MRCPRT010) were purchased from Millipore, Burlington, MA. Vivacon 10 kDa MWCO centrifugal filter unit with Hydrosart regenerated cellulose membrane was purchased from Sartorius, Gottingen, Germany (Cat. No. VN01H02). Rat plasma (7-week old female Sprague–Dawley, Charles River Laboratories, Willmington, MA) was collected fresh in K2EDTA tubes. HEPES buffer (1 M) was purchased from Gibco Laboratories, Gaithersburg, MD (Cat. No. 15630080). K2EDTA vacutainer tubes were purchased from Moore Medical, Richmond, VA (Cat. No. 87770). Test articles, Janssen’s Doxil (lot 600520P1), Sun Pharma’s doxorubicin HCl liposome (a generic liposomal doxorubicin) (lot JKR0865A), Abraxane (lot 6115194), and Gland Pharma Limited’s paclitaxel injection (a generic of Taxol) (lot PACCA1018) were supplied by the NIH pharmacy. Genexol-PM (lot GP31771), a paclitaxel formulation not approved or marketed in the United States, was a kind gift from Samyang Biopharmaceuticals Corporation (Seoul, Korea).
Stable Isotope Tracer Ultrafiltration Assay (SITUA)
The SITUA method of quantifying encapsulated, unencapsulated, and unbound plasma analytes was performed as described in Skoczen and Stern, 2018 (Figure 1).12 Briefly, for in vitro studies, Sprague–Dawley rat blood was collected in K2EDTA tubes and centrifuged at 2500g for 10 min to collect plasma. HEPES buffer (pH 7.4) was added at 50 μL/2 mL plasma to maintain a pH of 7.4. Plasma in glass vials was spiked with samples (solvent solubilized drug or nanomedicine) at final drug concentrations of 0.5–100 μg/mL DXR or PTX equivalent, in triplicate. All samples were then spiked with 0.1 μg/mL DXR_C13 or PTX_C13 (stable isotope tracer).
Samples were then incubated at 37 °C with agitation. At time points ranging from 10 min to 6 h, 50 μL of plasma was removed for protein precipitation and analyzed as described below; this sample was used to determine total reservoir drug concentration. A 400 μL sample of the incubate was then transferred to a prewarmed ultrafiltration tube (10 kDa Microcon for DXR, or 10 kDa Vivacon for PTX) and centrifuged at 6000g for 10 min. A 50 μL aliquot of the resulting filtrate was also taken for protein precipitation and analysis; this sample was used to determine unbound drug concentration.
The two 50 μL samples from above (noncentrifuged and centrifuged) were added to 200 μL of ice cold acetonitrile (ACN) with 0.1% formic acid (FA) containing either 25 ng/mL aclarubicin or PTX_d5 as an internal standard (ISTD) for DXR and PTX analysis, respectively. The samples were frozen at −80 °C for 10 min, thawed at room temperature, and then centrifuged at 18 000g, 4 °C, for 20 min. The supernatant was transferred to a clean Eppendorf tube and dried using either a nitrogen evaporator or a centrifuge speed vacuum for 25 min at 50 °C and 5 Torr. Following protein precipitation, samples were reconstituted in 150 μL of 25% ACN with 0.1% FA for DXR or 40% ACN with 0.1% FA for PTX. The reconstituted sample was transferred to a clean Eppendorf tube, centrifuged at 18 000g, 4 °C, for 10 min, and then transferred to an HPLC vial. Samples were analyzed on a Q-Orbitrap as described below.
For the analysis of samples from the in vivo PK study, 200 μL of plasma was spiked with 0.1 μg/mL PTX_C13 or DXR_C13 stable isotope tracers and incubated at 37 °C with agitation for 10 min. Following incubation, 25 μL of plasma was taken for total drug determination and 150 μL of the remaining plasma, instead of the 400 μL used above, was transferred to the ultrafiltration device and centrifuged to collect filtrate. No change in assay performance was noted with the reduced ultrafiltration sample volume. All other procedures were as described above. Control studies used to determine assay accuracy and evaluate the potential for sample processing artifacts were performed according to the methods described previously.12
The stable isotope tracer is used to calculate protein bound, unencapsulated, and encapsulated drug fractions, according to eqs 1, 2, and 3 below, respectively,
| 1 |
| 2 |
| 3 |
in which % bound D* is percentage of the stable isotope tracer that is bound to either protein or formulation components; [reservoir D*] is the concentration of stable isotope tracer in the ultrafiltration reservoir; [ultrafilterable D*] is the concentration of stable isotope tracer collected following filtration, also referred to as unbound concentration; [unencapsulated D] is the concentration of drug that is not encapsulated in the drug formulation; [ultrafilterable D] is the concentration of drug collected following filtration, also referred to as unbound concentration; [encapsulated D] is the concentration of drug encapsulated in the formulation; [reservoir D] is the concentration of drug in the ultrafiltration reservoir.
Q-Orbitrap Analysis
The LC-Orbitrap system consisted of a Q Exactive basic quadrupole-Orbitrap mass spectrometer, Vanquish UHPLC system, binary pump, and autosampler (Thermo Fisher Scientific). The LC conditions were 20 μL injection volume, 40 °C column oven, 10 °C autosampler, and a flow rate of 0.4 μL/min. The column was a Zorbax-SB-C18 RRHD 1.8 μm particle, 2.1 mm × 100 mm (Agilent technologies, Inc.) with a Zorbax-SB-C18 1.8 μm particle and 2.1 mm × 5 mm guard column (Agilent Technologies, Inc.).
For DXR analysis, mobile phase A consisted of water with 0.1% formic acid, and mobile phase B consisted of acetonitrile with 0.1% formic acid. The following gradient was used: hold at 25% B for 3 min, linear increase to 80% B from 3 to 7 min, linear increase to 95% B from 7.0 to 7.5 min, hold at 95% B for 0.5 min, and linear decrease to 25% B from 8.0 to 8.1 min, with a column regeneration time between injections of 4 min. The doxorubicin (DXR) and 13C2H3-doxorubicin (DXR_C13) elution times were 1.63 min, and the aclarubicin internal standard (ISTD) elution time was 5.56 min. The Orbitrap mass spectrometer was run in ESI positive mode, spray voltage was 3.5 kV, and the capillary and auxiliary gas temperatures were 270 and 290 °C, respectively. The collision energy was set at 13 AU for doxorubicin and 13C2H3-doxorubicin, and 15 AU for aclarubicin. Parallel reaction monitoring (PRM) of the following transitions were used: DXR 544.18 → 130.08, 379.07, 397.08; DXR_C13 548.20 → 130.08, 383.09, 401.11; and aclarubicin 812.34 → 570.23.
For PTX analysis, mobile phase A consisted of water with 0.1% formic acid, and mobile phase B consisted of acetonitrile with 0.1% formic acid. The following gradient was used: linear increase from 40% B to 100% B from 0 to 2 min, hold at 100% B for 2 min, and linear decrease from 100% B to 40% B in 0.5 min, with a column regeneration time between injections of 3.5 min. Paclitaxel (PTX), 13C6-paclitaxel (PTX_C13) tracer, and 2H5-paclitaxel (PTX_d5) internal standard (ISTD) elution times were all 2.03 min. The Orbitrap mass spectrometer was run in ESI positive mode, spray voltage was 3.5 kV, and the capillary and auxiliary gas temperatures were 150 and 400 °C, respectively. The collision energy was set at 13 AU for all three analytes. Parallel reaction monitoring (PRM) of the following transitions were used: PTX, 854.34 → 286.10, 509.21; PTX_C13, 860.36 → 286.10, 515.23; and PTX_d5, 859.37 → 291.13, 509.21.
Stock solutions of DXR and DXR_C13 were prepared in 25% ACN, and PTX and PTX_C13 were prepared in 100% ACN. These stocks were used to prepare calibration and quality control standards in plasma or protein-free plasma matrix. To prepare protein-free plasma, plasma was transferred to Amicon Ultra-4 centrifugal filter units with Ultracel 30 membranes, centrifuged at 5000g for 1 h, and the filtrate was collected.
DXR and DXR_C13 calibration standards were prepared in rat protein-free plasma, and rat plasma with concentrations ranging from 0.1 ng/mL to 300 μg/mL depending on the matrix and range needed for unknown samples. Aclarubicin was used as an internal standard at a concentration of 25 ng/mL. DXR and DXR_C13 high, medium, and low quality controls (QC) were also prepared in appropriate matrix at concentrations dependent on the range of the calibration curve.
PTX and PTX_C13 calibration standards were prepared in rat protein-free plasma, and rat plasma with concentrations ranging from 0.5 ng/mL to 10 μg/mL depending on the matrix and range needed for unknown samples. PTX_d5 was used as an internal standard at a concentration of 25 ng/mL. PTX and PTX_C13 low, medium, and high QC were also prepared in an appropriate matrix at concentrations dependent on the range of the calibration curve.
Fifty (50) microliters of standards and QC in matrix were processed similarly to the unknown samples described as above. Matrix blank, ISTD spiked matrix blank, and quality control samples were run with each calibration curve. The low, mid, and high QC standards met the acceptance criteria of accuracy deviation not exceeding 15% from the true value, and precision not exceeding 15%, in both plasma and protein-free plasma. The lower limit of quantitation (LLOQ) was established at 10 ng/mL in plasma and 0.1 ng/mL in protein-free plasma for DXR, and 10 ng/mL in plasma and 0.5 ng/mL in protein-free plasma for PTX.
Husbandry
Pharmacokinetic studies were conducted in double jugular catheterized 15-week-old male Sprague–Dawley rats (approximately weight of 400 g, Charles River Laboratories, Raleigh, N.C.). Animals arrived the day prior to study initiation. To keep the catheters patent, catheters were stored and flushed with 500 IU/mL heparin in PBS. Animal rooms were kept at 50% relative humidity, 68–72 °F with 12 h light/dark cycles. Rats were housed two animals/cage (rat polycarbonate cage type with 1/4 in. corncob bedding). Animals were allowed ad libitum access to Purina 5L79 and reverse osmosis water.
The Frederick National Laboratory for Cancer Research is accredited by Association for Assessment and Accreditation of Laboratory Animal Care International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals (Health Research Extension Act of 1985, Public Law 99-158, 1986). Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals (National Research Council, 1996; National Academy Press, Washington, DC.. All animal protocols were approved by the NCI at Fredrick Institutional Animal Care and Use Committee (ACUC).
Animal Study Designs
For the DXR pharmacokinetic study, rats were treated intravenously by left jugular catheter with 5 mg of DXR/5 mL/kg of Doxil or generic liposomal DXR (8 rats/treatment group). Blood samples (400 μL) were collected in K2EDTA tubes by the right jugular catheter at 0.25, 0.5, 1, 4, 8, 12, 24, 48, and 96 h.
For the PTX pharmacokinetic study, rats were treated intravenously by left jugular catheter with 6 mg PTX/3 mL/kg of Abraxane, Genexol-PM, or Taxol generic (8 rats/treatment group). Blood samples (400 μL) were collected in K2EDTA tubes by the right jugular catheter at 0.25, 0.5, 2, 4, 6, 8, 24, 48, and 72 h.
Blood was centrifuged at 2500g for 10 min, and plasma (∼200 μL) was collected in a glass vial. Analysis of the plasma samples was conducted as described in the analysis section above. For these in vivo studies, approximately 200 μL of plasma was the maximum obtainable volume for each time point, based on ACUC regulations. Therefore, plasma volumes were adjusted from those used in the in vitro drug release studies, as described in the SITUA section above.
Noncompartmental Pharmacokinetic Analysis
Noncompartmental pharmacokinetic parameters were determined using Phoenix WinNonlin version 6.3 software (Pharsight Corporation, Mountain View, CA). The area under the concentration–time curve including all time points (AUCall) was calculated using the linear trapezoidal rule without extrapolation; the area under the concentration–time curve to time infinity (AUCinf) was calculated using the linear trapezoidal rule with extrapolation to time infinity; the Cmax term is the maximum measured concentration; the Tmax term is the time of maximum concentration; clearance (CL) was determined by the equation CL = dose/AUCinf; the AUMC term is the area under the first moment curve; volume of distribution steady state (Vss) was determined by the equation Vss = (dose/AUCinf)×(AUMC/AUCinf); mean residence time (MRTinf) was determined by the equation MRTinf = AUMCinf/AUCinf; the λz term is the ln slope of the terminal elimination phase; half-life (T1/2), is determined by T1/2 = 0.693/λz; concentration time zero (C0) is the extrapolated concentration of the initial slope to the y-intercept; volume of distribution apparent (Vd) was determined by the equation Vd = dose/C0.
Statistics
Statistical differences were identified by Student’s t test or one-way ANOVA with Tukey’s posthoc test, with level of significance p ≤ 0.05.
Acknowledgments
The authors thank Samyang Biopharmaceuticals Corporation for generously providing the Genexol-PM formulation. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health under Contracts HHSN2612008000001E, and the Food and Drug Administration under task order HHSN26100024, Contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Author Contributions
Participated in research design: Kozak, Jiang, Liu, Stern. Conducted experiments: Skoczen, Snapp, Stern. Performed data analysis: Skoczen, Snapp, Stern. Contributed to the writing of the manuscript: Skoczen, Snapp, Crist, Kozak, Jiang, Liu, and Stern.
The authors declare no competing financial interest.
References
- European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP) (2013) Reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal product, European Medicines Agency.
- Food and Drug Administration (FDA), Office of Generic Drugs (2012) Draft guidance on paclitaxel, Food and Drug Administration.
- European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP) (2013) Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products, European Medicines Agency.
- Food and Drug Administration (FDA), Office of Generic Drugs (2013) Draft guidance on doxorubicin hydrochloride, Food and Drug Administration.
- Food and Drug Administration (FDA) (2014) Draft guidance on daunorubicin citrate, Food and Drug Administration
- Food and Drug Administration (FDA), Office of Generic Drugs (2016) Draft guidance on amphotericin B, Food and Drug Administration
- Ambardekar V. V., and Stern S. T. (2015) NBCD Pharmacokinetics and Drug Release Methods, in Non-Biological Complex Drugs; The Science and the Regulatory Landscape (Crommelin D. J. A., and de Vlieger J. S. B., Eds.) 1st ed., pp 261–287, Springer International Publishing.. [Google Scholar]
- Stern S. T.; Martinez M. N.; Stevens D. M. (2016) When Is It Important to Measure Unbound Drug in Evaluating Nanomedicine Pharmacokinetics?. Drug Metab. Dispos. 44, 1934–1939. 10.1124/dmd.116.073148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoczen S.; McNeil S. E.; Stern S. T. (2015) Stable isotope method to measure drug release from nanomedicines. J. Controlled Release 220, 169–174. 10.1016/j.jconrel.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A.; Tsay P. K.; Lin S. H.; Shau W. Y.; Chao D. Y. (2001) The applications of capture-recapture models to epidemiological data. Stat Med. 20, 3123–3157. 10.1002/sim.996. [DOI] [PubMed] [Google Scholar]
- Schellekens R. C.; Stellaard F.; Woerdenbag H. J.; Frijlink H. W.; Kosterink J. G. (2011) Applications of stable isotopes in clinical pharmacology. Br. J. Clin. Pharmacol. 72, 879–897. Erratum in: Br J Clin Pharmacol. (2013) 75, 1171.). 10.1111/j.1365-2125.2011.04071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoczen S. L.; Stern S. T. (2018) Improved Ultrafiltration Method to Measure Drug Release from Nanomedicines Utilizing a Stable Isotope Tracer. Methods Mol. Biol. 1682, 223–239. 10.1007/978-1-4939-7352-1_19. [DOI] [PubMed] [Google Scholar]
- Lee H. B.; Blaufox M. D. (1985) Blood volume in the rat. J. Nucl. Med. 26, 72–76. [PubMed] [Google Scholar]
- Siegal T.; Horowitz A.; Gabizon A. (1995) Doxorubicin encapsulated in sterically stabilized liposomes for the treatment of a brain tumor model: biodistribution and therapeutic efficacy. J. Neurosurg. 83, 1029–1037. 10.3171/jns.1995.83.6.1029. [DOI] [PubMed] [Google Scholar]
- Sorrento (2015) Next Generation Cancer Therapeutics, Sorrento, http://sorrentotherapeutics.com/wp-content/uploads/2013/10/SORRENTO-Apr-2015.pdf.
- Sparreboom A.; Scripture C. D.; Trieu V.; Williams P. J.; De T.; Yang A.; Beals B.; Figg W. D.; Hawkins M.; Desai N. (2005) Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin. Cancer Res. 11, 4136–4143. 10.1158/1078-0432.CCR-04-2291. [DOI] [PubMed] [Google Scholar]
- Deitchman A. N.; Singh R. S. P.; Derendorf H. (2018) Nonlinear Protein Binding: Not What You Think. J. Pharm. Sci. 107, 1754–1760. 10.1016/j.xphs.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousba A., Underbrink R., and Kruppa D. (2013) Comparison of Encapsulated and Non-encapsulated Doxorubicin Pharmacokinetics Following Single Intravenous Administration of ATI-0918, an Encapsulated Liposomal Formulation, and DOXIL in Female Rats, AAPS Annual Meeting, San Antonio, TX.
- Liu Z.; Bi Y.; Sun Y.; Hao F.; Lu J.; Meng Q.; Lee R. J.; Tian Y.; Xie J. (2017) Pharmacokinetics of a liposomal formulation of doxorubicin in rats. Saudi Pharm. J. 25, 531–536. 10.1016/j.jsps.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C. K.; Adamo B.; Karginova O.; Deal A. M.; Rawal S.; Darr D.; Schorzman A.; Santos C.; Bash R.; Kafri T.; Carey L.; Miller C. R.; Perou C. M.; Sharpless N.; Zamboni W. C. (2013) Pharmacokinetics and efficacy of PEGylated liposomal doxorubicin in an intracranial model of breast cancer. PLoS One 8, e61359. 10.1371/journal.pone.0061359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A.; Carmichael D.; Harris M.; Roh J. K. (1986) Comparative pharmacokinetics of free doxorubicin and doxorubicin entrapped in cardiolipin liposomes. Cancer Res. 46, 2295–2299. [PubMed] [Google Scholar]
- Schmidt S.; Gonzalez D.; Derendorf H. (2010) Significance of protein binding in pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 99, 1107–1122. 10.1002/jps.21916. [DOI] [PubMed] [Google Scholar]
- Jamis-Dow C. A.; Klecker R. W.; Katki A. G.; Collins J. M. (1995) Metabolism of taxol by human and rat liver in vitro: a screen for drug interactions and interspecies differences. Cancer Chemother. Pharmacol. 36, 107–114. 10.1007/BF00689193. [DOI] [PubMed] [Google Scholar]
- Kasai T.; Oka M.; Soda H.; Tsurutani J.; Fukuda M.; Nakamura Y.; Kawabata S.; Nakatomi K.; Nagashima S.; Takatani H.; Fukuda M.; Kinoshita A.; Kohno S. (2002) Phase I and pharmacokinetic study of paclitaxel and irinotecan for patients with advanced non-small cell lung cancer. Eur. J. Cancer 38, 1871–1878. 10.1016/S0959-8049(02)00231-9. [DOI] [PubMed] [Google Scholar]
- Vigano L.; Locatelli A.; Grasselli G.; Gianni L. (2001) Drug interactions of paclitaxel and docetaxel and their relevance for the design of combination therapy. Invest. New Drugs 19, 179–196. 10.1023/A:1010691218625. [DOI] [PubMed] [Google Scholar]
- Jameson G. S.; Borazanci E.; Babiker H. M.; Poplin E.; Niewiarowska A. A.; Gordon M. S.; Barrett M. T.; Rosenthal A.; Stoll-D’Astice A.; Crowley J.; Shemanski L.; Korn R. L.; Ansaldo K.; Lebron L.; Ramanathan R. K.; Von Hoff D. D. (2020) Response Rate Following Albumin-Bound Paclitaxel Plus Gemcitabine Plus Cisplatin Treatment Among Patients With Advanced Pancreatic Cancer: A Phase 1b/2 Pilot Clinical Trial. JAMA Oncol 6, 125–132. (Erratum in JAMA Oncol. 2019;5(11):1643. doi:10.1001/jamaoncol.2019.5486) 10.1001/jamaoncol.2019.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai N.; Trieu V.; Yao Z.; Louie L.; Ci S.; Yang A.; Tao C.; De T.; Beals B.; Dykes D.; Noker P.; Yao R.; Labao E.; Hawkins M.; Soon-Shiong P. (2006) Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 12, 1317–1324. 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- Giordano G.; Pancione M.; Olivieri N.; Parcesepe P.; Velocci M.; Di Raimo T.; Coppola L.; Toffoli G.; D’Andrea M. R. (2017) Nano albumin bound-paclitaxel in pancreatic cancer: Current evidences and future directions. World J. Gastroenterol 23, 5875–5886. 10.3748/wjg.v23.i32.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borga O.; Lilienberg E.; Bjermo H.; Hansson F.; Heldring N.; Dediu R. (2019) Pharmacokinetics of Total and Unbound Paclitaxel After Administration of Paclitaxel Micellar or Nab-Paclitaxel: An Open, Randomized, Cross-Over, Explorative Study in Breast Cancer Patients. Adv. Ther. 36, 2825–2837. 10.1007/s12325-019-01058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M.; Plaza C.; Musteanu M.; Illei P.; Brachmann C. B.; Heise C.; Pierce D.; Lopez-Casas P. P.; Menendez C.; Tabernero J.; Romano A.; Wei X.; Lopez-Rios F.; Von Hoff D. D. (2015) SPARC Expression Did Not Predict Efficacy of nab-Paclitaxel plus Gemcitabine or Gemcitabine Alone for Metastatic Pancreatic Cancer in an Exploratory Analysis of the Phase III MPACT Trial. Clin. Cancer Res. 21, 4811–4818. 10.1158/1078-0432.CCR-14-3222. [DOI] [PubMed] [Google Scholar]
- Kim H.; Samuel S.; Lopez-Casas P.; Grizzle W.; Hidalgo M.; Kovar J.; Oelschlager D.; Zinn K.; Warram J.; Buchsbaum D. (2016) SPARC-Independent Delivery of Nab-Paclitaxel without Depleting Tumor Stroma in Patient-Derived Pancreatic Cancer Xenografts. Mol. Cancer Ther. 15, 680–688. 10.1158/1535-7163.MCT-15-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubach J. M.; Kim E.; Yang K.; Cuccarese M.; Giedt R. J.; Meimetis L. G.; Vinegoni C.; Weissleder R. (2017) Quantitating drug-target engagement in single cells in vitro and in vivo. Nat. Chem. Biol. 13, 168–173. 10.1038/nchembio.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Zheng M.; Gao J.; Wang J.; Zhang Q.; Fawcett J. P.; He Y.; Gu J. (2020) Uptake and release profiles of PEGylated liposomal doxorubicin nanoparticles: A comprehensive picture based on separate determination of encapsulated and total drug concentrations in tissues of tumor-bearing mice. Talanta 208, 120358. 10.1016/j.talanta.2019.120358. [DOI] [PubMed] [Google Scholar]
- He H.; Yuan D.; Wu Y.; Cao Y. (2019) Pharmacokinetics and Pharmacodynamics Modeling and Simulation Systems to Support the Development and Regulation of Liposomal Drugs. Pharmaceutics 11, 110. 10.3390/pharmaceutics11030110. [DOI] [PMC free article] [PubMed] [Google Scholar]