

Abstract
CD137 modulates type 1 diabetes (T1D) progression in nonobese diabetic (NOD) mice. We previously showed that CD137 expression in CD4 T cells inhibits T1D, but its expression in CD8 T cells promotes disease development by intrinsically enhancing the accumulation of β-cell autoreactive CD8 T cells. CD137 is expressed on a subset of FOXP3+ regulatory CD4 T cells (Tregs), and CD137+ Tregs are the main source of soluble CD137 (sCD137). sCD137 suppresses T cells in vitro by binding to CD137 ligand (CD137L) upregulated on activated T cells. To further study how the opposing functions of CD137 are regulated, we successfully targeted Tnfsf9 (encoding CD137L) in NOD mice using the CRISPR/Cas9 system (designated NOD.Tnfsf9−/−). Relative to wildtype NOD mice, T1D development in the NOD.Tnfsf9−/− strain was significantly delayed, and mice developed less insulitis and had reduced frequencies of β-cell autoreactive CD8 T cells. Bone marrow chimera experiments showed that CD137L-deficient hematopoietic cells were able to confer T1D resistance. Adoptive T cell transfer experiments showed that CD137L deficiency on myeloid antigen-presenting cells was associated with T1D suppression. Conversely, lack of CD137L on T cells enhanced their diabetogenic activity. Furthermore, neither CD137 nor CD137L was required for the development and homeostasis of FOXP3+ Tregs. However, CD137 was critical for the in vivo T1D suppressive activity of FOXP3+ Tregs, suggesting that the interaction between CD137 and CD137L regulates their function. Collectively, our results provide new insights into the complex roles of CD137-CD137L interaction in T1D.
Introduction
Effective T cell activation requires signals provided by an array of co-stimulatory molecules. Among the known co-stimulatory receptors, CD137 (4–1BB) promotes immune responses against pathogens and cancers and modulates autoimmune diseases in animal models (1-3). CD137 is upregulated upon T cell activation and interacts with CD137 ligand (CD137L), mainly expressed on antigen-presenting cells (APCs) but also expressed on activated T cells (4). CD137-CD137L interaction can elicit bidirectional signaling pathways (5). The co-stimulatory activity of CD137 enhances CD8 T cell activation and survival, a functional characteristic that makes CD137 an important target for cancer immunotherapy (6). CD137L reverse signaling stimulates macrophages but it also exerts an inhibitory activity when expressed on activated T cells (7-9). Interestingly, CD137L can function independently of CD137. Interaction of the cytoplasmic domain of CD137L with toll-like receptor 4 (TLR4) leads to sustained tumor necrosis factor (TNF) production in activated macrophages (10).
Type 1 diabetes (T1D) is a chronic disorder of autoimmune-mediated destruction of insulin-producing pancreatic β cells (11, 12). The incidence of T1D has been increasing in the past two decades worldwide (13, 14). Both genetic predisposition and environmental triggering factors contribute to T1D development in humans as well as the nonobese diabetic (NOD) mouse model (11, 15-17). The C57BL/10 (B10)-derived Idd9.3 genetic locus located within the distal end of chromosome 4 confers T1D suppression when congenically introduced into NOD mice (18). Tnfrsf9 (encoding CD137) has been suggested as an underlying gene for the Idd9.3 susceptibility locus in NOD mice (19). CD137 is constitutively expressed on a subset of FOXP3+ regulatory CD4 T cells (Tregs) (20-24). B10 Tnfrsf9 encodes a more functional CD137 protein than the NOD allele and promotes accumulation of CD137+ FOXP3+ Tregs (25-27). Compared to CD137− Tregs, the CD137+ subset has enhanced ability to suppress proliferation of anti-CD3 and anti-CD28-stimulated T cells in vitro (26). This enhanced suppressive activity of CD137+ Tregs is at least partly due their increased ability to secrete soluble CD137 that binds CD137L expressed on activated T cells (8, 26).
Recently, we described opposing roles of CD137 in T1D with a disease promoting function mediated by CD137+ CD8 T cells and a suppressive activity performed by CD137+ CD4 T cells (28). Nevertheless, T1D development is inhibited in NOD mice with global knockout of CD137. It is assumed that CD137-CD137L interaction is important for the accumulation of β-cell autoreactive CD8 T cells and T1D development since CD137L is considered as the sole ligand for CD137. However, this interpretation needs to be further tested using CD137L-deficient NOD mice. Several matrix proteins such as laminin and fibronectin have been reported to interact with CD137 with unknown function (29). Additionally, galectin-9, a carbohydrate binding protein directly interacts with CD137 to facilitate its multimerization on the cell membrane for triggering downstream signaling in T cells (30). It is not known if the galectin-9/CD137 complex can have CD137L-independent activity. In addition, we do not know what cell types must express CD137L in order to engage CD137 on CD8 T cells for T1D development. We previously showed that CD137-deficient CD4 T cells were more diabetogenic than the wildtype counterparts (28). We speculated that this is due to the absence of CD137+ Tregs, although the T1D suppressive function of CD137+ Tregs has not been directly tested in vivo. It is also not known if CD137-CD137L interaction is important for the development or homeostasis of CD137+ Tregs. To address these questions, we generated and characterized CD137L-deficient NOD mice. Our results indicate that CD137L is critical for the survival of β-cell autoreactive CD8 T cells and T1D development likely through its expression on myeloid APCs. Additionally, we demonstrate that CD137 expression on Tregs plays an important role in T1D suppression in vivo but the development and homeostasis of total Tregs or the CD137+ subset are CD137L independent.
Materials and Methods
Mouse strains
NOD/ShiLtJ (NOD), NOD.129S7(B6)-Rag1tm1Mom/J (NOD.Rag1−/−), and NOD.B6-Ptprcb/6908MrkTacJ (NOD.Cd45.2) mice were purchased from The Jackson Laboratory and then maintained at the Medical College of Wisconsin (MCW) by brother-sister mating. NOD.Tnfsf9−/− mice were generated using the CRISPR/Cas9 technology as previously described (31). Briefly, NOD embryos were microinjected with 3 pL of a solution containing Cas9 mRNA and single guide RNA (sgRNA) at respective concentrations of 100 and 50 ng/mL. The sgRNA sequence (5’-GCGGATGCCAGACATCCAGC-3’) was designed to target the first coding exon of Tnfsf9. Genomic tail DNA was screened by Sanger sequencing. The genomic region around the targeted site was amplified by PCR with primers Tnfsf9-genoF (5’- AAGGGAAGAAGGACGGGCGGGCAC-3’) and Tnfsf9-genoR (5’-GCGGCGATCAGAAGCAGCAAAACC-3’). The resulting PCR product was purified and sequenced. During the screening of the N1 progeny, an allele with a 22 base-pair deletion was identified. If produced, the mutant protein is predicted to retain the first 17 amino acids of wildtype CD137L, followed by 49 aberrant amino acids prior to a stop codon. N1 mice were backcrossed to NOD mice followed by intercrossing to fix the mutation to homozygosity. NOD.Rag1−/−.Tnfsf9−/− mice were generated by outcrossing NOD.Tnfsf9−/− mice to the NOD.Rag1−/− strain followed by intercrossing. NOD.Tnfrsf9−/− mice have been previously reported (32). NOD.Foxp3-eGFP mice have been previously described (33). NOD.Tnfrsf9-/−.Foxp3-eGFP mice were generated by outcrossing NOD.Tnfrsf9−/− to NOD.Foxp3-eGFP mice followed by intercrossing. All mouse experimental protocols were carried out in accordance with the MCW Institutional Animal Care and Use Committee guidelines and approved by the committee.
Assessment of T1D and insulitis
T1D and insulitis development were assessed as previously described (28). Briefly, T1D development was monitored weekly using urine glucose strips (Diastix; Bayer) with onset defined by two consecutive readings of >250 mg/dL. For evaluation of insulitis, the pancreas was fixed in a 10% formalin solution and sectioned at four nonoverlapping levels. Granulated β cells were stained with aldehyde fuchsin dye and leukocytes with an H&E counterstain. Islets were individually scored as following: 0, no lesions; 1, peri-insular leukocytic aggregates; 2, < 25% islet destruction; 3, >25% islet destruction; and 4, complete islet destruction. For each mouse more than 30 islets were examined and used to calculate the mean insulitis score.
T cell adoptive transfer studies
To test the diabetogenic activity of CD137L-deficient T cells, total splenic T cells were isolated from 6–8-week-old female donor mice (NOD or NOD.Tnfsf9−/−) by negative selection using pan T cell isolation kit II (Miltenyi Biotec). A total of 5x106 T cells were injected intravenously into 4–6-week-old NOD.Rag1−/− female recipients. To test the diabetogenic function of CD137L in non-T and non-B cells, a total of 5x106 splenic T cells were isolated from 6–8-week or 13-week-old NOD females by negative selection using pan T cell isolation kit II (Miltenyi Biotec) and injected intravenously into 4–10-week old age-matched NOD.Rag1−/− or NOD.Rag1-/−.Tnfsf9−/− female. To test the suppression activity of CD137+ Tregs in vivo, FOXP3+ (GFP+) CD4 T cells were sorted from 7–10-week-old wildtype (NOD.Foxp3-eGFP) or CD137-deficient (NOD.Tnfrsf9−/−.Foxp3-eGFP) female mice using BD FACSAria II. CD25-depleted splenic T cells isolated from 12–15-week-old NOD females were transferred alone or with Tregs admixed at a 10:1 ratio into NOD.Rag1−/− female recipients. A total of 3–5x106 T cell mixtures were injected intravenously into 4–6-week old NOD.Rag1−/− female recipients. To test the homeostasis and stability of transferred CD137-deficient Tregs in vivo, FOXP3+ (GFP+) CD4 T cells were sorted from 7–10-week-old wildtype (NOD.Foxp3-eGFP) or CD137-deficient (NOD.Tnfrsf9−/−.Foxp3-eGFP) female mice. Non-FOXP3+ T cells (GFP−) sorted from 10-week-old wildtype (NOD.Cd45.2.Foxp3-eGFP) females were co-transferred with the sorted GFP+ Tregs admixed at a 10:1 ratio into NOD.Rag1−/− female recipients. A total of 5x106 T cells were injected intravenously into 4–6-week-old NOD.Rag1−/− female recipients. To test the response of CD137L-deficient T cells to Treg-mediated suppression in vivo, FOXP3+ (GFP+) CD4 T cells were sorted from 7–10-week-old wildtype (NOD.Foxp3-eGFP) female mice. CD25-depleted splenic T cells were isolated from 6–7-week-old NOD or NOD.Tnfsf9−/− females and co-transferred with sorted FOXP3+ Tregs admixed at a 10:1 ratio into 5–7-week-old NOD.Rag1−/− female recipients.
Generation of bone marrow (BM) chimeras
BM cells were collected from femurs and tibias of 7–10-week-old NOD, NOD.Cd45.2, and NOD.Tnfsf9−/− female mice. BM was depleted of T cells using anti-CD3e kit as described by the manufacturer (Miltenyi Biotec). Four to seven-week-old NOD, NOD.Cd45.2, NOD.Tnfsf9−/−, or (NOD x NOD.Cd45.2) F1 recipient mice were lethally irradiated (550 rads x 2) and reconstituted with T cell-depleted BM cells (5x106) isolated from the indicated donor strains. BM recipients were followed for T1D development or analyzed by flow cytometry.
Flow cytometry analysis
Fluorochrome-labeled antibodies specific for CD3ε (145–2C11), TCRβ (H57–597), CD4 (RM4–5), CD8α (53–6.7), CD44 (IM7.8.1), CD45.1 (A20), CD45.2 (104), FOXP3 (FJK-16s), CD25 (PC61), CD19 (ID3), B220 (RA3–6B2), CD137 (17B5/1AH2), CD11b (M1–70), CD11c (N418 or HL3), GR-1 (RB6–8C5), IFNγ (XMG1.2), PDCA-1 (eBio129c), and CD137L (TKS-1) were purchased from BD Biosciences (San Jose, CA), Bio-Legend (San Diego, CA), or eBioscience. Mouse MHC class I (Kd) tetramers loaded with an islet-specific glucose-6-phosphatase catalytic subunit–related protein (IGRP) mimotope peptide NRP-V7 (KYNKANVFL) was conjugated with BV421 and obtained from the National Institutes of Health Tetramer Core Facility (34). Single-cell suspension was prepared from the spleens, pancreatic lymph nodes (PLNs), thymi, and islets of mice at the indicated age. Red blood cells were lysed with the ACK lysis buffer then washed cells were suspended in FACS buffer. Cells were first blocked with Fc block (BD Biosciences) at room temperature for 10 min. For tetramer staining, cells were incubated with MHC class I tetramers and Fc block for 15 min at room temperature. For staining other surface markers, antibodies were then added to stain the cells for an additional 30 min at 4°C. To assess cell viability, stained islet cells were incubated with 200 ng/mL of Annexin V (Biolegend) and 2.5 μg/mL of 7-aminoactinomycin D (Sigma) for 15 min at room temperature. To analyze IFNγ production, cells were cultured at 37°C for 4 hours in the presence of phorbol myristate acetate (PMA, 20 ng/mL, Sigma), ionomycin (1 μg/mL, Sigma), and GolgiPlug (1 μL/mL, BD). For Treg staining, cells were stained for surface markers, washed with FACS buffer, and then fixed/permeabilized for 1 h using the FOXP3 staining buffer from eBioscience. Fixed cells were washed twice with permeabilization buffer and then stained with anti-FOXP3 antibody for 30 min at 4°C. Stained cells were washed once with the FACS buffer and acquired using the FACSCalibur or LSRFortessa flow cytometer (BD Biosciences). All flow cytometric data were analyzed with the FlowJo software (Tree Star, Ashland, OR).
Islet isolation and analysis of infiltrates
Islet infiltrating cells were prepared as previously described (35). Briefly, pancreata from 10–12-week-old NOD and NOD.Tnfsf9−/− female mice were inflated using a 30-gauge needle by injecting 3–5 mL of HBSS containing 0.5 U/mL collagenase P solution (Roche Diagnostics) and 10 μg/mL DNase (Sigma) into the bile duct. The inflated pancreas was digested at 37°C for 15 min. Next, digested pancreata were washed three times with 10 mL of HBSS containing 2% FBS. Each individual pancreas was then suspended in 5 mL complete RPMI 1640. Islets were visualized under a dissecting microscope and hand-picked. Pelleted islets were suspended in 200 μL of enzyme free cell dissociation buffer (Life Technologies). Cells were then washed with 500 μL of HBSS, resuspended in FACS buffer, and stained with the indicated antibodies.
Analysis of Tnfsf9 mRNA expression
To measure the expression of Tnfsf9 mRNA, spleens and PLNs (pooled from 5 mice) were harvested from 10-weeks-old NOD females and digested with collagenase D (Roche). After antibody staining, cells were sorted on BD FACSAria II into the following populations: CD3+ T cells, CD3− CD11chi conventional dendritic cells (cDCs), CD3− CD11clow PDCA-1+ plasmacytoid dendritic cells (pDCs), and CD3− CD11c− CD11b+ GR-1− macrophages. TRIzol reagent was added to the sorted cells, and RNA was extracted according to the manufacturer’s instructions (Invitrogen). cDNA was synthesized and amplified according to the Smart-Seq2 protocol described previously (36). Briefly, 10–20 ng of extracted RNA was used to synthesize the first strand of cDNA using Superscript II (Invitrogen), Betaine (sigma) for enhancing the reaction, and template switch oligo to unify the 3’ ends of the synthesized cDNA for the amplification step. Synthesized cDNA was subjected to an amplification step using the KAPA HiFi HotStart master mix (Kapa Biosystems) and ISPCR primer. Amplified cDNA was bound to Ampure XP beads (Beckman Coulter), washed with ethanol, and finally eluted in nucleases free water. About 22 ng of cDNA was used in a qRT-PCR reaction with a Taqman assay for Tnfsf9 (Mm00437154_m1) or Hprt1 (Mm01545399_m1). The results were analyzed with CFX Maestro software (Bio-Rad), and all data were normalized to Hprt1.
Statistical analysis
Mann-Whitney U test or Wilcoxon matched-pairs signed rank test was used for the comparative analysis between two groups as indicated. Log rank test was used for the analysis of T1D incidence. All statistical analyses were performed using the GraphPad Prism 8 software (La Jolla, CA).
Results
Generation of CD137L-deficient NOD mice
We previously demonstrated that CD137 plays dual roles in T1D. CD137 suppresses T1D development when expressed in CD4 T cells; conversely, it promotes T1D when expressed in CD8 T cells (28). Here, we aimed to assess the contribution of CD137L to the suppression and/or the enhancement of T1D mediated by CD137. To test the effect of CD137L deficiency on T1D, we used the CRISPR/Cas9 technology to directly target Tnfsf9 (encoding CD137L) for disruption in NOD mice. A 22 base pair deletion was introduced in the first coding exon of Tnfsf9 immediately downstream of the start codon (Figure 1A). We subsequently confirmed the lack of CD137L protein expression in the newly generated NOD.Tnfsf9−/− strain using flow cytometry. Upon LPS and anti-CD40 stimulation, splenic CD11c+ dendritic cells (DCs) from NOD but not NOD.Tnfsf9−/− mice expressed CD137L (Figure 1B).
Figure 1. Development of diabetes is suppressed in NOD.Tnfsf9−/− mice.

(A) CRISPR/Cas9-mediated modification of the Tnfsf9 gene in NOD mice. The structure of the Tnfsf9 gene is illustrated (boxes depict exons). Partial sequences of the first coding exon of the wildtype and mutant Tnfsf9 gene are shown. The 22-nucleotide deletion in the NOD.Tnfsf9−/− strain is indicated in red. (B) NOD.Tnfsf9−/− mice do not express CD137L. Total splenocytes harvested from NOD and NOD.Tnfsf9−/− mice were cultured overnight with 5 μg/mL anti-CD40 and 50 ng/mL LPS. Cells were stained with antibodies specific for CD137L (or isotype control), CD11c, and B220 and analyzed by flow cytometry. Representative histograms of CD137L expression on CD11c+ B220− cells are shown. Similar results were obtained from a separate set of NOD and NOD.Tnfsf9−/− mice. (C and D) T1D development in NOD.Tnfsf9−/− mice is suppressed. Female (C) and male (D) mice were monitored for the development of T1D every week over a course of 30 weeks. ***p < 0.001 by log-rank test. (E- H) Insulitis is reduced in NOD.Tnfsf9−/− mice. Insulitis was histologically analyzed for 10-week-old female (E-F) and male (G-H) NOD and NOD.Tnfsf9−/− mice. Representative islet images are shown on the left and summarized mean insulitis scores are shown on the right. Scale bars represent 100 μm. Pancreatic sections were scored for insulitis: 0, no lesions; 1, peri-insular leukocytic aggregates; 2, < 25% islet destruction; 3, > 25% islet destruction; and 4, complete islet destruction. Each symbol represents one mouse. *p < 0.05 by Mann-Whitney U test; NS: not significant.
CD137L deficiency suppresses T1D development
We previously showed that CD137 deficiency suppressed the onset of T1D in NOD mice (32). Moreover, CD137L blocking antibody treatment delayed the onset of T1D, indicating the significant contribution of the CD137-CD137L interaction to disease progression (28). To further investigate the role of CD137L in T1D development and pathogenesis, we first monitored NOD.Tnfsf9−/− mice for diabetes development. We observed that CD137L deficiency significantly reduced T1D incidence in both females and males (Figures 1C and 1D). Histological analysis of 10-week-old mice revealed significantly less insulitis in NOD.Tnfsf9−/− than in NOD females (Figures 1E and 1F). A reduction of insulitis was also observed in males although the difference was not statistically significant (Figures 1G and 1H). These results highlight a significant role of CD137L and further support the importance of the CD137-CD137L interaction in T1D development.
CD137L deficiency in hematopoietic cells confers T1D resistance
CD137L has been shown to function through its expression in non-immune cells (37). Thus, we next independently defined the impact of CD137L deficiency in hematopoietic and non-hematopoietic cells on T1D development. We transferred T cell-depleted BM cells from NOD or NOD.Tnfsf9−/− (both expressing CD45.1) into lethally irradiated wildtype NOD.Cd45.2 congenic mice and monitored the recipients for diabetes development. We observed that 55% of the NOD.Cd45.2 mice reconstituted with CD137L-deficient BM cells developed diabetes compared to 89% of those infused with wildtype BM cells (Figure 2A). While 55% of CD137L-deficient BM cell reconstituted NOD.Cd45.2 mice developed diabetes, the time of onset was significantly delayed compared to recipients of wildtype BM cells. Flow cytometry analysis performed at T1D onset or at the end of incidence study showed that NOD and NOD.Tnfsf9−/− donor BM cells reconstituted similarly in NOD.Cd45.2 recipients (data not shown). To ask if CD137L deficiency in radiation resistant cells can suppress T1D, we reconstituted lethally irradiated NOD and NOD.Tnfsf9−/− mice with T cell-depleted NOD.Cd45.2 BM cells. Compared to NOD recipients, NOD.Tnfsf9−/− mice reconstituted with NOD.Cd45.2 BM cells were not resistant to diabetes development (Figure 2B). Taken together, these results indicate that CD137L expression in hematopoietic cells is critical for T1D development.
Figure 2. CD137L deficiency in hematopoietic cells confers T1D resistance.
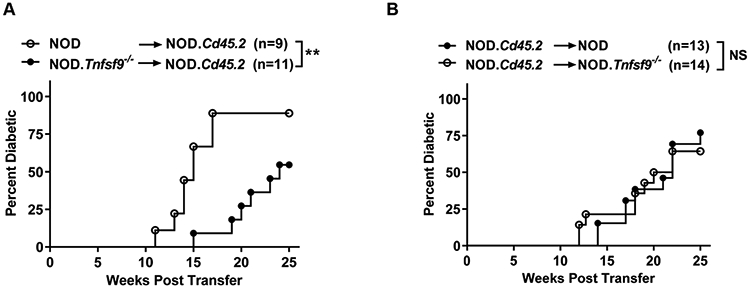
(A) T1D incidence study of lethally irradiated NOD.Cd45.2 females injected with 5x106 T cell-depleted sex-matched NOD or NOD.Tnfsf9−/− BM cells as indicated. **p < 0.01 by log-rank test. (B) T1D incidence study of lethally irradiated NOD or NOD.Tnfsf9−/− females injected with 5x106 T cell-depleted sex-matched NOD.Cd45.2 BM cells. NS: not significant by log-rank test. The mice were monitored for the development of T1D every week over the course of 25 weeks post-transfer. Combined results of 2-3 transfer experiments are shown.
Expression of CD137L on non-T and non-B cells promotes T1D development
The delayed onset of T1D in the absence of CD137L may be related to the loss of its expression on APCs and thus disruption of its interaction with CD137 on T cells. To test the role of CD137L on myeloid APCs, we transferred total splenic T cells from 6-week-old NOD mice into NOD.Rag1−/− or NOD.Rag1-/−.Tnfsf9−/− recipients. As expected, NOD.Rag1−/− recipients of NOD T cells developed T1D. In contrast, T1D development in the NOD.Rag1-/−.Tnfsf9−/− recipients was completely inhibited (Figure 3A). We next analyzed Tnfsf9 expression in DC and macrophage populations sorted from 10-week-old NOD females. T cells were also analyzed for comparison. The highest Tnfsf9 expression was found in cDCs isolated from PLNs, inferring that these myeloid APCs are important for providing CD137L to engage CD137 expressed on T cells (Figure 3B).
Figure 3. Expression of CD137L on non-T and non-B cells is important for the development of T1D.
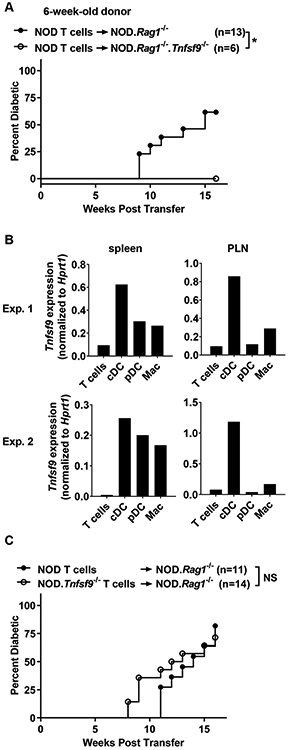
(A) T1D incidence study of NOD.Rag1−/− and NOD.Rag1−/−.Tnfsf9−/− mice adoptively infused with NOD T cells. Total splenic T cells (5x106) were isolated from 6-week-old NOD females and transferred into the indicated female mice. *p < 0.05 by log-rank test. (B) Tnfsf9 expression in T cells (CD3+), cDC (CD3− CD11chi), pDC (CD3− CD11clow PDCA-1+), and macrophages (Mac) (CD3− CD11c− CD11b+ GR-1−) sorted from spleens and PLNs of 10-week-old NOD females was analyzed by qRT-PCR in triplicates. The mean of the triplicates in each cell population is presented. Results from two independent experiments are shown. (C) Total splenic T cells (5x106) were isolated from 6-week-old NOD or NOD.Tnfsf9−/− females and transferred into sex-matched NOD.Rag1−/− recipients. NS: not significant by log-rank test. The mice were followed for the development of T1D every week over the course of 16 weeks post-transfer. Combined results of two independent transfer experiments are shown.
Expression of CD137L on activated T cells has also been reported (4, 9). Engagement of CD137L on the surface of T cells by soluble CD137 or plate-bound anti-CD137L inhibits their activation (8, 9). Hence, we asked if CD137L expression on T cells regulates T1D development. To test this, we transferred total splenic T cells isolated from NOD or NOD.Tnfsf9−/− mice into NOD.Rag1−/− recipients. Both recipient groups developed diabetes, indicating that CD137L expression on T cells is dispensable for their diabetogenic activity (Figure 3C). While the overall T1D incidence did not differ between NOD and NOD.Tnfsf9−/− T cell recipients, the absence of CD137L on T cells caused earlier diabetes onset. By 10 weeks post-transfer, 5/14 NOD.Tnsfs9−/− T cell recipients developed diabetes compared to 0/11 NOD T cell recipients (p=0.046, Fisher’s exact test). This observation is consistent with the idea that CD137L expression on T cells limits their effector activity (8, 9). To further test the hypothesis that CD137L on T cells suppresses their effector function, we transferred total wildtype or CD137L-deficient T cells into NOD.Rag1−/− mice and analyzed their expression of IFNγ and T-bet in the spleens, PLNs, and islets six weeks post-transfer. The frequencies of CD8 and CD4 (FOXP3−) T cells capable of producing IFNγ were identical in the spleens and PLNs between wildtype and CD137L-deficient T cell recipients (Supplementary Figures 1A and 1B). While not reaching statistical significance, on average the frequency of IFNγ+ CD8 T cells in islets was higher in the CD137L-deficient than the wildtype T cell recipients (32.9% and 24.9% respectively, Supplementary Figure 1C). It was also the case for the frequency of IFNγ+ CD4 (FOXP3−) T cells in the islets of CD137L-deficient and wildtype T cell recipients (31.3% and 23.2% respectively). Similar results were obtained when the proportions of T-bet+ IFNγ+ double positive T cells were analyzed (data not shown). The limitation of the analysis described above is that the ability to produce IFNγ+ upon in vitro PMA and ionomycin stimulation may not completely reflect the diabetogenic activity of T cells. Collectively, these results are in line with the idea that expression of CD137L on myeloid APCs drives T1D development while its presence on T cells may downregulate their diabetogenic activity.
Expression of CD137L on non-T and non-B cells contributes to the maintenance of β-cell autoreactive CD8 T cells
Activated β-cell autoreactive CD8 T cells are important effectors for T1D development and their frequency increases over time in lymphoid organs and islets in NOD mice (38, 39). Previously, we showed that NOD mice lacking the expression of CD137 had reduced β-cell autoreactive CD8 T cells (28). Thus, we asked whether CD137L deficiency also has a negative impact on the frequency of β-cell autoreactive CD8 T cells. Using Kd MHC class I tetramers loaded with a mimotope peptide (NRP-V7) (38), we assessed IGRP206–214-reactive CD8 T cells in the spleens, PLNs, and pancreatic islets of 10–12-week-old NOD and NOD.Tnfsf9−/− mice. Consistent with our previously published data of CD137-deficient NOD mice, we found significant reduction in the frequency and number of tetramer+ CD8 T cells in all tested tissues in NOD.Tnfsf9−/− mice (Figures 4A-4C). Next, we asked whether the absence of CD137-CD137L interaction affects the survival of islet IGRP206–214-reactive CD8 T cells by 7-AAD and Annexin V co-staining. Compared to the wildtype NOD control, NOD.Tnfsf9−/− mice had increased frequencies of dead (7-AAD+ Annexin V+) and reduced proportions of viable (7-AAD− Annexin V−) cells among total islet IGRP206–214-reactive CD8 T cells (Figure 4D). Our observation is in line with the previous studies where CD137-CD137L interaction promoted the survival of activated CD8 T cells and the memory response in non-autoimmune systems (40-43).
Figure 4. CD137L-deficient β-cell autoreactive CD8 T cells fail to accumulate in the spleens, PLNs, or islets of NOD.Tnfsf9−/− mice.
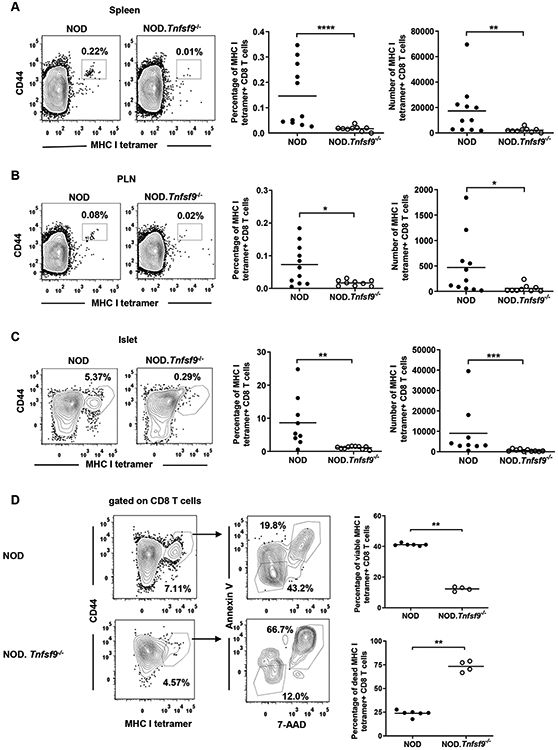
Cells isolated from the spleens (A), PLNs (B), and islets (C) of 10-12-week-old NOD and NOD.Tnfsf9−/− female mice were analyzed for the frequency and number of β-autoreactive CD8 T cells. Cells were stained with antibodies specific for CD45.1, CD3, CD8 and CD44, as well as the NRP-V7-loaded MHC class I tetramer. For (A-C), the left panels show the representative flow cytometry profiles (gated on CD8 T cells) and the right panels are the percentages and numbers of CD44high MHC class I tetramer+ cells summarized from three independent experiments. (D) Cells isolated from islets of 10-week-old NOD and NOD.Tnfsf9−/− female mice were co-stained with Annexin V and 7-AAD for the viability of MHC class I tetramer+ CD8 T cells. The percentages of viable (Annexin V− 7-AAD−) and dead (Annexin V+ 7-AAD+) are summarized on the right. Each symbol represents a mouse. *p < 0.05; **p< 0.01; ***p < 0.001; ****p < 0.0001 by Mann-Whitney U test.
We further tested if lack of CD137L expression on myeloid APCs affects the maintenance of β-cell autoreactive CD8 T cells. Splenic T cells from 13-week-old NOD donors were transferred into NOD.Rag1−/− and NOD.Rag1−/−.Tnfsf9−/− mice. At 13 weeks of age, antigen-experienced IGRP206–214-reactive CD8 T cells can be readily detected in the spleens of NOD mice, and these autoreactive CD8 T cells likely have received signals through CD137. While all NOD.Rag1−/− recipients developed diabetes, NOD.Rag1-/−.Tnfsf9−/− mice did not during the 16-week post-transfer period (Figure 5A). The frequency and number of IGRP206–214-reactive CD8 T cells were analyzed at T1D onset or at the end of the incidence study. Compared to the NOD.Rag1−/− recipients, the NOD.Rag1-/−.Tnfsf9−/− recipients of NOD T cells had greatly diminished IGRP206–214-reactive CD8 T cells although the overall reconstitution level of CD8 T cells was not impaired (Figures 5B-5D and data not shown). These results indicate that expression of CD137L on non-T and non-B cells, presumably myeloid APCs, is critical for the maintenance of β-cell autoreactive CD8 T cells. Our study also suggests that continuous CD137-CD137L interaction is important for the diabetogenic activity of previously activated β-cell autoreactive CD8 T cells.
Figure 5. CD137L expression on non-T and non-B cells is essential for efficient accumulation of β-cell autoreactive CD8 T cells.
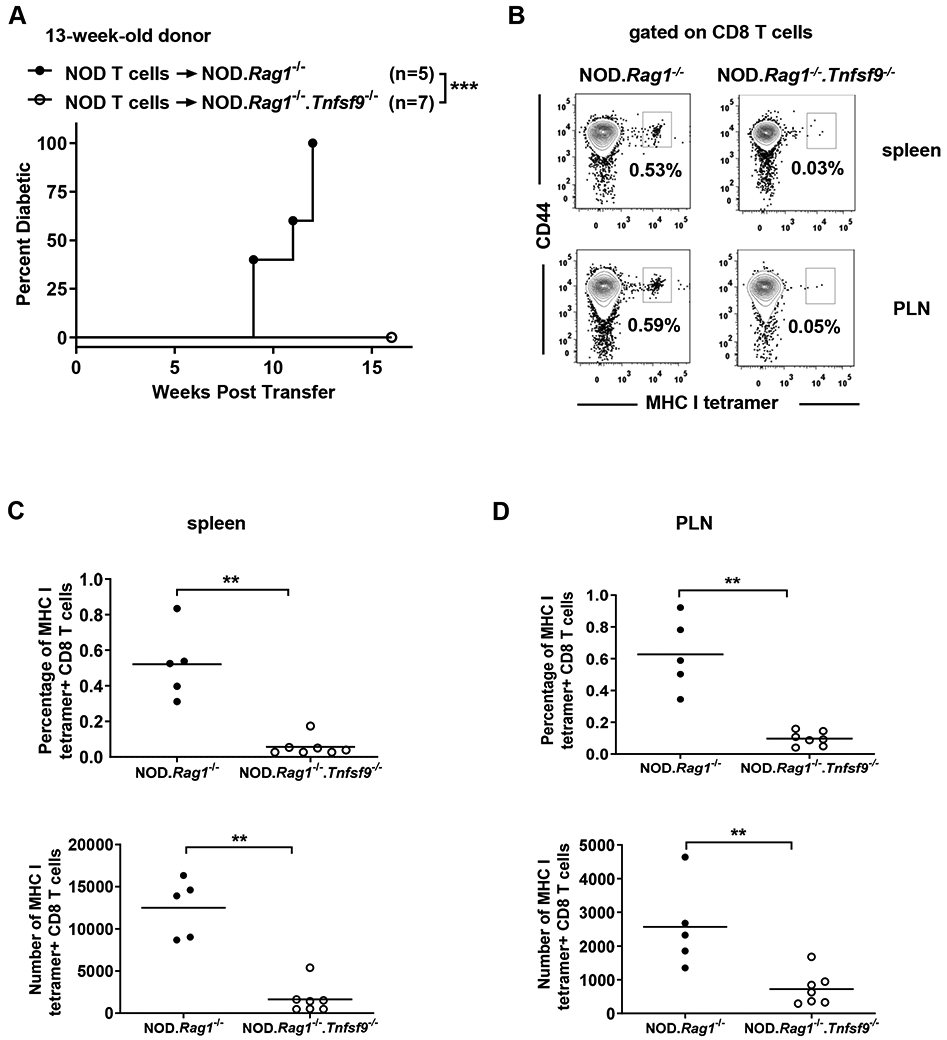
(A) T1D incidence study of NOD.Rag1−/− or NOD.Rag1−/−.Tnfsf9−/− mice adoptively infused with NOD T cells. Total splenic T cells (5x106) were isolated from 13-week-old NOD females and transferred into the indicated female recipients. ***p < 0.001 by log-rank test. (B-D) At diabetes onset or 16 weeks post-transfer, cells isolated from the spleens (C) and PLNs (D) were stained with antibodies specific for CD45.1, CD3, CD8 and CD44, as well as NRP-V7 loaded MHC class I tetramers. Representative flow cytometry plots of NRP-V7 MHC I tetramer staining are shown in (B). The summarized results for the percentages and numbers of NRP-V7 MHC class I tetramer positive CD8 T cells are shown in (C) and (D). Each symbol represents a mouse, **p< 0.01 by Mann-Whitney U test.
We previously demonstrated that CD137 intrinsically promotes the level of β-cell autoreactive CD8 T cells. Based on the results described above, we predicted that CD137L extrinsically controls the frequency of β-cell autoreactive CD8 T cells. To further test this, we reconstituted (NOD x NOD.Cd45.2) F1 recipients with equal proportion of BM cells isolated from NOD.Cd45.2 and NOD.Tnfsf9−/− (expressing CD45.1) donors. After 10–14 weeks of BM reconstitution, we analyzed the frequencies of IGRP206–214-reactive CD8 T cells in the spleens, PLNs, and pancreatic islets. The percentages of wildtype (CD45.2+) and Tnfsf9−/− (CD45.1+)-derived IGRP206–214 reactive CD8 T cells were similar (Supplementary Figure 2). Collectively, these results indicate that the significant reduction of β-cell autoreactive CD8 T cells detected in NOD.Tnfsf9−/− is a cell-extrinsic effect.
Disruption of CD137-CD137L interaction does not alter the frequency of FOXP3+ Tregs
FOXP3+ Tregs contribute significantly to the prevention of autoimmunity via suppressing the activation and effector functions of T cells (44). A subset of FOXP3+ Tregs constitutively express membrane bound CD137 and secrete a significant amount of the immunosuppressive soluble CD137 protein (26). CD137+ FOXP3+ Tregs can be found in the thymus and secondary lymphoid organs, but it is not known if CD137-CD137L interaction is critical for their development and homeostasis. Initially, we tested the impact of lacking CD137 expression on the homeostasis of FOXP3+ Tregs. We measured the frequency of FOXP3+ Tregs in different tissues harvested from NOD and NOD.Tnfrsf9-/−. The frequencies and numbers of FOXP3+ Tregs were comparable in the spleens, PLNs, and pancreatic islets of the two strains (Figures 6A-6C and data not shown). Although we did not observe any numerical difference of total FOXP3+ between NOD and NOD.Tnfrsf9−/− mice, it remained possible that the frequency of the CD137+ subset is affected in the absence of the CD137-CD137L interaction. The newly generated NOD.Tnfsf9−/− mice allowed us to assess the contribution of CD137L to the homeostasis of CD137+ FOXP3+ Tregs. Consistent with the observation in NOD.Tnfrsf9−/− mice, CD137L deficiency did not affect the frequencies and numbers of total FOXP3+ Tregs in the spleens, PLNs, and pancreatic islets of 10-weeks-old NOD.Tnfsf9−/− mice (data not shown). Next, we determined if CD137L deficiency affects the homeostasis of CD137+ FOXP3+ Tregs. The frequencies and numbers of the CD137+ subset were also comparable in the spleens, PLNs, and pancreatic islets of 10-week-old NOD and NOD.Tnfsf9−/− mice (Figures 6D-6F and data not shown). The frequencies and numbers of total or CD137+ FOXP3+ Tregs found in the spleens, PLNs, and thymi of 4-week-old NOD and NOD.Tnfsf9−/− mice were also comparable (data not shown). Taken together, these results indicate that the development and maintenance of total FOXP3+ Tregs or those expressing CD137 are independent of the CD137-CD137L interaction.
Figure 6. CD137-CD137L interaction is not required for the homeostasis of FOXP3+ Tregs.
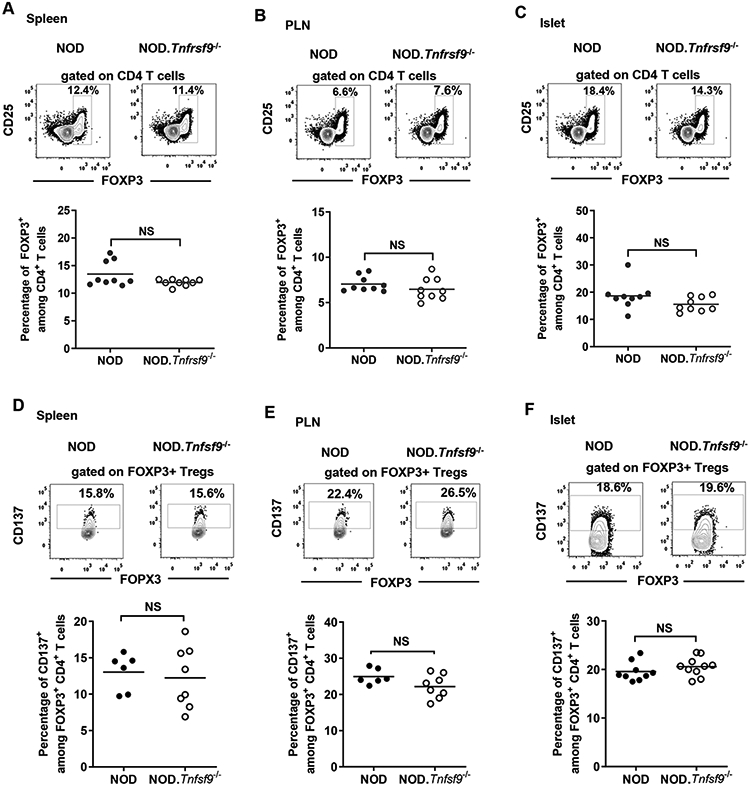
(A-C) CD137 deficiency (Tnfrsf9−/−) does not affect the homeostasis of FOXP3+ Tregs. Cells isolated from the spleens (A), PLNs (B), and islets (C) of 9-11-week-old NOD and NOD.Tnfrsf9−/− female mice were analyzed for the frequency of FOXP3+ Tregs. Cells were stained with antibodies specific for CD45.1, CD3, CD4, CD25, and FOXP3. The percentage of FOXP3+ Tregs among total CD4 T cells was analyzed by flow cytometry. Representative flow cytometry profiles are shown in the upper panels and summarized data from three independent experiments are presented in the lower panels. Each symbol represents a mouse. The horizontal bar depicts the mean. NS: Not significant by Mann-Whitney U test. (D-F) CD137L deficiency (Tnfsf9−/−) does not affect the frequency of CD137+ FOXP3+ Tregs. Cells isolated from the spleens (A), PLNs (B), and islets (C) of 10-week-old NOD and NOD.Tnfsf9−/− female mice were analyzed for the frequency of CD137+ FOXP3+ Tregs. Cells were stained with antibodies specific for CD45.1, CD3, CD4, CD25, CD137, and FOXP3. The percentage of FOXP3+ Tregs that expressed CD137 were analyzed by flow cytometry. Gating for CD137 staining is based on cells isolated from CD137-deficient NOD mice. Representative flow cytometry profiles are shown in the upper panels and summarized data from three independent experiments are presented in the lower panels. Each symbol represents a mouse. The horizontal bar depicts the mean. NS: Not significant by Mann-Whitney U test.
CD137 expression on FOXP3+ Tregs promotes their T1D suppressive function in vivo
Although CD137-CD137L interaction is dispensable for the maintenance of CD137+ Tregs, it may be critical for their T1D suppressive function. To test this hypothesis, we directly compared the in vivo T1D suppressive activity of wildtype and CD137-deficient FOXP3+ Tregs. To facilitate the isolation of Tregs, we first crossed NOD.Tnfrsf9−/− mice to the previously described NOD.Foxp3-eGFP strain to generate a new CD137-deficient NOD.Foxp3-eGFP stock (designated NOD.Tnfrsf9-/−.Foxp3-eGFP) (33). Consistent with our previous study showing decreased T1D in NOD.Tnfrsf9−/−mice and a diabetogenic role of CD137 in CD8 T cells (28), T1D development was significantly slower in NOD.Tnfrsf9-/−.Foxp3-eGFP than in NOD.Foxp3-eGFP mice (Supplementary Figure 3).
Next, we isolated wildtype and CD137-deficient FOXP3+ Tregs respectively from NOD.Foxp3-eGFP and NOD.Tnfrsf9−/−.Foxp3-eGFP mice. Sorted FOXP3+ Tregs were then co-transferred with CD25-depleted NOD (12–15-weeks old) splenic T cells at a 1:10 ratio into NOD.Rag1−/− recipients. As expected, the control group receiving only CD25-depleted NOD splenic T cells developed rapid onset of T1D (Figure 7A). The presence of wildtype FOXP3+ Tregs significantly delayed the onset of diabetes in NOD.Rag1−/− recipients (Figure 7A). In sharp contrast, Tnfrsf9−/− FOXP3+ Tregs failed to suppress T1D in NOD.Rag1−/− recipients (Figure 7A), indicating that CD137 is important for the in vivo suppressive activity of FOXP3+ Tregs.
Figure 7. CD137+ Tregs play a vital role in the suppression of T1D development in vivo.
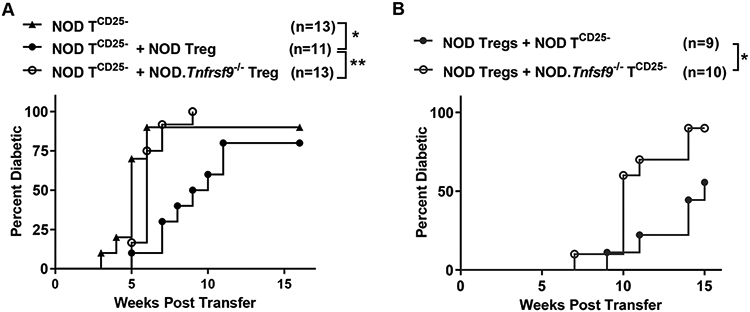
(A) The T1D suppressive function of CD137-deficient Tregs is impaired. CD25-depleted splenic T cells isolated from 12-15-week-old NOD females (NOD TCD25−) were transferred alone or at a 10:1 ratio with Tregs sorted from NOD.Foxp3-eGFP (NOD Treg) or NOD.Tnfrsf9−/−.Foxp3-eGFP (NOD.Tnfrsf9−/− Treg) female donors. A total of 3-5x106 cells were injected into 6-week-old NOD.Rag1−/− female mice. Recipients were analyzed weekly for the development of T1D over a course of 16 weeks post-transfer. The results are pooled from 5 experiments. *p <0.05; **p < 0.01 by log-rank test. (B) CD137L expression on conventional T cells is important for Treg-mediated T1D suppression in vivo. NOD Tregs sorted from 7-10-week-old NOD.Foxp3-eGFP female mice were co-transferred at a 10:1 ratio with CD25-depleted splenic T cells (TCD25−) isolated from 6-7-week-old NOD or NOD.Tnfsf9−/− females. A total of 5x106 cells were injected into 5-7-week-old NOD.Rag1−/− female mice. Recipients were analyzed weekly for the development of T1D over a course of 15 weeks post-transfer. The results are pooled from 3 experiments. *p <0.05 by log-rank test.
One mechanism that can explain the reduced T1D suppressive function of CD137-deficient Tregs is that they lack the ability to inhibit effector T cells through soluble CD137. Thus, we performed a different adoptive T cell transfer experiment to ask if CD137L-deficient T cells are more resistant than the wildtype counterparts to Treg-mediated suppression. We isolated CD25-depleted splenic wildtype and CD137L-deficient T cells from 6–7-week-old donors and co-transferred them with wildtype Tregs at a 10:1 ratio into NOD.Rag1−/− recipients. Recipients of CD137L-deficient T cells developed diabetes at a significantly higher rate compared to those infused with wildtype T cells (Figure 7B). At the time of T1D onset or the end of incidence study, we analyzed the levels of total CD4 and CD8 T cells as well as Tregs and found no difference between the recipient groups (data not shown). Collectively, these results support the idea that soluble CD137 produced by Tregs contributes to T1D suppression by interacting with CD137L expressed on pathogenic T cells.
Another non-mutually exclusive question was whether the observed impaired T1D suppressive activity of CD137-deficient Tregs was due to altered stability in NOD.Rag1−/− recipients. To test this, we co-transferred wildtype or CD137-deficient FOXP3+ Tregs (GFP+ CD4 T cells sorted from NOD.Foxp3-eGFP and NOD.Tnfrsf9−/−.Foxp3-eGFP, respectively) with non-Treg T cells (GFP− T cells sorted from NOD.Cd45.2.Foxp3-eGFP) in a ratio of 1:10 (Figure 8A). With this strategy, we were able to discriminate between the injected Tregs (GFP+ CD45.1+) and peripherally induced Tregs (GFP+ CD45.2+) originally from sorted GFP− non-Treg T cells. Five weeks post-transfer, we analyzed the spleens, PLNs, and islets and found that the percentages and numbers of total Tregs (GFP+ CD45.1+ and GFP+ CD45.2+) were similar in the two recipient groups (data not shown). The frequencies of the induced CD45.2+ Tregs were also comparable between the two recipient groups (data not shown). Importantly, the majority of transferred CD45.1+ wildtype and CD137-deficient Tregs remained FOXP3+ (GFP+) and the proportions of those that have lost FOXP3 expression (GFP−) were similar (Figures 8B-8D). Taken together, these results indicate that CD137 enhances FOXP3+ Treg-mediated T1D suppression but it is dispensable for their stability in NOD.Rag1−/− recipients.
Figure 8. CD137 deficiency does not alter the stability of FOXP3+ Tregs.
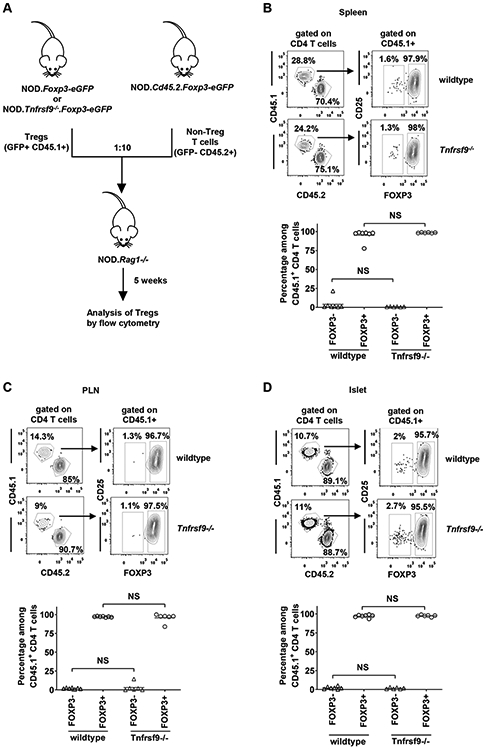
(A) Diagram shows the design for the adoptive transfer experiment. Splenic non-Tregs (GFP− CD45.2+) sorted from 10-week-old NOD.Cd45.2.Foxp3-eGFP females were transferred at a 10:1 ratio with Tregs (GFP+ CD45.1+) sorted from 7-10-week-old NOD.Foxp3-eGFP or NOD.Tnfrsf9−/−.Foxp3-eGFP female donors. A total of 5x106 cells were injected into 6-week-old NOD.Rag1−/− female mice. (B-D) Cells were isolated from the spleens (B), PLNs (C), and islets (D) of the NOD.Rag1−/− recipients 5 weeks post-transfer and stained with antibodies specific for CD45.1, CD45.2, CD3, CD4, and CD25. The percentage of GFP+ (FOXP3+) Tregs among total CD45.1+ CD4 T cells was analyzed by flow cytometry. Representative flow cytometry profiles are shown in the upper panels and summarized data from two independent transfer experiments are presented in the lower panels. Each symbol represents a mouse. The horizontal bar depicts the mean. NS: Not significant by Mann-Whitney U test.
Discussion
In the current study, we generated and characterized NOD.Tnfsf9−/− mice to determine the role of CD137L in T1D. We observed a similar partial T1D suppression phenotype in NOD.Tnfsf9−/− and the previously described NOD.Tnfrsf9−/− (CD137-deficient) mice (32). T1D resistance in both strains is associated with significantly reduced numbers of β-cell autoreactive CD8 T cells. These results confirm the importance of CD137-CD137L interaction in T1D development. We further show that CD137L-deficient hematopoietic cells confer T1D resistance and that CD137L expressed by non-T and non-B cells, presumably myeloid APCs, is important for the development of T1D. In addition, we show here that the development and homeostasis of CD137+ Tregs occur independent of CD137L. However, CD137-CD137L interaction is most likely important for the in vivo T1D suppressive function of Tregs.
CD137L is expressed in radiation resistant BM stromal cells where its function is to support the survival of memory CD8 T cells (45). We and others have shown that antigen-experienced β-cell autoreactive CD8 T cells are proportionally enriched in BM (28, 46). This raises the possibility that CD137-CD137L interaction can promote T1D development by maintaining the memory β-cell autoreactive CD8 T cell pool in BM. We addressed this possibility by using the BM chimera approach to determine if expression of CD137L in hematopoietic, non-hematopoietic, or both cell populations is important for T1D development. While lack of CD137L in hematopoietic cells inhibited T1D development, its absence in radiation resistant cells did not affect diabetes progression. Thus, maintaining the memory CD8 T cell pool in BM does not appear to be critical for T1D development. We subsequently demonstrated that CD137L expression on non-T and non-B cells sustains the pathogenic activity of diabetogenic T cells. Our results support the model that interaction between CD137 on β-cell autoreactive CD8 T cells and CD137L on myeloid APCs is important for T1D development. The frequency of activated β-cell autoreactive CD8 T cells is decreased in NOD.Tnfsf9−/− mice in part due to reduced survival. Our results are in line with the observation in non-autoimmune systems where CD137-CD137L interaction promotes survival signaling in T cells (40-43). For instance, CD137-CD137L signaling is required for the recall response of influenza infection where its ability of maintaining CD8 T cell survival supports this role (40). Consistently, CD137L deficiency decreased the accumulation of CD8 T cells in the lung during prolonged infection of the influenza virus and resulted in impaired viral clearance, compromised lung function, and reduced survival of mice (47).
CD137 is expressed on a subset of Tregs (24, 27). CD137 signaling elicited by agonistic anti-CD137 stimulation has been shown to expand Tregs (23, 24, 48, 49). Thus, we hypothesized that CD137-CD137L interaction is important for the accumulation of CD137+ Tregs. Unexpectedly, the development and homeostasis of CD137+ Tregs are independent of CD137L. Tnfrsf9 is a direct target of FOXP3 (50). However, this alone cannot fully explain why CD137 is only expressed on a subset of Tregs. In the islets of NOD mice, CD5 expression correlates with the strength of TCR signaling and CD5high Tregs express a higher level of Tnfrsf9 than that in the CD5low population (51). Therefore, CD137+ Tregs may receive stronger TCR signaling than that in CD137− Tregs. Nevertheless, our adoptive transfer study clearly demonstrates that CD137 expression on Tregs has a functional consequence and is not only a reflection of the TCR signaling strength received by these cells.
We previously showed that CD137-deficient CD4 T cells were more diabetogenic than their wildtype counterparts (28). We speculated that this is due to the absence of CD137+ Tregs. This hypothesis was based on our earlier observation that CD137+ Tregs had increased in vitro suppressive activity compared to the CD137− subset in part due to the ability to secrete soluble CD137 from the former population (26). Here, we used an adoptive transfer approach to directly compare the T1D suppressive function of wildtype and CD137-deficient Tregs. The results conclusively demonstrate that wildtype Tregs are superior to CD137-deficient Tregs in their ability to suppress diabetogenic T cells in vivo. We ruled out the possibility that CD137-CD137L interaction is important for the stability of Tregs. There are two other non-mutually exclusive mechanisms by which CD137-CD137L interaction could contribute to the suppressive function of Tregs. As discussed above, soluble CD137 produced by CD137+ Tregs can induce “reverse” signaling in CD137L-expressing T cells to suppress their activation (8, 9, 52). As CD137L-deficient conventional T cells were more resistant to Treg-mediated suppression, our results support the idea that impaired T1D suppressive activity of CD137-deficient Tregs is at least in part due to their inability to produce soluble CD137. Another possible explanation is that CD137 on the cell surface of Tregs enhances their suppressive function when engaged by CD137L expressed on APCs or by extracellular matrix proteins such as laminin and fibronectin (29). Additional experiments are required to further distinguish the contribution of these potential mechanisms.
Our present study provides important new insights into the roles of CD137-CD137L interaction in T1D. The complexity of the bi-directional signaling mediated by the two interacting partners necessitates dissecting the mechanisms mediated by each. Detailed understanding of the mechanisms mediated by CD137 and its ligand may open new avenues to identify novel therapeutic strategies to manipulate CD137-CD137L interaction for T1D prevention and treatment.
Supplementary Material
Key points.
CD137L deficiency in myeloid antigen-presenting cells suppresses type 1 diabetes
CD137L deficiency in pathogenic T cells enhances their diabetogenic activity
CD137 is important for the suppressive function of FOXP3+ regulatory CD4 T cells
Acknowledgments:
We thank Kevin Mueller and Stephanie Harleston for maintaining NOD and related mouse strains, and the staff at the Histology Core and Galina Petrova at the Flow Cytometry Core of the Children’s Hospital of Wisconsin Research Institute for excellent technical assistance. We thank The Jackson Laboratory Genetic Engineering Technologies group for technical support on this project. We thank the NIH Tetramer Core Facility for providing MHC class I tetramers.
This work was supported by NIH grants DK107541 (to Y.-G.C. and W.M.R.), DK097605 (to A.M.G. and Y.-G.C.), AI125879 (to Y.-G.C.), DK46266 (to D.V.S.), DK 95735 (to D.V.S.), and OD020351 (to D.V.S.), as well as a JDRF grant 2018–568 (to D.V.S.), an American Diabetes Association grant (to Y.-G.C. and W.M.R), the George and Ruth Leef Family and ITU AbsorbTech, and the Children’s Hospital of Wisconsin Foundation.
References
- 1.Wortzman ME, Clouthier DL, McPherson AJ, Lin GH, and Watts TH. 2013. The contextual role of TNFR family members in CD8(+) T-cell control of viral infections. Immunological reviews 255: 125–148. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez-Paulete AR, Labiano S, Rodriguez-Ruiz ME, Azpilikueta A, Etxeberria I, Bolanos E, Lang V, Rodriguez M, Aznar MA, Jure-Kunkel M, and Melero I. 2016. Deciphering CD137 (4–1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. European journal of immunology 46: 513–522. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, and Vignali DA. 2016. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity 44: 1034–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodwin RG, Din WS, Davis-Smith T, Anderson DM, Gimpel SD, Sato TA, Maliszewski CR, Brannan CI, Copeland NG, Jenkins NA, and et al. 1993. Molecular cloning of a ligand for the inducible T cell gene 4–1BB: a member of an emerging family of cytokines with homology to tumor necrosis factor. European journal of immunology 23: 2631–2641. [DOI] [PubMed] [Google Scholar]
- 5.Dharmadhikari B, Wu M, Abdullah NS, Rajendran S, Ishak ND, Nickles E, Harfuddin Z, and Schwarz H. 2016. CD137 and CD137L signals are main drivers of type 1, cell-mediated immune responses. Oncoimmunology 5: e1113367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yonezawa A, Dutt S, Chester C, Kim J, and Kohrt HE. 2015. Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin Cancer Res 21: 3113–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim DK, Lee SC, and Lee HW. 2009. CD137 ligand-mediated reverse signals increase cell viability and cytokine expression in murine myeloid cells: involvement of mTOR/p70S6 kinase and Akt. Eur J Immunol 39: 2617–2628. [DOI] [PubMed] [Google Scholar]
- 8.Kachapati K, Bednar KJ, Adams DE, Wu Y, Mittler RS, Jordan MB, Hinerman JM, Herr AB, and Ridgway WM. 2013. Recombinant soluble CD137 prevents type one diabetes in nonobese diabetic mice. J Autoimmun 47: 94–103. [DOI] [PubMed] [Google Scholar]
- 9.Eun SY, Lee SW, Xu Y, and Croft M. 2015. 4–1BB Ligand Signaling to T Cells Limits T Cell Activation. J Immunol 194: 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang YJ, Kim SO, Shimada S, Otsuka M, Seit-Nebi A, Kwon BS, Watts TH, and Han J. 2007. Cell surface 4–1BBL mediates sequential signaling pathways ‘downstream’ of TLR and is required for sustained TNF production in macrophages. Nature immunology 8: 601–609. [DOI] [PubMed] [Google Scholar]
- 11.Driver JP, Serreze DV, and Chen YG. 2011. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol 33: 67–87. [DOI] [PubMed] [Google Scholar]
- 12.Atkinson MA, Eisenbarth GS, and Michels AW. 2014. Type 1 diabetes. Lancet 383: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dabelea D, Mayer-Davis EJ, Saydah S, Imperatore G, Linder B, Divers J, Bell R, Badaru A, Talton JW, Crume T, Liese AD, Merchant AT, Lawrence JM, Reynolds K, Dolan L, Liu LL, and Hamman RF. 2014. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA 311: 1778–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patterson CC, Harjutsalo V, Rosenbauer J, Neu A, Cinek O, Skrivarhaug T, Rami-Merhar B, Soltesz G, Svensson J, Parslow RC, Castell C, Schoenle EJ, Bingley PJ, Dahlquist G, Jarosz-Chobot PK, Marciulionyte D, Roche EF, Rothe U, Bratina N, Ionescu-Tirgoviste C, Weets I, Kocova M, Cherubini V, Rojnic Putarek N, deBeaufort CE, Samardzic M, and Green A. 2019. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 1989–2013: a multicentre prospective registration study. Diabetologia 62: 408–417. [DOI] [PubMed] [Google Scholar]
- 15.Schneider DA, and von Herrath MG. 2014. Potential viral pathogenic mechanism in human type 1 diabetes. Diabetologia 57: 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pociot F, and Lernmark A. 2016. Genetic risk factors for type 1 diabetes. Lancet 387: 2331–2339. [DOI] [PubMed] [Google Scholar]
- 17.Paun A, Yau C, and Danska JS. 2017. The Influence of the Microbiome on Type 1 Diabetes. Journal of immunology 198: 590–595. [DOI] [PubMed] [Google Scholar]
- 18.Yamanouchi J, Puertas MC, Verdaguer J, Lyons PA, Rainbow DB, Chamberlain G, Hunter KM, Peterson LB, Wicker LS, and Santamaria P. 2010. Idd9.1 locus controls the suppressive activity of FoxP3+CD4+CD25+ regulatory T-cells. Diabetes 59: 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyons PA, Hancock WW, Denny P, Lord CJ, Hill NJ, Armitage N, Siegmund T, Todd JA, Phillips MS, Hess JF, Chen SL, Fischer PA, Peterson LB, and Wicker LS. 2000. The NOD Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity 13: 107–115. [DOI] [PubMed] [Google Scholar]
- 20.Gavin MA, Clarke SR, Negrou E, Gallegos A, and Rudensky A. 2002. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nature immunology 3: 33–41. [DOI] [PubMed] [Google Scholar]
- 21.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, and Byrne MC. 2002. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity 16: 311–323. [DOI] [PubMed] [Google Scholar]
- 22.Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS, and Kwon BS. 2004. 4–1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. Journal of leukocyte biology 75: 785–791. [DOI] [PubMed] [Google Scholar]
- 23.Zheng G, Wang B, and Chen A. 2004. The 4–1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. Journal of immunology 173: 2428–2434. [DOI] [PubMed] [Google Scholar]
- 24.Irie J, Wu Y, Kachapati K, Mittler RS, and Ridgway WM. 2007. Modulating protective and pathogenic CD4+ subsets via CD137 in type 1 diabetes. Diabetes 56: 186–196. [DOI] [PubMed] [Google Scholar]
- 25.Cannons JL, Chamberlain G, Howson J, Smink LJ, Todd JA, Peterson LB, Wicker LS, and Watts TH. 2005. Genetic and functional association of the immune signaling molecule 4–1BB (CD137/TNFRSF9) with type 1 diabetes. J Autoimmun 25: 13–20. [DOI] [PubMed] [Google Scholar]
- 26.Kachapati K, Adams DE, Wu Y, Steward CA, Rainbow DB, Wicker LS, Mittler RS, and Ridgway WM. 2012. The B10 Idd9.3 Locus Mediates Accumulation of Functionally Superior CD137+ Regulatory T Cells in the Nonobese Diabetic Type 1 Diabetes Model. J Immunol 189: 5001–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forsberg MH, Foda B, Serreze DV, and Chen YG. 2019. Combined congenic mapping and nuclease-based gene targeting for studying allele-specific effects of Tnfrsf9 within the Idd9.3 autoimmune diabetes locus. Sci Rep 9: 4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forsberg MH, Ciecko AE, Bednar KJ, Itoh A, Kachapati K, Ridgway WM, and Chen YG. 2017. CD137 Plays Both Pathogenic and Protective Roles in Type 1 Diabetes Development in NOD Mice. Journal of immunology 198: 3857–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chalupny NJ, Peach R, Hollenbaugh D, Ledbetter JA, Farr AG, and Aruffo A. 1992. T-cell activation molecule 4–1BB binds to extracellular matrix proteins. Proceedings of the National Academy of Sciences of the United States of America 89: 10360–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madireddi S, Eun SY, Lee SW, Nemcovicova I, Mehta AK, Zajonc DM, Nishi N, Niki T, Hirashima M, and Croft M. 2014. Galectin-9 controls the therapeutic activity of 4–1BB-targeting antibodies. The Journal of experimental medicine 211: 1433–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Presa M, Racine JJ, Dwyer JR, Lamont DJ, Ratiu JJ, Sarsani VK, Chen YG, Geurts A, Schmitz I, Stearns T, Allocco J, Chapman HD, and Serreze DV. 2018. A Hypermorphic Nfkbid Allele Contributes to Impaired Thymic Deletion of Autoreactive Diabetogenic CD8(+) T Cells in NOD Mice. Journal of immunology 201: 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen YG, Forsberg MH, Khaja S, Ciecko AE, Hessner MJ, and Geurts AM. 2014. Gene targeting in NOD mouse embryos using zinc-finger nucleases. Diabetes 63: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Presa M, Chen YG, Grier AE, Leiter EH, Brehm MA, Greiner DL, Shultz LD, and Serreze DV. 2015. The Presence and Preferential Activation of Regulatory T Cells Diminish Adoptive Transfer of Autoimmune Diabetes by Polyclonal Nonobese Diabetic (NOD) T Cell Effectors into NSG versus NOD-scid Mice. Journal of immunology 195: 3011–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han B, Serra P, Yamanouchi J, Amrani A, Elliott JF, Dickie P, Dilorenzo TP, and Santamaria P. 2005. Developmental control of CD8 T cell-avidity maturation in autoimmune diabetes. J Clin Invest 115: 1879–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serreze DV, Wasserfall C, Ottendorfer EW, Stalvey M, Pierce MA, Gauntt C, O’Donnell B, Flanagan JB, Campbell-Thompson M, Ellis TM, and Atkinson MA. 2005. Diabetes acceleration or prevention by a coxsackievirus B4 infection: critical requirements for both interleukin-4 and gamma interferon. Journal of virology 79: 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R. 2014. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9: 171–181. [DOI] [PubMed] [Google Scholar]
- 37.Wang C, Lin GH, McPherson AJ, and Watts TH. 2009. Immune regulation by 4–1BB and 4–1BBL: complexities and challenges. Immunol Rev 229: 192–215. [DOI] [PubMed] [Google Scholar]
- 38.Trudeau JD, Kelly-Smith C, Verchere CB, Elliott JF, Dutz JP, Finegood DT, Santamaria P, and Tan R. 2003. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 111: 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chee J, Ko HJ, Skowera A, Jhala G, Catterall T, Graham KL, Sutherland RM, Thomas HE, Lew AM, Peakman M, Kay TW, and Krishnamurthy B. 2014. Effector-memory T cells develop in islets and report islet pathology in type 1 diabetes. J Immunol 192: 572–580. [DOI] [PubMed] [Google Scholar]
- 40.Bertram EM, Lau P, and Watts TH. 2002. Temporal segregation of 4–1BB versus CD28-mediated costimulation: 4–1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol 168: 3777–3785. [DOI] [PubMed] [Google Scholar]
- 41.Cannons JL, Lau P, Ghumman B, DeBenedette MA, Yagita H, Okumura K, and Watts TH. 2001. 4–1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol 167: 1313–1324. [DOI] [PubMed] [Google Scholar]
- 42.Cooper D, Bansal-Pakala P, and Croft M. 2002. 4–1BB (CD137) controls the clonal expansion and survival of CD8 T cells in vivo but does not contribute to the development of cytotoxicity. Eur J Immunol 32: 521–529. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi C, Mittler RS, and Vella AT. 1999. Cutting edge: 4–1BB is a bona fide CD8 T cell survival signal. J Immunol 162: 5037–5040. [PubMed] [Google Scholar]
- 44.Buckner JH 2010. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nature reviews. Immunology 10: 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin GH, Edele F, Mbanwi AN, Wortzman ME, Snell LM, Vidric M, Roth K, Hauser AE, and Watts TH. 2012. Contribution of 4–1BBL on radioresistant cells in providing survival signals through 4–1BB expressed on CD8(+) memory T cells in the bone marrow. Eur J Immunol 42: 2861–2874. [DOI] [PubMed] [Google Scholar]
- 46.Li R, Perez N, Karumuthil-Melethil S, and Vasu C. 2007. Bone marrow is a preferential homing site for autoreactive T-cells in type 1 diabetes. Diabetes 56: 2251–2259. [DOI] [PubMed] [Google Scholar]
- 47.Lin GH, Sedgmen BJ, Moraes TJ, Snell LM, Topham DJ, and Watts TH. 2009. Endogenous 4–1BB ligand plays a critical role in protection from influenza-induced disease. J Immunol 182: 934–947. [DOI] [PubMed] [Google Scholar]
- 48.Elpek KG, Yolcu ES, Franke DD, Lacelle C, Schabowsky RH, and Shirwan H. 2007. Ex vivo expansion of CD4+CD25+FoxP3+ T regulatory cells based on synergy between IL-2 and 4–1BB signaling. Journal of immunology 179: 7295–7304. [DOI] [PubMed] [Google Scholar]
- 49.Zhang P, Gao F, Wang Q, Wang X, Zhu F, Ma C, Sun W, and Zhang L. 2007. Agonistic anti-4–1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand J Immunol 66: 435–440. [DOI] [PubMed] [Google Scholar]
- 50.Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, Levine SS, Fraenkel E, von Boehmer H, and Young RA. 2007. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 445: 931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sprouse ML, Scavuzzo MA, Blum S, Shevchenko I, Lee T, Makedonas G, Borowiak M, Bettini ML, and Bettini M. 2018. High self-reactivity drives T-bet and potentiates Treg function in tissue-specific autoimmunity. JCI Insight 3: e97322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Itoh A, Ortiz L, Kachapati K, Wu Y, Adams D, Bednar K, Mukherjee S, Chougnet C, Mittler RS, Chen YG, Dolan L, and Ridgway WM. 2019. Soluble CD137 Ameliorates Acute Type 1 Diabetes by Inducing T Cell Anergy. Front Immunol 10: 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.