

Abstract
A number of small, self-cleaving ribozyme classes have been identified including the hammerhead, hairpin, hepatitis delta virus (HDV), Varkud satellite (VS), glmS, twister, hatchet, pistol, and twister sister ribozymes. Within the active sites of these ribozymes, myriad functional groups contribute to catalysis. There has been extensive structure-function analysis of individual ribozymes, but the extent to which catalytic devices are shared across different ribozyme classes is unclear. As such, emergent catalytic principles for ribozymes may await discovery. Identification of conserved catalytic devices can deepen our understanding of RNA catalysis specifically and of enzymic catalysis generally. To probe similarities and differences amongst ribozyme classes, active sites from more than 80 high-resolution crystal structures of self-cleaving ribozymes were compared computationally. We identify commonalities amongst ribozyme classes pertaining to four classic catalytic devices: deprotonation of the 2′OH nucleophile (γ), neutralization of the non-bridging oxygens of the scissile phosphate (β), neutralization of the O5′ leaving group (δ), and in-line nucleophilic attack (α). In addition, we uncover conservation of two catalytic devices, each of which centers on the activation of the 2′OH nucleophile by a guanine: one to acidify the 2′OH by hydrogen bond donation to it (γ′) and one to acidify the 2′OH by releasing it from non-productive interactions by competitive hydrogen bonding (γ′′). Our findings reveal that the amidine functionalities of G, A, and C are especially important for these strategies, and help explain absence of U at ribozyme active sites. The identified γ′ and γ′′ catalytic strategies help unify the catalytic strategies shared amongst catalytic RNAs and may be important for large ribozymes, as well as protein enzymes that act on nucleic acids.
Keywords: catalytic strategy, catalytic device, general acid-base catalysis, computational, nucleophile activation
Graphical Abstract
INTRODUCTION
The discovery of RNA enzymes occurred thirty-five years ago when the Cech lab uncovered self-splicing of the Tetrahymena Group I intron and the Altman lab revealed tRNA-processing activity of the RNA subunit of ribonuclease P.1–2 To date, catalytic activity of many different RNAs has been characterized, including Group I and II introns, the ribosome, the spliceosome, and nine classes of small self-cleaving ribozymes: the hammerhead, hairpin, hepatitis delta virus (HDV), Varkud satellite (VS), glmS, twister, hatchet, pistol, and twister sister ribozymes.3–4 Although major advances in understanding the structure and mechanism of individual ribozymes have occurred, the field lacks a unified view of RNA catalysis. It is unclear, for example, if existing catalytic strategies are conserved and if new ones await discovery. These limitations cloud our understanding of RNA catalysis. A full understanding of such strategies could deepen insights into RNA catalysis in general and help elucidate related catalytic principles in proteins. In an effort to address this, we focus here on comparing the small, self-cleaving ribozymes, which use a 2′OH to attack the adjacent phosphorus and leave 2′,3′-cyclic phosphate and 5′-hydroxyl termini (Figure 1). These ribozymes, which range in size from ~50 to 150 nucleotides, rapidly self-cleave without the need for proteins.
Figure 1.
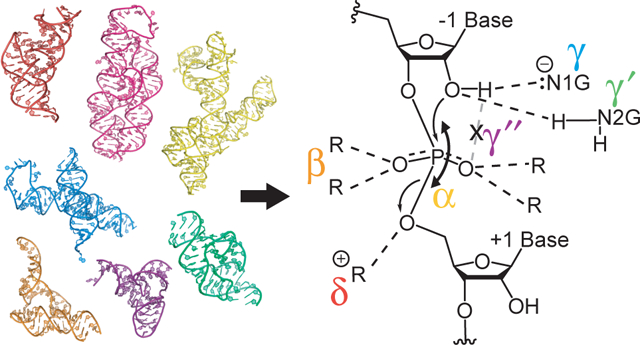
Intramolecular cleavage of the phosphodiester bond by nucleophilic attack of the O2′. The three states of phosphodiester bond cleavage are shown with the four previously reported catalytic strategies9 highlighted in the central structure; β contacts can be either a hydrogen bond donor or a metal ion, which are shown separately on one of the two NBOs for simplicity.
To identify commonalities and differences amongst the diverse set of small ribozymes, we designed a computational pipeline that procures active site information from available structural data and organizes it for subsequent analysis. This was necessary because the structures are too diverse for direct comparison by RMSD. This method was applied to 88 crystal or computationally minimized structures, the majority of which were crystal structures, beginning with the following four ribozyme classes: the hammerhead, hairpin, glmS, and twister ribozymes (see Table S1). Each ribozyme has nuances and features that separate it. For instance, the hairpin ribozyme plays an integral role in cellular processing of viral satellite RNA.5 The glmS ribozyme, which is also a riboswitch, occurs in many Gram positive bacteria where it uses the small molecule glucosamine-6-phosphate (GlcN6P) to downregulate synthesis of the glmS ribozyme-associated gene, which is involved in bacterial cell wall biosynthesis.6 The hammerhead and twister ribozymes are fairly widespread and are found in the domains of bacteria and eukarya.7–8
Protein and RNA enzymes catalyze some of the same reactions, including phosphodiester bond cleavage. It is thus not surprising to find that RNA enzyme catalysis shares similarities with protein enzyme catalysis. For instance, ribozymes and protein enzymes both use general acid-base, metal-ion, and electrostatic catalysis. Remarkably, whereas proteins can employ 20 diverse amino acids in catalysis, ribozymes use only four similar nucleobases to carry out similar catalytic strategies. Knowing how catalytic RNA nucleobases interact with other nucleobases and metal ions is key to understanding how RNA can catalyze some of the same reactions as proteins.
The Breaker lab articulated four classic strategies for how ribozymes perform chemistry: (1) Deprotonation of the O2′ nucleophile, termed ‘γ’, (2) Neutralization of the non-bridging oxygen (NBO) atoms of the scissile phosphate, termed ‘β’, (3) Protonation of the incipient oxyanion on the O5′, termed ‘δ’, and (4) In-line nucleophilic attack, termed ‘α’ (Figure 1).9–10 The γ strategy increases the nucleophilicity of the O2′ by increasing the energy of its lone pairs. The β strategy provides positive charge to developing negative charge on the NBOs in the transition state, via either concerted or stepwise reaction pathways.11 In the stepwise version of the reaction, the reaction proceeds through a pentacoordinate phosphorane intermediate that then decomposes to cleave the scissile phosphorus-O5′ bond. The δ strategy lowers the energy of the product alkoxide, which if not quenched, could attack the scissile phosphate and return the RNA back to its original, uncleaved state. Lastly, the α strategy involves optimization of the angle between the O2′, the phosphorus, and the O5′ towards 180°.
The aim of our study is to identify the functional groups and metal ions that carry out these catalytic roles within small self-cleaving ribozymes, including those that are not readily discerned by visual inspection, and to assess whether these functionalities and any catalytic roles assigned to them are conserved across small ribozyme classes. We are also especially interested in identifying any new catalytic strategies that are conserved amongst ribozymes. Furthermore, in an effort to see if interactions are unique, we seek to ascertain the extent to which these strategies are found at the scissile phosphate versus other phosphate linkages.
METHODS
Preparation of Published Crystal Structures for Data Analysis
To identify active site contacts conserved amongst diverse ribozyme classes we designed a contact-identifying pipeline. Pre-cleavage crystal structures for each ribozyme class were downloaded from the Protein Data Bank (Table S1). These structures have an average resolution of 2.7 ± 0.4 Å, and all had a resolution of 3.35 Å or better except one of 3.88 Å. All constructs used for crystallization were inhibited in some way to prevent self-cleavage during the crystallization process. This was usually achieved through modification of the nucleophilic 2′OH group to an inactive functional group, such as a deoxy, methoxy, amino, 2′,5′-linkage, or vanadate. We display contacts out to 5 Å to account for any deviation in crystal structures.
Several crystal structures contained a few nucleotides that adopted two different conformations, noted ‘A’ or ‘B’ in PyMOL. Conformation A exhibits an occupancy factor of 0.5 or greater in all such cases. When appropriate, a PyMOL plugin removed conformation B to preclude collecting two sets of data for a given contact atom. When analyzing crystal structures containing multiple copies of a ribozyme, data from only the first copy were acquired with the following two exceptions: data were collected from the second copy of 2NZ4 and 4QJD because the G1 on the first copy of 2NZ4 lacked an N3 and the first copy of 4QJD lacked two residues at the 5′ end.
γ, β, and δ Scissile Phosphate Plugin: Identification of Contact Atoms to Atoms of Interest at Scissile Phosphates (γ, β, and δ Strategies)
A custom PyMOL plugin was used to find all contact atoms (CAs) for each crystal structure within 5 Å of the atom of interest (AOI), which was a catalytic atom from either the −1 or +1 nucleotide (i.e. O2’, O5’, or NBO). All CAs of the AOI were output to a text file along with relevant information concerning these CAs, including their distance to the AOI, hydrogen bonding angle (see Supporting Methods and Figure S1), residue name and number (e.g. G33, U58, C49), atom name (e.g. O2’, N1, N4), and b-factor. The literature often contains different and conflicting numberings of nucleotides in ribozymes. We chose the most conventional ones, which came from the following references: glmS12, hammerhead,13 twister14, hairpin15, and pistol16 ribozymes. A Python script was used to sort and plot the data. Heteroatom candidates were filtered based on the following criteria: (1) carbon and phosphorus candidates were removed because they typically don’t participate in strong intermolecular interactions, and hydrogen was not counted because hydrogen bonding was inferred from heteroatom positioning; (2) candidates from the −1 and +1 nucleobases with a hydrogen bonding angle less than 140° were removed; and (3) candidates that were found in less than a quarter of the structures, where the number corresponding to a quarter of the structures was determined by integer division, were removed. Whenever present within 5 Å of the AOI, divalent and trivalent metal ions, including Mg2+, Mn2+, Ca2+, Co3+, and Tb3+, were grouped together as a single metal ion contact. After all contacts were filtered, they were ranked based on distance to the AOI.
Next, the top five CAs to an AOI were depicted on a distance plot for each catalytic strategy within a given ribozyme class along with their average distances and standard deviation when appropriate. As mentioned above, divalent or trivalent metal ions within 5 Å of the AOI were considered together and are labeled “M2+” on distance plots. Green bars indicate substitutions to certain nucleobases or functional groups. Purple bars indicate either an intermediate-mimic structure containing a vanadate or 2′,5′-linkage or a computationally modelled intermediate structure. Yellow bars indicate computationally modeled structures that are based on crystal structures. Orange bars indicate crystal structures that are deemed not catalytically relevant as discerned from the literature. One of these orange structures is a twister construct that has a disordered active site17 and the others are hammerhead structures that are of the minimal ribozyme (not the full-length)18. Data represented by black, purple, and yellow bars are included in the average distances. Data represented by green and orange bars, on the other hand, are excluded from all calculated averages but are still shown on plots for visualization. Averages are shown as horizontal red lines and standard deviation as vertical gray lines for each CA. The hydrogen bonding angle between the CA, the CA’s proton, and the AOI were determined for all nucleobase primary and secondary amines. Values within the plots above each CA represent the average angle for each CA (Supporting Methods and Figure S1). Suboptimal angles (< 140°) are colored red. Blue shading from 2.5 to 3.5 Å on the distance plots indicates ideal hydrogen bonding distances. For the γ strategy (deprotonation of the O2′), only crystal structures or simulated structures that have a heteroatom at the nucleophilic 2′OH position were included, except for the case of the pistol ribozyme. For pistol, only deoxy structures exist so a 2′O was modelled in so that information on the γ strategy could be obtained. Contacts within 4 Å of the AOI are inferred from the top five CAs and are depicted in ChemDraw structures to the right of each plot (γ and δ strategies) or set of plots (β strategy), displaying possible contacts within each ribozyme class. Putative contacts within 4 Å that are chemically unlikely are not shown in the ChemDraw structures and these instances are noted in the figure legends.
α Plugin: Identification of O2′-P-O5′ Angle at Scissile Phosphate (α Strategy)
A second custom PyMOL plugin was used to find the angle between the 2′ heteroatom, P, and O5′ of the scissile phosphate for all suitable crystal structures. Angles were output to a text file, the average was calculated for each ribozyme class, and the resulting values were plotted on an angle plot. In-line attack angle data for crystal structures where a deoxy substitution was made at the 2′ position were not included in the α analysis with the following exception: for the pistol ribozyme, only deoxy structures existed, so a 2′O was modelled in so that information on the α strategy could be obtained. Data bars in the angle plots were color coded as described above for the CA distance plots. In addition to non-catalytically relevant structures (orange bars), crystal structure intermediate-mimic and computationally modelled intermediate structures (purple bars) were not included in the average angle calculation since their angle is based on the molecular geometry about the phosphate (or vanadate), but these structures are still shown on the plots. Wild-type and nucleobase variant structures are both included in the averages and portrayed as black bars. Average angles are shown as horizontal red lines and standard deviation as vertical gray lines. Blue shading from 140° to 180° on the angle plot indicates an optimal in-line O2′-P-O5′ angle.
All-Phosphates Plugins: Comparison of Catalytic Strategies at Scissile versus all Non-Scissile Phosphates
Another custom PyMOL plugin was used to identify CAs within 5 Å from the O2′, NBOs, and O5′ AOIs at all phosphates within each ribozyme crystal structure. In addition, the O2′-P-O5′ angle was calculated at all phosphates using the a plugin from above. The angles and distances were output to text files, which were read by a Python script that organized the data and created plots for the α, γ, β, and δ strategies showing the distribution of angles and numbers of contacts at the scissile and non-scissile phosphates for each ribozyme class. Hydrogen, carbon, phosphorus, all oxygens except for O2′s, and nucleobase oxygens, and all atoms of the −1 and +1 nucleotides were omitted for the γ, β and δ strategy bar plots. Though structures with a deoxy substitution at the scissile phosphate were excluded for scissile phosphate γ and α analysis (with the exception of the pistol ribozyme), these structures were included in the non-scissile phosphate γ and α analyses since their non-scissile phosphates have adjacent 2′OHs.
RESULTS
Overview of Comparing Small Ribozymes
A primary goal of our study was to compare catalytic strategies amongst different classes of small naturally occurring ribozymes, focusing on the hammerhead, hairpin, glmS, and twister ribozymes. These ribozymes were chosen for study herein because in most cases many high-resolution structures are available for each, along with various extents of mechanistic investigation and biochemical validation. The hatchet ribozyme was omitted because no crystal structure was available at the time of writing, and the HDV19–20 and twister sister21–23 ribozymes were only included (see discussion) because they do not use the same mechanism to deprotonate the O2′ nucleophile. We analyzed the VS ribozyme but did not include it in our analysis because the recent crystal structure with the docked substrate has interactions between catalytic atoms and the general acid and base beyond the criteria of our pipeline. Comparison amongst these four ribozymes of interest is not straightforward, however, because their structures are very different from each other (Figure 2).12–14,24 The glmS ribozyme is fairly large (~150 nt), whereas the other self-cleaving ribozymes are relatively small (~50 nt). In addition, the glmS and hammerhead ribozymes are characterized by a flat “Y-shaped” three-dimensional architecture, whereas the others RNAs are more globular. All five of these ribozymes possess one or more pseudoknots (Figure 2). While this helps compact them to form the tightly packed active sites necessary for catalysis, the overall topology of these ribozymes, including the location and extent of pseudoknot formation, varies widely between ribozyme classes. Due to this diversity of secondary and tertiary structures—where insertion regions, flanking bases, and local structures differ—it is not possible to directly compare the different ribozyme classes using traditional structural comparison techniques such as root mean square deviation (RMSD). We thus sought to develop an alternative method to compare across the active sites of these diverse ribozyme classes.
Figure 2.
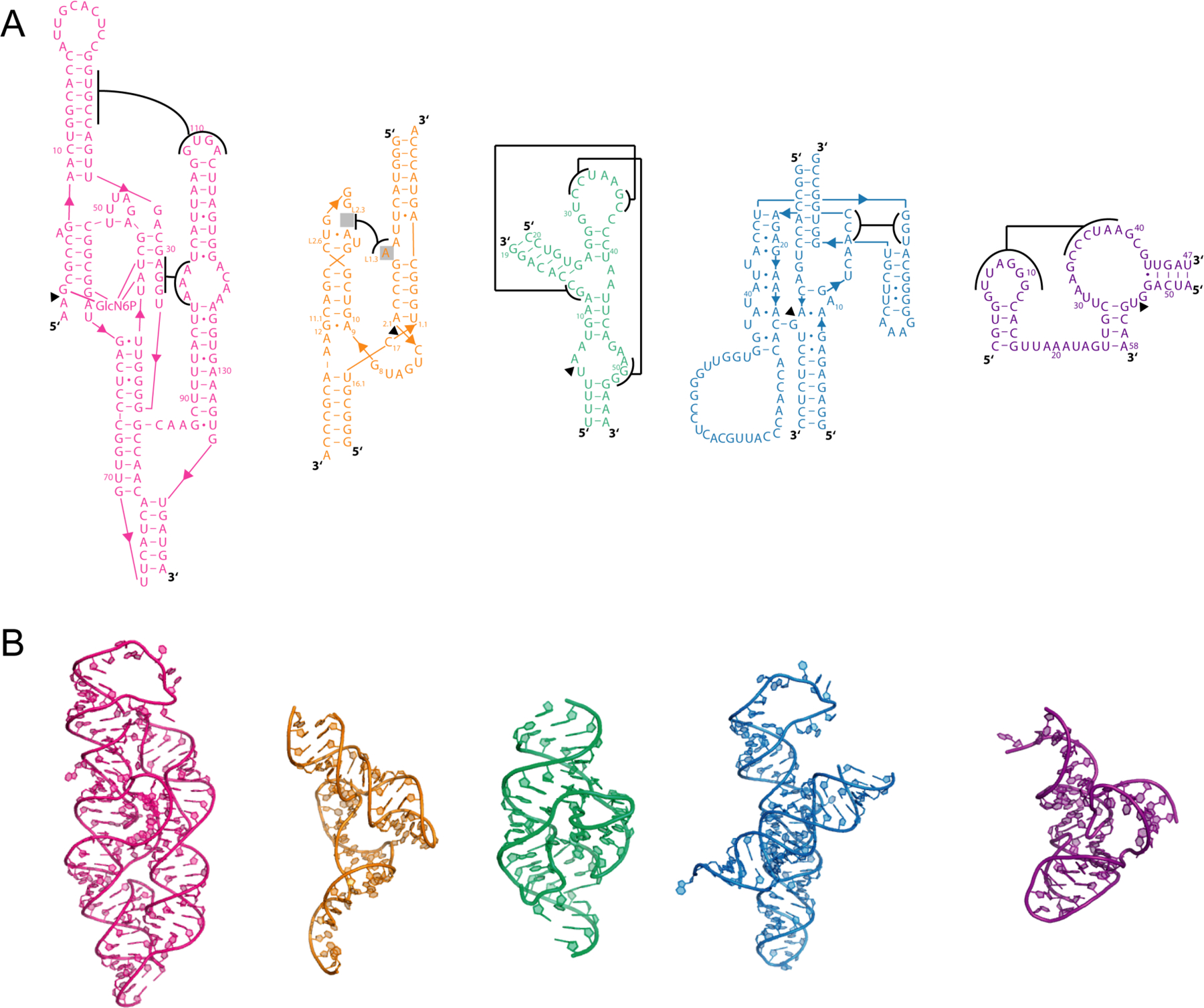
Structures of ribozymes analyzed in this study. (A) Secondary and (B) tertiary structures for glmS (pink)12, native hammerhead (orange)13, twister (green)14, hairpin (blue)24, and pistol (purple)16 ribozymes. In panel (A), pseudoknot interactions are depicted with brackets adjoined with lines. Tertiary structures were prepared using PyMOL.78
Python Pipeline for Identifying Common Contacts within Ribozyme Active Sites
To assess conservation of catalytic strategies amongst ribozymes, we created an automated contact identifying pipeline, depicted in Figure 3, to identify contact atoms (CA) to an atom of interest (AOI), typically of catalytic importance. In Step 1 of the pipeline, a custom PyMOL plugin identifies contacts between a user-defined AOI and CAs and exports relevant information concerning these contacts. In this study, the user-defined AOI is one of the key atoms of the catalytic strategies9: the nucleophilic O2′, scissile phosphate NBOs, or the leaving group O5′ (Figure 1). The custom PyMOL plugins find all CAs, which are defined as potential contacts, to the AOI within a user-defined radius, set to 5 Å herein, and output the distances to the AOI, hydrogen bonding angle, residue name and number, atom name (e.g. N1), and the b-factor (see Methods). These data are saved to a text file, making the datasets searchable. In Step 2 of the pipeline, a Python script organizes the resulting data generated by the plugins from Step 1. To identify contacts of potential catalytic importance, the hydrogen, carbon, and phosphorus atoms are filtered out. The contacts are subjected to an occurrence threshold, where they are required to occur in ≥ 25% of the crystal structures. The distances to an AOI for each of up to five CAs are plotted, with angles between the CA, the CA’s proton, and the AOI provided above each CA to indicate the likelihood of a CA to AOI hydrogen bond. In Step 3 of the pipeline, contacts amongst small ribozyme classes are unified to provide a general model of shared catalytic strategies.
Figure 3.
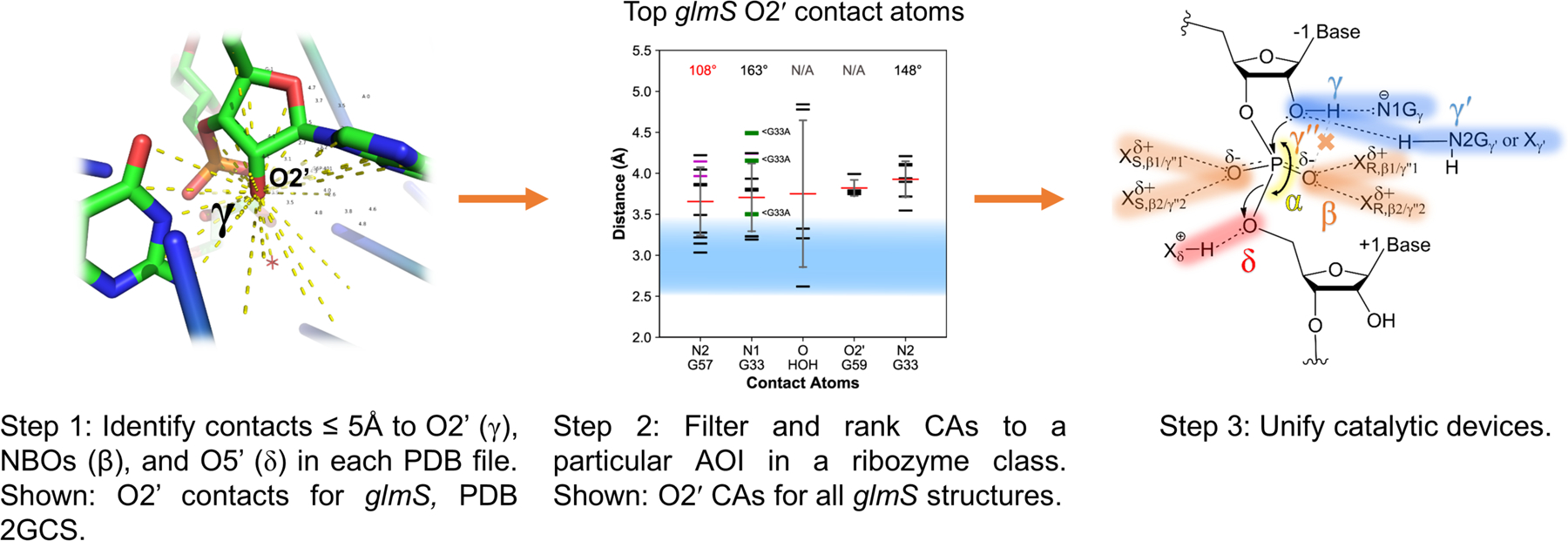
Automated pipeline to identify ribozyme contacts. Step 1: Contact atoms (CAs) within a specified distance (≤ 5Å) to the atom of interest (AOI), O2′ in this example, were identified and deemed to form a potential contact. Step 2: CAs were filtered according to the occurrence threshold (see Methods) and ranked (L to R) according to their average distance to the AOI; angles for hydrogen bonding are provided above each CA, with optimal hydrogen bonding distance shaded blue. Step 3: Identified contacts were unified to give a mechanistic model depicting strategies for small ribozyme self-cleavage.
In the next five sections, we describe analyses of different classes of ribozymes according to the established catalytic strategies of ribozymes (γ, β, δ, and α)9–10 and new catalytic strategies (γ′ and γ′′), one of which was recently reported (γ′′)25–26. Analyses on these six catalytic strategies are focused on the hammerhead, hairpin, glmS, and twister ribozymes, for which there is extensive crystallographic and mechanistic evidence, allowing us to apply more rigorous statistical analyses. We later compare the occurrence of these strategies at the scissile versus non-scissile phosphates to probe the uniqueness of the active site. We then consider the lesser-studied pistol ribozyme as an additional known ribozyme. In each section, we support observed proximities of the atoms with experimental tests of interactions from the literature.
γ - Deprotonation of the O2′ Nucleophile
The first interaction that we investigated was between the O2′ nucleophile and a possible base that could deprotonate the 2′ hydroxyl (γ; Figure 1). This catalytic strategy is important for increasing the nucleophilicity of the O2′. The γ catalytic strategy is probably the least understood, as nearly all ribozyme pre-cleaved structures have the 2′-hydroxyl of the −1 nucleotide modified, such as through a deoxy or methoxy substitution, to prevent self-cleavage during crystallization.
Upon searching for atoms within 5 Å of the O2′ of the −1 nucleotide, many conserved contacts were identified (Figure 4). Nearly all ribozymes have a guanine whose N1 is ~3 Å away from the O2′. For example, the glmS, hammerhead, twister, and hairpin ribozymes have guanine N1 contacts that are 3.7 ± 0.4, 3.0 ± 0.4, 2.9 ± 0.2, 3.4 ± 0.9 Å away from the O2′, respectively, where the values are averages and standard deviation amongst different crystal structures subjected to the criteria reported in the Methods. Furthermore, the N1-H-O2′ angle for the glmS, hammerhead, twister, and hairpin ribozymes is near the optimal for hydrogen bonding at 163, 148, 148, and 153°, respectively, where values for twister are from simulation-averaged structures.27 We note that there are other moieties in the vicinity of the O2′ of these four ribozyme classes; however, some of these acceptors are further than 4 Å from the O2′. We discuss potential hydrogen bond donors to the O2′ in the “Emergent strategies” section below.
Figure 4. Contacts for γ: Deprotonation of the O2′ nucleophile.
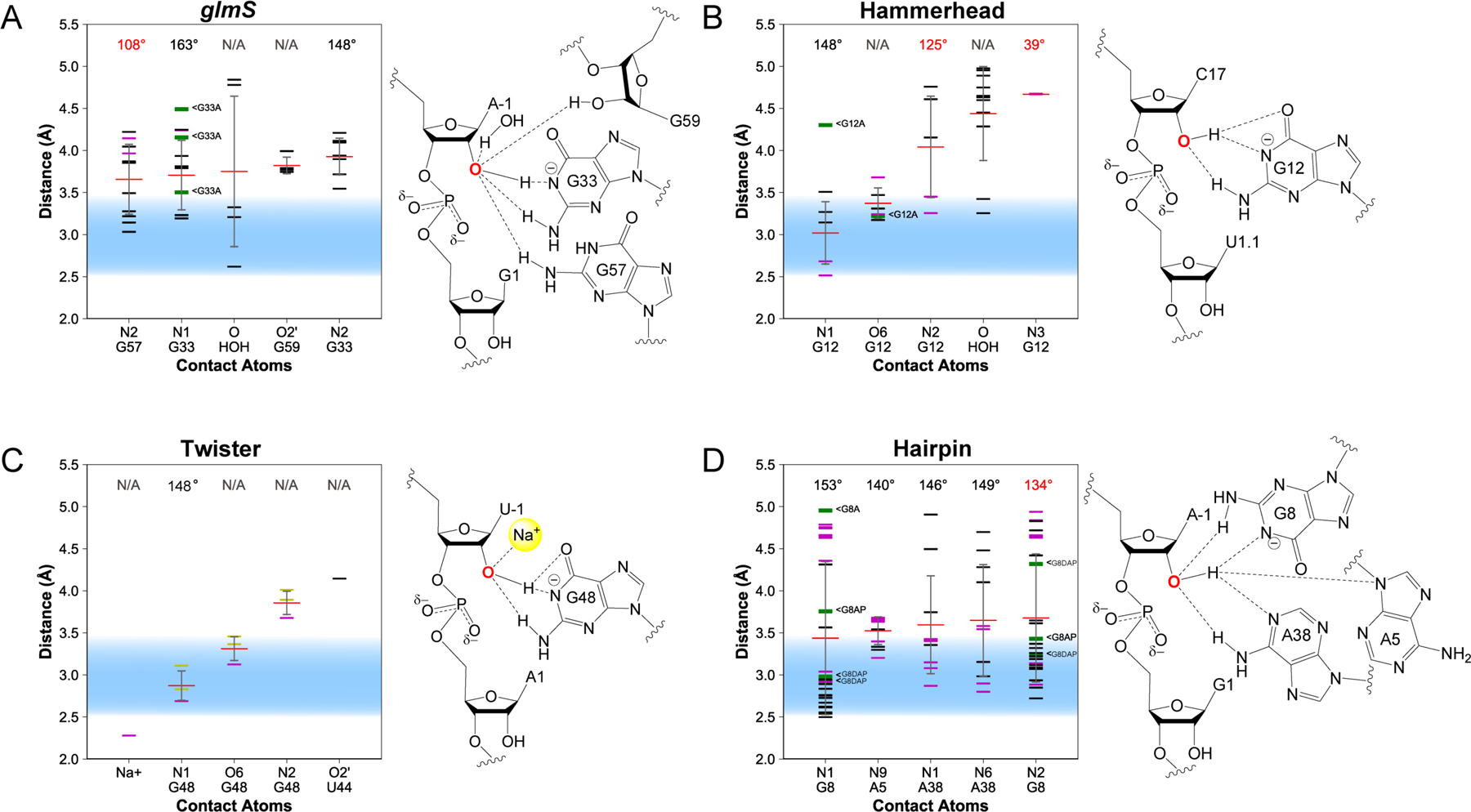
Distances between the O2′ and nearby atoms provided for the (A) glmS, (B) hammerhead, (C) twister, and (D) hairpin ribozymes. The distance observed in a given crystal structure is shown as horizontal black (WT), green (variant), purple (intermediate-mimic), yellow (simulation-averaged), or orange (not catalytically relevant) line; average distances are shown as horizontal red lines with standard deviation as a gray vertical line. Illustrations to the right of each plot visualize contacts within 4 Å of the O2′.
Deprotonation of the 2′OH by the N1 of a guanine is in agreement with previous biochemical experiments on the glmS, hammerhead, twister, and hairpin ribozymes. In the glmS ribozyme, mutations of the active-site guanine G33 to A, C, and U all significantly decreased the rate constant of self-cleavage (between 103- and 105-fold), supporting a critical role for the guanine in the self-cleavage mechanism.28–29 However, experimental evidence does not conclusively establish whether the guanine nucleobase acts in its protonated or deprotonated form, with some studies supporting an elevated pKa (i.e. shifted from 9.2 even further from neutrality) and even a role in a proton relay for the guanine.29–32
In the hammerhead ribozyme, the pH-rate profile is log-linear until pH ~8.5 to 9, after which the rate becomes independent of pH, supporting a role for guanine in the catalytic mechanism, which has an unshifted pKa value near this range.33–34 Substitution of the active-site guanine, G12, which has a pKa of 9.5, with a 2,6-diaminopurine analog (pKa of 5.1) or a 2-aminopurine analog (pKa of 3.8) alters the apparent pKa of the reaction profile in a fashion correlating with the base variant pKa without interfering with ribozyme folding, suggesting that G12 participates catalytically.35 Furthermore, crosslinking experiments reveal that G12 of the hammerhead ribozyme stacks with the C17 (–1 position) nucleotide that flanks the scissile phosphate, suggesting that it is found within the active site during ribozyme self-cleavage.36 In addition, an affinity-label experiment in which the C17 nucleobase possessed a 2′-bromoacetamide group resulted in alkylation of G12, suggesting that it is indeed the general base. This work also led to the conclusion that the pKa of G12 is perturbed towards neutrality to ~8.5, a shift that is commonly observed in catalytic RNA functional groups.37 The proximity of G12(O6) to the O2′ and the identification of the Hoogsteen face of G12 as a divalent metal ion binding site in two hammerhead structures38 is consistent with the divalent metal tuning the pKa of this G.
Experiments involving mutations to the active-site guanine of the twister ribozyme support its role as general base. A G48A mutation lowers the rate constant of self-cleavage by ~300-fold and decreases the apparent pKa values of the reaction to ~5 and 6.2, with a maximal rate around pH 5.39 Substitution of G48 with 2-aminopurine gives a similar low pH-shifted pH dependence as a G48A mutation, which is expected since A and 2AP have similar pKa values, whereas substitution of G48 with inosine results in a similar pH dependence compared to wild-type (WT) ribozyme, again expected since G and I have similar pKa values. The top contact for the O2′ in the twister ribozyme, however, is a Na+ ion that lies at a distance of 2.3 Å away from the O2′. However, the Na+ is present in only one of four structures whose data went into this plot, which is a computational transition-state simulation-averaged structure.27 In that study, this Na+ ion is also only 2.3 Å away from the pro-SP NBO and 2.5 Å away from the O6 of G48 (the putative general base). Consequently, the O6 of G48 also shows up as a top contact to the O2′ in the twister ribozyme. In addition, in the same study there is a second Na+ ion in proximity of G48(O6) in the transition-state simulation-averaged structure at 3.4 Å from the O6. Thus, it is possible that metal ions help to stabilize the deprotonated form of G48 and lower its pKa, facilitating its role as general base.
In the hairpin ribozyme, which has been suggested to utilize two nucleobases with varying pKa values in its general acid-base catalytic mechanism,40 deletion of the active-site G8 nucleobase results in a decrease in the rate as much as ~850-fold compared to the WT ribozyme.41–42 In addition, substitution of G8 with either uridine or imidazole (Im) slows self-cleavage activity, but does not affect the folding of the ribozyme.43–44 The G8Im substitution shows an altered pH-rate profile, where the profile becomes bell-shaped, consistent both with the lower pKa of imidazole compared to guanine and the idea that G8 acts catalytically.43 Substitution of G8 with either 2-aminopurine or 2,6-diaminopurine (pKa values of 3.8 and 5.1, respectively) also results in the emergence of a bell-shaped pH-rate profile with low activity at high pH values, consistent with the acidic pKa values of these two analogs.45–46
β – Neutralization of Charge on a Non-Bridging Oxygen
Moieties in the active site can stabilize the NBOs through Coulombic interactions, hydrogen bonding and dipole-dipole interactions, lowering the free energy of the transition state by mitigating charge build-up. For instance, in the Tetrahymena group I intron, two metal ions contact the pro-SP oxygen of the scissile phosphate.47 Ribonuclease A, which catalyzes the same reaction as the small ribozymes, makes contacts to the substrate NBO atoms via protonated amino acid side chains to achieve a similar effect in cleaving RNA.48–49 We asked how the NBO atoms of small ribozymes are stabilized. Because the two NBOs at the scissile phosphate of ribozymes are pro-chiral, we analyzed contacts for each NBO separately (Figure 5).
Figure 5. Contacts for β: Neutralization of the negative charge on the NBO atoms.
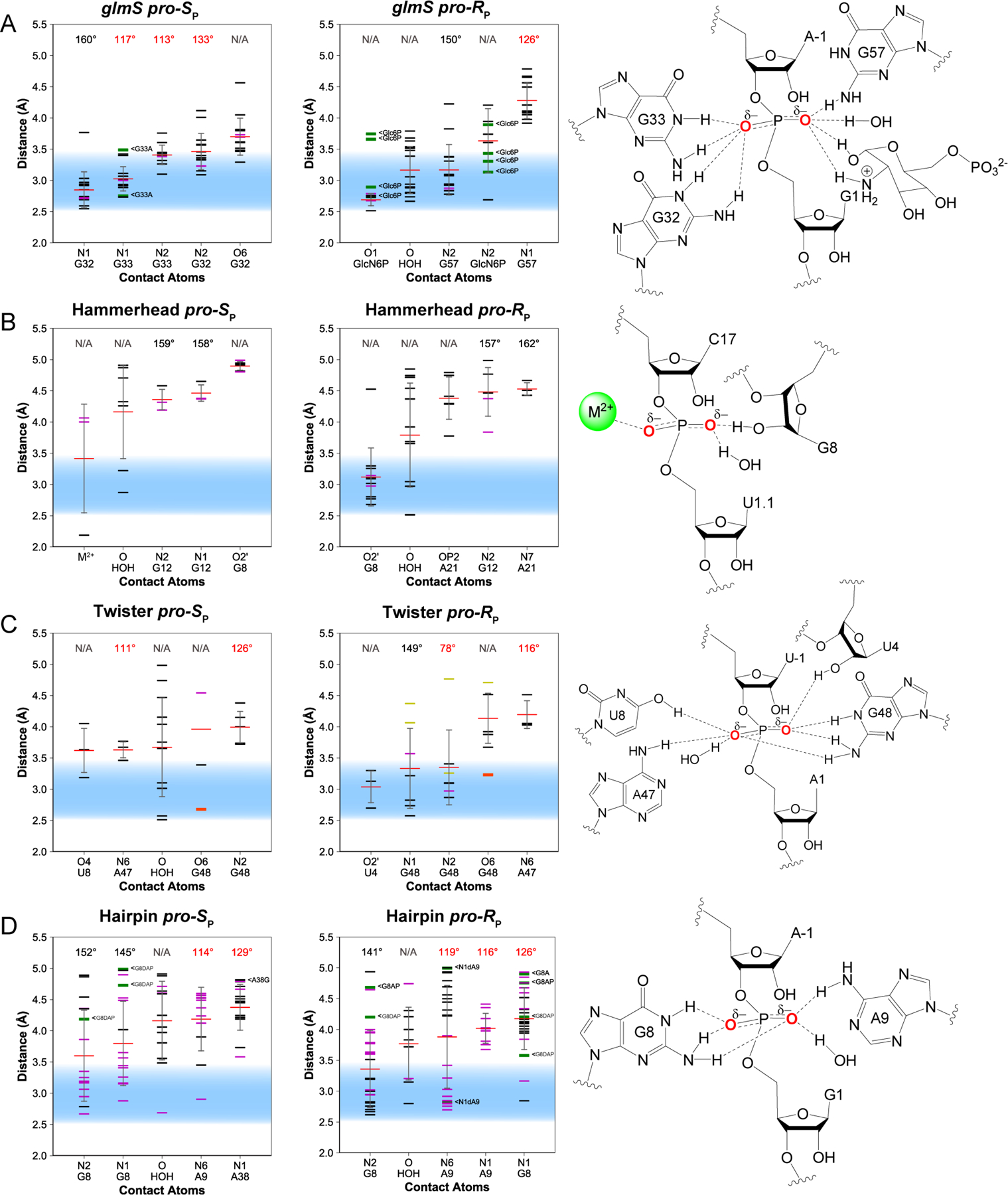
Distances between the pro-RP and pro-SP NBO atoms and nearby atoms are provided for the (A) glmS, (B) hammerhead, (C) twister, and (D) hairpin ribozymes. Distances observed in each crystal structure are shown as horizontal black (WT), green (variant), purple (intermediate-mimic), yellow (simulation-averaged), or orange (not catalytically relevant) lines; average distances are shown as horizontal red lines; standard deviation is shown as a gray vertical line. Illustrations to the right of each set of plots visualize contacts within 4 Å of the NBO atoms. Note that interactions between G32(O6) and G48(O6) and the pro-SP NBO of the glmS and twister ribozymes, respectively, are not shown, but are possible with an iminol tautomer.
The environment surrounding the NBOs in the glmS ribozyme show that the O1 of the cofactor is in close proximity to the pro-RP NBO (2.7 ± 0.1 Å), as is the N2 of the cofactor and the N2 of G57. Similarly, the pro-SP NBO is near quite a few contacts, primarily guanine N1 and N2s, including the N1 and N2 atoms of both G32 and G33 (Figure 5A). In support of these findings, experimental evidence suggests that the glmS ribozyme has an overdetermined set of hydrogen bonds to the NBO atoms that helps promote nucleophile activation.26 In addition, thio substitutions at the NBO atoms have been found to result in severe decreases in cleavage activity, especially at the pro-RP position.25 These thio effects are not rescuable by thiophilic metal ions, suggesting that the inhibition resulting from thio substitution is due to disruption of NBO contacts rather than disruption of metal ion binding at these NBO positions.6,25 Furthermore, mutations of G32 (pro-SP contact) to an adenine and especially of G57 (pro-RP contact) to both adenine and cytosine result in a lowering of the self-cleavage rate compared to WT ribozyme, suggesting that these NBO contacts are important for stabilization of the NBO oxygen atoms during self-cleavage.26,29,50 In addition, deletion of the O1 group of GlcN6P, which contacts the pro-RP oxygen of the scissile phosphate, reduces self-cleavage activity ~70-fold but still results in a full processing of ribozyme self-cleavage, with other mutations also resulting in decreased activity.51
In contrast to the glmS ribozyme, the NBO atoms of the hammerhead ribozyme are involved in relatively few hydrogen-bonding contacts (Figure 5B). The pro-RP NBO in the hammerhead ribozyme has an O2′ and a water molecule nearby with average distances of 3.1 ± 0.5 Å and 3.8 ± 0.8 Å, respectively. Previous thio substitution experiments where the NBO atoms of the scissile phosphate were individually probed with thio substitutions revealed a metal ion contact with the pro-RP NBO. This metal ion also contacts the P9/G10.1 phosphate, interacting with both of these sites in the transition state during self-cleavage.52 It is thought that a major conformational rearrangement occurs during self-cleavage, which brings this metal ion in contact with these two proposed binding sites. Recently, a structural study emerged showing divalent metal ion interaction with an NBO of the scissile phosphate in a vanadate structure, albeit the pro-SP NBO (Figure 5B and Figure S2).53
The twister ribozyme appears to be like the glmS ribozyme in that there are multiple contacts to both the pro-RP and pro-SP NBOs (Figure 5C). The pro-RP NBO in twister shows on average an O2′ at 3.0 ± 0.3 Å, a guanine N1 at 3.3 ± 0.6 Å, and a guanine N2 at 3.3 ± 0.6 Å. Mutations of G48 to adenine, the nucleobase responsible for the N1G and N2G contacts and also the putative general base, reduces self-cleavage drastically. In addition, mutation of G48 to inosine essentially results in the disappearance of a stereospecific RP thio effect that is seen for the WT ribozyme, suggesting that the guanine N2 is important for stabilization through hydrogen-bonding to the pro-RP position.14 The pro-SP NBO also has multiple possible contacts. Nearby are the O4 of U8 and the N6 of A47 at 3.6 ± 0.4 Å and 3.6 ± 0.1 Å, respectively. The O4 of U8 cannot act as a hydrogen bond donor in its canonical state; however, tautomerization with N3 could lead to a protonated O4, which could then donate to the pro-SP NBO. Mutation of A47 significantly reduces self-cleavage activity.14
The hairpin ribozyme exhibits the N2 of G8 only 3.4 ± 0.6 Å away from the pro-RP NBO (Figure 5D). The pro-RP NBO in hairpin is also 3.8 ± 0.6 Å from a water molecule that is observed nine times in 25 structures. The G8(N2) that contacts the pro-RP NBO is also 3.6 ± 0.7 Å from the pro-SP NBO. The N1 of this same guanine (G8) is the second top CA for the pro-SP NBO, strongly implicating G8 in neutralization of the NBO atoms. Mutation of G8 to inosine reduces the rate by ~50-fold, consistent with a role of its amino group.54
δ – Neutralization of Charge on the 5′-Oxygen
The general acid in the self-cleavage mechanism of small ribozymes neutralizes charge build-up on the leaving group oxygen, stabilizing the alkoxide generally through protonation or chelation. A diverse collection of general acids is observed for the ribozymes in this study (Figure 6).
Figure 6. Contacts for δ: Neutralization of charge on the O5′.
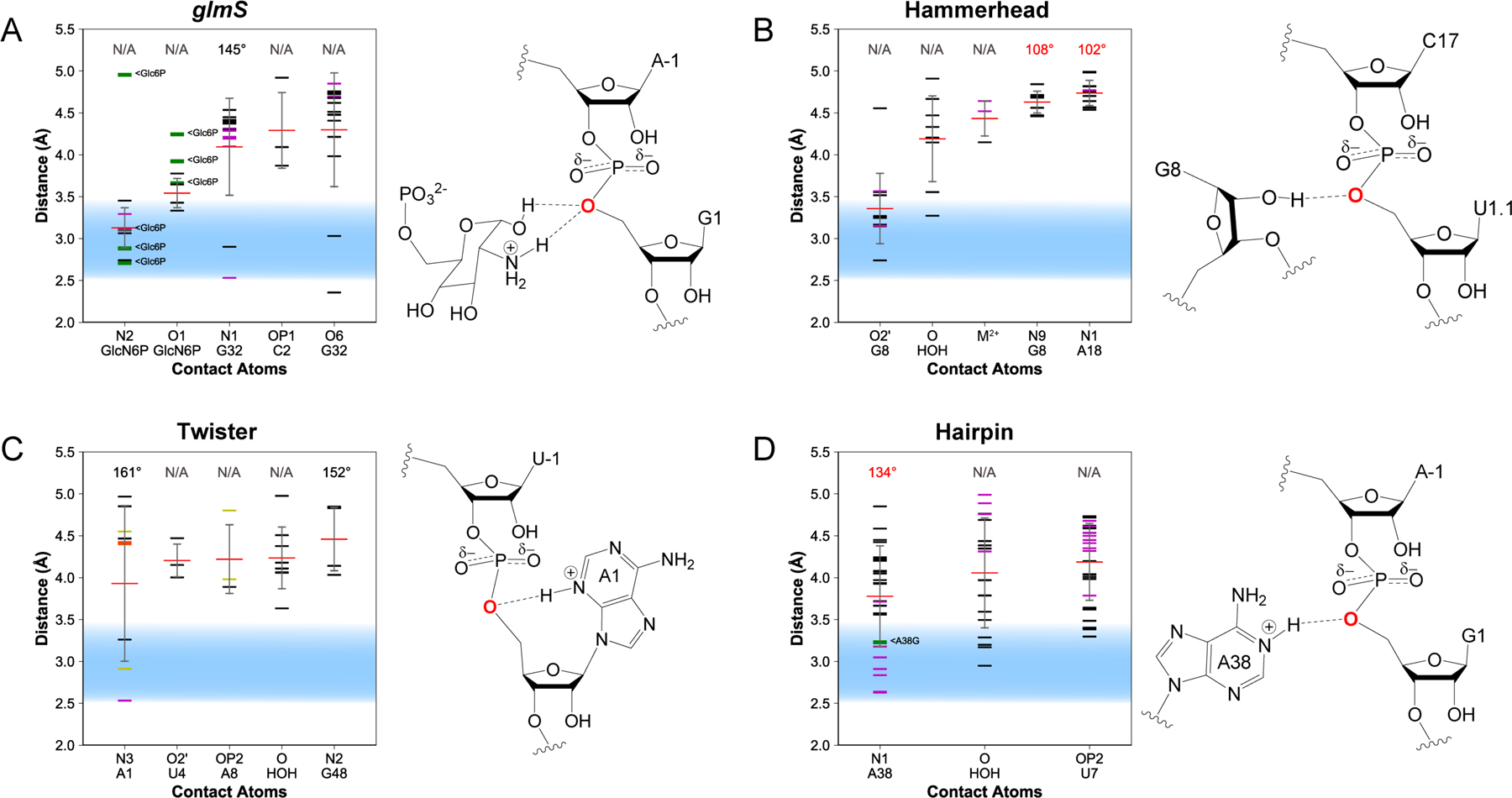
Distances between the O5′ and nearby atoms are shown for the (A) glmS, (B) hammerhead, (C) twister, and (D) hairpin ribozymes. Distances observed in each crystal structure are shown as horizontal black (WT), green (variant), purple (intermediate-mimic), yellow (simulation-averaged), or orange (not catalytically relevant) lines; average distances are shown as horizontal red lines; standard deviation is shown as a gray vertical line. Illustrations to the right of each plot visualize contacts within 4 Å of the O5′.
In the glmS ribozyme, the closest contact to the O5′ atom of the +1 nucleotide is the exocyclic amine of the general acid cofactor, GlcN6P (Figure 6A). The average distance between the O5′ and the N2 of the cofactor is 3.1 ± 0.2 Å. The second closest contact to the O5′ is the O1 of GlcN6P. The catalytic cofactor was observed in close proximity in every structure that was crystallized in its presence. The close proximity and high prevalence of the cofactor suggests a vital role in catalysis. This is in agreement with previous experiments in the literature. The presence of GlcN6P is found to accelerate glmS ribozyme self-cleavage as much as ~106-fold over the reaction in the absence of GlcN6P,6,25 and self-cleavage in the presence of a cofactor analog that is missing the exocyclic amine, Glc6P, is drastically reduced.6 Furthermore, nucleotide analog interference mapping and suppression experiments suggest GlcN6P binds in the active site,55 and hydroxyl radical footprinting and crosslinking experiments suggest that the ribozyme active site is pre-folded prior to GlcN6P binding,56 advocating a direct catalytic, rather than an allosteric, role for the cofactor. In addition to these findings, the pKa of GlcN6P is found to be perturbed towards neutrality, as would be expected for a moiety that participates in the chemical step of the reaction.57–60 Furthermore, self-cleavage activity is especially sensitive to mutations of the amine group of the cofactor,51 and rate constants and apparent pKa values of the reaction have been found to depend on the microscopic pKa value of various cofactor analogs that have been tested.57,61 These results all point to a role for GlcN6P in general acid-base catalysis, and paired with the observation that the cofactor is poised for protonation of the O5′ leaving group, GlcN6P likely serves as the general acid in glmS self-cleavage.
In the hammerhead ribozyme, the closest contact to the O5′ of the +1 nucleotide is the O2′ of G8, with an average distance of 3.4 ± 0.4 Å (Figure 6B). This O2′ was observed within 5 Å of the O5′ in every crystal structure. The close proximity and high prevalence of the O2′ suggests that it may play a role in catalytic enhancement, perhaps through hydrogen bonding as originally suggested by Martick and Scott.18 Crosslinking experiments from the literature reveal that G8 crosslinks to C17 and N1.1 at the cleavage site, with the G8-C17 crosslink retaining catalytic activity.36 Furthermore, an analog with a bridging phosphorothioate at the cleavage site O5′ leaving group position was not inhibited by mutation of the 2′OH group of G8 whereas the WT ribozyme was inhibited, which suggests that the G8 2′OH group is involved in proton transfer and general acid catalysis.62 This work also suggested that a metal ion may help to bring the pKa of the basic 2′OH group of G8 towards neutrality to better help it serve a catalytic role, which is also supported by MD and QM/MM calculations.63
The closest contact to the O5′ in the twister ribozyme was found to be the N3 of the A1 nucleotide (Figure 6C).27 This is in agreement with most biochemical experiments and computational studies on the ribozyme. Previous biochemical experiments demonstrated a 104-fold reduction in rate at pH 7.0 upon substitution of A1 with guanine.39 Furthermore, substitution of A1 with a variant that lacks the N1 and N3 atoms was found to be deleterious to self-cleavage, and the inhibition was found to be directly linked to removal of the N3.64–65 The pKa of this adenine was predicted from pH-dependent NMR experiments to be shifted from a pKa of 3.7 ± 0.1 towards neutrality to ~5.1 ± 0.1 upon folding of the twister ribozyme.64 Moreover, computational work suggests that the N3 is poised to act as the general acid in solution and that the pKa of this adenine could be shifted even more towards neutrality by a total of ~5 pKa units.27 Conversely, a different computational study found a Na+ ion between the O5′ and N3 of A1, suggesting that the Na+ ion could be stabilizing the O5′ leaving group and A1(N3) could just serve to position the catalytic metal ion.66 More experiments will be needed to discern between these mechanisms and whether one or both plays a significant role, but it is clear that A1(N3) somehow facilitates O5′ stabilization.
In the hairpin ribozyme, the closest contact to the O5′ was the N1 of A38, which has an average distance of 3.8 ± 0.6 Å (Figure 6D). This agrees with the idea that the hairpin mechanism uses two nucleobase moieties in general acid-base chemistry, one with a high pKa and one with a low pKa, where A38 would serve as the lower pKa species.40 Removal of the base at position 38 results in a ~10,000-fold loss in activity, signifying its importance.67 Substitution of this nucleobase with purine is detrimental to the rate but not in the background of a bridging thio substitution at the O5′, strongly suggesting that A38 is indeed the general acid in hairpin ribozyme catalysis.46
α – In-line Nucleophilic Attack
The in-line nucleophilic attack strategy is defined by the angle between the O2′ nucleophile, the phosphorus, and the O5′ leaving group. This angle should be near 180° for efficient overlap between the lone-pairs of the O2′ and the anti-bonding orbital of the P–O5′ bond. In addition, it has been shown in computational studies on nucleophilic attack of phosphodiesters that the O2′–P–O5′ angle can deviate significantly from 180° in the transition state itself to values as low as 159°, and even more in pre- and post-cleavage states.68 Measuring this angle in each crystal structure revealed many structures that were in this vicinity. Figure 7 provides angles for glmS, hammerhead, twister, and hairpin ribozymes. It should be noted that crystal structure and computationally modelled intermediate or intermediate-mimic structures are not included in the average angle calculation but are still shown in the plot. The average angles corresponding to α for the glmS and hammerhead ribozymes are closest to ideal, at 154 ± 9 and 157 ± 12°, respectively. The twister and hairpin ribozymes deviated slightly more, with average angles of 136 ± 34° and 133 ± 29°, respectively, but are still in the vicinity for in-line attack (Figure 7).
Figure 7. Contacts for α: In-line nucleophilic attack at the scissile phosphate.
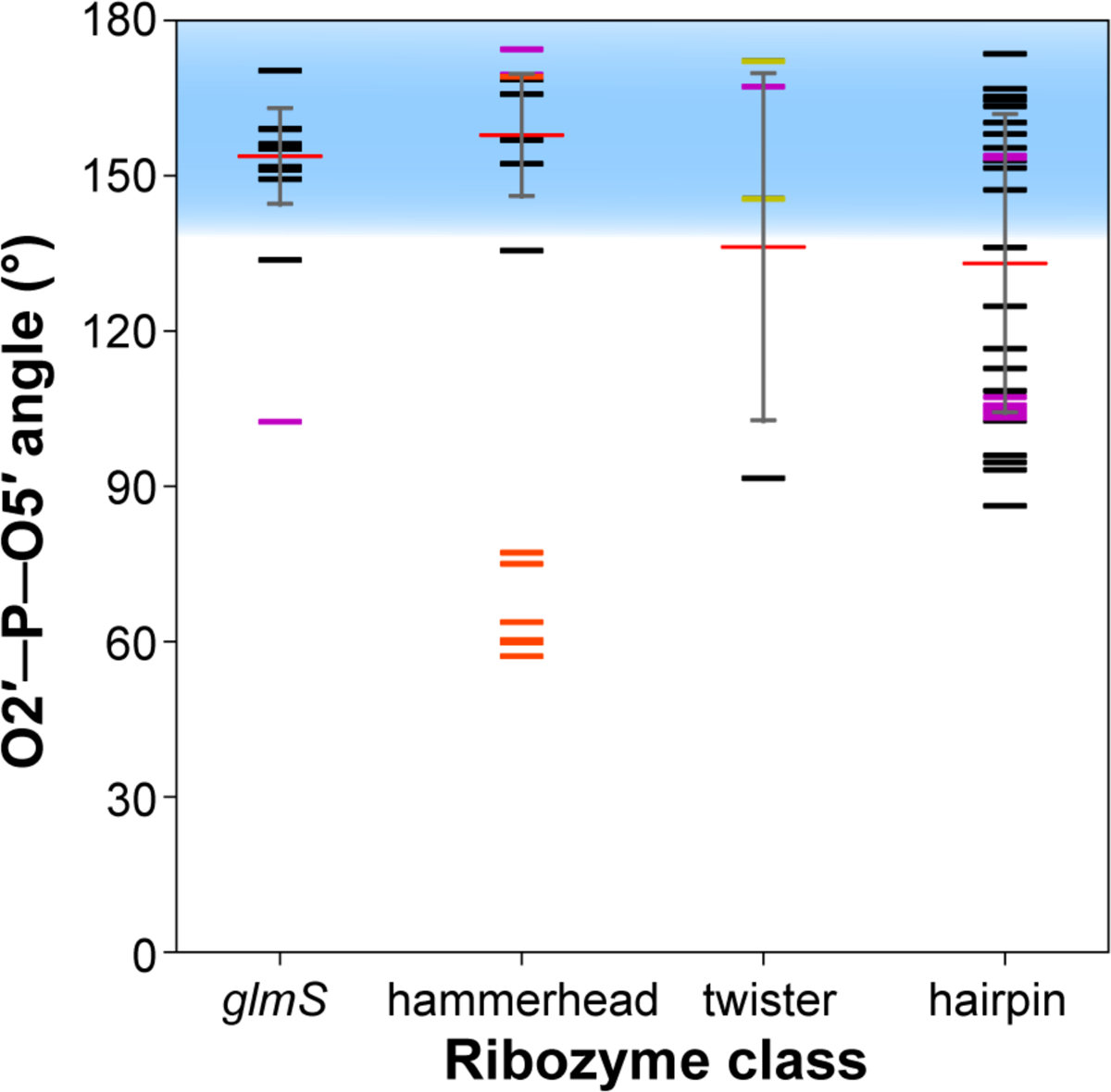
Values corresponding to the O2′─P─O5′ angle of the scissile phosphate are shown for the glmS, hammerhead, twister, and hairpin ribozymes. Angles observed in each crystal structure are shown as horizontal black (WT and nucleobase variant), purple (intermediate-mimic), yellow (simulation-averaged), or orange (not catalytically relevant) lines; average angles are shown as horizontal red lines; standard deviation is shown as a gray vertical line; and the optimal angles for in-line nucleophilic attack (140–180°) are shown as a blue shaded region. Note that structures represented by purple or orange bars are not included in determination of the average angle (see Methods).
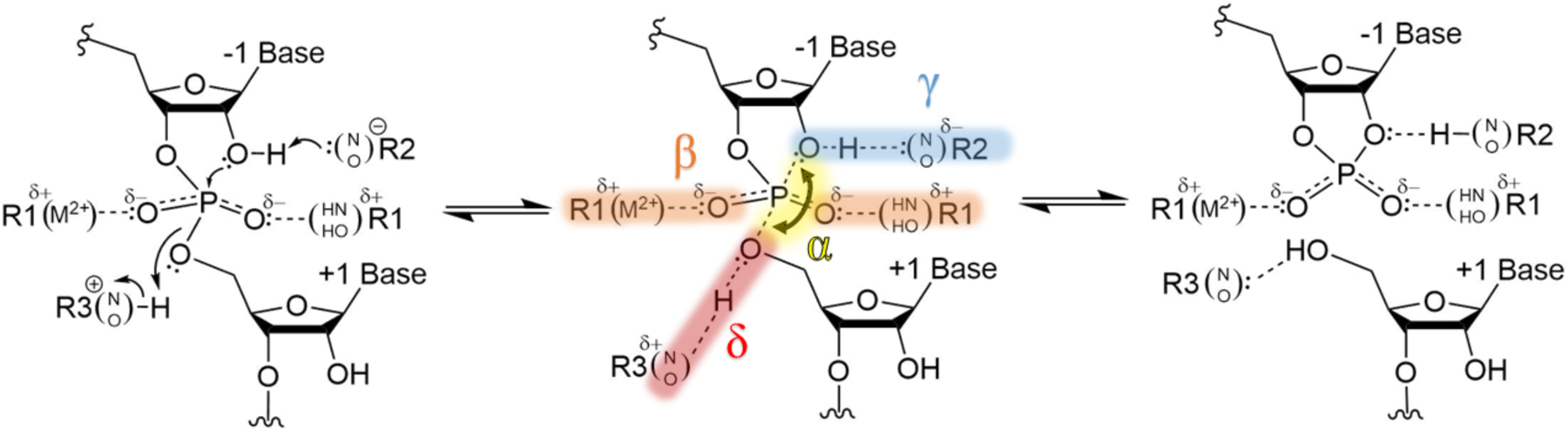
Emergent Catalytic Strategies: γ′ and γ′′ – Acidification and Release of the 2′OH
We described above the four classic strategies for ribozyme catalysis.9–10 Some interactions that carry out these strategies are conserved between ribozyme classes, such as for deprotonation of the O2′ nucleophile (γ), where all the ribozymes in this study show an active-site guanine positioned to act as the general base. However, other strategies are achieved through diverse interactions, including stabilization of the NBO atoms (β) and protonation of the 5′O leaving group (δ). In particular, CAs to the NBO atoms identified in the β strategy are diverse and include nucleobases, sugars, organic molecules, and metal ions. Remarkably, regardless of the level of CA conservation, evidence for each of the classic strategies is found in the crystal structures studied using our computational pipeline.
We were curious whether additional catalytic strategies might be uncovered by this approach. Our computational studies revealed one new strategy and the conservation of a recently identified strategy, each of which leads to activation of the nucleophile through its acidification. Nucleophile acidification allows for better pKa matching between the 2′OH nucleophile and the general base, which ultimately promotes proton abstraction by the general base (see Discussion). Since the general base strategy for deprotonating the O2′ nucleophile was termed γ by Breaker, we term these two new strategies γ′ and γ′′ since they also act at the O2′: γ′ has the potential to acidify the 2′OH through hydrogen bond donation to it, and γ′′ has the potential to acidify the 2′OH by releasing the nucleophile from an inhibitory interaction. We recently identified the latter of these two experimentally and computationally for the glmS ribozyme,25–26 and ask here whether this strategy is conserved.
The γ′ strategy, which is typically seen as an N2G/O2′ hydrogen bond, describes the acidification of the O2′ through donation of a hydrogen bond to the nucleophile. The contacts that perform this role are encompassed within the γ strategy figure (deprotonation of the O2′ nucleophile; Figure 4). All four ribozymes studied in detail herein (glmS, hammerhead, twister, and hairpin) support interaction between the N2 of the putative general base guanine or the N2 of a nearby guanine and the O2′, with average distances of 3.7 ± 0.4 Å, 4.0 ± 0.6 Å, 3.9 ± 0.1 Å, 3.7 ± 0.8 Å, respectively (Figure 4). For instance, the two top ranked N2 contacts in the glmS ribozyme come from G33, the putative general base, and G57.
There is experimental support for the importance of the N2 of guanine in the γ′ strategy. In the twister ribozyme, substitution of the G48 general base to inosine results in ~500-fold slower rate.39 Likewise, in the hammerhead ribozyme, substitution of the G12 general base with inosine results in 1000-fold slower rate,35 and in the hairpin ribozyme, substitution of the G8 general base with inosine results in a 50-fold slower rate.54 Together, these large rate decreases strongly support the broad importance of the general base guanine exocyclic amine to catalysis.
The γ′′ strategy, which involves contacts to the NBO atoms of the scissile phosphate, represents activation of the 2′OH nucleophile through competitive hydrogen bonding. The contacts that perform this role are encompassed within the β strategy figure (neutralization of negative charge on the NBOs; Figure 5). The γ′′ strategy is observed in all the ribozymes studied herein, and in many cases multiple contacts to each NBO atom are found. Previous work from our lab supports the notion that NBO contacts can activate the 2′OH nucleophile through competitive hydrogen bonding. Inverse thio effects in the glmS aporibozyme (i.e. lacking the general acid cofactor, GlcN6P), where sulfur substitution at the pro-RP NBO increases self-cleavage activity relative to WT, suggest that activation of the 2′OH nucleophile is facilitated by a sulfur atom at the pro-RP position, which is a poor hydrogen bond acceptor. From this result, it is reasoned that the WT GlcN6P(O1)-to-pro-RP NBO hydrogen bond enhances self-cleavage activity through competitive hydrogen bonding, activating the 2′OH.25 This is further supported by studies involving glmS ribozyme variants that selectively induce an inhibitory 2′OH to pro-RP interaction that is released by sulfur substitution at the pro-RP position leading to inverse thio effects.26 Similar arguments have been made for the HDV ribozyme.69
Scissile Phosphates have Unique Properties as Compared to all Phosphates
The above sections considered six catalytic strategies focused on the scissile phosphate. If these strategies are catalytic, then the majority of the contacts should be unique to the scissile phosphate. To compare use of the six catalytic strategies by the scissile phosphate versus all ribozyme phosphates, the O2′–P–O5′ angle (α strategy), number of CAs to the O2′ (γ and γ′ strategy), number of CAs to the NBO atoms (β and γ′′ strategy), and number of CAs to the O5′ (δ strategy) were calculated for all phosphates across more than 60 crystal structures of the four main ribozyme classes (Figure 8).
Figure 8. Angles and contacts across all phosphates: Scissile phosphate is unique in α, β, γ, and δ catalysis.
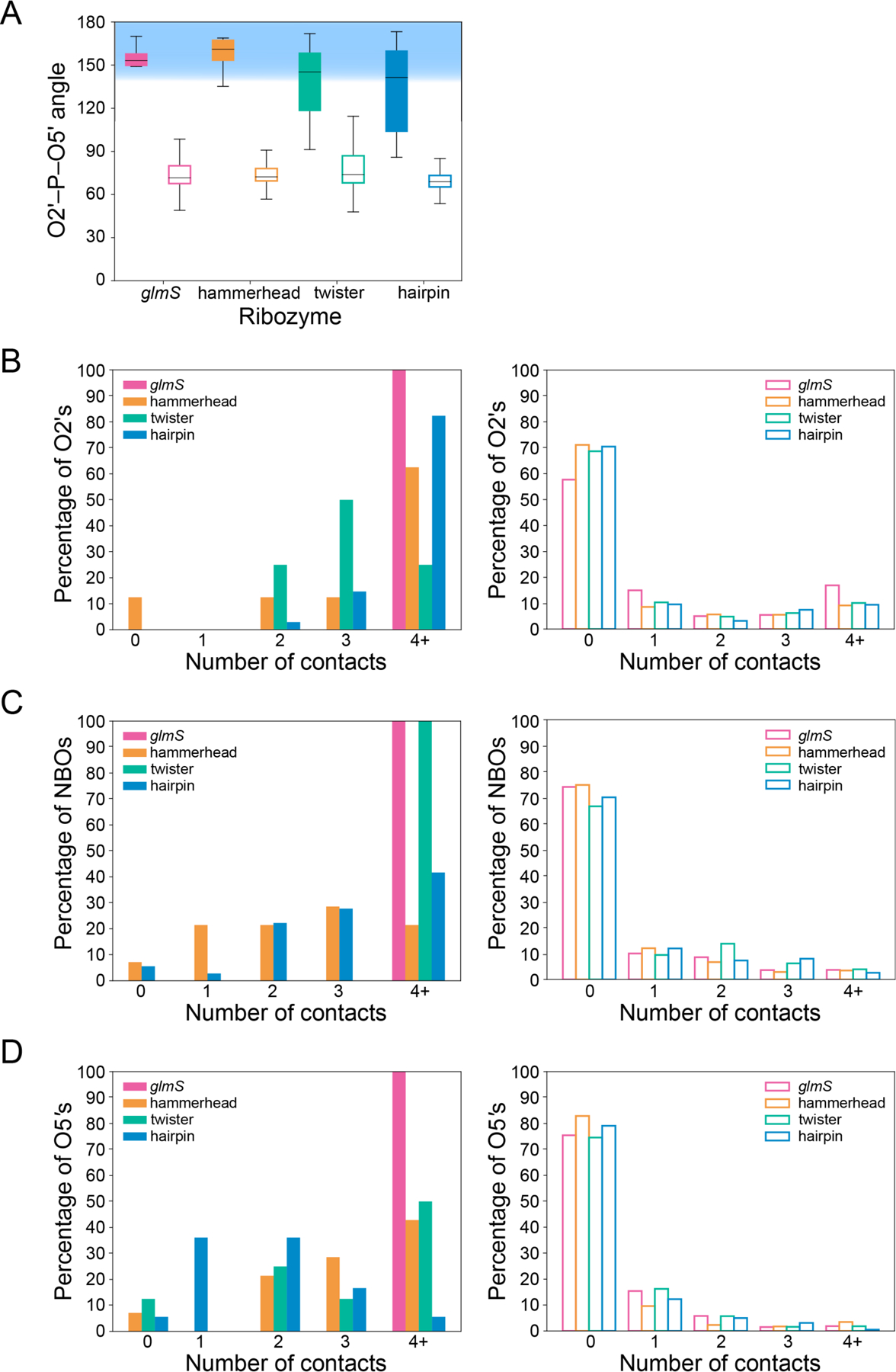
(A) In-line attack angles for scissile (filled bars) and non-scissile (open bars) phosphates in the glmS (pink), hammerhead (orange), twister (teal), and hairpin (blue), ribozymes (α strategy). (B,C,D) Number of contacts within 5 Å to the (B) O2′ (γ and γ′ strategy), (C) NBO atoms (β and γ′′ strategy), and (D) O5′ (δ strategy) at scissile (filled bars) and non-scissile (open bars) phosphates. Ribozyme coloring is the same as for (A).
Comparison of the O2′–P–O5′ angle (α strategy) at the scissile phosphate versus that at the non-scissile phosphates reveals a significantly greater angle for the scissile phosphate for all four of the main ribozyme classes: glmS, hammerhead, twister, and hairpin ribozymes (Figure 8A). The average angle for scissile phosphates across these four classes of ribozymes is 140 ± 27° compared to 76 ± 16° for non-scissile phosphates. Paired with the observation that the O2′–P–O5′ angle can deviate significantly from 180° even in the transition state,68 the finding that the scissile phosphate O2′–P–O5′ angles are much closer to 180° than non-scissile phosphates supports the significance of the conclusion that the scissile phosphate angles are in-line and catalytically relevant.
To determine whether the scissile phosphate was unique across all ribozyme phosphates in having a multitude of CAs to the O2′ atom (γ and γ′ strategy), the number of CAs within 5 Å of the O2′ atom was calculated for all phosphates in each ribozyme crystal structure. The average number of CAs to the active site O2′ nucleophile for all crystal structures of all ribozyme classes, 2.7 ± 1.1, significantly exceeds that of the non-scissile phosphates at 0.5 ± 0.1, with the majority of O2′ atoms at non-scissile phosphates having no contacts (Figure 8B). This finding strongly suggests that the scissile phosphate is unique in having a multitude of ribozyme moieties poised to acidify and deprotonate the O2′ nucleophile.
A similar analysis was performed across all ribozyme phosphates to look for enhancement of CAs at the NBO atoms of the scissile phosphate (β and γ′′ strategy). For this analysis, the number of CAs within 5 Å to the NBO atoms was calculated for all phosphates in each ribozyme crystal structure, where the number of CAs to each NBO atom was limited to two (Figure 8C), although we note that it is possible for greater than two CAs to be within our defined interaction distance. The average number of CAs to the scissile phosphate NBO atoms is significantly greater than for the non-scissile phosphate NBO atoms at 2.6 ± 1.4 and 0.14 ± 0.02, respectively. For instance, in all ribozyme classes, the majority of phosphates contain no contacts to either NBO atom, with only a few phosphates containing one or more contacts to the NBO atoms. This difference suggests that the scissile phosphate is indeed unique in its use of the β and γ′′ strategies. Apparently, ribozyme active sites have evolved to have high numbers of hydrogen bond donors to the NBO atoms, possibly both to prevent the 2′OH from engaging in inhibitory interactions with the NBO atoms and to promote stabilization of charge during self-cleavage.
Finally, to investigate the uniqueness of the cleavage site in the presence of CAs to the O5′ atom (δ strategy), the number of CAs within 5 Å to the O5′ atom was calculated for all nucleotides in each ribozyme crystal structure. The average number of CAs to the O5′ atom at the cleavage site, 2.0 ± 1.3, differed significantly from that at nucleotides away from the cleavage site, 0.08 ± 0.04, for all ribozyme crystal structures. Thus, similar to the finding for the β and γ′ strategies, ribozymes have a significantly higher presence of potential general acid moieties at the O5′ atom at the cleavage site.
In sum, ribozyme active sites are unique in their use of the six catalytic strategies. Comparison of the occurrence of CAs and the presence of an in-line attack angle versus all other phosphates supports the notion that these strategies are effective in enhancing self-cleavage at a site-specific position.
Extension of Catalytic Strategies to a New Ribozyme: The Pistol Ribozyme
We next wanted to test whether the six catalytic strategies are predictive for another ribozyme. Two papers recently described crystal structures for a newly discovered ribozyme, the pistol ribozyme.16,70 This ribozyme is found most commonly in the firmicutes phylum of bacteria and in environmental samples near genes associated with bacteriophages.71 We applied our pipeline to the four structures in these papers. To do so, we substituted a 2′ OH in place of the 2′ H present in all structures (Figure S3).
The contacts for the γ and γ′ strategies—general base and O2′ acidification, respectively—are provided in Figure S3A. Analysis of γ and γ′ support G40 as the general base since it has the closest N1 to the O2′, at 3.4 ± 0.2 Å. Additionally, the N2 of G40 is 3.0 ± 0.2 Å from the O2′, suggestive of roles in hydrogen bond donation to the two O2′ lone pairs that could acidify and help remove the O2′ proton. It thus appears that both γ and γ′ strategies are conserved in the pistol ribozyme. Moreover, participation of the N2 of the general base and N2 of another guanine in donating a hydrogen bond to the O2′ is reminiscent of the γ and γ′ strategies of the glmS ribozyme. Consistent with the importance of G40, mutation of G40 + C41 completely abolishes catalytic activity.71–72
The pistol ribozyme contacts for the β strategy—charge stabilization of the NBO atoms—are provided in Figure S3B. The pro-SP NBO atom of the scissile phosphate shows little contacts within 4 Å. The pro-RP NBO atom, on the other hand, has a few contacts in relatively close proximity. Specifically, there is the N1 from G40, a water molecule, an O2′ from A32, the N2 from G40, and an M2+ all within 4.0 Å of the pro-RP NBO atom. The relatively sparse but metal-mediated contacts to the NBO are reminiscent of the β strategy of the hammerhead ribozyme, where the pro-RP has been implicated in metal ion catalysis.52,63,73 These species could help to stabilize charge development at the pro-RP position through hydrogen bond donation and electrostatics and could also release the 2′OH through competitive interactions.
The pistol ribozyme contacts for the δ strategy—protonation of the O5′—are provided in Figure S3C. There are no CAs within hydrogen bonding distance of the O5′ atom, although all four crystal structures contain a polyvalent ion within 5 Å of the O5′. The G33N7 was shown to coordinate this hydrated metal ion close to the O5’, possibly acting as general acid.74 This most recent study suggests that the N3 of A32 does not play a catalytic role in the reaction.
Finally, the pistol ribozyme angles for the a strategy (in-line attack for O2′-P-O5′) are provided in Figure S3D and have an average of 151 ± 30°, which is near ideal for chemistry. This observation gives confidence that the above analysis is representative of an active site that is near optimally arranged for catalysis.
In sum, survey of the catalytic strategies for the pistol ribozyme reveals characteristics of two different ribozymes: Activation of the nucleophile is glmS-like, with N1G for γ and two N2Gs for γ′ and stabilization of charge build-up and release of the 2′OH from NBOs is hammerhead-like, with a metal ion at the pro-RP and pro-SP positions driving β and γ′′ catalysis.
DISCUSSION
While it is common to speculate on mechanisms of ribozymes by inspection of a given structure, the pipeline introduced here provides an objective means of identifying new and conserved catalytic strategies that can be tested by wet bench experiments and theoretical calculations. In this study, we described six catalytic devices, each of which is conserved across a variety of ribozymes. These include the four previously presented by Breaker and colleagues (α, β, γ, and δ)9–10 as well as the newly described γ′ and γ′′ (Figure 9)25–26. This study reveals that γ′ and γ′′ act in conjunction with α, β, γ, and δ to activate the 2′OH nucleophile and prepare it for base abstraction and subsequent attack on the scissile phosphate.
Figure 9.
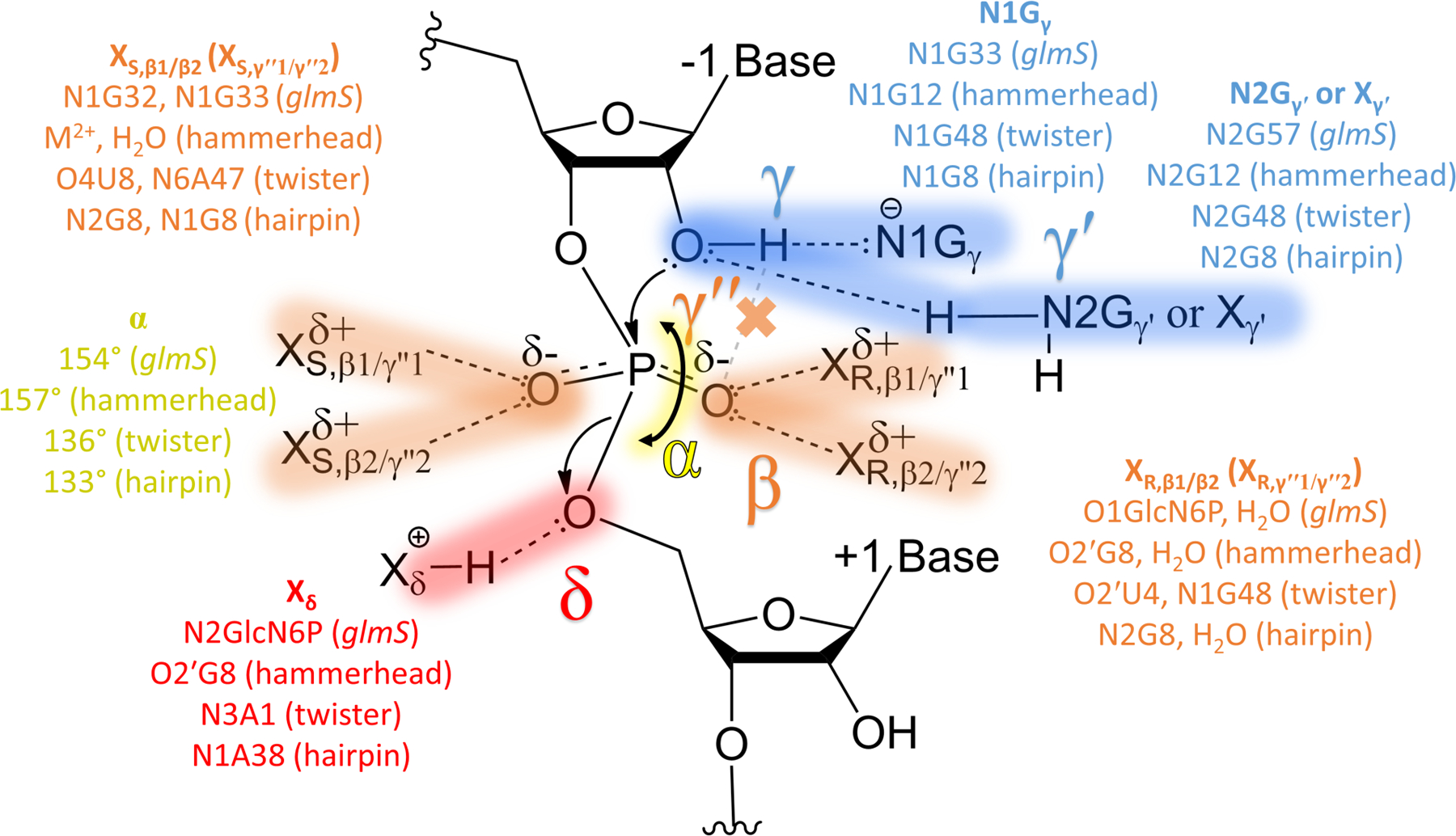
Unified mechanistic model depicting catalytic strategies of small, self-cleaving ribozymes. Contacts or angles for the four previously recognized catalytic strategies (α, γ, β, and δ) in each of the small self-cleaving ribozyme classes studied are provided (N1Gγ, XR,β1/β2 or XS,β1/β2, and Xδ, respectively), and contacts for new identified catalytic strategies (γ′ and γ′′) are noted (N2Gγ′ or Xγ′ and XR,γ′′1/γ′′2 or XS,γ′′1/γ′′2, respectively).
A phenomenon that plays a role in chemical catalysis is a high effective concentration of functional groups that results from positioning of side chains.75 An example of this found herein is the amidine functionality in ribozymes, which consists of an endocyclic nitrogen atom bonded to a carbon that has an exocyclic amine bonded to it. Amidines provide the potential for stabilization and positioning of the nucleobases by way of multiple contacts. The exocyclic amine can donate two hydrogen bonds, which could be important for tuning the pKa of the O2′ (γ′) (see below), stabilizing charge build up at the NBOs, freeing up the 2′OH (β and γ′′), and positioning atoms. The amidine functionality exists in adenine, guanine, and cytosine and plays important roles in the six catalytic strategies. Absence of the amidine in uracil provides a possible explanation for its absence in the active site of nearly all ribozymes. Guanine is found at the general base position likely because it has a high pKa for removing the proton from the basic 2′OH (see below). It is notable that many different types of nucleosides and metal ions contribute to the d and b strategies, supporting uniqueness of nucleobase identity at the general base position.
A central problem in ribozymes, as well as in protein enzymes that act on nucleic acids (e.g. polymerases, ligases, and nucleases), is activation of the nucleophile. The 2′OH, as a secondary alcohol, has a pKa of ~15 in the unperturbed state76; moreover, this value is raised even further from neutrality to ~20 if the 2′OH donates a hydrogen bond to an acceptor such as an NBO in glmS31. The pKa of a guanine is typically ~9, making it very unfavorable to deprotonate the 2′OH, and if the pKa of the guanine shifts toward neutrality to favor the functional deprotonated form of the N1 at biological pH, the problem is further exacerbated: a pKa of ~7 of G needs to accept from a donor with a pKa of ~20. One can envision two methods to better match the pKas of the O2′ and the general base and thus overcome this problem: (1) lowering the pKa of the 2′OH, by preventing its donation and promoting its acceptance of hydrogen bonds77 and (2) raising the pKa of the putative general base. Our findings herein suggest that method (1) is operative when guanine is the general base as inferred through the presence of both the γ′ and γ′′ strategies, although the HDV19–20 and twister sister21–23 ribozymes may also use method (2) in which the putative general base is a Mg2+-coordinated inner-sphere water molecule that has a pKa of ~11.419. The γ′′ strategy uses ribozyme functionalities to compete with the 2′OH for hydrogen bonding to the NBO, effectively lowering the pKa of the O2′ to its unperturbed value of ~15, while the γ′ strategy uses 1–2 guanine amine(s) to increase bond order at the O2′ and acidify it, possibly by another 5 units, assuming a similar contribution of hydrogen bonding. By bringing the pKa of the O2′ closer to neutrality, catalysis under biological conditions of pH ~7 would be greatly facilitated. Further experiments and calculations will be needed to refine and quantify these models. Now that two new catalytic strategies have been identified, future experimental and theoretical studies will be needed to evaluate the catalytic contribution and additivity of all six strategies to the overall cleavage rate of ribozymes. It will be especially intriguing to see if the strategies proposed here extend to large ribozymes and even protein enzymes that act on nucleic acids.
Supplementary Material
ACKNOWLEDGMENTS
We thank Giovanni Bussi for helpful conversations that helped motivate the comparative analysis methods developed in this manuscript. We also thank Darrin York for sharing coordinates of computational studies on the twister ribozyme and Joe Piccirilli for advice on the VS ribozyme. This work was supported by NSF grant CHE-1213667 and REU supplement, NASA grant 80NSSC17K0034-EXO, and NSF REU grant CHE-1659679.
Footnotes
ADDITIONAL INFORMATION
The Supporting Information files are available free of charge on the ACS Publications website.
Supporting Information 1: Supporting methods describing computational plugins and scripts, contact angle calculations (Figure S1), contacts for β for hammerhead structures 5EAQ and 5EAO (Figure S2), pistol scissile phosphate contacts and angles (Figure S3), and crystal structures studied herein (Table S1).
Supporting Information 2: Plugin and script codes.
REFERENCES
- 1.Cech TR; Zaug AJ; Grabowski PJ, Cell 1981, 27, 487–496. [DOI] [PubMed] [Google Scholar]
- 2.Guerrier-Takada C; Gardiner K; Marsh T; Pace N; Altman S, Cell 1983, 35, 849–857. [DOI] [PubMed] [Google Scholar]
- 3.Ward WL; Plakos K; DeRose VJ, Chem. Rev 2014, 114, 4318–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jimenez RM; Polanco JA; Lupták A, Trends Biochem. Sci 2015, 40, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldstein PA; Buzayan JM, Bruening G Gene 1989, 82, 53–61. [DOI] [PubMed] [Google Scholar]
- 6.Winkler WC; Nahvi A; Roth A; Collins JA; Breaker RR, Nature 2004, 428, 281–286. [DOI] [PubMed] [Google Scholar]
- 7.Perreault J; Weinberg Z; Roth A; Popescu O; Chartrand P; Ferbeyre G; Breaker RR, PLOS Comput. Biol 2011, 7, e1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roth A; Weinberg Z; Chen AGY; Kim PB; Ames TD; Breaker RR, Nat. Chem. Biol 2014, 10, 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breaker RR; Emilsson GM; Lazarev D; Nakamura S; Puskarz IJ; Roth A; Sudarsan N, RNA 2003, 9, 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emilsson GM; Nakamura S; Roth A; Breaker RR, RNA 2003, 9, 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganguly A; Thaplyal P; Rosta E; Bevilacqua PC; Hammes-Schiffer S, J. Am. Chem. Soc 2014, 136, 1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cochrane JC; Lipchock SV; Strobel SA, Chem. Biol 2007, 14, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mir A; Chen J; Robinson K; Lendy E; Goodman J; Neau D; Golden BL, Biochemistry 2015, 54, 6369–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren A; Košutić M; Rajashankar KR; Frener M; Santner T; Westhof E; Micura R; Patel DJ, Nat. Commun 2014, 5, 5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alam S; Grum-Tokars V; Krucinska J; Kundracik ML; Wedekind JE, Biochemistry 2005, 44, 14396–14408. [DOI] [PubMed] [Google Scholar]
- 16.Ren A; Vusurovic N; Gebetsberger J; Gao P; Juen M; Kreutz C; Micura R; Patel DJ, Nat. Chem. Biol 2016, 12, 702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eiler D; Wang J; Steitz TA, Proc. Natl. Acad. Sci. U.S.A 2014, 111, 13028–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martick M; Scott WG, Cell 2006, 126, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakano S; Chadalavada DM; Bevilacqua PC, Science 2000, 287, 1493–1497. [DOI] [PubMed] [Google Scholar]
- 20.Golden BL; Hammes-Schiffer S; Carey PR; Bevilacqua PC, An Integrated Picture of HDV Ribozyme Catalysis In Biophysics of RNA Folding, Russell R, Ed. Springer: New York: 2013; pp. 236. [Google Scholar]
- 21.Liu Y; Wilson TJ; Lilley DMJ, Nat. Chem. Biol 2017, 13, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaines CS; York DM, Angew. Chem. Int. Ed. Engl 2017, 56, 13392–13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng L; Mairhofer E; Teplova M; Zhang Y; Ma J; Patel DJ; Micura R; Ren A, Nat. Commun 2017, 8, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rupert PB; Massey AP; Sigurdsson ST; Ferré-D’Amaré AR, Science 2002, 298, 1421. [DOI] [PubMed] [Google Scholar]
- 25.Bingaman JL; Zhang S; Stevens DR; Yennawar NH; Hammes-Schiffer S; Bevilacqua PC, Nat. Chem. Biol 2017, 13, 439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bingaman JL; Gonzalez IY; Wang B; Bevilacqua PC, Biochemistry 2017, 56, 4313–4317. [DOI] [PubMed] [Google Scholar]
- 27.Gaines CS; York DM, J. Am. Chem. Soc 2016, 138, 3058–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein DJ; Been MD; Ferré-D’Amaré AR, J. Am. Chem. Soc 2007, 129, 14858–14859. [DOI] [PubMed] [Google Scholar]
- 29.Viladoms J; Scott LG; Fedor MJ, J. Am. Chem. Soc 2011, 133, 18388–18396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banáš P; Walter NG; Šponer J. i.; Otyepka M, J. Phys. Chem. B 2010, 114, 8701–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang S; Ganguly A; Goyal P; Bingaman JL; Bevilacqua PC; Hammes-Schiffer S, J. Am. Chem. Soc 2015, 137, 784–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soukup JK, The structural and functional uniqueness of the glmS ribozyme In Progress in Molecular Biology and Translational Science, Soukup GA, Ed. Academic Press: Oxford: 2013; Vol. 120, pp. 236. [DOI] [PubMed] [Google Scholar]
- 33.Canny MD; Jucker FM; Kellogg E; Khvorova A; Jayasena SD; Pardi A, J. Am. Chem. Soc 2004, 126, 10848–10849. [DOI] [PubMed] [Google Scholar]
- 34.Frankel EA; Strulson CA; Keating CD; Bevilacqua PC, Biochemistry 2017, 56, 2537–2548. [DOI] [PubMed] [Google Scholar]
- 35.Han J; Burke JM, Biochemistry 2005, 44, 7864–7870. [DOI] [PubMed] [Google Scholar]
- 36.Heckman JE; Lambert D; Burke JM, Biochemistry 2005, 44, 4148–4156. [DOI] [PubMed] [Google Scholar]
- 37.Thomas JM; Perrin DM, J. Am. Chem. Soc 2008, 130, 15467–15475. [DOI] [PubMed] [Google Scholar]
- 38.Chen H; Giese TJ; Golden BL; York DM, Biochemistry 2017, 56, 2985–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y; Wilson TJ; McPhee SA; Lilley DMJ, Nat. Chem. Biol 2014, 10, 739–744. [DOI] [PubMed] [Google Scholar]
- 40.Bevilacqua PC, Biochemistry 2003, 42, 2259–2265. [DOI] [PubMed] [Google Scholar]
- 41.Lebruska LL; Kuzmine II; Fedor MJ, Chem. Biol 2002, 9, 465–473. [DOI] [PubMed] [Google Scholar]
- 42.Kuzmin YI; Da Costa CP; Fedor MJ, J. Mol. Biol 2004, 340, 233–251. [DOI] [PubMed] [Google Scholar]
- 43.Wilson TJ; Ouellet J; Zhao Z-Y; Harusawa S; Araki L; Kurihara T; Lilley DMJ, RNA 2006, 12, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson TJ; Zhao Z-Y; Maxwell K; Kontogiannis L; Lilley DMJ, Biochemistry 2001, 40, 2291–2302. [DOI] [PubMed] [Google Scholar]
- 45.Pinard R; Hampel KJ; Heckman JE; Lambert D; Chan PA; Major F; Burke JM, EMBO J 2001, 20, 6434–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kath-Schorr S; Wilson TJ; Li N-S; Lu J; Piccirilli JA; Lilley DMJ, J. Am. Chem. Soc 2012, 134, 16717–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hougland JL; Kravchuk AV; Herschlag D; Piccirilli JA, PLOS Biol. 2005, 3, e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herschlag D, J. Am. Chem. Soc 1994, 116, 11631–11635. [Google Scholar]
- 49.Breslow R; Chapman WH, Proc. Natl. Acad. Sci. U.S.A 1996, 93, 10018–10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soukup GA, Nucleic Acids Res 2006, 34, 968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim J; Grove BC; Roth A; Breaker RR, Angew. Chem. Int. Ed. Engl 2006, 45, 6689–6693. [DOI] [PubMed] [Google Scholar]
- 52.Wang S; Karbstein K; Peracchi A; Beigelman L; Herschlag D, Biochemistry 1999, 38, 14363–14378. [DOI] [PubMed] [Google Scholar]
- 53.Mir A; Golden BL, Biochemistry 2016, 55, 633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grasby JA; Mersmann K; Singh M; Gait MJ, Biochemistry 1995, 34, 4068–4076. [DOI] [PubMed] [Google Scholar]
- 55.Jansen JA; McCarthy TJ; Soukup GA; Soukup JK, Nat. Struct. Mol. Biol 2006, 13, 517–523. [DOI] [PubMed] [Google Scholar]
- 56.Hampel KJ; Tinsley MM, Biochemistry 2006, 45, 7861–7871. [DOI] [PubMed] [Google Scholar]
- 57.McCarthy TJ; Plog MA; Floy SA; Jansen JA; Soukup JK; Soukup GA, Chem. Biol 2005, 12, 1221–1226. [DOI] [PubMed] [Google Scholar]
- 58.Xin Y; Hamelberg D, RNA 2010, 16, 2455–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis JH; Dunican BF; Strobel SA, Biochemistry 2011, 50, 7236–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gong B; Klein DJ; Ferré-D’Amaré AR; Carey PR, J. Am. Chem. Soc 2011, 133, 14188–14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viladoms J; Fedor MJ, J. Am. Chem. Soc 2012, 134, 19043–19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas JM; Perrin DM, J. Am. Chem. Soc 2009, 131, 1135–1143. [DOI] [PubMed] [Google Scholar]
- 63.Lee T-S; López CS; Giambaşu GM; Martick M; Scott WG; York DM, J. Am. Chem. Soc 2008, 130, 3053–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Košutić M; Neuner S; Ren A; Flür S; Wunderlich C; Mairhofer E; Vušurović N; Seikowski J; Breuker K; Höbartner C; Patel DJ; Kreutz C; Micura R, Angew. Chem. Int. Ed. Engl 2015, 54, 15128–15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson TJ; Liu Y; Domnick C; Kath-Schorr S; Lilley DMJ, J. Am. Chem. Soc 2016, 138, 6151–6162. [DOI] [PubMed] [Google Scholar]
- 66.Ucisik MN; Bevilacqua PC; Hammes-Schiffer S, Biochemistry 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuzmin YI; Da Costa CP; Cottrell JW; Fedor MJ, J. Mol. Biol 2005, 349, 989–1010. [DOI] [PubMed] [Google Scholar]
- 68.Lopez X; Dejaegere A; Leclerc F; York DM; Karplus M, J. Phys. Chem. B 2006, 110, 11525–11539. [DOI] [PubMed] [Google Scholar]
- 69.Thaplyal P; Ganguly A; Hammes-Schiffer S; Bevilacqua PC, Biochemistry 2015, 54, 2160–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nguyen LA; Wang J; Steitz TA, Proc. Natl. Acad. Sci. U.S.A 2017, 114, 1021–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harris KA; Lünse CE; Li S; Brewer KI; Breaker RR, RNA 2015, 21, 1852–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weinberg Z; Kim PB; Chen TH; Li S; Harris KA; Lunse CE; Breaker RR, Nat. Chem. Biol 2015, 11, 606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Rear JL; Wang S; Feig AL; Beigelman L; Uhlenbeck OC; Herschlag D, RNA 2001, 7, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neuner S; Falschlunger C; Fuchs E; Himmelstoss M; Ren A; Patel D; Micura R, Angew. Chem. Int. Ed. Engl doi: 10.1002/anie.201708679. [DOI] [PubMed] [Google Scholar]
- 75.Fersht A, Enzyme Structure and Mechanism. Second ed.; W. H. Freeman and Company: New York, 1985, pp. 475. [Google Scholar]
- 76.Lyne PD; Karplus M, J. Am. Chem. Soc 2000, 122, 166–167. [Google Scholar]
- 77.Legault P; Pardi A, J. Am. Chem. Soc 1997, 119, 6621–6628. [Google Scholar]
- 78.DeLano WL The PyMOL Molecular Graphics System, DeLano Scientific: San Carlos, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.