

Abstract
Manipulation of DNA by CRISPR-Cas enzymes requires the recognition of a protospacer adjacent motif (PAM), limiting target site recognition to a subset of sequences. To remove this constraint, we engineered variants of Streptococcus pyogenes Cas9 (SpCas9) to eliminate the NGG PAM requirement. We developed a variant named SpG capable of targeting an expanded set of NGN PAMs, and further optimized this enzyme to develop a near-PAMless SpCas9 variant named SpRY (NRN>NYN PAMs). SpRY nuclease and base-editor variants can target almost all PAMs, exhibiting robust activities on a wide range of sites with NRN PAMs in human cells and lower but substantial activity on those with NYN PAMs. Using SpG and SpRY, we generated previously inaccessible disease-relevant genetic variants, supporting the utility of high-resolution targeting across genome editing applications.
One Sentence Summary:
Engineered SpCas9 variants nearly eliminate the PAM requirement for DNA-targeting CRISPR enzymes, enabling precision nuclease and base editing applications.
The requirement for DNA-targeting CRISPR-Cas enzymes to recognize a short sequence motif adjacent to target sites in foreign DNA is a critical step for CRISPR systems to distinguish self from non-self(1, 2). For genome editing applications, however, the necessity of protospacer-adjacent motif(3-6) (PAM) recognition by Cas9 and Cas12a proteins constrains targeting and affects editing efficiency and flexibility. The prototypical Cas9 from Streptococcus pyogenes (SpCas9) naturally recognizes target sites with NGG PAMs(5, 7, 8), making it one of the most targetable CRISPR enzymes characterized to-date. While other naturally occurring Cas orthologs can in principle expand targeting by recognizing divergent non-canonical PAMs, the vast majority of Cas9 and Cas12a enzymes(9-12) require extended motifs that limit their utility for genome editing. Thus, the PAM requirement prevents the accurate positioning of CRISPR target sites and is a major barrier for genome editing applications that command high resolution target site positioning (e.g. targeting small genetic elements, base editing, generating efficient HDR-mediated alterations, performing tiling screens, etc.(13-19)).
One method to improve the targeting range of genome editing technologies is to purposefully engineer CRISPR enzymes that can target previously inaccessible PAMs. SpCas9 primarily recognizes its optimal NGG PAM by direct molecular readout of the guanine DNA bases via the amino acid side chains of R1333 and R1335(20) (Figs. 1A, 1B and S1A). Modification of either arginine alone ablates SpCas9 nuclease activity against sites with NGG, NAG, or NGA PAMs(8, 20), necessitating the use of molecular evolution to alter PAM preference by mutation of other amino acids in the PAM interacting (PI) domain. Several protein engineering strategies have been pursued towards expanding targeting with SpCas9, including using directed evolution or structure-guided engineering to develop variants with altered PAM profiles(8, 21) (e.g. SpCas9-VQR, VRQR, and VRER) or relaxed PAM preferences(22, 23) (e.g. SpCas9-NG and xCas9). While these variants expand the potential targeting space of SpCas9, target sites encoding the majority of non-canonical PAMs still remain inaccessible for genome editing.
Fig. 1. Engineering and characterization of SpCas9 variants capable of targeting NGN PAMs.
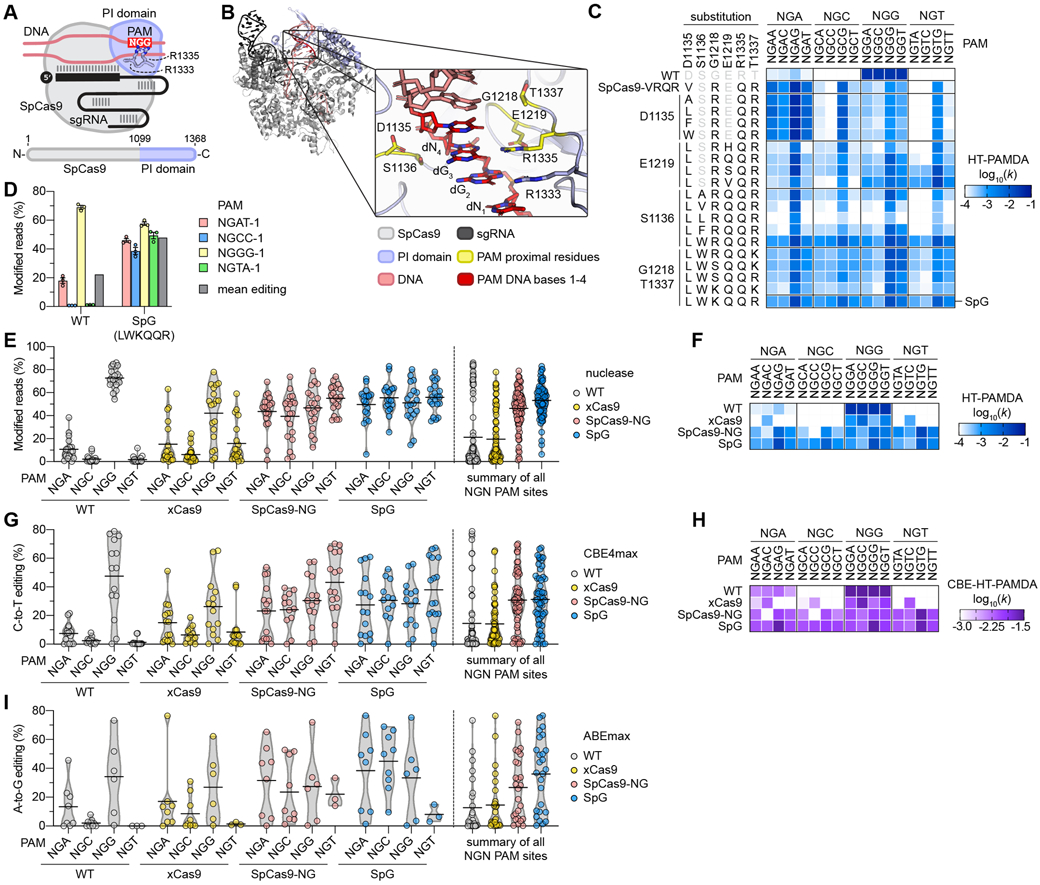
(A) Schematic of SpCas9, highlighting the PAM-interacting (PI) domain along with R1333 and R1335 that make base-specific contacts to the guanines of the NGG PAM. (B) Rendering of a crystal structure of SpCas9 with amino acid side chains proximal to the second guanine of the NGG PAM shown in yellow. In the zoomed image the non-target strand (NTS) is hidden for clarity. Image generated from PDB ID 4UN3(20). (C) HT-PAMDA characterization of wild-type (WT) SpCas9 and engineered variants to illustrate their NGNN PAM preferences. The log10 rate constants (k) are the mean of at least two replicates against two distinct spacer sequences (see also Figs. S2A-C, E). (D) Modification of endogenous sites in human cells bearing a canonical and noncanonical PAMs with WT SpCas9 and SpG. Editing assessed by targeted sequencing; mean, s.e.m., and individual data points shown for n = 3. (E) Mean nuclease activity plots for WT, xCas9(23), SpCas9-NG(22), and SpG on 78 sites with NGN PAMs in human cells. The black line represents the mean of 19-20 sites for each PAM class (see also Fig. S5A), and the grey outline is a violin plot. (F) HT-PAMDA characterization of WT, xCas9, SpCas9-NG, and SpG to illustrate their NGNN PAM preferences. The log10 rate constants (k) are the mean of at least two replicates against two distinct spacer sequences. (G) Mean C-to-T editing plots for WT, xCas9, SpCas9-NG, and SpG cytosine base editors (CBEs) on 57 cytosines within the editing windows (positions 3 through 9) of 20 target sites harboring NGN PAMs in human cells. The black line represents the mean of 12-16 cytosines for each PAM class (see also Fig. S6A), and the grey outline is a violin plot. (H) CBE-HT-PAMDA data for WT, xCas9, SpCas9-NG, and SpG to illustrate their NGNN PAM preferences. The log10 rate constants (k) are single replicates against one spacer sequence (see also Figs. S6C, D). (I) Mean A-to-G editing plots for WT, xCas9, SpCas9-NG, and SpG adenine base editors (ABEs) on 24 adenines within the editing windows (positions 5 through 7) of 21 target sites harboring NGN PAMs in human cells. The black line represents the mean of 3-9 adenines for each PAM class (see also Fig. S7A), and the grey outline is a violin plot.
Here we utilized structure-guided engineering to nearly completely relax the PAM requirement of SpCas9. This approach enabled the generation of a highly enzymatically active NGN PAM variant (named SpG), and subsequent optimization of SpG led to a variant capable of editing nearly all PAMs (named SpRY). We demonstrate that SpG and SpRY improve editing resolution and offer new genome editing capabilities. More broadly, the molecular strategy described herein should in principle be extensible to a wide diversity of Cas orthologs, paving a path towards the development of a suite of editing technologies no longer constrained by their inherent targeting limitations.
Results:
Structure-guided mutagenesis to relax SpCas9 PAM preference
Towards eliminating the PAM requirement of SpCas9, we first developed a variant capable of recognizing a reduced NGN PAM compared to the canonical NGG sequence. Our previous efforts to alter SpCas9 PAM preference illuminated several PAM-proximal residues important for PAM recognition(8) (Fig. 1B), observations supported by structural studies(20, 24, 25) (Figs. S1A-E and (26)). Characterizations of SpCas9-VQR (harboring D1135V/R1335Q/T1337R substitutions) and the derivative SpCas9-VRQR variant (that additionally encodes G1218R) suggests that these enzymes target an expanded number of non-canonical PAMs including those with variable bases in the 3rd position of the PAM (NGAN>NGNG)(8, 21). We therefore hypothesized that R1335Q-harboring variants with other PI domain substitutions could recognize an expanded number of PAMs (Figs. S1F-1I). Thus, we utilized SpCas9-VRQR as a molecular scaffold to further relax SpCas9 PAM preference.
To more thoroughly investigate the impacts of amino acid substitutions in PI domain residues, we developed a high-throughput PAM determination assay (HT-PAMDA) to comprehensively profile the PAM preferences of a large number of SpCas9 variants (Figs. S2A-2D and (26)). HT-PAMDA accurately replicated the PAM profiles of wild-type (WT) SpCas9 and other previously described variants (Figs. 1C and S2D). To engineer an SpCas9 variant capable of more relaxed targeting, we utilized HT-PAMDA to sequentially determine the contributions of substitutions at five PI-critical positions D1135, S1136, G1218, E1219, and T1337 in the context of SpCas9-VRQR (Fig. S2E and (26)). We identified several variants bearing combinations of rational substitutions at these five important residues that exhibited more balanced tolerances for any nucleotide at the 3rd and 4th PAM positions (Figs. 1C and S2E). One variant bearing D1135L/S1136W/G1218K/E1219Q/R1335Q/T1337R substitutions, henceforth named SpG, exhibited the most even targeting of NGA, NGC, NGG, and NGT PAMs (Figs. 1C and S2E).
We then compared the activities of WT SpCas9 to SpG and nearly all intermediate variants in human HEK 293T cells to corroborate our HT-PAMDA findings. Using an optimal nuclear localization signal (NLS)(27) (Fig. S3) and non-saturating nuclease expression conditions in HEK 293T cells, we examined the human cell editing activities of this large collection of variants on four sites with NGA, NGC, NGG, and NGT PAMs (Figs. S4A, S4B, and (26)). These experiments revealed high-activity editing on the four NGN PAM sites with SpG (Figs. 1D and S4B), results that were consistent with the PAM preference of SpG characterized using HT-PAMDA (Figs. 1C and S2F). We then sought to bolster the activity of SpG through the addition of non-specific contacts mediated by L1111R and A1322R substitutions, mutations that are necessary for the NGN PAM tolerance of SpCas9-NG(22) (Figs. S4C and S4D). However, the L1111R and A1322R substitutions were detrimental to the human cell editing activities of SpG, albeit without alteration of PAM preference (Figs. S4E-S4F and S2F-S2G, respectively, and see (26)).
SpG activities as a nuclease, CBE, and ABE
Given the broad compatibility of SpG with NGN PAMs as determined by HT-PAMDA but across only sixteen target sites in HEK 293T cells, we more thoroughly compared its nuclease activity in human cells against WT SpCas9, xCas9(3.7), and SpCas9-NG. We assessed the editing activities of SpG and these three nucleases on 78 sites bearing NGNN PAM sequences that encompassed an approximately even distribution of nucleotide identities in the 3rd and 4th positions of the PAM (Fig. S5A and Table S1). Our assessment recapitulated the PAM preference of WT SpCas9(7, 8), with a mean editing activity of 72.8% on sites with NGG PAMs and a reduced 4.7% mean editing across the remaining NGH sites (where H is A, C, or T; Fig. 1E). SpG exhibited the highest mean editing activities across all NGN PAM sites, averaging 51.2% on sites with NGG PAMs and 53.7% on sites with NGH PAMs (Fig. 1E). Finally, of the two variants reported to recognize sites with NGN PAMs, xCas9 displayed more modest editing (42.2% on NGG and 12.5% across NGH), while SpCas9-NG editing activities were even across NGN PAM sites (46.9% on NGG and 46.0% across NGH) but lower compared to SpG (Fig. 1E).
To better understand the PAM requirements of each of the NGN-PAM variants, we utilized HT-PAMDA to profile SpG, xCas9, and SpCas9-NG (Fig. 1F and Fig. S2F). These experiments demonstrated that SpG exhibited the most even and robust targeting of all NGN PAMs (ranking SpG > SpCas9-NG > xCas9; Fig. 1F), consistent with the results of our 78-site human cell experiment (Fig. 1E). Closer inspection of our HT-PAMDA and human cell data did not reveal substantial 1st PAM, 4th PAM, or 1st spacer position preferences for WT SpCas9, SpG, or SpCas9-NG (Figs. S5B-S5D and (26)). This analysis also attributed the decreased activities observed with xCas9 to a preference for a C in the 4th position of the PAM, which likely makes targeting of sites with NGND PAMs (where D is A, G, or T) less efficient (Fig. S5E; for a more complete analysis of xCas9 PAM preference, see (26)). For the four nucleases, HT-PAMDA values for each NGNN PAM class correlated with mean editing activities in HEK 293T cells (Fig. S5F). Together, these results indicate that SpG is an efficient and broadly targeting nuclease across sites bearing NGN PAMs.
Given the ubiquitous use of base editor (BE) technologies to mediate single nucleotide substitutions in various organisms(17, 18, 28), we investigated whether the improved activities of SpG could enhance BE activities across sites with NGN PAMs. We compared C-to-T editing with WT SpCas9, xCas9, SpCas9-NG, and SpG BE4max cytosine base editor(29) (CBE) constructs across 22 endogenous sites in human cells bearing NGNN PAMs (Fig. S6A). We observed that whereas WT- and xCas9-CBE exhibited mean C-to-T editing efficiencies above 15% only on sites with NGG PAMs, both SpG- and SpCas9-NG-CBE were capable of mean C-to-T editing above 23% across NGN sites (Fig. 1G) and displayed typical CBE substrate preferences (Fig. S6B). To ensure that the CBE versions of the PAM variants harbored the same PAM profiles as the nucleases, we developed a modified CBE high-throughput PAM determination assay (CBE-HT-PAMDA) (Fig. S6C and (26)). The PAM compatibilities of the CBEs were largely consistent with the preferences of the nucleases (compare Figs. 1H and 1F, respectively, and see Figs. S6D and S6E).
Beyond C-to-T editing, adenine base editor (ABE) constructs have also been developed that mediate A-to-G edits(18). Thus, we also compared the A-to-G editing potencies of WT SpCas9, xCas9, SpCas9-NG, and SpG in the ABEmax architecture(29) across 21 endogenous sites harboring NGNN PAMs (Fig. S7A). Similar to our observations for CBEs, WT- and xCas9-ABE could only efficiently perform A-to-G edits on target sites with NGG PAMs (Fig. 1I). However, both SpG- and SpCas9-NG-ABE efficiently edited target sites with NGNN PAMs, where SpG-ABE exhibited the most robust activity across all NGNN sites (Fig. 1I) with typical ABE substrate preferences (Fig. S7B).
Collectively, these results demonstrate that SpCas9 PAM preference can be relaxed to a single NGN nucleotide motif by designing a more tolerant PI domain, and that the SpG variant developed through this strategy exhibits robust nuclease, CBE, and ABE activities across NGN PAMs.
Engineering SpCas9 variants capable of targeting NRN PAMs
Notwithstanding the efficient modification of sites with NGN PAMs using SpG, many genomic regions remain inaccessible to genome editing. Because we observed efficient modification of sites bearing NGN PAMs with SpG, we speculated that SpG could be utilized as a molecular scaffold upon which to further relax PAM specificity. To alter recognition of the 2nd position of the PAM, we hypothesized that substitution of R1333 to glutamine might enable access to sites harboring NAN PAMs by forming a base specific contact with the adenine base in the second position of the PAM(8, 20, 24) (Fig. 2A). However, our initial tests of SpG(R1333Q) nearly abolished activity in human cells against four sites bearing NRN PAMs (where R is A or G) (Fig. 2B), revealing that the R1333Q alone was insufficient to enable highly active targeting of NAN PAMs (consistent with observations for WT SpCas9(8, 20)). Interestingly, contrary to our previous finding that L1111R and A1322R substitutions negatively impacted SpG activity (Fig. S4E and S4F), we observed that these non-specific DNA contacts were able to rescue some activity of SpG(R1333Q) across the four sites bearing NRN PAMs in human cells (Fig. 2B). Concurrent HT-PAMDA experiments to analyze the same variants corroborated a general relaxation of PAM specificity against NRN PAMs but with a lower overall activity (Fig. 2C).
Fig. 2. Engineering and characterization of SpCas9 variants capable of targeting NRN PAMs.
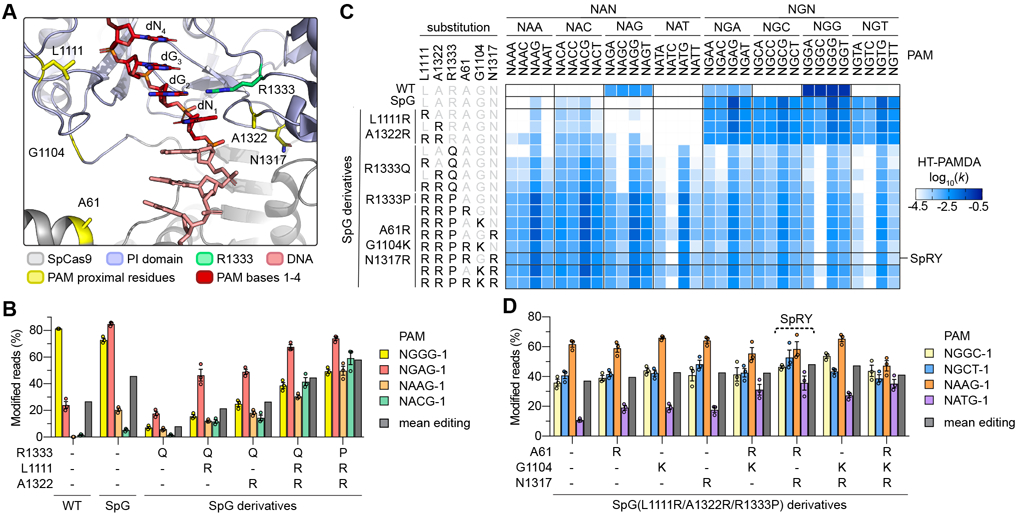
(A) Crystal structure of SpCas9 to illustrate amino acid side chains of R1333 and selected PAM-proximal residues. The non-target strand (NTS) is hidden for clarity. Image generated from PDB ID 4UN3(20). (B) Modification of endogenous sites in human cells bearing different NRN PAMs with WT SpCas9, SpG, and SpG derivatives. Editing assessed by targeted sequencing; mean, s.e.m., and individual data points shown for n = 3. (C) HT-PAMDA characterizations of WT SpCas9, SpG, and SpG derivatives to illustrate their NRNN PAM preferences. The log10 rate constants (k) are the mean of at least two replicates against two distinct spacer sequences (see also Figs. S2A-C). (D) Modification of endogenous sites in human cells bearing different NRN PAMs SpG(L1111R/A1322R/R1333P) and derivatives bearing additional substitutions. See Fig. S8C for all variants tested. Editing assessed by targeted sequencing; mean, s.e.m., and individual data points shown for n = 3.
Next, to determine whether the R1333Q substitution was the most permissive for recognition of an expanded number of PAMs, we utilized HT-PAMDA to investigate whether variants harboring other amino acid substitutions at residue 1333 might be more amenable to highly active and broad targeting of NRN PAMs. Systematic evaluation of SpG(L1111R/A1322R) variants harboring all 20 possible amino acids at residue 1333 revealed that the range substitutions at this position cause different 2nd PAM position preferences and overall levels of activity (Fig. S8A). Surprisingly, variants bearing R1333 substitutions to alanine, cysteine, or proline conferred the most efficient collective targeting of NRN PAMs. Experiments in HEK 293T cells against the same four sites harboring NRN PAMs demonstrated that one SpG(L1111R/A1322R) variant that also harbored the R1333P substitution exhibited greater activity on NRN PAMs compared to the precursor R1333Q-containing variants (Fig. 2B). HT-PAMDA experiments confirmed these observations (Fig. 2C).
Given that the addition of L1111R and A1322R to SpG-R1333Q improved on-target activity, we determined whether additional analogous substitutions could further enhance editing of sites with NRN PAMs. We utilized crystal structures of SpCas9(20, 24, 25) to identify other positions in the PI domain whose substitution to positively charged residues might be expected to increase activity by forming novel non-specific DNA contacts (Fig. 2A). We utilized HT-PAMDA to determine the single or combinatorial effects of three such substitutions, A61R, G1104K, or N1317R, in the context of SpG(L1111R/A1322R) variants also bearing R1333A, R1333C, or R1333P substitutions (Figs. 2C and S8B). This analysis revealed that different combinations of the three non-specific substitutions were well-tolerated by nearly all variants, and that the NRN PAM-preferences of variants harboring R1333A, C, or P substitutions were similar by HT-PAMDA.
To determine which variant exhibited the highest activities in human cells, we tested this large series of variants against four additional sites bearing NRN PAMs (Fig. S8C). We observed that the SpG(L1111R/A1322R) variant harboring the R1333P substitution and a combination of A61R/N1317R offered the greatest mean editing against NRN PAMs (Fig. 2D). Use of HT-PAMDA to examine the sequential effects of the substitutions encoded by this variant demonstrated a stepwise progression from NGN to NRN PAM preference, and also revealed the surprising finding that this variant may target some NYN PAMs (where Y is C or T; Fig. S8D). Together, our human cell and HT-PAMDA data suggest that the SpG(L1111R/A1322R) derivative containing A61R, N1317R, and R1333P substitutions (henceforth referred to as SpRY, for SpCas9 variant capable of targeting NRN>NYN PAMs) enables targeting of sites with NRN PAMs.
SpRY activities as a nuclease, CBE, and ABE in human cells
Having established the potential of SpRY to widely expand sequence targeting, we more thoroughly assessed its nuclease activities in HEK 293T cells. We compared the on-target editing of WT SpCas9 and SpRY across 64 sites, 32 each harboring NANN and NGNN PAMs (Figs. S9A and S9B, respectively). Consistent with prior reports(7, 8), we observed that WT SpCas9 preferred NGG>NAG>NGA PAMs with negligible targeting of the remaining NRN PAMs (Fig. 3A). In comparison, SpRY was more effective than WT at targeting sites encoding NRN PAMs, except for sites harboring canonical NGG PAMs (Fig. 3A). Across the 32 sites with NGN PAMs, SpRY often exhibited comparable activities to SpG, though SpG remained the most effective NGN PAM variant (Figs. S9B and S9C). Overall, SpRY was capable of efficiently targeting the majority of target sites with NRN PAMs, where the range of activities could not necessarily be explained by PAM preference alone. These results demonstrate the ability to effectively target a range of sites with NAN PAMs using a Cas9 variant.
Fig. 3. Comparison of WT SpCas9 and SpRY nuclease and base editor activities across NNN PAM sites in human cells.
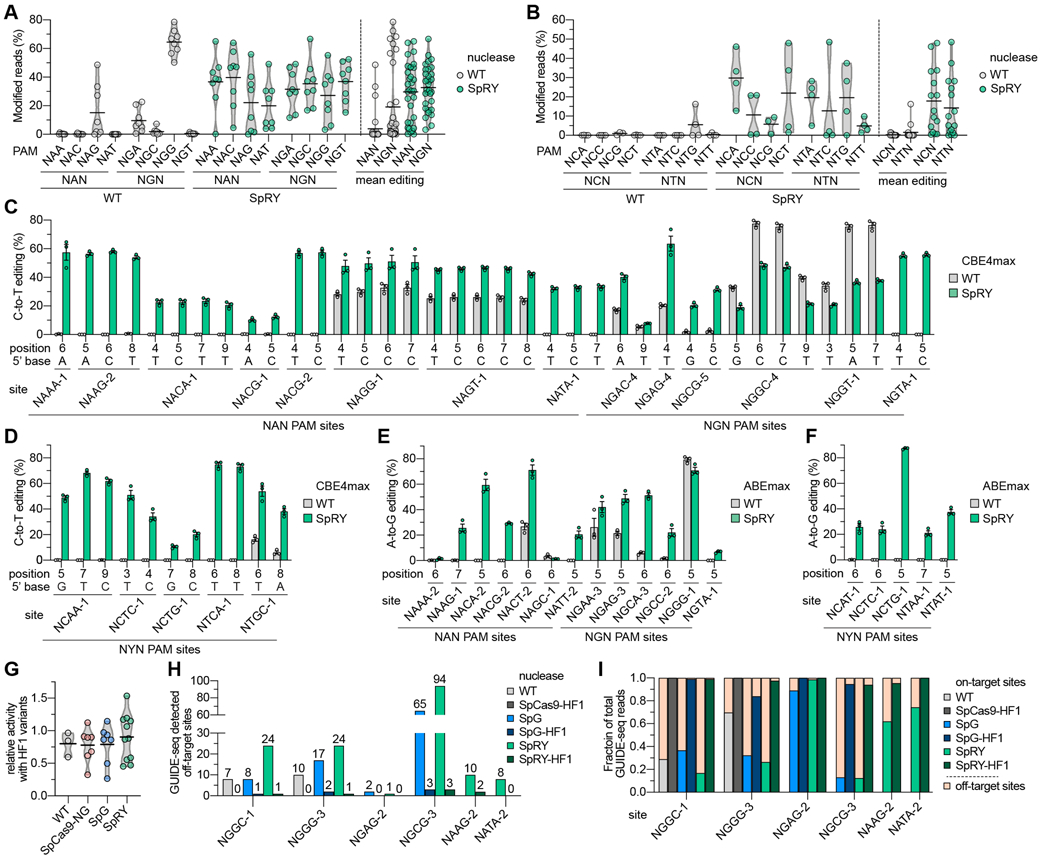
. (A, B) Mean nuclease activity plots for WT SpCas9 and SpRY on 64 sites with NRN PAMs (panel A) and 31 sites with NYN PAMs (panel B) in human cells. The black line represents the mean of 8 or 3-4 sites (panels a and b, respectively) for each PAM of the indicated class (see also Figs. S9A-C), and the grey outline is a violin plot. (C, D) C-to-T base editing of endogenous sites in human cells bearing NRN and NYN PAMs (panels C and D, respectively) with WT SpCas9 and SpRY-CBE4max constructs. Editing of cytosines in the edit window (positions 3 through 9) assessed by targeted sequencing; the five NYN PAM target sites were selected from high-activity sites in panel B; mean, s.e.m., and individual data points shown for n = 3. (E, F) A-to-G base editing of endogenous sites in human cells bearing NRN and NYN PAMs (panels E and F, respectively) with WT SpCas9 and SpRY-ABEmax constructs. Editing of adenines in the edit window assessed by targeted sequencing; the five NYN PAM target sites were selected from high-activity sites in panel B; mean, s.e.m. and individual data points shown for n = 3. For base editing data in panels C-F, see also Table S5. (G) Relative nuclease activity plots for SpCas9-HF1, SpCas9-NG-HF1, SpG-HF1, and SpRY-HF1 compared to their parental variants across 3-10 sites endogenous sites in HEK 293T cells. Mean modification from sites in Fig. S11A shown as dots, with black line representing the mean of those sites, and the grey outline is a violin plot. The HF1 variants additionally encode N497A, R661A, Q695A, and Q926A substitutions(21). (H) Histogram of the number of GUIDE-seq detected off-target sites for SpCas9 variants across sites with NGG, NGN, and NAN PAMs (see Figs. S12A-C, respectively). (I) Fraction of GUIDE-seq reads attributed to the on- and off-target sites for WT SpCas9, SpG, SpRY, and their respective HF1 variants across 2-6 targets (see also Figs. S12A-C and Table S6).
Combined with the observation of modest levels of NYN targeting with SpRY in HT-PAMDA (Fig. S8D), structural analysis of the R1333P substitution in SpRY led us to speculate that R1333P-containing variants might also enable targeting of any base in the 2nd PAM position (including thus far unexamined NYN PAMs; Fig. S9D). We examined the activities of WT SpCas9 and SpRY across 31 sites with NYN PAMs (15 NCNN and 16 NTNN sites; Fig. S9E). Surprisingly, SpRY was able to edit 13 of 31 sites (42%) with NYN PAMs to levels higher than 20% modification, compared to 0 sites with WT SpCas9 (Fig. 3B). While the mean editing activities on sites with NYN PAMs were approximately half of what we observed on sites with NRN PAMs, the activities were greater than the essentially negligible editing with WT SpCas9 (Fig. 3B). The PAM preference of SpRY as determined by HT-PAMDA was generally consistent with the mean nuclease editing levels for each PAM class (Fig. S9F and (26)), although additional experiments are needed to more thoroughly characterize the substrate requirements of SpRY (Figs. S9G-J). Collectively, these results demonstrate the ability to target sites with NRN PAMs and some NYN PAMs using an SpCas9 variant.
Because SpRY enables nuclease targeting of many sites with NNN PAMs in human cells, we examined its compatibility with base editors given their dependence on the availability of PAMs to appropriately position the CBE or ABE edit windows(28). Assessment of SpRY-CBE across 14 sites bearing NRN PAMs revealed mean C-to-T editing of 38.0% across all substrate cytosines (Fig. S10A), with SpRY-CBE achieving greater than 20% modification of at least one cytosine per site for all but one site (Fig. 3C). Comparatively, WT-CBE most efficiently modified sites bearing NGG PAMs, and was also capable of modifying sites bearing NAG and NGA PAMs, albeit at lower efficiency than SpRY (Figs. 3C and S10A). We also assessed the activities of SpRY-CBE on five high-activity NYN PAM sites from our nuclease datasets (see Fig. S9E). For these pre-selected high-activity sites, we observed robust levels of editing compared to negligible editing with WT-CBE (Figs. 3D and S10B). Similar to our observations for SpG-CBE, SpRY-CBE exhibited typical CBE substrate preferences (Fig. S10C).
We then examined the A-to-G editing activities of SpRY-ABE across 13 sites with NRN PAMs, and 5 high-activity sites with NYN PAMs (the latter from Fig. S9E). For the NRN PAM sites, we observed mean A-to-G editing activities of 34.7% with SpRY-ABE on substrate adenines (Fig. S10D) and achieved greater than 20% modification on at least one adenine for 10 of 13 sites (Fig. 3E). With WT-ABE, the most efficient editing was observed on the NGG PAM site and minor editing was detected on 3 sites with non-canonical PAMs (Fig. 3E). Across the five pre-selected high activity sites harboring NYN PAMs, A-to-G editing activities were generally more modest with SpRY but we did observe editing of one adenine to near 90%; no editing with WT ABEmax was observed on any of the five sites (Fig. 3F). Overall, SpRY-ABE exhibited greater mean A-to-G editing compared to WT-ABE across sites containing NRN and NYN PAMs with typical ABE substrate requirements (Figs. S10D-F).
An important consideration for genome editing is the ability to mitigate potential off-target effects. To reduce off-target editing observed with WT SpCas9, we and others previously engineered high-fidelity (HF) variants of SpCas9 with improved genome-wide specificities(21, 30). Since the relaxed PAM tolerances of SpG and SpRY can, in principle, lead to recognition of new off-target sites, we first tested whether our new variants were compatible with the fidelity-enhancing substitutions of SpCas9-HF1(21). Across several target sites bearing different PAMs, we observed that WT, SpCas9-NG, SpG, SpRY, and their HF1 derivatives exhibited comparable levels of on-target modification (Figs. 3G and S11A).
We then performed GUIDE-seq experiments(31) to analyze the genome-wide specificity profiles of these variants. In transfections containing the GUIDE-seq double-stranded oligodeoxynucleotide (dsODN) tag, we also observed similar levels of on-target editing between WT, SpG, SpRY, and their HF1 derivative variants (Figs. S11B-F). Analysis of GUIDE-seq experiments revealed that SpG and SpRY exhibited a somewhat increased propensity for off-target editing compared to WT SpCas9, albeit at similar absolute levels previously reported for WT SpCas9 (21, 31) (Fig. 3H). Nearly all novel off-targets for SpG and SpRY were attributable to the expanded PAM recognition by these variants (Figs. S12A-C). Importantly, the HF1 variants were able to eliminate nearly all off-target editing events (Fig. 3H) and substantially increased the fraction of total editing events observed at the on-target sites (Fig. 3I). These results demonstrate that SpG- and SpRY-HF1 offer improved fidelity for applications that necessitate high specificity.
Expanded targeting of SpG and SpRY enables the generation of protective genetic variants
The requirement of DNA-targeting CRISPR enzymes to recognize a PAM fundamentally limits precision targeting, a constraint that is exacerbated when using base editors whose activities are restricted to short ‘editing windows’ (17, 18, 28)(Fig. 4A). In a proof-of-concept application, we leveraged the expanded targeting of SpG and the near-PAMless qualities of SpRY to generate biologically relevant substitutions implicated to protect individuals against coronary heart disease, type 2 diabetes, osteoporosis, chronic pain, and others(32-38). We first selected two genetic variants (SOST W124X and MSTN IVS1) with nearby NGG PAMs that might appropriately position the CBE edit window (Fig. 4B). We assessed the activities of WT-, SpG-, and SpRY-CBE constructs against target sites with a variety of PAMs, and as expected observed the most robust editing with WT-CBE on sites with NGG PAMs. However, for MSTN IVS1 we also observed potentially deleterious bystander editing of a nearby cytosine with WT-CBE; this collateral edit that was avoided or reduced when using SpG- and SpRY-CBEs constructs targeted to nearby sites bearing an NGC and NAT PAM (Fig. 4B).
Fig. 4. Expanded capabilities of C-to-T base editors with SpG and SpRY to generate protective genetic variants.
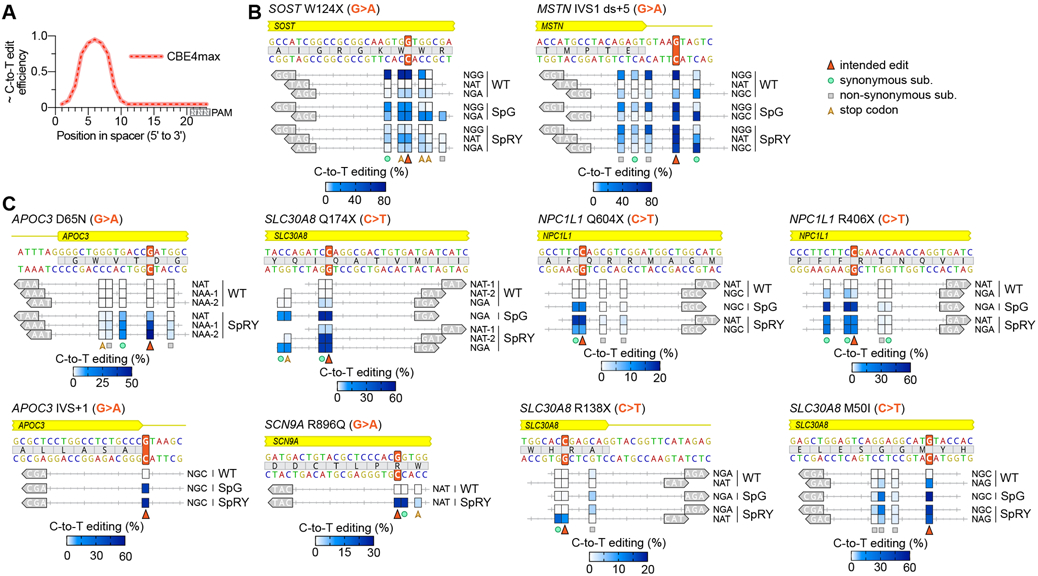
(A) Illustration of the relative C-to-T edit activity of CBE4max constructs depending on the position of the cytosine in the spacer (as reviewed in(28)) (B, C) Comparison of the C-to-T editing activities of WT SpCas9, SpG, and SpRY CBE4max constructs across 22 target sites covering ten previously described protective genetic variants accessible or inaccessible with target sites harboring NGG PAMs (panels B and C, respectively). The intended edit highlighted with an orange arrow; C-to-T editing for cytosines within the spacer that are edited above 1% by any variant are plotted for all appropriate variant/guide combinations; SpG was tested only on sites harboring NGN PAMs; editing of cytosines assessed by targeted sequencing with mean C-to-T editing shown for n = 3. The intended edit, bystander synonymous, non-synonymous, and stop codon C-to-T edits are indicated; the PAMs for each target site are shown in the grey arrow annotation; for raw data see Table S5.
Next, we systematically evaluated the capability to generate 8 additional protective variants that lack nearby canonical NGG PAMs (Fig. 4C). We examined target sites harboring NRN PAMs using WT-, SpG-, and SpRY-CBEs that would position the intended C-to-T edit within the high-activity CBE edit window (Fig. 4A). With WT-CBE we were unable to efficiently generate the intended edit for 7 of 8 substitutions, due the absence of nearby canonical PAMs, reinforcing the need for variants with expanded targeting capabilities. With SpG- and SpRY-CBEs, we screened additional target sites for each substitution and observed that we could efficiently introduce the intended C-to-T edit across all eight targets (Fig. 4C). For the majority of the genetic variants, we were able to avoid deleterious bystander editing with SpG and SpRY by selecting from multiple targetable sites that produce silent or tolerable collateral edits instead of those that cause non-synonymous C-to-T changes. These results demonstrate the utility of SpG and SpRY for higher resolution targeting of previously un-editable genomic sites and the capability to examine additional target sites to avoid detrimental bystander edits.
Discussion:
While the PAM requirement of CRISPR systems enables bacteria to distinguish self from non-self, for genome editing applications the necessity of PAM recognition constrains CRISPR-Cas systems for use across genomic loci that lack or sparsely encode PAMs. The SpG and SpRY variants circumvent this limitation by relaxing or almost entirely removing the dependence of SpCas9 on a requisite PAM, extending targeting to sites with NGN and NAN PAMs, and many sites with NCN or NTN PAMs albeit at a reduced relative efficiency. These variants should enable unconstrained targeting for a variety of applications that require precise DNA breaks, nicks, deamination, or binding events. Beyond the experiments presented herein in HEK 293T cells, the utility of SpG and SpRY across different applications and delivery contexts requires additional investigation (e.g. for tiling putative regulatory elements in various cell types).
In principle, the strategy we utilized to reduce or eliminate the PAM requirement should be applicable to other native or engineered CRISPR-Cas9 and -Cas12a orthologs for which structural information is available. While future studies are needed to more precisely elucidate the molecular roles of the amino acid substitutions in SpG and SpRY, we speculate that SpRY achieves its expanded targeting range through the removal of the canonical base-specific interactions, displacement of the PAM DNA to facilitate interactions in the major groove of the PAM, and energetic compensation by the addition of novel non-specific protein:DNA contacts. More practically, when contemplating which enzyme to utilize for experiments requiring on-target activity, we suggest utilizing WT SpCas9 for sites harboring NGG PAMs, SpG for NGH PAMs, and SpRY for targets encoding the remaining NHN PAMs (with NAN > NCN/NTN).
The potential for undesirable off-target effects requires methods to mitigate them. As observed when developing engineered CRISPR-Cas12a and -Cas9 enzymes with expanded PAM tolerances, relaxation of the PAM can reduce specificity(22, 39). However, both enAsCas12a and SpCas9-NG were compatible with substitutions that enhance genome-wide specificity. Similarly, SpG and SpRY are compatible with SpCas9-HF1 substitutions to eliminate nearly all detectable off-target effects as determined by GUIDE-seq, enabling applications that require higher fidelity.
By developing SpCas9 variants capable of high-resolution editing, we demonstrate that protein engineering can eliminate biological constraints that limit applications of CRISPR-Cas enzymes, . With SpRY supporting the editing of many sites containing NRN > NYN PAMs, the vast majority of the genome is now targetable.
Supplementary Material
Acknowledgments:
We thank S. Tsai and J. Lopez for advice on GUIDE-seq data analysis; J. Hsu for assistance with the PAMDA scripts; J. Grünewald for advice with NextSeq sequencing, Z. Hebert and the Molecular Biology Core Facilities (MBCF) at Dana-Farber Cancer Institute (DFCI) for support with NextSeq sequencing; K. Clement for guidance with CRISPResso2; S. Johnstone and B. Bernstein for assistance with and access to their Covaris E220; D. Gao for informatic support; M. Prew and M. Welch for providing gRNA plasmids; S. Katherisan for advice on protective variants; J.K. Joung for suggestions on the manuscript.
Funding: Research in the Kleinstiver Lab was supported by NIH R00-CA218870, NIH P01-HL142494, a Career Development Award from the American Society of Gene & Cell Therapy, and the Margaret Q. Landenberger Research Foundation.
Footnotes
Competing Interests: R.T.W. and B.P.K. are inventors on various patents and patent applications that describe gene editing and epigenetic editing technologies. B.P.K. is a consultant for Avectas Inc and an advisor for Acrigen Biosciences.
Data and Materials Availability: Plasmids generated in this study are available through Addgene (https://www.addgene.org/Benjamin_Kleinstiver/; see also Table S2). All data are available in the manuscript, in the supplementary material, or in the following databases: high-throughput sequencing data sets from nuclease, CBE, ABE, HT-PAMDA, HT-PAMDA-CBE, and GUIDE-seq experiments are available through the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) under BioProject ID PRJNA605711 (https://www.ncbi.nlm.nih.gov/bioproject/605711). The commands for visualizing crystal structures in PyMOL are available in(26); CRISPResso2(40) was used to analyze nuclease and base editor data (with non-standard parameters detailed in the Methods section); custom python scripts used to analyze HT-PAMDA and HT-PAMDA-CBE data are available through Zenodo (41).
References and Notes:
- 1.Marraffini LA, Sontheimer EJ, Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 463, 568–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, Marraffini LA, Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 519, 199–202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S, Phage Response to CRISPR-Encoded Resistance in Streptococcus thermophilus. J Bacteriol. 190, 1390–1400 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C, Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiol Read Engl. 155, 733–40 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA, DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507, 62–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA, RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 31, 233–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh J-RJ, Aryee MJ, Joung JK, Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 523, 481–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM, Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 10, 1116–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonfara I, Rhun AL, Chylinski K, Makarova KS, Lécrivain A-L, Bzdrenga J, Koonin EV, Charpentier E, Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 42, 2577–90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, Siksnys V, Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol. 16, 253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F, Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 163, 759–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M, Gene Conversion Tracts from Double-Strand Break Repair in Mammalian Cells. Mol Cell Biol. 18, 93–101 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J, Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 513, 120–3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, Luc S, Kurita R, Nakamura Y, Fujiwara Y, Maeda T, Yuan G-C, Zhang F, Orkin SH, Bauer DE, BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 527, 192–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR, Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 33, 661–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR, Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 533, 420–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR, Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kan Y, Ruis B, Takasugi T, Hendrickson EA, Mechanisms of precise genome editing using oligonucleotide donors. Genome Res. 27, 1099–1111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anders C, Niewoehner O, Duerst A, Jinek M, Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 513, 569–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK, High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 529, 490–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, Noda T, Abudayyeh OO, Gootenberg JS, Mori H, Oura S, Holmes B, Tanaka M, Seki M, Hirano H, Aburatani H, Ishitani R, Ikawa M, Yachie N, Zhang F, Nureki O, Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 361, 1259–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, Liu DR, Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anders C, Bargsten K, Jinek M, Structural Plasticity of PAM Recognition by Engineered Variants of the RNA-Guided Endonuclease Cas9. Mol Cell. 61, 895–902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirano S, Nishimasu H, Ishitani R, Nureki O, Structural Basis for the Altered PAM Specificities of Engineered CRISPR-Cas9. Mol Cell. 61, 886–94 (2016). [DOI] [PubMed] [Google Scholar]
- 26.See Supplementary Materials
- 27.Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z, Kurita M, Hishida T, Li M, Aizawa E, Guo S, Chen S, Goebl A, Soligalla RD, Qu J, Jiang T, Fu X, Jafari M, Esteban CR, Berggren WT, Lajara J, Nuñez-Delicado E, Guillen P, Campistol JM, Matsuzaki F, Liu G-H, Magistretti P, Zhang K, Callaway EM, Zhang K, Belmonte JCI, In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540, 144–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rees HA, Liu DR, Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koblan LW, Doman JL, Wilson C, Levy JM, Tay T, Newby GA, Maianti JP, Raguram A, Liu DR, Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol. 36, 843–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F, Rationally engineered Cas9 nucleases with improved specificity. Sci New York N Y. 351, 84–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, Joung JK, GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 33, 187–97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balemans W, Ebeling M, Patel N, Hul EV, Olson P, Dioszegi M, Lacza C, Wuyts W, Ende JVD, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Hul WV, Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 10, 537–543 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee S-J, Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. New Engl J Med. 350, 2682–2688 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Cox JJ, Sheynin J, Shorer Z, Reimann F, Nicholas AK, Zubovic L, Baralle M, Wraige E, Manor E, Levy J, Woods CG, Parvari R, Congenital insensitivity to pain: novel SCN9A missense and in-frame deletion mutations. Hum Mutat. 31, E1670–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flannick J, Thorleifsson G, Beer NL, Jacobs SBR, Grarup N, Burtt NP, Mahajan A, Fuchsberger C, Atzmon G, Benediktsson R, Blangero J, Bowden DW, Brandslund I, Brosnan J, Burslem F, Chambers J, Cho YS, Christensen C, Douglas DA, Duggirala R, Dymek Z, Farjoun Y, Fennell T, Fontanillas P, Forsén T, Gabriel S, Glaser B, Gudbjartsson DF, Hanis C, Hansen T, Hreidarsson AB, Hveem K, Ingelsson E, Isomaa B, Johansson S, Jørgensen T, Jørgensen ME, Kathiresan S, Kong A, Kooner J, Kravic J, Laakso M, Lee J-Y, Lind L, Lindgren CM, Linneberg A, Masson G, Meitinger T, Mohlke KL, Molven A, Morris AP, Potluri S, Rauramaa R, Ribel-Madsen R, Richard A-M, Rolph T, Salomaa V, Segrè AV, Skärstrand H, Steinthorsdottir V, Stringham HM, Sulem P, Tai ES, Teo YY, Teslovich T, Thorsteinsdottir U, Trimmer JK, Tuomi T, Tuomilehto J, Vaziri-Sani F, Voight BF, Wilson JG, Boehnke M, McCarthy MI, Njølstad PR, Pedersen O, Consortium G-T, Consortium T-G, Groop L, Cox DR, Stefansson K, Altshuler D, Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 46, 357–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.T. and H. W. G. of the E. S. P. Institute National Heart, Lung, and Blood, Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang Z, Zhang H, Hindy G, Masca N, Stirrups K, Kanoni S, Do R, Jun G, Hu Y, Kang HM, Xue C, Goel A, Farrall M, Duga S, Merlini PA, Asselta R, Girelli D, Olivieri O, Martinelli N, Yin W, Reilly D, Speliotes E, Fox CS, Hveem K, Holmen OL, Nikpay M, Farlow DN, Assimes TL, Franceschini N, Robinson J, North KE, Martin LW, DePristo M, Gupta N, Escher SA, Jansson J-H, Zuydam NV, Palmer CNA, Wareham N, Koch W, Meitinger T, Peters A, Lieb W, Erbel R, Konig IR, Kruppa J, Degenhardt F, Gottesman O, Bottinger EP, O’Donnell CJ, Psaty BM, Ballantyne CM, Abecasis G, Ordovas JM, Melander O, Watkins H, Orho-Melander M, Ardissino D, Loos RJF, McPherson R, Willer CJ, Erdmann J, Hall AS, Samani NJ, Deloukas P, Schunkert H, Wilson JG, Kooperberg C, Rich SS, Tracy RP, Lin D-Y, Altshuler D, Gabriel S, Nickerson DA, Jarvik GP, Cupples LA, Reiner AP, Boerwinkle E, Kathiresan S, Loss-of-function mutations in APOC3, triglycerides, and coronary disease. New Engl J Medicine. 371, 22–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harper AR, Nayee S, Topol EJ, Protective alleles and modifier variants in human health and disease. Nat Rev Genetics. 16, 689–701 (2015). [DOI] [PubMed] [Google Scholar]
- 38.M. I. G. C. Investigators, Stitziel NO, Won H-H, Morrison AC, Peloso GM, Do R, Lange LA, Fontanillas P, Gupta N, Duga S, Goel A, Farrall M, Saleheen D, Ferrario P, König I, Asselta R, Merlini PA, Marziliano N, Notarangelo MF, Schick U, Auer P, Assimes TL, Reilly M, Wilensky R, Rader DJ, Hovingh GK, Meitinger T, Kessler T, Kastrati A, Laugwitz K-L, Siscovick D, Rotter JI, Hazen SL, Tracy R, Cresci S, Spertus J, Jackson R, Schwartz SM, Natarajan P, Crosby J, Muzny D, Ballantyne C, Rich SS, O’Donnell CJ, Abecasis G, Sunaev S, Nickerson DA, Buring JE, Ridker PM, Chasman DI, Austin E, Kullo IJ, Weeke PE, Shaffer CM, Bastarache LA, Denny JC, Roden DM, Palmer C, Deloukas P, Lin D-Y, Tang Z, Erdmann J, Schunkert H, Danesh J, Marrugat J, Elosua R, Ardissino D, McPherson R, Watkins H, Reiner AP, Wilson JG, Altshuler D, Gibbs RA, Lander ES, Boerwinkle E, Gabriel S, Kathiresan S, Inactivating mutations in NPC1L1 and protection from coronary heart disease. New Engl J Medicine. 371, 2072–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfò I, Maus MV, Pinello L, Aryee MJ, Joung JK, Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat Biotechnol. 37, 276–282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, Pinello L, CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol. 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scripts for analyzing High-Throughput PAM Determination Assay (HT-PAMDA) experimental data for CRISPR enzymes; available through Zenodo, doi: 10.5281/zenodo.3710516. [DOI]
- 42.Rohland N, Reich D, Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao L, Cox DBT, Yan WX, Manteiga JC, Schneider MW, Yamano T, Nishimasu H, Nureki O, Crosetto N, Zhang F, Engineered Cpf1 variants with altered PAM specificities. Nat Biotechnol. 35, 789–792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK, Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 33, 1293–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo M, Ren K, Zhu Y, Tang Z, Wang Y, Zhang B, Huang Z, Structural insights into a high fidelity variant of SpCas9. Cell Res. 29, 183–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, Shen AH, Ren C, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA, Bauer DE, Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 25, 776–783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.