

SUMMARY
Plants are foundational to global ecological and economic systems, yet most plant proteins remain uncharacterized. Protein interaction networks often suggest protein functions and open new avenues to characterize genes and proteins. We therefore systematically determined protein complexes from 13 plant species of scientific and agricultural importance, greatly expanding the known repertoire of stable protein complexes in plants. Using co-fractionation mass spectrometry, we recovered known complexes, confirmed complexes predicted to occur in plants, and identified previously unknown interactions conserved over 1.1 billion years of green plant evolution. Several novel complexes are involved in vernalization and pathogen defense, traits critical to agriculture. We also observed plant analogs of animal complexes with distinct molecular assemblies, including a megadalton-scale tRNA multi-synthetase complex. The resulting map offers a cross-species view of conserved, stable protein assemblies shared across plant cells and provides a mechanistic, biochemical framework for interpreting plant genetics and mutant phenotypes.
Keywords: Plants, protein interactions, evolution, protein complexes, co-fractionation mass spectrometry (CF-MS), comparative proteomics, cross-linking/mass spectrometry (CL-MS), interaction-to-phenotype, pathogen defense
In brief
This massive plant proteomics project, using co-fractionation-mass spectrometry to measure the amounts and associations of over two million proteins from 13 diverse plant species, reveals stable protein complexes shared across plant cells and provides a framework for interpreting plant genetics and mutant phenotypes.
Graphical Abstarct
INTRODUCTION
Plants make up most of the planet’s biomass and sustain global economic and environmental systems. Despite increasing numbers of sequenced plant genomes, biochemical characterization of encoded plant proteins captures a relatively small portion of the expected breadth of biological functions. The best characterized dicotyledonous plant, Arabidopsis thaliana, encodes approximately 35,000 protein-coding genes, but the functions of the majority of these proteins remain uncharacterized, even by homology (Rhee and Mutwil, 2014). This trend is similar for Oryza sativa (rice), a critical food crop, and the best characterized monocotyledonous plant (Kawahara et al., 2013). In marked contrast to other model organisms, it is estimated that only 5% of Arabidopsis proteins and considerably fewer proteins in other plants have had biochemical activity, localization, and biological roles determined by direct experimentation (Niehaus et al., 2015; Rhee and Mutwil, 2014; Swarbreck et al., 2008). Extraordinary experimental efforts are needed to define the core expressed proteome and molecular machinery of plants.
The active multiprotein assemblies in plant cells have not yet been systematically defined, in stark contrast to recent progress in humans (Havugimana et al., 2012; Hein et al., 2015; Huttlin et al., 2015, 2017; Kirkwood et al., 2013; Kristensen et al., 2012; Malovannaya et al., 2011; Wan et al., 2015). Determining protein-protein interactions is a key step for discovering gene and protein function and frequently opens new avenues to study and manipulate critical cellular processes (Eisenberg et al., 2000; Hartwell et al., 1999; Schwikowski et al., 2000; Walhout and Vidal, 2001). In model organisms, such as yeast or Drosophila, systematic mapping of protein complexes led to critical functional insights, facilitated understanding of conserved and disease-related pathways (Vidal et al., 2011), and helped characterize uncharacterized proteins via guilt by association (Hu et al., 2009; Marcotte et al., 1999; Oliver, 2000). Revealing this class of biochemical information for plants will dramatically advance efforts in fundamental plant research, as well as guide practical applications such as improvements in crop yields, disease/stress resistance, and biofuel production.
Unfortunately, many of the techniques that have been used to discover protein complexes at scale in animals and yeast (e.g., high-throughput affinity purification (AP-MS)) are prohibitively expensive or even intractable at large-scale in plants due to issues of complex genomes, polyploidy, and transformation efficiency. Consequently, AP-MS experiments in plants have been limited to targeted protein families, primarily in Arabidopsis and rice (Bassel et al., 2012; Rohila et al., 2009; Van Leene et al., 2008, 2010, 2011; Zhang et al., 2010). Such approaches for studying protein-protein interactions are disproportionately difficult in less well-characterized plant species necessitating new strategies for comparative studies.
Co-fractionation mass spectrometry (CF-MS) is a high-throughput method to detect interacting proteins, applicable to any organism, without the need for antibodies or transgenic epitope tagging of individual proteins. CF-MS involves chromatographically separating a native protein extract, then identifying proteins in each biochemical fraction using mass spectrometry. Co-elution (co-fractionation) of proteins in a separation serves as evidence of physical association, which, when measured over multiple distinct separations is a rigorous signal of protein interaction (Wan et al., 2015). Observation of repeated coelution in multiple different species reduces species-specific artifacts and adds power to discover conserved—and thus more likely functional—interactions. Furthermore, the use of machine learning methods allows for strong control over false discovery rates of protein interactions.
Several papers have classified candidate Arabidopsis protein complexes using native (non-denaturing) separations such as gels (Takabayashi et al., 2017) or co-fractionation (Aryal et al., 2017; Gilbert and Schulze, 2019; McBride et al., 2019). However, in a single or small set of experiments, two proteins can coincidentally co-elute without a physical association. By comparison, repeated co-elution using multiple distinct species, tissues, and biochemical separations provides increasing confidence in a protein interaction (Wan et al., 2015). As with genome-wide association studies (GWAS) or recombination mapping of a single mutation by phenotype, large amounts of data are required to build a statistically strong observation.
In this largest survey to date of expressed plant proteins as well as their physical assemblies, we collected a massive and diverse plant co-elution dataset to define high confidence protein-protein interactions in a statistical computational framework, recovering known complexes and identifying novel complexes conserved across plants for over a billion years. Multiple newly discovered complexes have direct relevance to agronomically important traits. Collectively, the resulting set of protein abundances and map of stable protein interactions will help interpret plant gene functions in a mechanistic, evolutionary, and biochemical framework.
RESULTS AND DISCUSSION
A massive data set of protein abundances and co-purification from 13 plant species
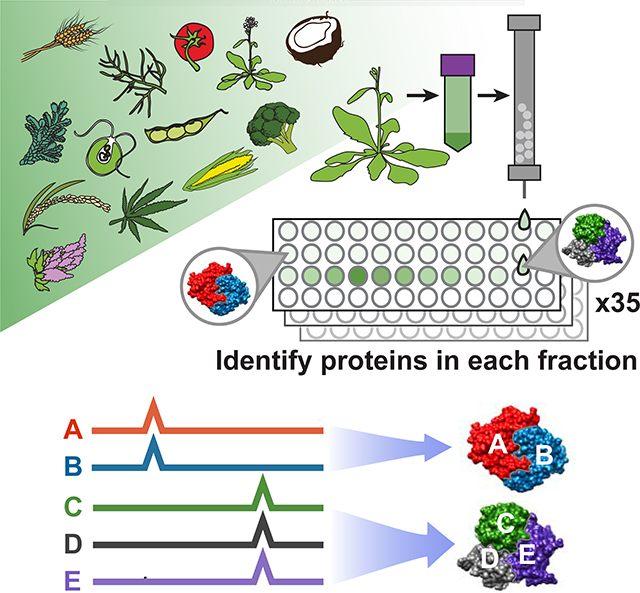
We generated a large, diverse, and representative proteomics dataset from 13 species spanning 1.1 billion years of green plant evolution (Figure 1A). Our compendium incorporates proteomic data from Arabidopsis thaliana, Brassica oleracea (broccoli), Glycine max (soy), Cannabis sativa (hemp), Solanum lycopersicum (tomato), Chenopodium quinoa (quinoa), Zea mays (maize), Oryza sativa ssp. japonica (rice), Triticum aestivum (wheat), Cocos nucifera (coconut), Ceratopteris richardii (fern), Selaginella moellendorffii (spikemoss), and the green algae Chlamydomonas reinhardtii. We chose crop and model plants important to the research community, including species that overcome specific technical challenges (e.g. high yields of nuclei from broccoli and coconut; embryonic tissue from wheat and hemp). We included vascular plants that branched off before the split of seed-bearing plants (fern, spikemoss) and single-celled green algae (Chlamydomonas) for their ancestral characteristics. Our dataset combines multiple species and cell types to give a broad view of expressed proteins across Viridiplantae.
Figure 1. Integrative Co-fractionation Mass Spectrometry (CF-MS) Workflow Used to Determine Stable Plant Protein Complexes.

(A) The selected species represent a broad range of evolutionary time (MYA, million years ago (Kumar et al., 2017)).
(B) Native extracts are chromatographically separated and the proteins in each fraction identified by mass spectrometry.
(C) Co-fractionation of proteins is evidence of physical association.
(D) Proteins from each species’ proteome are first assigned to orthologous groups (OGs).
(E) Peptides that match more than one orthogroup (light gray text) are not used, however peptides that uniquely match a single orthogroup are used to quantify the abundance of an orthogroup in individual chromatographic fractions. Elution profiles for each orthogroup are shown here as ridgelines or heat maps (blue showing normalized abundance).
(F) Heat map of the full dataset of abundance measurements for each of the 23,896 detected orthogroups across all fractionations for the thirteen species. Dashes under heat map delineate each fractionation experiment.
(G) Enlarged portions of (F) showing observed strong co-elution for subunits (names at left, see Table S1) of six well-known protein complexes (names at right). Color intensity (blue is positive signal) depicts measured abundances for each orthogroup (labeled at left) in two distinct chromatographic separations (labeled at top) out of the 35 total separations.
We separated each native, non-denatured protein extract by some biophysical property, either size exclusion chromatography (SEC), ion exchange chromatography (IEX), or isoelectric focusing (IEF) (Figure 1B–C), and analyzed each biochemical fraction by mass spectrometry. In all, we collected 14,520,970 interpretable peptide mass spectra from 2,111 individual fractions, each capturing distinct subsets of native plant proteins and protein assemblies. This deep proteome profiling will be valuable for addressing diverse questions of plant biochemistry and function, including protein modifications, expression, and determining which species or tissue contains high abundance of any particular protein.
An evolution-informed strategy improves proteomics coverage and enables comparisons across species with different ploidy levels
Integrating protein observations from different organisms is complicated due to orthology mapping. This long-standing problem is even more extreme in plants due to their often complex and polyploid genomes, as well as past whole-genome duplications (Jiao et al., 2011). For example, most farmed wheat is hexaploid and contains over 100,000 genes, which complicates comparison to model diploids such as Arabidopsis, with its approximately 35,000 genes. The existence of multiple near-identical proteins also reduces the number of peptides that uniquely match a single protein, reducing protein recovery by standard proteomics methods. Current protein-grouping statistical methods for assigning peptides to proteins tend to perform erratically for highly redundant genomes, in practice allocating shared peptides semi-randomly across similar proteins. We thus developed an evolution-informed protein-grouping approach which is generally applicable to proteomic data from any arbitrary number of different species.
Our strategy was to interpret mass spectral observations in terms of orthogroups, rather than individual proteins. An orthogroup (OG) is a set of genes in modern organisms that derive from the same original gene in those organisms’ last common ancestor. We began by assigning all protein-coding genes in each plant species to predetermined orthogroups (Huerta-Cepas et al., 2017), thereby organizing highly related protein sequences into sets (Figure 1D). We then considered mass spectra from any orthogroup member protein as evidence for the abundance of their orthogroup (Figure 1E), which allows peptides shared by multiple proteins in an orthogroup (but not unique to a single protein) to now contribute to quantification. Importantly, orthogroups, unlike proteins, have consistent identifiers that can be used as a key to integrate data from multiple species. Thus, we collapse the three proteins in the Ribosomal Protein L36 orthogroup in Arabidopsis to a set, directly comparable to the behavior of that orthogroup in other species, for example the set of seven proteins in this orthogroup in wheat. (We refer to orthogroups by available Arabidopsis common gene names throughout; Table S1 provides additional identifiers.) Figure 1F visually summarizes our over 2 million protein abundance measurements across plant species integrated into this comparative phylogenetic framework (described in Table S2, data at DOI:10.5281/zenodo.3666942) and Figure 1G highlights specific examples of complexes from the data in Figure 1F.
Consistent with ploidy levels (Figure 2A), diploid organisms such as Arabidopsis and Chlamydomonas exhibited a peak of one protein per orthogroup. In contrast, tetraploid quinoa and soy each showed a peak of two proteins per orthogroup (one from each of two subgenomes) and in the same manner, hexaploid wheat showed a peak of three proteins. The spikemoss (Selaginella moellendorfii) genome, a greenhouse hybrid (Banks et al., 2011), interestingly has a peak of two proteins per orthogroup, or one protein per parent genome.
Figure 2. Proteomics in High-Ploidy Species Enhanced by Assignment of Proteins to Orthogroups. Also Figure S1.

(A) Number of proteins assigned per orthogroup for each plant species in our study colored by ploidy. Shaded ovals at left represent subgenome organization.
(B) Fold increase (x-axis) in peptide spectral matches that identify unique orthogroups vs. unique proteins. Each bar represents a single fractionation experiment conducted on the species named at left and color-coded by ploidy as in (A).
(C) The number of observed proteins (left plot) or orthogroups (right plot) experimentally observed (in blue) compared to the possible total in the proteome (gray). Note that relative coverage per species is a function of the amount of data collected from that species in this study.
(D) Our data set is sufficient to identify the majority of orthogroups possible by this method. Each dot represents the number of orthogroups identified (y-axis) in a subsample of n experiments (x-axis), with sampling repeated ten times per each n.
(E) Orthogroups with more than two proteins were approximately equally likely to be represented by a single dominant protein as not, regardless of ploidy.
(F) Orthogroups observed by mass spectra (green) represent those with higher mRNA abundances (TPM, transcripts per million, log scale; data from (Panchy et al., 2014)), as shown for Chlamydomonas. Gray represents orthogroups not observed in our study.
(G) Log-scale protein abundances (y-axis) show expected correlation with RNA abundances (x-axis, Transcripts per million; same as in (F)) in Chlamydomonas, however with numerous outliers, notably, RuBisCo (green dot).
This orthogroup-based proteomics interpretation strategy increased the recovery of unique spectral counts for the highly redundant proteome of hexaploid wheat by over 300%, while not strongly impacting organisms with small diploid genomes, e.g., Chlamydomonas (Figure 2B). Similarly, coverage of observed orthogroups is less variable than coverage of observed proteins across species. (Figure 2C). Collapsing sets of evolutionarily-related proteins into orthogroups is thus a flexible and broadly applicable solution for cross-species proteomics analyses, especially across plants with varying ploidy levels.
Characteristics of expressed plant proteomes
Our data represent over 141,910 unique proteins and 23,896 orthogroups from diverse species and extracts, the largest proteomic survey of plants performed to date, covering broad areas of function (Figure S1). We sufficiently capture the proteome observable by native fractionation mass spectrometry, as evidenced by saturating the number of new orthogroups observed per additional experiment (Figure 2D), suggesting that more samples are unlikely to markedly improve conserved proteome coverage. In all, we observed 96.7% of the 11,339 most conserved Viridiplantae orthogroups (details in STAR Methods). Our dataset, therefore, provides a meaningful snapshot of the conserved and expressed proteome of green plants.
Collapsing sets of proteins to orthogroups masks individual protein behavior, so we examined the behavior of paralogs within orthogroups. We found that about half of orthogroups with three or more member proteins contain one dominant protein that is highly expressed relative to other members (Figure 2E). An approximately equal set have protein expression more evenly shared across members. These trends were consistent across multiple species with widely varying genome sizes (Figure 2E). Thus, some degree of functional divergence, as measured by differential expression, was evident in about 50% of multi-gene orthogroups.
The proteins we observed also tended to be products of more abundant mRNAs (Figure 2F). Work in other organisms has shown that there is an imperfect correlation between protein abundances and RNA transcript levels, largely attributable to post-transcriptional, translational, and degradation rate effects on steady-state protein levels (Vogel and Marcotte, 2012). Our data here similarly illustrate this point (Spearman r = 0.45, Figure 2G). One notable outlier is the enzyme RuBisCo, the most abundant protein on earth (Feller et al., 2007) and the most abundant protein in 16/35 of our experiments. RuBisCo is substantially more abundant at the protein level than would be expected based on its transcript levels, e.g. in Chlamydomonas (Figure 2G) or in Arabidopsis tissues (Figure S1).
Systematic identification and scoring of stable protein-protein interactions
In many cases, subunits of known complexes show co-elution patterns easily detected by eye, as for subunits of the 20S proteasome, prefoldin, and TSET complexes that respectively coelute with distinct, complex-specific, elution patterns (Figure 1G). However, a computational framework is necessary to identify co-eluting proteins systematically and at high-throughput.
To quantitatively score co-elution behavior indicative of stably interacting (not transient) proteins, we employed a supervised machine learning approach based upon observed data for known complexes. Protein-protein interactions were derived solely from the coordinated separation behavior of proteins over multiple, orthogonal biochemical fractionation experiments. The classifier assigned a probabilistic CF-MS score (Co-Fractionation-Mass Spectrometry score) between 0 and 1 to each potential protein-protein interaction, with 1 indicating a high likelihood of physical association based on observing strongly coordinated protein elution profiles, and 0 indicating no evidence for interaction (details in STAR Methods, scores at DOI:10.5281/zenodo.3666940).
We next aimed to rigorously assess statistical confidence for physical protein-protein interactions, evaluating against a fully withheld test set of 886 known protein interactions that were not used at any point during model training. Ranking protein interactions by their CF-MS scores accurately recapitulated this withheld test set (Figure 3A) and allowed us to measure classifier error rates: For interactions with CF-MS scores over 0.50, we observed 90% precision (i.e. ≤10% false positive interactions) and 23% recall. Interactions with a CF-MS score above 0.2 exhibited 50% precision at 51% recall, and thus were still informative in many cases (Figure 3B). Interactions tended to be highly conserved, but known interactions were less likely to be found if either partner had <200 spectral counts (Figure S2). Nonetheless, our performance compares well to human MS complex maps, which range from <5% to 38% recall at 90% precision (Drew et al., 2017).
Figure 3. Derivation and Global Validation of Protein Co-Complex Interactions. Also Figure S2.

(A) Precision-Recall of CF-MS scored protein-protein interactions (PPIs) on 886 known interactions withheld from training.
(B) False Discovery Rate (FDR) vs. CF-MS scores for the same withheld set as in (A).
(C) PPIs with high CF-MS scores (FDR < 10%) are highly correlated in a species withheld from training (maize).
(D) Protein interactions with higher CF-MS scores were more likely to have been identified by affinity purification, 2-hybrid in Arabidopsis, and were more likely to be co-expressed in Arabidopsis and rice.
(E) Agreement of the CF-MS protein-protein interactions (yellow) with affinity purification (blue) and 2-hybrid interactions (red) for three protein complexes.
Confirming CF-MS interactions by independent assays and chemical cross-linking
Our measured high-confidence protein interactions agree with independent protein interaction observations. As a direct test of our method, we asked if CF-MS-supported interactions were concordant with an independent biochemical separation of maize, a species that was not used to train the model. Ortholog pairs with a high CF-MS score (>0.5, derived from non-maize plants) co-elute strongly in maize shoots (Figure 3C). We also compared our observed interactions to other independent plant protein interaction datasets, finding that protein pairs with higher CF-MS scores were more likely to affinity purify together, to interact by yeast-2-hybrid, and to exhibit coordinated mRNA expression both in Arabidopsis and in rice (Figure 3D). Our high confidence scores align with and provide orthogonal support for biologically interesting interactions previously discovered by AP-MS and yeast-2-hybrid assays. Figure 3E illustrates three such cases for interaction of 1) TPLATE complex proteins (Gadeyne et al., 2014), 2) SWI/SNF components with LEAF AND FLOWER RELATED (LFR) and the uncharacterized protein AT5G17510 (Vercruyssen et al., 2014), and 3) two uncharacterized pentatricopeptide repeat proteins PPR59 and PPR351 (Arabidopsis Interactome Mapping, 2011).
To further independently validate our derived protein complexes using unbiased orthogonal methods (and thus avoid “cherry-picking” promising test cases), we implemented two untargeted, large-scale biochemical approaches. For the first method, we plotted expected monomeric mass against observed mass in a representative Arabidopsis size exclusion experiment and demonstrated that a substantial proportion of proteins eluted with a mass considerably larger than their monomeric masses, supporting that endogenous complexes remained intact in our experimental conditions (Figure 4A, STAR methods).
Figure 4. Protein Complexes Validated by Calibrated Molecular Mass Determination and Direct Chemical Cross-linking.

(A) Observed mass vs. predicted monomeric mass in a representative Arabidopsis size exclusion chromatography (SEC) fractionation. Shading reflects the number of orthogroups per hexagonal bin.
(B) Cross-linked proteins from soy and Chlamydomonas are more likely (green line, log-likelihood) to have high CF-MS scores compared to non-cross-linked observed proteins.
(C-D) Inter-subunit cross-links only appear in fractions where complex subunits co-elute. Elution profile and inter-subunit crosslinks for soy T-complex chaperonin (CCT) shown in (C) and Chlamydomonas Photosystem II (PSB) shown in (D)
(E-F) 3D homology models of complexes (see STAR methods) with observed inter-subunit cross-links (black lines). Soy CCT (E) is colored by subunit, and Chlamydomonas
Photosystem II (E) highlights PSBB, PSBC, and PSBO as blue, red, and yellow, respectively.
For the second validation method, we performed global chemical cross-linking to covalently tether interacting proteins in fractionated soy sprout and Chlamydomonas protein extracts. We identified 194 heteromeric protein-protein interactions from soy and 228 from Chlamydomonas, at a false discovery rate of 1% and ΔXlinkX score ≥70 (Table S3). Cross-linking recovered 31 withheld test set positive interactions and one negative interaction, empirically estimating the false discovery rate at 3%. Protein pairs with high CF-MS scores were considerably more likely to be cross-linked (Figure 4B).
Furthermore, our observed cross-links were consistent with physical constraints of known protein interactions (Figure 4C–F). For example, intersubunit cross-links in the soy CCT chaperonin complex were exclusively identified in the same fractions as co-eluting CCT subunits (Figure 4C) and occurred between residues appropriate to the cross-linker length (<30 Å Cɑ-Cɑ) (Figure 4E). While this complex is well documented in animals, a 22S complex containing CCT subunits was only reported in 2019 in Arabidopsis (Ahn et al., 2019). Our data confirm that the CCT complex is conserved across plants and indicate that the 3D subunit organization in plants resembles that in animals. Similarly, co-fractionation and cross-linking of the Photosystem II complex was observed in Chlamydomonas, with the observed inter-protein cross-links located at appropriate adjacent solvent-accessible subunit interfaces (Figure 4D, F) as expected for the native conformation of this complex. The recovery of multiple structurally coherent inter-subunit cross-links between cofractionating subunits provides experimental evidence that CF-MS faithfully captured protein assemblies.
Thus, a combination of direct experimental validation using independent biochemical methods (co-fractionation in an independent organism, calibrated size exclusion chromatography, and chemical cross-linking) and comparison to independently determined protein interactions from the literature all indicate that the CF-MS measurements provide strong evidence for physical assemblies.
Identification of multiprotein complexes confirms those inferred by gene content and reveals additional assemblies
As our CF-MS datasets faithfully captured many large multiprotein assemblies (Figures 1G, 4A, C–F), we next sought to systematically define higher-order plant protein complexes by clustering the proteins based on the measured pairwise interactions at FDR < 10% (Figure 5). Instead of choosing a single clustering cutoff to define discrete complexes, we selected multiple cutoffs to reflect hierarchies of interacting proteins and precision recall trade-offs (Figure S3). For example, one cutoff defined the 80S ribosome, while a finer cutoff differentiated its 40S and 60S subcomplexes (Figure 5). Orthogroups in well-characterized complexes are annotated dark green; however, we identified several previously unreported subunits and interactors with the potential to enrich our understanding of how these known complexes function in plants. Excitingly, we observed many complexes comprised of novel associations (Figure 5, yellow) as well as proteins that remain uncharacterized in plants (Figure 5, bold circles). Our clustered and annotated set of high confidence protein complexes is available as Table S4.
Figure 5. Overview of Evolutionarily Conserved Plant Protein Complexes. Also Figure S3.

Thin concentric circles show the clustering hierarchy of protein-protein interactions into complexes for each of four clustering thresholds. (Table S4 lists complexes and annotations.) Protein orthogroups (filled circles) are colored green for associations previously reported in any species or yellow for those first reported in this study. Bold outlines denote proteins uncharacterized in plants, defined as uncharacterized if all proteins in the orthogroup lack an Arabidopsis gene symbol and a Uniprot Function annotation. Bold complex labels are discussed in the text.
As internal positive controls, we identified 117 complexes that are reported in the literature. Some of these eukaryotic complexes, such as the Conserved Oligomeric Golgi (COG) complex, the SRP68/72 heterodimer, and the TRAPP and BRISC complexes have to our knowledge only previously been inferred in plants by gene content. Similarly, we find orthologs of complex members that have only been reported in non-plant species, such as MAA3 (a plant ortholog of the yeast protein Sen1 and human Senataxin) interacting with RNA polymerase III, an interaction recently found in yeast to regulate RNA polymerase III termination (Rivosecchi et al., 2019).
Likewise, while homologs to the yeast and mammalian oligosaccharyltransferase (OST) complex subunits exist in plants, the full complex has not been biochemically isolated (Strasser, 2016). We observe a plant OST complex with overlapping membership to the yeast and mammalian OST complex, and detect cross-links between HAP6 and OST48 in soy, suggesting the plant OST complex resembles that of other eukaryotes. We identify potential OST components Stomatin-like protein 1 (SLP1), SPC25, and EMC1. We also identified components of the initiation factor (eIF)2B complex in which the eIF2Bγ/ε and eIF2Bβ/δ dimers were clearly co-purifying with each other (Figure 1G), but the eIF2Bα subunit appeared to be less stably associated (Table S5). The existence of the eIF2B complex in plants has been speculative as this complex has not been characterized and isolated from plants, in contrast to its well-established observation and characterization in other eukaryotes (Browning and Bailey-Serres, 2015).
Protein interactions provide a means to characterize, corroborate, and predict protein functions via guilt-by-association. We found several instances where a top-scoring interaction to an uncharacterized protein could be confirmed in the literature, serving as additional positive controls. For example, the most confident interactor with the Arabidopsis protein AT5G14910 is the chloroplast ribosome protein RPS1; this interaction was independently confirmed in a recent study (Pulido et al., 2018). We also observed instances of interactions between proteins catalyzing consecutive enzymatic reactions, such as a novel interaction between OXP1 and GEP, enzymes catalyzing the last two steps of glutathione degradation. In cases where complexes identified at a stringent FDR lack known members, using the core subunits to query scored interactions often recovers the expected subunits and potential novel interactors. Stable interactions are better recovered, as for recovering the COP9 signalosome core but not its transient COP1 association. As protein interactions can reveal function, we provide a tool to query the map at http://plants.proteincomplexes.org.
Alternative multiprotein assemblies are apparent in the plant lineage
Analysis of protein complexes has revealed that homologous gene products are not always assembled in the same way (Marsh and Teichmann, 2015). We found many cases in which plants appear to have alternate arrangements of interacting proteins relative to other lineages, including the adoption of plant-specific subunits. Furthermore, we identified cases in which plants exhibited analogous assemblies to those in other lineages, achieving similar architectures or functions, but through distinct molecular interactions. In both cases, protein sequence homology alone was inadequate to predict protein complex structure.
One prominent example is a conserved tRNA multi-synthetase complex (MSC). While functional assemblies of essential aminoacyl tRNA synthetases seem ubiquitous across all life forms, distinct architectures, memberships, and accessory proteins have been cataloged in diverse organisms including animals, yeast, archaea, and bacteria (Laporte et al., 2014). They are frequently loosely associated and condition-dependent, making complete composition difficult to define. We observed a conserved megadalton-scale MSC with architecture and accessory subunits distinct from those in animals, fungi, and bacteria but with notable parallels (Figure 6A). Our plant MSC contains the ortholog of ARC1, the central scaffold of the yeast MSC, but lacks the p38, p43, and p18 scaffolds used in the human MSC. It is consistent with a candidate MSC containing ARC1, lysine and isoleucine tRNA synthetases recently reported in Arabidopsis (McBride et al., 2019). Conservatively, however, our plant MSC complex appears to contain ARC1, Ybak (a member of the trypanosome MSC (Cestari et al., 2013)), Clustered Mitochondria Homolog (CLU), the WD40 scaffold protein VIP3/SKI8, and the glutamate, isoleucine, and tryptophan-tRNA ligases (E, I, and W). Peripheral members may include valine, tyrosine, histidine, aspartate, proline, threonine, leucine, glutamine, lysine, and methionine-tRNA ligases (V, Y, H, D, P, T, L, Q, K, and M) (Figure 6A). Of the 20 eukaryotic tRNA ligases, there may be an advantage to assembling this particular set, as 8/9 of the human MSC ligases appear in the plant MSC.
Figure 6. Alternative Assemblies in Plant Analogs of Animal Multi-Protein Complexes.

(A) Plant Multi-tRNA Synthetase Complex (MSC). Top left, elution profiles of proteins observed in large molecular weight complexes containing aminoacyl tRNA synthetases in soy and wheat size exclusion fractionations. Top right, the observed molecular mass (circles) for each protein at left in all plant size exclusion separations in our data set compared to the predicted monomeric mass (triangles). Bottom, schematic of the domain structure and organization of MSC proteins in representative eukaryotic lineages.
(B) A plant proteasome assembly chaperone complex, with orthology of plant PAC2 to the human analog PAC2 indicated with double-headed black arrow. Right, the CF-MS score for the PAC2-PAC2L interaction (blue arrow) far exceeds that of any other protein interaction score with either PAC2 or PAC2L. Gray bars are binned CF-MS interaction scores for all other protein interactions.
(C) A plant transcriptional response module, with orthology of RZ1B/C to the human analog RBMX indicated with double-headed black arrow. Right, the CF-MS score for the RZ1B/C-VRN1 interaction (blue arrow), gray bars as in (B).
(D) Novel subunits of chloroplast NADH dehydrogenase-like complex (NDH). Left, heatmap shows coelution (purple) of known NDH subunits along with three novel interactors in specific plant extracts (arrows below). Middle, network diagram with proteins (circles) connected by interaction lines where line thickness reflects CF-MS score. Right, illustration of conserved molecular architecture and use of rhodanese sulfurtransferase subunit modules in electron transport complexes - two plant-specific (NDH, FNR) and one conserved mitochondrial (Complex I). Bottom, median CF-MS scores to all NDH subunits shown for known NDH subunits and all rhodanese-like domain proteins in plants.
Just as the presence of orthologs could not predict the plant MSC complex, the genetic absence of orthologs does not predict the absence of functionally similar complexes. One example is the complex of proteasome assembly chaperones. In humans, a stable heterodimer of PAC1 and PAC2 aids the assembly of the proteasome alpha subunit ring (Hirano et al., 2005). While plants lack a PAC1 gene, we found a plant-specific PAC2-like protein associated with PAC2 (Figure 6B). No proteasome assembly chaperone complex has been previously described in plants, suggesting that the PAC2/PAC2-like complex likely performs this function.
We also found several instances where plants utilize lineage-specific subunits to co-opt known molecular modules to serve plant-specific functions. One example is a heterodimer of a conserved eukaryotic transcription factor. In humans, RBMX interacts with SAFB to bind the promoter of the SREBP1 gene to regulate sterols in the liver (Omura et al., 2009). We found the plant ortholog of the RBMX transcription factor (RZ1B/C) interacting with the plant-specific protein VERNALIZATION1 (VRN1) (Figure 6C), both of which are known to regulate the FLOWERING LOCUS C (FLC) gene (Levy et al., 2002; Wu et al., 2016) and together control a crucial plant-specific event: rapid flowering at the appropriate seasonal time.
Finally, the chloroplast NADH dehydrogenase-like (NDH) complex provides a more intricate example of how plants have adapted conserved architectural modules with plant-specific proteins to serve plant-specific purposes. NDH is a chloroplast complex that shares architecture with the respiratory Complex I of mitochondria, with both complexes functioning in electron flow (Shikanai, 2016). We identified known NDH subunits and found three additional subunits, EGY1, EGY2, and STR4A (Figure 6D, left). These new subunits have particularly high CF-MS scores for interaction with NDH subcomplex B and L members, and in some cases score higher than interactions among known members of the NDH complex (Figure 6D, network). EGY1 and EGY2 are chloroplast-localized intramembrane metalloproteases, but their specific functions remain unknown (Adamiec et al., 2017). The specific enrichment of EGY1, EGY2, and the NDH complex in Bundle Sheath chloroplasts of C4 plants supports the association of these metalloproteases with NDH in vivo (Majeran et al., 2008). The third new subunit, STR4A, is a rhodanese-like protein of unknown function. While mitochondrial Complex I is known to interact with an accessory rhodanese domain sulfurtransferase (Abdrakhmanova et al., 2005), no such subunit has yet been reported for the architecturally similar plant NDH complex. STR4A is one of 6 rhodanese-like domain proteins in Arabidopsis; however, the CF-MS scores indicate that the association of STR4A with the NDH complex is unique among those six (Figure 6D, bottom right). One clue to the role of STR4A may come from the related protein, STR4, required for localization of the photosynthetic FNR complex to the thylakoid membrane of chloroplasts (Lintala et al., 2014). However, because NDH, FNR, and Complex I are all electron transport complexes, it is also possible that sulfurtransferase activity is important for some shared function in electron transport.
These observations highlight how shared features such as orthology and protein complex architecture can be informative, but as plants have alternatively purposed many proteins and complexes, directly measuring specific protein interactions and assemblies is necessary to understand the functional roles of plant proteins.
Interaction-to-phenotype: Discovering protein functions and phenotypes from protein interactions
Ultimately, the utility of large-scale datasets is their ability to drive biological discovery. Interacting proteins are more likely to share phenotypes (Fraser and Plotkin, 2007; Lage et al., 2007; McGary et al., 2007) (Table S6) and thus our data should provide a basis for linking genotype to phenotype and gaining biochemical insights into shared phenotypes. We therefore interrogated our dataset for cases where the derived interactions might suggest new hypotheses regarding protein function and phenotypes.
We found several links between pathogen defense and immunity genes through protein interactions. Plant pathogen resistance mechanisms are an area of intense research, as pathogens cause billions of dollars in crop losses annually (Savary et al., 2019). We discovered two complexes related to plant-pathogen interactions. The first is comprised of Basic endochitinase B (CHIB) and Osmotin-like protein 34 (OSM34) (Figure 7A), representing two different protein families, Pathogenesis-Related protein group 3 (PR3) and Pathogenesis-Related protein group 5 (PR5) respectively. Each protein has been individually reported to target fungal cell walls, and both are highly co-expressed following infection by the gray mold fungus B. cinerea (Dhawan et al., 2009). Our observations support a stable protein complex, which was unexpected among members of different pathogenesis-related protein classes. Mechanistic characterization of this protein complex could aid the development of strategies to prevent devastating crop losses from fungal infection.
Figure 7. Connecting Plant Genes to Phenotypes via Their Interactions.

The top section of each lettered panel shows sparklines with sample species and tissues indicated above. Bottom left panel of each lettered panel shows the complex interaction score (CF-MS) between subunits (blue arrow) is far greater than the interaction of either subunit with any other observed protein (gray bars representing binned scores).
(A) OSM34 and CHIB form a complex, consistent with co-expression evidence in response to fungal infection (diagram bottom right).
(B) PIP and NUDT3 form a complex in plants. Bacterial members of the PIP and NUDT families are injected into plant cells by a Type III secretion system.
(C) DOMINO1 and LA1 form a plant-specific ribosomal RNA-binding complex and heterozygotes of each have a similar Arabidopsis T-DNA insertion mutant phenotype of abnormal white seeds containing arrested embryos. Bottom left, representative portions of siliques from genotypes as labeled. Lower right, quantification of visually abnormal seeds in three siliques of each genotype. Ratio of normal to abnormal seeds reflects variable penetrance of the mutant phenotype and presence of homozygous and heterozygous embryos in each silique.
(D) Arabidopsis plants homozygous for VDAC2/5 or 3βHSD/D T-DNA insertion mutants show delayed flowering and reduced number of fertile siliques compared to wild type plants of the same stage. Lower panels illustrate fertility defects with main inflorescences at end of flowering. While vdac2 homozygotes produce almost no seeds, 3βhsd/d mutants show a range of fertility levels, ranging from plants with almost no seed-containing siliques to plants in which only early siliques show fertility defects. An enlarged view of wild type and early infertile siliques from plants of the genotypes is shown.
A second novel pathogen-related protein complex contains proline iminopeptidase (PIP) and Nudix hydrolase 3 (NUDT3) (Figure 7B). The native role of these proteins in plants is unclear, but intriguingly, bacterial versions of both PIP (Kan et al., 2018) and Nudix hydrolase (Dong and Wang, 2016; Mukaihara et al., 2004) are injected by bacterial type III secretion systems into plant cells in order to suppress plant immunity. While work remains to determine the native role of plant PIP and NUDT3, the information that these proteins form a stable endogenous complex will guide future efforts. The direct observation of known pathogen resistance proteins in complexes creates a concrete framework for interpreting previous results and examining the mechanism by which they impact plant health.
We also considered a new interaction between the DOMINO1 and LA1 proteins which served to confirm a joint role for the interaction partners in embryonic development. Loss of function of either of these genes is known to produce a nucleolar hypertrophy phenotype and nonviable embryos (Fleurdépine et al., 2007; Lahmy et al., 2004). While LA1 is an RNA-binding protein found in diverse eukaryotes, DOMINO1 is a plant-specific protein with mutations linked to ribosome biogenesis defects and slow embryonic growth (Lahmy et al., 2004). We observed a stable DOMINO1/LA1 complex in multiple plants and tissues (Figure 7C) and confirmed that both subunits affect the same biological process by comparing the phenotypes of individual domino1 or la1 insertional mutant lines of Arabidopsis. Heterozygous domino1 or la1 mutant plant lines produced siliques with many abnormal clear embryos (Figure 7C). These phenotypic similarities support a functional complex of the DOMINO1 and LA1 proteins in vivo with a proposed role in ribosome biogenesis.
Protein interactions also provide a basis to predict entirely new phenotypes. We exploited this trend for a new interaction between the mitochondrial outer membrane porin VDAC2/5 and 3β-hydroxysteroid dehydrogenase/C-4 decarboxylase (3βHSD/D). As vdac2 mutants have a well-characterized late flowering and sterility phenotype (Tateda et al., 2011), we predicted by interaction that 3βhsd/d mutants would show similar defects. We directly compared the phenotypes of Arabidopsis bearing loss of function T-DNA insertions in VDAC2 or 3βHSD/D2. Disruption of either gene delayed flowering, induced wavy leaves, and reduced fertility. (Figure 7D). The underlying defect of these shared phenotypes is likely related to transport and modification of plant sterols, as 3βHSD/D is a sterol modifying enzyme (Kriechbaumer et al., 2018), and VDAC2 is essential in mice for steroidogenesis (Prasad et al., 2015).
CONCLUSIONS
By using mass spectrometry proteomics to define the major protein complexes shared across plants, we have constructed a reference map of the basic biochemical ‘wiring diagram’ of a plant cell. Our deep proteomics data capture over two million protein abundance measurements from multiple tissues and diverse species, revealing stable, abundant protein complexes conserved over more than a billion years of plant evolution. The resulting map thus provides a global snapshot of protein organization in plants. While we have presented examples of paths to connect gene products with phenotypes and to test specific functional hypotheses, this rich dataset can be mined in myriad additional ways, laying the foundations for extensive basic and applied research across the vast landscape of plant biology.
STAR METHODS
LEAD CONTACT AND METHODS AVAILABILITY
Further information and requests for resources and reagents should be directed to the Lead Contact, Edward Marcotte (marcotte@icmb.utexas.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The following samples were obtained directly from producers and collaborators. Growth conditions are unknown or not applicable.
Broccoli heads (Brassica oleracea var. italica), Central Market H-E-B (Austin, TX)
Quinoa (Chenopodium quinoa) seed (husk removed), Central Market H-E-B (Austin, TX)
Hemp hearts “Raw Shelled Hemp Seed” (Cannabis sativa), Trader Joe’s (Austin, TX)
Broccoli leaves (Brassica oleracea var. italica), Barton Springs Nursery (Austin, TX)
Tomato leaves (Solanum lycopersicum), Barton Springs Nursery (Austin, TX)
Selaginella moellendorfii plants, Plant Delights Nursery, Inc., (Raleigh, NC)
Cocos nucifera 4 month old coconuts, Coconut Fields Forever (Davie, FL)
Arabidopsis thaliana ttg1–1 mucilage-less mutant seeds ((Koornneef, 1981), provided by Dr. Alan Lloyd), wheat (Triticum aestivum) germ extract (provided by Dr. Karen Browning)
Chlamydomonas reinhardtii UTEX90
Chlamydomonas was grown by the UTEX Culture Collection of Algae in UTEX Modified Bold 3N Medium on orbital shakers (100–120 rpm) under fluorescent lights with a 12/12 light cycle at 23–25°C.
Maize sprouts (Zea mays)
Maize seeds were planted in Pro-Mix Bx Biofungicide + Mycorrhizae soil with wetting agent supplemented with 1g Miracle-Gro Plant Food/gallon water, and one teaspoon Gnatrol Biological Larvicide (Valent Biosciences LLC). Plants were grown 8 days to V2 stage under an illumination cycle of 16 hours 22°C days and 8 hour 20°C nights.
Arabidopsis thaliana sprouts
Arabidopsis seeds were sterilized for 10 minutes in 20% Clorox bleach and washed five times with 1 mL diH2O. Sterilized seeds were plated on 0.5X Murashige and Skoog agar plates and incubated in darkness at 4°C for 3–5 days, followed by incubation in a lighted growth chamber for 7–10 days, under an illumination cycle of 16 hour 22°C days and 8 hour 20°C nights.
Arabidopsis thaliana (Phenotyping)
50 Arabidopsis seeds per genotype were sterilized and plated as above. Seedlings were transplanted to a soil mix of 75% Pro-Mix Biofungicide with wetting agent/25% Profile Field and Fairway calcined clay supplemented with 1g Miracle-Gro Plant Food/gallon water, and one teaspoon Gnatrol Biological Larvicide (Valent Biosciences LLC). Bonide copper sulfate was sprayed weekly to prevent fungus.
Arabidopsis thaliana Col-0 light and dark grown seedlings
Arabidopsis seeds were sterilized for 15 minutes in 50% Clorox bleach with 0.01% Triton X-100 and washed five times with diH2O. Sterilized seeds were plated on 1X Murashige and Skoog agar plates and incubated at 22°C for 3 days in the dark, followed by growth for 5 days with a cycle of 16 hours light and 8 hours dark, or 3 days in the dark.
Rice leaves (Oryza sativa ssp japonica, Kitaake cultivar)
Dehusked seeds were sterilized for 30 minutes in 30% bleach and rinsed three times with sterile water. Sterilized seeds were plated on 0.5X Murashige and Skoog plates, and incubated with a cycle of 14 hours fluorescent light at 28°C and 8 hours dark at 26°C. After a week, seedlings were translated to a sandy soil mix of 80% sand and 20% Peat (Redi-Gro, Sacramento, CA).
Wheat grass
Unroasted wheat berries (H-E-B, Austin, TX) were sprouted on wet filter paper and grown to approximately 5 cm in height.
Fern (Ceratopteris richardii, Hn-n) fronds
Sporophytes were greenhouse-grown in quart-sized pots on MetroMix 902 (Sun Gro, Agawam, MA), a peat-based medium. Mature green fronds were harvested.
Soy sprouts
Soybeans were planted on nutrient agar as described in (Veerappa et al., 2019), and the aerial portions of the plants were harvested after seven days of growth.
METHOD DETAILS
Native protein extraction
Unless otherwise stated, samples were quick-frozen and ground to a fine powder in liquid nitrogen using a chilled mortar and pestle. Powder was thawed to 4°C, and resuspended in approximately an equal volume of the specified lysis buffer plus protease and phosphatase inhibitors followed by nutation 30 minutes at 4°C. All subsequent steps were at 4°C. The crude homogenate was clarified by a low-speed spin (~3000 × g, 10 minutes), and the supernatant further clarified by a high-speed spin (~14,000 × g, 10 minutes). Protein concentration was determined by Bio-Rad Protein Assay or Bio-Rad DC Protein Assay, and 1–4 mg of extract was 0.45μm filtered (Ultrafree-MC HV Durapore PVDF, Millipore) and fractionated by HPLC chromatography. In general, detergent and salt were minimized where possible to avoid perturbing protein assemblies. Because the highly abundant plant protein RuBisCo dominates green tissues, we included etiolated seedlings (depleted for photosynthetic proteins) and two non-green tissues: germ tissues (enriched in proteins related to core cell biology), and isolated nuclei (enriched for DNA/transcription-related proteins) (Figure S1).
Green plant tissue extracts
Liquid nitrogen powders of leaves and/or sprouts of Arabidopsis, rice, wheat, broccoli, soy, tomato, fern, and Selaginella moellendorfii were lysed in Plant Lysis Buffer: 50 mM Tris-HCl pH 7.5 at room temperature, 150 mM NaCl, 5 mM EGTA, 10% glycerol, 1% NP40, 1 mM DTT, plant-specific protease inhibitors (Sigma) and phosphatase inhibitors (PhosSTOP EASY, Roche).
Embryonic plant tissue/seed extracts
Liquid nitrogen powders of quinoa, hemp seed, or Arabidopsis thaliana mucilage-less mutant ttg1–1 seeds were lysed in Wheat Germ Lysis Buffer: 20 mM HEPES pH 7.6, 130 mM K-acetate, 5 mM Mg acetate, 0.1 mM EDTA, 10 % glycerol, 1 mM DTT, protease and phosphatase inhibitors as above. In some cases, the crude homogenate was prefiltered through Miracloth (Millipore) to remove debris, or 14,000 × g supernatant required additional clarifying centrifugation of 10 minutes at 40,000 × g. Wheat germ extract was prepared as in (Browning and Mayberry, 2006).
Nuclear extracts
Nuclei were prepared from fresh broccoli using the CelLytic PN kit (Sigma-Aldrich). Briefly, fresh floret tips were shaved using a sharp knife into a beaker on ice and homogenized in a chilled mortar and pestle on ice in the Nuclear Isolation Buffer provided. The homogenate was further processed by brief treatment on ice with a Tissue Tearor (BioSpec Products, Inc.). Manufacturer’s instructions were followed for lysis using 1.0 % Triton X-100 and the “Semi-pure Preparation of Nuclei” protocol with 1.5 M sucrose. The resulting nuclear pellet was extracted twice with Nuclear Extraction Buffer, and the two extraction supernatants were pooled.
Coconut (Cocos nucifera) nuclei were purified using a composite method based on (Cutter et al., 1952; Matzke et al., 1992; Mondal et al., 1972). A young (5 month old, ~ 1.5 kg) green coconut supplied fresh from Coconut Fields Forever (Davie, FL) was opened, the liquid was drained, and the gelatinous endosperm removed by gentle scraping. Four volumes of ice-cold 4% sucrose were added to the endosperm and the mixture was homogenized 15 strokes with a loose pestle. All subsequent steps were on ice or at 4°C. The homogenate was filtered through cheesecloth to remove debris, and the nuclei were recovered from the filtrate by centrifugation at 250 × g, 10 min, 4°C. Nuclei were washed once with 15 ml Nuclear Isolation Buffer from the CelLytic PN kit (Sigma-Aldrich) and re-pelleted. After a second wash in 0.5 ml Nuclear Isolation Buffer, the recovered nuclear pellet was extracted as for broccoli nuclei using Nuclear Extraction Buffer (CelLytic PN kit, Sigma-Aldrich).
Chlamydomonas reinhardtii culture (2.5 liters of UTEX90) was pelleted by centrifugation 10 minutes at 1,100 × g with no brake and washed with 900 ml 10 mM HEPES pH 7.6 at room temperature. Washed cells were collected by centrifugation 10 minutes 1,800 × g room temperature with no brake and the pellet was frozen in liquid nitrogen, thawed, refrozen and ground in a liquid nitrogen-chilled mortar and pestle. Powder was thawed and refrozen for a second grinding. After resuspension in Plant Lysis Buffer, cells still appeared largely intact so the homogenate was sonicated on ice with a probe tip 6 × 10 sec, amplitude 28%, and 6 × 10 sec amplitude 70%. This lysate was incubated 30 minutes 4°C rotating end-over-end with subsequent steps as for other green tissues.
Fern transcriptome sequencing
As Ceratopteris richardii currently lacks an annotated genome, we de novo assembled the fern transcriptome from mRNA sequencing data collected from fronds, mature gametophytes, and spores as follows:
RNA sequencing
Spores were collected from adult plants and cultured on agar to gametophyte stage. Fronds were cut from healthy adult plants and flash-frozen in liquid nitrogen. Both fronds and gametophytes were ground to a fine powder prior to RNA extraction. Total RNA was extracted from spores by phenol-chloroform extraction, as previously described (Salmi and Roux, 2008). Total RNA was extracted from gametophytes, and fronds with the Spectrum Plant Total RNA Kit (Sigma-Aldrich) according to manufacturer’s instructions. Total RNA from each sample was diluted to 10–100 ng/μl and further prepared and sequenced by the Genome Sequence and Analysis Facility at UT Austin as follows: Total RNA was Poly-A-selected using the Poly(A)Purist Magnetic Kit (Life Technologies AM1919) and fragmented to ~200 bp. First strand synthesis was performed using the NEBNext First Strand Synthesis Module (NEB E7525L) with random primers and reverse transcriptase in the presence of Actinomycin D and Murine RNase Inhibitor. Second strand synthesis was performed using the NEBNExt Ultra Directional RNA Second Strand Synthesis Module (NEB E7750L) for 1 hour 16°C, and DNA was purified on AMPURE XP Beads (Beckman Coulter A63881). Double-stranded DNA was subjected to end repair and dA-tailing using the NEBNext End Repair and dA-Tailing Modules (NEB E6050L and E6053L) followed by ligation of barcode adapters (IDT) with T4 ligase (NEB). Following PCR amplification, the quality of the final library was confirmed by Bioanalyzer (Agilent). We collected 2×150 bp RNA-seq reads from fern spore, mature gametophyte, and adult frond on an Illumina HiSeq 4000 with a target of 30 million reads per tissue.
Fern transcriptome assembly
After removing low-quality reads (reads lacking all four nucleotides or with a no-call), we assembled transcripts with Velvet (version 1.2.06) and Oases (version 0.2.06) using each of five k-mer values (k=45, 55, 65, 75, 85). Also, we converted .fastq file to a non-redundant .fasta file and performed separate de novo transcriptome assembly with k=35, 45, 55, 65, and 75. Assembled transcripts were combined for each tissue, then redundant or fragmented sequences were removed based on BLASTN analysis. We determined the translational reading frame and corresponding peptide sequences from each assembled transcript based on BLASTP mapping results (after 6-frame translation in silico) to four plant reference proteome databases (Creinhardtii_169, Osativa_193_pep, Smoellendorffii_91_pep, TAIR10). Sequences lacking high-scoring BLASTP scores to the reference proteomes were considered to be non-coding and omitted from the resulting fern proteome database. The resulting protein sequences derived from the three tissues were combined and a non-redundant protein sequence set computed based on clustering with UCLUST (version 4.2.66), requiring >97% amino acid identity. The supporting code is available from the NuevoTx repository (https://github.com/taejoonlab/NuevoTx). Fern transcriptome and proteome assembly statistics are summarized in Table S7.
HPLC chromatography
Lysates were fractionated on a Dionex UltiMate3000 HPLC system consisting of an RS pump, Diode Array Detector, PCM-3000 pH and Conductivity Monitor, Automated Fraction Collector (Thermo Scientific, CA, USA) and a Rheodyne MXII Valve (IDEX Health & Science LLC, Rohnert Park, CA) using biocompatible PEEK tubing and either size exclusion chromatography or one of two ion exchange separations (mixed bed, or triple-phase WAXWAXCAT, see below). The sample loaded was 1–4 mg protein as measured by the BioRad Protein Assay or DC Protein Assay as appropriate to the sample buffer. Fractions were collected into 96-deep well plates. Select support ribs in the base were notched with a single-edged razor blade prior to fraction collection to accommodate subsequent use of the Life Technologies magnetic plate for bead-based mass spec preparation of samples.
Size exclusion
BioSep-SEC-s4000 600 × 7.8 mm ID, particle diameter 5 μm, pore diameter 500 Å (Phenomenex, Torrance, CA). Unless otherwise specified sample was 200 μl, flow rate 0.5 ml/min, with fraction collection every 45 seconds, and mobile phase 50 mM Tris-HCl pH 7.5, 50 mM NaCl. For the maize experiment the mobile phase was PBS pH 7.2 (Gibco) and for all nuclear extract experiments, Buffer S-NE (20 mM HEPES-KOH pH 7.5, 300 mM KCl, 1.5 mM MgCl2, and 0.2 mM EDTA). For molecular weight estimation, molecular weight standards (Sigma -Aldrich, MWGF1000, 2–5 ug each of Thyroglobulin (T9145), β-amylase (A8781), and bovine serum albumin (A8531)) were added to 1.2 mg soy extract prior to fractionation with Buffer S-NE as mobile phase.
Mixed bed ion exchange
Poly CATWAX A (PolyLC Mixed-Bed WAX-WCX) 200 × 4.6 mm ID, Particle diameter 5 μm, pore diameter 100 Å (PolyLC Inc., Columbia, MD). The bed contains the cation-exchange (PolyCAT A) and anion-exchange (PolyWAX LP) materials in equal amounts. A 200–250 μl sample was loaded at ≤ 40 mM NaCl, and eluted with a 1-hour salt gradient at 0.5 ml/min with collection of 0.5 ml fractions. Gradient elution was performed with Buffer A (10 mM Tris-HCl pH 7.5, 5% glycerol, 0.01% NaN3), and 0–70% Buffer B (1.5 M NaCl in Buffer A).
Triple phase ion exchange
Three columns, each 200 × 4.6 mm ID, particle diameter 5 μm, pore diameter 100 Å, were connected in series in the following order: two PolyWAX LP columns followed by a single PolyCAT A (PolyLC, Inc, Columbia, MD). Loading, buffers, and fraction collection were as for mixed bed ion exchange above with slight modifications in flow rate and elution from the methods of (Havugimana et al., 2012). The flow rate was 0.25 ml/min with a 120 min gradient from 5–100% Buffer B. For the separation of nuclear extracts, the gradient was modified to a 115-minute multiphasic elution from 5–100% Buffer B.
Isoelectric focusing
Isoelectric focusing was performed using the Agilent 3100 OFFGEL Fractionator (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s application note for native protein separation (Babu CV and Palaniswamy, 2014). Samples were separated into 24 fractions using the 24 cm IPG strips pH 3–10 NL (GE HealthCare, Chicago, IL). To achieve the required low salt concentration 2.5 mg wheat germ extract was diluted with water and centrifuged 10 min., 10,000 × g 4°C prior to dilution with OFFGEL Stock Solution. Broccoli nuclear extract was dialyzed in a Slide-A-Lyzer Cassette G2 with a 10,000 MWCO (Thermo Scientific, CA, USA) against 2 changes, 2 hours each, of 5.0 mM HEPES-KOH pH 7.5, 25 mM NaCl, 0.2 mM DTT followed by one change of 0.5 mM HEPES-KOH pH 7.9 overnight. A substantial precipitate formed so the solution was clarified by centrifugation 12,000 × g 10 min, 10°C, and the soluble portion further concentrated in an Amicon Ultra filter unit with a 10 kD MWCO (MilliporeSigma, Burlington, MA). BioRad Protein Assay determined that this soluble portion retained 1.5 mg (~ 35% of the predialysis amount) which was diluted with OFFGEL Stock Solution for focusing.
Mass spectrometry sample preparation
Samples were prepared for mass spectrometry in 96-well plate format using either ultrafiltration and in-solution digestion or SP3 bead-binding of proteins and on-bead digestion ((Hughes et al., 2014) with modifications as described below). Plates were sealed with transparent film during incubation steps.
Ultrafiltration was performed with an AcroPrep Advance 96-filter plate,3k MWCO (Pall) using a vacuum manifold (QIAvac 96 or Multiwell, Qiagen) at −0.75 Bar. Prior to the application of the samples, preservatives were removed from the membranes by sequential filtration of 100 μl LC/MS quality water and 100 μl trypsin digestion buffer (50 mM Tris-HCl, pH 8.0, 2 mM CaCl2). Samples were concentrated to 100 μl, diluted 2 fold with trypsin digestion buffer, and concentrated again to a final volume of 50–100 μl before being transferred back to a 96-deep well plate. 50 μl 2,2,2,-trifluoroethanol (TFE) was added and samples were reduced with TCEP (Bond-Breaker, Thermo,) at a final concentration of 5 mM for 30 min. 37°C. Iodoacetamide was added to 15 mM and plates were incubated in the dark 30 min. at room temperature. Alkylation was quenched by the addition of DTT to 7.5 mM. TFE was diluted to <5% by the addition of trypsin digestion buffer. 1 μg of trypsin was added to each fraction and the sealed plate was incubated 37°C overnight. Digestion was stopped by bringing samples to 0.1% formic acid, and peptides were desalted using a 5–7 μl C18 Filter Plate (Glygen Corp) with a vacuum manifold, dried, and resuspended for mass spectrometry in 3% acetonitrile, 0.1% formic acid.
Bead-based sample preparation was limited primarily to SEC fractionation samples due to interference of salt concentrations greater than 300 mM. Beads used were an equal mixture (vol:vol ) of SpeedBead Magnetic Carboxylate Modified Particles #45152105050250 and #65152105050250 (GE Healthcare, UK). Fractions were first adjusted to 20% TFE and reduced with 5mM TCEP, 45 min, 37°C. Samples were alkylated by the addition of Iodoacetamide to 25mM final concentration, 30 minutes in the dark at room temperature, and then the reaction was quenched with 15 mM DTT. 4 μl of the mixed bead suspension (5 μg/μl of each bead type) was added to each fraction. Protein binding was initiated by the addition of a premix of formic acid (to 20% of the alkylated sample) and acetonitrile (to equal 50% of final bead incubation volume. After 30 min. at room temperature with gentle rocking, the beads were pelleted by centrifugation (1000 × g, 5 min., room temperature). The deep well plate was placed on a magnetic plate (Life technologies) and all but 300 μl of the supernatant removed and discarded. After removing the sample plate from the magnet the beads were resuspended in the remaining 300 μl and transferred to a conical bottomed 450 μl 96-well plate and placed on the magnetic plate for all wash steps.
After beads were collected, the supernatant was removed and discarded and beads were washed rapidly and sequentially with 2 × 100 μl aliquots of 70% ethanol and 2 × 100 μl aliquots of acetonitrile. The plate was removed from the magnet and beads were allowed to air dry briefly before resuspension in 25 μl of 10% TFE/90% trypsin digestion buffer. 25 μl of trypsin digestion buffer containing 0.25 μg trypsin was then added to each resuspended sample for digestion overnight, 37°C. The digestion plate was placed on the magnet and the supernatant containing digested peptides was transferred to a fresh 450 μl 96-well plate and digestion stopped by bringing samples to 1% formic acid. Further peptides were recovered from the beads with two successive elution steps using 50 μl 2% DMSO and pooled with the original peptide supernatant. During the first DMSO elution, the plate was sealed and sonicated in a bath for 10 min. Peptides in the 150 μl total eluates were desalted as above.
Chemical cross-linking
Extract (2.5 mg soy sprouts, or 2.1 mg Chlamydomonas) was fractionated by size exclusion as above but with PBS pH 7.2 (Gibco) as the mobile phase. DSSO (Thermo Scientific) was dissolved immediately before use in dry dimethylformamide (stored under nitrogen) at a concentration of 50 mM and then further diluted in PBS for a working stock. Immediately after fractionation working stock DSSO was added to fractions to a final concentration of 0.5 mM and samples were incubated 1 hour at room temperature. Cross-linking was quenched by the addition of 1 M Tris pH 8.0 to a concentration of 24 mM. Cross-linked fractions were immediately frozen and stored −80°C until prepared for mass spectrometry using the ultrafiltration and in-solution digestion method. Soy samples were digested as usual with trypsin, while Chlamydomonas samples were split after reduction/alkylation and treated as follows. One set was digested with trypsin under standard conditions while the other set was diluted 8 fold with 50 mM Tris-HCl pH 8.0, 10 mM CaCl2 and digested with 0.5 μg chymotrypsin overnight 37°C. Both digests were stopped by adjusting samples to 0.1% formic acid. Peptides were desalted as above and dissolved in 20 μl 5% acetonitrile, 0.1% formic acid for mass spectrometry.
Mass spectrometry data acquisition and processing
Acquisition
Mass spectra were acquired using one of three Thermo mass spectrometers: Orbitrap Elite, Orbitrap Fusion, or Orbitrap Fusion Lumos. In all cases, peptides were separated using reverse phase chromatography on a Dionex Ultimate 3000 RSLCnano UHPLC system (Thermo Scientific) with a C18 trap to Acclaim C18 PepMap RSLC column (Dionex; Thermo Scientific) configuration. Peptides were eluted using a 5–40% acetonitrile gradient in 0.1% formic acid over 120 min. (for Orbitrap Elite), or a 3–45% gradient over 60 min. (for Fusion and Lumos) and directly injected into the mass spectrometer using nano-electrospray for data-dependent tandem mass spectrometry.
Data acquisition methods were as described below for each machine:
Orbitrap Elite: top 20 CID with full precursor ion scans (MS1) collected at 60,000 m/z resolution. Monoisotopic precursor selection and charge-state screening were enabled, with ions of charge > + 1 selected for collision-induced dissociation (CID). Up to 20 fragmentation scans (MS2) were collected per MS1. Dynamic exclusion was active with 45 s exclusion for ions selected twice within a 30 s window.
Orbitrap Fusion: top speed CID with full precursor ion scans (MS1) collected at 120,000 m/z resolution and a cycle time of 3 sec. Monoisotopic precursor selection and charge-state screening were enabled, with ions of charge > + 1 selected for collision-induced dissociation (CID). Dynamic exclusion was active with 60 s exclusion for ions selected once within a 60 s window. For some experiments, a similar top speed method was used with dynamic exclusion of 30 s for ions selected once within a 30 s window and high energy-induced dissociation (HCD) collision energy 31% stepped +/−4%. All MS2 scans were centroid and done in rapid mode.
Orbitrap Lumos: top speed HCD with full precursor ion scans (MS1) collected at 120,000 m/z resolution. Monoisotopic precursor selection and charge-state screening were enabled using Advanced Peak Determination (APD), with ions of charge > + 1 selected for high energy-induced dissociation (HCD) with collision energy 30% stepped +/− 3%. Dynamic exclusion was active with 20 s exclusion for ions selected twice within a 20 s window. All MS2 scans were centroid and done in rapid mode.
For identification of DSSO cross-linked peptides, peptides were resolved using a reverse phase nanoflow chromatography system with a 115 min 3–42% acetonitrile gradient in 0.1% formic acid. The top speed method collected full precursor ion scans (MS1) in the Orbitrap at 120,000 m/z resolution for peptides of charge 4–8 and with dynamic exclusion of 60 sec after selecting once, and a cycle time of 5 sec. CID dissociation (25% energy 10 msec) of the cross-linker was followed by MS2 scans collected in the orbitrap at 30,000 m/z resolution for charge states 2–6 using an isolation window of 1.6. Peptide pairs with a targeted mass difference of 31.9721 were selected for HCD (30% energy) and collection of rapid scan rate centroid MS3 spectra in the ion trap.
Computational analyses
Proteomes
Reference proteomes for Arabidopsis thaliana (ARATH), Brassica oleracea (BRAOL), Solanum lycopersicum (SOLLC), Glycine max (SOYBN), Oryza sativa var. Japonica (ORYSJ), Triticum aestivum (WHEAT), Zea mays (MAIZE), Selaginella moellendorffii (SELML), Chlamydomonas reinhardtii (CHLRE), were downloaded from Uniprot.org (UniProt Consortium, 2019) in August 2018. The Chenopodium quinoa (CHQUI) proteome was the Cquinoa_392_v1.0 assembly (Jarvis et al., 2017) downloaded from https://phytozome.jgi.doe.gov (Goodstein et al., 2012), the Cocos nucifera (COCNU) proteome (Armero et al., 2017) was downloaded from http://palm-comparomics.southgreen.fr, and the Cannabis sativa (CANSA) proteome was the canSat3 Purple Kush assembly (van Bakel et al., 2011) downloaded from http://genome.ccbr.utoronto.ca). The Ceratopteris richardii (CERRI) proteome was assembled de novo as described above.
Initial assignment of peptide mass spectra
Peptide inference was performed with MSGF+, X!Tandem, and Comet-2013020, each run with 10ppm precursor tolerance, and allowing for fixed cysteine carbamidomethylation (+57.021464) and optional methionine oxidation (+15.9949). Peptide search results were integrated with MSBlender (Kwon et al., 2011), https://github.com/marcottelab/msblender, https://github.com/marcottelab/run_msblender). For DSSO cross-linked experiments, inter-protein cross-links were identified using the XlinkX (Klykov et al., 2018) node in ProteomeDiscover 2.2 (ThermoScientific).
Orthogroup assignment
Each plant proteome was searched against EggNOG viridiplantae (virNOG) orthogroup HMMs using eggNOG-mapper v1 (Huerta-Cepas et al., 2017). Additionally, human and Arabidopsis proteomes were searched with the eggNOG-mapper against eukaryote (euNOG) orthogroup HMMs to allow annotation transfer of known human protein complexes. The set of proteins from all species assigned to the same orthogroup HMM were considered to belong to the same orthogroup.
Analysis of peptide-spectral matches at the level of orthogroups
All proteomes were in silico trypsin digested to peptides, allowing for two missed cleavages and a missed cleavage when lysine or arginine are followed by proline. Within each proteome, only peptides from proteins within the same orthogroup were retained, and peptides matching proteins in multiple orthogroups discarded. This gave a list of orthogroup-unique peptides sometimes deriving from multiple proteins. Experimentally-observed peptides were compared to these orthogroup unique peptides with allowance for leucine/isoleucine ambiguity to identify orthogroups present in each biochemical fraction.
Peptide spectral matches (PSMS) of peptides in the same orthogroup were summed to get orthogroup PSM counts, as follows: For a given mass spectrometry experiment, for each orthogroup, we summed the PSMs that could be uniquely attributed to that orthogroup. In this way, we avoided double-counting PSMs across near duplicate proteins, and we did not otherwise have to consider the number of proteins or their relative lengths within each orthogroup in a species.
Analysis of overall trends in protein recovery
We observed in at least one experiment 96.7% of the 11,339 Viridiplantae orthogroups that are most highly conserved i.e. conserved across at least half (7) of our 13 plant species. Of the 378 unobserved yet conserved orthogroups, 170 are membrane proteins suggesting their lack of detection may stem from non-ideal solubilization conditions for this class of proteins. The high coverage of conserved proteins was in marked contrast to the remaining 22,144 less conserved orthogroups, of which we only observed 58.4%. Observed proteins also tend to be longer than unobserved proteins, with a median length of 383 vs. 196 amino acids. Our observations mirror those for the extensively studied human proteome where notably 1,482/20,055 proteins have thus far evaded detection by all available proteomics technologies (Baker et al., 2017). We observed consistent tissue-specific functional trends among the observed proteins (Figure S1), supporting the idea that the not-yet-observed proteins, whether in plants or humans, may tend to exhibit strongly tissue or temporal specific expression or universally low abundance. Orthogroup abundances were highly reproducible across experiments (Figure S4).
Assembly of features for scoring putative protein interactions
For each experiment, we assembled an elution matrix of orthogroups by fractions as follows: We normalized each orthogroup’s elution profile on a per fractionation experiment basis by rescaling the maximum PSM count to one. This allowed us to compare elution profiles across orthogroups within an experiment in spite of e.g. different numbers of genes in different orthogroups. Across species, this also had the effect of normalizing observations from members of the same orthogroup, correcting automatically for potential length differences or differing orthogroup expansions.
We additionally concatenated our training set of 32 experiments into sets by species and taxonomic group, i.e. all Arabidopsis experiments joined into one matrix and likewise for all eudicot, monocot, angiosperm, vascular plant, and green plant experiments. Maize was withheld from these concatenations as a holdout species. An elution matrix of all experiments was also made (DOI:10.5281/zenodo.3666942). Cross-linked soy and cross-linked Chlamydomonas experiments were not used in training. We retained only orthogroups with at least 32 total PSMs observed across the 32 combined training experiments.
We next calculated a series of all-by-all pairwise scores between orthogroup elution profiles for all 46 training matrices – Pearson’s r, Spearman’s rho, Euclidean distance, Bray-Curtis similarity, and stationary cross-correlation, all with added Poisson noise. Euclidean distance and Bray-Curtis similarity scores were inverted and normalized to a max score of 1. We calculated a hypergeometric score for the co-occurrence of proteins in fractions with repeated sampling of fractions (Drew et al., 2017). Prior to building a feature matrix of these scores for machine learning, we removed orthogroup pairs that did not correlate with at least a Pearson r > 0.3 in at least three experiments spanning at least two species, thus explicitly requiring potential interactions to exhibit some degree of reproducibility and evolutionary conservation. (Thus, high-confidence interactions present in only one species would not be captured. Also, we made no attempt to identify differential interactions across plants or tissues.) All scores were joined to a 3,076,998 row feature matrix of orthogroup-orthogroup similarity scores, and missing values filled with zeros.
Construction of the gold standard protein complex training and test sets
We used known human protein complexes from the CORUM database (Giurgiu et al., 2019) as a gold standard set of positive stable protein-protein interactions. Human protein identifiers were converted to virNOG orthogroup identifiers via orthology to Arabidopsis proteins. 397 CORUM protein complexes were supplemented with 6 well-characterized plant protein complexes. Known complexes were divided into positive training and test complexes according to the scheme from (Drew et al., 2017) and complexes with over 30 members removed. We additionally supplemented 8,662 CORUM pairwise interactions with 2,562 pairs of plant proteins with direct evidence of stable protein-protein interaction, e.g. co-crystallization, co-immunoprecipitation, collected from the TAIR (Swarbreck et al., 2008) protein-protein interactions repository (ftp://ftp.arabidopsis.org/home/tair/Proteins). 20,000 negative training and test interactions were drawn from feature matrix rows, removing any interactions present in the positive gold standard set. 1240 positive training interactions and 884 positive test interactions were present in the feature matrix.
Identification of interacting proteins by supervised machine learning
We first used the scikit-learn ExtraTreesClassifier feature selection to reduce the dimensionality of the feature matrix to the top 100 features based on declining feature importance (Figure S2). We used the TPOT (Olson and Moore, 2016) AutoML wrapper of scikit-learn machine learning functions for all subsequent training steps. We discovered optimal hyperparameters for an ExtraTreesClassifier with 5-fold cross-validation, with an area under the precision-recall curve of 0.64. We then trained an Extra Trees Classifier with TPOT discovered hyperparameters. This model was applied to the entire feature matrix to give a Co-Fractionation Mass Spectrometry (CF-MS) score to each pair of orthogroups, with higher scores corresponding to higher co-elution. Precision, recall, and false discovery rates were calculated from 886 positive and 20,000 negative test set interactions. Although we only required high correlation in two species, a majority of discovered interactions show high correlation in at least ten species (Figure S2).
Clustering of interacting protein pairs to define multiprotein assemblies
Interaction scores above a 10% false discovery rate threshold (CF-MS score ≥ 0.509) were input into R igraph cluster_walktrap to define coherent protein complexes. Walktrap reweighted edges between orthogroups were reformatted to a dendrogram and cut at intervals to obtain a nested hierarchy of complexes. Cuts closer to the root of the dendrogram result in larger complexes and cuts closer to the tips further define subcomplexes. A portion of these clusters are homodimers or heterodimers of closely related proteins. Examples include two 16 kDa, pI 4–5 ferredoxins (FD3 and FD1/2) that have a CF-MS score of 0.6 and two 43–46 kDa, pI 9 NTF2-like proteins (NTF2L2 and NTF2L1) that have a CF-MS score of 0.6. The similarity of both size and charge makes it difficult to discern whether these proteins form homomeric or heteromeric complexes.
Size calibration
Based on known molecular weight size standards spiked into one soy size exclusion experiment, we fit a linear model of log10 molecular weight ~ fraction number. To transfer this calibration to other size exclusion experiments, we selected a series of internal standards complexes and proteins with known native molecular weights and consistent single peak elution patterns. A model derived from molecular weight standard spike-ins of 667, 443, 200 and 66 kDa was able to predict molecular weight values for our internal standards close to their known elution positions (Figure S5). We fit a new linear model for derived weights of internal standards and applied this model to all size exclusion experiments to obtain a molecular weight for each fraction.
External datasets
For comparison purposes, known plant protein-protein interactions were downloaded from the HitPredict database (López et al., 2015) and mRNA co-expression linkages from AraNet and RiceNet (Lee et al., 2010, 2011). RNA expression Transcripts per Million (TPM) for Chlamydomonas and Arabidopsis were downloaded from the Expression Atlas (Papatheodorou et al., 2018), experiment codes E-GEOD-62671, E-GEOD-38612, E-GEOD-55866, and E-GEOD-30720. Loss-of-function annotations were assembled from (Lloyd and Meinke, 2012), Uniprot, and TAIR, and phenotype ontology obtained from the Plant PhenomeNET project (Oellrich et al., 2015). Additionally, Arabidopsis protein molecular weights, GO annotations, functions, unipathway, BioCyc, Reactome, BRENDA, enzyme commission, and tissue were downloaded from Uniprot as annotations to guide interpretation.
Protein Parts Per Million (PPM) calculation
We calculated protein parts per million (PPM) following (Weiss et al., 2010), with scripts stored at github.com/marcottelab/MS_grouped_lookup/ppm_utils. Briefly, unique tryptic peptides were filtered to peptides between 7 and 40 amino acids long, and a correction factor calculated from the sum of the total length of peptides in this range per orthogroup. Observed peptide PSMs were multiplied by the peptide length, summed by orthogroup, divided by the orthogroup correction factor, multiplied by 1,000,000 and divided by the experiment total to get parts per million.
3D homology modeling
Precomputed 3D homology models from SWISS-MODEL (Bienert et al., 2017) were used to evaluate consistency with cross-linking data as follows: SWISS-MODEL 3D models of Arabidopsis CCT subunits were aligned to the known experimental Saccharomyces cerevisiae CCT molecular assembly (PDB: 4V94), then soy CCT subunit cross-links positioned as guided by sequence alignment to the corresponding Arabidopsis subunits. Chlamydomonas cross-links were evaluated using SWISS-MODEL 3D models of Chlamydomonas Photosystem II subunits that were aligned to the Arabidopsis Photosystem II (PDB: 5MDX). 3D models were visualized using Chimera (Pettersen et al., 2004).
Arabidopsis phenotyping
T-DNA mutant insertion line seeds from the SALK T-DNA insertion collection (O’Malley et al., 2015) were obtained from the Arabidopsis Biological Resource Center ( https://abrc.osu.edu), and wild-type Col-0 seeds were provided by the Z.J. Chen lab. Purchased strains were SALK_056025 (3βhsd/d2, AT2G26260), CS832348/SAIL_726_H02 (vdac2, AT5G67500), CS1002787/SK15485(domino1, AT5G62440), CS823259/SAIL_548_H11 (la1, AT4G32720). All mutant lines except CS832348 were grown an additional generation prior to phenotyping and quantitation. Days to flower were marked from the introduction of plates to light to the first visible petal, and siliques were counted at the end of flowering.
Genotyping
A portion of rosette leaf from each plant was flash-frozen in Eppendorf tubes in liquid nitrogen and ground to a fine powder with sterile rods. 400 μl extraction buffer (200 mM Tris-Cl pH 7.5, 250 mM NaCl, 25 mM ETDA, 0.5% SDS) was added to each tube and mixed briefly. Tubes were centrifuged at 3000 rcf for 7 minutes, then 350 μl supernatant transferred to a 96 well 2 mL plate prefilled with 350 μl isopropanol per well, mixed by pipette, and incubated at room temperature for 10 minutes. Following centrifuging for 35 minutes at 3000 rcf, supernatant was removed by quickly inverting the plate. 150 μl ethanol was added to each well, plates pulse spun, and supernatant again poured off with quick inversion of the plate. After air-drying for at least 15 minutes DNA was resuspended in 150 μl H2O.
PCR reactions to confirm the genotyping were set up as two separate reactions per mutant, one for wild type with a gene specific left primer (LP) and right primer (RP), and a second with the gene specific right primer (RP) and a left border primer for the T-DNA insert (L3 or LBb1.3), according to the instructions at http://signal.salk.edu/tdnaprimers.2.html. PCR cycle conditions were: denature 98°C, anneal 55°C, extend 72°C for 35 cycles.
Primers
SALK T-DNA insertion line primers
LP_SALK_056025_500maxn TGACAATAGGTGGAGTGGTCC
RP_SALK_056025_500maxn AGCTGCCATATGAAACACCAC
LBb1.3 ATTTTGCCGATTTCGGAAC
Syngenta T-DNA insertion line primers:
LP_SAIL_726_H02_VDAC CCATCAGGAGCTAGGCCTAAC
RP_SAIL_726_H02_VDAC TAAGCAGCGCACCTAAAGAAG
LP_SAIL_548_H11 GTCTTTGCTGGTCAGGAGTTG
RP_SAIL_548_H11 CTTCTGAGATTTGTTCCAGCG
LP_SK15485 AATCCGAATACCGAATATCGG
RP_SK15485 TAAATTGGACTCCTTTGCAGC
L3 TAGCATCTGAATTTCATAACCAATCTCGATACAC
QUANTIFICATION AND STATISTICAL ANALYSES
All statistical analyses were performed using the statistical software R, with code provided in DOI:10.5281/zenodo.3466034. Numbers of independent experimental analyses of each species, including counts of proteins observed and peptide-spectral counts, are provided in Table S2.
DATA AND CODE AVAILABILITY
The interaction data are publicly accessible via a dedicated web portal (http://plants.proteincomplexes.org). All raw and interpreted mass spectrometry data were deposited to the ProteomeXchange http://www.proteomexchange.org/ via the PRIDE (Perez-Riverol et al., 2019) partner repository with identifiers (PXD012810, PXD012865, PXD012969, PXD013004, PXD013080, PXD013093, PXD013198, PXD013213, PXD013214, PXD013264, PXD013280, PXD013281, PXD013282, PXD013300, PXD013320, PXD013321, PXD013322, PXD013369 PXD013704, PXD013735, PXD014617). Full documentation of computational analyses, non-external analysis scripts, and project data files are deposited at Zenodo (Full analysis: DOI:10.5281/zenodo.3466034, Elution data: DOI:10.5281/zenodo.3666942, Protein interactions: DOI:10.5281/zenodo.3666940). Analyses made use of the following github.com/marcottelab repositories: MS_grouped_lookup, protein_complex_maps, run_TPOT, MSblender, and run_MSblender. Fern (Ceratopteris richardii) transcriptome data and assemblies were deposited into the European Nucleotide Archive http://www.ebi.ac.uk/ena/ with accession number PRJEB33372, and proteome deposited at Zenodo (DOI:10.5281/zenodo.3467770).
Supplementary Material
Figure S1. Proteome properties, Related to Figure 2. (Top) Tissue enrichment of COG functional categories by tissue. Proportion of total spectral counts for each tissue annotated with each high-level COG category. Nuclear samples show enrichment for information and depletion for metabolism. 15-20% of spectral counts from all tissues derive from orthogroups with no COG annotation. (Bottom) Arabidopsis mRNA vs protein abundances. RuBisCo is observed to have relatively moderate mRNA transcript levels across Arabidopsis tissues but is frequently the most abundant protein in our experiments. The median protein abundance of RuBisCo protein in Arabidopsis leaves was plotted against RNA abundance for three separate green Arabidopsis tissues (Gan et al., 2011; Liu et al., 2012, 2016). Titles indicate accession numbers from the ExpressionAtlas database (Petryszak et al., 2016).
Figure S2. Interaction properties, Related to Figure 3. (A) Feature selection for protein-protein interaction scoring, as measured by scikit-learn Feature Importance. The top-ranked 100 features were selected for cross-validated model training and testing. (B) The observed protein-protein interactions tend to be broadly conserved across plant species. While we required a Pearson r > 0.3 in at least two species, a majority of interactions recovered exhibited high correlations at least ten species. (C) Gold standard interactions were far less likely to be found if either of the interacting proteins had fewer than ~200 peptide spectral matches (denoted by dashed lines) across the 1,942 mass spectrometry experiments employed for machine learning. Thus, CF/MS requires robust observation of both proteins in a protein interaction.
Figure S3. Precision-recall of protein complexes defined by different thresholds of unsupervised hierarchical clustering, Related to Figure 5. As a guide for interpreting clustering thresholds in Figure 5, precision and recall were measured against the 403 combined test and training gold standard complexes and computed directly from 2×2 confusion matrices comparing observed pairwise co-cluster memberships to the augmented CORUM gold standard co-cluster memberships. Labels refer to specific clustering threshold choices (Table S6). Lower numbered levels are more restrictive while higher numbered levels are more inclusive. The hierarchical levels displayed in Figure 5 are shown in red.
Figure S4. Reproducibility of protein abundance measurements per CF-MS fractionation experiment, Related to STAR Methods. Hierarchically clustered all-by-all Pearson correlation of total orthogroup abundances for each experiment (obtained by summing PPMs across fractions per orthogroup). Experiments performed on the same tissue have highly repeatable orthogroup abundances, e.g. wheat germ and Chlamydomonas experiments. Of the 5 repeat experiments (same species, tissue, and fractionation method), we found Pearson correlations of 0.89 for soybn_sprout1/2, 0.84 for orysj_leaf1/3, 0.71 for wheat_germ2/4, 0.90 for arath_leaf1/2, and 0.69 for arath_dark1/2.
Figure S5. Calibration of molecular weight estimation by size exclusion chromatography, Related to STAR methods. Proteins previously known to elute at particular molecular weights in size exclusion chromatography were selected as internal reference standards, for example, members of the COP9 signalosome complex, which are known to elute at a position corresponding to approximately 350 kDa. These literature-annotated elution positions (x-axis) were compared to observed elution positions (y-axis). We observe generally excellent agreement between expected and observed molecular weights (Spearman r = 0.97), with one marked outlier (an Hsp70) consistently observed above 70kDa, showing that this protein participates in higher-order assemblies in plants. (combine with corrs)
HIGHLIGHTS.
A global snapshot of protein organization in plants from deep proteomic profiling
Biochemical fractionation reveals stable protein complexes conserved across plants
Many observed complexes have previously only been inferred in plants by gene content
Known molecular modules are elaborated in plants with novel subunits and organization
ACKNOWLEDGMENTS
The authors thank Angel Syrett for plant illustrations, Ben Liebeskind for fern transcriptome and feature aid, Anna Battenhouse for data deposition aid, Claire Palmer for early orthology calculations, Hong Qiao, Alan Lloyd, and the University of Texas UTEX Culture Collection of Algae for plant and algal samples, Dan Boutz for mass spectrometry aid, Riddhiman Garge and Momo Sae-Lee for coconut prep, Andrew Emili for early discussions, and John Wallingford and Dannie Durand for feedback. Research was funded by grants from the Welch Foundation (F-1515 to E.M.M.), N.S.F. (1237975 to P.C.R. and E.M.M.), Army Research Office (W911NF-12-1-0390), and N.I.H. (GM123683 to C.D.M., K99 HD092613 to K.D., R01 GM109076 to Z. J. C., and R35 GM122480 to E. M. M.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
SUPPLEMENTAL INFORMATION
Supplemental Information includes 5 figures and 7 tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdrakhmanova A, Dobrynin K, Zwicker K, Kerscher S, and Brandt U (2005). Functional sulfurtransferase is associated with mitochondrial complex I from Yarrowia lipolytica, but is not required for assembly of its iron–sulfur clusters. FEBS Lett. 579, 6781–6785. [DOI] [PubMed] [Google Scholar]
- Adamiec M, Ciesielska M, Zalaś P, and Luciński R (2017). Arabidopsis thaliana intramembrane proteases. Acta Physiol. Plant. 39, 146. [Google Scholar]
- Ahn H-K, Yoon J-T, Choi I, Kim S, Lee H-S, and Pai H-S (2019). Functional characterization of chaperonin containing T-complex polypeptide-1 and its conserved and novel substrates in Arabidopsis. J. Exp. Bot. 70, 2741–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Interactome Mapping, C. (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armero A, Baudouin L, Bocs S, and This D (2017). Improving transcriptome de novo assembly by using a reference genome of a related species: Translational genomics from oil palm to coconut. PloS One 12, e0173300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal UK, McBride Z, Chen D, Xie J, and Szymanski DB (2017). Analysis of protein complexes in Arabidopsis leaves using size exclusion chromatography and label-free protein correlation profiling. J. Proteomics 166, 8–18. [DOI] [PubMed] [Google Scholar]
- Babu CV S, and Palaniswamy MS (2014). Agilent Application Note: Separation of Native Monoclonal Antibodies and Identification of Charge Variants. [Google Scholar]
- van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, and Page JE (2011). The draft genome and transcriptome of Cannabis sativa. Genome Biol. 12, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MS, Ahn SB, Mohamedali A, Islam MT, Cantor D, Verhaert PD, Fanayan S, Sharma S, Nice EC, Connor M, et al. (2017). Accelerating the search for the missing proteins in the human proteome. Nat. Commun. 8, 14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks JA, Nishiyama T, Hasebe M, Bowman JL, Gribskov M, dePamphilis C, Albert VA, Aono N, Aoyama T, Ambrose BA, et al. (2011). The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332, 960–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel GW, Gaudinier A, Brady SM, Hennig L, Rhee SY, and De Smet I (2012). Systems analysis of plant functional, transcriptional, physical interaction, and metabolic networks. Plant Cell 24, 3859–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert S, Waterhouse A, de Beer TAP, Tauriello G, Studer G, Bordoli L, and Schwede T (2017). The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 45, D313–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KS, and Bailey-Serres J (2015). Mechanism of cytoplasmic mRNA translation. Arab. Book 13, e0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KS, and Mayberry L (2006). In vitro translation of plant viral RNA. Curr. Protoc. Microbiol. Chapter 16, 16K.1.1–16K.1.13. [DOI] [PubMed] [Google Scholar]
- Cestari I, Kalidas S, Monnerat S, Anupama A, Phillips MA, and Stuart K (2013). A multiple aminoacyl-tRNA synthetase complex that enhances tRNA-aminoacylation in African trypanosomes. Mol. Cell. Biol. 33, 4872–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter VM, Wilson KS, and Dube GR (1952). The isolation of living nuclei from the endosperm of Cocos nucifera. Science 115, 58–59. [DOI] [PubMed] [Google Scholar]
- Dhawan R, Luo H, Foerster AM, Abuqamar S, Du H-N, Briggs SD, Mittelsten Scheid O, and Mengiste T (2009). HISTONE MONOUBIQUITINATION1 interacts with a subunit of the mediator complex and regulates defense against necrotrophic fungal pathogens in Arabidopsis. Plant Cell 21, 1000–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S, and Wang Y (2016). Nudix Effectors: A Common Weapon in the Arsenal of Plant Pathogens. PLoS Pathog. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K, Lee C, Huizar RL, Tu F, Borgeson B, McWhite CD, Ma Y, Wallingford JB, and Marcotte EM (2017). Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 13, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Marcotte EM, Xenarios I, and Yeates TO (2000). Protein function in the post-genomic era. Nature 405, 823–826. [DOI] [PubMed] [Google Scholar]
- Feller U, Anders I, and Mae T (2007). Rubiscolytics: fate of Rubisco after its enzymatic function in a cell is terminated. J. Exp. Bot. 59, 1615–1624. [DOI] [PubMed] [Google Scholar]
- Fleurdépine S, Deragon J-M, Devic M, Guilleminot J, and Bousquet-Antonelli C (2007). A bona fide La protein is required for embryogenesis in Arabidopsis thaliana. Nucleic Acids Res. 35, 3306–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, and Plotkin JB (2007). Using protein complexes to predict phenotypic effects of gene mutation. Genome Biol 8, R252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadeyne A, Sánchez-Rodríguez C, Vanneste S, Di Rubbo S, Zauber H, Vanneste K, Van Leene J, De Winne N, Eeckhout D, Persiau G, et al. (2014). The TPLATE adaptor complex drives clathrin-mediated endocytosis in plants. Cell 156, 691–704. [DOI] [PubMed] [Google Scholar]
- Gilbert M, and Schulze WX (2019). Global Identification of Protein Complexes within the Membrane Proteome of Arabidopsis Roots Using a SEC-MS Approach. J. Proteome Res. 18, 107–119. [DOI] [PubMed] [Google Scholar]
- Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 47, D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Hopfield JJ, Leibler S, and Murray AW (1999). From molecular to modular cell biology. Nature 402, C47–52. [DOI] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. (2012). A census of human soluble protein complexes. Cell 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. (2015). A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 163, 712–723. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Hendil KB, Yashiroda H, Iemura S, Nagane R, Hioki Y, Natsume T, Tanaka K, and Murata S (2005). A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437, 1381–1385. [DOI] [PubMed] [Google Scholar]
- Hu P, Janga SC, Babu M, Díaz-Mejía JJ, Butland G, Yang W, Pogoutse O, Guo X, Phanse S, Wong P, et al. (2009). Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 7, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, and Bork P (2017). Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, and Krijgsveld J (2014). Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis DE, Ho YS, Lightfoot DJ, Schmöckel SM, Li B, Borm TJA, Ohyanagi H, Mineta K, Michell CT, Saber N, et al. (2017). The genome of Chenopodium quinoa. Nature 542, 307–312. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100. [DOI] [PubMed] [Google Scholar]
- Kan J, An L, Wu Y, Long J, Song L, Fang R, and Jia Y (2018). A dual role for proline iminopeptidase in the regulation of bacterial motility and host immunity. Mol. Plant Pathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood KJ, Ahmad Y, Larance M, and Lamond AI (2013). Characterization of native protein complexes and protein isoform variation using size-fractionation-based quantitative proteomics. Mol Cell Proteomics 12, 3851–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klykov O, Steigenberger B, Pektas S, Fasci D, Heck AJR, and Scheltema RA (2018). Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat Protoc 13, 2964–2990. [DOI] [PubMed] [Google Scholar]
- Koornneef M (1981). The complex syndrome of ttg mutants. Arab. Inf. Serv. 18, 45–51. [Google Scholar]
- Kriechbaumer V, Maneta-Peyret L, Fouillen L, Botchway SW, Upson J, Hughes L, Richardson J, Kittelmann M, Moreau P, and Hawes C (2018). The odd one out: Arabidopsis reticulon 20 does not bend ER membranes but has a role in lipid regulation. Sci. Rep. 8, 2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AR, Gsponer J, and Foster LJ (2012). A high-throughput approach for measuring temporal changes in the interactome. Nat Methods 9, 907–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, and Hedges SB (2017). TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34, 1812–1819. [DOI] [PubMed] [Google Scholar]
- Kwon T, Choi H, Vogel C, Nesvizhskii AI, and Marcotte EM (2011). MSblender: A probabilistic approach for integrating peptide identifications from multiple database search engines. J. Proteome Res. 10, 2949–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tumer Z, Pociot F, Tommerup N, et al. (2007). A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 25, 309–316. [DOI] [PubMed] [Google Scholar]
- Lahmy S, Guilleminot J, Cheng C-M, Bechtold N, Albert S, Pelletier G, Delseny M, and Devic M (2004). DOMINO1, a member of a small plant-specific gene family, encodes a protein essential for nuclear and nucleolar functions. Plant J. Cell Mol. Biol. 39, 809–820. [DOI] [PubMed] [Google Scholar]
- Laporte D, Huot JL, Bader G, Enkler L, Senger B, and Becker HD (2014). Exploring the evolutionary diversity and assembly modes of multi-aminoacyl-tRNA synthetase complexes: Lessons from unicellular organisms. FEBS Lett. 588, 4268–4278. [DOI] [PubMed] [Google Scholar]
- Lee I, Ambaru B, Thakkar P, Marcotte EM, and Rhee SY (2010). Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat Biotechnol 28, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Seo YS, Coltrane D, Hwang S, Oh T, Marcotte EM, and Ronald PC (2011). Genetic dissection of the biotic stress response using a genome-scale gene network for rice. Proc Natl Acad Sci U A 108, 18548–18553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy YY, Mesnage S, Mylne JS, Gendall AR, and Dean C (2002). Multiple roles of Arabidopsis VRN1 in vernalization and flowering time control. Science 297, 243–246. [DOI] [PubMed] [Google Scholar]
- Lintala M, Schuck N, Thormählen I, Jungfer A, Weber KL, Weber APM, Geigenberger P, Soll J, Bölter B, and Mulo P (2014). Arabidopsis tic62 trol mutant lacking thylakoid-bound ferredoxin-NADP+ oxidoreductase shows distinct metabolic phenotype. Mol. Plant 7, 45–57. [DOI] [PubMed] [Google Scholar]
- Lloyd J, and Meinke D (2012). A Comprehensive Dataset of Genes with a Loss-of-Function Mutant Phenotype in Arabidopsis. Plant Physiol. 158, 1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López Y, Nakai K, and Patil A (2015). HitPredict version 4: comprehensive reliability scoring of physical protein–protein interactions from more than 100 species. Database J. Biol. Databases Curation 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W, Zybailov B, Ytterberg AJ, Dunsmore J, Sun Q, and van Wijk KJ (2008). Consequences of C4 Differentiation for Chloroplast Membrane Proteomes in Maize Mesophyll and Bundle Sheath Cells. Mol. Cell. Proteomics MCP 7, 1609–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, Ding C, Shi Y, Yucer N, Krenciute G, et al. (2011). Analysis of the human endogenous coregulator complexome. Cell 145, 787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, and Eisenberg D (1999). A combined algorithm for genome-wide prediction of protein function. Nature 402, 83–86. [DOI] [PubMed] [Google Scholar]
- Marsh JA, and Teichmann SA (2015). Structure, Dynamics, Assembly, and Evolution of Protein Complexes. Annu. Rev. Biochem. 84, 551–575. [DOI] [PubMed] [Google Scholar]
- Matzke AJ, Behensky C, Weiger T, and Matzke MA (1992). A large conductance ion channel in the nuclear envelope of a higher plant cell. FEBS Lett. 302, 81–85. [DOI] [PubMed] [Google Scholar]
- McBride Z, Chen D, Lee Y, Aryal UK, Xie J, and Szymanski DB (2019). A Label-Free Mass Spectrometry Method to Predict Endogenous Protein Complex Composition. Mol. Cell. Proteomics mcp.RA119.001400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary KL, Lee I, and Marcotte EM (2007). Broad network-based predictability of Saccharomyces cerevisiae gene loss-of-function phenotypes. Genome Biol 8, R258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal H, Mandal RK, and Biswas BB (1972). RNA polymerase from eukaryotic cells. Isolation and purification of enzymes and factors from chromatin of coconut nuclei. Eur. J. Biochem. 25, 463–470. [DOI] [PubMed] [Google Scholar]
- Mukaihara T, Tamura N, Murata Y, and Iwabuchi M (2004). Genetic screening of Hrp type III-related pathogenicity genes controlled by the HrpB transcriptional activator in Ralstonia solanacearum. Mol. Microbiol. 54, 863–875. [DOI] [PubMed] [Google Scholar]
- Niehaus TD, Thamm AM, de Crecy-Lagard V, and Hanson AD (2015). Proteins of Unknown Biochemical Function: A Persistent Problem and a Roadmap to Help Overcome It. Plant Physiol 169, 1436–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oellrich A, Walls RL, Cannon EK, Cannon SB, Cooper L, Gardiner J, Gkoutos GV, Harper L, He M, Hoehndorf R, et al. (2015). An ontology approach to comparative phenomics in plants. Plant Methods 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver S (2000). Guilt-by-association goes global. Nature 403, 601–603. [DOI] [PubMed] [Google Scholar]
- Olson RS, and Moore JH (2016). TPOT: A Tree-based Pipeline Optimization Tool for Automating Machine Learning. In JMLR: Workshop and Conference Proceedings, pp. 66–74. [Google Scholar]
- O’Malley RC, Barragan CC, and Ecker JR (2015). A user’s guide to the Arabidopsis T-DNA insertion mutant collections. Methods Mol. Biol. Clifton NJ 1284, 323–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura Y, Nishio Y, Takemoto T, Ikeuchi C, Sekine O, Morino K, Maeno Y, Obata T, Ugi S, Maegawa H, et al. (2009). SAFB1, an RBMX-binding protein, is a newly identified regulator of hepatic SREBP-1c gene. BMB Rep. 42, 232–237. [DOI] [PubMed] [Google Scholar]
- Panchy N, Wu G, Newton L, Tsai C-H, Chen J, Benning C, Farré EM, and Shiu S-H (2014). Prevalence, evolution, and cis-regulation of diel transcription in Chlamydomonas reinhardtii. G3 Bethesda Md 4, 2461–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatheodorou I, Fonseca NA, Keays M, Tang YA, Barrera E, Bazant W, Burke M, Füllgrabe A, Fuentes AM-P, George N, et al. (2018). Expression Atlas: gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 46, D246–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, and Bose HS (2015). Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J. Biol. Chem. 290, 2604–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido P, Zagari N, Manavski N, Gawronski P, Matthes A, Scharff LB, Meurer J, and Leister D (2018). CHLOROPLAST RIBOSOME ASSOCIATED Supports Translation under Stress and Interacts with the Ribosomal 30S Subunit. Plant Physiol. 177, 1539–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, and Mutwil M (2014). Towards revealing the functions of all genes in plants. Trends Plant Sci 19, 212–221. [DOI] [PubMed] [Google Scholar]
- Rivosecchi J, Larochelle M, Teste C, Grenier F, Malapert A, Ricci EP, Bernard P, Bachand F, and Vanoosthuyse V (2019). Senataxin homologue Sen1 is required for efficient termination of RNA polymerase III transcription. EMBO J. 38, e101955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohila JS, Chen M, Chen S, Chen J, Cerny RL, Dardick C, Canlas P, Fujii H, Gribskov M, Kanrar S, et al. (2009). Protein-protein interactions of tandem affinity purified protein kinases from rice. PLoS One 4, e6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmi ML, and Roux SJ (2008). Gene expression changes induced by space flight in single-cells of the fern Ceratopteris richardii. Planta 229, 151–159. [DOI] [PubMed] [Google Scholar]
- Savary S, Willocquet L, Pethybridge SJ, Esker P, McRoberts N, and Nelson A (2019). The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439. [DOI] [PubMed] [Google Scholar]
- Schwikowski B, Uetz P, and Fields S (2000). A network of protein-protein interactions in yeast. Nat Biotechnol 18, 1257–1261. [DOI] [PubMed] [Google Scholar]
- Shikanai T (2016). Chloroplast NDH: A different enzyme with a structure similar to that of respiratory NADH dehydrogenase. Biochim. Biophys. Acta 1857, 1015–1022. [DOI] [PubMed] [Google Scholar]
- Strasser R (2016). Plant protein glycosylation. Glycobiology 26, 926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, Foerster H, Li D, Meyer T, Muller R, Ploetz L, et al. (2008). The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res 36, D1009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabayashi A, Takabayashi S, Takahashi K, Watanabe M, Uchida H, Murakami A, Fujita T, Ikeuchi M, and Tanaka A (2017). PCoM-DB Update: A Protein Co-Migration Database for Photosynthetic Organisms. Plant Cell Physiol. 58, e10. [DOI] [PubMed] [Google Scholar]
- Tateda C, Watanabe K, Kusano T, and Takahashi Y (2011). Molecular and genetic characterization of the gene family encoding the voltage-dependent anion channel in Arabidopsis. J. Exp. Bot. 62, 4773–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium UniProt (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leene J, Witters E, Inze D, and De Jaeger G (2008). Boosting tandem affinity purification of plant protein complexes. Trends Plant Sci 13, 517–520. [DOI] [PubMed] [Google Scholar]
- Van Leene J, Hollunder J, Eeckhout D, Persiau G, Van De Slijke E, Stals H, Van Isterdael G, Verkest A, Neirynck S, Buffel Y, et al. (2010). Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol Syst Biol 6, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leene J, Boruc J, De Jaeger G, Russinova E, and De Veylder L (2011). A kaleidoscopic view of the Arabidopsis core cell cycle interactome. Trends Plant Sci 16, 141–150. [DOI] [PubMed] [Google Scholar]
- Veerappa R, Slocum RD, Siegenthaler A, Wang J, Clark G, and Roux SJ (2019). Ectopic expression of a pea apyrase enhances root system architecture and drought survival in Arabidopsis and soybean. Plant Cell Environ. 42, 337–353. [DOI] [PubMed] [Google Scholar]
- Vercruyssen L, Verkest A, Gonzalez N, Heyndrickx KS, Eeckhout D, Han S-K, Jégu T, Archacki R, Van Leene J, Andriankaja M, et al. (2014). ANGUSTIFOLIA3 binds to SWI/SNF chromatin remodeling complexes to regulate transcription during Arabidopsis leaf development. Plant Cell 26, 210–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Cusick ME, and Barabasi AL (2011). Interactome networks and human disease. Cell 144, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout AJ, and Vidal M (2001). Protein interaction maps for model organisms. Nat. Rev. Mol. Cell Biol. 2, 55–62. [DOI] [PubMed] [Google Scholar]
- Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, et al. (2015). Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M, Schrimpf S, Hengartner MO, Lercher MJ, and von Mering C (2010). Shotgun proteomics data from multiple organisms reveals remarkable quantitative conservation of the eukaryotic core proteome. Proteomics 10, 1297–1306. [DOI] [PubMed] [Google Scholar]
- Wu Z, Zhu D, Lin X, Miao J, Gu L, Deng X, Yang Q, Sun K, Zhu D, Cao X, et al. (2016). RNA Binding Proteins RZ-1B and RZ-1C Play Critical Roles in Regulating Pre-mRNA Splicing and Gene Expression during Development in Arabidopsis. Plant Cell 28, 55–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao P, and Yuan JS (2010). Plant protein-protein interaction network and interactome. Curr Genomics 11, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Proteome properties, Related to Figure 2. (Top) Tissue enrichment of COG functional categories by tissue. Proportion of total spectral counts for each tissue annotated with each high-level COG category. Nuclear samples show enrichment for information and depletion for metabolism. 15-20% of spectral counts from all tissues derive from orthogroups with no COG annotation. (Bottom) Arabidopsis mRNA vs protein abundances. RuBisCo is observed to have relatively moderate mRNA transcript levels across Arabidopsis tissues but is frequently the most abundant protein in our experiments. The median protein abundance of RuBisCo protein in Arabidopsis leaves was plotted against RNA abundance for three separate green Arabidopsis tissues (Gan et al., 2011; Liu et al., 2012, 2016). Titles indicate accession numbers from the ExpressionAtlas database (Petryszak et al., 2016).
Figure S2. Interaction properties, Related to Figure 3. (A) Feature selection for protein-protein interaction scoring, as measured by scikit-learn Feature Importance. The top-ranked 100 features were selected for cross-validated model training and testing. (B) The observed protein-protein interactions tend to be broadly conserved across plant species. While we required a Pearson r > 0.3 in at least two species, a majority of interactions recovered exhibited high correlations at least ten species. (C) Gold standard interactions were far less likely to be found if either of the interacting proteins had fewer than ~200 peptide spectral matches (denoted by dashed lines) across the 1,942 mass spectrometry experiments employed for machine learning. Thus, CF/MS requires robust observation of both proteins in a protein interaction.
Figure S3. Precision-recall of protein complexes defined by different thresholds of unsupervised hierarchical clustering, Related to Figure 5. As a guide for interpreting clustering thresholds in Figure 5, precision and recall were measured against the 403 combined test and training gold standard complexes and computed directly from 2×2 confusion matrices comparing observed pairwise co-cluster memberships to the augmented CORUM gold standard co-cluster memberships. Labels refer to specific clustering threshold choices (Table S6). Lower numbered levels are more restrictive while higher numbered levels are more inclusive. The hierarchical levels displayed in Figure 5 are shown in red.
Figure S4. Reproducibility of protein abundance measurements per CF-MS fractionation experiment, Related to STAR Methods. Hierarchically clustered all-by-all Pearson correlation of total orthogroup abundances for each experiment (obtained by summing PPMs across fractions per orthogroup). Experiments performed on the same tissue have highly repeatable orthogroup abundances, e.g. wheat germ and Chlamydomonas experiments. Of the 5 repeat experiments (same species, tissue, and fractionation method), we found Pearson correlations of 0.89 for soybn_sprout1/2, 0.84 for orysj_leaf1/3, 0.71 for wheat_germ2/4, 0.90 for arath_leaf1/2, and 0.69 for arath_dark1/2.
Figure S5. Calibration of molecular weight estimation by size exclusion chromatography, Related to STAR methods. Proteins previously known to elute at particular molecular weights in size exclusion chromatography were selected as internal reference standards, for example, members of the COP9 signalosome complex, which are known to elute at a position corresponding to approximately 350 kDa. These literature-annotated elution positions (x-axis) were compared to observed elution positions (y-axis). We observe generally excellent agreement between expected and observed molecular weights (Spearman r = 0.97), with one marked outlier (an Hsp70) consistently observed above 70kDa, showing that this protein participates in higher-order assemblies in plants. (combine with corrs)
Data Availability Statement
The interaction data are publicly accessible via a dedicated web portal (http://plants.proteincomplexes.org). All raw and interpreted mass spectrometry data were deposited to the ProteomeXchange http://www.proteomexchange.org/ via the PRIDE (Perez-Riverol et al., 2019) partner repository with identifiers (PXD012810, PXD012865, PXD012969, PXD013004, PXD013080, PXD013093, PXD013198, PXD013213, PXD013214, PXD013264, PXD013280, PXD013281, PXD013282, PXD013300, PXD013320, PXD013321, PXD013322, PXD013369 PXD013704, PXD013735, PXD014617). Full documentation of computational analyses, non-external analysis scripts, and project data files are deposited at Zenodo (Full analysis: DOI:10.5281/zenodo.3466034, Elution data: DOI:10.5281/zenodo.3666942, Protein interactions: DOI:10.5281/zenodo.3666940). Analyses made use of the following github.com/marcottelab repositories: MS_grouped_lookup, protein_complex_maps, run_TPOT, MSblender, and run_MSblender. Fern (Ceratopteris richardii) transcriptome data and assemblies were deposited into the European Nucleotide Archive http://www.ebi.ac.uk/ena/ with accession number PRJEB33372, and proteome deposited at Zenodo (DOI:10.5281/zenodo.3467770).