

Abstract
Gliosarcoma is a rare histopathologic variant of glioblastoma traditionally associated with a poor prognosis. While gliosarcoma may represent a distinct clinical entity given its unique histologic composition and molecular features, its relative prognostic significance remains uncertain. While treatment of gliosarcoma generally encompasses the same standardized approach used in glioblastoma, supporting evidence is limited given its rarity. Here, we characterized thirty-two cases of gliosarcoma and retrospectively evaluated survival relative to four hundred and fifty-one glioblastoma patients diagnosed during the same era within the same institution. Overall, we identified twenty-two primary gliosarcomas, representing 4.7% of WHO Grade IV primary glioblastomas, and ten secondary gliosarcomas. With median age of 62, patients were predominately Caucasian (87.5%) and male (65.6%). Tumors with available molecular profiling were primarily MGMT-unmethylated (87.5%), IDH-1-preserved (100%) and EGFR wild-type (100%). Interestingly, while no significant median survival difference between primary gliosarcoma and glioblastoma was observed across the entire cohort (11.0 vs. 14.8 months, p=0.269), median survival was worse for gliosarcoma specifically among patients who received modern temozolomide-based (TMZ) chemoradiotherapy (11.0 vs. 17.3 months, p=0.006). Matched-pair analysis also trended toward worse median survival among gliosarcomas (11.0 vs. 19.6 months, log-rank p=0.177, Breslow p=0.010). While adjuvant radiotherapy (HR 0.206, p=0.035) and TMZ-based chemotherapy (HR 0.531, p=0.000) appeared protective, gliosarcoma emerged as a significantly poor prognostic factor on multivariate analysis (HR 3.27, p=0.012). Collectively, our results suggest that gliosarcoma may still portend worse prognosis even with modern trimodality therapy.
Keywords: Gliosarcoma, Glioblastoma, Neuro-oncology, Radiotherapy, Temozolomide
Glioblastoma represents the most common adult primary malignant brain tumor, with over twelve thousand estimated cases diagnosed during 2016 within the United States alone [1]. Even despite the current standard of care encompassing maximal-safe surgical resection, adjuvant radiotherapy and concurrent and adjuvant temozolomide-based (TMZ) chemotherapy, clinical outcomes remain poor with five-year survival rates below 10% [2,3]. Gliosarcomas are an uncommon histopathologic variant of glioblastoma characterized by biphasic histopathologic compositions displaying adjacent regions of gliomatous and sarcomatous differentiation, with mesenchymal components commonly resembling fibrosarcoma but also sometimes including lipomatous, osteoid, chondral, osteochondral or myomatous elements [4]. Interestingly, while its pathogenesis remains poorly understood, multiple studies have identified shared mutations and cytogenetic abnormalities (including PTEN and p53 mutations, CDK4 and MDM2 amplifications and p16 deletion) between gliomatous and sarcomatous elements of individual tumors, supporting a monoclonal origin involving inappropriate mesenchymal differentiation of gliomagenic cells [5–10].
Across the United States, gliosarcomas account for approximately 2–3% of glioblastomas nationwide [11–13]. Historically, gliosarcomas were viewed similarly to traditional WHO Grade IV glioblastomas based on early studies suggesting similar demographic patterns, symptomatic presentations and survival trends [14–18]. During the past fifteen years, however, published reports regarding relative prognosis of gliosarcoma have remained conflicting. Moreover, fewer studies have addressed comparative outcomes of gliosarcoma and GBM during the modern era and efficacy of current multimodality therapy since widespread implementation of the Stupp protocol. Generally, while gliosarcoma patients also receive TMZ, supporting evidence remains limited. Encouragingly, a retrospective single-institutional German analysis found significant survival benefit among patients who received combined TMZ chemoradiotherapy over radiotherapy alone [19]. Most recently, a U.S. registry-based retrospective study analyzing gliosarcoma patients diagnosed during the TMZ era demonstrated apparent benefit of trimodality therapy but found no median survival difference between gliosarcoma and GBM [13].
Given ongoing debate, the purpose of this study was to retrospectively characterize demographic trends, treatment patterns and eventual outcomes of histopathologically diagnosed gliosarcomas from our institution in order to better understand the natural history and clinical course of the disease. Given remaining questions regarding prognostic significance particularly in the modern era, we also compared survival for primary gliosarcoma relative to conventional WHO Grade IV glioblastomas from our institution during the same period, as well as on matched-pair analysis and subset analysis among patients who received modern TMZ-based chemoradiotherapy.
Methods:
This study was approved by our Institutional Review Board and conducted in accordance with ethical standards of the 1964 Helsinki Declaration and its later amendments. We retrospectively reviewed institutional charts from patients aged ≥18 who received histopathologic diagnoses of WHO Grade IV gliomas between 1995–2016 through centralized internal pathologic review. Histopathologic diagnosis of gliosarcoma was based on standardized WHO Classification guidelines specifying a biphasic growth pattern on hematoxylin- and eosin-stained sections demonstrating both glial fibrillary acidic protein (GFAP)-positive gliomatous regions and GFAP-negative spindle-shaped cell-containing sarcomatous regions with foci of vascular proliferation and necrosis. All patients with ambiguity regarding final histopathologic diagnosis of gliosarcoma at initial diagnosis were excluded to minimize potential bias. We defined primary gliosarcomas as de novo tumors in patients without prior history and secondary gliosarcomas based on histopathologic diagnoses of GSM made upon reresection following earlier diagnosis with high-grade glioma [20].
Using internal and available outside records, we collected baseline demographic information and treatment-related variables including age, race, gender, Karnofsky Performance Status (KPS), anatomic localization, laterality, multicentricity, extent of resection (EoR), molecular characteristics including MGMT promoter methylation, IDH-1 status, EGFR amplification, EGFRvIII (EGFRr.89_889del) status, FISH 1p/19q, immunohistochemistry for p53, PTEN, PDGFR-A and Olig-2, adjuvant radiotherapy, RT dose, TMZ, reresection and salvage therapies. Anatomic localization was based on perioperative contrast-enhanced MRI and CT scans at initial diagnosis. EoR was defined based on operative reports and postoperative radiographic findings. Overall survival was calculated as the time from initial surgery yielding histopathologic diagnosis until death or last follow-up. Progression-free survival was calculated from initial surgery until date of first radiographic recurrence prompting reresection or salvage therapy. Overall survival for secondary gliosarcomas was calculated from the date of GSM diagnosis until death or last follow-up. Median transformation time was calculated from time of initial surgical diagnosis of glioma until subsequent resection histopathologically diagnosed as gliosarcoma. Twenty-two glioblastomas were selected randomly matched to all twenty-two primary gliosarcoma patients based on age, gender, KPS, resection and receipt of adjuvant radiation and TMZ for matched-pair analysis.
Descriptive statistics were used for baseline characteristics. Primary versus secondary gliosarcoma and primary gliosarcoma versus glioblastoma were compared using contingency tables with Pearson’s χ2 and two-sided Fisher’s exact tests for categorical variables with Bonferroni correction at the 0.05 level and independent samples t-tests for continuous variables. Survival curves were generated using the Kaplan-Meier method. Survival differences between groups were evaluated using Mantel-Cox log-rank, Breslow and Tarone-Ware tests. Patients with unknown dates of death were censored as of last known follow-up date. Cox proportional hazards models were used to estimate hazard ratios with 95% confidence intervals. Univariate Cox regression with threshold P value <0.3 was used to identify potential covariates for multivariate analysis, which was performed using stepwise inclusion. P values <0.05 were considered significant on univariate and multivariate analyses without adjustment for multiple comparisons. Data analysis was performed using IBM SPSS, Version 24.
Results:
Cumulatively, we identified twenty-two primary gliosarcomas and ten secondary gliosarcomas (Table 1). Median age at diagnosis was 61.5 years (IQR: 49.75–67.75 years). Patients were predominantly male (65.6%) and Caucasian (85.7%). Median pretreatment KPS was 80%. Overall, primary gliosarcoma represented 4.7% of histopathologically diagnosed WHO Grade IV glioblastomas. Primary and secondary gliosarcomas demonstrated different patterns of anatomic localization (p=0.045], with apparent temporal and frontal lobe predominances for primary and secondary gliosarcomas, respectively. Cumulatively, including multilobar disease, the most frequently affected anatomic site was the temporal lobe (40% all GSM; 50% among primary GSM), followed by the frontal lobe (36.7% all GSM; 25% of primary GSM). Interestingly, one patient presented with a meningeal tumor confined to the cerebral falx who ultimately survived for over five years, consistent with earlier descriptions of a rare meningioma-like subtype conveying better prognosis [21]. While infrequent, bilateral involvement (9.4% of all GSMs) was significantly more common among secondary gliosarcomas. No patients had evidence of multicentricity. Ultimately, one primary gliosarcoma (4.5%) developed leptomeningeal disease, and one secondary gliosarcoma (10%) presented with pathologically confirmed upper thoracic vertebral metastasis six months after diagnosis. No patients presented with known distant metastatic disease.
Table 1 |.
Patient Characteristics of Primary and Secondary Gliosarcomas
| Patient Characteristics | Total Gliosarcoma | Primary Gliosarcoma | Secondary Gliosarcoma | P-value |
|---|---|---|---|---|
| Baseline Demographics | 32 patients | 22 patients | 10 patients | |
| Median Age, years (range) | 61.5 (33–77) | 62 (33–77) | 60.5 (39–70) | |
| <50 | 25.0% | 27.3% | 20.0% | 0.307 |
| ≥50–≤70 | 40.6% | 31.8% | 60.0% | |
| <70 | 34.4% | 40.9% | 20.0% | |
| Median KPS (range) | 80% (60–100%) | 80% (60–90%) | 80% (70–100%) | - |
| RTOG RPA Class | 0.559 | |||
| III | - | - | - | |
| IV | 76.9% | 85.7% | 66.7% | |
| V/VI | 23.1% | 14.3% | 33.3% | |
| Gender | 0.703 | |||
| Male | 65.6% | 68.2% | 60% | |
| Female | 34.4% | 31.8% | 40% | |
| Ethnicity | 1.000 | |||
| Caucasian | 85.7% | 83.3% | 90% | |
| Hispanic | 14.3% | 16.7% | 10% | |
| African-American | - | - | - | |
| Asian | - | - | - | |
| Tumor Characteristics | ||||
| Neuroanatomic Localization | 0.045 | |||
| Multilobar | 33.3% | 40.5% | 10.0% | |
| Temporal | 23.3% | 30.0% | 10.0% | |
| Frontal | 26.7% | 10.0% | 60.0% | |
| Parietal | 10.0% | 10.0% | 10.0% | |
| Occipital | 6.7% | 5.0% | 10.0% | |
| Meningeal | 3.1% | 4.5% | 0.0% | |
| Laterality | ||||
| Unilateral | 90.6% | 100.0% | 70.0% | 0.024 |
| Bilateral | 9.4% | - | 30.0% | |
| Treatment Characteristics | ||||
| Surgical Extent of Resection | ||||
| Gross total resection | 56.7% | 63.6% | 30.0% | |
| Near-total resection/Subtotal resection | 43.3% | 27.3% | 70.0% | |
| Biopsy | - | - | - | |
| Adjuvant Radiotherapy | ||||
| Yes | 84.4% | 77.3% | 100.0%* | |
| No | 15.6% | 22.7% | - | |
| Median RT Dose, Gy (IQR) | 60 (40.5–60) | 59.4 (40.5–60) | 60 (45–60) | |
| Temozolomide | ||||
| Yes | 65.6% | 63.6% | 70.0%* | |
| No | 34.4% | 36.4% | 30.0% | |
| Salvage Treatments | ||||
| Reresection | 41.9% | 47.6% | 30.0% | |
| Stereotactic radiosurgery | 3.1% | 4.5% | 0.0% | |
| Systemic therapy | 37.5% | 36.4% | 40.0% | |
| Re-RT/Immunotherapy | 6.3% | 4.5% | 10.0% | |
Refers to treatment following initial diagnosis of high-grade glioma prior to developing secondary gliosarcoma.
Among tumors with available molecular profiling, molecular characterization of gliosarcomas revealed unfavorable prognostic features (Table 2). Tumors were mostly MGMT-unmethylated (87.5%, n=8) and IDH-1 wildtype (100%, n=10). Consistent with prior characterizations, gliosarcomas were EGFR amplification-negative (100%, n=9) and EGFRvIII mutation-negative (100%, n=7). While potential prognostic significance remains unknown, several gliosarcomas were 1p-deleted/19q-preserved (57%, n=7). All patients underwent maximal-safe surgical resection with apparent gross total (56.7%) or subtotal (43.3%) resection. The majority of primary gliosarcoma patients received adjuvant radiotherapy (77%) combined with TMZ (63.6%).
Table 2 |.
Molecular and Histopathologic Characteristics of Primary and Secondary Gliosarcomas
| Total Gliosarcoma | Primary Gliosarcoma | Secondary Gliosarcoma | |
|---|---|---|---|
| O6-MGMT promoter status | |||
| Unmethylated (% of available) | 7 patients (87.5%) | 5 patients (83.3%) | 2 patients (100%) |
| Methylated (% of available) | 1 patient (12.5%) | 1 patient (16.7%) | - |
| IDH-1 status (IHC) | |||
| Wild-type (% of available) | 10 patients (100%) | 7 patients (100%) | 3 patients (100%) |
| R132 mutation (% of available) | - | - | - |
| EGFR 7p12 amplification status | |||
| No (% of available) | 9 patients (100%) | 8 patients (100%) | 1 patient (100%) |
| Yes (% of available) | - | - | - |
| EGFRvIII (EGFRr.89_889del) status | |||
| Negative (% of available) | 7 patients (100.0%) | 5 patients (100.0%) | 2 patients (100.0%) |
| Positive (% of available) | - | - | - |
| Chromosomal 1p/19q status (FISH) | |||
| Preserved (% of available) | 3 patients (42.9%) | 3 patients (60.0%) | - |
| Codeleted (% of available) | - | - | - |
| 1p-preserved/19q-deleted (% of available) | - | - | - |
| 1p-deleted/19q-preserved (% of available) | 4 patients (57.1%) | 2 patients (40.0%) | 2 patients (40.0%) |
| p53 overexpression (IHC) | |||
| Negative (% of available) | 2 patients (13.3%) | 1 patient (10.0%) | 1 patient (20.0%) |
| Rare, few or <10% (% of available) | 6 patients (40.0%) | 3 patients (30.0%) | 3 patients (60.0%) |
| Positive (% of available) | 7 patients (46.7%) | 6 patients (60.07%) | 1 patient (20.0%) |
| PDGFR-α expression (IHC) | |||
| Negative (% of available) | 1 patient (14.3%) | 1 patient (20.0%) | - |
| Rare, few or <10% (% of available) |
1 patient (14.3%) | 1 patient (20.0%) | - |
| Positive (% of available) | 5 patients (71.4%) | 3 patients (60.0%) | 2 patients (100%) |
| Olig-2 expression (IHC) | |||
| Negative (% of available) | 1 patient (14.3%) | - | 1 patient (50%) |
| Positive (% of available) | 6 patients (85.7%) | 5 patients (100%) | 1 patient (50%) |
| PTEN expression (IHC) | |||
| Loss/Negative (% of available) | 6 patients (50.0%) | 5 patients (62.5%) | 1 patient (25.0%) |
| Weak (% of available) | 3 patients (25%) | 2 patients (25%) | 1 patient (25.0%) |
| Positive (% of available) | 3 patients (25%) | 1 patient (12.5%) | 2 patients (50.0%) |
| Median Ki-67 | 12 patients | 16 patients | 4 patients |
| % (range) | 45.5% (5.0–90.0%) | 45.0% (17.5–75%) | 45.5% (5.0–90.0%) |
For primary gliosarcoma, median overall survival and progression-free survival were 11.0 months (IQR: 7.2–17.2) and 5.6 months (IQR: 4.7–6.8), respectively. Extent of surgical resection was a significant prognostic factor on univariate analysis (HR: 3.574 for STR vs. GTR, p=0.025) but failed to reach significance on multivariate analysis (Supplementary Table 1). Approximately half of primary gliosarcoma patients (47.6%) underwent reresection for recurrent disease, with one patient also receiving stereotactic radiosurgery. Systemic salvage therapies included bevacizumab (27.2%, with one patient receiving concurrent irinotecan), nivolumab and reirradiation (35 Gy in 10 fractions) (n=1), carmustine (n=2), TMZ (n=1), TM-601 (n=1) and procarbazine (n=1).
All ten secondary gliosarcoma patients had previously received radiation therapy (Nine patients were previously diagnosed with glioblastoma and one with anaplastic oligodendroglioma). Median transformation time to secondary gliosarcoma was 15.1 months, and median survival following diagnosis of secondary gliosarcoma was 3.5 months. Ultimately, three patients (30%) underwent reresection for recurrent secondary GSM. Multiple received systemic salvage therapies including bevacizumab (n=1), TMZ (n=1) and combination experimental TMZ and veliparib (n=1). One patient was treated with combined nivolumab and reirradiation (35 Gy in 10 fractions) but progressed approximately 3.5 months later, subsequently undergoing reresection, adjuvant Novo-TTF and lomustine salvage therapy.
Subsequently, we compared median survival for primary gliosarcoma against four hundred and fifty-one conventional WHO Grade IV glioblastomas from our institution diagnosed during the same period (Figure 1A) to better understand relative prognostic implications of gliosarcoma. Generally, baseline characteristics were comparable between groups including age, gender and pretreatment functional status (Supplementary Table 2). However, fewer GSM patients had received radiotherapy. While unavailable for the majority of patients, approximately 40% of glioblastomas with available data were MGMT-methylated. Across the entire cohort, we observed no significant survival difference between gliosarcoma and GBM (11.0 months GSM vs. 14.8 months GBM, p=0.269). However, gliosarcoma demonstrated worse median survival on subset analysis of patients who had received TMZ-based chemoradiotherapy (11.0 months GSM vs. 17.3 months GBM; log-rank p=0.006) (Figure 1B). We also performed matched-pair analysis of primary gliosarcoma and glioblastoma. Again, gliosarcoma trended toward worse survival (11.0 months GSM vs. 19.6 months GBM, log-rank p=0.177, Breslow p=0.010) (Figure 2). Univariate analysis identified histopathologic diagnosis of gliosarcoma, age, KPS, RPA Class, adjuvant radiation, RT dose, TMZ, extent of resection and reresection as potential prognostic factors (Table 3). Multivariate analysis revealed histopathologic diagnosis of gliosarcoma over glioblastoma as a significant poor prognostic factor (HR: 3.267, 95% CI: 1.291–8.264, p=0.012), with other significant covariates including age, KPS, adjuvant radiotherapy, RT dose and TMZ-based chemotherapy (Table 3).
Figure 1|.
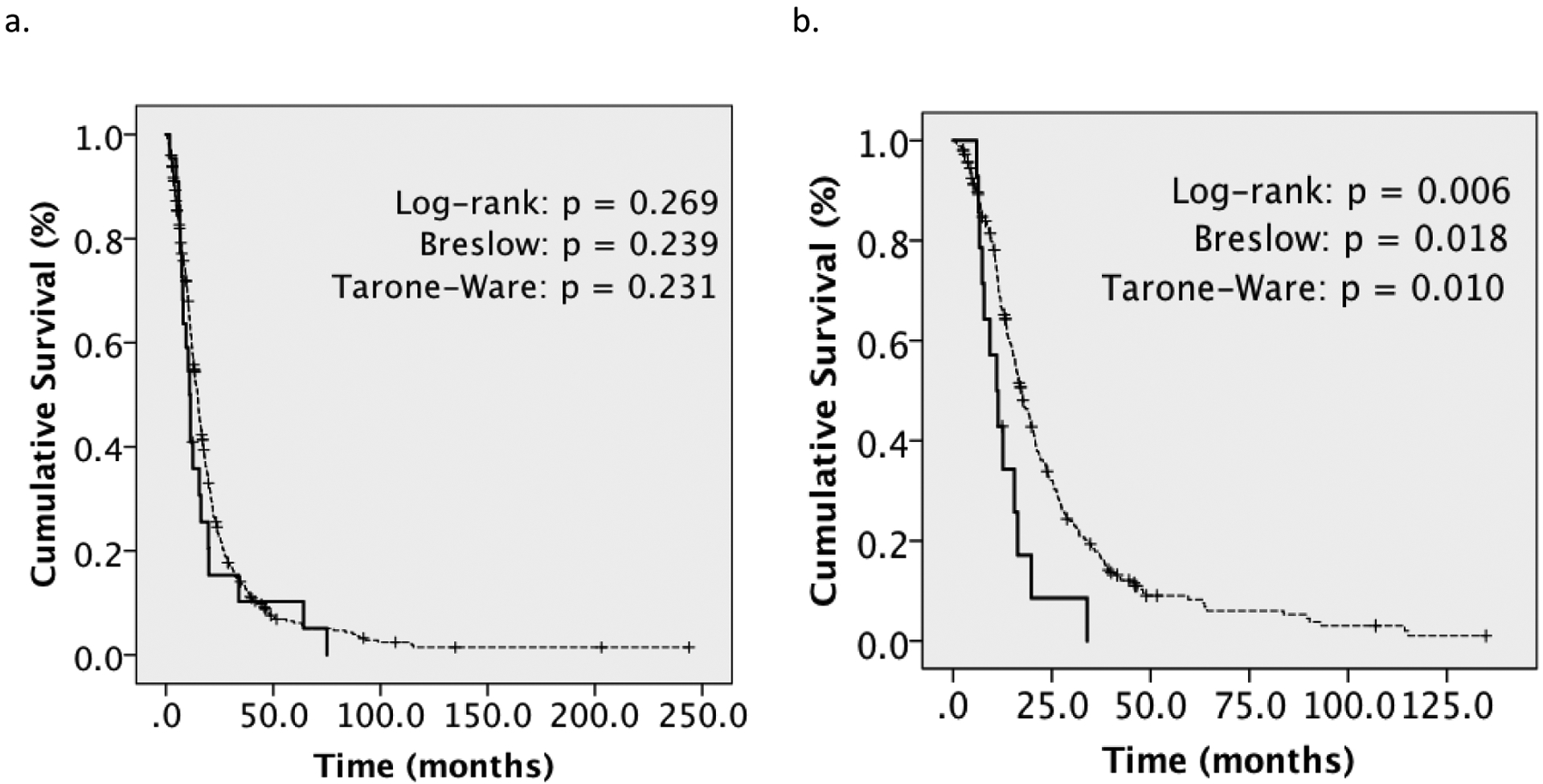
Kaplan-Meier Overall Survival Analysis for Primary Gliosarcoma versus Glioblastoma
A. mOS for primary gliosarcoma (n=22) versus GBM (n=451) (11.0 versus 14.8 months, log-rank p=NS). B. mOS among patients treated with temozolomide-based chemoradiotherapy (11.0 months GSM, n=14 versus 17.3 months GBM, n=256) (log-rank p=0.006)
Figure 2|.
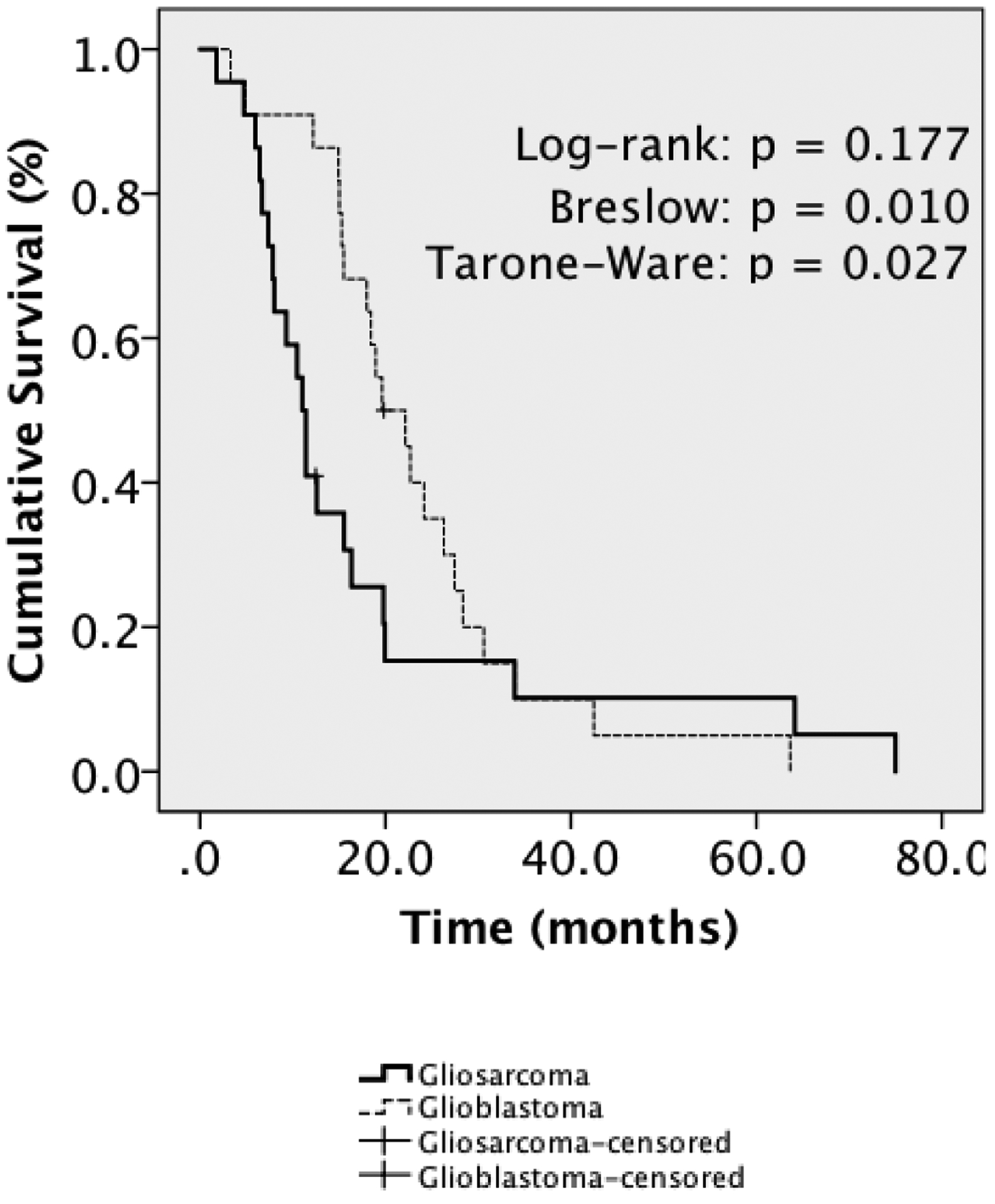
Matched-Pair Kaplan-Meier Analysis for Primary Gliosarcoma versus Glioblastoma
mOS was 11.0 months for primary gliosarcoma (n=22) versus 19.6 months for GBM (n=22), log-rank p=0.177, Breslow p=0.010, Tarone-Ware p=0.027.
Table 3 |.
Univariate and Multivariate Cox Regression
| Factors | Univariate Analysis Overall Survival | Multivariate Analysis Overall Survival | ||
|---|---|---|---|---|
| Hazard Ratio (95% CI) | P-value | Hazard Ratio (95% CI) | P-value | |
| Histopathology | 0.271 | 0.012 | ||
| Gliosarcoma | 1.280 (0.825–1.988) | 3.267 (1.291–8.264) | ||
| GBM | Reference | Reference | ||
| Demographics | ||||
| Age at Diagnosis | 1.029 (1.021–1.037) | 0.000 | 1.027 (1.016–1.038) | 0.000 |
| KPS (Karnofsky Performance Status) | 0.966 (0.958–0.973) | 0.000 | 0.975 (0.965–0.986) | 0.000 |
| RTOG RPA Class | 0.000 | 0.899 | ||
| III | Reference | Reference | ||
| IV | 1.650 (1.162–2.345) | 1.090 (0.697–1.705) | ||
| V/VI | 4.542 (3.096–6.662) | 1.045 (0.583–1.872) | ||
| Gender | 0.555 | |||
| Female | Reference | |||
| Male | 1.060 (0.873–1.287) | |||
| Ethnicity | 0.538 | |||
| Caucasian | Reference | |||
| Hispanic | 1.027 (0.775–1.361) | |||
| African-American | 0.969 (0.609–1.453) | |||
| Asian | 0.656 (0.368–1.169) | |||
| Treatment Characteristics | ||||
| Surgical EoR | 0.000 | 0.000 | ||
| Biopsy | Reference | Reference | ||
| STR/NTR | 0.393 (0.297–0.518) | 0.372 (0.262–0.528) | ||
| GTR | 0.231 (0.159–0.338) | 0.229 (0.142–0.368) | ||
| Adjuvant Radiotherapy | 0.017 | 0.035 | ||
| No | Reference | Reference | ||
| Yes | 0.616 (0.426–0.919) | 0.206 (0.047–0.894) | ||
| RT Dose | 0.000 | 0.042 | ||
| 59.4–60 Gy | Reference | Reference | ||
| <59.4 Gy | 2.753 (2.093–3.621) | 1.396 (1.013–1.925) | ||
| Temozolomide | 0.000 | 0.000 | ||
| Yes | 0.643 (0.530–0.781) | 0.531 (0.420–0.672) | ||
| No | Reference | Reference | ||
| Reresection | 0.000 | 0.136 | ||
| Yes | 0.570 (0.459–0.708) | 0.819 (0.630–1.065) | ||
| No | Reference | Reference | ||
Discussion:
Gliosarcoma is a rare clinicopathologic variant of glioblastoma traditionally associated with a dismal prognosis [11]. While modern-era treatment generally adapts our standardized approach toward glioblastoma per the Stupp protocol encompassing surgical resection, adjuvant radiotherapy and TMZ-based chemotherapy, no prospective randomized evidence exists to support efficacy toward gliosarcoma. Given its rarity, our current understanding relies mostly on retrospective series. Further, most existing knowledge on gliosarcoma derives from studies published before widespread adaptation of modern TMZ-based trimodality therapy. Here, we describe our institutional experience, comparing survival among primary gliosarcoma patients against that of glioblastoma patients during the same era.
Consistent with prior published estimates of 1.8–8% [11,14,17,18], primary gliosarcomas accounted for 4.7% of WHO Grade IV glioblastomas. Demographically, our cohort appeared generally consistent with prior epidemiologic descriptions of gliosarcoma from nationwide cancer registry-based studies, where patients were predominantly middle-aged Caucasian men (specifically, 60–67.4% male and 85–88.9% non-Hispanic Caucasian with median age of 61–63) [11–13,22,23]. Hispanic patients may have accounted for a higher percentage of our cohort (14.3% versus 5.0–8.7% nationwide), reflecting institutional trends for glioblastoma and presumably the unique patient population surrounding our medical center [12,13,22,24]. However, potential prognostic significance remains unknown.
Among primary gliosarcoma patients, estimated median survival was 11.0 months. During the past fifteen years, nearly all retrospective studies examining adult primary gliosarcomas have reported similar estimates ranging from 8.3–16.7 months [11,12,21,25–29], although notably, one study from a major academic center where most patients were enrolled on experimental trials reported 24.7 months [30]. During the past five years, several nationwide cancer registry-based studies reported median survivals of 7.0–10.7 months, although each potentially also included secondary gliosarcomas given current registry-based classification guidelines and apparent inclusion of all known adult gliosarcomas independent of prior cancer diagnoses [12,13,22]. Possible explanations for our potentially shorter observed median survival time compared to select recent studies (11.0 months vs. 13.4–16.7 months) [21,26,29] include an older median age [26,29] and apparently lower frequency of MGMT promoter methylation [26,31], both previously associated with worse outcomes for gliosarcoma [11,29]. We also identified only one meningioma-like gliosarcoma within our cohort, previously associated with better survival [21]. While greater EoR may correlate with improved outcomes [11,13], most primary gliosarcomas in our series (63.6%) underwent apparent GTR.
Without treatment, gliosarcoma carries an incredibly poor prognosis with median survival of approximately four months [11]. Despite inherent retrospective biases, multiple studies have shown that adjuvant radiotherapy was associated with improved survival (6.9–10.6 months with RT vs. 3–4.3 months without) [11,12,13,16]. Despite limited statistical power given sample size to detect therapeutic benefit among primary gliosarcomas alone, multivariate analysis of both gliosarcoma and GBM found adjuvant radiotherapy to be significantly protective (Table 3). One recent study found that higher total radiotherapy dose (at least 54 Gy) was associated with improved survival (6 versus 14 months) [25].
Within our study, TMZ-based chemotherapy failed to reach significance as a positive prognostic factor for primary gliosarcoma. However, TMZ appeared protective on multivariate analysis of gliosarcoma and GBM (Table 3). Despite limited sample size, while evidence regarding optimal first-line management remains conflicting, current literature suggests that TMZ-based chemotherapy provides significant therapeutic benefit. Two earlier studies found no significant survival benefit with TMZ [12] or TMZ-based chemoradiotherapy [21], raising questions regarding efficacy of TMZ toward gliosarcoma [32]. More recently, Castelli et al. also found no significant benefit with combined TMZ-based chemoradiotherapy over radiation alone [25]. Within the last two years, however, two studies indeed found that TMZ-based chemotherapy was associated with significant survival benefit and beneficial prognostic significance (9.9 months with RT alone vs. 13.9 months with TMZ/RT [19]; 11.9 without vs. 21.2 months with TMZ [26]). Similarly, despite unavailability of information regarding specific chemotherapy administered, Walker et al. demonstrated significantly improved two-year survival of gliosarcoma patients diagnosed during the TMZ era compared to the preceding era using a large nationwide cohort [12]. Recently, a nationwide hospital-based cohort analysis found that chemoradiotherapy was associated with significantly improved survival over adjuvant radiotherapy alone in the TMZ era (6.9 months vs. 13.5 months, p<0.001), although only efficacy of trimodality therapy over all other treatments combined was compared on propensity score-matched analysis [13].
Here, no significant survival discrepancy between gliosarcoma and GBM was observed on cohort-wide Kaplan-Meier analysis. However, only approximately 70% of GSMs and 60% of GBMs were diagnosed during the TMZ era (2004 onward), and adjuvant radiotherapy was significantly more common among GBM patients (Supplementary Table 1). Despite apparent survival benefits using TMZ for gliosarcoma evident from existing literature, we found that among patients who had received TMZ-based chemoradiotherapy, gliosarcomas nevertheless fared worse than GBMs (11.0 months vs. 17.3 months; log-rank p=0.006). Similarly, matched-pair analysis trended toward significantly worse outcomes for gliosarcoma (Figure 2). Multivariate analysis also demonstrated that histopathologic diagnosis of gliosarcoma was a significantly poor prognostic factor after adjustment for receipt of radiotherapy and TMZ (Table 3).
Over time, various studies addressing relative aggressiveness of gliosarcoma compared to conventional GBM have remained conflicting. While early data from two studies that retrospectively evaluated gliosarcoma patients enrolled on prospective Phase II/III RTOG and NCCTG clinical trials suggested that gliosarcoma might portend worse prognosis, neither demonstrated significant survival differences [15,17]. Subsequently, however, a large nationwide registry-based analysis evaluating cases diagnosed before introduction of TMZ found that gliosarcoma conveyed poor prognostic significance [11]. Importantly, few studies have evaluated relative prognostic significance during the modern era. Recently, one multicenter Australian study in which most patients had received TMZ-based chemoradiotherapy trended toward worse outcomes among gliosarcomas (9.7 months vs. 12.2 months GBM) [27], while a retrospective Chinese analysis demonstrated a short but statistically significant survival difference (13 months GSM vs. 14 months GBM, p=0.001) [31]. However, all patients apparently received nitrosoureas rather than TMZ as first-line therapy, presumably reflecting more gradual nationwide adaption of the Stupp protocol. Finally, Frandsen et al. found no significant survival difference between gliosarcoma and glioblastoma (11.9 months vs. 10.7 months, p=NS). In this study, consistent with early findings of Kozak and colleagues before introduction of TMZ [11], we found that gliosarcoma still emerged as a poor prognostic factor compared to conventional GBM, even amongst a more modern cohort where most patients had received TMZ-based chemoradiotherapy.
While our results are intriguing, several limitations exist, most notably including our retrospective study design and limited statistical power given sample size. For these reasons, potential conclusions regarding optimal treatment recommendations remain limited, and we caution against major conclusions regarding molecular features of gliosarcoma based on this study alone given limited availability of molecular data. Regarding study design, along with potential effects of unknown confounders, retrospective analyses are intrinsically predisposed toward recall bias, observer bias (here, for example, implicit physician preconceptions regarding relative clinical aggressiveness of gliosarcoma may have ultimately prompted more/less aggressive regimens or differential recruitment onto experimental trials compared to GBM that may have impacted survival), selection bias (e.g., patients with higher baseline pretreatment functional status more likely receiving trimodality therapy) and misclassification bias (for example, regarding apparent EoR and ultimate histopathologic diagnosis). Other limitations include lack of blinded repeat histopathologic review of cases for inclusion in our study, inability to incorporate mutational status including MGMT methylation into prognostication given limited availability of molecular profiling, inherent difficulties in assessing EoR given the diffusely-infiltrative nature of high-grade gliomas and our reliance on subset Kaplan-Meier analysis given baseline differences between GSM and GBM patients, although matched-pair analysis and Cox regression demonstrated supportive findings.
Despite these shortcomings, our results nevertheless provide important contributions toward prognostication of gliosarcoma during the modern era. Unfortunately, gliosarcoma was associated with poor prognostic significance (HR: 3.27) and shortened median survival relative to GBM among patients receiving TMZ-based trimodality therapy (11.0 months vs. 17.3 months). Reassuringly, despite limited statistical power in our study, current evidence from existing literature does suggest that both radiotherapy and TMZ-based chemotherapy per the Stupp protocol appear to provide meaningful therapeutic benefits for patients diagnosed with gliosarcoma. However, while further studies evaluating larger cohorts will be essential, our results indicate that gliosarcoma nevertheless may still portend worse prognosis even in the modern era of TMZ-based trimodality therapy.
Supplementary Material
Conflict of Interest:
Dr. Wang reports personal fees and non-financial support from AbbVie, non-financial support from Merck, personal fees from AstraZeneca, personal fees from Doximity, non-financial support from Novocure, personal fees and non-financial support from Elekta and personal fees from Wolthers Kluwer, outside the submitted work.
References:
- 1.Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: A state of the science review. Neuro Oncol. 2014;16(7):896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005;35210:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 5.Paulus W, Bayas A, Ott G, Roggendorf W. Interphase cytogenetics of glioblastoma and gliosarcoma. Acta Neuropathol. 1994;88(5):420–425. doi: 10.1007/BF00389493. [DOI] [PubMed] [Google Scholar]
- 6.Biernat W, Aguzzi A, Sure U, Grant JW, Kleihues P, Hegi ME. Identical Mutations of the p53 Tumor Suppressor Gene in the Gliomatous and the Sarcomatous Components of Gliosarcomas Suggest a Common Origin from Glial Cells. J Neuropathol Exp Neurol. 1995;54(5):651–656. [DOI] [PubMed] [Google Scholar]
- 7.Reis RM, Könü-Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H. Genetic profile of gliosarcomas. Am J Pathol. 2000;156(2):425–432. doi: 10.1016/S0002-9440(10)64746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker C, Joyce KA, Thompson-Hehir J, et al. Characterisation of molecular alterations in microdissected archival gliomas. Acta Neuropathol. 2001;101(4):321–333. doi: 10.1007/s004010000259. [DOI] [PubMed] [Google Scholar]
- 9.Actor B, Ludwig Cobbers JMJ, Büschges R, et al. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosom Cancer. 2002;34(4):416–427. doi: 10.1002/gcc.10087. [DOI] [PubMed] [Google Scholar]
- 10.Boerman RH, Anderi K, Herath J, et al. The Glial and Mesenchymal Elements of Gliosarcomas Share Similar Genetic Alterations. J Neuropathol Exp Neurol. 1996;55(9):973–981. [DOI] [PubMed] [Google Scholar]
- 11.Kozak KR, Mahadevan A, Moody JS. Adult gliosarcoma: epidemiology, natural history and factors associated with outcome. April. 2009;Cancer:183–191. doi: 10.1215/15228517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker GV, Gilbert MR, Prabhu SS, Brown PD, McAleer MF. Temozolomide use in adult patients with gliosarcoma: An evolving clinical practice. J Neurooncol. 2013;112(1):83–89. doi: 10.1007/s11060-012-1029-7. [DOI] [PubMed] [Google Scholar]
- 13.Frandsen J, Orton A, Jensen R, et al. Patterns of care and outcomes in gliosarcoma: an analysis of the National Cancer Database. ©AANS J Neurosurg. 2017:1–6. doi: 10.3171/2016.12.JNS162291. [DOI] [PubMed] [Google Scholar]
- 14.Morantz R a, Feigin I, Ransohoff J. Clinical and pathological study of 24 cases of gliosarcoma. J Neurosurg. 1976;45(4):398–408. doi: 10.3171/jns.1976.45.4.0398. [DOI] [PubMed] [Google Scholar]
- 15.Meis JM, Martz KL, Nelson JS. Mixed Glioblastoma Mulfiforme and Sarcoma. 1991:2342–2349. [DOI] [PubMed]
- 16.Perry JR, Ang LC, Bilbao JM, Muller PJ. Clinicopathologic features of primary and postirradiation cerebral gliosarcoma. Cancer. 1995;75(12):2910–2918. doi:. [DOI] [PubMed] [Google Scholar]
- 17.Galanis E, Buckner JC, Dinapoli RP, et al. Clinical outcome of gliosarcoma compared with glioblastoma multiforme: North Central Cancer Treatment Group results. J Neurosurg. 1998;89(3):425–430. doi: 10.3171/jns.1998.89.3.0425. [DOI] [PubMed] [Google Scholar]
- 18.Lutterbach J, Guttenberger R, Pagenstecher A. Gliosarcoma : a clinical study. 2001;61:57–64. [DOI] [PubMed] [Google Scholar]
- 19.Adeberg S, Bernhardt D, Ben Harrabi S, et al. Radiotherapy plus concomitant temozolomide in primary gliosarcoma. J Neurooncol. 2016;128(2):341–348. doi: 10.1007/s11060-016-2117-x. [DOI] [PubMed] [Google Scholar]
- 20.Han SJ, Yang I, Tihan T, Chang SM, Parsa AT. Secondary gliosarcoma: a review of clinical features and pathological diagnosis. J Neurosurg. 2010;112(1):26–32. doi: 10.3171/2009.3.JNS081081. [DOI] [PubMed] [Google Scholar]
- 21.Han SJ, Yang I, Ahn BJ, et al. Clinical characteristics and outcomes for a modern series of primary gliosarcoma patients. Cancer. 2010. doi: 10.1002/cncr.24857. [DOI] [PubMed] [Google Scholar]
- 22.Ortega A, Nuño M, Walia S, Mukherjee D, Black KL, Patil CG. Treatment and survival of patients harboring histological variants of glioblastoma. J Clin Neurosci. 2014;21(10):1709–1713. doi: 10.1016/j.jocn.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Thakkar JP, Dolecek TA, Horbinski C, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. 2014;23(10):1985–1996. doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu C-C, Wang TJC, Jani A, et al. A Modern Radiotherapy Series of Survival in Hispanic Patients with Glioblastoma. World Neurosurg. 2016;88:260–269. doi: 10.1016/j.wneu.2015.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castelli J, Feuvret L, Haoming QC, et al. Prognostic and therapeutic factors of gliosarcoma from a multi-institutional series. J Neurooncol. 2016;129(1):85–92. doi: 10.1007/s11060-016-2142-9. [DOI] [PubMed] [Google Scholar]
- 26.Rath G, Sharma D, Mallick S, et al. Clinical outcome of patients with primary gliosarcoma treated with concomitant and adjuvant temozolomide: A single institutional analysis of 27 cases. Indian J Cancer. 2015;52(4):599. doi: 10.4103/0019-509X.178407. [DOI] [PubMed] [Google Scholar]
- 27.Damodaran O, Van Heerden J, Nowak AK, et al. Clinical management and survival outcomes of gliosarcomas in the era of multimodality therapy. J Clin Neurosci. 2014;21(3):478–481. doi: 10.1016/j.jocn.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 28.Biswas A, Kumar N, Kumar P, et al. Primary gliosarcoma--clinical experience from a regional cancer centre in north India. Br J Neurosurg. 2011;25(6):723–729. doi: 10.3109/02688697.2011.570881. [DOI] [PubMed] [Google Scholar]
- 29.Kang SH, Park KJ, Kim CY, et al. O6-methylguanine DNA methyltransferase status determined by promoter methylation and immunohistochemistry in gliosarcoma and their clinical implications. J Neurooncol. 2011;101(3):477–486. doi: 10.1007/s11060-010-0267-9. [DOI] [PubMed] [Google Scholar]
- 30.Cachia D, Kamiya-Matsuoka C, Mandel JJ, et al. Primary and secondary gliosarcomas: clinical, molecular and survival characteristics. J Neurooncol. 2015;125(2):401–410. doi: 10.1007/s11060-015-1930-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang G, Huang S, Zhang J, Wu Z, Lin S, Wang Y. Clinical outcome of gliosarcoma compared with glioblastoma multiforme: a clinical study in Chinese patients. J Neurooncol. 2016;127(2):355–362. doi: 10.1007/s11060-015-2046-0. [DOI] [PubMed] [Google Scholar]
- 32.Lee D, Kang SY, Suh YL, Jeong JY, Lee J Il, Nam DH. Clinicopathologic and genomic features of gliosarcomas. J Neurooncol. 2012;107(3):643–650. doi: 10.1007/s11060-011-0790-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.