

Abstract
Motivation: The SEABED web server integrates a variety of docking and QSAR techniques in a user-friendly environment. SEABED goes beyond the basic docking and QSAR web tools and implements extended functionalities like receptor preparation, library editing, flexible ensemble docking, hybrid docking/QSAR experiments or virtual screening on protein mutants. SEABED is not a monolithic workflow tool but Software as a Service platform.
Availability and implementation: SEABED is a free web server available athttp://www.bsc.es/SEABED. No registration is required.
Contact: ramon.goni@bsc.es
Supplementary information: Supplementary data are available atBioinformatics online.
1 Introduction
In silico methods such as molecular docking and quantitative-activity relationship (QSAR) are gaining an increased role in drug discovery (Alvarez, 2004;Bajorath, 2002). QSAR approaches are predictive models trained to distinguish compounds from molecular descriptors. Molecular docking tries to recover active ligands by finding the best possible fitting of compounds to a 3D-binding site. QSAR is faster and often more robust than docking approaches, but is limited to suggesting molecules close to those of proven activity. Docking requires knowledge of the structure of biological target, is computationally very demanding, and is less robust than QSAR, but can make suggestions for new drug scaffolds. We are then looking at two complementary techniques, and it is not a surprise that during daily practice drug designers are continuously using both approaches, which is largely facilitated by commercial programs, and a non-negligible list of open source software and web services (Colemanet al., 2013;Grosdidieret al. 2011;O’Boyleet al., 2011;Tetkoet al., 2005;Trott and Olson, 2010). SEABED is a web server that automates different tools for virtual screening. This includes classical QSAR (with a large variety of machine learning tools) or molecular docking (with many features for target and ligand-binding characterization). The server allows a unique integration of molecular dynamics information in flexible (ensemble) docking (Carlson, 2002;Chaudhuriet al., 2012;Ruedaet al., 2009) enriched with essential dynamics MD (EDMD) sampling (Carrilloet al., 2012). It also enables us to combine in a single project docking and QSAR (following the ideas inYoonet al., 2005), and to incorporate genetic variability into the docking process. The modular nature of SEABED (seeSupplementary Fig. S1 andSupplementary Data) allows an easy extension to incorporate additional modules. Users can automate docking, QSAR, structure-ensemble docking, hybrid docking/QSAR and docking of protein mutants (Supplementary Table S1 provides an estimated processing time for each feature). The server is complemented with other functionalities to prepare the receptor and the compound libraries. Receptor and compounds can be uploaded by the user or extracted from public databases.
2 Methods
2.1 Basic capabilities
SEABED provides a default representative subset of molecules. Users can use similarity measures to retrieve chemical entities similar to a lead using Tanimoto score based on Daylight fingerprints and can derive a series of descriptors for potential binders, such as Lipinski’s, logP contributions or atomic charges (seeSupplementary Data). Macromolecular receptors can be explored using our quality control (seeSupplementary Table S2). Users can also explore the titration of a protein for a given pH (Liet al., 2005) to generate realistic electrostatic distributions, to compute and to visualize interaction potentials determined by means of our cMIP algorithm, (Gelpíet al., 2001). SEABED can automatically locate the binding site of the receptor from holo structures, or predict cavities from apo forms using FPocket (Le Guillouxet al., 2009). Receptor and ligand preparation, docking and scoring is implemented using Autodock Vina tools (Trott and Olson 2010). Docking results are shown as a dynamic table of 2D compounds ranked by docking score. For each docked ligand, users can explore a 3D view of the atom and amino acid contributions to the binding energy based on Van der Waalls and electrostatic potentials (Orozco and Luque, 2000), information that can be useful for mutation design and for lead optimization.
2.2 Advanced capabilities
To account for receptor flexibility SEABED supports ensemble docking. The receptor can be either multiple rigid structures taken from PDB, homology models or snapshots from a MD simulation. Our server is integrated into INB’s FlexPortal (http://mmb.irbbarcelona.org/FlexPortal/) and links out to MoDEL (Meyeret al., 2010), MDWeb (Hospitalet al., 2012) and FlexServ (Campset al., 2009) to access existing trajectories or calculate an ensemble of 3D structures from MD simulations. The supported formats are PDB, Charmm, Amber, Gromacs or NAMD, among others (seeSupplementary Table S3). By default, the server processes the dynamic information on the receptor or receptor-ligand trajectory and selects up to 10 representative snapshots from a K-means snapshot clustering, based on all protein/pocket atom RMSd (pocket can be selected manually by the user, or guessed by SEABED, seeSupplementary Data). Docking results for ensemble docking are presented in a similar way than those of rigid docking, but the page adds now both the best and the average scores among the MD ensemble.
SEABED supports QSAR studies using a wide list of 1D and 2D fingerprints (Supplementary Table S4). The server implements several machine learning methods, with some configurable parameters including Random Forest, Support Vector Machines or Naive Bayes, among others—see full list inSupplementary Table S5. For trained models, the ROC curve and Area Under the Curve (AUC) score is computed with 10-fold cross-validation. Predictive QSAR models are then stored and can be applied later to other extended small molecules datasets. It is also possible to scan drug-like Zinc (15M of compounds).
Hybrid docking/QSAR is run providing the receptor’s 3D structure to screen a library. First, a docking experiment for a short list of molecules is run to define one set of ‘actives’ (molecules docked with good scores) and another set of ‘decoys’ (bad dockings). These two sets are then used to train machine learning algorithms to derive predictive QSAR models. As shown by others, this hybrid approach increases performance by some orders of magnitude with respect to massive docking and shows good prediction rates (Cherkasovet al., 2006;Fukunishiet al., 2008, Goñiet al., in preparation;Yoonet al., 2005).
SEABED can automatically search for known annotated protein mutants as those deposited in the 1000 Genomes database (McVeanet al., 2012). Non-synonymous SNPs are flagged on the protein’s 3D structure. The predicted pathological nature of the mutation is determined using PMUT (Ferrer-Costaet al., 2005). The server allows the user to obtain 3D models of the mutants by running PELE refinements (Borrelliet al., 2005;Madadkar-Sobhani and Guallar, 2013).
2.3 How to use SEABED
SEABED provides a virtual workspace to scan for active compounds of a given target. This section describes how to test the prediction capability of SEABED on MAP kinase (MK14) p38 alpha (see full details inSupplementary Data and SEABED online Tutorial/Getting Started sections). MK14 is a protein involved on a wide variety of cellular processes such as proliferation, differentiation, transcription regulation and development.Figure 1 shows the basic schema of SEABED basic protocol to run a rigid docking, an ensemble docking and a docking on known mutants of MK14.
Fig. 1.
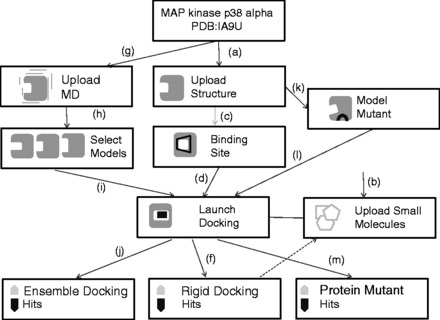
Workflows available to SEABED. For rigid docking, users (a) upload a protein structure, (b) upload a set of small molecules, (c) select or predict binding site, (d) launch docking and (f) get small molecules ranked by docking score. A sub set of this list can be saved and stored in the workspace for other experiments. For MD ensemble docking the workflow is similar but users need to (g) upload a MD trajectory instead of a rigid PDB structure and then (h) choose a set of snapshots before (i) launch and (j) get docking results. For mutants, users need to first (k) model protein variant and then (l) launch docking and (m) get results
On SEABED’s landing page, user needs to create and name a ‘New project’ (see details athttp://www.bsc.es/SEABED/static/NewProject.html). For a rigid docking, user needs to first upload the target structure, upload a set of small molecules and select a binding site before launching the docking (seeFig. 1a–d) and full description athttp://www.bsc.es/SEABED/static/RigidDocking.html). For this example we select 1A9U (one of the structures of MK14) from ‘Upload PDB/traj’. SEABED pre-loads a set of drug-like compounds on every project by default, but for this example we will upload (through ‘Upload SDF’ button) a set of known active/decoy compounds in SDF format that can be downloaded from DUD-E database (Mysingeret al., 2012). Then, we will run ‘New Docking’ and choose to automatically predict binding site region and run all dockings in background. Registered users (registration is optional) will be notified by email once the job is completed. Docking results (Fig. 1f) are available in a table format and can be downloaded as an SDF file. Results can be filtered by ranking score and re-loaded as a new subset on the workspace. The expected results (AUC and ROC curves) of this test are available onSupplementary Data.
Ensemble docking adds a few steps to the workflow, as the user needs to upload a MD trajectory and choose a number of representative snapshots to be used in docking (Fig. 1g–i and full description at (http://www.bsc.es/SEABED/static/EnsembleDocking.html). To run Ensemble docking, select ‘New Docking’ from the bottom menu and select ‘Molecular Dynamics’ on the Select target panel. SEABED will expect two files: topology (Reference file) and coordinates (Trajectory file). Then we will choose the number of representative snapshots (up to 10) based on K-means clustering and RMSd score of atoms from pocket. The expected results (Fig. 1j) are also available onSupplementary Data. As reported in a previous work (Chaudhuriet al., 2012) for 1A9U, MD ensemble can significantly improve AUC score, from 0.64 for rigid docking to 0.75 for ensemble docking.
The last workflow of this example explains how to use SEABED to run docking on known mutants of this protein. Users will need to search and model mutants by following the workflow steps onFigure 1k–m. A full description is available athttp://www.bsc.es/SEABED/static/MutantDocking.html). SEABED is able to identify two mutants from MK14 found in 1000 Genomes database (P6A and D343G). From the ‘Structures’ panel go to 1a9u and select ‘Info’. Then select ‘Mutation Information’ from the drop down menu. Then select ‘Model the mutation’ and the two new structures will be available on the ‘Structures’ main panel. Then run a rigid docking workflow. Docking these mutants using the same dataset (see expected results onSupplementary Data) we do not see big differences compared with the wild type. Thus, the performance of the mutants does not differ significantly. For a more detailed description of this example seeSupplementary Data and SEABED online Tutorial and Getting Started sections.
Supplementary Material
Acknowledgements
We thank technical assistance of Víctor Guallar (PELE), Josep Lluís Gelpí and Laia Codó (cMIP), Santiago Villalba, Manuel Rueda. We also thank Robert Soliva, Floriane Montanari and Lucía Díaz for their valuable feedback.
Funding
Spanish Ministry of Science and Competitiveness (MINECO; Bio2012-32868; SEV-2011-00067); National Institute of Bioinformatics (INB) Marató de TV3, and the Catalan AGAUR (Grup Consolidat; MO). MO is an ICREA Academia Fellow.
Conflict of Interest: none declared.
References
- Alvarez J.C. (2004)High-throughput docking as a source of novel drug leads.Curr. Opin. Chem. Biol.,8,365–370. [DOI] [PubMed] [Google Scholar]
- Bajorath J. (2002)Integration of virtual and high-throughput screening.Nat. Rev. Drug Discov.,1,882–894. [DOI] [PubMed] [Google Scholar]
- Borrelli K.W., et al. (2005)PELE: protein energy landscape exploration. A novel Monte Carlo based technique.J. Chem. Theory Comput.,1,1304–1311. [DOI] [PubMed] [Google Scholar]
- Camps J., et al. (2009)FlexServ: an integrated tool for the analysis of protein flexibility.Bioinformatics,25,1709–1710. [DOI] [PubMed] [Google Scholar]
- Carlson H.A. (2002).Protein flexibility is an important component of structure-based drug discovery.Curr. Pharm. Des.,8,1571–1578. [DOI] [PubMed] [Google Scholar]
- Carrillo O., et al. (2012).Fast atomistic molecular dynamics simulations from essential dynamics samplings.J. Chem. Theory Comput.,8,792–799. [DOI] [PubMed] [Google Scholar]
- Chaudhuri R., et al. (2012)Application of drug-perturbed essential dynamics/molecular dynamics (ED/MD) to virtual screening and rational drug design.J. Chem. Theory Comput. 8,2204–2214. [DOI] [PubMed] [Google Scholar]
- Cherkasov A., et al. (2006)Progressive docking: a hybrid QSAR/docking approach for accelerating in silico high throughput screening.J. Med. Chem. 49,7466–7478. [DOI] [PubMed] [Google Scholar]
- Coleman R.G., et al. (2013)Ligand pose and orientational sampling in molecular docking.PLoS One,8,e75992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Costa C., et al. (2005).PMUT: a web-based tool for the annotation of pathological mutations on proteins.Bioinformatics,21,3176–3178. [DOI] [PubMed] [Google Scholar]
- Fukunishi H., et al. (2008)Hidden active information in a random compound library: extraction using a pseudo-structure-activity relationship model.J. Chem. Inf. Model.,48,575–582. [DOI] [PubMed] [Google Scholar]
- Gelpí J.L., et al. (2001)Classical molecular interaction potentials: improved setup procedure in molecular dynamics simulations of proteins.Proteins,45,428–437. [DOI] [PubMed] [Google Scholar]
- Grosdidier A., et al. (2011)SwissDock, a protein-small molecule docking web service based on EADock DSS.Nucleic Acids Res.,39,W270–W277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital A., et al. (2012)MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations.Bioinformatics,28,1278–1279. [DOI] [PubMed] [Google Scholar]
- Le Guilloux V., et al. (2009)Fpocket: an open source platform for ligand pocket detection.BMC Bioinform.,10,168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. (2005).Very fast empirical prediction and interpretation of protein pKa values.Proteins,61,704–721. [DOI] [PubMed] [Google Scholar]
- Madadkar-Sobhani A., Guallar V. (2013)PELE web server: atomistic study of biomolecular systems at your fingertips.Nucleic Acids Res.,41,W322–W328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G.A., et al. (2012)An integrated map of genetic variation from 1,092 human genomes.Nature,491,56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T., et al. (2010)MoDEL (molecular dynamics extended library): a database of atomistic molecular dynamics trajectories.Structure,18,1399–1409. [DOI] [PubMed] [Google Scholar]
- Mysinger M.M., et al. (2012)Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking.J. Med. Chem.,55,6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N.M., et al. (2011)Open Babel: an open chemical toolbox.J. Cheminform. 3,33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco M., Luque F.J. (2000)Theoretical methods for the description of the solvent effect in biomolecular systems.Chem. Rev.,100,4187–4226. [DOI] [PubMed] [Google Scholar]
- Rueda M., et al. (2009)Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes.J. Chem. Inform. Model.,49,716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetko I.V., et al. (2005)Virtual computational chemistry laboratory—design and description.J. Comput. Aided Mol. Des.,19,453–463. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. (2010)AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading.J. Comput. Chem. 31,455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S., et al. (2005)Surrogate docking: structure-based virtual screening at high throughput speed.J. Comp. Aided Mol. Des.,19,483–497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.