

SUMMARY
Comprehensive sequencing approaches have allowed for the identification of the most frequent contributors to cancer, known as drivers. They have also revealed a class of mutations in understudied, infrequently altered genes, referred to as “long tail” (LT) drivers. A key challenge has been to find clinically relevant LT drivers and to understand how they cooperate to drive disease. Here, we identified far upstream binding protein 1 (FUBP1) as an LT driver using an in vivo CRISPR screen. FUBP1 cooperates with other tumor suppressor genes to transform mammary epithelial cells by disrupting cellular differentiation and tissue architecture. Mechanistically, FUBP1 participates in regulating N6-methyladeno-sine (m6A) RNA methylation, and its loss leads to global changes in RNA splicing and widespread expression of aberrant driver isoforms. These findings suggest that somatic alteration of a single gene involved in RNA splicing and m6A methylation can produce the necessary panoply of contributors for neoplastic transformation.
Graphical Abstract
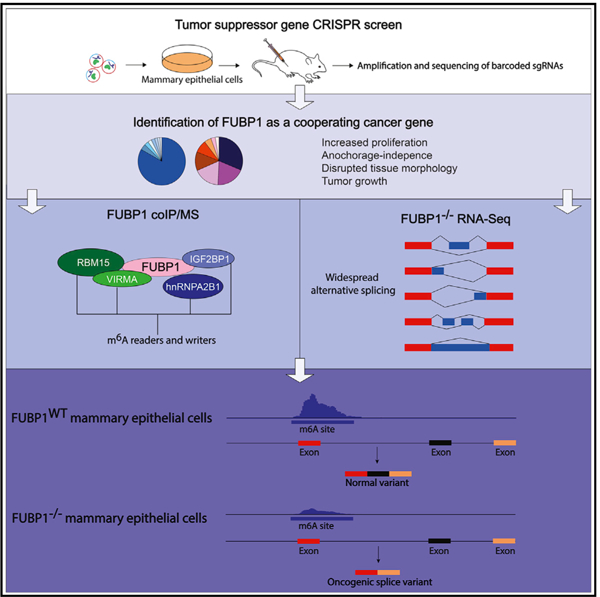
In Brief
Elman et al. identify FUBP1 as a long tail cancer driver using a combinatorial CRISPR/Cas9 screen. FUBP1 and PTEN cooperate to transform mammary epithelial cells by disrupting cellular differentiation and tissue architecture. FUBP1−/− cells exhibit decreased m6A RNA methylation, leading to widespread expression of aberrant driver isoforms.
INTRODUCTION
Within the past decade, tremendous efforts have focused on systematic, genome-wide surveys to identify the most frequently modified genes to distinguish those that drive carcinogenesis from those that are modified because of generalized genomic instability. Results from these tumor sequencing studies indicate that many cancers harbor alterations in only 3–4 known drivers, and 15% of tumors lack alterations in even a single known driver (Davoli et al., 2013; Garraway and Lander, 2013; Imielinski et al., 2012). In addition, solid tumors arise because of oncogenic cooperation between alterations of multiple drivers (Fearon and Vogelstein, 1990; Hanahan and Weinberg, 2000). These considerations strongly suggest that there are many drivers yet to be identified that are altered at low frequencies in cancer (Bailey et al., 2018; Lawrence et al., 2014; Liu et al., 2006; Merid et al., 2014; Tokheim et al., 2016). Although these long tail (LT) driver genes are difficult to identify because they are mutated at frequencies only slightly greater than those for passenger genes, knowledge of how they affect major signaling pathways controlling growth and proliferation is critical for understanding and treating cancer based on its genetic underpinnings.
It has been challenging to identify cooperating cancer genes (CCGs) from analysis of existing collections of tumor sequence data. They are underpowered because of the relatively few tumors for many tumor types (Bailey et al., 2018; Lawrence et al., 2014; Liu et al., 2006; Merid et al., 2014; Tokheim et al., 2016). In addition, extreme levels of tissue-specificity exist with respect to drivers of cell proliferation and survival regulation in cells (Haigis et al., 2019; Sack et al., 2018; Schneider et al., 2017); therefore, the precise tissue of interest must be examined in the organism of interest. Because of this, functional approaches, such as insertional mutagenesis, using transposon-based screens in mice, have been used to identify CCGs. For example, genes that may cooperate with PTEN, SMAD4, or BRAF for tumorigenesis have been identified with Sleeping Beauty or piggyBac mutator systems (Ni et al., 2013; de la Rosa et al., 2017; Takeda et al., 2016). However, for breast cancer, mouse and human tissues differ markedly in their genetic requirements for transformation (Perlman, 2016), which has limited the extent to which insertional mutagenesis studies directly model human breast cancer and can be used functionally identify CCGs. A method to functionally identify cooperating genetic lesions in human breast cancers is needed. Therefore, we have developed a combinatorial loss-of-function screen to identify putative tumor suppressor genes (TSGs) whose cooperation is selected for by promoting tumorigenesis in vivo. We show that a top hit, FUBP1, is an LT gene that cooperates with PTEN as a powerful driver of neoplastic transformation. Furthermore, via regulation of RNA N6-methyladenosine (m6A) methylation, FUBP1 globally affects the landscape of alternative splicing to create aberrant proteins that drive malignant transformation.
RESULTS
Identification of Cooperating TSGs in an In vivo Loss-of-Function CRISPR-Cas9 Screen
We created a unique resource to screen for functionally relevant LT drivers in an unbiased manner: a small genetic library targeting genes that are clinically altered in cancer and thus strongly enriched for high-priority candidates. For loss-of-function screening, a “TSG” library, comprising the 500 genes that are most significantly affected by loss of function in human cancers according to the Tumor Suppressor and Oncogene (TUSON) Explorer algorithm (Davoli et al., 2013) in a lentiviral CRISPR-Cas9 format, was developed. This library has already been used in conjunction with an oncogene open reading frame (ORF) library to investigate how cancer drivers interact in an in vitro screen, revealing a spectrum of TSGs and oncogenes that can genetically interact and substitute for one another to modify the behavior of cancer cells with disrupted EGFR signaling (Liao et al., 2017). Additionally, the same libraries have been used in vitro to identify TSGs and oncogenes that may similarly substitute for BRD4-NUT, the complex of an epigenetic factor BRD4 and the protein NUT that cause oncogenic signaling and tumor growth, particularly in NUT midline carcinoma (Liao et al., 2018).
To screen for preferentially cooperating LT drivers, we created a sub-library of 100 TSGs with 10 single guide RNAs (sgRNAs) per each targeted gene (Figure 1A; Table S1). MCF10F cells (immortalized, non-transformed human mammary epithelial cells) were transduced with the pooled library at a multiplicity of infection (MOI) of 3, to ensure that most cells received at least two sgRNAs. This strategy enabled us to interrogate approximately 1 × 106 different sgRNA combinations, targeting an average of three putative TSGs in each cell. Infected cells were orthotopically implanted into the inguinal mammary glands of immunocompromised non-obese diabetic/and severe combined immunodeficiency (NOD-SCID) mice and monitored for tumor growth. All mice developed tumors, and tumors larger than 5 cm3 were randomly sampled for histology, whereas the remainder of the tumor was sequenced. Numerous growth patterns were observed within and across the H&E-stained tumors, including squamous, metaplastic, and papillary carcinomas (Figure 1B). We also detected variable expression of biomarkers and hormone-receptor expression common in human tumors, recapitulating clinical presentations of heterogeneous human breast cancers (Figures 1C and S1). This intra- and inter-tumor heterogeneity mirrors clinical cancers and is consistent with different combinations of genetic drivers influencing tumor phenotype (Marusyk et al., 2012).
Figure 1. Identification of Cooperating TSGs in an In vivo Loss-of-Function CRISPR-Cas9 Screen.

(A) Experimental schema for CRISPR-Cas9 library in vivo screen for cooperating TSGs. The library was packaged in lentivirus used to infect MCF10F cells at an MOI = 3. Cells were selected for expression of the library and implanted into NOD-SCID mammary fat pads at 1 × 106 cells per gland. Tumors were excised and sequenced for analyses.
(B) H&E staining of tumors for identification of histological phenotypes: squamous (i and v), metaplastic (ii), papillary (iii and vi), and adenocarcinoma (iv).
(C) Immunohistochemistry (IHC) staining of tumors for identification of specific epithelial and proliferation markers: EpCAM (i), Ki67 (ii), PR(203) (iii), PR(403) (iv), E-cadherin (v), p53 (vi). Scale bars represent 100 μm. Magnification = 203 for microscope images.
(D) Pie charts representing the clonal heterogeneity and dominant contributing and/or cooperating TSGs in the tumors. Each chart represents one tumor; each slice of each pie chart represents a subclone.
(E) Mutual exclusivity analysis of FUBP1 (gene A) and tumor suppressor genes that cooperated with FUBP1 in the screen (gene B) across 69,310 human cancer samples in 233 studies using cBioPortal. “Neither” represents the number of samples in which neither gene A nor B was altered. “A” represents the number of samples in which only gene A is altered. “B” represents the number of samples in which only gene B was altered. “A&B” represents the number of samples in which both genes A and B were altered.
Sequencing the recovered sgRNAs from the tumors’ genomic DNA revealed that the tumors were clonally diverse and identified expected as well as unexpected CCGs (Figure 1D; Table 1). We noted that well-established cancer drivers clinically validated by sequencing of patient breast cancers (PTEN, RB, p53, NF2, SMAD4, WT1, RUNX1, GATA3, MAP3K1, MLLT4, NCOR1) cooperated with genes that have yet to be shown to be functional cancer drivers (FUBP1, TRIP12, KDM5C, ARID1B) in the breast. Among the unexpected CCGs, far upstream binding protein 1 (FUBP1), a gene previously uncharacterized in breast cancer, was a recurring hit in multiple breast tumors and appeared to cooperate with multiple established drivers (PTEN, RB, p53). Given its stark contribution to tumor formation in the screen and its understudied role in breast cancer, we focused on elucidating the role of FUBP1 as an LT driver and how concurrent deletion of PTEN may enhance its role in cancer.
Table 1.
Co-occurring sgRNAs Identified in Tumor Subclones
| Tumor 1 | Tumor 2 | Tumor 3 | Tumor 4 | Tumor 5 | Tumor 6 | |
|---|---|---|---|---|---|---|
| Subclone 1 | FUBP1, PTEN | PTEN, KDM5C | FUBP1, PTEN | PTEN, ARID1B | SMAD4, PTEN | NF2, SMARCB1, GATA3, RB1 |
| Subclone 2 | PTEN, NF2 | MLLT4, ACVR1B | PTEN, PIK3R1, TP53 | PTEN, RASA1 | PTEN, CDKN2A | SMAD4, PTEN |
| Subclone 3 | FUBP1, TP53, CDH1 | ZNF318, NF2 | PTEN, NCOR1 | STAG2, SMARCB1, PTEN | PIK3R1, NF2 | WT1, GATA3, RUNX1 |
| Subclone 4 | ATM,SMAD4 | GATA3, CDKN1A | RB, PTEN, FUBP1 | KDM5C | BAP1, RASA1, TRIP12 | SMAD2, TRIP12, ZNF318 |
| Subclone 5 | NF2, PIK3R1 | APC, RNP43 | RNX1, MLLT4 | KDM6A, ZNF318 | HLA-B, RBM10 | NF2, TGFBP2 |
| Subclone 6 | CDKN2A1, TGFBR2, DMK5C | CRIPAK, ASXL1, AJUBA | TRIP12, ZNF318 | FBXW7, TP53, PTEN | SMARCA4, ARHGAP35 | |
| Subclone 7 | CREBBP, STK11, SMAD4 | FUBP, KDM5C, PTEN | PIK3R1, MLLT4, NCOR1 | ARID2, RB1 |
To examine the clinical relevance of concurrent loss of FUBP1 and PTEN, as well as other CCGs from the screen, we interrogated public tumor datasets (Cerami et al., 2012; Gao et al., 2013). We found that FUBP1 alterations tend to significantly (p < 0.001) co-occur with PTEN alterations, as well as alterations in RB1, TP53, CDH1, and KDM5C, across many cancer types, including glioblastoma and lung squamous cell carcinoma, as well as breast-invasive carcinoma (Figure 1E). These correlations lend further validity to the screen as a method of identifying TSGs that preferentially cooperate to drive cancer.
FUBP1 is a single-stranded DNA and RNA-binding protein best known for its role as a positive regulator of c-MYC in normal hematopoiesis (Duncan et al., 1994; Zhou et al., 2016). FUBP1 is also a regulator of post-transcriptional events, such as translation, mRNA stability, and splicing (Hwang et al., 2018; Jacob et al., 2014; Zhang and Chen, 2013). FUBP1 has been isolated in association with spliceosomal complexes (Hwang et al., 2018; Irwin et al., 1997; Jacob et al., 2014; Li et al., 2013; Lin et al., 2009; Miro et al., 2015) and includes four K homology domains, which are homologous to heterogeneous nuclear ribonucleoprotein K, a component of the spliceosomal H complexes (Benjamin et al., 2008; Braddock et al., 2002). Its role in splicing has only recently begun to be understood. Depending on context, FUBP1 can either enhance or suppress splicing (Hwang et al., 2018; Jacob et al., 2014; Li et al., 2013). In patients, FUBP1 missense, nonsense, and silent mutations, as well as whole-gene deletions, frameshift deletions, and insertions are observed in cancers, such as those of the central nervous system and intestinal cancer (Bailey et al., 2018; Bettegowda et al., 2011; Malz et al., 2009; Rabenhorst et al., 2009; Sahm et al., 2012; Singer et al., 2009; Zhang et al., 2013). Despite the biochemical roles of FUBP1, it is unclear how its alteration may affect transformation or cancer pathogenesis.
FUBP1 Loss Drives Several Characteristic Features of Transformation and Cooperates with PTEN Loss to Promote Tumor Growth In vivo
We used CRISPR-Cas9 to generate FUBP1-, PTEN-, and PTEN/FUBP1-null MCF10F cells using two different sgRNAs targeting FUBP1 to assess how loss of the genes individually or in combination might affect features of transformation. Western blot analysis validated complete loss of FUBP1 and/or PTEN in MCF10F cells (Figures 2A and S2). Loss of FUBP1 or PTEN alone or in combination significantly increased cellular proliferation compared with the non-targeting control (NTC) (Figures 2B and S2). Although there was occasional colony formation of PTEN null cells on soft agar, loss of FUBP1 in combination with PTEN led to the most significant increase in anchorage-independent colony formation on soft agar (Figures 2C, 2D and S2).
Figure 2. FUBP1 Loss Drives Several Characteristic Features of Transformation and, with PTEN Loss, Promotes Tumor Growth In vivo.

(A) Western blot of lysates from MCF10F cells transduced with NTC, PTEN, FUBP1, or PTEN+FUBP1 CRISPR-Cas9 sgRNA to show knockout of corresponding genes.
(B) Proliferation of the indicated cell lines over 7 days, measured by MTS assay, analyzed with an ANOVA with a multiple-comparisons test.
(C) Soft agar growth assays for the indicated cell lines. Images show representative soft agar fields for the indicated cell lines after 2 weeks. Scale bar represents 50 μm. Analyzed with one-way ANOVA with a multiple comparisons test against sgNTC.
(D) Quantification of soft agar colonies in the indicated cell lines after 2 weeks.
(E) Representative immunofluorescent (IF) images of the indicated cell lines after 10 days in hydrogels. Green, CK14+; red, CK8/18+. Nuclei stained with Hoechst (blue). Scale bars represent 100 μm. Pie charts represent quantification of 3D tissue morphology, cellular polarity, and expression. Blue sections represent T1 structures, red represent T2, green represent T3, and white represent T4.
(F) Bioluminescence imaging was used to detect tumor growth in NOD-SCID mice injected with the indicated cell lines (n = 5 mice per cell line, 5 × 106 cells injected per gland).
(G) Quantification of bioluminescence emitted from each injected gland at 2 weeks after injection.
(H) Gross and microscopic detection of tumor growth in NOD-SCID gland injected with PTEN/FUBP1-null MCF10F cells. Images depict highly vascularized tumors (i) with angiogenesis and inflammation (ii and iii), as well as an abnormally mitotic cell (iv). Scale bars represent 50 μm. Data are presented as means ± SEM, n = 3 biological replicates per cell line. *p < 0.05, ***p < 0.0005 (two-tailed Student’s t tests unless otherwise indicated).
Because loss of tissue architecture is a hallmark of transformation, we seeded cells in 3D hydrogels to assess the role of FUBP1 on tissue morphogenesis and differentiation. FUBP1-, PTEN-, and PTEN/FUBP1-null MCF10F cells were grown in collagen-based hydrogels (Miller et al., 2017; Sokol et al., 2016) supplemented with extracellular matrix components and allowed to grow for 10 days (Figures 2E and S2). In this assay, the cells grew into different types of 3D tissues with varying degrees of structural complexity. We classified the growths based on morphology, presence of CK14+ basal and CK8/18+ luminal mammary epithelial cells, and integrity of cellular polarity (basal cells surrounding luminal cells). The 3D outgrowths were classified into the following morphologically distinct groups: normal glandular tissue with conserved organization of cells (T1), normal structure but loss of polarity (T2), disrupted structure and loss of either basal cells or luminal cells (T3), and complete loss of structure with significantly diminished numbers of basal cells (T4) (Figures 2E and S2). We found that most control cells grow as normal T1 and T2 structures, whereas the loss of PTEN, FUBP1, or the combination of PTEN/FUBP1 caused a significant increase in the number of abnormal T3 and T4 structures. PTEN loss led to the preferential formation of T3 structures, whereas FUBP1 loss alone or in combination with PTEN was associated with the development of disrupted T4 structures (Figures 2E and S2). Thus, FUBP1 loss affected 3D tissue organization, basal differentiation, and cellular polarity, including a significant loss of basal cells.
To determine whether loss of FUBP1 alone or combined with the loss of PTEN promotes tumorigenic behavior in vivo, FUBP1-, PTEN-, or FUBP1/PTEN-null MCF10F cells were implanted into the inguinal mammary glands of NOD-SCID mice. Within 2 weeks, all mice implanted with FUBP1/PTEN-deficient cells had formed tumors, whereas FUBP1-null or PTEN-null cells did not (Figures 2F and 2G). Gross and microscopic examination of the PTEN/FUBP1-deficient tumors revealed expansive, angiogenic, cystic tumors containing widespread inflammation and abnormal mitoses (Figure 2H). Collectively, these findings demonstrate that cells with the loss of FUBP1 alone and in combination with PTEN exhibit classic hallmarks of transformation, including increased proliferation, anchorage-independent growth, and loss of tissue architecture, and they form tumors in vivo.
FUBP1 Loss Causes Widespread Alternative Splicing and Processing of Cancer Driver Genes
To determine how FUPB1 loss contributes to neoplastic transformation, gene expression profiling was performed by RNA sequencing (RNA-seq) in cells deficient for FUBP1 (Figure S3). There were a number of differentially expressed genes between the FUBP1-null and NTC cells, ~25% of which were noncoding RNA transcripts (ncRNA). Gene set enrichment analysis (GSEA) of differentially expressed genes revealed a strong negative enrichment for the epithelial-to-mesenchymal transition (EMT)-related genes (TGFB1, SNAI2, MFAP5), consistent with the loss of basal cells in FUBP1-null cells. Given previous findings showing that FUBP1 can promote MYC activation (Duncan et al., 1994; He et al., 2000), we also examined whether FUBP1 deficiency might be affecting MYC expression and/or downstream MYC signaling. However, consistent with a recent report (Seiler et al., 2018), we did not see a significant reduction in MYC expression levels in cells lacking FUBP1.
Accumulating evidence shows that alternative splicing has a key role in cancer pathogenesis; we and others (Lee et al., 2018; Ni and Kuperwasser, 2016) have shown that that mRNA splicing and processing creates genes and proteins that act as functional cancer drivers. Given that previous work has implicated FUBP1 as a splicing regulator (Hwang et al., 2018; Jacob et al., 2014), we were interested in determining whether FUBP1 loss might be affecting alternative splicing and processing (AS) events that would be leading to upregulation of variant mRNA transcripts. Applying replicative multivariate analysis of splicing (rMATS 4.0.1) to the RNA-seq data, we found more than 10,000 differential AS events between the NTC and FUBP1-null MCF10F cells, ~60% of which are unannotated splicing events. Across the differential splice events, 70% were skipped exons (SEs), 16% mutually exclusive exons (MXEs), 7% alternative 3′-splice sites (A3SS), 6% alternative 5′-splice sites (A5SS), and 1% retained intron events (RIs) (Figure 3A). We used the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (Huang et al., 2009a, 2009b) to functionally cluster the AS transcripts most significantly expressed differentially to begin to understand the global consequences of these AS events. Many of the alternatively spliced genes in FUBP1-null cells were involved in mRNA processing, cell cycle, and DNA damage repair (Figure 3B), which might signal potential vulnerabilities of tumors bearing FUBP1 mutations.
Figure 3. FUBP1 Promotes Alternative Splicing of Cancer Driver Genes.

(A) rMATS 4.0.1 used for detection of alternative splicing events in FUBP1-null cells. Pie chart shows distribution of A5′-splice site (A5SS), A3′-splice site (A3SS), skipped exon (SE), retained intron (RI), and mutually exclusive exon (MXE) splice events.
(B) Enrichment of functions of alternatively spliced genes detected by rMATS, performed by DAVID functional annotation analysis.
(C) Summary of cancer genes that are alternatively spliced in FUBP1-null cells, indicating gene name, biotype, and summary of function.
(D–F) Western blots of NTC and FUBP1-null cell lysates for CASP8 (D), BRCA1 (E), and MAGI3 (F) and corresponding sashimi plot of alternative splicing. Blot for BRCA1 was stripped and re-probed for MAGI3. In sashimi plots, y axis represents a modified reads per kilobase of transcript (RPKM), per a million mapped reads. Peaks report number of junction reads. Below each, cartoon representations of alternative isoforms: exons and introns are not drawn to scale and represented as black rectangles and lines, respectively.
There is increasing interest in understanding how mRNA events, such as AS, can be as potent as DNA mutation events in driving tumorigenesis. In human breast cancer and chronic lymphocytic leukemia (CLL), recurrent, truncated mRNAs caused by intronic polyadenylation affect genes with tumor-suppressive functions and can give rise to oncoproteins (Lee et al., 2018; Ni and Kuperwasser, 2016; Ni et al., 2018). In FUBP1-null cells, 11 genes with tumor-suppressive functions were found to be truncated by AS (Figure 3C). Three FUBP1 AS targets were the same recurrent variants found in human CLL: DICER, MGA, and ZMYM5 (Lee et al., 2018) (Figures S4A–S4C). Our analyses revealed that 11% of DICER1 transcripts, 61.5% of MGA transcripts, and 28.5% of ZMYM5 transcripts undergo AS to generate aberrantly A3′-spliced isoforms. The prematurely truncated form of DICER1 encodes for a protein that is unable to process microRNA (miRNA), leading to reduced functional DICER protein and altered miRNA processing (Lee et al., 2018). The prematurely truncated form of MGA creates a dominant-negative regulator of full-length MGA to act as an oncoprotein (Lee et al., 2018). Additionally, in concordance with previous reports showing that FUBP1 acts as a splicing regulatory factor for the oncogene MDM2, we also detected that FUBP1-null cells exhibit AS of MDM2, leading to a 4-fold increase in the expression of MDM2-ALT2, an alternative variant reported to increase cyclin D1 to promote malignancy (Jacob et al., 2014) (Figure S4D). Other FUBP1 targets include Caspase8 (CASP8), which undergoes alternative A3′ splicing, resulting in a non-functional transcript (Figure 3D). CASP8 is an important regulator of apoptosis, and the loss of functional CASP8 may result in a classic hallmark of cancer, evasion of apoptosis (Stupack, 2013). Western blotting for CASP8 revealed a complete loss of CASP8 protein in FUBP1-null cells, and the corresponding Sashimi plot shows that, although there are 18 raw junction read counts for the A3′-spliced CASP8 transcript in the FUBP1-null samples, there are only four in the NTC (Figure 3D).
FUBP1 also targets the tumor suppressor BRCA1, a gene notoriously linked to breast and ovarian cancers (Figure 3E). FUBP1-null cells strongly overexpress a BRCA1 variant, BRCA1Δ11b, which lacks most of exon 11, the large internal exon that contains the nuclear localization signal (NLS) and the RAD51 binding domain, and it is the mutational hotspot in a large fraction of BRCA1 mutation carriers (Miki et al., 1994; Wilson et al., 1997). Previous work has shown that BRCA1Δ11b-expressing cells have diminished double-stranded break repair (Huber et al., 2001; Westermark et al., 2003). We used immunoblotting to validate that the ~100-kDa BRCA1Δ11b is expressed in the FUBP1-null, but not the NTC, cells.
Additionally, the oncogenic, prematurely truncated MAGI3 transcript MAGI3pPA is strongly expressed in the FUBP1-null, but not the NTC, cells (Figure 3F). The full-length MAGI3FL is a TSG and a regulator of the Hippo signaling pathway that normally binds and inactivates YAP to prevent tissue overgrowth and other behaviors characteristic of cancer (Ni and Kuperwasser, 2016). The prematurely truncated form, MAGI3pPA, is unable to bind YAP, thus promoting classic oncogenic behavior (Ni and Kuperwasser, 2016). Validation of CASP8, MAGI3, and BRCA1 AS events was also performed with FUBP1-null cells generated with an alternative sgRNA, confirming those changes were not off-target events (Figures S2F–S2H).
We also assessed the effects of FUBP1 loss on AS in an additional nontumorigenic mammary epithelial cell line, MCF10A (Figure S5A). Similar to the FUBP1-null MCF10F cells, FUBP1-null MCF10A cells showed a complete loss of CASP8 protein, compared with the NTC (Figure S5B). Unlike the MCF10F cells, however, the FUBP1-null MCF10A cells showed no difference in protein expression or splicing of MAGI3 or BRCA1 (Figures S5C and S5D). Interestingly, the NTC MCF10A cells exhibited a striking baseline expression of the aberrant isoforms MAGI3pPA and BRCA1Δ11b. This may be because the MCF10A cell line is more stem-like (Qu et al., 2015) or perhaps because they already contain mutations in other genes that are important for splicing regulation.
To further expand our investigation beyond the mammary epithelium, we surveyed splicing data from cancers that harbor FUBP1 loss-of-function (LoF) mutations as well as splicing changes in the human glioma cell line U87MG, in which FUBP1 was inhibited with small interfering RNA (siRNA) (Seiler et al., 2018). We found an overlap of FUBP1 splicing targets in primary human brain cancers as well as in FUBP1-knockdown U87MG cells with FUBP1-null MCF10F. Of the 307 conserved alternatively spliced genes between the FUBP1-null MCF10F cells, primary glioma samples and siFUBP1 U87MG cells, 26 were significant (Table 2). Several of those overlapping genes were A3SS, A5SS, and SE splicing events (false discovery rate [FDR] < 0.05). Taken together, these results support FUBP1 as an important regulator of AS and indicate that FUBP1 targets include cancer genes that are truncated or otherwise altered to drive malignant transformation.
Table 2.
FUBP1 Splicing Targets Conserved across Breast and Brain
| Splice Event | Gene Names |
|---|---|
| A3SS | SLC43A3, FUS, ZBED5, RESP1, KIAA1529, ABI2 |
| A5SS | C6orf48, TPM1, IARS, SEC31A, FASTK, SLC43A3, ITF81, KCNK2, SKA2, ALDH18A1, UBAP2 |
| SE | MYL6, FUS, MYL6B, CAPN2, UBC, SCARB1, DALRD3, FAM72D, BATS |
| RI | None |
| MXE | Data Not Available |
FUBP1 Loss Leads to a Reduction in m6A Modification Upstream of FUBP1-Regulated Splice Sites
To further investigate the mechanism by which FUBP1 controls AS events, we next performed co-immunoprecipitation (coIP) of V5-tagged FUBP1 followed by mass spectrometry (MS) to identify binding partners of FUBP1 that may participate in the regulation of mRNA processing (Table S2). We found significant enrichment for proteins involved in RNA splicing, mRNA processing and transport, as well as other regulators of translation (p ≤ 0.01) (Figure S6A; Table S3) with clusterProfiler (Yu et al., 2012) on the proteins that uniquely co-immunoprecipitated with FUBP1. This is consistent with previous reports of FUBP1 as a nucleic acid-binding protein and regulator of AS (Hwang et al., 2018; Jacob et al., 2014; Li et al., 2013; Miro et al., 2015).
Among the set of FUBP1-interacting proteins identified by coIP/MS, we also identified several regulators of the mRNA methylation modification m6A, including RBM15, IGF2BP1, hnRNPA2B1, and VIRMA (Figure S6B). RNA binding motif 15 (RBM15) has been implicated as an RNA-binding protein that is involved with m6A machinery important in recruiting m6A core catalytic proteins (Patil et al., 2016). Protein Virilizer Homolog (VIRMA) is similarly a key component of the m6A methyltransferase that has a role in recruiting the core catalytic components METTL3, METTL14, and WTAP (Yue et al., 2018). We confirmed the presence of a protein-protein interaction between RBM15 and VIRMA, as well as other members of the m6A reactome with FUBP1 (Figure 4A). We additionally probed for the m6A methyltransferase (methyltransferase-like protein 3 [METTL3]), and found that it co-immunoprecipitates with V5-tagged FUBP1, although not as strongly as with RBM15 and VIRMA (Figure 4A). Given these findings, we hypothesized that FUBP1 recruits RBM15 and/or VIRMA, which then recruit METTL3 to form the methyltransferase complex that ultimately methylates target mRNA sites.
Figure 4. Exons Upstream of FUBP1-Regulated Splice Sites Exhibit Diminished m6A Levels in FUBP1-Null Cells.

(A) Western blot validation of significant proteins from IP/MS experiment.
(B and C) Dot blot measuring global m6A levels in mRNA of indicated cell lines (B), quantified in (C).
(D) RNA-IP with m6A-modified or non-modified RNA bait followed by immunoblotting for a bona fide m6A reader, YTHDF2, and FUBP1.
(E) Distribution of m6A-seq peaks across the CASP8, BRCA1, and MAGI3 loci, based on analysis of previously published m6A-seq data in HepG2 cells. The locations of the putative m6A sites are indicated within exons directly upstream of splice sites yielding AS transcripts found in FUBP1-null cells.
(F) Relative m6A levels at m6A consensus sites of CASP8, BRCA1, and MAGI3 in exons upstream of splice sites that yield alternative variants, determined by m6A RIP-qPCR in NTC and FUBP1-null MCF10F cells.
(G) CASP8, BRCA1, and MAGI3 mRNA levels relative to GAPDH determined by quantitative real-time PCR in NTC and FUBP1-null MCF10F cells (n = 2) using primers flanking the regions distal from splice sites, not surrounding m6A consensus sites. Data are presented as means ± SEM, n = 3 biological replicates per cell line. *p < 0.05 (two-tailed Student’s t tests), unless otherwise stated.
To determine whether expression of FUBP1 affects mRNA m6A levels, we performed dot-blot assays with mRNA from NTC and FUBP1-null cells. Interestingly, we found that loss of FUBP1 results in a modest, but reproducible, decrease in global m6A-modified mRNA, compared with the NTC (Figures 4B and 4C). Given that finding, we next investigated whether FUBP1 might also be recognizing and binding m6A-modified mRNA. We performed immunoprecipitations with biotinylated RNA baits with or without m6A modification, followed by probing for FUBP1 as well as a bona fide m6A-binding protein, YTHDF2 (Wang et al., 2015). Our results show that FUBP1 binds to both m6A-methylated and non-methylated RNAs (Figure 4D), suggesting that FUBP1 may participate in bringing the m6A machinery to its intended site to promote methylation, but not that FUBP1 is specifically recognizing and binding methylated sites.
Given these findings, we next investigated whether FUBP1 acts as an m6A effector of AS. Recently, we reported that m6A has a role in regulating the formation of AS isoforms of various TSGs, including MAGI3, BRCA1, and LATS1 (Ni et al., 2018). If FUBP1 is acting as an m6A effector of AS, then m6A marks might be enriched within large internal exons of the FUBP1 splicing targets. To test that, we analyzed an available transcriptome-wide m6A-sequencing dataset (GEO Database: GSE37005) generated in the human hepatocellular carcinoma HepG2 cell line to identify enrichment of m6A peaks in exons upstream of splice sites in FUBP1-regulated transcripts (Dominissini et al., 2013a). We indeed found a concordance between m6A peak and splice-site localization (Figures 4E and S7). We found significant enrichment for m6A in exon 8 of CASP8, upstream of the splice site that results in the formation of the non-coding CASP8 transcript upregulated in FUBP1-null cells. Examination of m6A-seq peaks in BRCA1 also revealed strong enrichment in exon 10, upstream of exon 11, which is skipped in the BRCA1Δ11b isoform. There was also a strong m6A signal in exon 10 of MAGI3, which we previously showed to be related to the formation of the oncogenic, truncated MAGI3pPA (Ni et al., 2018). Given that FUBP1-null cells exhibit a global decrease in m6A deposition and increased AS, these data suggest that it is possible that the same splice variants affected by FUBP1 loss may be regulated by m6A deposition in exons directly upstream of their splice sites.
To investigate whether the splice variants affected by FUBP1 loss are indeed regulated by m6A modifications in exons upstream of their splice sites, we used an m6A antibody and FUBP1-null MCF10F cells. Relative methylation levels of exons upstream of FUBP1-regulated splice sites were determined by quantitative real-time PCR (Figure 4F). We found significantly decreased m6A methylation levels in two m6A consensus sites in exon 8 of CASP8, two m6A sites in exon 10 of BRCA1, and two m6A sites in exon 10 of MAGI3 in FUBP1-null cells, compared with the NTC cells. Additionally, we performed quantitative real-time PCR on total RNA from NTC and FUBP1-null cells with primers flanking regions without m6A sites that were distal from the exons of interest for each gene (Figure 4G). We detected that the NTC and FUBP1-null cells did not express significantly different levels of CASP8, BRCA1, or MAGI3 mRNA, indicating that the decreases in m6A in the exons upstream of the splice sites of interest were not simply due to a decrease in expression of the genes overall. Altogether, these data suggest that the loss of FUBP1 results in decreased m6A in regions that may be important for the regulation of mRNA splicing. Further work to examine how inhibiting or genetically deleting other components of the m6A complex affects the splicing of these and other genes is warranted to solidify the significance of these proteins in AS.
FUBP1 and Other m6A-Associated Proteins Are Altered in Human Breast Cancers
We next assessed the mutational frequency and copy number variations (CNVs) of FUBP1 in ~3,000 human breast cancer clinical samples using the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) dataset, generated from a long-term study of 2,509 breast tumors and 548 matched normal samples (Cerami et al., 2012; Gao et al., 2013; Pereira et al., 2016). Not surprisingly, ~6% of human breast tumors assessed have alterations in FUBP1, in concordance with the low alteration rate of LT drivers (Figure 5A). We found that, in contrast to canonical drivers like PTEN, which are mostly affected by deep deletions in cancer, FUBP1 alterations in these breast tumor samples include a range of alterations, including amplifications, as well as decreased mRNA levels. This may be due to the multiple roles FUBP1 has been shown to have in cancer, ranging from both activating MYC and negatively regulating its expression (Hsiao et al., 2010) to participating in a variety of post-transcriptional events.
Figure 5. FUBP1 and Other m6A-Associated Proteins that Are Altered in Human Breast Cancers.

(A and B) Percentage of breast cancer samples with (A) low (left) or high (right) copy number or mRNA alterations in FUBP1 or (B) other m6A-related genes, reported by METABRIC (2,509 samples).
(C and D) Schematic representation of FUBP1 mechanism in regulating alternative splicing: FUBP1 binds VIRMA and RBM15 to help recruit the rest of the m6A complex to target mRNA sites that affect splicing of cancer drivers (C). In the context of FUBP1 loss (D), there are fewer m6A modifications, thus preventing the interaction of normal m6A-binding proteins with modified sites and their downstream effects, i.e., AS of cancer driver genes.
Since our findings suggest that FUBP1 contributes to tumorigenicity by participating in m6A RNA methylation to regulate AS, we speculated that other genes involved in this modification might also be altered in breast cancer. Thus, we examined the frequency of mRNA and CNVs in FUBP1 binding partners as well as other m6A regulators (Figure 5B). VIRMA has copy number or significant changes to mRNA levels in more than 20% of the human breast tumors assessed, whereas other FUBP1 binding partners, RBM15 and IGFBP2, are altered in breast cancers at frequencies similar to FUBP1. Given those findings, we extended the analysis to other m6A modifiers. Indeed, alterations in other m6A readers, writers, and erasers are present in breast cancer at frequencies similar to FUBP1, suggesting they too may represent LT drivers. Like FUBP1, these genes undergo both amplifications and deep deletions, suggesting that their alteration in either direction may alter a homeostatic balance needed for the complex to function properly. The significance of such alterations in these m6A regulators will require further investigation to determine the extent of their contribution to malignant transformation. Cumulatively, however, these data reveal that alterations in the effectors of the m6A modification are clinically relevant in human breast cancers, strongly supporting the theory that disrupting this process is a key driver of human breast cancer.
DISCUSSION
Tumor sequencing studies have enabled the identification of the most potent drivers of cancer, which have been the focus of intense study and represent major therapeutic targets in cancer medicine. The strategies used to discover these drivers are limited, however, because they focus on identifying genes based on their frequency of alteration. Here, we set out to discover understudied, less-frequently mutated drivers, which have been referred to as the “long tail” cancer drivers (Armenia et al., 2018; Cho et al., 2016; Leiserson et al., 2015; Wood et al., 2007). We employed an unbiased combinatorial screen for preferentially CCGs in a mouse xenograft model of breast cancer using a TSG sgRNA library at a high MOI to ensure multiple alterations per cell. This created an environment in which cells with multiple genetic alterations competed for clonal dominance in vivo after orthotopic implantation into murine inguinal fat pads. This study uncovered multiple TSGs that showed genetic cooperativity with drivers such as PTEN, including FUBP1, a regulator of RNA modification and alternative splicing that affected multiple cancer-driver genes.
RNA splicing presents an opportunity for the expression of differentially configured transcripts that may be exploited by the cell to affect proliferation and survival. Examples of the dysregulation of splicing from cancer-promoting mutations in splicing factors, such as SF3B1 (Brinkman, 2004; Climente-González et al., 2017; Seiler et al., 2018; Venables, 2006), has captured the attention of the cancer researchers and brought the role of splicing in cancer to the forefront of cancer research. Modifications of mRNA with the potential to influence splicing patterns are also gaining attention. Research on the most abundant RNA modification, m6A, has intensified in the past decade (Dominissini et al., 2013a; Meyer and Jaffrey, 2014, 2017; Meyer et al., 2015). The effect of m6A levels on translation, degradation, and AS is far-reaching and has consequences in processes ranging from circadian rhythms to cancer (Dai et al., 2018; Fustin et al., 2013; Hastings, 2013; Jaffrey and Kharas, 2017; Pan et al., 2018). Most frequently found in large internal exons and the 3′ UTR, the m6A marks are well positioned for regulating alternative polyadenylation and AS (Meyer et al., 2012). There have been specific examples of m6A binding proteins directly regulating splicing. For instance, through association with SRSF3 and SRSF10, YTHDC1 binds m6A-modified mRNA to influence AS (Xiao et al., 2016). In addition, we have previously shown that reduced m6A marks are associated with the shortening of the MAGI3 (Ni et al., 2018).
In this study, we discovered FUBP1 among the most significant hits in the screen in cooperation with other TSGs, most frequently PTEN. We validated that loss of FUBP1, alone and in combination with PTEN, promotes classic malignant phenotypes, including hyperproliferation, anchorage-independent growth, and alteration of 3D tissue architecture. Furthermore, concurrent loss of PTEN and FUBP1 induced in vivo tumor growth. Analysis of human tumor datasets across many cancer types revealed that FUBP1 significantly co-occurs with PTEN as do the other PTEN cooperating genes from our screen, further validating our genetic approach.
We found that loss of FUBP1 resulted in the generation of ~10,000 AS events affecting a variety of cancer-related genes, such as BRCA1, MAGI3, CASP8. The FUBP1-regulated spliced form of BRCA1, BRCA1Δ11b, is missing most of exon 11 (Raponi et al., 2014; Tammaro et al., 2012; Wang et al., 2016), an alternatively spliced isoform that results in the functional loss of BRCA1. Therefore, creating this variant has significant implications for DNA repair, accumulation of mutational burden, and potential therapeutic approaches, such as the use of PARP inhibitors, which are known to be toxic to BRCA1 mutant cells (Deng and Scott, 2000; Hill et al., 2014; Nacson et al., 2018; Wang et al., 2016; Xu et al., 1999). FUBP1-null MCF10F cells also express a variant form of MAGI3, MAGI3pPA, which we previously showed codes for an active oncoprotein that inhibits its full-length form (Ni and Kuperwasser, 2016). When present in excess, MAGI3pPA binds MAGI3FL and promotes the nuclear translocation of YAP, a Hippo pathway transcription factor that drives tissue overgrowth and malignant transformation. The MAGI3pPA variant is recurrent and physiologically relevant in human breast cancers because it occurs in 7.2% of primary breast tumors (Ni and Kuperwasser, 2016). We also identified that FUBP1-null cells generate an aberrant form of CASP8 mRNA that is noncoding, resulting in loss of CASP8 protein. Dysregulated apoptosis is a classic hallmark of human cancer (Hanahan and Weinberg, 2000), so the implications of losing a regulator of apoptosis are widespread. In breast cancer, especially, loss of CASP8 has been shown to contribute to tumorigenicity and correlates with unfavorable outcomes (Mistry et al., 2006; Pu et al., 2017; Yin et al., 2010). As a tumor suppressor, FUBP1 acts analogously to classical epigenetic TGSs that alter the overall architecture of the transcriptional landscape. In this case, however, FUBP1 alters the post-transcriptional landscape in a profound manner to cooperate to drive tumorigenesis.
Although FUBP1 loss led to cell-type-specific AS of target genes, we observed that 8.5% of FUBP1-target genes are conserved among the AS events in both mammary epithelium and brain. This frequency is similar to the rate of FUBP1-target genes conserved between siFUBP1-U87MG cell line and primary glioma tumors with FUBP1 LoF (7.1%). As such, this suggests that the loss of FUBP1 results in consistent and generalizable AS changes across different tissue types but also shows profound tissue-specific differences. Further studies are needed to understand the significance of individual FUBP1-regulated AS events to tumorigenesis in a tumor- and tissue-specific manner.
In addition, we demonstrated that FUBP1 co-immunoprecipitates with members of the m6A complex and that FUBP1 loss results in decreased m6A levels. In parallel with these findings, we commissioned previously published m6A-seq databases to identify enrichment of m6A levels in exons directly upstream of FUBP1-regulated AS sites. Based on our results, we propose a mechanism for FUBP1 in cancer: through binding RBM15 and VIRMA, FUBP1 helps to recruit the rest of the methyltransferase complex to allow for m6A deposition on intended RNA sites (Figure 5C). Because of FUBP1 loss, there are fewer m6A modifications, thus preventing the interaction of normal m6A-binding proteins with modified sites and their downstream effects, i.e., AS of cancer driver genes (Figure 5D). In addition, we found that several members of the m6A complex, including FUBP1 binding partners, are altered in human breast cancer. Although, individually, these are also rarely mutated and represent LT genes, collectively, they represent a significant genetic alteration that is present across many breast cancers at rates comparable to major drivers. The consequences of these copy number alterations with respect to tumorigenesis will need to be explored in the future.
In sum, our findings demonstrate FUBP1 loss significantly alters a large collection of genes that contribute to cancer pathogenesis. We have identified FUBP1 to be an LT driver of breast cancer and have shown that manipulating it alone has powerful implications for the role of AS factors as the gatekeepers to the hallmarks of cancer. Our analyses suggest that genes involved in AS encompass an underappreciated mechanism of oncogenic transformation, and with deeper understanding of their targets and role in cancer, such genes might represent prominent clinical targets.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charlotte Kuperwasser (charlotte.kuperwasser@tufts.edu). Plasmids and cell lines generated in this study are available upon request via a material transfer agreement (MTA).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Lines and Tissue Culture
The cell lines used in this study (MCF10F, MCF10A and HEK293T) were purchased from ATCC, are originally derived from female tissue (Manassas, VA), and have been authenticated by shorten tandem repeat DNA profiling and validation that they are free of mycoplasma contamination. Cells were grown at 37°C with 5% CO2 and cultured in DMEM/F12 supplemented with 5% horse serum, 100 ng/ml cholera toxin, 5 μg/ml insulin, 0.5 μg/ml hydrocortisone, 50 ng/ml EGF, and 1% antibiotic/antimycotic (Corning, Corning, NY). Transfections were performed using FuGENE HD (Promega, Madison, WI). For lentivirus production, HEK293T cells were co-transfected with pCMV-VSV-G, pCMV-D8.2-Δvpr, and lentiviral expression vectors. For the generation of cell lines, MCF10F, MCF10A and HEK293T cells were incubated overnight in viral supernatants supplemented with 8 μl/ml protamine sulfate and subsequently selected for with antibiotics. Cell lines harboring multiple genetic manipulations were created by serial transductions. Clonal cell populations were created from single cells sorted into 96-well plates.
Mice and Orthotopic Tumor Growth Studies
A colony of immunocompromised NOD-SCID mice (RRID: IMSUR_JAX:001303) was maintained in-house under aseptic sterile conditions. Mice were administered autoclaved food and water ad libitum. Surgeries were performed under sterile conditions, and animals received analgesic subcutaneously before surgical procedures and antibiotics in the drinking water up to 2 weeks after all surgical procedures. Before surgery, 6-week old female NOD-SCID mice were anesthetized by isoflurane vapor. An incision was made along the right and left flanks to expose the inguinal mammary glands, and cells were injected in a total volume of 30 μL 1:1 Matrigel:phosphate-buffered saline were injected into the gland. Post-operative analgesic, antibiotic and monitoring were provided. Animals were sacrificed when tumors reached burden limit (~2 cm), and tumors were dissected and measured. In experiments in which injected cells expressed luciferin, cell dissemination was monitored weekly using an IVIS Spectrum (PerkinElmer, Waltham, MA). Image acquisition was performed 5 minutes after intraperitoneal injection of 75 mg/kg D-Luciferin (Gold Biotechnology, St. Louis, MO) with mice dorsal side up. The signals were quantified using the LivingImage software (PerkinElmer) by drawing a region of interest around each mammary gland to determine the radiance (in photons) emitted for a given time.
METHOD DETAILS
CRISPR-Cas9 Tumor Suppressor Library Screen
DNA oligonucleotides encoding sgRNA sequences designed to target the top 100 TUSON-predicted TSGs were synthesized on a custom microarray (Agilent). These oligonucleotides were PCR-amplified separately with specific sets of primers. PCR-amplified gRNA libraries were digested with BbsI and purified on a 10% TBE PAGE gel. Purified, digested fragments were cloned into BsmBI-digested pLentiCRISPRv2 (Addgene Plasmid #52961). A negative control non-cutting gRNA library comprised of 500 gRNAs targeting the E. coli genome was designed and cloned in parallel. Gel purified digestion products were cloned into a XhoI/EcoRI digested pHAGE-pInducer10-miRE-pheS(ΔEcoRI) plasmid. To create the pHAGE-pInducer10miRE-pheS(ΔEcoRI) vector, the pInducer10 mir30 shRNA construct was moved to the pHAGE backbone, with pertinent mir30 elements being replaced by miR-E elements by PCR. In addition, the sole EcoRI restriction site in the pHAGE backbone was mutated to facilitate cloning. To produce lentivirus, HEK293T cells were seeded in tissue culture dishes at 6×105 cells per 0.9 cm2 of tissue culture surface area. Plasmid DNA was diluted into serum-free medium with a lentiviral packaging plasmid mixture of SV40 VSVg, Gag/Pol, Tat, and Rev, and transfected with PolyJet (SignaGen). Cell culture media was changed 24 hours later. After 48 hours, the supernatant was harvested, filtered through a low-protein-binding HT Tuffryn® membrane with 0.45 μm pores (Pall, cat #4184), aliquoted, and stored at −80°C. Lentiviral titer was determined by transducing U2-OS cells plated at clonogenic density with serial dilutions of virus in the presence of 4 μg/mL polybrene. After selecting with puromycin, colonies were stained with methylene blue and counted manually to determine viral titer.
MCF10F cells were incubated overnight with the lentiviral libraries, at a multiplicity of infection (MOI) of 3 viral particles per cell. After 24 hours, the media was replaced with regular growth media, and the following day the library-infected cells were selected with puromycin. Surviving cells were expanded over 7 days. A fraction of cells, a “pre-screen pellet” for analyses of library representation in the cells pre-injection, was set aside and frozen, and the remaining cells were used for orthotopic xenografts, in which 1*106 cells were injected per gland. Tumors grew over a span of 10 weeks, at the end of which animals were sacrificed. Tumors larger than 5 cm3 were randomly sampled for histology, and the remainder of the tumor was digested and processed for sequencing. If the tumor was smaller than 5 cm3, it was entirely used for sequencing. Tumors were manually diced with a razor blade: for histology, sections were fixed in 10% neutral buffered formalin for 24 hours before storage in 70% ethanol prior to paraffin embedding and sectioning. Remaining parts of tumors were digested overnight at 55°C in 10mM Tris pH 8.0, 10mM EDTA, 0.5% SDS, and 0.5mg/mL Proteinase K. Subsequently, genomic DNA was isolated by two rounds of phenol: chloroform extraction using Phaselock tubes (5 PRIME), followed by two rounds of chloroform extraction. RNase A was added at a final concentration of 25 μg/mL and incubated for at least 4 hours at 37°C before two additional rounds of phenol: chloroform and one additional round of chloroform extraction. DNA was ethanol-precipitated, recovered by centrifugation, washed three times with 70% ethanol, and resuspended in 10mM Tris-Cl pH 8.5. sgRNA sequences were PCR-amplified and adapted for Illumina sequencing. This adaptation involved the addition of a P5 adaptor and a stagger sequence of variable length 5′ to the variable sgRNA, followed by a 3′ Illumina index sequence primer binding site, a 7 base pair index sequence, and a P7 adaptor. The relative representation of library sgRNA in each sample was determined by Illumina sequencing on a HiSeq2000 system.
To identify the cooperating TSGs promoting tumor growth, we recovered the barcoded sgRNAs from genomic DNA via PCR, followed by high-throughput sequencing of the PCR amplicons. An enrichment score for each gRNA was calculated by dividing the number of reads for each TSG in the screened cells by that of the pre-screen cells (transduced but not injected cells) and of cells that were transduced with non-targeting sgRNAs (control). The sgRNAs in cooperative subclones were grouped together based on read count similarity. The differences in read counts were calculated, and a cluster of two or more sgRNAs with very small differences followed by a steep drop off indicated a higher likelihood that the sgRNAs appeared in the same tumor subclone.
Cellular proliferation and soft agar assays
To measure cellular proliferation, 200 cells were plated per well in a 96-well plate, and after 2 days, cell viability was measured using CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI) every day for the following week. Colorimetric readout was measured on a SpectraMax (San Jose, CA) plate reader. For soft agar assay to detect anchorage independent growth, 1*104 cells were suspended in a 0.4% Seaplaque (Lonza, Basel, Switzerland) agarose:culture media mixture and layered onto a 0.8% agarose in DMEM. Fresh growth medium was applied every 7 days and colonies were allowed to grow for 2 weeks. Colonies were stained with 0.005% crystal violet and imaged for quantification (colonies ≥ 50 μm) with Fiji ImageJ software.
3D Hydrogel Seeding and Analyses
Protocol for seeding of hydrogels (750 cells per gel) and immunofluorescence staining was followed as previously described (Miller et al., 2017; Sokol et al., 2016). Fixed, permeabilized, and stained gels were mounted on glass slides and coverslipped, and images were acquired on a Zeiss 510 confocal microscope. Confocal images were first processed to adjust color palette, balance, and contrast using the Fiji ImageJ software applied to the entire image before figure assembly in Adobe Illustrator.
Protein isolation for immunoblotting, immunoprecipitation, and mass spectrometry
To isolate protein from whole-cell lysates for immunoblotting, cells were scraped and lysed in RIPA buffer (Cell Signaling Technology, Danvers, MA) supplemented with protease inhibitors (Roche, Boston, MA) and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). Protein samples were separated by SDS-PAGE according to standard procedures, transferred onto nitrocellulose membranes and blocked with 5% milk in Tris-buffered saline, 0.05% Tween-20. Immunoblotting was performed according to standard procedures and protein detection was visualized using enhanced chemiluminescence (Thermo Fisher Scientific). For immunoprecipitation for detecting FUBP1-interacting proteins, HEK293T cells were stably transfected with a pLenti6.2/V5-DEST expression vector containing full-length FUBP1, kindly gifted to us from the La Baer lab (Arizona State University). Control cells were transfected with an empty vector pLenti6.2/V5-DEST lentivirus (ThermoFisher). Cells were selected for blasticydin resistance and cellular lysates were pre-blocked with Protein A Magnetic Beads (Pierce, Waltham, MA) and subsequently incubated with anti-V5 antibody. Protein A Magnetic Beads were used to immunoprecipitate V5-bound proteins, and samples were eluted in NuPAGE LDS Sample Buffer (Thermofisher Scientific, Waltham, MA), followed by SDS-PAGE and standard immunoblotting procedure. For protein mass spectrometry to identify FUBP1-interacting proteins, FUBP1-V5 IP and empty vector (EV)-V5 lysates were separated by SDS-PAGE, fixed in the gel, stained with a 0.3% Coomassie Blue R250 solution, then destained overnight. Large (50–250kD), medium (30–50kD) and small (10–30kD) bands were excised, digested, and analyzed by liquid chromatography-tandem-mass spectrometry (Taplin Mass Spectrometry Facility, Harvard Medical School). The accepted list of interacting proteins was obtained by subtracting common contaminants (CRAPome), and only including proteins that uniquely appeared with ≥ 6 unique peptides in at least 2/3 experiments, compared to empty vector control.
Dot blot assays, and m6A IP-IB assays
For dot blot assays, poly(A) RNA was purified from total RNA using DynaBeads mRNA Purification Kit (ThermoFisher, Waltham, MA). Poly(A) RNA was serially diluted to 180 ng/μl, 45 ng/μl, 11.25 ng/μl. Each dilution was dotted (2.5ml) on a BrightStar-Plus positively charged nylon membrane (Invitrogen, Carlsbad, CA) in duplicate. The poly(A) RNA was crosslinked to the membrane in a Stratalinker 2400 Crosslinker twice (1,200 μJ) and the membrane was washed for 5 min in wash buffer (PBS, 0.02% Tween-20) before blocking for 1 hr (PBS, 5% Milk, 0.02% Tween-20). The membrane was incubated overnight at 4°C in polyclonal rabbit anti-m6A antibody diluted in blocking buffer. Treatment with secondary antibody was performed according to standard immunoblotting procedures and m6A detection was visualized using enhanced chemiluminescence. Levels of m6A were quantified by measuring density of dots using Fiji ImageJ. To immunoprecipitate proteins that bind m6A-modified and unmodified RNA, protocol was followed as described by Dominissini et al. (2012). Samples were separated by SDS-PAGE and immunoblotting by standard procedures followed.
RNA isolation and RNA-seq
Total RNA was extracted using the MiRNeasy Maxi Kit (QIAGEN). Sequencing libraries were prepared using the TruSeq Stranded mRNA kit and samples were sequenced by the Illumina HiSeq 2500 system. For paired end sequencing, the Rapid PE150 flow cell was used.
m6A-RIP qPCR
Poly(A) RNA was purified from total RNA using DynaBeads mRNA Purification Kit (ThermoFisher Scientific). Poly(A) RNA was fragmented using the NEBNext Magnesium RNA Fragmentation Module (NEB) according to manufacturer’s protocol. The fragmentation reaction was stopped with 50 mM Fragmentation Stop Solution (NEB) and one round of ethanol precipitation was used for RNA cleanup: fragmented RNA was combined with sodium acetate, linear acrylamide, and 100% ethanol. Fragmented poly(A) RNA was incubated with 1 μl EpiMark anti-m6A antibody (NEB) pre-bound to pre-washed Protein G magnetic beads (NEB) in reaction buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40). m6A-bound beads were washed twice in reaction buffer, then twice in low salt reaction buffer (50 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40) and then two washes in high salt reaction buffer (500 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40). Immunoprecipitated RNA was eluted in 30 μl Buffer RLT (QIAGEN), then cleaned and concentrated with Dynabeads MyOne Silane (ThermoFisher) according to manufacturer’s protocol. Bound RNA was eluted in DEPC water and used for first-strand cDNA synthesis using ProtoScript II First Strand cDNA Synthesis Kit (NEB). cDNA was also generated from total RNA, representing the input for the RIP.
Three-step qPCR amplification was performed using SYBR Green Supermix (BioRad) on a CFX96 Real-time Thermal Cycler (BioRad). Threshold cycle numbers were converted to relative gene expression values using the 2−ΔΔCt method.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Seq Analyses
Raw sequencing data were aligned using RSEM using a human hg38 library and differential expression was performed with EBSeq (Leng et al., 2013; Leng et al., 2013). STAR (Dobin et al., 2013) was used to align raw seq files with the most current reference human genome (hg38). To identify differential alternative splicing events between the control and FUBP1 null samples tested, STAR output was then used to run rMATS4.0.1 (Shen et al., 2014), which generated 5 individual output files that annotated and statistically analyzed the five different kinds of splicing events (A5SS, A3SS, SE, MXE, and RI). Only splicing events with a p value < 0.001 were considered statistically significant. Sashimi plots were generated using rmats2sashimiplot (https://github.com/Xinglab/rmats2sashimiplot).
EBSeq output was used for functional clustering and universal gene enrichment analyses of differentially expressed genes in sequenced sgNTC and sgFUBP1 RNA. The R package clusterProfiler (Yu et al., 2012, 2017) was used with the MSigDB collection of annotated gene sets (Leng et al., 2013) to detect the most significantly enriched functions among the gene list of interest, which contained the top up and downregulated (FDR ≤ 0.05, log2 fold change ≥ j1.5) genes between the sgNTC and sgFUBP1 samples. GO and KEGG annotation data for Homo sapiens was downloaded from the R package Bioconductor (Gentleman et al., 2004; Huber et al., 2015) and the groupGO function in clusterProfiler was used to functionally cluster the gene list. Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) was performed with the same gene list that was used for clusterProfiler, generated in a ranked .rnk format.
For bioinformatic analysis and identification of putative m6A peaks, sequence data were downloaded from the Gene Expression Omnibus (GEO), dataset GSE37005. Alignment data were obtained by following a previously published protocol for m6A-Seq analysis (Dominissini et al., 2013a).
Clinical Data Analysis
For detection of alteration frequency of FUBP1 and other hits from the screen across various human cancers, all 233 studies in cBioPortal (cbioportal.org) encompassing 69,310 samples were used. For analysis of alteration frequency of FUBP1 and other m6A regulators in breast cancer, the METABRIC Nature Communications study encompassing 2,509 breast invasive carcinoma samples was selected (Pereira et al., 2016) and the indicated genes were searched for detection of frequency of copy number and mRNA level alterations.
Statistical Analysis
The data are presented as mean and SEM from n cells or animals. Comparisons between two groups were performed using a t test. Analyses with three or more groups were performed with a one-way or two-way analysis of variance (ANOVA). Data were analyzed and compared between groups with GraphPad Prism (GraphPad Software, Inc) versions 7 and 8. p < 0.05 was considered statistically significant.
DATA AND CODE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE122416. Raw IP/MS data are shown in Table S2.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |||
|---|---|---|---|---|---|
| Antibodies | |||||
| Mouse monoclonal: β-actin | Abcam | Cat# ab6276; RRID: AB_2223210 | |||
| Rabbit polyclonal: m6A | Synaptic Systems | Cat# 202–003; RRID: AB_2279214 | |||
| Rabbit monoclonal: FUBP1 | Abcam | Cat# ab181111; EPR12327 | |||
| Rabbit monoclonal: PTEN | Cell Signaling | Cat# 9559; RRID: AB_390810 | |||
| Mouse monoclonal: V5 | Thermofisher | Cat# R960–25; RRID: AB_2556564 | |||
| Rabbit polyclonal: MATR3 | Bethyl Labs | Cat# A300–591A; RRID: AB_495514 | |||
| Rabbit polyclonal: RBM15 | Abcam | Cat# ab96544; RRID: AB_10680900 | |||
| Rabbit monoclonal: HuR | Cell Signaling | Cat# 12582S; D9W7E | |||
| Rabbit polyclonal: hnRNPA2B1 | Thermofisher | Cat# PA5–30061; RRID: AB_2547535 | |||
| Rabbit polyclonal: METTL3 | Fisher | Cat# 15073–1AP; RRID: AB_2142033 | |||
| Rabbit polyclonal: YTHDF2 | Abcam | Cat# ab99080; RRID: AB_10675362 | |||
| Rabbit polyclonal: CK14 | Vector Labs | Cat# 9020-P; RRID: AB_149727 | |||
| Mouse monoclonal: CK8/18 | Thermofisher | Cat# MA5 12281; RRID: AB_10986863 | |||
| Rabbit polyclonal: VIRMA | Bethyl Labs | Cat# A302–123A; RRID: AB_1720420 | |||
| Rabbit polyclonal: CASP8 | Genetex | Cat# GTX110723; RRID: AB_2036448 | |||
| Rabbit polyclonal: BRCA1 | Santa Cruz | Cat# sc-642; RRID: AB_630944 | |||
| Rabbit polyclonal: MAGI3 | Novus Biologicals | Cat# NBP2–17210 | |||
| Mouse: AF555- | Thermofisher | Cat# A21424; RRID: AB_141780 | |||
| Rabbit: AF488- | Thermofisher | Cat# R37116; RRID: AB_2556544 | |||
| Mouse: HRP- | Cell Signaling | Cat# 7076; RRID: AB_330924 | |||
| Rabbit: HRP- | Cell Signaling | Cat# 7074; RRID: AB_2099233 | |||
| Mouse monoclonal: MDM2 | Santa cruz | Cat# sc-965; RRID: AB_627920 | |||
| Rabbit monoclonal: CASP8 | Abcam | Cat#ab32125; RRID: AB_2068469 | |||
| Rabbit monoclonal: FUBP1 | Abcam | Cat#ab213525; EPR19208 | |||
| Bacterial and Virus Strains | |||||
| pLentiCRISPRv2-blast | Addgene | RRID: Addgene_83480 | |||
| pLentiCRISPRv2-puro | Addgene | RRID: Addgene_52961 | |||
| pLenti6.2/V5-DEST-FUBP1 | DNASU Repository | HsCD00329438 | |||
| pLenti6.2/V5-DEST Empty Vector | ThermoFisher | V36820 | |||
| pLenti-PGK V5-LUC Neo | Addgene | RRID: Addgene_21471 | |||
| pHAGE-pInducer10-miRE-pheS(ΔEcoRI) | Elledge Lab | RRID: Addgene_44011 | |||
| One shot Stbl3 competent E. coli | ThermoFisher | C737303 | |||
| Critical Commercial Assays | |||||
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) | Promega | G3582 | |||
| TruSeq mRNA Stranded Library Prep | Illumina | 20020594 | |||
| NEBNext Magnesium RNA Fragmentation Module | NEB | E6150S | |||
| DynaBeads mRNA Purification Kit | ThermoFisher | 61006 | |||
| DynaBeads MyOne Silane | ThermoFisher | 37002D | |||
| protoScript II First Strand cDNA Synthesis Kit | NEB | E6360S | |||
| Deposited Data | |||||
| Raw and analyzed RNaseq data | This paper | GEO: GSE122416 | |||
| FUBP1-V5 and Empty Vector-V5 Immunoprecipitation/Mass spectrometry data | This paper | Table S3 | |||
| m6A Seq data | Dominissini et al., 2012 | GEO: GSE37005 | |||
| Experimental Models: Cell Lines | |||||
| Human: MCF10F cells | ATCC | Cat# CRL-10318; RRID: CVCL_3633 | |||
| Human: MCF10A cells | ATCC | Cat#CRL-10317; RRID: CVCL_0598 | |||
| Mouse: HEK293T cells | ATCC | Cat# CRL-3216; RRID: CVCL_0063 | |||
| Experimental Models: Organisms/Strains | |||||
| Mouse: NOD.CB17-Prkdcscid/J | The Jackson Laboratory | Cat# 001303; RRID: IMSR_JAX:001303 | |||
| Oligonucleotides | |||||
| Primers for m6A RIP-qPCR: See Table S4 | This paper | Table S4 | |||
| sgRNA for FUBP1.3: GTTTGCTGCTGATGCATCGG | This paper | N/A | |||
| sgRNA for FUBP1.10: GCAGCCCCATATGCTCCCCA | This paper | N/A | |||
| sgRNA for PTEN: GCATCTGGATTATAGACCAG | This paper | N/A | |||
| Methylated RNA bait: biotin-AUGGGCCGUUCAUCUGCU AAAAGG-m6A- CUGCUUUUGGGGCUUGU | Dominissini et al., 2012 | N/A | |||
| Non-methylated RNA bait for RIP: biotin-AUGGGCCGUU CAUCUGCUAAAAGGACUGCUUUUGGGGCUUGU | Dominissini et al., 2012 | N/A | |||
| Recombinant DNA | |||||
| Plasmid: pLentiCRISPRv2-blast-FUBP1.3 | This paper | N/A | |||
| Plasmid: pLentiCRISPRv2-blast-FUBP1.10 | This paper | N/A | |||
| Plasmid: pLentiCRISPRv2-puro-PTEN | This paper | N/A | |||
| Plasmid: pLenti6.2/V5-DEST-FUBP1 | DNASU Repository | HsCD00329438 | |||
| Plasmid: pLenti-PGK V5-LUC Neo | Campeau et al., 2009 | Addgene Plasmid #21471 | |||
| Software and Algorithms | |||||
| RSEM | Li and Dewey, 2011 | https://github.com/deweylab/RSEM | |||
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ | |||
| EBSeq | Leng and Kendziorski, 2019 | http://bioconductor.org/packages/release/bioc/html/EBSeq.html | |||
| rMATS.4.0.1 | Shen et al., 2012, 2014; Park et al., 2013 | http://rnaseq-mats.sourceforge.net/ | |||
| Rmats2sashimiplot | Gohr and Irimia, 2019 | https://github.com/Xinglab/rmats2sashimiplot | |||
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR | |||
| ClusterProfiler | Yu et al., 2012 | http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | |||
| Sene Set Enrichment Analysis (GSEA) | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp | |||
| Database for Annotation, Visualization, and Integrated Discovery (DAVID) | Huang et al., 2009a; Huang et al., 2009b | https://david.ncifcrf.gov | |||
Highlights.
A combinatorial CRISPR screen identifies clinically relevant cooperating cancer genes
FUBP1 loss cooperates with PTEN loss to promote tumor growth
FUBP1−/− cells undergo widespread alternative splicing and reduced m6A methylation
Loss of one gene can alter the transcriptional landscape to promote tumorigenicity
ACKNOWLEDGMENTS
This work was supported by funding from the Raymond & Beverly Sackler Convergence Laboratory and grants from the FTC Breast Cancer Foundation (to C.K.), the Breast Cancer Research Foundation (to C.K.), and the NIH/NICHD HD073035 and NIH/NCI CA170851 grants (to C.K.). S.J.E. was funded by a grant from the Cancer Research UK Grand Challenge and the Mark Foundation For Cancer Research to the SPECIFICANCER team and a grant from the Ludwig Foundation. S.J.E. is an investigator with the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.060.
DECLARATION OF INTERESTS
C.K. is a shareholder of Naveris, Inc., and a member of its scientific board of advisors.
REFERENCES
- Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, et al. ; PCF/SU2C International Prostate Cancer Dream Team (2018). The long tail of oncogenic drivers in prostate cancer. Nat. Genet 50, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. ; MC3 Working Group; Cancer Genome Atlas Research Network (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin LR, Chung HJ, Sanford S, Kouzine F, Liu J, and Levens D (2008). Hierarchical mechanisms build the DNA-binding specificity of FUSE binding protein. Proc. Natl. Acad. Sci. USA 105, 18296–18301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, et al. (2011). Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333, 1453–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braddock DT, Louis JM, Baber JL, Levens D, and Clore GM (2002). Structure and dynamics of KH domains from FBP bound to single-stranded DNA. Nature 415, 1051–1056. [DOI] [PubMed] [Google Scholar]
- Brinkman BMN (2004). Splice variants as cancer biomarkers. Clin. Biochem 37, 584–594. [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho A, Shim JE, Kim E, Supek F, Lehner B, and Lee I (2016). MUFFINN: cancer gene discovery via network analysis of somatic mutation data. Genome Biol. 17, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climente-González H, Porta-Pardo E, Godzik A, and Eyras E (2017). The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 20, 2215–2226. [DOI] [PubMed] [Google Scholar]
- Dai D, Wang H, Zhu L, Jin H, and Wang X (2018). N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 9, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, and Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa J, Weber J, Friedrich MJ, Li Y, Rad L, Ponstingl H, Liang Q, de Quirós SB, Noorani I, Metzakopian E, et al. (2017). A single-copy Sleeping Beauty transposon mutagenesis screen identifies new PTEN-cooperating tumor suppressor genes. Nat. Genet 49, 730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng CX, and Scott F (2000). Role of the tumor suppressor gene Brca1 in genetic stability and mammary gland tumor formation. Oncogene 19, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, and Rechavi G (2013a). Transcriptome-wide mapping of N(6)-methyladeno-sine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc 8, 176–189. [DOI] [PubMed] [Google Scholar]
- Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, and Levens D (1994). A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 8, 465–480. [DOI] [PubMed] [Google Scholar]
- Fearon ER, and Vogelstein B (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, and Okamura H (2013). RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155, 793–806. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, and Lander ES (2013). Lessons from the cancer genome. Cell 153, 17–37. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohr A, and Irimia M (2019). Matt: Unix tools for alternative splicing analysis. Bioinformatics 35, 130–132. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Cichowski K, and Elledge SJ (2019). Tissue-specificity in cancer: The rule, not the exception. Science 363, 1150–1151. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hastings MH (2013). m(6)A mRNA methylation: a new circadian pacesetter. Cell 155, 740–741. [DOI] [PubMed] [Google Scholar]
- He L, Liu J, Collins I, Sanford S, O’Connell B, Benham CJ, and Levens D (2000). Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 19, 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Clark AP, Silver DP, and Livingston DM (2014). BRCA1 pathway function in basal-like breast cancer cells. Mol. Cell. Biol 34, 3828–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao HH, Nath A, Lin CY, Folta-Stogniew EJ, Rhoades E, and Braddock DT (2010). Quantitative characterization of the interactions among c-myc transcriptional regulators FUSE, FBP, and FIR. Biochemistry 49, 4620–4634. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huber LJ, Yang TW, Sarkisian CJ, Master SR, Deng CX, and Chodosh LA (2001). Impaired DNA damage response in cells expressing an exon 11-deleted murine Brca1 variant that localizes to nuclear foci. Mol. Cell. Biol 21, 4005–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I, Cao D, Na Y, Kim DY, Zhang T, Yao J, Oh H, Hu J, Zheng H, Yao Y, and Paik J (2018). Far Upstream Element-Binding Protein 1 Regulates LSD1 Alternative Splicing to Promote Terminal Differentiation of Neural Progenitors. Stem Cell Reports 10, 1208–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin N, Baekelandt V, Goritchenko L, and Benowitz LI (1997). Identification of two proteins that bind to a pyrimidine-rich sequence in the 3′-untranslated region of GAP-43 mRNA. Nucleic Acids Res. 25, 1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob AG, Singh RK, Mohammad F, Bebee TW, and Chandler DS (2014). The splicing factor FUBP1 is required for the efficient splicing of oncogene MDM2 pre-mRNA. J. Biol. Chem 289, 17350–17364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, and Kharas MG (2017). Emerging links between m6A and misregulated mRNA methylation in cancer. Genome Med. 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Singh I, Tisdale S, Abdel-Wahab O, Leslie CS, and Mayr C (2018). Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 561, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MDM, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. (2015). Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet 47, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, Haag JD, Gould MN, Stewart RM, and Kendziorski C (2013). EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, and Kendziorski C (2019). EBSeq: An R package for gene and isoform differential expression analysis of RNA-seq data. R package version 1.24.0. http://bioconductor.org/packages/release/bioc/html/EBSeq.html. [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang Z, Zhou X, Cheng Y, Xie Z, Manley JL, and Feng Y (2013). Far upstream element-binding protein 1 and RNA secondary structure both mediate second-step splicing repression. Proc. Natl. Acad. Sci. USA 110, E2687–E2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Davoli T, Leng Y, Li MZ, Xu Q, and Elledge SJ (2017). A genetic interaction analysis identifies cancer drivers that modify EGFR dependency. Genes Dev 31, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Maertens O, Cichowski K, and Elledge SJ (2018). Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes Dev 32, 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Li ML, and Shih SR (2009). Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res 37, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kouzine F, Nie Z, Chung HJ, Elisha-Feil Z, Weber A, Zhao K, and Levens D (2006). The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J 25, 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malz M, Weber A, Singer S, Riehmer V, Bissinger M, Riener MO, Longerich T, Soll C, Vogel A, Angel P, et al. (2009). Overexpression of far upstream element binding proteins: a mechanism regulating proliferation and migration in liver cancer cells. Hepatology 50, 1130–1139. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Almendro V, and Polyak K (2012). Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer 12, 323–334. [DOI] [PubMed] [Google Scholar]
- Merid SK, Goranskaya D, and Alexeyenko A (2014). Distinguishing between driver and passenger mutations in individual cancer genomes by network enrichment analysis. BMC Bioinformatics 15, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, and Jaffrey SR (2014). The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol 15, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, and Jaffrey SR (2017). Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol 33, 319–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. (1994). A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71. [DOI] [PubMed] [Google Scholar]
- Miller DH, Sokol ES, and Gupta PB (2017). 3D Primary Culture Model to Study Human Mammary Development. Methods Mol. Biol 1612, 139–147. [DOI] [PubMed] [Google Scholar]
- Miro J, Laaref AM, Rofidal V, Lagrafeuille R, Hem S, Thorel D, Méchin D, Mamchaoui K, Mouly V, Claustres M, and Tuffery-Giraud S (2015). FUBP1: a new protagonist in splicing regulation of the DMD gene. Nucleic Acids Res. 43, 2378–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry S, Rafii S, Shippen A, MacPherson G, Balasubramanian S, Reed M, and Cox A (2006). Role of CASP8 D302H and other apoptosis gene variants in breast cancer. Breast Cancer Res. 8, 11. [Google Scholar]
- Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, et al. (2018). BRCA1 mutation-specific responses to 53bp1 loss-induced homologous recombination and PARP inhibitor resistance. Cell Rep. 24, 3513–3527.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni TK, and Kuperwasser C (2016). Premature polyadenylation of MAGI3 produces a dominantly-acting oncogene in human breast cancer. eLife 5, e14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni TK, Landrette SF, Bjornson RD, Bosenberg MW, and Xu T (2013). Low-copy piggyBac transposon mutagenesis in mice identifies genes driving melanoma. Proc. Natl. Acad. Sci. USA 110, E3640–E3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni TK, Elman JS, Jin DX, Gupta PB, and Kuperwasser C (2018). Premature polyadenylation of MAGI3 is associated with diminished N6-methyladenosine in its large internal exon. Sci. Rep 8, 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Ma P, Liu Y, Li W, and Shu Y (2018). Multiple functions of m6A RNA methylation in cancer. J. Hematol. Oncol 11, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Tokheim C, Shen S, and Xing Y (2013). Identifying Differential Alternative Splicing Events from RNA Sequencing Data Using RNASeq-MATS In Deep Sequencing Data Analysis, Shomron N, ed. (Humana Press; ), pp. 171–179. [DOI] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, and Jaffrey SR (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira B, Chin SF, Rueda OM, Vollan HKM, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, et al. (2016). Erratum: The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun 7, 11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman RL (2016). Mouse models of human disease: An evolutionary perspective. Evol. Med. Public Health 2016, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu X, Storr SJ, Zhang Y, Rakha EA, Green AR, Ellis IO, and Martin SG (2017). Caspase-3 and caspase-8 expression in breast cancer: caspase-3 is associated with survival. Apoptosis 22, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Han B, Yu Y, Yao W, Bose S, Karlan BY, Giuliano AE, and Cui X (2015). Evaluation of MCF10A as a Reliable Model for Normal Human Mammary Epithelial Cells. PLoS ONE 10, e0131285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenhorst U, Beinoraviciute-Kellner R, Brezniceanu ML, Joos S, Devens F, Lichter P, Rieker RJ, Trojan J, Chung HJ, Levens DL, and Zörnig M (2009). Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology 50, 1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raponi M, Smith LD, Silipo M, Stuani C, Buratti E, and Baralle D (2014). BRCA1 exon 11 a model of long exon splicing regulation. RNA Biol. 11, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. (2018). Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 173, 499–514.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahm F, Koelsche C, Meyer J, Pusch S, Lindenberg K, Mueller W, Herold-Mende C, von Deimling A, and Hartmann C (2012). CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 123, 853–860. [DOI] [PubMed] [Google Scholar]
- Schneider G, Schmidt-Supprian M, Rad R, and Saur D (2017). Tissue-specific tumorigenesis: context matters. Nat. Rev. Cancer 17, 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Huang J, Dittmar KA, Lu Z, Zhou Q, Carstens RP, and Xing Y (2012). MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res 40, e61–e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, Smith PG, Buonamici S, Yu L, Caesar-Johnson SJ, et al. ; Cancer Genome Atlas Research Network (2018). Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 23, 282–296.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer S, Malz M, Herpel E, Warth A, Bissinger M, Keith M, Muley T, Meister M, Hoffmann H, Penzel R, et al. (2009). Coordinated expression of stathmin family members by far upstream sequence element-binding protein-1 increases motility in non-small cell lung cancer. Cancer Res. 69, 2234–2243. [DOI] [PubMed] [Google Scholar]
- Sokol ES, Miller DH, Breggia A, Spencer KC, Arendt LM, and Gupta PB (2016). Growth of human breast tissues from patient cells in 3D hydrogel scaffolds. Breast Cancer Res. 18, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupack DG (2013). Caspase-8 as a therapeutic target in cancer. Cancer Lett. 332, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H, Rust AG, Ward JM, Yew CCK, Jenkins NA, and Copeland NG (2016). Sleeping Beauty transposon mutagenesis identifies genes that cooperate with mutant Smad4 in gastric cancer development. Proc. Natl. Acad. Sci. USA 113, E2057–E2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammaro C, Raponi M, Wilson DI, and Baralle D (2012). BRCA1 exon 11 alternative splicing, multiple functions and the association with cancer. Biochem. Soc. Trans 40, 768–772. [DOI] [PubMed] [Google Scholar]
- Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, and Karchin R (2016). Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 113, 14330–14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP (2006). Unbalanced alternative splicing and its significance in cancer. BioEssays 28, 378–386. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O’Brien SW, et al. (2016). The BRCA1-D11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 76, 2778–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark UK, Reyngold M, Olshen AB, Baer R, Jasin M, and Moynahan ME (2003). BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol. Cell. Biol 23, 7926–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, Payton MN, Elliott GS, Buaas FW, Cajulis EE, Grosshans D, Ramos L, Reese DM, Slamon DJ, and Calzone FJ (1997). Differential subcellular localization, expression and biological toxicity of BRCA1 and the splice variant BRCA1-delta11b. Oncogene 14, 1–16. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. (2007). The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113. [DOI] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. (2016). Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519. [DOI] [PubMed] [Google Scholar]
- Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, and Deng CX (1999). Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 3, 389–395. [DOI] [PubMed] [Google Scholar]
- Yin M, Yan J, Wei S, and Wei Q (2010). CASP8 polymorphisms contribute to cancer susceptibility: evidence from a meta-analysis of 23 publications with 55 individual studies. Carcinogenesis 31, 850–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and Dall’Olio G (2017). Statistical analysis and visualization of functional profiles for genes and gene clusters. Bioconductor. https://bioconductor.statistik.tu-dortmund.de/packages/3.6/bioc/vignettes/clusterProfiler/inst/doc/clusterProfiler.html. [Google Scholar]
- Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. (2018). VIRMA mediates preferential m6A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Chen QM (2013). Far upstream element binding protein 1: a commander of transcription, translation and beyond. Oncogene 32, 2907–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Tian Q, and Wang Y (2013). Far upstream element-binding protein 1 (FUBP1) is overexpressed in human gastric cancer tissue compared to non-cancerous tissue. Onkologie 36, 650–655. [DOI] [PubMed] [Google Scholar]
- Zhou W, Chung YJ, Parrilla Castellar ER, Zheng Y, Chung HJ, Bandle R, Liu J, Tessarollo L, Batchelor E, Aplan PD, and Levens D (2016). Far upstream element binding protein plays a crucial role in embryonic development, hematopoiesis, and stabilizing Myc expression levels. Am. J. Pathol 186, 701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO: GSE122416. Raw IP/MS data are shown in Table S2.