

Abstract
Breast tumors display tremendous heterogeneity in part due to varying molecular alterations, divergent cells of origin, and differentiation. Understanding where and how this heterogeneity develops is likely important for effective breast cancers eradication. Insulin-like growth factor (IGF) signaling is critical for normal mammary gland development and function, and has an established role in tumor development and resistance to therapy. Here we demonstrate that constitutive activation of the IGF1 receptor (IGF1R) influences lineage differentiation during mammary tumorigenesis. Transgenic IGF1R constitutive activation promotes tumors with mixed histologies, multiple cell lineages and an expanded bi-progenitor population. In these tumors, IGF1R expands the luminal-progenitor population while influencing myoepithelial differentiation. Mammary gland transplantation with IGF1R-infected mammary epithelial cells (MECs) resulted in hyperplastic, highly differentiated outgrowths and attenuated reconstitution. Restricting IGF1R constitutive activation to luminal versus myoepithelial lineage-sorted MECs resulted in ductal reconstitutions co-expressing high IGF1R levels in the opposite lineage of origin. Using in vitro models, IGF1R constitutively activated MCF10A cells showed increased mammosphere formation and CD44+/CD24− population, which was dependent upon Snail and NFκB signaling. These results suggest that IGF1R expands luminal progenitor populations while also stimulating myoepithelial cell differentiation. This ability to influence lineage differentiation may promote heterogeneous mammary tumors, and have implications for clinical treatment.
Keywords: IGF1R, lineages, differentiation, breast cancer
Background
The mammary gland, which undergoes extensive remodeling throughout a lifetime, is a highly dynamic organ comprised of a complex hierarchy of cell lineage differentiation, from stem and progenitor cells, to the luminal-based lineage that line the duct, and the myoepithelial lineage that contracts to push milk through the ducts. Breast tumors show tremendous heterogeneity in both lineage cell type and molecular markers; there are 18 histological and at least six molecular subtypes currently recognized [1–5]. The capability of cancer cells to evolve and alter cell fate may depend upon the cell of origin, and/or the type of molecular alterations. Additionally, the cell of origin may determine the extent by which a genetic modification can re-direct cell differentiation. Importantly, a tumor’s molecular subtype(s) and potential to change cell fate may greatly affect how a breast tumor cell responds to treatment [6–8]. The complexity of cell of origin versus molecular alteration is perhaps best exemplified in the basal/triple-negative subset of breast cancers. These tumors have characteristics of both the luminal ductal and myoepithelial basal lineage [9]. BRCA1-mutated tumors, which typically have a basal-like phenotype, arise from committed luminal progenitor cells, not basal or myoepithelial lineages as originally believed [10, 11]. These findings underline the importance in defining and categorizing the cell of origin by examining both histological and molecular subtypes. Understanding cell of origin, including cell plasticity and cell fate potential, will aid in the understanding of the pathways and therapies necessary to target mixed lineage tumors.
Insulin-like growth factor 1 (IGF1) and IGF1 receptor (IGF1R) are critical for mammary gland development and function [12–15]. Mammary gland development and homeostasis relies on stem cells for the production and maintenance of the myoepithelial and luminal lineages [8]. The terminal end bud (TEB), where lineage differentiation and ductal morphogenesis occurs [12, 16], is promoted by IGF1 signaling. The IGF pathway is also important in pregnancy and lactation where luminal differentiation is vital [17, 18]. IGF1R regulates expression of genes which regulate cell cycle, survival, motility, attachment and as such is important for tumorigenesis [19, 20]. The activated form of IGF1R is expressed in up to 50% of breast tumors[21] where it plays key roles in cancer promotion [20, 22–26], resistance [27–31], and recurrence [8]. Although IGF1R is overexpressed and/or amplified across all breast cancer subtypes, it has specific expression and function in each [8]. Additionally, IGF1R expression is an important prognostic factor in some subtypes but not others [8, 32–34]. Together, these studies suggest that IGF1R may play differential roles in tumorigenesis depending on the cellular and molecular context.
Our laboratory previously demonstrated that constitutive activation of IGF1R [25], or the downstream signaling intermediates IRS1 or IRS2 [35], results in mammary tumorigenesis. The purpose of this study was to evaluate the ability of IGF1R to affect mammary cell lineages during development and tumorigenesis. We demonstrate that IGF1R-induced mammary tumors show multiple histological subtypes, cell lineages, and contain an increase in a putative bi-progenitor population. Herein, our data suggests IGF1R promotes self-renewal and formation of differentiated progeny, expanding the luminal progenitor population while also stimulating myoepithelial cell differentiation.
Methods
Cell culture
MCF10A-Ctrl, MCF10A-IGF1R, and MCF10A-IGF1R-SnailDN cells [20] were cultured per ATCC guidelines. For mammosphere assays, MCF10A cells were trypsinized and re-plated at 20,000 cells per 2mls in 6-well, ultra-low attachment plates (Fisher-Scientific) in 2 mls of serum-free DMEM/F12 media (Hyclone), supplemented with 20ng/ml bFGF (BD Biosciences), 20ng/ml EGF (BD Biosciences), 4 μg/ml heparin (Sigma), penicillin-streptomycin (Hyclone), and B27 (Gibco). For inhibitor sphere assays, 10 μM of IKK II inhibitor (Calbiochem) or DMSO control was added to the media. 1 ml of media was added every 2–3 days. On day 10, all cells/spheres were collected, separated using 0.05% trypsin-EDTA (Invitrogen), and single cells plated as above at 2,000 cells per well to perform secondary mammosphere assays. Mammospheres were counted on day 8 of the second passage. Tertiary sphere formation followed the same process as secondary spheres. Primary mouse cells were plated at a density of 100,000 cells/2mls for primary sphere formation and 2,000 cells/2mls for secondary and tertiary sphere formation.
For differentiation assays, MMTV-CD8-IGF1R transgenic mouse tumors and wild-type mammary glands from 10wk FVB/N mice (Jackson Laboratory) were minced, digested with Collagenase Type 3 (285 units/mg) (Worthington) for 1.5 hours, differentially centrifuged four times at 0.3g for five seconds to remove the majority of lineage positive single cells, trypsinized into single cells for 12 minutes, and strained through a 70 micron filter to dissociate into single cells and plated into primary and then secondary and tertiary tumorspheres. Differentiation assays were performed following Bachelard-Cascales et al. [36]. The day before sphere dissociation, 40,000 irradiated NIH 3T3-fibroblasts (30gamma) were plated on coverslips in 12 well plates with 10%FBS DMEM. Secondary and tertiary spheres were dissociated and the media was removed from the irradiated fibroblasts followed by a 1xPBS wash. The dissociated spheres were plated onto the irradiated fibroblasts as 3,000 or 6,000 single cells per well in 1ml EpiCult-B media (Stemcell Technologies) with 5% FBS and grown for 10 days with 50% media changes every 2–3 days. Of note, EpiCult-B media contains insulin, but no additional IGF ligands above those present in FBS. Differentiated colonies were fixed with 100% cold methanol, stained with 0.05% crystal violet and counted, distinguishing between luminal, myoepithelial and mixed phenotypes by morphological differences.
qPCR
Total RNA from MCF10A cells was isolated using an Illustra RNAspin Mini RNA Isolation kit (GE). Total RNA was isolated from frozen mouse tissues by homogenizing 200ug of crushed sample in 350ul lysis buffer and following the Illustra RNAspin Mini RNA Isolation Kit (GE). RNA was reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). qPCR was performed using primers shown in Supplementary Table 1. β-actin was used to calculate relative expression.
Flow Cytometry
To analyze CD44/CD24, 5×105 MCF10A-Ctrl and MCF10A-IGF1R cells were plated in 10cm plates and grown for 48 hours. For inhibitor sphere assays, 0, 1, 5, 10, or 20 uM of IKK II inhibitor was added with DMSO as a control at the time of plating. Cells were trypsinized, washed with 0.5%BSA-PBS, stained with anti-CD44-PE-Cy7 (cat#560569) and anti-CD24-PerCP (#561647) (BD Biosciences) 1:50 for 30 minutes, washed with PBS, then resuspended in 400 μl of 0.5% BSA-PBS. Fluorescence was detected with the BD LSR2 Flow Cytometer (BD Biosciences). To analyze mammary lineages, normal mammary glands or tumors were harvested from FVB/N wildtype mice (Jackson Labs) or MMTV-CD8-IGF1R transgenic FVB/N mice. Allografts of p53-null tumors [37, 38] grown in BalbC mice or MMTV-Her2/Neu tumors [39] in FVB/N mice were used for lineage controls. Tissues were minced, digested with Collagenase Type 3 (285 units/mg) (Worthington) for 1.5 hours, differentially centrifuged four times at 0.3g for five seconds to remove the majority of lineage positive single cells, trypsinized into single cells for 12 minutes, and strained through a 70 micron filter. Single cells were then labeled with the Biotin Mouse Lineage Panel (BD Pharmigen) as well as anti-mouse CD140a-Biotin, washed with 2% FBS-HBSS, labeled with MACS anti-Biotin Microbeads (Milteny Biotec), and then lineage positive cells were depleted from the cell solution through an LD Column using the QuadroMACS separator (Miltenyi Biotec) following manufacturer’s protocol. Flow-through was collected and centrifuged. The cell pellet was incubated for 30 min with anti-CD24-APC (cat#562349), anti-CD49f-PE (cat#562474), anti-CD29-FITC (cat#561796), anti-CD61-BV421 (cat#562917) (BD Biosciences), and Sytox Blue Dead Cell stain (Invitrogen, cat#S34857). Cells were then analyzed on a BD FACSAria II (BD Biosciences) for mammary lineage populations.
Lentiviral Production
Lentivirus was produced as described previously [40]. Briefly, twenty 10cm plates of 293T cells were PEI-transfected with pHIV-ZSGreen empty vector (Addgene, #18121) or pHIV-ZSGreen-CD8-IGF1R. Supernatant was collected at 48, 64, and 72 hours, virus was pelleted, resuspended in media, combined, aliquoted, and frozen. Virus was titered by serial dilution and then FACS analysis.
Transgenic Mouse Models
All procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the IACUC at the University of Pittsburgh. MMTV-CD8-IGF1R mice have been previously reported [25]. MMTV-ErbB2 [39] and p53-null claudin-low [37, 38] mammary tumor allografts, passage 1 to 3, were grown in the mammary glands of FVB/N or BalbC mice, respectively. Mice were maintained on a 12-h light, 12-h dark schedule with ad libitum access to laboratory chow and water.
Histology
Five micron serial tumor sections were deparaffinized, gradually hydrated, and stained for hematoxylin and eosin (H&E) and then examined microscopically. 20 MMTV-CD8-IGF1R tumors and 22 MMTV-ErbB2 control tumors were examined by H&E. Tumors were stained by immunohistochemistry with Troma1-Keratin 8 (1:200; Developmental Studies Hybridoma Bank), Keratin 14 (1:200; Covance), Keratin 6 (1:200, Covance). Trichome staining was performed using the Masson Trichome Stain kit (Sigma, HT15–1KT) according to manufacturer’s instructions.
Immunofluorescence
Mouse tissue samples were fixed with 4% PFA overnight and paraffin embedded. Paraffin embedded sections were processed with xylene, ethanol dehydration series, rinsed in PBS then dH20, incubated 20 min at 100°C in 1x in citrate buffer pH 6.0 (citrate buffer: 321mM sodium citrate, 79.84mM citric acid monohydrate), cooled, and incubated in 1xPBS-100mM Glycine 3×10 min. Sections were circled with a PAP pen (Dako), rinsed in dH20, incubated in primary block (IF buffer:1xPBS, 7.7mM NaN3, 0.1%BSA, 0.2% TritonX-100, 0.05% Tween20 containing) [41] and 10% goat serum for 1 hour, then incubated in combination of mouse, rabbit, and chicken primary antibodies in IF buffer,10% goat serum for 1 hour. Slides were washed 3×10min in IF buffer, incubated in Alexa Fluor secondary antibody (1:500, ThermoFisher), IF buffer,10% goat serum, for 45 min. Slides were mounted with Prolong Diamond Antifade Mountant with Dapi (ThermoFisher). Primary antibodies included Troma1-Keratin 8 (1:200; Developmental Studies Hybridoma Bank, cat#TROMA-I-c), Keratin 14 (1:400; Covance, cat#906001), Keratin 5 (1:400, Covance, cat#PRB-160P-100) and IGF1R (1:200; Ventana, cat#7904346). 488, 546, and 647 goat secondary Alexa Fluor antibodies were used against mouse, rat, rabbit, and chicken.
Reconstitution Mouse Model
Mammary glands from 5 to 15 (depending on the experiment) wild type 8–10 week old FVB/N mice (Jackson Laboratory) were harvested, minced, digested, trypsinized, and lineage depleted as detailed in the flow cytometry methods section. For total population reconstitution, lineage positive depleted cells were infected for 2 hours with an MOI of 20 of either pHIV-ZSG empty vector control (Addgene, #18121) or pHIV-ZSG CD8-IGF1R containing virus under centrifugation in ultra low attachment plates and implanted, same-day, into cleared fat pads of 23 day old recipient mice as previously described [40]. For reconstitution of sorted populations, cells were sorted for mammary lineages as described in flow cytometry methods and then infected overnight at 37° in ultra-low attachment plates. The next morning, cells were collected. Fat pads were cleared from 23-day old FVB/N mice (Jackson Laboratory) [40]. Using a Hamilton 25ul syringe, 10ul of media containing 4,000 to 15,000 cells, depending on the experiment, was injected into the cleared fat pad. Mice were left for 8 weeks to allow the gland to reconstitute. Glands were harvested, analyzed for green fluorescence and either carmine stained for analysis of reconstitution and/or prepared into FFPE blocks.
Results
Expression of constitutively active IGF1R causes mammary tumors with multiple cell lineages and mixed histologies.
We previously reported generation of transgenic mice expressing constitutively active IGF1R (CD8-IGF1R) in the mammary gland using a standard MMTV-LTR. Expression of constitutive IGF1R, confirmed by immunoblotting, resulted in abnormal mammary gland development as early as 4 weeks of age, and mammary tumors developed at 8 weeks of age in virgin mice [25]. This data is consistent with MMTV-LTR expression in early mammary gland development as seen by others and consistent with the known role of the IGF system in development of the embryonic bud and TEB. To examine their histology and cell composition, we compared MMTV-CD8-IGF1R mammary tumors to well-defined luminal-like MMTV-ErbB2 mammary tumors. MMTV-ErbB2 tumors were predominately stroma-poor, solid adenocarcinomas which lack gland formation, myoepithelial cells, keratinization or squamous metaplasia (Figure 1A). In contrast, expression of CD8-IGF1R resulted in tumors with various histological phenotypes. Tumor phenotypes included solid adenocarcinomas similar to that observed for MMTV-ErbB2 tumors as well as tumors exhibiting squamous metaplasia and associated keratin swirls. Other tumors were well-differentiated adenocarcinomas with clearly defined ductal structures and occasional lactational change, while some were less well-differentiated and showed only rudimentary ductal structures or not at all. Finally, IGF1R-expressing tumors frequently displayed abundant dense stroma with lymphocytic infiltrates (Figure 1B). Previous studies have shown that the type of transgene overexpressed can predict tumor phenotype [42]. Consistent with this, tumors that express constitutively active IGF1R were similar to tumors we previously reported from IRS1 and 2 overexpression, [43, 44] which often show multiple differentiated cell lineages.
Figure 1: Mammary glands from MMTV-CD8-IGF1R transgenic mice exhibit mixed histologies and multi-lineage tumors.
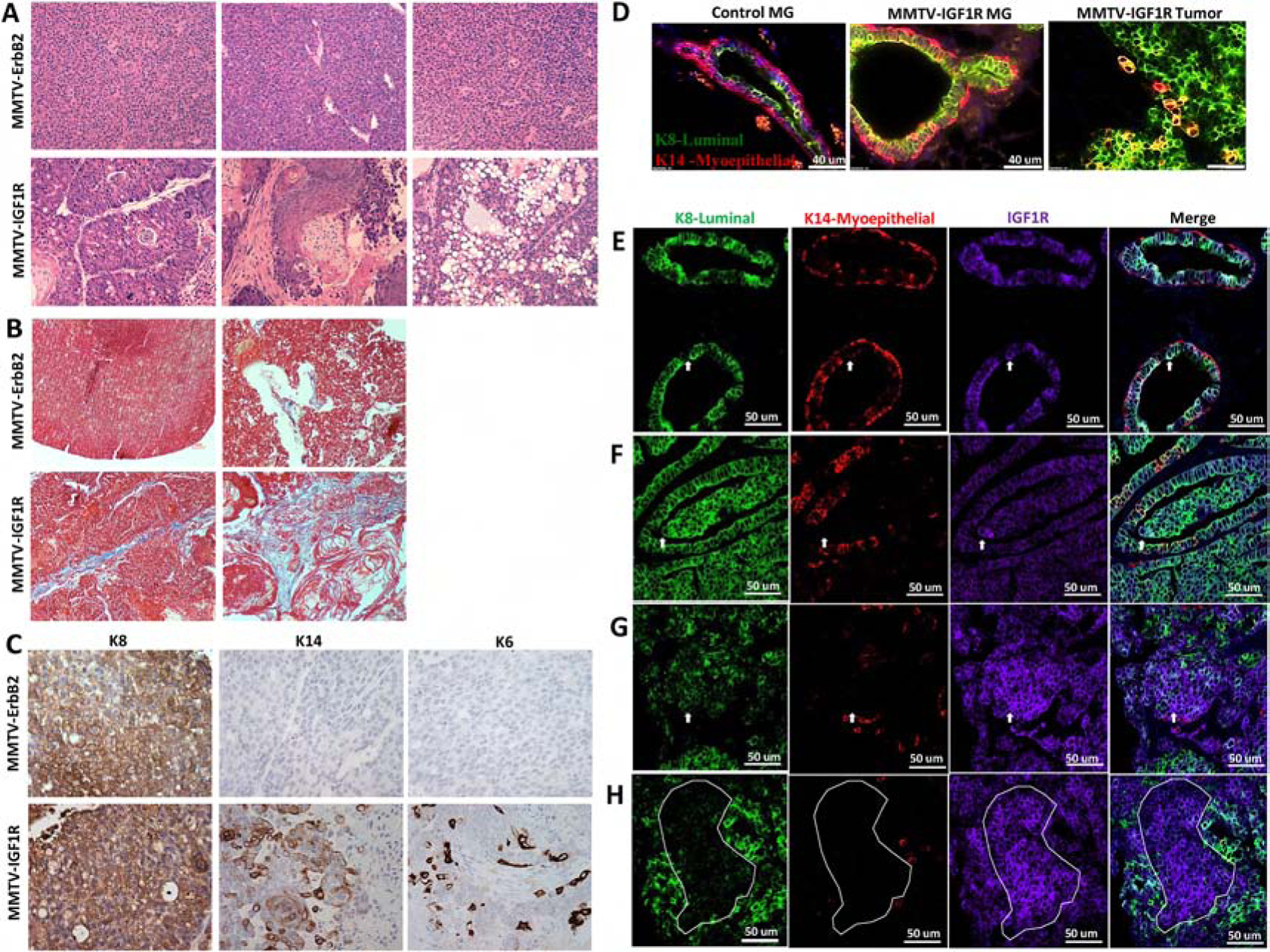
A) H&E and Masson’s Trichrome staining of MMTV-ErbB2 tumors and MMTV-CD8-IGF1R mammary tumors. 20x magnification. B) Masson’s Trichrome staining of MMTV-ErbB2 tumors and MMTV-CD8-IGF1R mammary tumors. C) IHC of luminal keratin 8 (K8), myoepithelial keratin 14 (K14), and progenitor keratin 6 (K6) markers in MMTV-ErbB2 and MMTV-CD8-IGF1R mammary tumors. Representative images taken at 20x magnification. D) IF of wild type control mammary gland (MG), MMTV-CD8-IGF1R pre-neoplastic mammary gland, and MMTV-CD8-IGF1R mammary tumor co-stained with K8 (green) and K14 (red) (n=5). E) IF of MMTV-CD8-IGF1R mammary gland (n=4) and F-H) IF of MMTV-CD8-IGF1FR tumors (n=5) co-stained with K8 (green), K14 (red), and IGF1R (purple). In (E-F) white arrow indicate example co-stained K8 and IGF1R cell, in (G) white arrow indicates example cell highly expressing IGF1R but with weak K8 expression, and in (H) white dots outline differential staining areas.
To further characterize the spectrum of tumors observed in MMTV-CD8-IGF1R transgenic mice, immunohistochemistry was performed for lineage-specific markers. As expected from previous reports, MMTV-ErbB2 mammary tumors stained positive for the luminal marker cytokeratin 8 (K8) and were negative for both the myoepithelial marker cytokeratin 14 (K14) and the putative progenitor cell marker cytokeratin 6 (K6) (Figure 1C). This is consistent with reports that ErbB2/Neu initiates and promotes cancer in either fully differentiated luminal cells or that ErbB2/Neu drives progenitor cells towards a differentiated luminal lineage [45]. In contrast, MMTV-CD8-IGF1R tumors stained positive for all three lineage markers, luminal epithelial (K8), myoepithelial (K14), and putative progenitor (K6) (Figure 1C). These lineage results are consistent with IGF1R driving highly differentiated tumor phenotypes.
To more comprehensively characterize the lineages, we used immunofluorescence to co-stain markers in pre-neoplastic mammary glands and tumors, comparing MMTV-CD8-IGF1R mice to wildtype FVB/N. CD8-IGF1R mammary glands showed a similar composition as wild type glands of littermates: The K8 luminal marker present in the inner layer of the duct, and the K14 myoepithelial marker present along the basal edge (Figure 1D). Interestingly, in CD8-IGF1R mammary glands we observed K14 positive cells among the luminal layer, including a few more rounded cells which resided on the inside of the luminal layer (Figure 1D). In MMTV-CD8-IGF1R tumors, we observed large areas of cells staining positive for both K8 and K14. This large dual positive population of cells is not common in other mammary gland tumor models and may suggest IGF1R is driving the expansion of a bi-progenitor cell or that IGF1R is promoting differentiated MMTV-IGF1R expressing cells to gain both luminal and myoepithelial properties.
To determine what cell types overexpress IGF1R, we co-stained mammary glands and tumors with K8, K14, and IGF1R. In wild-type glands, IGF1R is lowly expressed across both luminal and myoepithelial cell types. In MMTV-CD8-IGF1R glands and tumors, we observed IGF1R expression in K8-positive luminal cells (Figure 1E–F), especially in more ‘normal-like’ areas. Interestingly, we also observed more cell-dense regions where IGF1R expression is higher while K8 is lower (Figure 1F–H). It is unclear if these regions are two distinct populations that have grown around each other or if the IGF1R expressing cells have lost K8 marker expression. Together, these results suggest that IGF1R is affecting myoepithelial and luminal cell behavior with possible effects on cellular differentiation.
IGF1R expands a luminal-progenitor population in vivo, but enhances myoepithelial differentiation in vitro.
To determine if IGF1R constitutive activation alters mammary lineage composition, we analyzed mammary cell populations of MMTV-CD8-IGF1R transgenic mice [25]. We analyzed cell lineages by mRNA expression and by FACS. Gene expression microarrays previously identified gene signatures of mammary epithelial lineages [46]. mRNA from wild-type glands and IGF1R transgenic tumors, as well as ErbB2 luminal and p53-null claudin low basal tumor allografts [37, 38], were analyzed for luminal progenitor or basal cell associated genes (Figure 2A). MMTV-CD8-IGF1R expressing tumors showed elevated expression of luminal progenitor-associated genes. Interestingly, IGF1R tumors also demonstrated elevated basal-associated transcription factors as compared to wildtype gland, but this increase was less dramatic than that observed in p53-null basal-like tumors.
Figure 2: MMTV-CD8-IGF1R expression expands the luminal-progenitor population and drives myoepithelial differentiation.
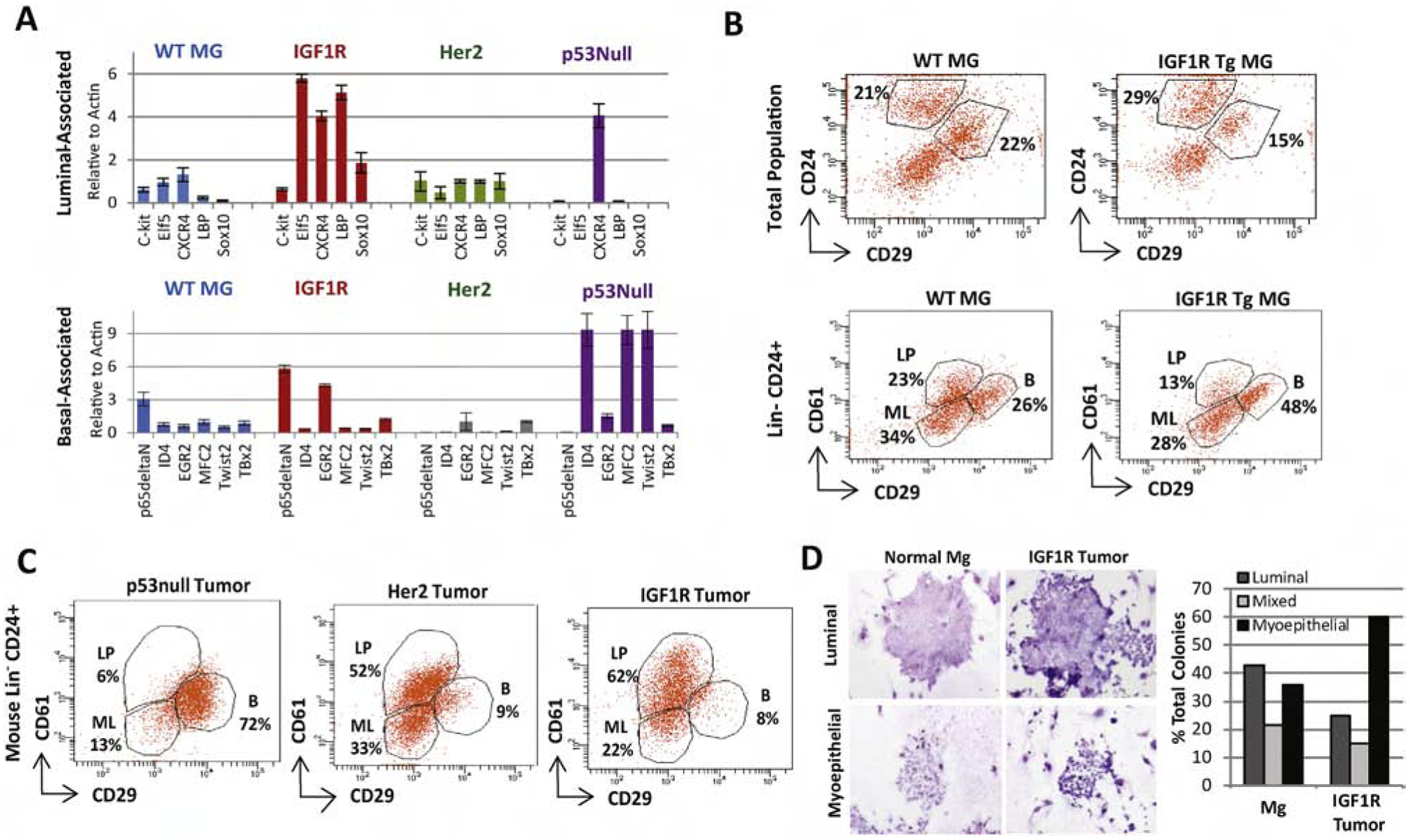
A) qRT-PCR was performed on MMTV-CD8-IGF1R, MMTV-Her2-Neu, and p53null frozen tumors or control WT mammary glands using a lineage-associated gene panel. n=2 biological replicates. B) Control and pre-neoplastic mammary glands or C) tumors were dissociated, and FACS analyzed for CD24, CD49f, CD29, and CD61 markers to separate the luminal progenitor (LP)(CD61 high,CD29 low), mature luminal (ML)(CD61 low, CD29 low), and basal/myoepithelial populations(CD29+). D) Tumorspheres from MMTV-CD8-IGF1R tumors were plated into differentiation assays as outlined in materials and methods. Biological replicates n=3, mean+/− s.e.m. Representative of 2 experiments. Images taken at 11.5x.
FACS analysis of lineage-associated cell surface markers [47] showed an increase in the basal population in IGF1R pre-neoplastic mammary glands (48%) compared to wild-type controls (26%) (Figure 2B). However, IGF1R transgenic tumors were composed primarily of luminal lineages (84% luminal progenitor and mature luminal), compared to p53-null basal tumors (19% luminal progenitor and mature luminal) (Figure 2C). IGF1R tumors closely mirrored ErbB2 tumors, 85% combined luminal markers; however, IGF1R tumors contained a larger luminal progenitor population (CD61 positive), 62% as compared to 52%, with higher CD61 expression not present in the ErbB2 luminal tumors.
Interestingly, when cells from IGF1R transgenic tumors were tested for differentiation in vitro, cells formed approximately 2.5 fold more myoepithelial-like colonies (Figure 2D), suggesting that, although IGF1R tumors have luminal progenitor markers, these cells preferentially differentiate in a myoepithelial direction. All together, these in vivo lineage analyses suggest that IGF1R alters lineage differentiation in the mammary gland, promoting formation of the luminal progenitor population and maintaining the myoepithelial population.
Constitutive activation of IGF1R in mammary epithelial cells causes attenuated mammary gland reconstitution associated with hyperplastic, highly differentiated outgrowths.
Our studies of MMTV-CD8-IGF1R mice include mammary gland IGF1R constitutive activation from fetal development onwards. To analyze how IGF1R affects lineage outgrowth and the differentiation potential of adult mammary cells, we performed mammary gland reconstitution assays using mouse mammary epithelial cells (MECs). MECs were harvested from wild-type FVB/N mice, infected for 2 hours with either constitutively active IGF1R or empty vector control, and injected (same day as harvesting) into cleared mammary fat pads of 23 day old FVB/N mice. Mammary glands reconstituted with MECs expressing constitutively active IGF1R and harvested between 4 and 8 weeks demonstrated a hyperplastic phenotype and greatly attenuated reconstitution as compared to the vector-infected controls (Figure 3A). Vector-infected control mouse MECs exhibited normal ductal outgrowth (Figure 3B). The elongation of IGF1R-infected ducts was greatly stunted (Figure 3B) and the cellularity was greatly increased with cells assembling lobule-like formations (Figure 3C). This phenotype is very similar to the hyperplastic glands we previously reported in IGF1R transgenic mice [25]; however, unlike the transgenic mice, IGF1R expression is observed only regionally throughout the reconstituted gland (Supplemental Figure 1). All together, these results suggest IGF1R constitutive activation promotes mammary gland differentiation, reducing ductal outgrowth.
Figure 3: IGF1R-infected primary mouse MECs demonstrate hyperplastic, highly differentiated outgrowths resulting in attenuated reconstitution.
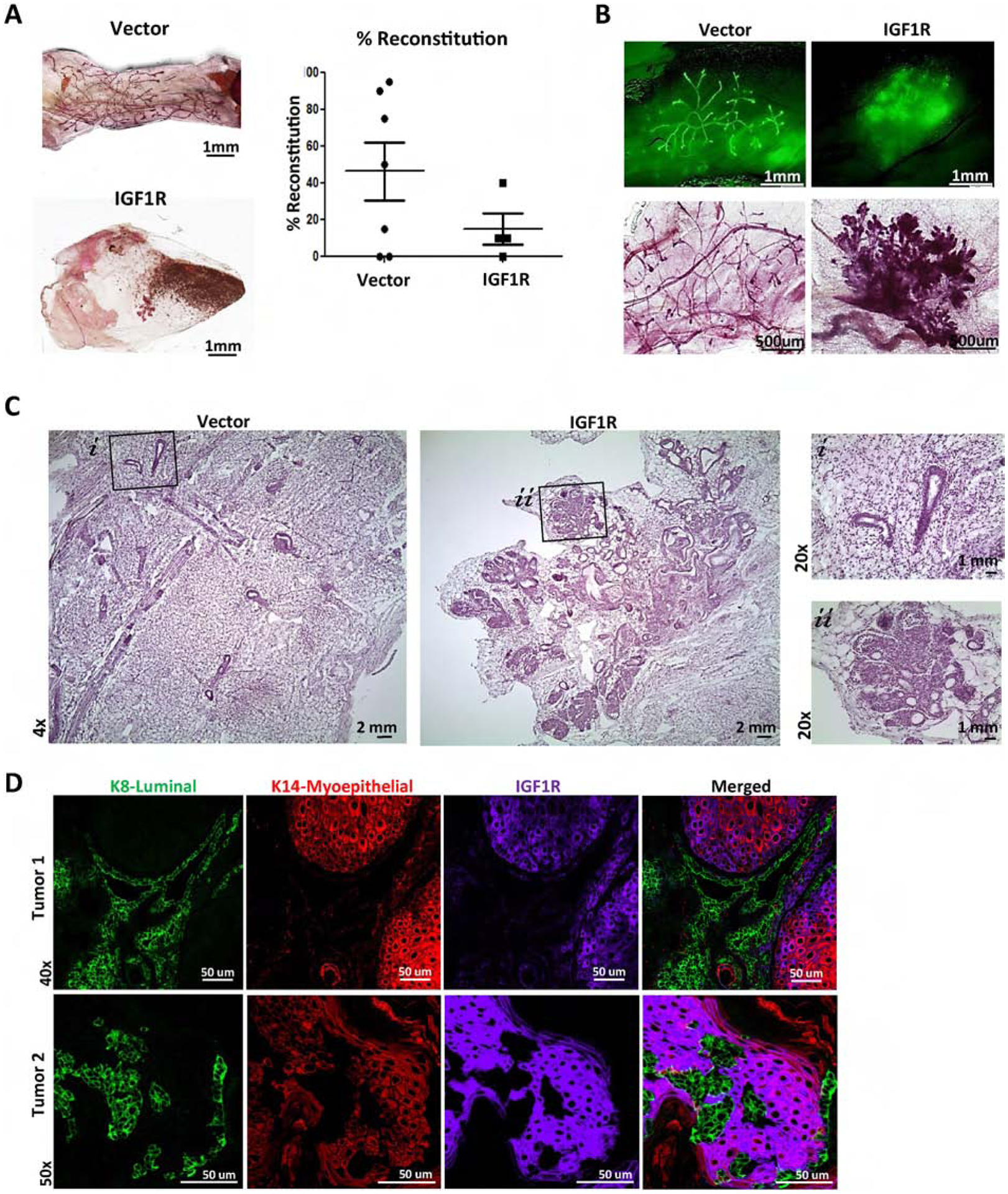
A) Reconstituted glands of wild-type MECs infected with empty vector control (n=7) or CD8-IGF1R lentivirus (n=6). B) Demonstrating ZSGreen and carmine stained mammary outgrowths; 4x and 10x, respectively. C) H&E staining; 4x with 20x insets. D) IF of 15,000 cell reconstituted tumors (n=2) co-stained with K8 (green), K14 (red), and IGF1R (purple).
Mammary gland reconstitutions of transplanted MECs resulted in tumor outgrowth as early as 8 weeks post-transplantation. Two tumors were collected for analysis. Tumor 1 showed highly organized lineage-specific cellular patterns with K8 positive cells forming duct-like formations among K14 positive clusters, with tumor 2 closely resembling the unstructured cell mass observed in the transgenic mice shown in Figure 1 (Figure 3D). Interestingly, in both instances, IGF1R co-expressed with the K14 myoepithelial lineage marker while expression was very low in, or completely excluded from, the K8 luminal lineage. The exclusivity of IGF1R and the K8 luminal marker across these tumors recapitulates the restricted regions observed in the transgenic mice above in Figure 1G. These results suggest that IGF1R influences lineage differentiation, with outgrowth features varying across reconstitutions and tumor models.
Lineage-restricted IGF1R alters both ductal outgrowth and lineage-marker co-expression depending upon the cell of origin.
As observed above, IGF1R tumors derived from both transgenic mice and reconstituted mammary glands have various cell lineages, across multiple tumors as well as within the same tumor. To determine whether these diverse cell lineages derive from IGF1R’s ability to promote lineage differentiation, we sorted mouse MECs into luminal and myoepithelial lineages as described previously [47] (Supplemental Figure 2), expressed CD8-IGF1R, and analyzed mammary gland reconstitution. Similar to previous observations, control-infected control-sorted total populations contained normal gland structures and low levels of homogenously-expressed IGF1R (Figure 4A and Supplemental Figure 3A). Reconstitution of IGF1R-infected control-sorted total populations also mirrored previous observations, resulting in areas of normal-like ducts as well as cellular-dense tumor-like areas (Figure 4A lower panel and Supplemental Figure 3B, 3C). These results suggest the control-sorted total populations, with and without IGF1R, act similarly to previous models outlined above.
Figure 4: Lineage-restricted IGF1R-infected primary mouse MECs demonstrate differential ductal outgrowths dependent on lineage cell of origin.
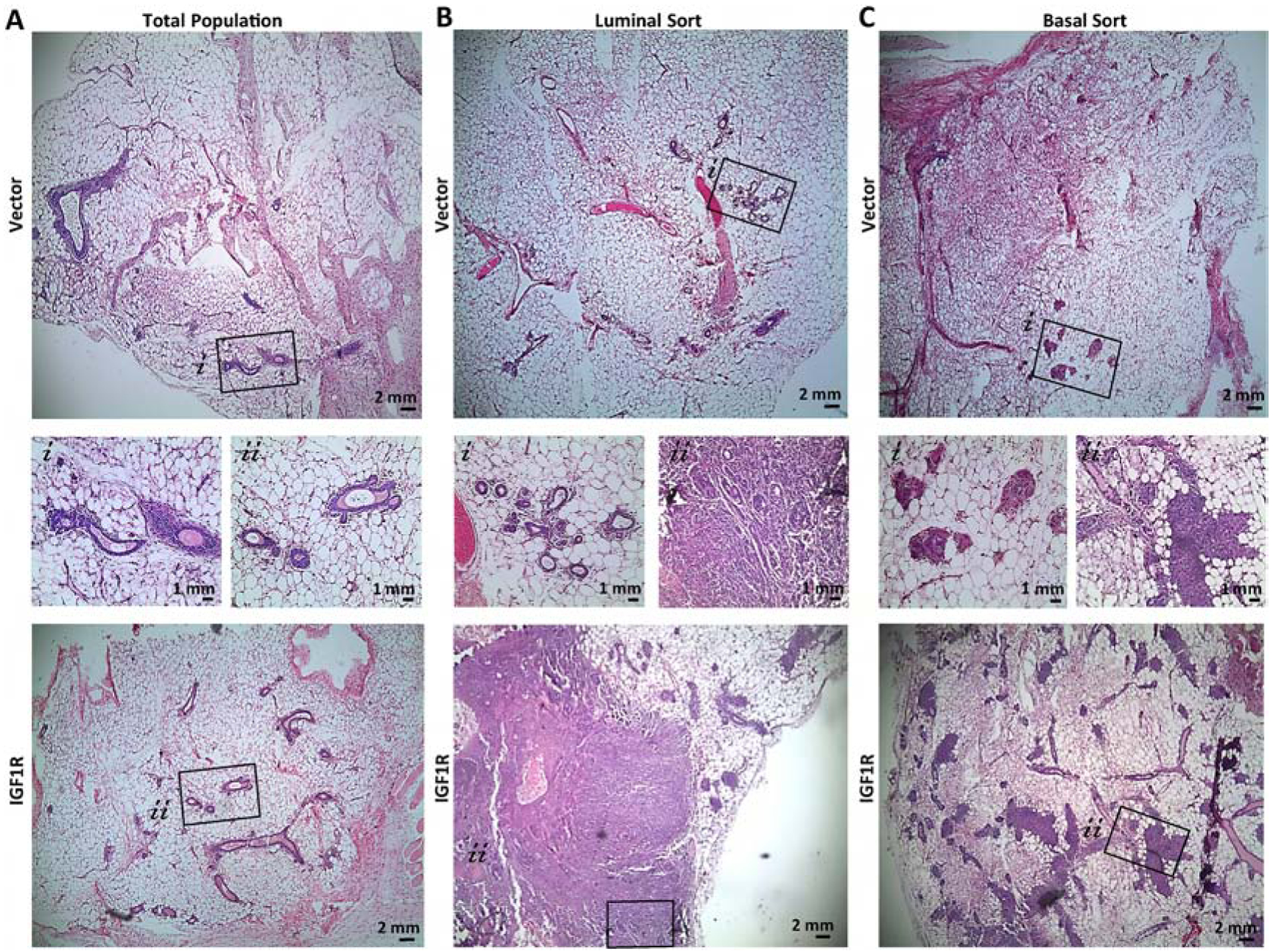
Representative H&E of reconstituted glands from A) Total population, B) luminal, or C) basal sorted cells. Average of 5 mice implanted per group. MEC populations infected with CD8-IGF1R or empty vector control.
Control-infected, luminal-sorted populations had normal outgrowths (Figure 4B) while the basal-sorted population had limited outgrowths that did not consist of the normal hollow ductal structure (Figure 4C). Interestingly, luminal-sorted populations infected with IGF1R generated a normal-like gland which led to a cell-dense tumor area, palpable at removal (Figure 4B bottom panel). In the normal-like area, K8 and K14 expression recapitulates normal staining with IGF1R co-expressing in the K14 myoepithelial layer (Figure 5A). Interestingly, some luminal layer cells of these normal-like ducts expressed K8 at very high levels and others at lower more typical levels. While these areas of high and low K8 expression can be observed in normal tissues, it is not observed often nor nearly to this same extent as it is in the IGF1R-luminal sorted reconstituted mammary glands, suggesting that IGF1R signaling in nearby cells may be affecting the transitional K8 marker and perhaps affecting the luminal state. The K14 and IGF1R co-expression found in normal areas was observed much more dramatically in the solid tumor area of the luminal reconstitution where K14 and IGF1R were much more strongly expressed. In these regions, smaller pockets of K8-only expressing cells were intermittently present (Figure 5B). These observations strongly mirrored the co-expression and K8 exclusion of the reconstituted tumors discussed in Figure 1G. We observed that cells expressing lower levels of IGF1R were present in the K8-specific regions (Figure 5B bottom panel). These low-expressing IGF1R cells within the K8 positive regions had much lower K8 expression than surrounding cells (Supplemental Figure 4). Similar phenotypes were sparingly observed in the IGF1R transgenic tumor. The IGF1R-expressing cells could be infiltrating and overgrowing these K8 positive areas, or, more likely, gaining IGF1R expression through paracrine signaling of highly expressive IGF1R cells. As staining was performed on serial sections, it is unclear if these IGF1R-low K8-low regions co-express K14, but observation of few K14-positive cells amongst the K8 pocket suggest this is the case. Overall, these results suggest luminal-sorted IGF1R-infected reconstituted MECs promote tumor formation with IGF1R overexpression in the myoepithelial layer and exclusion of K8 positive cells regions.
Figure 5: Lineage-restricted co-expression of IGF1R is dependent on the lineage cell of origin.
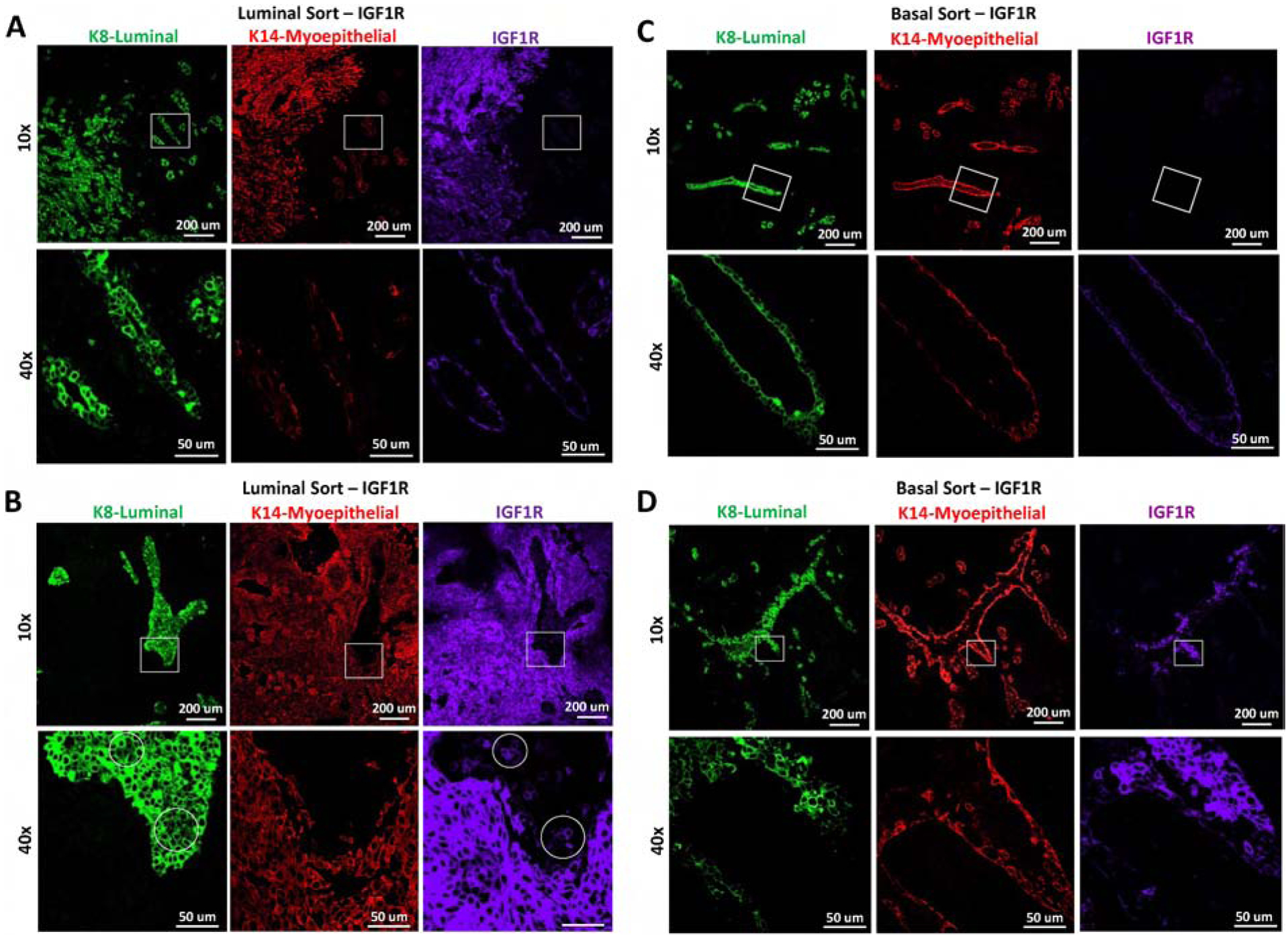
Representative IF of reconstituted mammary glands from A-B) luminal or C-D) basal MEC populations infected with CD8-IGF1R or empty vector control and stained with K8 (green), K14 (red), IGF1R (purple). A and C demonstrate areas which are normal appearing, B and D demonstrate abnormal regions. Circles represent differential staining area. The IF represents co-staining of K8 and IGF1R with a serial section staining of K14. All samples were stained and imaged together and thus expression levels are comparable across all panels.
The IGF1R-infected basal MEC reconstituted glands consisted of normal-like ductal areas mixed with dense lumen-filled cellular areas (Figure 4C lower panel and Figure 5C–D). In these dense cell-filled ducts, IGF1R is overexpressed and high IGF1R co-expresses with the K8 luminal lineage marker while excluding K14 to the outer myoepithelial layer (Figure 5D). While this co-expression of IGF1R and K8 was commonly observed in the transgenic tumors, IGF1R co-expressed mostly with K14 in the total and luminal-sorted MEC reconstituted glands. Only in the basal-sorted reconstitution did IGF1R expression overlap with K8, suggesting that the transgenic tumors may be originating from basal cells which have the capacity to gain luminal characteristics such as K8 expression.
IGF1R expands tumor initiating phenotypes.
We have previously shown that IGF1R promotes epithelial to mesenchymal transition (EMT) via upregulation of the NFκB pathway and Snail [20]. Accumulation of mesenchymal traits is linked to the acquisition of stem-like properties [48]. Additionally, IGF1R expression is linked to stem cell and tumor initiating phenotypes in numerous tissues [49–52]. To extend our in vivo work, we examined the effect of IGF1R on stem-like features in the MCF10A immortalized but non-transformed mammary epithelial cell line. We examined CD44/CD24 populations by FACS and mammosphere formation in serum-free low attachment conditions, both assays having previously been used to assess stem-like characteristics [53–55]. Expression of CD8-IGF1R increased the population of CD44+/CD24− cells (Figure 6A). Additionally, CD8-IGF1R increased primary and secondary mammosphere formation (Figure 6B).
Figure 6: IGF1R constitutive activation expands tumor initiation/stem cell-like characteristics through NFκB signaling and Snail.
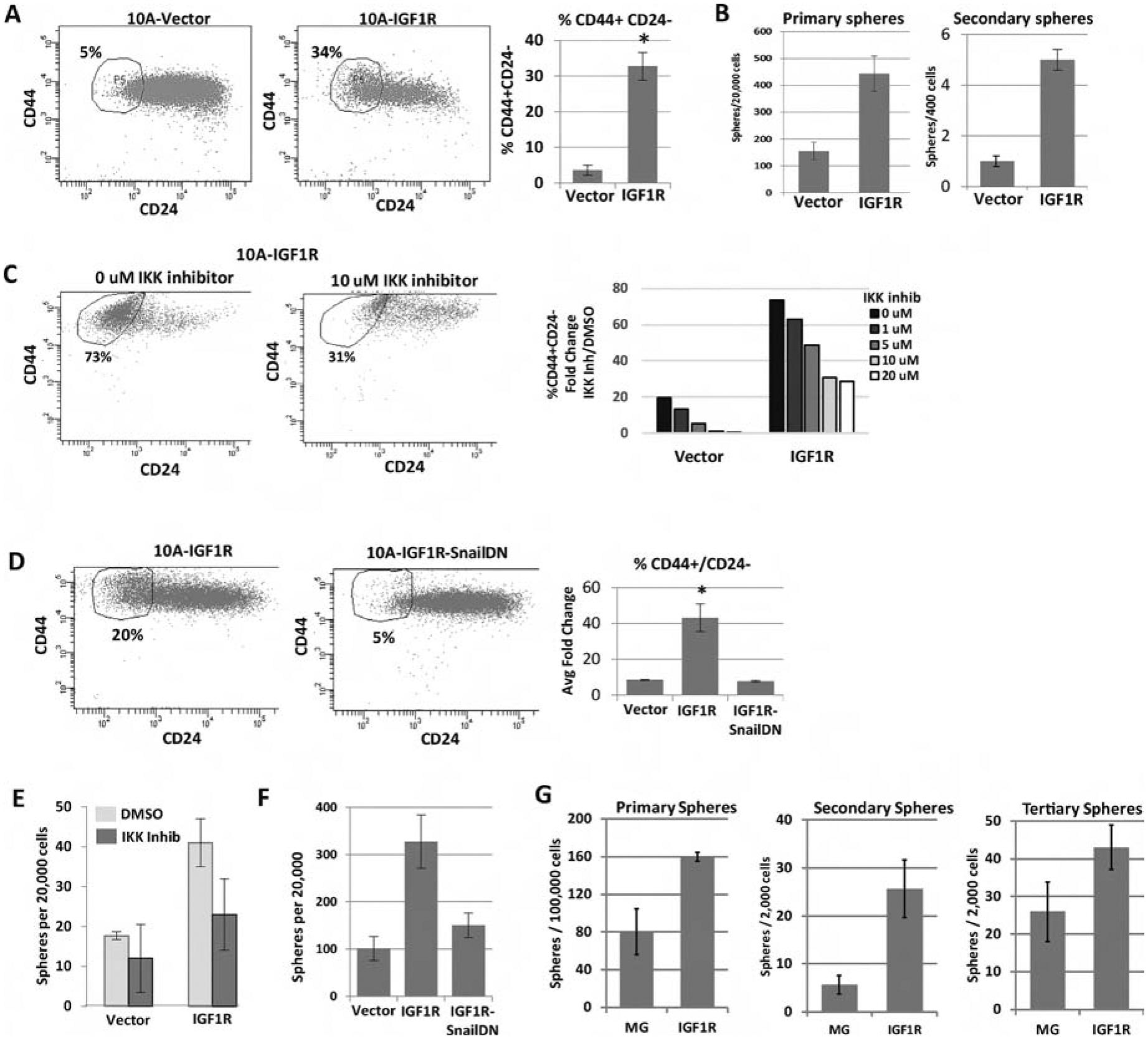
A) FACS analysis of the CD44 and CD24 cell surface markers in MCF10A cells overexpressing CD8-IGF1R or empty vector control. Average of 4 experiments with 1 to 2 biological replicates each: t-test p<0.01. B) Sphere formation of MCF10A control and MCF10A-CD8-IGF1R cells grown in serum-free low attachment conditions. 3 independent experiments: t-test p <0.05. C) Representative FACS analysis of CD44 and CD24 with IKK II inhibitor. D) Cells expressing dominant-negative Snail. Quantitative analysis of all inhibitor concentrations is graphed. Representative of 2 (IKK inhibitor) or 3 (SnailDN) experiments. E) Sphere formation of MCF10A-IGF1R cells with IKKII Inhibitor or F) dominant-negative Snail. Representative of 3 independent experiments. G) Sphere formation of MMTV-CD8-IGF1R dissociated tumor cells (see materials and methods). Representative of 3 experiments.
We next determined if IGF1R-induced NFκB and Snail, previously reported to be required for IGF1R induced EMT, are also critical for the increase in CD44+/CD24− cells by inhibition of IKK and expression of dominant negative Snail, respectively. We focused our studies on a role for NFκB and Snail as we found these previously to be key for IGF1R-induced changes in EMT [20] and analysis of E-cadherin and vimentin showed inconsistent changes following expression of CD8-IGF1R (data not shown). Blocking either NFκB- or Snail-mediated gene transcription resulted in more than a 10-fold decrease in the IGF1R-induced CD44+/CD24− population (Figure 6C–D) and inhibited mammosphere formation (Figure 6E–6G). These results suggest that IGF1R not only induces an EMT through regulation of NFκB and Snail but also expands stem-like properties.
Discussion
The IGF1R pathway is critical for mammary development, where it plays significant roles in cell growth, survival, and migration. In this study, we report that IGF1R-induced mammary tumors exhibit multiple histologies and cell lineages. Multiple models of mammary ductal outgrowth and tumorigenesis were used to establish if IGF1R influenced lineage differentiation. We demonstrate that IGF1R-driven tumors express luminal-progenitor-associated characteristics with myoepithelial differentiation. IGF1R forms highly differentiated glands and tumors, underscored by a hyperplastic and attenuated phenotype. Limiting IGF1R expression to either the luminal or basal compartment influences not only glandular structure but also the co-expression of IGF1R between the lineage compartments. We demonstrate that, in these model systems, IGF1R affects mammary stem cell-associated phenotypes, and does so through the upregulation of NFκB and Snail. Interestingly, mammary cells expressing tumor initiating characteristics such as CD44+/CD24− have been shown to not only initiate tumorigenesis but also form phenotypically diverse and mixed progeny populations [54, 55]. Herein, we demonstrate the ability of IGF1R constitutive activation to alter lineage outgrowth by expanding the luminal progenitor population and producing mixed lineage tumors.
Malaguarnera and Belfiore recently summarized the growing literature supporting a role for the IGF1R pathway in stem cell-related processes across several tissue types, both normal and cancerous [56]. IGF1R is not only required for stem cell maintenance in many normal tissues, including the neural stem cell niche, hematopoietic and muscular systems, but is also required for differentiation. For example, activation of the IGF pathway is linked with both thyroid [49] and neural cell lineage differentiation. In the neural system, stem cells display mostly IGF2 and IR-A signaling; however, lineage restricted neural progenitors primarily express IGF1R [57]. In the mammary gland, IGF1R is not only critical for maintaining the stem cell niche of the terminal end bud (TEB) [16], where lineage differentiation and ductal morphogenesis occurs but, importantly, is also critical for luminal alveolar differentiation [17, 18], suggesting a role for IGF1R in maintaining the luminal mammary compartment and luminal cell fate. Interestingly, Rota et al. showed that loss of IGF1R activity, resulting from transgenic overexpression of a dominant-negative IGF1R, causes expansion of a luminal progenitor population, and this has been shown to promote Wnt-induced mammary tumorigenesis [58, 59]. Surprisingly, we find that constitutive activation of IGF1R also results in expansion of a luminal progenitor population. It is possible that IGF1R naturally promotes luminal differentiation, thus loss causes a blockade of differentiation and an increase in the luminal progenitor population as seen by Rota et al., but expression in our system sequesters activators required for differentiation and similarly results in the expansion of the luminal progenitor population. It is also possible that IGF1R expression actually limits growth or causes apoptosis of certain cell lineages to then cause an apparent lineage change. Our experimental setup did not allow us to examine this possibility.
In breast cancer, IGF1R expression strongly correlates with the estrogen receptor (ER) positive luminal subtype [60, 61]; however, this subset of IGF1R expressing tumors has the best prognosis [33], in part due to the success of ER-targeted therapies. Importantly, IGF1R is also expressed and active in triple-negative breast cancers (TNBC), where expression correlates with poor prognosis [34] and patient xenograft samples are responsive to IGF inhibition [62]. TNBCs often have dysregulation of TP53 and BRCA1. Loss of these tumor suppressor genes results in increased expression and activation of several IGF pathway members [63–65]. Importantly, while BRCA1-null tumors present as TNBC, BRCA1-null mammary glands show expansion of luminal progenitor cells [10, 11, 66–69], suggesting that the TNBC tumors arise from this ER+ progenitor. Our results show that expression of a constitutive IGF1R results in expansion of a luminal progenitor population, yet also promoting myoepithelial differentiation and results in heterogeneous tumors with multiple cell lineages. Our model mimics IGF1R-driven TNBC. We note however that constitutive activation of CD8-IGF1R may have caused changes not seen by simple overexpression of IGF1R in human breast cancer.
Recent in vivo lineage tracing work with activation of either ErbB2 or Polyoma Middle T antigen (PyMT) signaling demonstrates that luminal cells are able to generate basal cells in their progression to breast cancer [70]. This study underscores luminal cell plasticity during tumorigenesis and provides an explanation for cellular heterogeneity within cancers. IGF1R and the IGF pathway are known signaling intermediates for these proteins, especially critical for PYMT oncogenesis [71], further supporting a relationship between IGF1R and luminal to myoepithelial cell fate.
PIK3CA was demonstrated as a driver of altered mammary cell fate and mammary intratumor heterogeneity [72, 73]. The cell of origin containing the constitutively active PIK3CA-H1047R mutation dictates tumor formation and frequency by promoting committed basal and luminal cells into a multipotent stem-like state. Deletion of p53 enhances PIK3CA-driven phenotypes [72]. These cells undergo substantial oncogene-induced reprogramming, resulting in significant cell fate gene signature exchanges. Interestingly, expression of mutated PIK3CA in unipotent basal cells resulted in luminal-like cells while expression in unipotent luminal cells gave rise to basal-like cells. We observe a similar phenotype in our IGF1R-induced MEC reconstitutions. Mammary glands developed from IGF1R-infected luminal cells gave rise to K14 positive IGF1R-expressing cells while IGF1R-infected myoepithelial cells gave rise to luminal K8 positive IGF1R-expressing cells. This parallel further supports IGF1R as a driver of mammary cell fate alteration.
In conclusion, we show that IGF1R promotes multi-lineage tumors and drives phenotypically different outgrowths and tumors depending on the origin of the cell in which it is overexpressed. Additionally, we demonstrate that IGF1R may control lineage differentiation and induce multipotency in a similar manner as BRCA1 and PIK3CA mutations, promoting luminal progenitor expansion and basal differentiation. Understanding where and how this heterogeneity develops may be critical for treating the entirety of the tumor.
Supplementary Material
Supplemental Table 1: qPCR primer sequences
Supplemental Figure 1: IGF1R-infected primary mouse MEC gland reconstitutions demonstrate regional IGF1R expression. A) IHC of CD8-IGF1R-reconstituted glands with anti-IGF1R; 4x with 20x inserts.
Supplemental Figure 2: FACS populations of lineage-specific sort. Wild type mammary glands were dissociated, and FACS analyzed for CD24, CD49f, CD29, and CD61 markers to separate the luminal and basal populations.
Supplemental Figure 3: Total-sorted controls of IGF1R-infected primary mouse MECs have similar lineage markers as observed in total un-sorted reconstitutions and transgenic mice. IF of glands from total population sorted cells infected with A) vector control or B-C) CD8-IGF1R co-stained with K8 (green), K14 (red), and IGF1R (purple); 10x with 40x insets.
Supplemental Figure 4: Luminal-sorted IGF1R-infected primary mouse MECs have differential co-expression of IGF1R with K8 lineage marker in reconstituted glands. IF of glands reconstituted with luminal-sorted MECs infected with CD8-IGF1R. Sections are co-stained with K8 (green) or IGF1R (purple); 40x. Circled sections highlight areas of high IGF1R expression and minimal to no lineage marker expression.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-Cy™7 Rat Anti-Mouse CD44 | BD Biosciences | cat#560569 |
| PerCP-Cy™5.5 Mouse Anti-Human CD24 | BD Biosciences | cat#561647 |
| Biotin Mouse Lineage Panel | BD Pharmigen / BD Biosciences | cat#559971 |
| anti-mouse CD140a-Biotin (APA5), eBioscience | Invitrogen/Thermo Fisher | cat#13-1401-82 |
| MACS anti-Biotin Microbeads | Milteny Biotec | cat#130-090-485 |
| APC Rat Anti-Mouse CD24 | BD Biosciences | cat#562349 |
| FITC Hamster Anti-Rat CD29 | BD Biosciences | cat#561796 |
| BV421 Hamster Anti-Mouse CD61 | BD Biosciences | cat#562917 |
| Sytox Blue Dead Cell stain | Invitrogen | cat#S34857 |
| Anti-Troma1-Keratin 8 | Developmental Studies Hybridoma Bank | cat#TROMA-I-c |
| Chicken Anti-Keratin 14 polyclonal | Covance | cat#906001 |
| Anti-Keratin 5 polyclonal, purified | Covance | cat#PRB-160P-100 |
| Rabbit anti-IGF1R | Ventana | cat#7904346 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| IKK II inhibitor | Calbiochem | CAS 354812-17-2 |
| Critical Commercial Assays | ||
| N/A | ||
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| MCF10A-Ctrl, MCF10A-IGF1R, and MCF10A-IGF1R-SnailDN cells | Kim, H.J., et al 2007 | NA |
| Experimental Models: Organisms/Strains | ||
| Mouse: FVB/N | The Jackson Laboratory | JAX: 001800 |
| MMTV-CD8-IGF1R transgenic FVB/N mice | Carboni, J.M et al. 2005 | N/A |
| Allografts of p53-null claudin-low tumors | Zhang, M. et al. 2008 | N/A |
| Mouse: BalbC | The Jackson Laboratory | JAX: 000651 |
| Allografts of MMTV-Her2/Neu tumors | Muller, W.J. | N/A |
| Oligonucleotides | ||
| Table S1 for qPCR primers | This paper | N/A |
| Recombinant DNA | ||
| pHIV-ZSGreen empty vector | Addgene | cat#18121 |
| pHIV-ZSGreen-CD8-IGF1R | This paper | N/A |
| Software and Algorithms | ||
| N/A | ||
| Other | ||
| N/A | ||
Highlights:
IGF1R constitutive activation causes mammary tumors with mixed lineages and histologies
IGF1R expands luminal-progenitors in vivo with myoepithelial differentiation in vitro
IGF1R constitutive activation causes attenuated, hyperplastic, differentiated reconstitution
IGF1R alters outgrowth and lineage co-expression depending upon the cell of origin
IGF1R expands tumor initiating phenotypes
Acknowledgements
We would like to thank Dr. Mei Zhang for the p53null tumors and Dr. Shivendra Singh for the MMTV-ErbB2 tumors. FACS analysis was carried out at Magee Women’s Research Institute (MWRI) and University of Pittsburgh Cancer Institute core facilities. We would like to thank Dr Judith Yanowitz at MWRI for confocal microscopy support. All animal procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, were approved by the IACUC at the University of Pittsburgh, and cared for at the MWRI Animal Facility.
Funding: Research reported in this publication was supported in part by a Department of Defense Breast Cancer Research Postdoctoral Research Fellowship award to Susan Farabaugh (W81XWH-14-1-0063) and the National Cancer Institute of the National Institutes of Health under award numbers R01CA94118 and P30CA047904. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. AVL is a recipient of a Susan G. Komen Scholar award (SAC150021). The authors acknowledge support from the UPMC Hillman Cancer Center and UPMC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare that the research was conducted in the absence of any commercial, financial, or non-financial interests in the subject matter or materials discussed in this manuscript that could be construed as a potential conflict of interest.
References
- 1.Santagata S, et al. , Taxonomy of breast cancer based on normal cell phenotype predicts outcome. J Clin Invest, 2014. 124(2): p. 859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malhotra GK, et al. , Histological, molecular and functional subtypes of breast cancers. Cancer Biol Ther, 2010. 10(10): p. 955–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makki J, Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin Med Insights Pathol, 2015. 8: p. 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Network CGA, Comprehensive molecular portraits of human breast tumours. Nature, 2012. 490(7418): p. 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perou CM, et al. , Molecular portraits of human breast tumours. Nature, 2000. 406(6797): p. 747–52. [DOI] [PubMed] [Google Scholar]
- 6.Priedigkeit N, et al. , Intrinsic Subtype Switching and Acquired ERBB2/HER2 Amplifications and Mutations in Breast Cancer Brain Metastases. JAMA Oncol, 2017. 3(5): p. 666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haque W, et al. , Response rates and pathologic complete response by breast cancer molecular subtype following neoadjuvant chemotherapy. Breast Cancer Res Treat, 2018. 170(3): p. 559–567. [DOI] [PubMed] [Google Scholar]
- 8.Farabaugh SM, Boone DN, and Lee AV, Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Front Endocrinol (Lausanne), 2015. 6: p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Amato NC, et al. , Evidence for phenotypic plasticity in aggressive triple-negative breast cancer: human biology is recapitulated by a novel model system. PLoS One, 2012. 7(9): p. e45684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim E, et al. , Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med, 2009. 15(8): p. 907–13. [DOI] [PubMed] [Google Scholar]
- 11.Molyneux G, et al. , BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell, 2010. 7(3): p. 403–17. [DOI] [PubMed] [Google Scholar]
- 12.Bonnette SG and Hadsell DL, Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology, 2001. 142(11): p. 4937–45. [DOI] [PubMed] [Google Scholar]
- 13.Hadsell DL, Bonnette SG, and Lee AV, Genetic manipulation of the IGF-I axis to regulate mammary gland development and function. J Dairy Sci, 2002. 85(2): p. 365–77. [DOI] [PubMed] [Google Scholar]
- 14.Lee AV, et al. , Developmental and hormonal signals dramatically alter the localization and abundance of insulin receptor substrate proteins in the mammary gland. Endocrinology, 2003. 144(6): p. 2683–94. [DOI] [PubMed] [Google Scholar]
- 15.Stull MA, et al. , Growth factor regulation of cell cycle progression in mammary epithelial cells. J Mammary Gland Biol Neoplasia, 2004. 9(1): p. 15–26. [DOI] [PubMed] [Google Scholar]
- 16.Ruan W and Kleinberg DL, Insulin-like growth factor I is essential for terminal end bud formation and ductal morphogenesis during mammary development. Endocrinology, 1999. 140(11): p. 5075–81. [DOI] [PubMed] [Google Scholar]
- 17.Loladze AV, et al. , Epithelial-specific and stage-specific functions of insulin-like growth factor-I during postnatal mammary development. Endocrinology, 2006. 147(11): p. 5412–23. [DOI] [PubMed] [Google Scholar]
- 18.Sun Z, et al. , Decreased IGF type 1 receptor signaling in mammary epithelium during pregnancy leads to reduced proliferation, alveolar differentiation, and expression of insulin receptor substrate (IRS)-1 and IRS-2. Endocrinology, 2011. 152(8): p. 3233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casa AJ, et al. , Estrogen and insulin-like growth factor-I (IGF-I) independently down-regulate critical repressors of breast cancer growth. Breast Cancer Res Treat, 2012. 132(1): p. 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HJ, et al. , Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol, 2007. 27(8): p. 3165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Law JH, et al. , Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res, 2008. 68(24): p. 10238–46. [DOI] [PubMed] [Google Scholar]
- 22.Sell C, et al. , Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol, 1994. 14(6): p. 3604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaleko M, Rutter WJ, and Miller AD, Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol Cell Biol, 1990. 10(2): p. 464–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sell C, et al. , Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A, 1993. 90(23): p. 11217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carboni JM, et al. , Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res, 2005. 65(9): p. 3781–7. [DOI] [PubMed] [Google Scholar]
- 26.Jones RA, et al. , Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene, 2007. 26(11): p. 1636–44. [DOI] [PubMed] [Google Scholar]
- 27.Jerome L, et al. , Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates herceptin activity in vivo. Cancer Res, 2006. 66(14): p. 7245–52. [DOI] [PubMed] [Google Scholar]
- 28.Oliveras-Ferraros C, et al. , Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotype: new insights into de novo resistance to trastuzumab (Herceptin). Int J Oncol, 2010. 37(3): p. 669–78. [DOI] [PubMed] [Google Scholar]
- 29.Nahta R, Deciphering the role of insulin-like growth factor-I receptor in trastuzumab resistance. Chemother Res Pract, 2012. 2012: p. 648965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller TW, Endocrine resistance: what do we know? Am Soc Clin Oncol Educ Book, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Massarweh S, et al. , Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res, 2008. 68(3): p. 826–33. [DOI] [PubMed] [Google Scholar]
- 32.Browne BC, et al. , Evaluation of IGF1R and phosphorylated IGF1R as targets in HER2-positive breast cancer cell lines and tumours. Breast Cancer Res Treat, 2012. 136(3): p. 717–27. [DOI] [PubMed] [Google Scholar]
- 33.Yerushalmi R, et al. , Insulin-like growth factor receptor (IGF-1R) in breast cancer subtypes. Breast Cancer Res Treat, 2012. 132(1): p. 131–42. [DOI] [PubMed] [Google Scholar]
- 34.Hartog H, et al. , Divergent effects of insulin-like growth factor-1 receptor expression on prognosis of estrogen receptor positive versus triple negative invasive ductal breast carcinoma. Breast Cancer Res Treat, 2011. 129(3): p. 725–36. [DOI] [PubMed] [Google Scholar]
- 35.Dearth RK, et al. , Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1(IRS-1) or IRS-2. Mol Cell Biol, 2006. 26(24): p. 9302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachelard-Cascales E, et al. , A protocol to quantify mammary early common progenitors from long-term mammosphere culture. Curr Protoc Stem Cell Biol, 2012. Chapter 1: p. Unit 1E 7. [DOI] [PubMed] [Google Scholar]
- 37.Herschkowitz JI, et al. , Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A, 2012. 109(8): p. 2778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, et al. , Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res, 2008. 68(12): p. 4674–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller WJ, et al. , Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell, 1988. 54(1): p. 105–15. [DOI] [PubMed] [Google Scholar]
- 40.DeRose YS, et al. , Patient-derived models of human breast cancer: protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr Protoc Pharmacol, 2013. Chapter 14: p. Unit14 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Debnath J, Muthuswamy SK, and Brugge JS, Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods, 2003. 30(3): p. 256–68. [DOI] [PubMed] [Google Scholar]
- 42.Cardiff RD, et al. , Transgenic oncogene mice. Tumor phenotype predicts genotype. Am J Pathol, 1991. 139(3): p. 495–501. [PMC free article] [PubMed] [Google Scholar]
- 43.Michaelson JS and Leder P, beta-catenin is a downstream effector of Wnt-mediated tumorigenesis in the mammary gland. Oncogene, 2001. 20(37): p. 5093–9. [DOI] [PubMed] [Google Scholar]
- 44.Satyamoorthy K, et al. , Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and beta-catenin pathways. Cancer Res, 2001. 61(19): p. 7318–24. [PubMed] [Google Scholar]
- 45.Li Y and Rosen JM, Stem/Progenitor Cells in Mouse Mammary Gland Development and Breast Cancer. Journal of Mammary Gland Biology and Neoplasia, 2005. 10(1): p. 17–24. [DOI] [PubMed] [Google Scholar]
- 46.Lim E, et al. , Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res, 2010. 12(2): p. R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Visvader JE, Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev, 2009. 23(22): p. 2563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mani SA, et al. , The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 2008. 133(4): p. 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malaguarnera R, et al. , Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab, 2011. 96(3): p. 766–74. [DOI] [PubMed] [Google Scholar]
- 50.Dallas NA, et al. , Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res, 2009. 69(5): p. 1951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bartling B, et al. , Insulin-like growth factor binding proteins-2 and −4 enhance the migration of human CD34-/CD133+ hematopoietic stem and progenitor cells. Int J Mol Med, 2010. 25(1): p. 89–96. [PubMed] [Google Scholar]
- 52.Gualco E, et al. , IGF-IR-dependent expression of Survivin is required for T-antigen-mediated protection from apoptosis and proliferation of neural progenitors. Cell Death Differ, 2010. 17(3): p. 439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dontu G, et al. , In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev, 2003. 17(10): p. 1253–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Al-Hajj M, et al. , Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A, 2003. 100(7): p. 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ginestier C, et al. , ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell, 2007. 1(5): p. 555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malaguarnera R and Belfiore A, The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne), 2014. 5: p. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ziegler AN, et al. , Insulin-like growth factor-II (IGF-II) and IGF-II analogs with enhanced insulin receptor-a binding affinity promote neural stem cell expansion. J Biol Chem, 2014. 289(8): p. 4626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rota LM, et al. , IGF1R inhibition in mammary epithelia promotes canonical Wnt signaling and Wnt1-driven tumors. Cancer Res, 2014. 74(19): p. 5668–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rota LM and Wood TL, Crosstalk of the Insulin-Like Growth Factor Receptor with the Wnt Signaling Pathway in Breast Cancer. Front Endocrinol (Lausanne), 2015. 6: p. 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bhargava R, et al. , Insulin-like growth factor receptor-1 (IGF-1R) expression in normal breast, proliferative breast lesions, and breast carcinoma. Appl Immunohistochem Mol Morphol, 2011. 19(3): p. 218–25. [DOI] [PubMed] [Google Scholar]
- 61.Kolacinska A, et al. , Apoptosis-, proliferation, immune function-, and drug resistance- related genes in ER positive, HER2 positive and triple negative breast cancer. Neoplasma, 2012. 59(4): p. 424–32. [DOI] [PubMed] [Google Scholar]
- 62.Litzenburger BC, et al. , BMS-536924 reverses IGF-IR-induced transformation of mammary epithelial cells and causes growth inhibition and polarization of MCF7 cells. Clin Cancer Res, 2009. 15(1): p. 226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maor S, et al. , Elevated insulin-like growth factor-I receptor (IGF-IR) levels in primary breast tumors associated with BRCA1 mutations. Cancer Lett, 2007. 257(2): p. 236–43. [DOI] [PubMed] [Google Scholar]
- 64.Abramovitch S and Werner H, Functional and physical interactions between BRCA1 and p53 in transcriptional regulation of the IGF-IR gene. Horm Metab Res, 2003. 35(11–12): p. 758–62. [DOI] [PubMed] [Google Scholar]
- 65.Hudelist G, et al. , Intratumoral IGF-I protein expression is selectively upregulated in breast cancer patients with BRCA1/2 mutations. Endocr Relat Cancer, 2007. 14(4): p. 1053–62. [DOI] [PubMed] [Google Scholar]
- 66.Cheang MC, et al. , Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res, 2008. 14(5): p. 1368–76. [DOI] [PubMed] [Google Scholar]
- 67.Cheang MC, van de Rijn M, and Nielsen TO, Gene expression profiling of breast cancer. Annu Rev Pathol, 2008. 3: p. 67–97. [DOI] [PubMed] [Google Scholar]
- 68.Prat A, et al. , Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res, 2010. 12(5): p. R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Perou CM and Borresen-Dale AL, Systems biology and genomics of breast cancer. Cold Spring Harb Perspect Biol, 2011. 3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hein SM, et al. , Luminal epithelial cells within the mammary gland can produce basal cells upon oncogenic stress. Oncogene, 2016. 35(11): p. 1461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Novosyadlyy R, et al. , Physical and functional interaction between polyoma virus middle T antigen and insulin and IGF-I receptors is required for oncogene activation and tumour initiation. Oncogene, 2009. 28(39): p. 3477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Keymeulen A, et al. , Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature, 2015. 525(7567): p. 119–23. [DOI] [PubMed] [Google Scholar]
- 73.Koren S, et al. , PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature, 2015. 525(7567): p. 114–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: qPCR primer sequences
Supplemental Figure 1: IGF1R-infected primary mouse MEC gland reconstitutions demonstrate regional IGF1R expression. A) IHC of CD8-IGF1R-reconstituted glands with anti-IGF1R; 4x with 20x inserts.
Supplemental Figure 2: FACS populations of lineage-specific sort. Wild type mammary glands were dissociated, and FACS analyzed for CD24, CD49f, CD29, and CD61 markers to separate the luminal and basal populations.
Supplemental Figure 3: Total-sorted controls of IGF1R-infected primary mouse MECs have similar lineage markers as observed in total un-sorted reconstitutions and transgenic mice. IF of glands from total population sorted cells infected with A) vector control or B-C) CD8-IGF1R co-stained with K8 (green), K14 (red), and IGF1R (purple); 10x with 40x insets.
Supplemental Figure 4: Luminal-sorted IGF1R-infected primary mouse MECs have differential co-expression of IGF1R with K8 lineage marker in reconstituted glands. IF of glands reconstituted with luminal-sorted MECs infected with CD8-IGF1R. Sections are co-stained with K8 (green) or IGF1R (purple); 40x. Circled sections highlight areas of high IGF1R expression and minimal to no lineage marker expression.