

Abstract
Background
The rapidly enlarging COVID-19 pandemic caused by the novel SARS-corona virus-2 is a global public health emergency of an unprecedented level. Unfortunately no treatment therapy or vaccine is yet available to counter the SARS-CoV-2 infection, which substantiates the need to expand research efforts in this direction. The indispensable function of the main protease in virus replication makes this enzyme a promising target for inhibitors screening and drug discovery to treat novel coronavirus infection. The recently concluded α-ketoamide ligand-bound X-ray crystal structure of SARS-CoV-2 Mpro (PDB ID: 6Y2F) from Zhang et al. has revealed the potential inhibitor binding mechanism and the molecular determinants responsible for substrate binding.
Methods
For the study, we have targeted the SARS-CoV-2 Mpro for the screening of FDA approved antiviral drugs and carried out molecular docking based virtual screening. Further molecular dynamic simulation studies of the top three selected drugs carried out to investigated for their binding affinity and stability in the SARS-CoV-2 Mpro active site. The phylogenetic analysis was also performed to know the relatedness between the SARS-CoV-2 genomes isolated from different countries.
Results
The phylogenetic analysis of the SARS-CoV-2 genome reveals that the virus is closely related to the Bat-SL-CoV and does not exhibit any divergence at the genomic level. Molecular docking studies revealed that among the 77 drugs, screened top ten drugs shows good binding affinities, whereas the top three drugs: Lopinavir–Ritonavir, Tipranavir, and Raltegravir were undergone for molecular dynamics simulation studies for their conformational stability in the active site of the SARS-CoV-2 Mpro protein.
Conclusions
In the present study among the library of FDA approved antiviral drugs, the top three inhibitors Lopinavir–Ritonavir, Tipranavir, and Raltegravir show the best molecular interaction with the main protease of SARS-CoV-2. However, the in-vitro efficacy of the drug molecules screened in this study further needs to be corroborated by carrying out a biochemical and structural investigation.
Keywords: Coronavirus, SARS-CoV-2, COVID-19, Drug repurposing, Docking
Introduction
The highly contagious and pathogenic novel Severe Acute Respiratory Syndrome — Coronavirus-2 (SARS-CoV-2), causative agent ongoing Covid-19 pandemic, has spread rapidly and posed a health threat of unprecedented magnitude on the global population. The novel SARS-CoV-2 was first reported in December 2019 to have emerged in the live wildlife market in the Wuhan region of Hubei province, where it has caused mystic pneumonia-like respiratory illnesses in the human population of the affected area [1]. According to data presented by the COVID-19 situation report from World Health Organization (WHO), as of May 15, 2020, the virus has infected more than 4,347,935 people around the world including a staggering 297,241 deaths, with a cumulative mortality rate of >6.8% and exponentially increased in between March and April [2]. Despite the instantaneous and monumental research efforts from the scientific community around the globe at present, no effective antiviral treatment or vaccine is available for COVID-19. However, significant efforts have been made to the development of vaccines and therapeutic drugs, which were under small scale clinical trials [3,4]. Presently the SARS-CoV-2 infected patient’s treatments have been limited to the use of prophylactic and symptomatic management like mild symptoms such as dry cough, sore throat, and fever, and various fatal complications including organ failure, septic shock, pulmonary edema, severe pneumonia, and Acute Respiratory Distress Syndrome (ARDS) [5]. Therefore, there is an urgent need for the discovery of a potential treatment therapy to check and control the SARS-CoV-2 pandemic. Coronaviruses (CoVs), belong to family coronaviridae of viruses, constitute an important class of pathogens for humans and other vertebrates [6].
Before the current SARS-CoV-2, only six of the CoVs were known to cause mild to severe illnesses in humans. The novel human coronavirus: HCoV-229E and HCoV-NL63, fall in genera alpha-coronavirus, cause milder upper respiratory disease in adults, and sometimes can also cause severe infection in infants and young children. Whereas the beta-coronaviruses like HCoV-OC43, HKU1, SARS-CoV (severe acute respiratory syndrome coronavirus; which has triggered an epidemic in China during 2002–03) and MERS-CoV (Middle East Respiratory Syndrome Coronavirus; an etiological agent of middle East coronavirus epidemic of 2012) have potential to cause infection in lower respiratory tract along with cough & fever and triggers severe respiratory illness in humans [7]. The causative agent of the current outbreak SARS-CoV-2 also belongs to beta coronaviruses [8] and is closely related to SARS-CoV with an overall genomic sequence similarity of >79%. All of these CoVs belong to the Coronaviridae, a family of viruses that possess a positive-sense single-stranded RNA genome [9].
The virion of SARS-CoV-2 is consists of crown-shaped peplomers, 80–160 nm in diameter, and consists of a ∼30 kb long single-stranded RNA molecule of positive polarity with 5′ cap and 3′ Poly-A tail [10].
The RNA genome is composed of at least six open reading frames (ORFs) of which the first ORF (ORF1a/b) makes up the 5′ two-third and encodes two polypeptides pp1a and pp1ab both of which furthermore leads to the production of 16 nonstructural proteins (nsPs). Other ORFs that make up the remaining one-third of the viral genome give rise to the production of four main structural factors of the virion: Spike protein (S), Envelope protein (E), Membrane protein (M) and Nucleocapsid protein (N) [11].
The SARS-CoV-2 virus uses the heterotrimeric Spike (S) protein, which consists of S1 and S2 subunit, on its surface to interacts with the ACE2 (angiotensin-converting enzyme 2) cellular receptor, abundantly expressed on many cell types in human tissues [12]. Upon internalization into the cell, genomic RNA is used as a template for direct translation of two polyprotein pp1a and pp1ab which encodes several crucial nonstructural proteins (nsPs) including two proteases; Chymotrypsin-like protease (3CLpro) or main protease (Mpro)-nsP5 and papain-like a protease (Ppro)-nsP3, both of which processes the polypeptide pp1a and pp1ab in a sequence-specific manner to produce 16 different nsPs [13,14]. The papain protease processes the polyprotein to generate nsP1-4. At the same time, the Mpro operates at as many as 11 cleavage sites by specifically recognizing the sequence Leu-Gln*Ser-Ala-Gly (* marks the cleavage site) to generate rest of the critical nsPs including helicase, methyltransferase, and RNA dependent RNA polymerase (RdRp) all of which play a critical role in the viral infection cycle by forming a replication-transcription complex (RTC) [15]. Therefore, the main protease constitutes a major and attractive drug target to block the production of nonstructural viral components and thereby to hamper the replication event of the virus life cycle. Additionally, no human protease with similar cleavage specificity is known to rule out the possibility of cellular toxicity upon the potential inhibition of the main viral protease [16].
In recent years drug repurposing methodology has emerged as a resourceful alternative to fasten the drug development process against rapidly spreading emerging infections such as the one of SARS-CoV-2 [17]. The approach of drug repurposing has successfully led to the discoveries of potential drug candidates against several diseases such as Ebola disease, hepatitis C virus, and zika virus infection [18,19]. Recently several repurposing studies on SARS-CoV-2 have been performed using clinically approved drugs [20], among which a very new study comes out on a clinical trial of Lopinavir–Ritonavir drug for COVID-19, which was indicated on top of our drug repurposing study [21].
In the present study, we have performed in silico based drug repurposing method using molecular docking studies on the spectrum of Food and Drug Administration (FDA) — approved antiviral drugs against SARS-CoV-2 Mpro. To this end, a recently elucidated X-ray crystal structure of SARS-CoV-2 Mpro (PDB ID: 6Y2F) which have been shown with an α-ketoamide as a potent inhibitor in the enzyme’s active site, was chosen and screened for several FDA approved antiviral drugs to simulate the Mpro–α-ketoamide interactions and thereby blocking the active pocket [16]. The crystal structure of SARS-CoV-2 Mpro in the apo form (PDB ID: 6Y2E) and α-ketoamide bound form (PDB ID: 6Y2F) shows that the protein makes a crystallographic dimer composed of two monomers of identical conformations. Each protomer furthermore is made up of three domains. The interface of domain I and domain II form the active site of the protein, which is composed of a Cys145–His41 dyad where α-ketoamide derivative 13b is bound (Fig. 1 A). The uniquely globular domain III is linked to domain II through a linker region and deemed essential for the catalytic activity of these chymotrypsin-like proteas [22]. The α-ketoamide derivative 13b is shown bound in the active site. It is stabilized by several interactions with the active site residues His41 & Cys145 and adjacent residues in substrate binding cleft such as Gly143 and Ser144 [16] (Fig. 1B).
Fig. 1.

(A) Structural features of the main protease of SARS-CoV-2 monomer. A SARS-CoV-2 main protease consists of three domains. The active site of protein lies at the interface of domain I and domain II and composed of a characteristic Cys–His dyad. A linker joins domain II to domain III, which is critical for the dimerization of protein. (B) Sphere representation of Main protease monomer, α-ketoamide 13b, is shown bound in the active site groove.
We have selected several existing FDA approved drugs, most of which are reported to be used in humans for countering certain viral infections and screened them for binding in the active cleft of Mpro. Our results have shown that some of the drugs occupied the active site of Mpro with even increased binding affinity than that of the bound α-ketoamide 13b. While the rest of the compounds has shown appreciable binding while holding most of the crucial active site determinants. We envisage that further in vitro examination of the inhibitory potential of these drugs on the catalytic activity of the main protease could lead the way to repurpose one or more of the tested FDA approved drugs in this study as a treatment therapy for SARS-CoV-2 induced disease.
Materials and methods
Phylogenetic analysis of SARS-CoV-2 genome
To understand the evolutionary relationship between the previously known human coronavirus and the novel SARS-CoV-2, we have performed the phylogenetic analysis. For analysis, all the closely related and complete reference genome sequences of SARS-CoV-2 were downloaded from the NCBI GenBank database. A total of 50 genomes were considered for the study. MEGA 6.0 was used for multiple sequence alignment and construction of a phylogenetic tree, and 1000 bootstrap replicates performed using the Neighbor-joining method [23].
Molecular docking
The recently elucidated X-ray crystal structure coordinates of SAR-CoV-2 Mpro was downloaded from RCSB PDB (PDB ID: 6Y2F), having 1.75 Å resolution. In this structure, the Mpro was co-crystallized with the improved bound α-ketoamide (13b) inhibitor, and multiple intermolecular interactions of the ligand with the active site residues are characterized [16]. To further identify the potent inhibitors for SAR-CoV-2 Mpro among the FDA approved antiviral drugs, we have downloaded more than 75 drug compounds from the PubChem chemical database [24]. For molecular docking based drug repurposing, the download 3D structures of compounds and protein were prepared. The docking study was performed by AutoDock Vina [25], which uses a Lamarckian genetic algorithm (GA) in combination with grid-based energy estimation, to check the docking accuracy of software we have performed re-docking to the co-crystal bound ligand. The main aim of this molecular interaction study was to identify the highly interacting drug with SAR-CoV-2 protein crystal structure and to propose the drug by in-silico repurposing method. All the interaction visualization analysis studies were performed by DiscoveryStudio Visualizer (DS), PyMol molecular visualization tool, and LIGPLOT+ [26,27].
Molecular dynamics simulations
The molecular dynamics (MD) simulations were performed by YASARA version 19.12.14.W.64 (Yet Another Scientific Artificial Reality Application) commercial Package [28], throughout 10 ns with 101 snapshots and the AMBER14 force field. By MD, we computationally see the physical movement of atoms and molecules, which provides the structural level integrity and conformational changes that occur in the protein-ligand docked complex. In the present study, the docked complexes of virtually screened top three drugs Lopinavir–Ritonavir, Tipranavir, and Raltegravir with the X-ray crystal structure of SARS-CoV-2 Mpro(PDB ID: 6Y2F) were analyzed through MD simulations. The MD simulations parameters were kept as follows, where the temperature is kept 298 K, the pressure at the bar, coulomb electrostatics at the cutoff of 7.86, 0.9% NaCl, pH 7, solvent density 0.997, 1-femtosecond (fs) time steps, periodic boundaries in one simulation box [29]. The conformational changes in the structural level integrity of docked complexes were analyzed using root mean square deviation (RMSD) and root mean square fluctuation (RMSF) evaluations [30].
Results
Genome sequence alignment and phylogenetic analysis of SAR-CoV-2
The sequence alignment of the SAR-CoV-2 genome shows high similarity with the closely related reference genomes of other coronaviruses. The Blastn search of the complete genome of SAR-CoV-2 reveals that the most closely related virus available in GenBank is SL-CoVZXC45 (MG772933.1) (Bat SARS-like coronavirus) showing 95% query coverage and 89.11% identity. In contrast, another bat SARS-CoV genome SL-CoVZXC21 (MG772934.1) showed 94% query coverage and 88.65% sequence identity both isolated from china. Majorly phylogenetic tree was clustered into three clades I, II, and clade III; Clade I consists of 25 SARS-CoV and Bat-SL-CoV complete genome and share sequence identity range from 88.18% to 100% when sequence was aligned using Blastn tool. Whereas Clade II consists of total 12 complete genomes of SARS-CoV-2 and Bat-SL-CoV, in which ten genomes are of SARS-CoV-2 which were isolated from patients in different countries [China (MN988668.1, NC_045512.2, MN938384.1, MN975262.1), USA (MN994467.1, MN994468.1, MN985325.1, MN997409.1, MN988713.1) and Nepal (MT072688.1)]. The other two genomes of Bat-SL-CoV were isolated from China (MG772933.1, MG772934.1). In Clade III, there are two complete genomes of Bat coronavirus isolated from Germany (GU190215.1) and Kenyan Bat (KY352407.1). The rest of the 11 complete genomes of viruses are from Hibicovirus, Nobecovirus, Merbecovirus, and Embecovirus. Importantly, phylogenetic analysis revealed that there is no divergence in the SAR-CoV-2 genome sequence of different SAR-CoV-2 viruses isolated from different countries during the ongoing outbreak (Fig. 2 ).
Fig. 2.

The phylogenetic tree generated for the SARS-CoV-2 complete genome, with different neighboring complete genomes of MERS-CoV, SARS-CoV, and Bat-SL-CoV. The tree is majorly showing three clades; clade I, II, and Clade III.
Inhibitor binding cleft of Mpro
Coronaviruses use a chymotrypsin-like a protease along with papain protease to process and cleaves its long polyprotein precursor into individually functional nsPs. Multiple sequence analysis of the main protease of SARS-CoV-2 with that of SARS-CoV reveals that amino acid sequence is conserved with a sequence identity of 96% (Fig. 3 ). The active site residues are thoroughly conserved and make a catalytic Cys145–His41 dyad. Additionally, there are substrate-binding subsites positioned in the active site groove of the protease. The specific subsite residues located in the enzyme active site are named as S1’, S1, S2, S3, and S4 depending on their relative position to the cleavage site and subsites P1’, P1, P2, P3 and P4 in the polyprotein. Subsite P1 corresponds to the amino acid just before the cleavage site, and position P1’ corresponds to the residue immediately after the cleavage site [31,32].
Fig. 3.

Multiple sequence alignment analysis of the amino acid sequence of SARS_CoV-2 Mpro. Amino acids marked underneath with * represent the catalytic residues and residues marked underneath with # represent substrate-binding residues of various subsites.
In the Mpro of SARS-CoV-2 active site region, the S1’ residues are contributed by Cys145, Gly143 and Ser144 which also serve as the oxyanion hole. The S1 residue is His163, while Glu166 & Gln189 located at the S2 position. Bulky Gln189 and Pro168 make the S4 site [16] (Fig. 4 A). The main protease recognizes and binds specific residues at each subsite of the peptide substrate to determine the initiation of proteolysis and production of nsPs for the formation of the replication-transcription complex.
Fig. 4.

(A) Different S1’, S1, S2, S3 & S4 subsites groups in the substrate-binding subsites of SARS-CoV-2MPro(PDB ID: 6Y2F). (B) Re-docked α-ketoamide 13b in the active site of MPro (purple) and crystallized α-ketoamide (orange).
Docking analysis
The molecular docking based virtual screening of FDA approved antiviral drugs against the SARS-CoV-2 Mpro revealed the strong interaction with higher docking energy and binding affinities. All the potential drugs docked with the independent confirmation in the active site of protein, where the co-crystal structure ligand (improved α-ketoamide 13b) bound. Molecular docking binding affinity of all the docked and analyzed drugs with their binding energy ranking is shown in (Table S1). The molecular re-docking was also performed to check the docking accuracy of the software AutoDock Vina, and it was observed that the co-crystal bound ligand, and re-docked ligand shows RMSD value of 0.51 Å, suggesting the high fidelity of docking method (Fig. 4B). In the present study, we focused on the top 10 docking results for further analysis as these drug compounds showing higher binding affinity as Lopinavir–Ritonavir (−10.6 kcal/mol), Tipranavir (−8.7 kcal/mol), Raltegravir (−8.3 kcal/mol), α-ketoamide13b (−8.3 kcal/mol), Nelfinavir (−8.2 kcal/mol), Dolutegravir (−8.1 kcal/mol), Tenofovir-disoproxil (−8.1 kcal/mol), Baloxavir-marboxil (−8.1 kcal/mol), Letermovir (−8.0 kcal/mol), and Maraviroc (−8.0 kcal/mol). Although among the top 10 drugs, the top three drug compounds were showing binding affinity even higher than that of the improved α-ketoamide 13b compound (Fig. 5 A–D) (Table 1 ). All the screened top 10 drugs were reported for their antiviral activity against SARS-CoV, influenza A & B, Hepatitis B, Human immune deficiency virus, and cytomegalovirus [[33], [34], [35], [36], [37], [38], [39], [40], [41], [42]].
Fig. 5.

Molecular docking interaction of docked antiviral drugs with SARS-CoV-2 Mpro. (A) Lopinavir–Ritonavir. (B) Tipranavir. (C) Raltegravir and (D) Improvedα-ketoamide (13b). These top three drug compounds show a higher binding affinity than the bound α-ketoamide compound.
Table 1.
Showing the top10 drug compounds 2-dimensional representation of docking poses interacting with amino acids of target SAR-CoV-2 Mpro (COVID-19) X-ray crystal structure, including co-crystal bound ligand (improved α-ketoamide).
| S. no. | Ligand with a binding affinity (kcal/mol) | Schematic of intermolecular interactions |
|---|---|---|
| 1. | Lopinavir–Ritonavir (−10.6) | 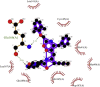 |
| 2. | Tipranavir (−8.7) | 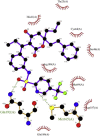 |
| 3. | Raltegravir (−8.3) | 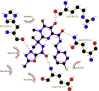 |
| 4. | α-Ketoamide13b (−8.3) | 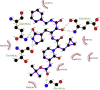 |
| 5. | Nelfinavir (−8.2) | 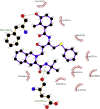 |
| 6. | Dolutegravir (−8.1) | 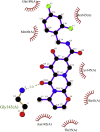 |
| 7. | Tenofovir-disoproxil (−8.1) | 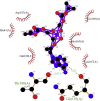 |
| 8. | Baloxavir-marboxil (−8.1) | 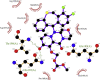 |
| 9. | Letermovir (−8.0) | 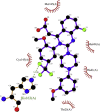 |
| 10. | Maraviroc (−8.0) | 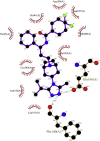 |
Molecular dynamics simulation of top three drug-protein complexes
After the docking studies, the molecular dynamics was performed of screened top three drugs (Lopinavir–Ritonavir, Tipranavir, and Raltegravir), to know the binding stability of docked complexes. The simulation was performed for 10 ns to study the conformational stability of the complexes.
The information retrieved through trajectory was used to investigate the stability of the secondary structure of the complexes by plotting Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuations (RMSF). Fig. 6 showing the RMSD values of bound and unbound ligands in different time interval summarizing the conformational changes of the ligands in 10 ns, the procedure delivers information about the movement of the ligand in its binding pocket. Fig. 7 showing the total energy in kJ/mol versus time interval of all the complexes atoms with the distribution of energy between −1,280,648 to −1,692,992 kJ/mol in 10 ns time interval. Whereas in Fig. 8 , the RMSF per solute amino acid residues calculated from the average RMSF constituting the residues. Fig. S1 is showing the hydrogen bond interaction between the ligands atoms and protein and also representing the number of hydrogen bonds formed between solute and solvent for all three selected complexes.
Fig. 6.

RMSD calculations showing the conformational deviation of drugs-protein complexes: the drugs were represented in different colors as (Lopinavir–Ritonavir (blue), Raltegravir (red), and Tipranavir (green).
Fig. 7.

The molecular motions of the SARS-COV-2 Mpro protein structure: the drugs interaction energy (kJ/mol) was represented in different colors as a function of time (Lopinavir–Ritonavir (blue), Raltegravir (red), and Tipranavir (green)).
Fig. 8.

RMSF calculation of complexes (A) Lopinavir–Ritonavir, (B) Tipranavir, and (C) Raltegravir.
Discussion
The rapidly spreading disease caused by the novel SARS-CoV-2 is now called COVID-19 [43]. World Health Organization (WHO) has declared the outbreak a pandemic, which has been increasing form the second week of March 2020 and has affected nearly all countries around the globe [2]. Although the phylogenetic analysis of different isolates of SARS-CoV-2 samples across the world clearly shows that, the SARS-CoV-2 is evolutionarily closely related to the genomes of (SARS-Like Coronavirus) Bat-SL-CoV (the coronavirus present in the bat in China), identified [44]. Our study also reveals that it might be possible that SARS-CoV-2 has been originated from Bat-SL-CoV-2 with few mutations because they share 89.11% genome identity.
Till now, there is no potent drug or vaccine that has been reported or approved to treat the SARS-CoV-2 infected individuals; only symptomatic treatment was given to the sever patients [45]. However, the efforts from the scientific community have been exceptional in advancing research effort towards the development of therapeutic intervention and finding viral drug targets. To that end, crystal structure of a few of the important viral proteins such as Spike (S) protein and viral papain protease & chymotrypsin-like protease have been deduced [46]. From the recently published studies for SARS-CoV-2, it was observed that virus binds with angiotensin-converting enzymes 2 (ACE2) receptors in the lower respiratory tracts of infected patients to gain entry into the lungs. The study reveals that SARS-CoV-2 main protease (Mpro) is the best drug target among coronaviruses [47].
Interestingly, one of the most characterized and promising drug targeting against coronavirus infection is the main protease (Mpro, also known as 3CLpro), which has been co-crystallized with a bound ligand ‘improved α-ketoamide 13b’ in case of SARS-Cov-2 main protease [16]. This crystal structure reveals that the α-ketoamide 13b is occupying the active site of the protein and making several hydrogen bonds and hydrophobic interactions with the active site residues as well as other substrate-binding residues of the binding pocket. In the present study, we have screened more than 75 antiviral, anticancer, and anti-malarial drugs for the identification of potential drug molecules using drug repurposing virtual screening methods. Molecular docking studies have revealed that the maximum of the screened drug compounds interact with SARS-CoV-2 Mpro protein and share the same binding pocket with similar interacting amino acid residues. The SARS-CoV-2 bound ligand (improved-α-ketoamide) shows strong bond interactions with surrounding amino acids within the region of 4 Å at different subsites with His164, Glu166, Gly143, His163, Cys145, His41, and Phe140 where it forms Hydrogen bonds with active site His41 and also accept hydrogen bond from the backbone amides of Gly143, Cys145, and Ser144. This protein-ligand interaction reveals a strong inhibition of virus protease (Fig. 5D) [16]. The screening and molecular docking of at least 75 preexisting drugs we have carried out have shown to fit in the active site of protease in independent conformation and appreciable binding energy score (Fig. 6A–D).
Further, we have analyzed and repurposing the top 10 drugs, which showed higher or similar binding affinity as compared to the co-crystal bound ligand of SARS-CoV-2. The top 3 drugs that are exhibiting the interaction with same amino acid residues as of the α-ketoamide with the main proteases are Lopinavir–Ritonavir showing binding affinity of (−10.6 kcal/mol) and Tipranavir (−8.7 kcal/mol), whereas Raltegravirhas binding affinity of (−8.3 kcal/mol), which is similar to improved-α-ketoamide 13b (−8.3 kcal/mol). While the rest of the drug compounds have also shown good binding energy score, as presented in Table 1.
The drug Lopinavir–Ritonavir is a combination product contains two medications lopinavir and ritonavir. This drug is mainly used for HIV-AIDS to control HIV infection by inhibiting the protease and help to decrease the amount of HIV in the body by promoting the function of the body’s natural immune system to work better [48]. The enzyme SARS-CoV-2 Mpro along with the papain-like proteases, is essential for processing the polyproteins into various nonstructural proteins by cleaving at specific sites that are translated from the viral RNA. The interacting amino acids in the Mpro enzyme active site were reported to be Leu, Gln, Ser, Ala, Gly along with the Cys–His dyad which marks the cleavage site, similarly our in silico docking study shows that top screened drug Lopinavir–Ritonavir combination interacts with Glu 166 (also form strong hydrogen bonding), Gln 189, Leu 167, Met 165, Asp 187, Met 49, His 41, Cys 145, and Leu 141 (Figs. 5A and 9 B).
Fig. 9.

Substrate binding cleft of SARS-CoV-2 Mpro harboring the docked inhibitors. Top three docked inhibitors. (A) Tipranavir, (B) Lopinavir–Ritonavir, and (C) Raltegravir occupy the active site region with independent confirmation as the originally (D) bound α-ketoamide 13b ligand in the co-crystallized structure (PDB ID: 6Y2F).
Interestingly, the binding energy score of Lopinavir–Ritonavir in protein-ligand docking was found to be even better than that of the docked α-ketoamide, and the in silico inhibition constant (Ki) was obtained to be 16 nM. In silico inhibition constant (Ki), as obtained by docking, is given in (Table 2 ) for the top 10 drugs.
Table 2.
In silico inhibition constant (Ki) obtained by molecular docking for the top 10 drugs.
| S. no. | Ligands | In silico inhibition constant in (Ki) value in Molar |
|---|---|---|
| 1. | Lopinavir–Ritonavir | 1.6754 × 10−8 |
| 2. | Tipranavir | 4.1487 × 10−7 |
| 3. | Raltegravir | 8.1265 × 10−7 |
| 4. | Improved-α-ketoamide 13b | 8.1535 × 10−7 |
| 5. | Nelfinavir | 9.6539 × 10−7 |
| 6. | Dolutegravir | 1.1230 × 10−6 |
| 7. | Tenofovir-disoproxil | 1.1430 × 10−6 |
| 8. | Baloxavir-marboxil | 1.1435 × 10−6 |
| 9. | Letermovir | 1.3533 × 10−6 |
| 10. | Maraviroc | 1.3236 × 10−6 |
Drug tipranavir or tipranavir disodium is another nonpeptidic protease inhibitor used in combination with ritonavir to treat HIV infection [49]. In our study, the drug shows interaction with Gln192, Met165 (both form hydrogen bonding), Gln189, Asp187, Met49, Arg188, Ser46, Cys44, Thr25, and His41 in different conformation from that of α-ketoamide inhibitor (Figs. 5B and 9 D). We hypothesize that tipranavir or its other derivatives with even improved binding affinity in combination with ritonavir could serve as the potential protease inhibitor to counter SRAS-CoV-2 multiplication in cell-based assay.
Another drug that has shown comparable binding affinity and binding energy with that of the docked α-ketoamide in Mpro active site, the raltegraviris, a characterized antiretroviral medication which works by inhibiting the integrase strand transfer and is used in combination with other drugs to relieve the HIV infection [42]. In the present study, the raltegravir drug shows interaction with His164, Arg188, Gln192, Glu166 (all residues were bonded with strong hydrogen bond), Met49, Met165, Phe140, Pro168, and Leu167. The drug shows four H-bonds with nearest interacting amino acids of the SARS-CoV-2 Mpro enzyme, which indicates good inhibition (Figs. 5C and 9 C). This drug could also be used with other combinations like Raltegravir and Lopinavir for the treatment of COVID-19, if found producing desirable inhibitory effect against SARS-CoV-2 protease in biochemical activity assay or cell-based assays. Additionally, other drugs that were screened and docked in the substrate-binding cleft of the Mpro, has shown good binding energy score, which is comparably similar to the original compound in the protein crystal structure. Many of these drugs such as Dolutegravir, Letermovir & Nelfinavir, are commonly used for treating different infections ranging from HIV to cytomegalovirus by employing the different mechanisms of action [[50], [51], [52]]. The identified repurposed drug and their interaction with binding amino acids in the Mpro active site have been shown in Table 1.
After screening the different FDA approved drugs, the present study enabled us to understand the mode of interaction of approved antiretroviral drugs with new coronavirus SARS-Cov-2 main protease enzyme. The top three drugs (Lopinavir–Ritonavir, Raltegravir, and Tipranavir) were further run for MD simulations studies for the period of 10 ns. In the results, we see that all three ligands are intact and bound to its binding site. Later the protein backbone RMSD analysis of all the complexes was performed, which reveals that nearly all three complexes were stable after 4 ns and showing RMSD calculations within the range of 1.5–2.458 Å after starting from 0.5 Å Fig. 6. The total binding energies were showing that drug Lopinavir–Ritonavir was showing more stable energy values, Fig. 7. In order to know the effect of ligands binding on the SARS-CoV-2 Mpro, we performed the RMSF analysis from the average RMSF of protein constituting residues atoms since all trajectories of all complexes become stable with minor fluctuations between range 1.0–2 Å Fig. 8. From the analysis, it was observed that Lopinavir–Ritonavir shows all hydrogen bonds made with nine acceptors and two donors, whereas a total number of 20 hydrogen bonds are possible Fig S1. In Raltegravir, 11 acceptors and three donors, H-bonds were formed, whereas a total number of 25 hydrogen bonds are possible, similarly drug Tipranavir formed eight acceptors and two donors H-bonds. In contrast, a total number of 18 hydrogen bonds are possible Fig S1. This MD simulation analysis shows the promising binding stability of the drug compounds with the binding pocket of CoV-2 Mpro (PDB ID: 6Y2F).
However, we believe that all the drugs studied and screened for repurposing against COVID-19 in this study should furthermore be tested, and their in vitro inhibitory potential needs to be investigated through robust biochemical proteolytic activity assays and other biophysical & structural studies.
Conclusion
In conclusion, this study reveals the potential of repurposed antiviral drugs to bind in the active site of SARS-CoV-2 main protease in a highly specific binding pattern similar to that of the crystal bound α-ketoamide Mpro structure. Three of the screened drugs Lopinavir–Ritonavir, Raltegravir, and Tipranavir have shown the strongest binding and that MD simulation study confirmed the stability and conformational flexibility of these drugs in the enzyme active site. Since all the drugs identified in this study are of known pharmacokinetics standards and approved by FDA for human use for the treatment of respective illneses, so it may be possible to move straight to clinical trials as per standard and approved by FDA for new indication, and therefore can fasten up the process of the therapeutics development against SARS-CoV-2 infection. The molecular dynamics studies performed in this study for screened top three drugs also proving the binding conformational stability with CoV-2 Mpro. Our phylogenetic analysis of the available genomes of SARS-CoV-2 isolated from different sources also reveals that the virus is not showing any sign of severing mutation or diversification rapidly. Therefore the repurposed drug combinations could be used against SARS-CoV-2 on the pan-community level. Furthermore we suggest that the efficacy of the repurposed drugs in this study needs to be experimentally confirmed by carrying out the biochemical and structural studies for the prevention and treatment of Covid-19.
Funding
No funding sources.
Competing interests
None declared.
Ethical approval
Not required.
Acknowledgments
YK acknowledges CSIR-CIMAP, Lucknow, UKE Hamburg, Germany, and thanks Priyanka Kumari for her useful suggestions. HS thanks Petrus Roelofs & Anjali Malik for insightful discussions. YK also acknowledges Dr. Om Prakash (IMSc. Chennai) for formatting a manuscript and give special thanks to Gujarat University, department of bioinformatics for providing a supercomputing facility. YK and HS both thank and appreciate all the researchers and scientists who are working round the clock to deal with the COVID-19 pandemic.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.jiph.2020.06.016.
Appendix A. Supplementary data
The following are Supplementary data to this article:

References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO coronavirus disease (COVID-19) dashboard. n.d. https://covid19.who.int/. [Accessed 15 May 2020].
- 3.Hung I.F.-N., Lung K.-C., Tso E.Y.-K., Liu R., Chung T.W.-H., Chu M.-Y. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020;0 doi: 10.1016/S0140-6736(20)31042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reuters; 2020. Gilead to end coronavirus drug trials, adding to access worry — researchers. [Google Scholar]
- 5.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.CDC Coronavirus disease 2019 (COVID-19) Cent Dis Control Prev. 2020 https://www.cdc.gov/coronavirus/2019-ncov/index.html [Accessed 14 May 2020] [Google Scholar]
- 8.Hui D.S., Azhar E.I., Madani T.A., Ntoumi F., Kock R., Dar O. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health — the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:1–18. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo P.C.Y., Huang Y., Lau S.K.P., Yuen K.-Y. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2:1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hussain S., Pan J., Chen Y., Yang Y., Xu J., Peng Y. Identification of novel subgenomic RNAs and noncanonical transcription initiation signals of severe acute respiratory syndrome coronavirus. J Virol. 2005;79:5288–5295. doi: 10.1128/JVI.79.9.5288-5295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y.M., Gupta S.K., Kim K.J., Powers B.E., Cerqueira A., Wainger B.J. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 2013;12:713–726. doi: 10.1016/j.stem.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun W., Sanderson P.E., Zheng W. Drug combination therapy increases successful drug repositioning. Drug Discov Today. 2016;21:1189–1195. doi: 10.1016/j.drudis.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kouznetsova J., Sun W., Martínez-Romero C., Tawa G., Shinn P., Chen C.Z. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg Microbes Infect. 2014;3:1–7. doi: 10.1038/emi.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He S., Lin B., Chu V., Hu Z., Hu X., Xiao J. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.3010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrows N.J., Campos R.K., Powell S.T., Prasanth K.R., Schott-Lerner G., Soto-Acosta R. A screen of FDA-approved drugs for inhibitors of zika virus infection. Cell Host Microbe. 2016;20:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan H.-H., Wang L.-Q., Liu W.-L., An X.-P., Liu Z.-D., He X.-Q. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin Med J (Engl) 2020;133:1051–1056. doi: 10.1097/CM9.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. 2020;382:1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci U S A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.PubChem. PubChem. n.d. https://pubchem.ncbi.nlm.nih.gov/. [Accessed 15 May 2020].
- 25.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 27.PyMOL | pymol.org. n.d. https://pymol.org/2/. [Accessed 15 May 2020].
- 28.Krieger E., Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krieger E., Darden T., Nabuurs S.B., Finkelstein A., Vriend G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 30.Desai V.H., Kumar S.P., Pandya H.A., Solanki H.A. Receptor-guided de novo design of dengue envelope protein inhibitors. Appl Biochem Biotechnol. 2015;177:861–878. doi: 10.1007/s12010-015-1784-y. [DOI] [PubMed] [Google Scholar]
- 31.Kiemer L., Lund O., Brunak S., Blom N. Coronavirus 3CLproproteinase cleavage sites: possible relevance to SARS virus pathology. BMC Bioinformatics. 2004;5 doi: 10.1186/1471-2105-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 33.De Luca A., Pezzotti P., Boucher C., Döring M., Incardona F., Kaiser R. Clinical use, efficacy, and durability of maraviroc for antiretroviral therapy in routine care: a European survey. PLoS One. 2019;14 doi: 10.1371/journal.pone.0225381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foolad F., Aitken S.L., Chemaly R.F. Letermovir for the prevention of cytomegalovirus infection in adult cytomegalovirus-seropositive hematopoietic stem cell transplant recipients. Expert Rev Clin Pharmacol. 2018;11:931–941. doi: 10.1080/17512433.2018.1500897. [DOI] [PubMed] [Google Scholar]
- 35.Hayden F.G., Sugaya N., Hirotsu N., Lee N., de Jong M.D., Hurt A.C. Baloxavir Marboxil for uncomplicated influenza in adults and adolescents. N Engl J Med. 2018;379:913–923. doi: 10.1056/NEJMoa1716197. [DOI] [PubMed] [Google Scholar]
- 36.Agarwal K., Brunetto M., Seto W.K., Lim Y.-S., Fung S., Marcellin P. 96 weeks treatment of tenofovir alafenamide vs. tenofovir disoproxil fumarate for hepatitis B virus infection. J Hepatol. 2018;68:672–681. doi: 10.1016/j.jhep.2017.11.039. [DOI] [PubMed] [Google Scholar]
- 37.Blair H.A. Dolutegravir/rilpivirine: a review in HIV-1 infection. Drugs. 2018;78:1741–1750. doi: 10.1007/s40265-018-1005-4. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem Biophys Res Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del Puente F., Berruti M., Riccardi N., di Biagio A. Comment on: dual therapy combining raltegravir with etravirine maintains a high level of viral suppression over 96 weeks in long-term experienced HIV-infected individuals over 45 years on a PI-based regimen: results from the Phase II ANRS 163 ETRAL study. J Antimicrob Chemother. 2020 doi: 10.1093/jac/dkaa120. [DOI] [PubMed] [Google Scholar]
- 41.Plosker G.L., Figgitt D.P. Tipranavir. Drugs. 2003;63:1611–1618. doi: 10.2165/00003495-200363150-00009. [DOI] [PubMed] [Google Scholar]
- 42.Brites C., Nóbrega I., Luz E., Travassos A.G., Lorenzo C., Netto E.M. Raltegravir versus lopinavir/ritonavir for treatment of HIV-infected late-presenting pregnant women. HIV Clin Trials. 2018;19:94–100. doi: 10.1080/15284336.2018.1459343. [DOI] [PubMed] [Google Scholar]
- 43.WHO Director-General’s remarks at the media briefing on 2019-nCoV on 11 February 2020. n.d. https://www.who.int/dg/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020. [Accessed 15 May 2020].
- 44.Forster P., Forster L., Renfrew C., Forster M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci U S A. 2020;117:9241–9243. doi: 10.1073/pnas.2004999117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coronavirus outbreak: top coronavirus drugs and vaccines in development. Clin Trials Arena. 2020 https://www.clinicaltrialsarena.com/analysis/coronavirus-mers-cov-drugs/ [Accessed 15 May 2020] [Google Scholar]
- 46.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181 doi: 10.1016/j.cell.2020.02.058. 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y., Guo Y., Pan Y., Zhao Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020;525:135–140. doi: 10.1016/j.bbrc.2020.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doyon L., Tremblay S., Bourgon L., Wardrop E., Cordingley M.G. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral Res. 2005;68:27–35. doi: 10.1016/j.antiviral.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 49.Larder B.A., Hertogs K., Bloor S., van den Eynde C.H., DeCian W., Wang Y. Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples. AIDS Lond Engl. 2000;14:1943–1948. doi: 10.1097/00002030-200009080-00009. [DOI] [PubMed] [Google Scholar]
- 50.Walmsley S.L., Antela A., Clumeck N., Duiculescu D., Eberhard A., Gutiérrez F. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807–1818. doi: 10.1056/NEJMoa1215541. [DOI] [PubMed] [Google Scholar]
- 51.Cottrell M.L., Hadzic T., Kashuba A.D.M. Clinical pharmacokinetic, pharmacodynamic and drug-interaction profile of the integrase inhibitor dolutegravir. Clin Pharmacokinet. 2013;52:981–994. doi: 10.1007/s40262-013-0093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marty F.M., Ljungman P., Chemaly R.F., Maertens J., Dadwal S.S., Duarte R.F. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017;377:2433–2444. doi: 10.1056/NEJMoa1706640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.