

Abstract
A single, subanesthetic dose of (R,S)-ketamine (ketamine) exerts rapid and robust antidepressant effects. Several groups previously reported that (2S,6S;2R,6R)-hydroxynorketamine (HNK) had antidepressant effects in rodents, and that (2R,6R)-HNK increased cortical electroencephalographic gamma power. This exploratory study examined the relationship between ketamine metabolites, clinical response, psychotomimetic symptoms, and gamma power changes in 34 individuals (ages 18–65) with treatment-resistant depression (TRD) who received a single ketamine infusion (0.5 mg/kg) over 40 min. Plasma concentrations of ketamine, norketamine, and HNKs were measured at 40, 80, 120, and 230 min and at 1, 2, and 3 days post-infusion. Linear mixed models evaluated ketamine metabolites as mediators of antidepressant and psychotomimetic effects and their relationship to resting-state whole-brain magnetoencephalography (MEG) gamma power 6–9 h post-infusion. Three salient findings emerged. First, ketamine concentration positively predicted distal antidepressant response at Day 11 post-infusion, and an inverse relationship was observed between (2S,6S;2R,6R)-HNK concentration and antidepressant response at 3 and 7 days post-infusion. Norketamine concentration was not associated with antidepressant response. Second, ketamine, norketamine, and (2S,6S;2R,6R)-HNK concentrations at 40 min were positively associated with contemporaneous psychotomimetic symptoms; post-hoc analysis revealed that ketamine was the predominant contributor. Third, increased (2S,6S;2R,6R)-HNK maximum observed concentration (Cmax) was associated with increased MEG gamma power. While contrary to preclinical observations and our a priori hypotheses, these exploratory results replicate those of a recently published study documenting a relationship between higher (2S,6S;2R,6R)-HNK concentrations and weaker antidepressant response in humans and provide further rationale for studying gamma power changes as potential biomarkers of antidepressant response.
Subject terms: Biochemistry, Depression
Introduction
(R,S)-ketamine (ketamine), a racemic mixture comprising (S)- and (R)-ketamine, produces antidepressant effects within a few hours in individuals with major depressive disorder (MDD) [1], bipolar depression [2, 3], and treatment-resistant depression (TRD) [2–8] but is also associated with psychotomimetic side effects, neuronal loss, and abuse liability [9–11]. As a result, recent research efforts have focused on identifying safer forms of ketamine by exploring its enantiomers and metabolites as well as alternate routes of administration [12–14].
Ketamine is rapidly and stereoselectively metabolized into (R,S)-norketamine (NK), (R,S)-dehydronorketamine, hydroxyketamines, and hydroxynorketamines (HNKs) [15–18], among others. A number of HNK metabolites have been described, including the (2,4)-, (2,5)-, and (2,6)-HNKs [17–19]. (2R,6R;2S,6S)-HNK is the most prevalent ketamine metabolite in human plasma [20] and in the plasma and brain of rodents [12, 21, 22]. Although early studies classified (2R,6R;2S,6S)-HNK as an inactive metabolite due to its lack of anesthetic effects [23], more recent studies have established its biological activity [12, 24–41]. Notably, (2R,6R)-HNK was found to exert rapid and sustained antidepressant-relevant behavioral effects in mice and rats [12, 25, 28, 32, 37, 39, 41] while lacking the adverse effects and abuse potential associated with ketamine [12, 41]. These antidepressant actions occurred independently of direct N-methyl-D-aspartate receptor (NMDAR) inhibition but involved early and sustained activation of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs) by presynaptically released glutamate [12, 38] via a mechanism convergent with metabotropic glutamate receptor 2/3 signaling [39].
Both animal [42–44] and human [45–47] studies found that subanesthetic-dose ketamine infusion robustly increases gamma power. In addition to being a putative measure of cortical disinhibition, this finding indicates activation of fast ionotropic excitatory receptors, including AMPARs [48–50]. The decreased activity in gamma aminobutyric acid-ergic interneurons and the resulting disinhibition of excitatory pyramidal neurons [51] seemingly provides the mechanism for increased gamma oscillations in response to ketamine [52, 53]; thus, increases in stimulus-evoked somatosensory cortical gamma power might, by extension, be a biomarker of synaptic plasticity and of antidepressant response to ketamine [53, 54]. Previous findings suggested that depressed participants who experienced robust increases from very low baseline levels of resting-state gamma power also experienced better clinical response to ketamine compared with those with higher baseline gamma [47]. In addition, previous studies observed that (2R,6R)-HNK increased cortical electroencephalographic (EEG) gamma power when administered to rodents [12, 39]. Such high-frequency oscillations, in vivo, have parallels to many forms of activity-dependent plasticity, such as long-term potentiation, in which high-frequency stimuli induce sustained strengthening of excitatory synapses via enhanced synchrony between limbic-connected brain regions [55]. The finding that (2R,6R)-HNK enhances cortical gamma EEG oscillations suggests that this metabolite engages endogenous processes that promote synaptic strengthening. This, in turn, suggests that enhanced gamma EEG oscillations may be directly involved in rapid antidepressant response [39].
This exploratory analysis sought to assess the relationship of ketamine’s primary metabolites to their antidepressant and psychotomimetic effects as well as their effects on gamma oscillations. The primary hypothesis was that increased ketamine and NK plasma concentrations would be preferentially related to improved acute response (i.e., at 230 min), while increased (2S,6S;2R,6R)-HNK concentrations would be related to improved extended response (i.e., at Day 7). As a secondary goal, the relationship between ketamine concentration and the concentrations of its metabolites to increases in magnetoencephalography (MEG) gamma power was also explored. Because prior preclinical work indicated that (2R,6R)-HNK significantly and acutely increases gamma power, we hypothesized that (2S,6S;2R,6R)-HNK—but not ketamine or NK—concentrations would positively correlate with greater increases in resting-state gamma power measured during an antidepressant response window 6–9 h post-ketamine administration. A tertiary goal explored how ketamine’s primary metabolites were related to its dissociative side effect profile. We hypothesized that at 40 min post-infusion, concentrations of ketamine and NK would be positively related to dissociative side effects, but that no such association would be observed for the HNK metabolites.
Materials and methods
Participants
Men and women (18–65 years old) participated in an inpatient, randomized, placebo-controlled, crossover trial of ketamine conducted on the Mood Disorders Research Unit of the National Institutes of Health (NIH) in Bethesda, MD from 2011 to 2017. Thirty-four participants with TRD and 23 healthy controls (HCs) for whom plasma levels of ketamine and its metabolites were obtained as part of a larger study (NCT00088699, NIH Protocol No. 04-M-0222, substudy 4) were included in the current analysis. The goal of the original study from which the TRD and HC data were drawn was to investigate ketamine’s mechanism of action; the current analysis is exploratory. In the current study, HC data were used only to assess the effects of ketamine and its metabolites on dissociative symptoms. All participants provided written consent prior to study entry, and this study was approved by the Combined Neuroscience Institutional Review Board of the NIH. Most of the participants in this sample have been previously described [47], but an additional three unmedicated participants with bipolar disorder were included in this analysis. All participants were free of psychotropic medications for at least 2 weeks before randomization (5 weeks for fluoxetine and 3 weeks for aripiprazole) and remained drug-free for the duration of study. Participant demographics appear in Table 1. Full eligibility criteria and a CONSORT diagram can be found in the Supplementary materials.
Table 1.
Participant demographics.
| TRD (N = 34) | HC (N = 23) | |
|---|---|---|
| n (%) | n (%) | |
| Female sex | 22 (65%) | 16 (70%) |
| Diagnosis | ||
| Major depressive disorder | 31 (91%) | – |
| Bipolar disorder | 3 (9%) | – |
| M ± SD | M ± SD | |
| Age (years) | 36.12 ± 10.4 | 34.65 ± 10.82 |
| BMI | 27.28 ± 6.83 | 27.53 ± 4.59 |
| MADRS total score at study baseline | 33.65 ± 4.83 | 1.57 ± 1.67 |
TRD treatment-resistant depression, HC healthy control, BMI body mass index, MADRS Montgomery-Asberg Depression Rating Scale.
Procedures
This was a two-arm, two-period, crossover design. In randomized (1:1) order, 40-min intravenous infusions of 0.5 mg/kg of ketamine hydrochloride or saline were administered 2 weeks apart in an inpatient setting.
Clinical ratings
Blinded clinical ratings in the TRD group were conducted 60 min prior to each infusion, at 40, 80, 120, and 230 min post-infusion, and at Days 1, 2, 3, 7, and 11 post-infusion. Montgomery-Asberg Depression Rating Scale (MADRS) [56] total score was the primary outcome measure. Timepoints prior to 230 min post-infusion were excluded from the primary efficacy analysis. Dissociative symptoms were assessed using the Clinician Administered Dissociative States Scale (CADSS) [57] total score at 40 min post-infusion.
Bioanalytical methods
Plasma samples were obtained during both study arms at baseline, at 40, 80, 120, and 230 min post-infusion, and at Days 1, 2, and 3 post-infusion. HCs did not provide plasma samples at Days 2 and 3. Whole blood samples were collected using BD vacutainer tubes with sodium heparin and centrifuged at 3000 rpm at 4 °C for 10 min; separated plasma samples were aliquoted and stored at −80 °C until assay. Plasma concentrations of ketamine, NK, and HNK were determined as previously described; additional methods can be found in the Supplementary materials [17, 20] (assignment standards are found in Supplementary Figs. S1–S3). While circulating levels of ketamine, NK, and (2S,6S;2R,6R)-HNK were quantitative, only relative concentrations of (2R,4R;2S,4S-2S,6R;2R,6S)-HNK and (2R,4S;2S,4R-2S,5S;2R,5R)-HNK are reported.
Pharmacokinetic methods
Pharmacokinetic parameters for ketamine, NK, (2S,6S;2R,6R)-HNK, (2R,4R;2S,4S-2S,6R;2R,6S)-HNK, and (2R,4S;2S,4R-2S,5S;2R,5R)-HNK were calculated by direct observation and by noncompartmental analysis using Phoenix WinNonlin (version 8, Certara USA, Inc.). The area under the plasma concentration-time curve (AUC) was determined using the “linear up, log down” trapezoidal rule; data through Day 1 were available for all participants, so AUC0–1440 min was used for analysis. Other parameters of interest included plasma elimination half-life (T1/2) and the maximum observed concentration (Cmax). Additional information can be found in the Supplementary materials.
MEG
Resting-state neuromagnetic data were collected from all TRD participants in one or two sessions ~6–9 h after ketamine infusion. Data were acquired at 1200 Hz with a bandwidth of 0–300 Hz using a 275-channel CTF system with SQUID-based axial gradiometers (VSM MedTech Ltd, Couquitlam, BC, Canada) housed in a magnetically shielded room (Vacuumschmelze, Germany).
Statistical analysis
Clinical outcomes
Linear mixed models were used to evaluate the effects of time, drug, mean-centered metabolite value, and the full factorial expression of their three-way interaction on MADRS total score. Model details are provided in the Supplementary materials. Mediation was defined using the MacArthur guidelines [58]; the mediation hypotheses were evaluated using contrast statements to test the degree of difference from zero in the mediator-by-drug interaction at each timepoint.
A linear model was used to evaluate whether metabolite concentrations predicted CADSS total scores at 40 min post-ketamine. Both TRD and HC participants were included in this analysis, so diagnostic group was added as a covariate. Because there was no a priori hypothesis regarding potential relationships between metabolites and dissociative side effects based on diagnosis, the parameter of interest for the secondary hypothesis was the main effect of the mediator on CADSS score within the ketamine condition, averaged across diagnostic groups.
Restricted maximum likelihood estimation was used for all analyses; this method accommodates missing data, so no imputation of missing values was performed. Because this was an exploratory analysis, no a priori power calculation was performed. Consistent with the current recommendations of the American Statistical Association [59], specific uncorrected p values are provided, and parameter estimates and their 95% confidence intervals are interpreted rather than employing a threshold for “statistical significance.” The SAS/STAT 9.4 syntax for these analyses and complete results are provided in the Supplementary materials.
MEG data analysis
Data were processed using CTF software (http://www.ctf.com), MNE-python [60], Analysis of Functional NeuroImages (AFNI) [61], and routines developed in-house. Each MEG dataset was automatically filtered using a high pass filter of 2 Hz and visually inspected to identify and mark time periods with significant muscular, ocular, or movement artifacts. Up to 10 segments lasting 15 s outside marked artifacts were identified in an automated fashion. Datasets were discarded if at least five 15-s artifact-free segments could not be defined. The final analysis included 25 TRD participants with adequate, artifact-free data. All further analyses were carried out on these clean epoched datasets.
Data were localized to source space on a 5 mm grid using synthetic aperture magnetometry [62] (see Supplementary materials for more information), and a multisphere head model was calculated from co-registered MRI scans. Beamformer weights were calculated using a bandpass frequency of 2–100 Hz, and power was normalized by the projected noise floor of the virtual sensor. Data were then projected in the gamma (30–50 Hz) band, normalized by the square root of the sum of squared images for the six canonical bands between 2 and 100 Hz. All images were subsequently warped to Talairach space using AFNI and masked to remove nonbrain matter and cerebellum. From this point forward, “gamma power” refers to the normalized root mean square gamma power images.
A linear mixed-effects model implemented in the AFNI routine 3dLME [63] was used to measure associations between gamma power during the ketamine study arm and ketamine, NK, and HNK concentrations. Separate models were constructed for Cmax and AUC values. Gamma power was modeled by metabolite (a within-subject factor with values for ketamine, NK, and the HNK metabolites) and MADRS total score. Post-hoc tests were performed within the 3dLME routine to assess individual contrasts for ketamine, NK, and HNK concentrations. Images are shown at a voxel-corrected threshold of pFDR < 0.05.
Results
Descriptive statistics for all metabolites are shown in Supplementary Table S1; raw concentration curves appear in Supplementary Fig. S4. Because results of the exploratory data analysis indicated that some pharmacokinetic parameters were not sufficiently variable, several were excluded from further analysis; in particular, Cmax, T1/2, and AUC were analyzed for both ketamine and NK, but only Cmax and AUC0–1440 were analyzed for the HNKs.
Mediation of ketamine’s antidepressant effects in TRD
The average antidepressant response to ketamine versus placebo at Day 1 was −7.11 (95% CI: −11.04, −3.19) points on MADRS total score. The results for all mediational models of this effect are illustrated in Fig. 1 (full model results are provided in Supplementary Tables S2 and S3). While ketamine concentration did not mediate antidepressant response at the acute timepoints, it was positively related to antidepressant response at Day 11 post-infusion [AUC0–1440: t(51.60) = −3.22, p = 0.002; Cmax: t(54.40) = −2.69, p = 0.009], such that higher ketamine concentration was associated with better antidepressant response. Using model parameters to generate an example, TRD participants with ketamine AUC0–1440 that was one standard deviation above average experienced an average 10.13-point [95% CI: 5.40, 14.85] better antidepressant response at Day 11 than participants with sample-average ketamine AUC0–1440. No meaningful relationship to response was observed for NK concentration, nor for (S)-NK or (R)-NK (Supplementary Tables S4 and S5).
Fig. 1. Relationship between pharmacokinetic parameter value and treatment effects.

The figure plots the results of specific contrasts testing the parameter*drug interaction at each timepoint; slopes are multiplied by the standard deviation of the parameter to yield change in treatment effect associated with a one standard deviation increase in pharmacokinetic parameter. K ketamine, NK norketamine, HNK (2S,6S;2R,6R)-hydroxynorketamine, AUC area under the curve from 0 to 1440 min, CMAX maximum concentration, HL half-life.
Similar results were obtained for (2S,6S;2R,6R)-HNK Cmax and HNK AUC0–1440 (Supplementary Tables S2 and S3), in that lower (2S,6S;2R,6R)-HNK concentration was associated with better antidepressant response and higher (2S,6S;2R,6R)-HNK concentration was associated with attenuated response; this relationship was stronger at the middle timepoints (Days 3 and 7) but was not observed at Day 11 (Fig. 2). This result was most robust for Cmax; using model parameters to compare participants with sample average (2S,6S;2R,6R)-HNK Cmax to those with a one standard deviation decrease in Cmax, the decrease in Cmax was associated with a 7.97-point [95% CI: 3.65, 12.30] better antidepressant response at Day 7 [t(39.3) = 2.99, p = 0.005].
Fig. 2. Relationship between (2S,6S;2R,6R)-hydroxynorketamine (HNK) Cmax and antidepressant response to ketamine.

The figure plots the difference in MADRS total score between ketamine and placebo arms (response; placebo minus ketamine) by (2S,6S;2R,6R)-HNK Cmax, at each timepoint. The slope for each relationship (with 95% confidence interval) is provided (see also Supplementary Table S2). Increased (2S,6S;2R,6R)-HNK maximum concentration was associated with less clinical response, but this relationship was most robust at later timepoints. MADRS: Montgomery-Asberg Depression Rating Scale.
Given previous preclinical (2R,6R)-HNK findings, an initial pilot analysis of 10 participants who were extreme examples of either antidepressant response or nonresponse found trends suggesting that (2R,6R)-HNK and (2S,6S)-HNK concentrations measured at 230 min correlated with antidepressant response. Based on this, the analysis was repeated in the full sample; however, in the full sample, the correlation with antidepressant response for (2R,6R)-HNK and (2S,6S)-HNK was lost. The remaining HNKs—(2R,4R;2S,4S-2S,6R;2R,6S)-HNK and (2R,4S;2S,4R-2S,5S;2R,5R)-HNK—also did not mediate ketamine’s antidepressant effects (Supplementary Tables S4 and S5).
Mediation of ketamine’s dissociative side effects
Higher concentrations of ketamine [t(48) = 3.36, p = 0.002], NK [t(48) = 1.89, p = 0.07], and (2S,6S;2R,6R)-HNK [t(48) = 2.09, p = 0.042] at 40 min post-infusion were related to higher CADSS total scores (Supplementary Tables S6 and S7 and Supplementary Fig. S5). Given the similar trend observed across ketamine, NK, and (2S,6S;2R,6R)-HNK concentrations, a post-hoc analysis was conducted to determine whether one was more important than the others. After confirming sufficient lack of collinearity, ketamine, NK, and (2S,6S;2R,6R)-HNK concentrations were contemporaneously entered into a model predicting CADSS total score. Ketamine concentration remained positively related to dissociation (B = 0.11 [95% CI: 0.02, 0.21], p = 0.018) but (2S,6S;2R,6R)-HNK did not (B = −0.02 [95% CI: −0.31, 0.28], p = 0.91). While the point estimate for NK remained positive, the confidence interval was wide and included zero (B = 0.46 [95% CI: −0.29, 1.22], p = 0.22). When the relationship between dissociation and the additional HNKs were evaluated in TRD participants, only (2R,4R;2S,4S-2S,6R;2R,6S)-HNK was found to be related (Supplementary Table S8).
Associations with gamma power
The linear mixed-effects model of ketamine-arm data identified a network of brain regions showing significant (pFDR < 0.05) associations between gamma power and Cmax metabolite levels but not AUC metabolite levels (Fig. 3). In particular, (2S,6S;2R,6R)-HNK Cmax values were found to be positively associated with gamma power in parts of the occipital, temporal, and frontal cortices, though no interaction with MADRS total score was observed. In contrast, NK Cmax values were negatively associated with gamma power in the same core regions. No meaningful associations between ketamine Cmax values and gamma power were noted.
Fig. 3. Associations between gamma power and (2S,6S;2R,6R)-hydroxynorketamine (HNK) and norketamine (NK) Cmax values.
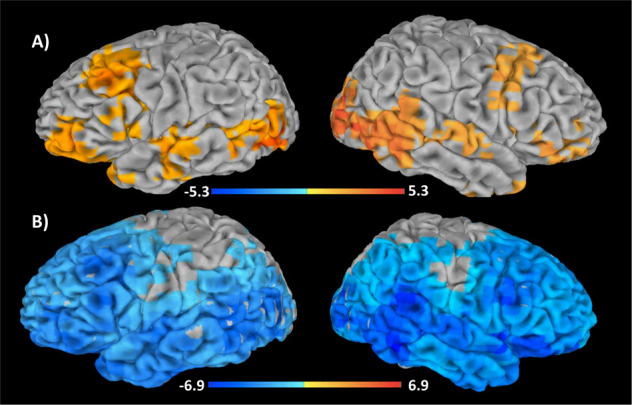
Images of normalized, root mean square gamma power (30–50 Hz) estimates associated with (a) (2S,6S;2R,6R)-HNK Cmax levels and (b) NK Cmax levels. Images are superimposed on a high-resolution structural scan and thresholded at pFDR < 0.05. Reddish colors represent regions where metabolite levels were positively associated with gamma power, while bluish colors represent regions where metabolite levels were negatively associated with gamma power.
Discussion
This study examined the relationship between ketamine and its metabolites with clinical response and gamma power changes in individuals with TRD and with psychotomimetic symptoms in HCs and TRD participants. In direct contrast with our a priori hypothesis, lower (2S,6S;2R,6R)-HNK concentrations were associated with better antidepressant response at Days 3 and 7 post-ketamine infusion. Our exploratory efforts to localize the effects of (2S,6S;2R,6R)-HNK to (2R,6R)-HNK or (2S,6S)-HNK did not provide clarity. This finding replicates a recently published clinical study that found that individuals with suicidal depression who had higher (2S,6S;2R,6R)-HNK concentrations immediately after ketamine infusion experienced less improvement in both the short and long term [64]; neither ketamine concentration nor NK concentration were related to clinical improvement.
Notably, both the work by Grunebaum et al. and the present study produced similar results despite major methodological differences; Grunebaum et al. had a larger sample that included participants with significant suicidal ideation, sampled metabolite concentrations only once, and pooled both open-label and randomized data [64]. In contrast, an earlier study found no relationship between (2S,6S;2R,6R)-HNK concentration and clinical response rate at 230 min post-ketamine infusion [20]. However, that study included TRD participants with either MDD or bipolar disorder, some of whom concomitantly received riluzole, valproate, or lithium, and response was defined categorically based on change at 230 min post-ketamine (i.e., degree of placebo response was not considered). Furthermore, in that study, only (2S,6S;2R,6R)-HNK and (2R,5R;2S,5S)-HNK were correctly assigned based on presently available analytical standards, limiting comparison with more recent studies [20].
Thus, both the current study and a previous clinical one [64] appear to contrast with preclinical findings, namely that, in rodents, (2R,6R)-HNK exerts antidepressant-like effects [12, 37]. Other differences between preclinical and clinical ketamine data have been reported in clinical pharmacological studies, which can be largely explained by species differences, metabolite formation, dosing, and route differences [65]. In the current study, ketamine and its metabolites were quantified in the plasma compartment, but the target of interest is the CNS. Perhaps, as suggested by others [64], (2S,6S;2R,6R)-HNK has preferential distribution in humans outside of the plasma compartment. Any prospective study of HNK’s putative antidepressant effects should quantify HNK in CSF. It should be noted that the finding of a relationship between ketamine concentration and better long-term antidepressant response (here, Day 11) was not observed in prior clinical studies. As described above, differences between our participant sample and those used in previous studies may account for this discrepant finding; nevertheless, this work needs replication.
The present results support our second hypothesis that (2S,6S;2R,6R)-HNK concentrations would positively correlate with greater increases in resting-state gamma power. Unexpectedly, the Cmax of NK was also found to be associated with lower levels of gamma power throughout the cortex. The significance of this second finding is unclear, though it is important to note that the gamma power measurement occurred 6–9 h post-ketamine infusion. Nevertheless, the direction of the association between (2S,6S;2R,6R)-HNK and gamma power observed here supports previous preclinical findings [12, 37], suggesting that (2S,6S;2R,6R)-HNK is critical to ketamine’s antidepressant effects because of its association with increased cortical excitation. While this contrasts with the findings of an inverse relationship between HNK and clinical response reported here, recent findings have nevertheless demonstrated that baseline gamma power moderates the relationship between ketamine-mediated increases in gamma power and antidepressant response [47]. It is possible that baseline gamma levels might also influence the relationship between HNK and antidepressant response, a hypothesis that merits further study. Taken together, the findings suggest that gamma power is associated with (2S,6S;2R,6R)-HNK concentration measured during an antidepressant response window not influenced by acute dissociative effects. It remains to be seen whether similar findings occur at the time of ketamine administration or whether these findings extend to task-mediated, evoked gamma responses.
Finally, individual concentrations of ketamine, NK, and (2S,6S;2R,6R)-HNK at 40 min post-ketamine infusion were all found to be positively related to ketamine’s dissociative effects. However, a post-hoc analysis demonstrated that ketamine and NK—but not (2S,6S;2R,6R)-HNK—were uniquely related to dissociation. This finding is consistent with prior findings that ketamine and NK, but not (2R,6R)-HNK, functionally inhibit the NMDAR at exposure levels observed in our participants [12, 37]. This supports the conclusion that NMDAR inhibition mediates ketamine’s dissociative effects [13] and is also consistent with recent clinical data reporting that NK, but not HNK, was associated with dissociative symptoms immediately after ketamine infusion [64]. It should be noted, however, that these results are inconsistent with a previous clinical study in individuals with bipolar disorder who were concomitantly treated with valproate or lithium; that study found that ketamine and NK were not related to dissociative symptoms and that (2R,5R;2S,5S)-HNK was negatively correlated with CADSS score [20].
Interestingly, recent preclinical work has suggested that (S)-NK may be a safer, though perhaps less effective, alternative to ketamine and esketamine, with less abuse liability and fewer side effects [66, 67]. However, while the current study found no relationship between (S)-NK or (R)-NK and dissociative side effects, it also found no relationship between (S)-NK and antidepressant response. Given that (S)-NK resulted in antidepressant-like effects when administered directly to mice [66, 67], additional prospective work in humans is needed to clarify this issue.
This study had several strengths, including use of placebo-controlled clinical data, estimation of the treatment effect as a continuous variable, and a detailed pharmacokinetic analysis of multiple timepoints. The inclusion of HC data in the analysis of psychotomimetic symptoms is also a strength. Nevertheless, the study is also associated with several notable limitations. First, ketamine metabolism is dynamic, in that this agent is extensively metabolized to several pharmacologically active metabolites. Therefore, correlating antidepressant response to any individual metabolite may not provide an accurate window into ketamine’s effects. Second, the sample size was relatively small, and some data were missing (see Supplementary Fig. S6 (CONSORT diagram)); particularly given the large number of comparisons in this exploratory study, Type I errors are a risk. All results of this exploratory study must therefore be confirmed prospectively. Third, the plasma sampling strategy was not ideally placed to measure both early and late ketamine metabolites and prevented the precise calculation of some pharmacokinetic parameters. As a result, T1/2 of the HNK metabolites was not quantifiable for most participants, and we were unable to analyze parameters other than AUC0–1440, Cmax, and T1/2. Fourth, the metabolites were measured peripherally, which does not necessarily reflect their activity in the CNS. Finally, gamma power changes were measured 6–9 h post-ketamine infusion rather than contemporaneously with administration or when metabolites were measured.
Despite these limitations, the current exploratory analysis builds on previous work to provide valuable information regarding the relationship between ketamine metabolites and clinical response, psychotomimetic symptoms, and gamma power changes among individuals with TRD. Although the finding that (2S,6S;2R,6R)-HNK was inversely associated with extended antidepressant response to ketamine was not predicted by previous preclinical work in mice, it is consistent with a recent independent finding in humans [64]. (2S,6S;2R,6R)-HNK was also positively associated with increases in gamma power in the TRD group, and ketamine was the analyte most robustly associated with dissociative side effects for both TRD and HC participants. Future research is needed to better characterize the antidepressant and side effect profiles of (2S,6S;2R,6R)-HNK, particularly with regard to CNS levels of the metabolite and their relationship to gamma power changes proximal to drug administration.
Funding and disclosure
Funding for this work was supported in part by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; ZIAMH002857), by the National Institute on Aging (NIA; ZIAAG000297), by a NARSAD Independent Investigator Award to Dr CAZ, by a Brain and Behavior Mood Disorders Research Award to Dr CAZ, and by an NIH Bench-to-Bedside award to Drs CAZ and TDG (NIH; MH107615). Dr CAZ is listed as a coinventor on a patent for the use of ketamine in major depression and suicidal ideation. Drs CAZ and RM are listed as coinventors on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydro and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain. Drs CAZ, RM, and TDG are coinventors on a patent application for the crystal forms and methods of synthesis of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine and the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post-traumatic stress disorders. Drs CAZ and RM have assigned their patent rights to the U.S. government but will share a percentage of any royalties that may be received by the government. Dr TDG has assigned his patent rights to the University of Maryland. All other authors have no conflict of interest to disclose, financial or otherwise.
Supplementary information
Acknowledgements
The authors thank the 7SE research unit and staff for their support. Ioline Henter (NIMH) provided invaluable editorial assistance. This work used the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Author contributions
CAF: drafted and revised the paper; conducted the statistical analysis; helped interpret the statistical analysis; approved the final version of the paper. JRG: revised the paper; collected the data; assisted in statistical design, analysis, and interpretation; approved the final version of the paper. RM: edited the paper for critical intellectual content; helped interpret the statistical analysis; approved the final version of the paper. JG: edited the paper for critical intellectual content; collected the data; approved the final version of the paper. LA: collected the data; revised the paper; approved the final version of the paper. JL: collected the data; revised the paper; approved the final version of the paper. ACN: Helped conceptualize the study; edited the paper for critical intellectual content; approved the final version of the paper. BK: edited the paper for critical intellectual content; helped interpret the statistical analysis; approved the final version of the paper. PY: collected the data; revised the paper; approved the final version of the paper. TDG: edited the paper for critical intellectual content; helped interpret the statistical analysis; approved the final version of the paper. LTP: helped conceptualize the study; revised the paper; provided research supervision; approved the final version of the paper. CAZ: conceptualized the study; designed the study; edited the paper for critical intellectual content; helped interpret the statistical analysis; provided research supervision; approved the final version of the paper.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
5/6/2020
A Correction to this paper has been published: 10.1038/s41386-020-0699-7
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41386-020-0663-6).
References
- 1.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 2.Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zarate CA, Jr, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. 2012;71:939–46. doi: 10.1016/j.biopsych.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 5.Fava M, Freeman MP, Flynn M, Judge H, Hoeppner BB, Cusin C, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry. 2018. 10.1038/s41380-018-0311-2. [DOI] [PMC free article] [PubMed]
- 6.Singh JB, Fedgchin M, Daly EJ, De Boer P, Cooper K, Lim P, et al. A double-blind, randomized, placebo-controlled, dose-frequency study of intravenous ketamine in patients with treatment-resistant depression. Am J Psychiatry. 2016;173:816–26. doi: 10.1176/appi.ajp.2016.16010037. [DOI] [PubMed] [Google Scholar]
- 7.Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–42. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67:139–45. doi: 10.1016/j.biopsych.2009.08.038. [DOI] [PubMed] [Google Scholar]
- 9.Henter ID, de Sousa RT, Zarate CA., Jr Glutamatergic modulators in depression. Harv Rev Psychiatry. 2018;26:307–19. doi: 10.1097/HRP.0000000000000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu W, Ding Z, Zhang Y, Shi J, Hashimoto K, Lu L. Risks associated with misuse of ketamine as a rapid-acting antidepressant. Neurosci Bull. 2016;32:557–64. doi: 10.1007/s12264-016-0081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C, Zheng D, Xu J, Lam W, Yew DT. Brain damages in ketaime addicts as revealed by magnetic resonance imaging. Front Neuroanat. 2013;7:23. doi: 10.3389/fnana.2013.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533:481–6. doi: 10.1038/nature17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, et al. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev. 2018;70:621–60. doi: 10.1124/pr.117.015198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leal GC, Bandeira ID, Correia-Melo FS, Telles M, Mello RP, Vieira F, et al. Intravenous arketamine for treatment-resistant depression: open-label pilot study. Eur Arch Psychiatry Clin Neurosci. 2020. 10.1007/s00406-020-01110-5. [DOI] [PubMed]
- 15.Adams JD, Jr, Baillie TA, Trevor AJ, Castagnoli N., Jr Studies on the biotransformation of ketamine. 1-Identification of metabolites produced in vitro from rat liver microsomal preparations. Biomed Mass Spectrom. 1981;8:527–38. doi: 10.1002/bms.1200081103. [DOI] [PubMed] [Google Scholar]
- 16.Chang T, Glazko AJ. Biotransformation and disposition of ketamine. Int Anesthesiol Clin. 1974;12:157–78. doi: 10.1097/00004311-197412020-00018. [DOI] [PubMed] [Google Scholar]
- 17.Moaddel R, Venkata SLV, Tanga MJ, Bupp JE, Green CE, Iyer L, et al. A parallel chiral–achiral liquid chromatographic method for the determination of the stereoisomers of ketamine and ketamine metabolites in the plasma and urine of patients with complex regional pain syndrome. Talanta. 2010;82:1892–904. doi: 10.1016/j.talanta.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woolf TF, Adams JD. Biotransformation of ketamine, (Z)-6-hydroxyketamine, and (E)-6-hydroxyketamine by rat, rabbit, and human liver microsomal preparations. Xenobiotica. 1987;17:839–47. doi: 10.3109/00498258709043993. [DOI] [PubMed] [Google Scholar]
- 19.Desta Z, Moaddel R, Ogburn ET, Xu C, Ramamoorthy A, Venkata SL, et al. Stereoselective and regiospecific hydroxylation of ketamine and norketamine. Xenobiotica. 2012;42:1076–87. doi: 10.3109/00498254.2012.685777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zarate CA, Jr, Brutsche N, Laje G, Luckenbaugh DA, Venkata SL, Ramamoorthy A, et al. Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72:331–8. doi: 10.1016/j.biopsych.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Can A, Zanos P, Moaddel R, Kang HJ, Dossou KS, Wainer IW, et al. Effects of ketamine and ketamine metabolites on evoked striatal dopamine release, dopamine receptors, and monoamine transporters. J Pharmacol Exp Ther. 2016;359:159–70. doi: 10.1124/jpet.116.235838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moaddel R, Sanghvi M, Ramamoorthy A, Jozwiak K, Singh N, Green C, et al. Subchronic administration of (R,S)-ketamine induces ketamine ring hydroxylation in Wistar rats. J Pharm Biomed Anal. 2016;127:3–8. doi: 10.1016/j.jpba.2016.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem. 1986;29:2396–9. doi: 10.1021/jm00161a043. [DOI] [PubMed] [Google Scholar]
- 24.Cavalleri L, Merlo Pich E, Millan MJ, Chiamulera C, Kunath T, Spano PF, et al. Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol Psychiatry. 2018;23:812–23. doi: 10.1038/mp.2017.241. [DOI] [PubMed] [Google Scholar]
- 25.Chou D, Peng HY, Lin TB, Lai CY, Hsieh MC, Wen YC, et al. (2R,6R)-hydroxynorketamine rescues chronic stress-induced depression-like behavior through its actions in the midbrain periaqueductal gray. Neuropharmacology. 2018;139:1–12. doi: 10.1016/j.neuropharm.2018.06.033. [DOI] [PubMed] [Google Scholar]
- 26.Collo G, Cavalleri L, Chiamulera C, Merlo Pich E. (2R,6R)-Hydroxynorketamine promotes dendrite outgrowth in human inducible pluripotent stem cell-derived neurons through AMPA receptor with timing and exposure compatible with ketamine infusion pharmacokinetics in humans. Neuroreport. 2018;29:1425–30. doi: 10.1097/WNR.0000000000001131. [DOI] [PubMed] [Google Scholar]
- 27.Faccio AT, Ruperez FJ, Singh NS, Angulo S, Tavares MFM, Bernier M, et al. Stereochemical and structural effects of (2R,6R)-hydroxynorketamine on the mitochondrial metabolome in PC-12 cells. Biochim Biophys Acta Gen Subj. 2018;1862:1505–15. doi: 10.1016/j.bbagen.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukumoto K, Fogaca MV, Liu RJ, Duman C, Kato T, Li XY, et al. Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci USA. 2019;116:297–302. doi: 10.1073/pnas.1814709116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho MF, Correia C, Ingle JN, Kaddurah-Daouk R, Wang L, Kaufmann SH, et al. Ketamine and ketamine metabolites as novel estrogen receptor ligands: induction of cytochrome P450 and AMPA glutamate receptor gene expression. Biochem Pharmacol. 2018;152:279–92. doi: 10.1016/j.bcp.2018.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, Jimenez L, et al. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in alpha7 nicotinic acetylcholine receptors. Eur J Pharmacol. 2013;698:228–34. doi: 10.1016/j.ejphar.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paul RK, Singh NS, Khadeer M, Moaddel R, Sanghvi M, Green CE, et al. (R,S)-Ketamine metabolites (R,S)-norketamine and (2S,6S)-hydroxynorketamine increase the mammalian target of rapamycin function. Anesthesiology. 2014;121:149–59. doi: 10.1097/ALN.0000000000000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, Alvarez JC, et al. Common neurotransmission recruited in (R,S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol Psychiatry. 2017;84:e3–e6. doi: 10.1016/j.biopsych.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 33.Wray NH, Schappi JM, Singh H, Senese NB, Rasenick MM. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol Psychiatry. 2018;24:1833–43. doi: 10.1038/s41380-018-0083-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao N, Skiteva O, Zhang X, Svenningsson P, Chergui K. Ketamine and its metabolite (2R,6R)-hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Mol Psychiatry. 2018;23:2066–77. doi: 10.1038/mp.2017.239. [DOI] [PubMed] [Google Scholar]
- 35.Ye L, Ko CY, Huang Y, Zheng C, Zheng Y, Chou D. Ketamine metabolite (2R,6R)-hydroxynorketamine enhances aggression via periaqueductal gray glutamatergic transmission. Neuropharmacology. 2019;157:107667. doi: 10.1016/j.neuropharm.2019.107667. [DOI] [PubMed] [Google Scholar]
- 36.Kroin JS, Dav V, Moric M, Buvanendran A. Efficacy of the ketamine metabolite (2R,6R)-hydroxynorketamine in mice models of pain. Reg Anesth Pain Med. 2019;44:111–7. doi: 10.1136/rapm-2018-000013. [DOI] [PubMed] [Google Scholar]
- 37.Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, et al. Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci USA. 2019;116:5160–9. doi: 10.1073/pnas.1816071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riggs LM, Aracava Y, Zanos P, Fischell J, Albuquerque EX, Pereira EFR, et al. (2R,6R)-hydroxynorketamine rapidly potentiates hippocampal glutamatergic transmission through a synapse-specific presynaptic mechanism. Neuropsychopharmacology. 2020;45:426–36. doi: 10.1038/s41386-019-0443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zanos P, Highland JN, Stewart BW, Georgiou P, Jenne CE, Lovett J, et al. (2R,6R)-hydroxynorketamine exerts mGlu2 receptor-dependent antidepressant actions. Proc Natl Acad Sci USA. 2019;116:6441–50. doi: 10.1073/pnas.1819540116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zanos P, Highland JN, Liu X, Troppoli TA, Georgiou P, Lovett J, et al. (R)-Ketamine exerts antidepressant actions partly via conversion to (2R,6R)-hydroxynorketamine, while causeing adverse effects at sub-anesthetic doses. Br J Pharmacol. 2019;176:2573–92. doi: 10.1111/bph.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Highland JN, Morris PJ, Zanos P, Lovett J, Ghosh S, Wang AQ, et al. Mouse, rat, and dog bioavailability and mouse oral antidepressant efficacy of (2R,6R)-hydroxynorketamine. J Psychopharmacol. 2018;29:269881118812095. doi: 10.1177/0269881118812095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinault D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry. 2008;63:730–5. doi: 10.1016/j.biopsych.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 43.Anderson PM, Pinault D, O’Brien TJ, Jones NC. Chronic administration of antipsychotics attenuates ongoing and ketamine-induced increases in cortical gamma oscillations. Int J Neuropsychopharmacol. 2014;17:1895–904. doi: 10.1017/S1461145714000959. [DOI] [PubMed] [Google Scholar]
- 44.Jones NC, Anderson P, Rind G, Sullivan C, van den Buuse M, O’Brien TJ. Effects of aberrant gamma frequency oscillations on prepulse inhibition. Int J Neuropsychopharmacol. 2014;17:1671–81. doi: 10.1017/S1461145714000492. [DOI] [PubMed] [Google Scholar]
- 45.Rivolta D, Heidegger T, Scheller B, Sauer A, Schaum M, Birkner K, et al. Ketamine dysregulates the amplitude and connectivity of high-frequency oscillations in cortical-subcortical networks in humans: evidence from resting-state magnetoencephalography-recordings. Schizophr Bull. 2015;41:1105–14. doi: 10.1093/schbul/sbv051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw AD, Saxena N, L EJ, Hall JE, Singh KD, Muthukumaraswamy SD. Ketamine amplifies induced gamma frequency oscillations in the human cerebral cortex. Eur Neuropsychopharmacol. 2015;25:1136–46. doi: 10.1016/j.euroneuro.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 47.Nugent AC, Ballard ED, Gould TD, Park LT, Moaddel R, Brutsche NE, et al. Ketamine has distinct electrophysiological and behavioral effects in depressed and healthy subjects. Mol Psychiatry. 2019;24:1040–52. doi: 10.1038/s41380-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH. Inhibition-based rhythms: experimental and mathematical observations on network dynamics. Int J Psychophysiol. 2000;38:315–36. doi: 10.1016/s0167-8760(00)00173-2. [DOI] [PubMed] [Google Scholar]
- 49.Cunningham MO, Davies CH, Buhl EH, Kopell N, Whittington MA. Gamma oscillations induced by kainate receptor activation in the entorhinal cortex in vitro. J Neurosci. 2003;23:9761–9. doi: 10.1523/JNEUROSCI.23-30-09761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muthukumaraswamy SD, Shaw AD, Jackson LE, Hall J, Moran R, Saxena N. Evidence that subanesthetic doses of ketamine cause sustained disruptions of NMDA and AMPA-mediated frontoparietal connectivity in humans. J Neurosci. 2015;35:11694–706. doi: 10.1523/JNEUROSCI.0903-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–48. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cornwell BR, Salvadore G, Furey M, Marquardt CA, Brutsche NE, Grillon C, et al. Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry. 2012;72:555–61. doi: 10.1016/j.biopsych.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nugent AC, Wills KE, Gilbert JR, Zarate CA., Jr Synaptic potentiation and rapid antidepressant response to ketamine in treatment-resistant major depression: a replication study. Psychiatry Res Neuroimaging. 2019;283:64–6. doi: 10.1016/j.pscychresns.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gould TD, Zarate CAJ, Thompson SM. Molecular pharmacology and neurobiology of rapid-acting antidepressants. Annu Rev Pharmacol Toxicol. 2019;59:213–26. doi: 10.1146/annurev-pharmtox-010617-052811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 57.Bremner JD, Krystal JH, Putnam FW, Southwick SM, Marmar C, Charney DS, et al. Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS) J Trauma Stress. 1998;11:125–36. doi: 10.1023/A:1024465317902. [DOI] [PubMed] [Google Scholar]
- 58.Kraemer HC, Wilson GT, Fairburn CG, Agras WS. Mediators and moderators of treatment effects in randomized clinical trials. Arch Gen Psychiatry. 2002;59:877–83. doi: 10.1001/archpsyc.59.10.877. [DOI] [PubMed] [Google Scholar]
- 59.Wasserstein RL, Schirm AL, Lazar NA. Moving to a world beyond “p < 0.05”. Am Stat. 2019;73(Suppl 1):1–19. [Google Scholar]
- 60.Gramfort A, Luessi M, Larson E, Engemann DA, Strohmeier D, Brodbeck C, et al. MEG and EEG data analysis with MNE-Python. Front Neurosci. 2013;7:267. doi: 10.3389/fnins.2013.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162–73. doi: 10.1006/cbmr.1996.0014. [DOI] [PubMed] [Google Scholar]
- 62.Vrba J, Robinson SE. Signal processing in magnetoencephalography. Methods. 2001;25:249–71. doi: 10.1006/meth.2001.1238. [DOI] [PubMed] [Google Scholar]
- 63.Chen G, Saad ZS, Britton JC, Pine DS, Cox RW. Linear mixed-effects modeling approach to FMRI group analysis. Neuroimage. 2013;73:176–90. doi: 10.1016/j.neuroimage.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grunebaum MF, Galfalvy HC, Choo TH, Parris MS, Burke AK, Suckow RF, et al. Ketamine metabolite pilot study in a suicidal depression trial. J Psychiatr Res. 2019;117:129–34. doi: 10.1016/j.jpsychires.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Radford KD, Park TY, Jaiswal S, Pan H, Knutsen A, Zhang M, et al. Enhanced fear memories and brain glucose metabolism (18F-FDG-PET) following sub-anesthetic intravenous ketamine infusion in Sprague-Dawley rats. Transl Psychiatry. 2018;8:263. doi: 10.1038/s41398-018-0310-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashimoto K, Yang C. Is (S)-norketamine an alternative antidepressant for esketamine? Eur Arch Psychiatry Clin Neurosci. 2019;269:867–8. doi: 10.1007/s00406-018-0922-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang C, Kobayashi S, Nakao K, Dong C, Han M, Qu Y, et al. AMPA receptor activation–independent antidepressant actions of ketamine metabolite (S)-norketamine. Biol Psychiatry. 2018;84:591–600. doi: 10.1016/j.biopsych.2018.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.