

Abstract
Long non‐coding RNAs (lncRNAs) are emerging regulators of genomic stability and human disease. However, the molecular mechanisms by which nuclear lncRNAs directly contribute to DNA damage responses remain largely unknown. Using RNA antisense purification coupled with quantitative mass spectrometry (RAP‐qMS), we found that the lncRNA BGL3 binds to PARP1 and BARD1, exhibiting unexpected roles in homologous recombination. Mechanistically, BGL3 is recruited to DNA double‐strand breaks (DSBs) by PARP1 at an early time point, which requires its interaction with the DNA‐binding domain of PARP1. BGL3 also binds the C‐terminal BRCT domain and an internal region (amino acids 127–424) of BARD1, which mediates interaction of the BRCA1/BARD1 complex with its binding partners such as HP1γ and RAD51, resulting in BRCA1/BARD1 retention at DSBs. Cells depleted for BGL3 displayed genomic instability and were sensitive to DNA‐damaging reagents. Overall, our findings underscore the biochemical versatility of RNA as a mediator molecule in the DNA damage response pathway, which affects the accumulation of BRCA1/BARD1 at DSBs.
Keywords: BRCA1/BARD1, BGL3, homologous recombination repair, LncRNA
Subject Categories: DNA Replication, Repair & Recombination; RNA Biology
A PARP1‐recruited ncRNA is involved in interactions between BRCA1, RAD51 and HP1 during homologous recombination repair of DNA double strand breaks.
Introduction
BRCA1 is a key player in homologous recombination (HR), which is frequently mutated in familial breast and ovarian cancers (Roy et al, 2011; Zhao et al, 2017). Poly‐(ADP‐ribose) polymerase (PARP) inhibitors selectively kill breast and ovarian cancers with BRCA1 mutations (Tangutoori et al, 2015; Lord & Ashworth, 2017; D'Andrea, 2018). Accumulation of BRCA1 at sites of DNA damage is required for the proper response to DSBs (Jackson & Bartek, 2009; Ciccia & Elledge, 2010). BRCA1 forms three distinct complexes through its C‐terminal BRCT repeats domain, which associates with different adaptor proteins: ABRAXAS, BACH1, and CtIP (Cantor et al, 2001; Yu & Chen, 2004; Yun & Hiom, 2009; Huen et al, 2010; Coleman & Greenberg, 2011). In addition to the C‐terminal BRCT domain, BRCA1 contains an N‐terminal RING domain that binds BARD1 to form a RING heterodimer core complex (Brzovic et al, 2001). The BRCA1/BARD1 complex is proposed to function in two distinct steps in the HR: (i) 5′–3′ resection of DSBs to generate 3′ ssDNA overhangs, and (ii) binding and loading RAD51 recombinase onto the ssDNA (Greenberg et al, 2006; Huen et al, 2010; Zhao et al, 2017). Although compelling evidence suggests that BRCA1/BARD1 functions in HR and tumorigenesis (Huen et al, 2010; Jiang & Greenberg, 2015), the regulation of BRCA1/BARD1 following DNA damage remains not fully understood. In addition to proteins, whether RNA molecules are directly involved in the BRCA1/BARD1 signaling axis is largely unknown.
Long non‐coding RNAs (lncRNAs) are RNA transcripts that do not code for protein. These are arbitrarily considered to be at least 200 nucleotides in length, based on a convenient practical cut‐off in RNA purification protocols that distinguishes lncRNAs from short non‐coding RNAs such as microRNAs (Mercer et al, 2009; Fatica & Bozzoni, 2014). The human genome contains thousands of lncRNAs (Mercer et al, 2009; Ponting et al, 2009; Kung et al, 2013), but the specific biological functions and biochemical mechanisms of most lncRNAs are still largely unknown (Mercer et al, 2009; Quinn & Chang, 2016). Small RNAs generated at DSBs are implicated in mammalian DNA repair (Wei et al, 2012; Keskin et al, 2014). Recent studies show that following formation of DSBs, bidirectional transcription events adjacent to the breaks generate small RNAs that trigger the DNA damage response by local RNA: RNA interactions (Di Micco et al, 2006; Francia et al, 2012). LncRNAs are also emerging as regulators of genome stability (Khanduja et al, 2016; Munschauer et al, 2018). LncRNAs can be broadly induced by DNA damage and regulate DNA repair processes (Zhang & Peng, 2015), but the precise mechanism of action for individual lncRNAs in the DNA damage response (DDR) is generally unknown. Previous studies reported that lncRNA BGL3 critically regulates Bcr‐Abl‐mediated cellular transformation by acting as a competitive endogenous RNA (Guo et al, 2015). However, the functions of lncRNA‐BGL3 in genome stability and cancer remain a conundrum.
In this study, we used RNA antisense purification coupled with quantitative mass spectrometry (RAP‐qMS) technology to identify lncRNA‐BGL3 binding proteins. We found that lncRNA‐BGL3 binds to PARP1 and BARD1, exhibiting unexpected roles in HR. Furthermore, BGL3 is recruited to DSBs at an early time point. We also explored the functions and underlying mechanisms of BGL3 in genome integrity.
Results
BGL3 directly binds BARD1/PARP1 in cells
To identify proteins that directly interact with BGL3 in living cells, we captured and characterized BGL3‐interacting proteins in 293T cells by using RNA antisense purification coupled with quantitative mass spectrometry (RAP‐qMS) (Engreitz et al, 2013; McHugh et al, 2015; McHugh & Guttman, 2018; Munschauer et al, 2018; Fig EV1A; Dataset EV1). Specifically, 4‐thiouridine‐labeled 293T cells were treated with 365‐nm UV light, which allowed covalently crosslinking proteins to RNA instead of other proteins (Baltz et al, 2012). LncRNA–protein complexes were purified by RNA hybrid selection with antisense oligonucleotides that target BGL3 under denaturing and reducing conditions at high temperature, to minimize the contamination of indirectly bound proteins (McHugh et al, 2015; Fig EV1A). We employed a label‐free quantitative proteomics approach to compare the resulting proteins to those captured in purifications with antisense oligonucleotides that target the well‐characterized “RNA component of mitochondrial RNA processing endoribonuclease” (RMRP) (Esakova & Krasilnikov, 2010), which is not expected to interact with the same proteins as BGL3 (Munschauer et al, 2018). The RT–PCR product sequencing analysis confirmed that the lncRNA being pulled down by the RNA antisense pull‐down experiment is indeed BGL3 (Fig EV1B and C, and sequencing files shown as Dataset EV3). In three biological replicates, 224, 234, and 214 proteins were quantified, and 217 proteins were quantifiable in at least two replicates (Fig EV1D). We also achieved good reproducibility in protein quantification in three replicates, with Pearson correlation coefficients from 0.83 to 0.86 (Fig EV1E), demonstrating our ability to perform differential analyses of lncRNA‐interacting proteins under various conditions. We quantified more than 100 BGL3‐interacting proteins in triplicate; these are involved in the DDR, DNA metabolism, cell cycle regulation, and other pathways (Figs 1A and EV1F; Dataset EV2).
Figure EV1. BGL3 directly binds BARD1 and PARP1 in living cells.
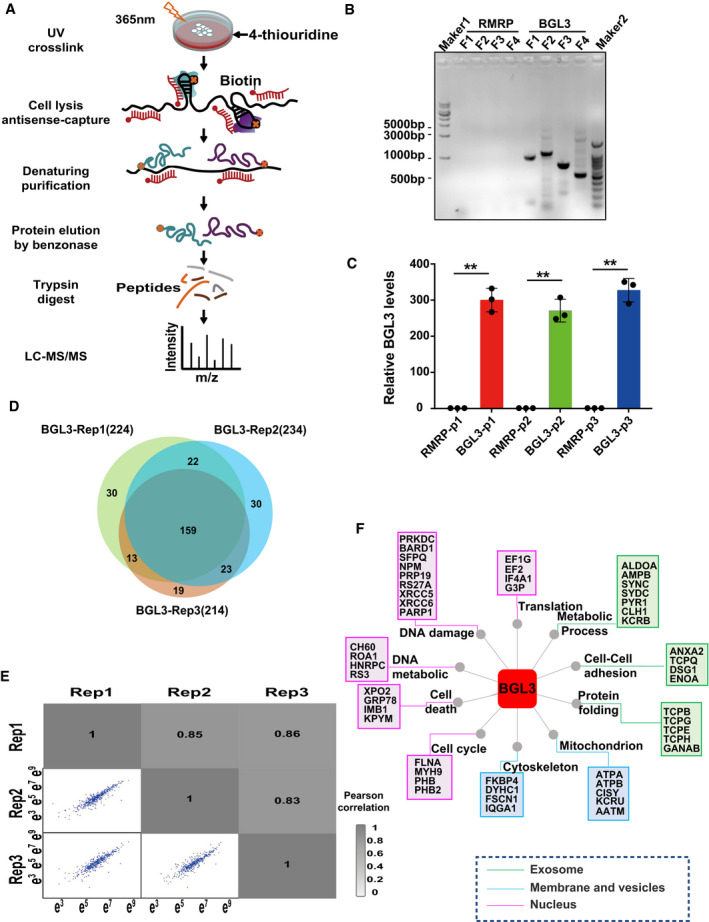
- Schematic overview of RAP‐MS.
- RT–PCR products of RNA antisense captured lncRNAs were analyzed on an agarose gel. Ladder, 1 kb (Maker1) and 100 bp (Maker2) DNA ladder. F1: BGL3 DNA fragment 1–970 bp, F2: BGL3 DNA fragment 970–2,016 bp, F3: BGL3 DNA fragment 1,905–2,650 bp, F4: BGL3 DNA fragment 3,181–3,696 bp.
- LncRNA being pulled down in the “BGL3 antisense group” (BGL3 group) and the control group (RMRP group) was examined by qRT–PCR using three different primers. P1: primer1, P2: primer2, P3: primer3. Data were presented as mean ± SD of three biological replicates and analyzed by two‐tailed Student's t‐test, **P < 0.01.
- Overlap of non‐redundant quantified proteins captured by BGL3 in three biological replicates.
- Pairwise correlations of protein intensity between replicate experiments. The Pearson correlation coefficient is indicated.
- BGL3‐interacting proteins were grouped based on cellular component and biological process pathways by GO analysis.
Source data are available online for this figure.
Figure 1. BGL3 directly binds BARD1/PARP1.
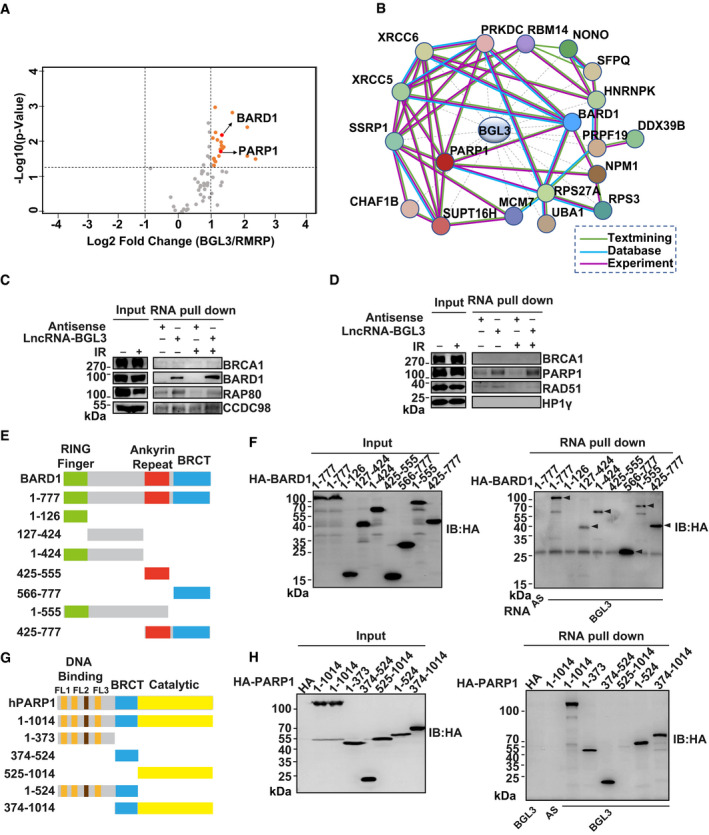
-
AProteins captured by BGL3 and RMRP antisense purifications. Proteins with an absolute log2 (fold change) > 1 and −log10 (P‐value) > 1.3 (compared with control purification) were considered as proteins enriched by BGL3 specifically.
-
BProtein–protein interaction network of the DDR‐related proteins captured by lncRNA‐BGL3.
-
C, DRNA pull‐down assay for BGL3‐BARD1/PARP1 interactions before and after irradiation. Biotinylated in vitro‐transcribed lncRNA‐BGL3 sense or antisense transcripts were incubated with HEK‐293T cell lysates with or without ionizing radiation (10 Gy) with 1‐h recovery for in vitro streptavidin RNA pull‐down assays, followed by Western blots using the indicated antibodies.
-
E–HSchematic representation of BARD1 (E)‐ or PARP1(G)‐truncated mutants used in this study. HA‐tagged full‐length or deletion mutants of BARD1 (F) or PARP1 (H) were transfected into 293T cells. Forty‐eight hours later, BGL3 RNA pull‐down assay was performed, and arrowheads indicate the bands for HA‐tagged full‐length or deletion mutants of BARD1 (F).
Source data are available online for this figure.
GO enrichment analysis showed that 19 of the BGL3‐interacting proteins are involved in the DDR pathway (Fig 1B; Dataset EV2). We focused on BARD1 and PARP1, which are the key regulators in DDR (Ray Chaudhuri & Nussenzweig, 2017; Fig 1A and B). We used RNA pull‐down assays and RNA immunoprecipitation (RIP) to validate these interactions. Both assays showed that BGL3 binds to BARD1 and PARP1 in cells and in vitro (Figs 1C–H and EV2A–F). We confirmed these interactions in a non‐denaturing pull‐down assay (Fig EV2B); BRCA1 was also pulled down with BGL3 through BARD1. BGL3‐BARD1 interactions increased upon DNA damage, but BGL3‐PARP1 interactions did not (Fig 1C). To further explore this interaction, we mapped BGL3 binding sites on BARD1 and PARP1. As shown in Figs 1E and F, and EV2C and D, both RNA pull‐down and RIP results indicated that BARD1 binds to BGL3 through its C‐terminal BRCT domain (amino acids 566–777) and an internal region (amino acids 127–424). Both the DNA‐binding domain (DBD, amino acids 1–373) and BRCT domain (amino acids 374–524) of PARP1 bind to BGL3 (Figs 1G and H, and EV2E and F). We could not identify a specific region in BGL3 responsible for PARP1/BARD1 binding (data not shown), perhaps because the full‐length BGL3 structure is essential for this binding. To test the specificity of this interaction, we examined BRCA1, HP1γ, and other proteins in the HR pathway; we observed no interactions with BGL3 (Fig 1C and D).
Figure EV2. Mapping the interaction regions between BGL3 and BARD1/PARP1.
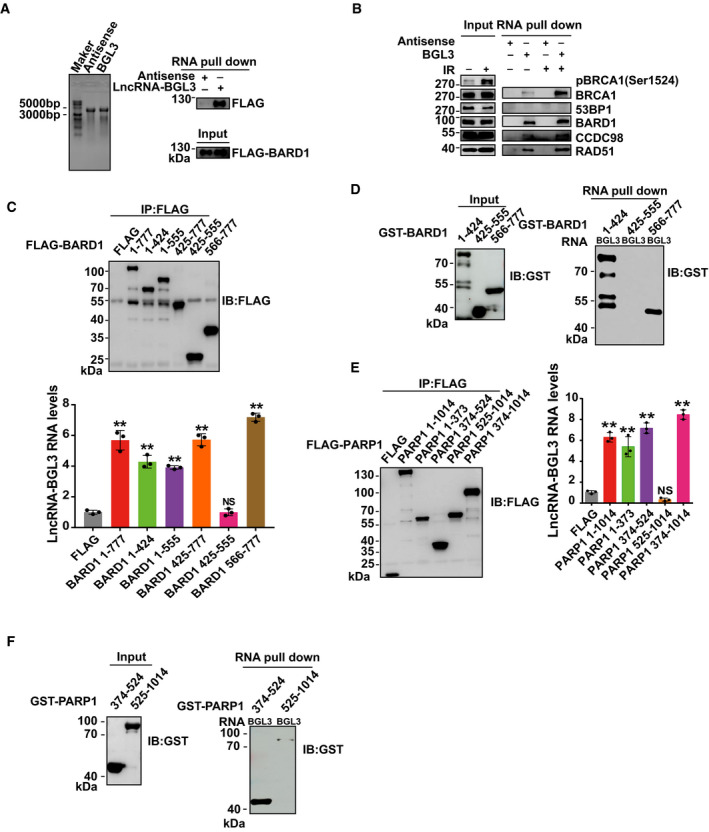
-
ABGL3 and antisense were transcribed in vitro and analyzed on an agarose gel. Ladder, 10 kb ssRNA ladder (left panel). FLAG‐BARD1 was transfected into 293T cells. Forty‐eight hours later, BGL3 RNA pull‐down assays were performed. Samples were separated by sodium dodecyl sulfate (SDS) gel and blotted with indicated antibodies (right panel).
-
BA non‐denaturing pull‐down assay for BGL3‐BARD1 interaction. 293T cells were treated with or without IR (10 Gy) and recovered for 1 h. Biotinylated in vitro‐transcribed LncRNA‐BGL3 sense or antisense transcripts were incubated with cell lysates by non‐denaturing lysis buffer, followed by Western blots using the indicated antibodies.
-
C–FFLAG‐tagged full‐length or deletion mutants of BARD1 (C) or PARP1 (E) were transfected into 293T cells. BGL3 RNA immunoprecipitation assays were performed as described in the Materials and Methods. Data reported as the average of three independent experiments. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference. GST‐ BARD1 (D) or PARP1 (F) deletion mutants were purified from Escherichia coli, and in vitro RNA pull‐down assays were performed, as described in the Materials and Methods.
Source data are available online for this figure.
BGL3 is recruited to DNA damage sites
To investigate the possible roles of BGL3 in DDR, we performed fluorescence in situ hybridization (FISH) assays to examine the subcellular localization of BGL3 upon exposure to DNA damage. Endogenous lncRNA‐BGL3 accumulated at DNA damage sites generated by laser micro‐irradiation (Fig 2A), suggesting that lncRNA‐BGL3 functions in proximity to DNA lesions. BGL3 co‐localized with endogenous or overexpressed BARD1 in cells following DNA damage (Fig EV3A). The kinetics of BGL3 recruitment showed that BGL3 is recruited to DSBs at a very early time point, with a peak at 5 min (Fig 2A). To rule out possible off‐target effects of the FISH probe, we depleted BGL3 using siRNA; BGL3 accumulation disappeared, demonstrating the specificity of our assay (Fig EV3B). We also found that DNA damage induced BGL3 translocation from the cytoplasm to the nucleus and promoted its binding to chromatin (Fig EV3C).
Figure 2. BGL3 is recruited to DNA damage sites and regulates genome stability.
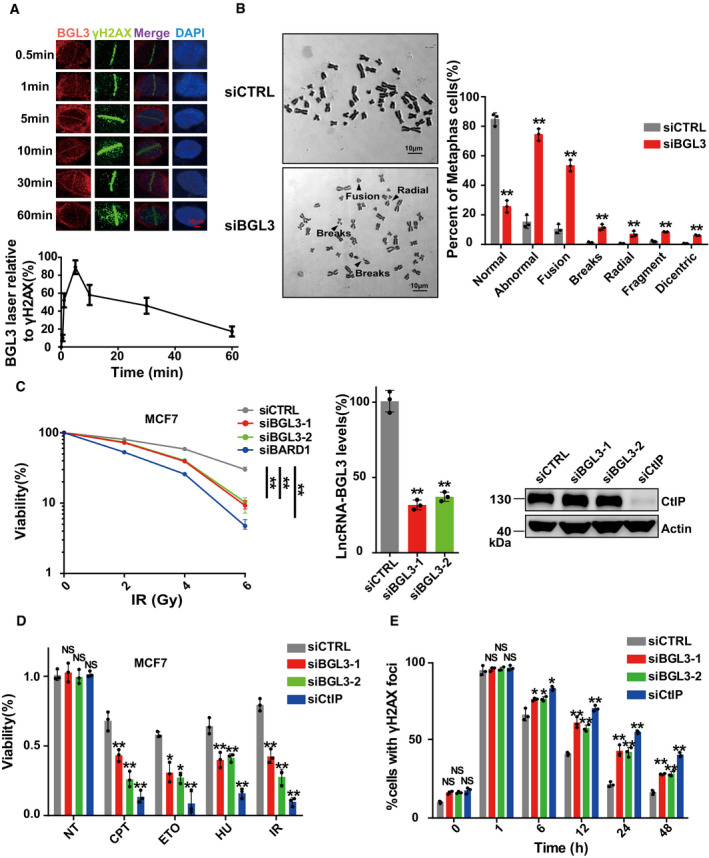
- BGL3 is recruited to DNA damage sites. U2OS cells were subjected to laser micro‐irradiation to generate DSBs in a line pattern. The relocation kinetics of BGL3 to DSBs was monitored by RNA fluorescence in situ hybridization (FISH) in a time course as indicated. BGL3 intensities at the laser line were normalized into a numerical value using Nikon NIS‐Elements AR software (version 4.40.00). Data are presented as mean ± SD of four biological replicates.
- Chromosome aberrations induced by BGL3 depletion. Wild‐type or BGL3 depleted BJ‐5ta cells were used in the metaphase spread analysis for spontaneous DNA breaks. Representative image (left panel) and the percentage of cells containing at least one DNA break (right panel). Data are presented as mean ± SD of three biological replicates. Two‐tailed Student's t‐test, **P < 0.01.
- MCF‐7 cells were treated as indicated, and cell response to ionizing radiation was analyzed by colony formation assays (left panel). Data are presented as mean ± SEM of four independent experiments and analyzed by two‐way analysis of variance (ANOVA), **P < 0.01. Knockdown efficiency of BGL3 siRNA was examined by qRT–PCR and normalized to β‐actin (middle panel); data are presented as mean ± SD of three biological replicates. Two‐tailed Student's t‐test, **P < 0.01. Knockdown efficiency of CtIP siRNA was examined by blotting (right panel).
- MCF‐7 cells were transfected with the indicated siRNAs. Cell sensitivity to camptothecin (CPT), hydroxyurea (HU), etoposide (ETO), or ionizing radiation was determined by MTS assays. Data are presented as mean ± SD of three biological replicates. Two‐tailed Student's t‐test, *P < 0.05, **P < 0.01, NS: no significant difference.
- BGL3 deficiency inhibits DNA damage repair. Quantification of γ‐H2AX foci at indicated times after irradiation (2 Gy) is presented. Data shown are results of three independent experiments (100 cells for each experiment), presented as mean ± SD, two‐tailed Student's t‐test, *P < 0.05, **P < 0.01, NS: no significant difference.
Source data are available online for this figure.
Figure EV3. BGL3 regulates DNA damage response.
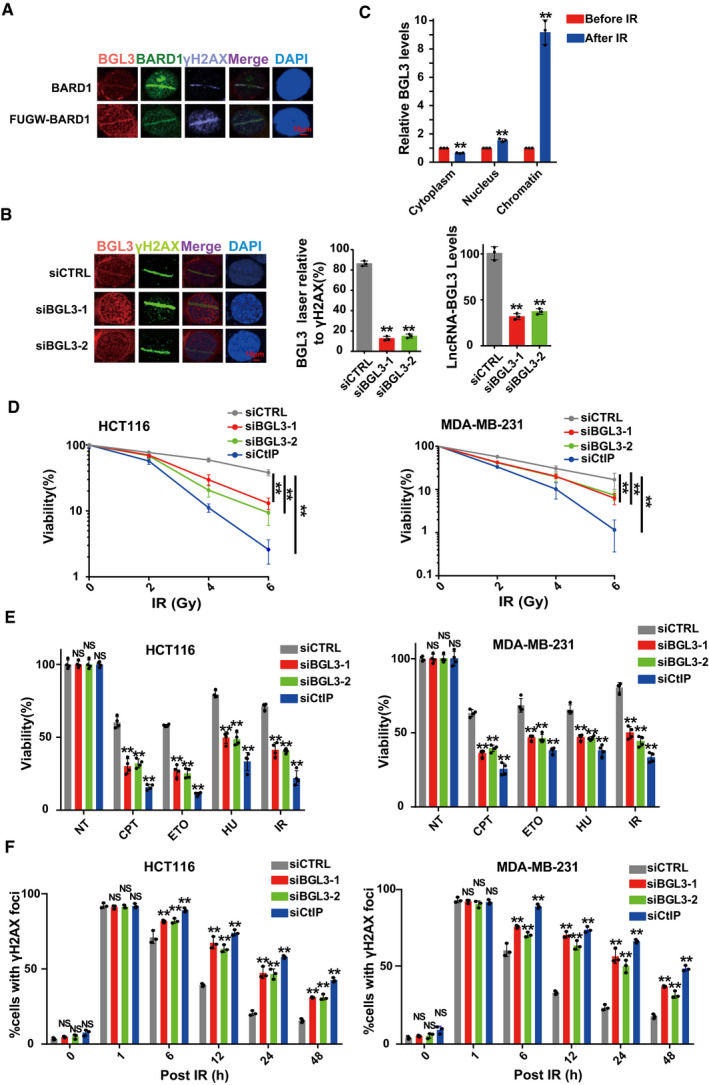
- U2OS cells transfected with or without FUGW‐GFP‐BARD1 were subjected to laser micro‐irradiation to generate DSBs, and 10 min later, BGL3 and BARD1 recruitment was examined as described in the Materials and Methods.
- U2OS cells were transfected with the indicated siRNAs, and BGL3 (RNA FISH) and γ H2A.X (IF) recruitment to DNA damage sites was examined. Data shown are the average of three independent experiments (middle panel), and 100 cells were counted for each experiment, two‐tailed Student's t‐test, **P < 0.01. Knockdown efficiency of BGL3 siRNA was examined by qRT–PCR (right panel). Data are presented as mean ± SD of three independent experiments, two‐tailed Student's t‐test, **P < 0.01.
- RT–qPCRs to examine the BGL3 RNA levels of cytosolic, nuclear, and chromatin fractions before and following 6 Gy IR in 293T cells. Data are presented as mean ± SD of three biological replicates, two‐tailed Student's t‐test, **P < 0.01.
- HCT116 cells (left panel) and MDA‐MB‐231 cells (right panel) were treated as described in the Materials and Methods section, and cell response to ionizing radiation (IR) was analyzed by colony formation assays. Data are presented as mean ± SEM of four independent experiments and analyzed by two‐way analysis of variance (ANOVA), **P < 0.01.
- HCT116 cells (left panel) and MDA‐MB‐231 cells (right panel) were transfected with the indicated siRNAs. Cell sensitivity to camptothecin (CPT), hydroxyurea (HU), etoposide (ETO), or IR was determined by MTS assays. Data were presented as mean ± SD of three biological replicates. Two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
- BGL3 deficiency inhibits DNA damage repair. HCT116 cells (left panel) and MDA‐MB‐231 cells (right panel) were transfected with the indicated siRNA. Quantification of γ‐H2AX foci at indicated times after irradiation (2 Gy) is presented. Data shown are results of three independent experiments (100 cells for each experiment), presented as mean ± SD, and analyzed by two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
BGL3 regulates genome stability
To further monitor the effects of BGL3 deficiency on genome stability, we prepared metaphase chromosome spreads from wild‐type and BGL3‐depleted cells. Untreated BGL3‐depleted cells showed a nearly threefold increase of spontaneous single chromatid breaks compared to wild‐type cells (Fig 2B), suggesting an intrinsic defect in genome stability in BGL3‐deficient cells. In support of this result, downregulation of BGL3 resulted in cellular hypersensitivity to ionizing radiation and DNA‐damaging reagents (Figs 2C and D, and EV3D and E), with decreased DSB repair efficiency as indicated by delayed γH2AX disappearance (Figs 2E and EV3F). Consistent with these results, the Trypan Blue dye exclusion test showed that BGL3 knockdown promoted cell death following DNA damage (Fig EV4D).
Figure EV4. BGL3 is required for HR .
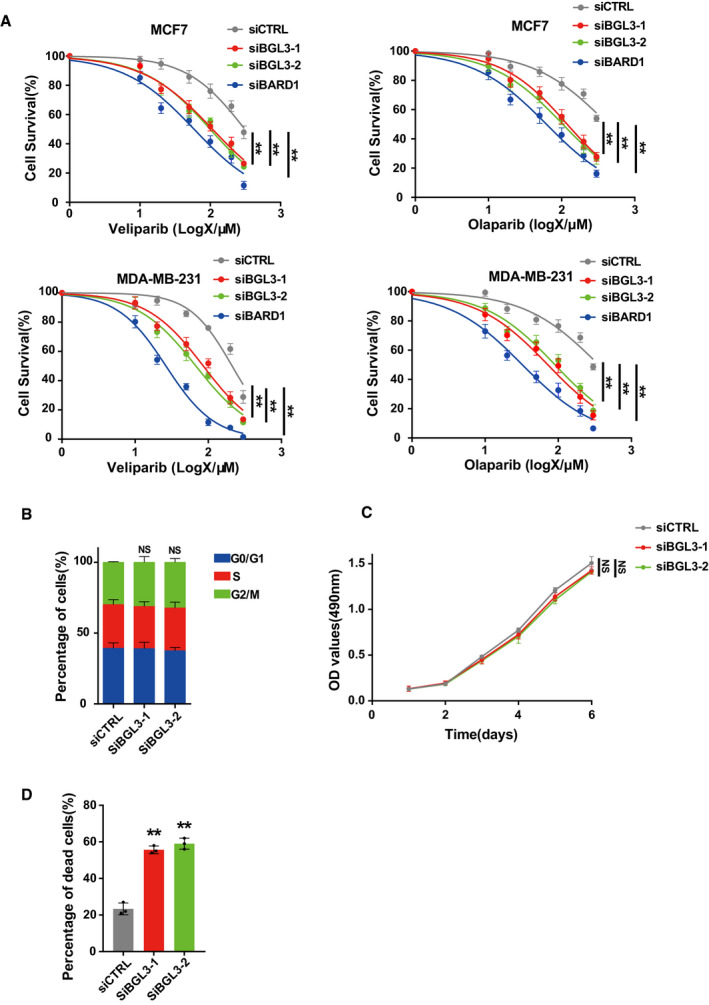
- MCF7 cells (upper panel) and MDA‐MB‐231 cells (lower panel) were transfected with the indicated siRNA, and 48 h later, cell responses to the PARP inhibitors veliparib (left panel) or olaparib (right panel) were measured by MTS assay. Data are presented as mean ± SEM of three independent experiments and analyzed by two‐way analysis of variance (ANOVA), **P < 0.01.
- Cell cycle profile of wild‐type or BGL3‐depleted U2OS cells. Data shown are averages of three independent experiments, presented as mean ± SD, NS: no significant difference.
- Effects of BGL3 expression on cell growth. OD values of MCF7 cells expressing either control siRNA or different BGL3‐specific siRNAs were monitored for 6 days. Data are presented as mean ± SEM of four independent experiments and analyzed by two‐way analysis of variance (ANOVA), NS: no significant difference.
- MCF7 cells were transfected with the indicated siRNA. Quantification of dead cells at 12 h after irradiation (6 Gy) is presented. Data shown are results from three independent experiments, presented as mean ± SD, and analyzed by two‐tailed Student's t‐test, **P < 0.01
BGL3 is required for HR and BRCA1/BARD1 accumulation at DSBs
Next, we examined whether BGL3 regulates DNA repair using well‐established reporter assays for HR and NHEJ. As shown in Fig 3A, BGL3 depletion decreased HR frequency similar to the effect achieved by depleting the key HR factor CtIP (Sartori et al, 2007; Yun & Hiom, 2009). Conversely, we did not observe a significant change in NHEJ frequency in BGL3 knockdown cells (Fig 3B). BGL3 depletion also rendered cells hypersensitive to PARP inhibitors (veliparib and olaparib) (Fig EV4A), also supporting BGL3's role in HR. Importantly, knockdown of BGL3 had no significant effect on cell cycle profile or cell growth without DNA damage, indicating that the effects of BGL3 knockdown on HR were not caused by cell cycle change or cell proliferation deficiency (Fig EV4B and C).
Figure 3. BGL3 regulates HR and BRCA1/BARD1 accumulation at DSBs.
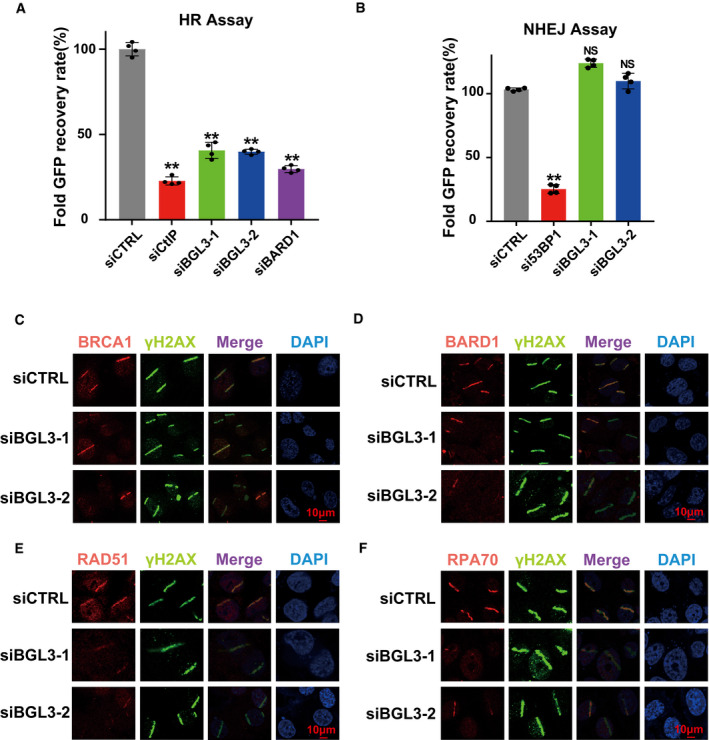
-
A, BU2OS cells integrated with HR or NHEJ reporter were transfected with the indicated siRNAs and subjected to the HR (A) or NHEJ assay (B) as described in Materials and Methods. Data presented as mean ± SD of four biological replicates and positive cell percentages as compared with the control group. Two‐tailed Student's t‐test, **P < 0.01. NS: no significant difference.
-
C–FWild‐type (CTRL) or BGL3 knockdown U2OS cells were subjected to micro‐irradiation; 1 h later, cells were fixed and processed for immunostaining with indicated antibodies. Representative images of BRCA1 (C), BARD1 (D), RAD51 (E), and RPA70 (F) accumulation at sites of laser‐induced DNA damage are shown.
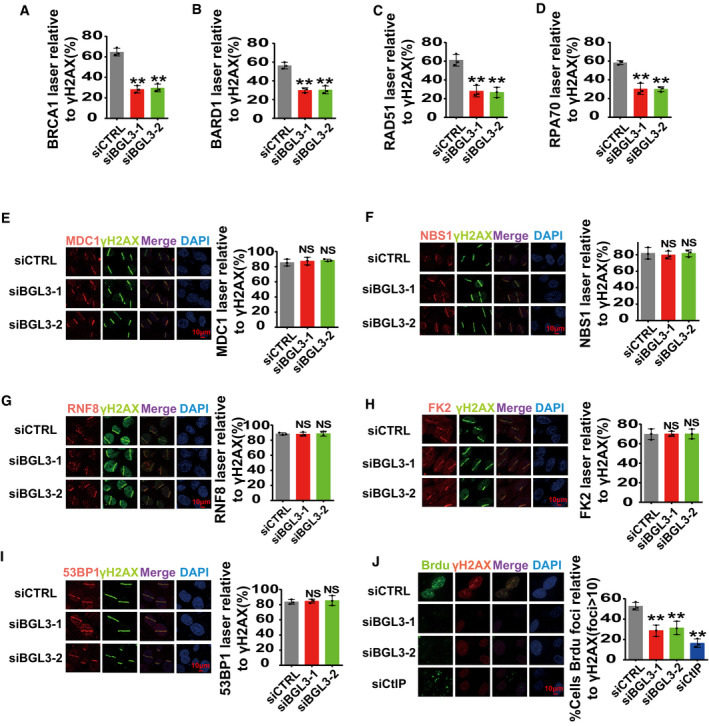
To ascertain the detailed functions of BGL3 in HR, we examined the accumulation of several DDR factors at DNA lesions induced by ultraviolet laser micro‐irradiation. BGL3 deficiency resulted in compromised accumulation of BRCA1, BARD1 (Fig 3C and D; quantification shown in Fig EV5A and B), and downstream factors, such as CtIP, RAD51, and RPA70 (Figs 3E and F, and 4A; quantification for RAD51 and RPA70 shown in Fig EV5C and D). However, BGL3 deficiency did not affect accumulation of upstream regulators of BARD1, such as γH2AX, MDC1, NBS1, RNF8, and ubiquitination signals (FK2) (Fig EV5E–H). BGL3 depletion also had no effect on 53BP1 and Ku80 recruitment to DNA damage sites (Fig EV5I and data not shown). Based on these results, we hypothesized that lncRNA‐BGL3 regulated HR through its effect on DNA end resection. Using an anti‐BrdU antibody staining technique that only detects DNA in single‐stranded form (Broderick et al, 2016), we consistently found that camptothecin triggered substantial ssDNA formation in control cells but not in BGL3‐depleted cells (Fig EV5J).
Figure EV5. BGL3 regulates BRCA1/BARD1 retention at DSBs.
-
A–DQuantification of Fig 3C–F. Data shown are the average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
-
E–JParental or BGL3‐deficient U2OS cells were subjected to laser micro‐irradiation to generate DSBs in a line pattern. DDR factor recruitment was examined 1 h later. Data shown are the average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
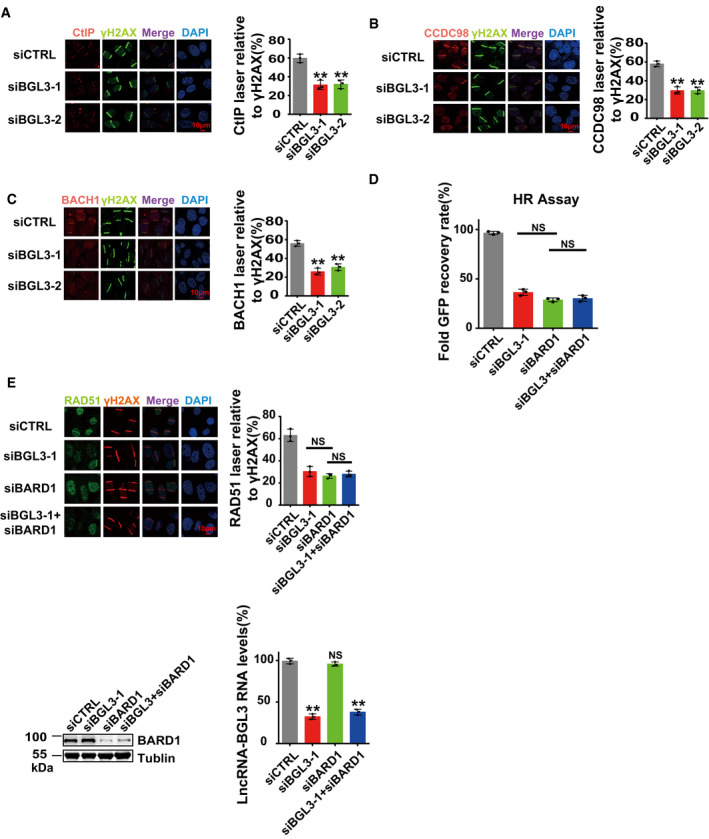
Figure 4. BGL3 and BARD1 function together to regulate HR.
-
A–CParental or BGL3‐deficient U2OS cells were subjected to laser micro‐irradiation to generate DSBs in a line pattern. DDR factor recruitment was examined 2 h later. Data shown are the average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, **P < 0.01.
-
DU2OS cells were transfected with the indicated siRNAs, and 48 h later, HR efficiency was determined using the reporter assay, as described in the Materials and Methods. Data shown are the average of three independent experiments. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, NS: no significant difference.
-
EU2OS cells were transfected with the indicated siRNA, and RAD51 accumulation at sites of laser‐induced DNA damage was examined. Data shown are the average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, NS: no significant difference. Knockdown efficiency of BARD1 siRNA was examined by blotting (bottom left panel), and knockdown efficiency of BGL3 siRNA was examined by qRT–PCR and normalized to β‐actin (bottom right panel), data are presented as mean ± SD of three biological replicates. Two‐tailed Student's t‐test, **P < 0.01.
Source data are available online for this figure.
BRCA1 forms three distinct complexes through its C‐terminal BRCT repeats, associating with three different adaptor proteins: ABRAXAS, BACH1, and CtIP (Cantor et al, 2001; Wang et al, 2007; Huen et al, 2010; Coleman & Greenberg, 2011; Wang, 2012). Depletion of BGL3 also affected CCDC98, BACH1, and CtIP recruitment to DNA damage sites (Fig 4A–C). In BARD1 recruitment kinetics experiments, BGL3 mainly regulated BARD1 retention at DSBs (Fig EV6A). Knockdown of BGL3 had no effect on total levels of BARD1, BRCA1, and other DDR proteins (Fig EV6B), and BGL3 levels also did not change after DNA damage (Fig EV6C). Given that both BGL3 and BARD1 regulated HR and that BGL3 affected BARD1 recruitment, it is proposed that BGL3 functions through BARD1. To test this hypothesis, we knocked down both BGL3 and BARD1, and then examined HR efficiency. As shown in Fig 4D and E, double knockdown of BGL3 and BARD1 showed similar HR efficiency and RAD51 recruitment compared to single knockdown of each. These results indicate that BGL3 and BARD1 function in the same pathway.
Figure EV6. Both BGL3 and BARD1 function in HR .
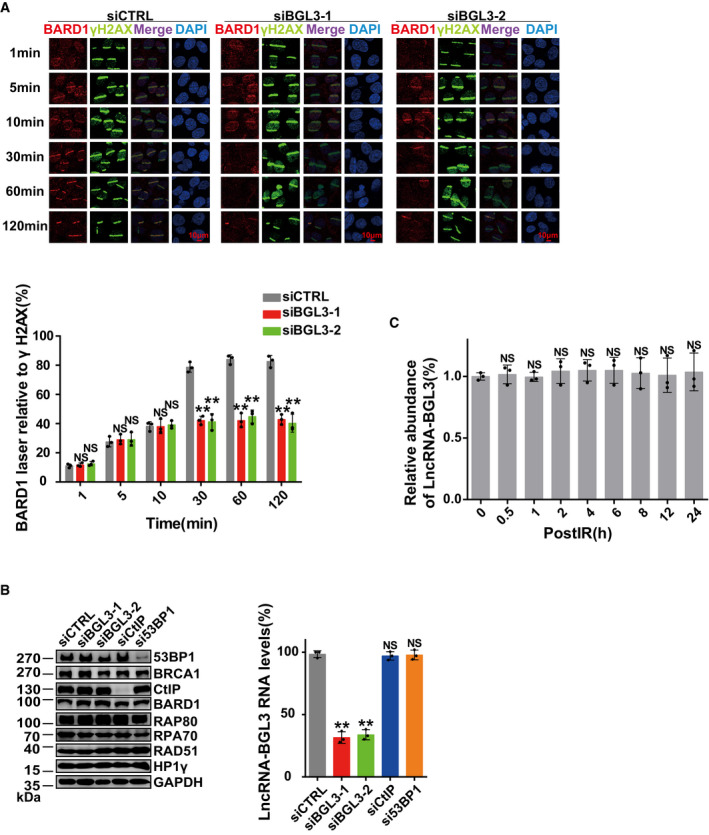
- U2OS cells were transfected with BGL3 or control siRNA, and BARD1 accumulation at sites of laser‐induced DNA damage was examined at the indicated time points. Data shown are the average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
- 293T cells were transfected with the indicated siRNAs, and cells were lysed. Samples were blotted with indicated antibodies (left panel). Knockdown efficiency of BGL3 siRNA was examined by qRT–PCR (right panel). Data shown are the average of three independent experiments, presented as mean ± SD, and analyzed by two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
- BGL3 expression levels were examined by qRT–PCR at the indicated time points after irradiation (2 Gy). Data shown are the average of three independent experiments, presented as mean ± SD, and analyzed by two‐tailed Student's t‐test, NS: no significant difference.
Source data are available online for this figure.
BGL3 enhances molecular interactions between BARD1 and its partners
Our results suggest that BGL3 interacts with and affects BARD1 function but not protein levels, perhaps serving as a scaffold to enhance molecular interactions between BARD1 and other DNA damage proteins. In support of this notion, knockdown of BGL3 markedly decreased BARD1‐HP1 interaction, which is important for BARD1 retention at DSBs (Lee et al, 2013; Wu et al, 2015; Peng et al, 2019) (Fig 5A). In response to DNA damage, BARD1 interacts with Lys9‐dimethylated histone H3 (H3K9me2) (Fukuda et al, 2016). This interaction is mediated by HP1 through direct binding of the chromo shadow domain of HP1 to a conserved PxVxL motif in the BRCT domain of BARD1 (Bannister et al, 2001; Lachner et al, 2001; Wu et al, 2015; Fukuda et al, 2016). BARD1‐HP1 interaction regulates BRCA1/BARD1 retention at DSBs. BGL3 also affected binding of RAP80 (Kim et al, 2007; Huen et al, 2010; Hu et al, 2011; Wang, 2012; Wu et al, 2012; Jiang & Greenberg, 2015), a subunit of the BRCA1 A complex, to K63‐linked ubiquitin (Fig 5B), which mediates BRCA1 complex recruitment (Sobhian et al, 2007). However, BRCA1‐BARD1 heterodimer formation was not affected (Fig 5A). The precise mechanism or mechanisms by which BGL3 promotes RAP80 binding to ubiquitin remain to be elucidated, but might include inducing a conformational change in the BRCA1 A complex.
Figure 5. BGL3 enhances molecular interactions between BARD1 and its partners.
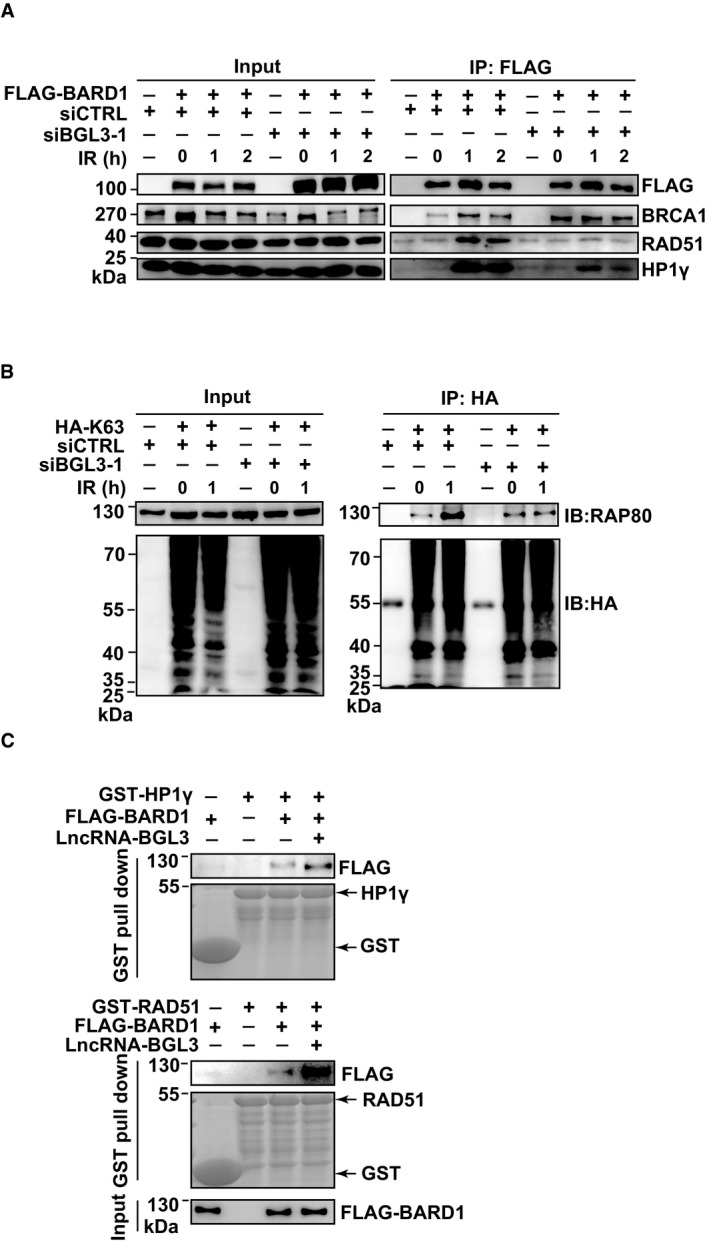
- BGL3 promotes BARD1‐HP1γ interactions. 293T cells expressing the indicated siRNAs were irradiated (10 Gy) and BARD1–HP1γ interactions examined at the indicated time points.
- BGL3 promotes RAP80 binding to K63 ubiquitin chain in cells. 293T cells expressing the indicated siRNAs and plasmids were irradiated (10 Gy). Cell lysates were immunoprecipitated with HA beads and subjected to immunoblot with the indicated antibodies.
- BGL3 promotes BARD1–HP1γ (up panel) and BARD1‐RAD51 (middle panel) interactions in vitro. GST‐HP1γ, GST‐RAD51, and FLAG‐BARD1 were overexpressed and purified from cells as indicated in Materials and Methods. FLAG‐BARD1 was divided into two equal parts, one part was used for BARD1‐HP1γ interaction analysis (up panel), and another part was used for BARD1‐RAD51 interaction analysis (middle panel). BARD1‐HP1γ (up panel) and BARD1‐RAD51 (middle panel) interactions with or without BGL3 were, respectively, detected by GST pull‐down assays; here, GST was used as a pull‐down control.
Source data are available online for this figure.
Previous studies also showed that both BRCA1 and BARD1 interact with RAD51 and that BRCA1‐BARD1 enhances recombinase activity of RAD51 (Scully et al, 1997; Zhao et al, 2017). This process is critical for BRCA1/BARD1 functions in HR. Therefore, we determined whether BGL3 affected BARD1‐RAD51 interaction. As shown in Fig 5A, depletion of BGL3 significantly affected BARD1 binding to RAD51. To directly assess these mechanisms, we purified HP1γ, RAD51, BARD1, and BGL3, and then examined BARD1‐HP1 and BARD1‐RAD51 interactions with or without BGL3, respectively. As shown in Fig 5C, in vitro biochemical experiments show that BGL3 promotes BARD1 binding to HP1γ and RAD51. Thus, BGL3 binds BARD1, serving as a scaffold to enhance the molecular interaction between BARD1 and HP1/RAD51.
BGL3 is recruited to DNA damage sites at an early time point by PARP1
Following DNA damage, BGL3 is recruited to DNA damage sites (Fig 2A). Although recruitment of certain repair complexes depends on specific upstream signaling kinases, e.g., ATM (ataxia telangiectasia‐mutated) and ATR (ataxia telangiectasia and Rad3‐related) (Falck et al, 2005; Mu et al, 2007; Blackford & Jackson, 2017), depletion of these two kinases had no detectable impact on BGL3 recruitment (Fig 6A). Caffeine treatment showed the similar results (Fig 6A), implying that ATM or ATR is not the upstream regulator of BGL3. Among critical early regulators of the DDR (Li & Yu, 2013; Ray Chaudhuri & Nussenzweig, 2017), only PARP was required for BGL3 accumulation after ultraviolet laser micro‐irradiation (Figs 6A and B, and EV7A and B). PARP inhibitor treatment or PARP1 knockdown diminished BGL3 accumulation at DNA damage sites (Figs 6A and B, and EV7A and B). PARP1 affected BGL3 recruitment only at an early time point and had no effect thereafter (Figs 6C, and EV7A and B).
Figure 6. BGL3 is recruited to DNA damage sites at an early time point by PARP1.
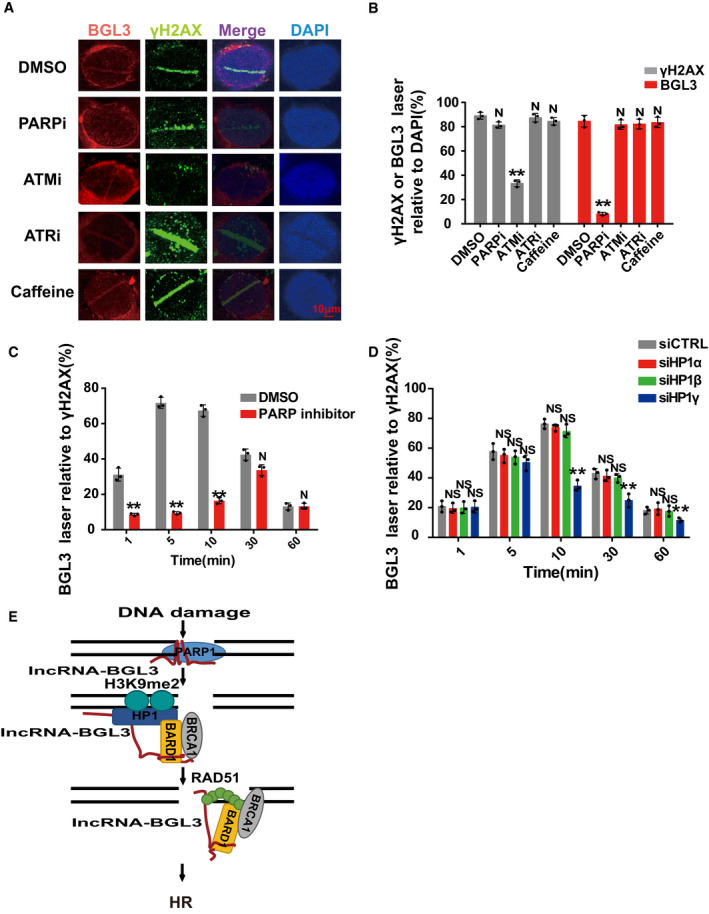
- PARP1 inhibitor treatment abolished BGL3 recruitment to DSBs. U2OS cells were treated with DMSO, PARP inhibitor (olaparib), ATM inhibitor (KU‐55933), ATR inhibitor (VE‐821), or caffeine. BGL3 recruitment to DNA damage sites was assessed by RNA FISH assays.
- Quantification of the positive cells. For each group, 100 randomly selected cells were counted. Data are presented as mean ± SD of three biological replicates. Two‐tailed Student's t‐test, **P < 0.01, N: no significant difference.
- PARP inhibitor mainly affected BGL3 recruitment at an early time point. U2OS cells were treated with DMSO or olaparib, and BGL3 recruitment was examined following laser micro‐irradiation at indicated time point. Data presented are the average of three independent experiments, and 100 cells were counted for each experiment. Two‐tailed Student's t‐test, **P < 0.01, N: no significant difference.
- U2OS cells were transfected with the indicated siRNAs, and BGL3 recruitment to DSBs was monitored at the indicated time point by RNA FISH. Data presented are the average of three independent experiments, and 100 cells were counted for each experiment. Two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
- A model demonstrating how the BGL3 lncRNA mediates BRCA1/BARD1 complex retention at DNA damage sites.
Figure EV7. PARP1 and HP1γ affect BGL3 recruitment to DNA damage sites.
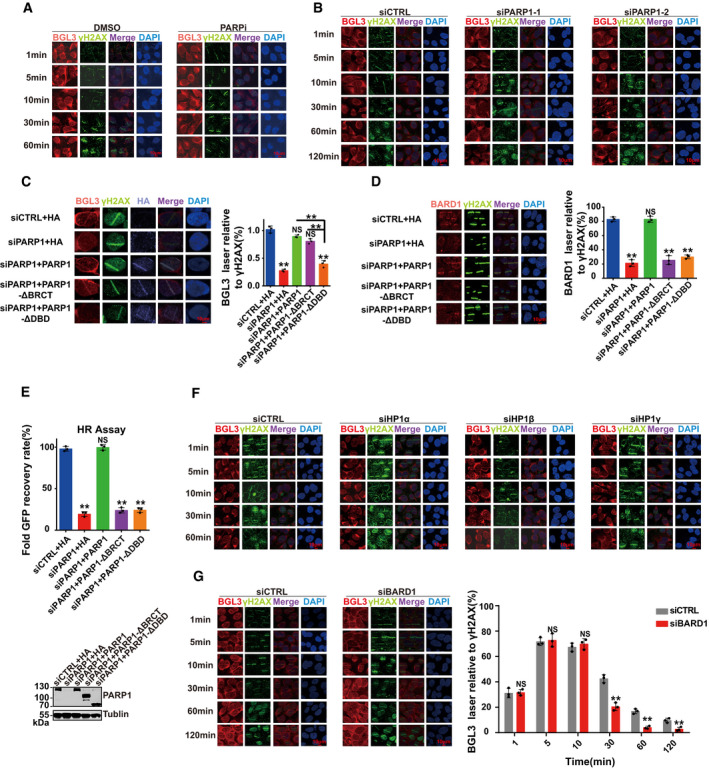
- Representative micrographs of Fig 6C.
- U2OS cells were transfected with indicated siRNAs, and BGL3 recruitment at the indicated time points was monitored.
- U2OS cells were transfected with the indicated siRNAs or plasmids; then, BGL3 (RNA FISH) and γ H2A.X (IF) were assessed following laser micro‐irradiation and a 10‐min recovery. Data shown are the average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD, **P < 0.01, NS: no significant difference.
- U2OS cells were transfected with the indicated siRNAs or plasmids; then, BARD1 and γ‐H2AX accumulation at sites of laser‐induced DNA damage were examined. Data shown are the average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD, **P < 0.01, NS: no significant difference.
- U2OS cells were transfected with the indicated siRNAs or plasmids, and the HR efficiency was determined using the reporter assay. Data are presented as mean ± SD of three biological replicates, **P < 0.01, NS: no significant difference.
- Representative micrographs of Fig 6D.
- U2OS cells were transfected with BARD1 or control siRNA; then, BGL3 (RNA FISH) and γ H2A.X (IF) accumulation at sites of laser‐induced DNA damage was examined at the indicated time point. Quantification was average of three independent experiments, and 100 cells were counted for each experiment. Data are presented as mean ± SD and analyzed by two‐tailed Student's t‐test, **P < 0.01, NS: no significant difference.
Source data are available online for this figure.
We reason that PARP1 interacts with BGL3 and acts as a recruiter of BGL3 at an early time point. Both the DBD (amino acids 1–373) and BRCT domain (amino acids 374–524) of PARP1 bind to BGL3 (Figs 1G and H, and EV2E and F). Thus, we transfected U2OS cells with PARP1 siRNA and reconstituted these cells with siRNA‐resistant WT PARP1 or the BGL3 binding‐deficient mutant. As shown in Fig EV7C, BGL3 recruitment to DSBs was rescued in both WT PARP1 and BRCT domain‐deficient cells, but not in cells expressing the DBD deletion mutant. We concluded that the DBD of PARP1 interacts with and recruits BGL3 to DNA damage sites. Although the BRCT domain of PARP1 also binds BGL3, this domain is not essential for BGL3 recruitment to DNA damage sites at an early time point (Fig EV7C). The DBD‐BGL3 interaction is also important for BARD1 recruitment and HR efficiency (Fig EV7D and E), further supporting our notions.
We next analyzed BGL3 retention at DSBs at a late time point. BGL3 interacts with and works together with BARD1, and BARD1 retention at DSBs requires BARD1 BRCT domain–HP1γ interactions at a late time point (Lee et al, 2013; Wu et al, 2015). So we investigated whether HP1 affects BGL3 retention at DNA damage sites at a late time point. As shown in Figs 6D and EV7F, HP1γ, neither HP1α nor HP1β, affected BGL3 retention at a late time point. But BGL3 did not directly bind HP1γ (Fig 1D). Perhaps BGL3 forms a complex with BARD1, promoting BARD1‐HP1 interaction, and BGL3 retention at DSBs depends on BARD1‐HP1 interaction. In support of this concept, depletion of BARD1 affected BGL3 retention at a late time point but had no effect at an early time point (Fig EV7G).
Discussion
Most studies about DNA damage response have focused on proteins involved in these signaling pathways. However, whether RNA molecules other than proteins are directly involved in DNA repair machinery is still largely unknown. Here, for the first time, we report that BGL3 lncRNA is involved in HR (Fig 6E). Specifically, BGL3 serves as an RNA mediator in HR, regulates the retention of BRCA1/BARD1 complex at DNA lesions, and controls DNA end resection.
BRCA1 binds BARD1 to form a heterodimer in cells, which interacts with different proteins to execute their functions in HR (Huen et al, 2010). For example, BRCA1 binds ABRAXAS, BACH1, or CtIP to form three different complexes (Huen et al, 2010). Previous studies reported that BARD1 binds HP1γ to promote BRCA1/BARD1 retention at DSBs (Wu et al, 2015; Peng et al, 2019). Previous studies also showed that both BRCA1 and BARD1 interact with RAD51 and that BARD1/BRCA1 enhances RAD51 recombinase activity (Zhao et al, 2017). This effect is important for BARD1/BRCA1 functions in HR. But how these interactions are regulated upon DNA damage is still largely unknown. Here, we show that BGL3 lncRNA is an important scaffold molecule to promote BARD1 binding to its target proteins. Perhaps DNA damage‐induced BGL3‐BARD1 interaction promotes BRCA1/BARD1 conformational change and enhances its affinity to its partners. But how this occurs still needs to be further studied.
BARD1 is a multi‐domain protein that binds to BGL3 through its C‐terminal BRCT domain (amino acids 566–777) and an internal region (amino acids 127–424). The BARD1 C‐terminal BRCT domain, ankyrin repeat region, and N‐terminal RING domain bind different proteins (Brzovic et al, 2001; Irminger‐Finger et al, 2016), and all these interactions are essential for BARD1 functions in HR (Irminger‐Finger et al, 2016). Here, we also confirm that the internal region (amino acids 127–424) of BARD1 binds BGL3, which is essential for DNA end resection and HR. Perhaps four different regions of BARD1 bind different proteins and/or RNA, which coordinate together to execute HR. Furthermore, a canonical role for the BRCT domain of DNA damage elements is to bind phospho‐protein and/or poly‐(ADP‐ribose) (PAR) (Manke et al, 2003; Yu et al, 2003; Li & Yu, 2013). Thus, lncRNA‐BGL3 joins a growing number of modules known to recognize the BRCT domain of BARD1. The structural basis for this recognition still needs to be investigated.
Many lncRNAs are responsive to DNA damage signaling, and their expression levels are up‐ or downregulated upon DNA damage (Liu & Lu, 2012). For example, GUARDIN is a p53‐responsive lncRNA triggered by genotoxic stress (Hu et al, 2018). However, BGL3 levels did not change after DNA damage (Fig EV6C). On the other hand, knockdown of BGL3 had no effect on the total levels of BARD1, BRCA1, and other DDR proteins (Fig EV6B). BGL3 re‐localized to DSBs at an early time point by direct interactions with the DBD of PARP1. Our FISH assays showed that BGL3 is recruited to DNA damage sites by PARP1, and the interaction between the DBD and BGL3 is essential for BGL3 recruitment (Fig EV7C). Previous studies implied that the DBD of PARP1 binds both DNA and RNA (Ali et al, 2012; Melikishvili et al, 2017), and our findings support this notion. We also found that PARP inhibitor treatment diminished BGL3 recruitment (Fig 6A). PARP inhibitors block the enzymatic activity of PARP and trap PARP1 on the damaged DNA. Perhaps PARP1‐mediated another protein or itself poly(ADP‐ribosyl)ation (PARylation) at DNA damage sites affects BGL3 recruitment. PARP1 trapping by PARP inhibitors also may induce chromatin conformation change, which also affects BGL3 recruitment, but the underlying mechanism is still unclear.
In summary, our study has identified lncRNA‐BGL3 as a new regulator of the HR pathway, which impacts genome instability, a major hallmark of cancer. BGL3 levels are negatively correlated with overall survival in patients with breast cancer (http://bioinformatics.mdanderson.org/main/TANRIC: Overview) (Li et al, 2015). Perhaps overexpression of BGL3 enhances DNA repair capability, promoting tumor progression and drug resistance, but this still needs to be further investigated. As with BGL3, RAD51 is essential for HR repair, but overexpression of RAD51 occurs in several cancer types, such as breast and pancreatic cancers, and is associated with enhanced tumor progression and drug resistance (Henning & Sturzbecher, 2003; Klein, 2008). It will be interesting to explore new ways to sensitize cancer cells to radiation and chemotherapy based on our results. For example, small molecules that block the interaction of BGL3 with BARD1/PARP1 could be useful in developing new cancer treatments.
Materials and Methods
Cell lines
MCF7 cells were cultured in Eagle's minimum essential medium (EMEM) (ATCC 30‐2003). U2OS cells, HEK‐293T cells, MDA‐MB‐231 cells, and BJ‐5ta cells were cultured in Dulbecco's modified Eagle medium (DMEM; high glucose, GIBCO 11‐965‐118) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C with 5% CO2. HCT116 cells were grown in McCoy's 5A medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. All cells were monitored by mycoplasma PCR testing and maintained in mycoplasma‐free conditions.
Antibodies and plasmids
Antibodies used in this study were as follows: BRCA1 (D‐9, Santa Cruz, WB 1:500, IF 1:50) and (C‐20, Santa Cruz, WB 1:500, IF 1:50), HA (H9658, Sigma, WB 1:2,000), FLAG (F3165, Sigma, WB 1:2,000), γH2AX (05‐636, Millipore, WB 1:5,000, IF 1:1,000) and (A300‐081A, Bethyl Laboratories, WB 1:1,000, IF 1:200), CtIP (61141, Active Motif, WB 1:1,000, IF 1:200), MDC1 (ab11171, Abcam, WB 1:1,000, IF 1:200), RIF1 (A300569A, Bethyl Laboratories, WB 1:1,000, IF 1:200), RAD51 (GTX70230, GeneTex, WB 1:1,000, IF 1:100), 53BP1 (NB100‐304, Novus, WB 1:1,000, IF 1:100), BARD1 (A300‐263A, Bethyl Laboratories, WB 1:1,000, IF 1:200), RAP80 (A300‐764A, Bethyl Laboratories, WB 1:1,000, IF 1:200), HP1γ (MABE656, Millipore, WB 1:1,000, IF 1:200), RPA 70 (#2267, Cell Signaling Technology, WB 1:1,000, IF 1:100), RNF8 (14112‐1‐AP, Proteintech, WB 1:1,000, IF 1:200), CCDC98 (ab139191, Abcam, WB 1:1,000, IF 1:200), BACH1 (ab49657, Abcam, WB 1:1,000, IF 1:200), ATM (sc‐23921, Santa Cruz, WB 1:1,000, IF 1:500), and BrdU (347580, BD, IF 1:50).
Full‐length BARD1 was cloned into the S‐SBP‐FLAG‐tagged vector (pIRES2‐EGFP), HA‐tagged vector (pCMV‐HA), or GFP‐tagged vector (pFUGW). PARP1 and BRCA1 full‐length were cloned into the S‐SBP‐FLAG‐tagged vector (pIRES2‐EGFP) or HA‐tagged vector (pCMV‐HA). BGL3 full‐length and truncated mutants were cloned into the pLVX‐Puro and pcDNA3.1 vector. RAD51 and HP1γ full‐length were cloned into the GST‐tagged vector (pGEX‐4T‐2). BARD1‐ and PARP1‐truncated mutants were cloned into the GST‐tagged vector (pGEX‐4T‐2). A K63‐only ubiquitin was cloned into a HA‐tagged vector (pCMV‐HA).
RNAi target sequences
For siRNA transfection, cells were transfected twice at 24‐h intervals with the indicated siRNA using Lipofectamine® RNAiMAX (Invitrogen) according to the manufacturer's instructions. The target sequences of siRNAs against human BGL3 were as follows: GGACUACAGACCAUGUUUAdTdT (siBGL3‐1) and GUGCCCGUGUUUAUAUCAUdTdT (siBGL3‐2). The other siRNA sequences were as follows: PARP1: GUUGCUGAUGGGUAGUACCdTdT (siPARP1‐1) and GCGCUUCUGCACCAACUCAdTdT (siPARP1‐2), BARD1: AGCUGAAUAUUAUACCAGAdTdT, MDC1: UCCAGUGAAUCCUUGAGGUdTdT, RAP80: AGAAGGAAGUAGCUAUUUCdTdT, Hp1α: CCUGAGAAAAACUUGGAUUdTdT, Hp1β: CCCGACCUCAUUGCUGAGUdTdT, Hp1γ: AGACAGCAGUGGAGAAUUGdTdT, CtIP: GCUAAAACAGGAACGAAUCdTdT, 53BP1: GAGCUGGGAAGUAUAAAUUdTdT, H2AFX: ACAAGAAGACGCGAAUCAUdTdT, ATM: GCUAUUUACGGAGCUGAUUdTdT.
Laser micro‐irradiation and imaging of cells
Cells were cultured in glass‐bottom dishes and pre‐treated with 2 μM Hoechst 33342 Solution (Thermo Scientific, catalogue number 62249) for 15 min. UVA laser (50 mW) irradiation was conducted using an inverted microscope (A1R, Nikon) with a laser microdissection workstation. Then, cells were recovered at 37°C for the indicated times, washed once with cold PBS, and then fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were then washed three times with cold PBS and permeabilized with 1% Triton X‐100 for 7 min. After permeabilization, cells were washed extensively with immunofluorescence blocking buffer (PBS containing 1% goat serum, 0.1% NaN3) and then incubated with the indicated antibodies at 4°C overnight. Staining was conducted with fluorescent secondary antibodies. Images were captured on a Nikon Eclipse TiE microscope.
In vitro transcription and RNA pull‐down assays
The DNA template used for in vitro synthetization of biotinylated lncRNA‐BGL3 was generated by plasmid linearization. The linearized plasmid was purified using the DNA Gel Extraction Kit (Omega Bio‐Tek), and in vitro transcription was performed using the T7 High Yield RNA Transcription Kit (Vazyme) according to the manufacturer's instructions.
Cell lysates were prepared by sonication in RIP buffer (150 mM KCl, 25 mM Tris (pH 7.4), 0.5 mM dithiothreitol, 0.5% NP‐40) or non‐denaturing lysis buffer (20 mM Tris–HCl pH 8, 137 mM NaCl, 1% Nonidet P‐40 (NP‐40), 2 mM EDTA), with complete protease inhibitor cocktail and RNase inhibitors and pre‐cleared against streptavidin magnetic beads (both, Invitrogen). In vitro‐transcribed RNA adsorbed to streptavidin magnetic beads was then incubated with cell lysate at 4°C for 2 h before washing five times in RIP buffer and elution in the loading sample buffer. Eluted proteins were separated on SDS–PAGE columns for Western blotting.
RAP‐MS
To capture endogenous BGL3, 5′ biotinylated 90‐mer DNA oligonucleotides antisense to the target RNA sequence were synthesized. Twenty probes were designed to cover the full‐length BGL3. For BGL3 antisense purifications, 200 million HEK‐293T cells were treated with 200 μM 4‐thiouridine for 8 h. Cells were washed once with ice‐cold PBS and then crosslinked on ice using 0.8 J/cm2 of 365‐nm ultraviolet light in a Stratalinker. Cells were scraped from the culture dishes, washed once with PBS, and pelleted by centrifugation at 450 g for 5 min at 4°C. The samples were flash‐frozen in liquid nitrogen for storage at −80°C. Preparation of total cell lysates and antisense purification of crosslinked RNA–protein complexes were performed as previously described (McHugh et al, 2015; Munschauer et al, 2018). Twenty micrograms of mixed antisense probes was used for 200 million lysed cells. For pre‐clearance of lysates and capture of RNA/DNA hybrids, 1.2 ml streptavidin Dynabeads MyOne C1 magnetic beads (Thermo Fisher Scientific) were used for 200 million cells. Captured proteins were eluted from streptavidin beads by digestion with nucleic acids using 125 U of benzonase (Millipore) for 2 h at 37°C. The captured proteins were analyzed by Western blots and LC‐MS/MS analysis (Dataset EV1). A sample of beads was removed between 0.5 and 1% of the total volume and transfer to a PCR strip tube as the RNA elution sample before eluting of captured proteins. RNA was further extracted from beads collected by TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed using the cDNA synthesis kit (TIANGEN), and abundance of BGL3 was detected by qPCR using SYBR Green Real‐Time PCR Master Mix (TOYOBO) according to the manufacturer's instructions. Because captured RNA was usually of sufficient quantity to be detected by RT–qPCR analysis but perhaps not enough to detect on a standard agarose gel, cDNA was amplified by RT–PCR and confirmed by sequencing.
Fluorescent in situ hybridization with immunofluorescence staining
RNA fluorescence in situ hybridization (FISH) was performed with a FISH kit (Ribobio Co.) according to the manufacturer's instructions with minor modifications. Briefly, cells grown in glass‐bottom dishes were treated with 2 μM Hoechst 33342 Solution (Thermo Scientific, catalogue number 62249) for 15 min. UVA laser (50 mW) irradiation was conducted using an inverted microscope (A1R, Nikon) with a laser microdissection workstation. After irradiation, cells were incubated at 37°C for the indicated times, washed three times with cold PBS, and fixed with PBS containing 4% paraformaldehyde at room temperature for 15 min. Next, cells were permeabilized in 1% Triton X‐100 at room temperature for 7 min and washed three times with cold PBS. The fixed cells were further treated with Pre‐hybridization Buffer at 42°C for 30 min. The pre‐hybridized cells were subjected to incubation with a 1 μM FISH probe (Ribobio Co. cy3 labeling probe mix) in hybridization buffer. The hybridization was performed at 42°C overnight and dishes washed with Hybridization Wash Buffer I (4 × SSC, 0.1% Tween‐20) three times at 47°C for 5 min, and then was washed with Hybridization Wash Buffer II (2 × SSC) at 47°C for 5 min and Hybridization Wash Buffer III (1 × SSC) at 47°C for 5 min. Lastly, dishes were washed with PBS at room temperature for 5 min and incubated with the indicated antibodies at 37°C for 1 h. Secondary antibodies were labeled with Alexa Fluor 488. Samples were visualized by a Nikon ECLIPSE E800 fluorescence microscope.
Cell lysis, immunoprecipitation, and immunoblotting
Cells were homogenized in RIPA buffer supplemented with Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail, Panobinostat, and Methylstat on ice for 30 min, and then sonicated for 15 s. Lysates were cleared by centrifugation at 13,000 g for 10 min at 4°C. Supernatants were collected for immunoblotting or immunoprecipitation.
For immunoprecipitation, cell lysates or cell fraction lysates were incubated with 10 μl protein G or protein A agarose beads coupled with antibody against the indicated proteins at 4°C for 8 h. Beads were washed three times with RIPA buffer and analyzed by Western blots.
RNA fractionation assays
RNA fractionation assays were performed as described previously (Diamant et al, 2016). Briefly, MCF7 cells were treated as indicated and harvested after washing twice with PBS, pelleted by centrifugation at 450 g for 5 min at 4°C, resuspended in 200 μl cytosol lysis buffer (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1.5 mM MgCl2, 0.5% NP‐40), gently vortexed for 10 s, and centrifuged for 2 min at 850 g. The supernatant was transferred to a new tube, and cytosolic RNA was extracted with four volumes of TRIzol reagent. For isolation of nucleosol (nuclear soluble fraction) and chromatin‐associated RNA, nuclear pellets were resuspended in a glycerol buffer (20 mM Tris, pH7, 75 mM NaCl, 0.5 mM EDTA, 50% glycerol, 0.85 mM DTT, 0.5 mM PMSF) by gentle finger flicking. One volume of nuclear lysis buffer was then added (10 mM HEPES, pH 7.6, 300 mM NaCl, 0.2 mM EDTA, 7.5 mM MgCl2, 1 M urea, 1% NP‐40, 1 mM DTT). Nuclei were immediately vortexed for 2 s twice, incubated on ice for 2 min, and centrifuged for 10 min at 12,000 rpm. The nucleosol supernatant was transferred to a new tube and harvested with four volumes of TRIzol reagent. Chromatin pellets were washed once in PBS with 1 mM EDTA, and RNA was extracted with TRIzol reagent. RNA was treated with DNase using an RNA‐free TURBO DNase Kit (Ambion). Reverse transcription was performed using the cDNA synthesis kit (TIANGEN), and abundance of BGL3 was detected by qPCR using SYBR Green Real‐Time PCR Master Mix (TOYOBO) according to the manufacturer's instructions.
RNA immunoprecipitation (RIP) and RNA extraction
Anti‐FLAG M2 Affinity beads (Sigma) were incubated in 5 ml 1× RIPA buffer supplemented with Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail, and RNase inhibitor at 4°C for 1 h. Then, the beads were washed twice with RIPA buffer and kept on ice. We prepared the pre‐cleared cell lysates as described above in the Cell Lysis, Immunoprecipitation, and Immunoblotting section. Pre‐cleared lysates were transferred to tubes containing anti‐FLAG M2 Affinity beads (Sigma) and then rotated at 4°C for 3–5 h. The protein‐captured beads were washed with RIPA buffer for three times. RNA was further extracted from beads by TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed using the cDNA synthesis kit (TIANGEN), and abundance of BGL3 was detected by qPCR using SYBR Green Real‐Time PCR Master Mix (TOYOBO) according to the manufacturer's instructions.
GST pull‐down assays
For BARD1 or PARP1 GST pull‐down assays, GST fusion proteins were purified from BL21 Escherichia coli cells and immobilized onto glutathione Sepharose 4B columns at 4°C overnight. HEK‐293T cells transfected with indicated constructs were treated as indicated. Then, cells were lysed in the NETN buffer with the protease inhibitor and incubated with the Sepharose immobilized with indicated proteins and RNAs at 4°C for 8 h. Sepharose was then washed three times with the NETN buffer and boiled in 2 × SDS loading buffer. Samples were subjected to immunoblot with indicated antibodies.
HR and NHEJ reporter assays
Reporter assays were performed as described previously (Zhao et al, 2017). U2OS cells integrated with DR‐GFP or NHEJ‐GFP cassettes were used for HR or NHEJ analyses. Cells transfected with indicated plasmids or siRNA were then transfected with I‐Scel expression plasmid pCBA‐Scel. Forty‐eight hours after transfection, percentages of GFP‐positive cells were analyzed by flow cytometry. HR or NHEJ efficiency is presented as the percentage of control cells. HR or NHEJ frequencies presented in figures are the mean ± SD of three independent experiments.
Colony formation assays
Colony formation assays were performed as we have described previously (Pei et al, 2011).
Flow cytometry and cell cycle analysis
Cells were trypsinized and washed with PBS twice and centrifuged (800 rpm) at 4°C. Cells were fixed in 500 μl ice‐cold 70% ethanol overnight at 4°C. Fixed cells were centrifuged at 500 rpm at 4°C for 10 min and suspended in 500 μl PBS containing 50 μg/ml propidium iodide (Sigma) and 10 μg/ml RNase for 30 min at 4°C. FACS analysis was performed with a FACS Calibur instrument (BD Biosciences). ModFit (version 2.0) software was used to analyze cell cycles (G1, G2, and S phases).
BrdU assays
Detection of BrdU incorporation in the native conditions was performed. Cells were incubated with BrdU (10 μM) for 24 h and then treated with or without CPT (1 μM) for 1 h before fixation. Immunostaining was performed with anti‐BrdU and γ‐H2AX antibody without DNA denaturing. Slides were mounted in medium containing DAPI (Sigma) and analyzed under a fluorescence microscope.
Sensitivity to DNA‐damaging agents
BGL3‐silenced and BGL3‐nontargeting siRNA‐transfected HCT116 cells were plated onto 96‐well plates and treated with etoposide, camptothecin, hydroxyurea, or ionizing radiation as indicated. Two days later, viability of the cells was determined using the CellTiter‐Blue reagent (Promega) and the average of four experiments was plotted. Data are presented as mean ± SD of three independent experiments.
Mass spectrometry analysis
Sample preparation
Captured proteins were reduced with 15 mM dithiothreitol at 56°C for 30 min, followed by alkylation with 45 mM iodoacetamide in the dark at room temperature for 20 min. The proteins were resolved on SDS–PAGE gels, the gels were stained with Coomassie Blue, and destained with 20% ethanol/10% acetic acid. The gel lanes were cut into cubes (about 1 × 1 × 1 mm), and gel cubes were washed with 50 mM ammonium bicarbonate twice. Gel cubes were subsequently subjected to in‐gel digestion with 15 ng/μl sequencing‐grade trypsin (Promega) at 37°C for 12 h. Peptides were sequentially extracted by 50% ACN/0.1% TFA and 80% ACN/0.1% TFA at 37°C for 30 min. The peptides were desalted using C18 Zip Tips (Millipore) and evaporated on a vacuum centrifuge to dryness.
LC‐MS/MS analysis
Peptides were analyzed using Orbitrap Fusion Tribrid (Thermo Scientific) coupled to an EASY‐nano‐LC 1000 system (Thermo Scientific). Lyophilized peptides were re‐dissolved in solvent A (solvent A, 0.1% FA in water; solvent B, 95% ACN/0.1% FA) and automatically loaded onto a customized trap column (100 μm × 2 cm; particle size, 3 μm; pore size, 120 Å; SunChrom, USA) at a flow rate of 10 μl/min. The sample was subsequently separated with a homemade analytical column at a flow rate of 600 nl/min (150 μm × 12 cm; particle size, 1.9 μm; pore size, 120 Å; SunChrom, USA). The mobile phase changed linearly from 5 to 7% of solvent B over 2 min, from 7 to 10% of solvent B over 8 min, from 10 to 20% of solvent B over 40 min, from 20 to 30% of solvent B over 20 min, from 30 to 90% of solvent B over 1 min, and maintained at 90% solvent B for 7 min. The electrospray voltage of 2.3 kV versus the inlet of the mass spectrometer was used. Survey full scans were acquired from 300 to 1,400 m/z at a resolution of 120,000, and the maximum injection time was set to 100 ms or an automatic gain control target of 5e5. The 20 most intense precursors were selected for fragmentation per cycle with dynamic exclusion time of 18 s. Fragment mass spectra were acquired in centroid mode with a normalized collision energy of 32%, and ion trap scan rate was rapid. The activation type was higher energy collision dissociation, and the maximum injection time was set to 35 ms or an AGC target of 5e3.
MS data analysis
The raw data files were searched with MaxQuant (v 1.5.2.8). Spectra were searched against the human proteome obtained from the UniProtKB dataset (downloaded December 10, 2017). The following parameters were used for searching. Fixed modification: carbamidomethyl (C, +57.022 Da); variable modifications: oxidation (M, +15.995 Da); enzyme: trypsin; missed cleavages: 2; precursor mass tolerance: 6 ppm; fragment mass tolerance: 0.5 Da; false discovery rate < 1%. Intensity‐based absolute quantification was performed on the identified peptides to quantify protein abundance. Protein groups exported from MaxQuant were filtered to remove “Potential contaminant” or “Reverse” decoy database entries, and entries with “Only identified by site”. Proteins detected with at least two unique peptides were used for further analysis (Dataset EV1). Protein–protein interaction networks were identified using STRING (Search Tool for the Retrieval of Interacting Genes/Proteins).
Analysis of metaphase chromosomes
BJ‐5ta cells were seeded to obtain approximately 70% confluence. Cells were treated with 20 ng/ml colcemid (Invitrogen) for 2 h, and then harvested and incubated in hypotonic solution (0.06 M KCl) at 37°C for 30 min. Next, cells were fixed in 10 ml of Carnoy's fixative and were spread onto pre‐cold glass slides and dried. Slides were stained with Giemsa, and 100 metaphase spreads were calculated for each aberration. Data are reported as mean ± SD of three independent experiments.
Data analysis
The statistical results were derived from three biological replicates. No samples were excluded from the analysis. Statistical analysis was performed by Student's t‐test for two groups and by analysis of variance for multiple groups. P < 0.05 was considered significant.
Author contributions
ZH, SM, TZ, CP, LC, and YP performed the experiments and analyzed the data. CJ, YY, QS, and WZ contributed to the critical reagents and the experimental design. HP, CJ, XZL, and XPL designed the study, analyzed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
H. Pei was supported by a Research Scholar Grant, RSG‐19‐032‐01‐DMC from the American Cancer Society. We thank Dr. Jeremy Stark at City of Hope, Duarte, CA, USA, for providing the U2OS DR‐GFP and U2OS EJ5‐GFP cell lines.
EMBO Journal (2020) 39: e104133
Contributor Information
Chenxi Jia, Email: cjia@mail.ncpsb.org.
Huadong Pei, Email: huadongpei@gwu.edu.
Data availability
The mass spectrometry data from this publication have been deposited to the PRIDE database http://www.ebi.ac.uk/pride/archive/projects/PXD018186 and assigned the identifier PXD018186.
References
- Ali AAE, Timinszky G, Arribas‐Bosacoma R, Kozlowski M, Hassa PO, Hassler M, Ladurner AG, Pearl LH, Oliver AW (2012) The zinc‐finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat Struct Mol Biol 19: 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold‐Brown D, Drew K, Milek M et al (2012) The mRNA‐bound proteome and its global occupancy profile on protein‐coding transcripts. Mol Cell 46: 674–690 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell 66: 801–817 [DOI] [PubMed] [Google Scholar]
- Broderick R, Nieminuszczy J, Baddock HT, Deshpande R, Gileadi O, Paull TT, McHugh PJ, Niedzwiedz W (2016) EXD2 promotes homologous recombination by facilitating DNA end resection. Nat Cell Biol 18: 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE (2001) Structure of a BRCA1‐BARD1 heterodimeric RING‐RING complex. Nat Struct Biol 8: 833–837 [DOI] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA et al (2001) BACH1, a novel helicase‐like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105: 149–160 [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA (2011) The BRCA1‐RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem 286: 13669–13680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea AD (2018) Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 71: 172–176 [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A et al (2006) Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature 444: 638–642 [DOI] [PubMed] [Google Scholar]
- Diamant G, Eisenbaum T, Leshkowitz D, Dikstein R (2016) Analysis of subcellular RNA fractions revealed a transcription‐independent effect of tumor necrosis factor alpha on splicing, mediated by Spt5. Mol Cell Biol 36: 1342–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya‐Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES et al (2013) The Xist lncRNA exploits three‐dimensional genome architecture to spread across the X chromosome. Science 341: 1237973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O, Krasilnikov AS (2010) Of proteins and RNA: the RNase P/MRP family. RNA 16: 1725–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA‐PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I (2014) Long non‐coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15: 7–21 [DOI] [PubMed] [Google Scholar]
- Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d'Adda di Fagagna F (2012) Site‐specific DICER and DROSHA RNA products control the DNA‐damage response. Nature 488: 231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Tsuruga T, Kuroda T, Takeuchi J, Wu W, Ohta T (2016) The BARD1/HP1 interaction: another clue to heterochromatin involvement in homologous recombination. Mol Cell Oncol 3: e1030535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM (2006) Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1‐containing complexes. Genes Dev 20: 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Kang Q, Zhu X, Chen Q, Wang X, Chen Y, Ouyang J, Zhang L, Tan H, Chen R et al (2015) A long noncoding RNA critically regulates Bcr‐Abl‐mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene 34: 1768–1779 [DOI] [PubMed] [Google Scholar]
- Henning W, Sturzbecher HW (2003) Homologous recombination and cell cycle checkpoints: Rad51 in tumour progression and therapy resistance. Toxicology 193: 91–109 [DOI] [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM (2011) RAP80‐directed tuning of BRCA1 homologous recombination function at ionizing radiation‐induced nuclear foci. Genes Dev 25: 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WL, Jin L, Xu A, Wang YF, Thorne RF, Zhang XD, Wu M (2018) GUARDIN is a p53‐responsive long non‐coding RNA that is essential for genomic stability. Nat Cell Biol 20: 492–502 [DOI] [PubMed] [Google Scholar]
- Huen MS, Sy SM, Chen J (2010) BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol 11: 138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irminger‐Finger I, Ratajska M, Pilyugin M (2016) New concepts on BARD1: regulator of BRCA pathways and beyond. Int J Biochem Cell Biol 72: 1–17 [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Greenberg RA (2015) Deciphering the BRCA1 tumor suppressor network. J Biol Chem 290: 17724–17732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F (2014) Transcript‐RNA‐templated DNA recombination and repair. Nature 515: 436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanduja JS, Calvo IA, Joh RI, Hill IT, Motamedi M (2016) Nuclear noncoding RNAs and genome stability. Mol Cell 63: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X (2007) Ubiquitin‐binding protein RAP80 mediates BRCA1‐dependent DNA damage response. Science 316: 1202–1205 [DOI] [PubMed] [Google Scholar]
- Klein HL (2008) The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 7: 686–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JT, Colognori D, Lee JT (2013) Long noncoding RNAs: past, present, and future. Genetics 193: 651–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120 [DOI] [PubMed] [Google Scholar]
- Lee YH, Kuo CY, Stark JM, Shih HM, Ann DK (2013) HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Res 41: 5784–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Yu X (2013) Function of BRCA1 in the DNA damage response is mediated by ADP‐ribosylation. Cancer Cell 23: 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, Weinstein JN, Liang H (2015) TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Can Res 75: 3728–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu X (2012) Non‐coding RNAs in DNA damage response. Am J Cancer Re 2: 658–675 [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A (2017) PARP inhibitors: synthetic lethality in the clinic. Science 355: 1152–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, Yaffe MB (2003) BRCT repeats as phosphopeptide‐binding modules involved in protein targeting. Science 302: 636–639 [DOI] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya‐Jones A, Blanco M, Burghard C, Moradian A et al (2015) The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA, Guttman M (2018) RAP‐MS: a method to identify proteins that interact directly with a specific RNA molecule in cells. Methods Mol Biol 1649: 473–488 [DOI] [PubMed] [Google Scholar]
- Melikishvili M, Chariker JH, Rouchka EC, Fondufe‐Mittendorf YN (2017) Transcriptome‐wide identification of the RNA‐binding landscape of the chromatin‐associated protein PARP1 reveals functions in RNA biogenesis. Cell Discov 3: 17043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS (2009) Long non‐coding RNAs: insights into functions. Nat Rev Genet 10: 155–159 [DOI] [PubMed] [Google Scholar]
- Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, Besusso D, Jung SY, Qin J (2007) A proteomic analysis of ataxia telangiectasia‐mutated (ATM)/ATM‐Rad3‐related (ATR) substrates identifies the ubiquitin‐proteasome system as a regulator for DNA damage checkpoints. J Biol Chem 282: 17330–17334 [DOI] [PubMed] [Google Scholar]
- Munschauer M, Nguyen CT, Sirokman K, Hartigan CR, Hogstrom L, Engreitz JM, Ulirsch JC, Fulco CP, Subramanian V, Chen J et al (2018) The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 561: 132–136 [DOI] [PubMed] [Google Scholar]
- Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z (2011) MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470: 124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Liao Q, Tan W, Peng C, Hu Z, Chen Y, Li Z, Li J, Zhen B, Zhu W et al (2019) The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat Commun 10: 1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136: 629–641 [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Chang HY (2016) Unique features of long non‐coding RNA biogenesis and function. Nat Rev Genet 17: 47–62 [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Nussenzweig A (2017) The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18: 610–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN (2011) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 12: 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450: 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM (1997) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265–275 [DOI] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA (2007) RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316: 1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangutoori S, Baldwin P, Sridhar S (2015) PARP inhibitors: a new era of targeted therapy. Maturitas 81: 5–9 [DOI] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ (2007) Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316: 1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B (2012) BRCA1 tumor suppressor network: focusing on its tail. Cell Biosci 2: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y (2012) A role for small RNAs in DNA double‐strand break repair. Cell 149: 101–112 [DOI] [PubMed] [Google Scholar]
- Wu J, Liu C, Chen J, Yu X (2012) RAP80 protein is important for genomic stability and is required for stabilizing BRCA1‐A complex at DNA damage sites in vivo . J Biol Chem 287: 22919–22926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Nishikawa H, Fukuda T, Vittal V, Asano M, Miyoshi Y, Klevit RE, Ohta T (2015) Interaction of BARD1 and HP1 Is required for BRCA1 retention at sites of DNA damage. Can Res 75: 1311–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, Chen J (2003) The BRCT domain is a phospho‐protein binding domain. Science 302: 639–642 [DOI] [PubMed] [Google Scholar]
- Yu X, Chen J (2004) DNA damage‐induced cell cycle checkpoint control requires CtIP, a phosphorylation‐dependent binding partner of BRCA1 C‐terminal domains. Mol Cell Biol 24: 9478–9486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun MH, Hiom K (2009) CtIP‐BRCA1 modulates the choice of DNA double‐strand‐break repair pathway throughout the cell cycle. Nature 459: 460–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Peng G (2015) Non‐coding RNAs: an emerging player in DNA damage response. Mutat Res Rev Mutat Res 763: 202–211 [DOI] [PubMed] [Google Scholar]
- Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, Jian Ma C, Kwon Y, Rao T, Wang W, Sheng C et al (2017) BRCA1‐BARD1 promotes RAD51‐mediated homologous DNA pairing. Nature 550: 360–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The mass spectrometry data from this publication have been deposited to the PRIDE database http://www.ebi.ac.uk/pride/archive/projects/PXD018186 and assigned the identifier PXD018186.