

Abstract
During angiogenesis, VEGF acts as an attractive cue for endothelial cells (ECs), while Sema3E mediates repulsive cues. Here, we show that the small GTPase RhoJ integrates these opposing signals in directional EC migration. In the GTP‐bound state, RhoJ interacts with the cytoplasmic domain of PlexinD1. Upon Sema3E stimulation, RhoJ released from PlexinD1 induces cell contraction. PlexinD1‐bound RhoJ further facilitates Sema3E‐induced PlexinD1‐VEGFR2 association, VEGFR2 transphosphorylation at Y1214, and p38 MAPK activation, leading to reverse EC migration. Upon VEGF stimulation, RhoJ is required for the formation of the holoreceptor complex comprising VEGFR2, PlexinD1, and neuropilin‐1, thereby preventing degradation of internalized VEGFR2, prolonging downstream signal transductions via PLCγ, Erk, and Akt, and promoting forward EC migration. After conversion to the GDP‐bound state, RhoJ shifts from PlexinD1 to VEGFR2, which then terminates the VEGFR2 signals. RhoJ deficiency in ECs efficiently suppressed aberrant angiogenesis in ischemic retina. These findings suggest that distinct Rho GTPases may act as context‐dependent integrators of chemotactic cues in directional cell migration and may serve as candidate therapeutic targets to manipulate cell motility in disease or tissue regeneration.
Keywords: directional cell migration, endothelial cell, RhoJ, Sema3E, VEGF
Subject Categories: Signal Transduction, Vascular Biology & Angiogenesis
RhoJ GTPase regulates angiogenesis and endothelial cell migration in a context‐dependent fashion by mediating ligand‐dependent interaction between VEGFR2 and PlexinD1.

Introduction
During development, organ morphogenesis largely depends on the directional migration of constituent cells, which is cooperatively controlled by attractive and repulsive cues and cell surface receptors (Friedl & Gilmour, 2009). These chemotactic signals also contribute to the initiation and progression of various diseases (Friedl & Gilmour, 2009). During vascular morphogenesis, endothelial cells (ECs) migrate both forward and backward within growing blood vessels, changing their respective positions (Jakobsson et al, 2010; Arima et al, 2011). Upon reaching the leading vascular tip, ECs dynamically rearrange their cytoskeleton and cell‐matrix adhesions in response to the microenvironment, thereby determining the sprouting directions of new blood vessels (Gerhardt et al, 2003). The tip ECs then migrate back to the stalk positions, resulting in a shuffling of tip and stalk ECs at the leading vascular front (Jakobsson et al, 2010; Arima et al, 2011). Such directional EC migration ensures efficient vascular growth for delivering blood flow to hypoxic tissues (Potente et al, 2011).
Vascular endothelial growth factor A (VEGF‐A, herein VEGF) acts as the major chemotactic attractant for migrating ECs (Potente et al, 2011; Walchli et al, 2015). Binding of VEGF to the tyrosine kinase VEGF receptor 2 (VEGFR2) induces its autophosphorylation at multiple tyrosine residues including Y1175 and Y1124, thereby activating a series of intracellular signaling cascades (Olsson et al, 2006; Koch & Claesson‐Welsh, 2012). Full activation of the phospholipase C gamma (PLCγ)‐extracellular signal‐regulated kinase (Erk) pathway and the phosphoinositide 3‐kinase (PI3K)‐Akt pathway requires VEGFR2 phosphorylation at Y1175 followed by VEGFR2 internalization (Olsson et al, 2006; Koch & Claesson‐Welsh, 2012). These signals, from the intracellular compartments, are then terminated by VEGFR2 degradation (Koch & Claesson‐Welsh, 2012; Simons et al, 2016). The endocytic trafficking of internalized VEGFR2 is largely influenced by neuropilin‐1 (Nrp1), which associates with VEGFR2 upon VEGF stimulation (Koch & Claesson‐Welsh, 2012; Simons et al, 2016). In contrast, p38 mitogen‐activated protein kinase (MAPK) can be activated by surface VEGFR2 following phosphorylation at Y1214 (Chen et al, 2010).
Semaphorin 3E (Sema3E) binding to the transmembrane receptor PlexinD1 mediates repulsive signals in ECs by inducing PlexinD1 internalization, actin depolymerization, and focal adhesion disassembly (Gu et al, 2005; Sakurai et al, 2010; Fukushima et al, 2011; Burk et al, 2017). Notably, both VEGFR2 and Nrp1 can directly associate with PlexinD1 (Gitler et al, 2004; Bellon et al, 2010). In HEK293T cells co‐transfected with VEGFR2 and PlexinD1, binding of Sema3E to PlexinD1 led VEGFR2 transphosphorylation at Y1175 and Y1214 which was enhanced by Nrp1 co‐expression (Bellon et al, 2010). In neurons, PlexinD1, VEGFR2, and Nrp1 form a trimeric receptor complex, whereby Sema3E binding to PlexinD1 results in VEGFR2 transphosphorylation and Akt activation, which subsequently converts Sema3E to the attractant (Chauvet et al, 2007; Bellon et al, 2010). However, the precise crosstalk among these receptors in ECs remains unclear.
In response to diverse extrinsic signals, the Rho family of small GTPases act as molecular switches to control the migration of various types of cells, by cycling between the GTP‐ and GDP‐bound states (Heasman & Ridley, 2008). GTP‐bound Rho GTPases are the active forms that bind to specific effector molecules (Heasman & Ridley, 2008). In growing blood vessels, ECs strongly express RhoJ, which shares 55% homology to Cdc42 by amino acid sequence (Fukushima et al, 2011; Kaur et al, 2011; Yuan et al, 2011). Both RhoJ and Cdc42 bind to the Cdc42/Rac‐interactive binding domains of their effector proteins, such as p21‐activated kinase and neural Wiskott–Aldrich syndrome protein (Vignal et al, 2000; Abe et al, 2003). In contrast to Cdc42, RhoJ induces actin depolymerization, focal adhesion disassembly, and subsequent EC contraction (Fukushima et al, 2011; Kusuhara et al, 2012; Wilson et al, 2014). As RhoJ depletion abolished Sema3E‐induced EC contraction, we previously proposed that RhoJ mediates repulsive actions of Sema3E by competing with Cdc42 for their common effectors (Fukushima et al, 2011). Notably, VEGF can convert RhoJ from a GTP‐bound state to a GDP‐bound state (Fukushima et al, 2011). Furthermore, RhoJ deficiency impaired VEGF‐induced EC migration, retarded retinal vascular development, and suppressed tumor angiogenesis (Takase et al, 2012; Kim et al, 2014; Wilson et al, 2014). However, the mechanism by which RhoJ simultaneously mediates the chemotactic actions of VEGF and Sema3E in migrating ECs has remained obscure.
Here, we show pleiotropic roles of RhoJ in integrating the VEGF and Sema3E signals by controlling cytoskeletal rearrangements, VEGFR2‐PlexinD1 association, and VEGFR2 degradation. These RhoJ activities observed in ECs suggest that distinct Rho GTPases may also play modulatory roles in transducing growth factor signals into directional migration of other cell types.
Results
RhoJ integrates chemotactic actions of VEGF and Sema3E on directional EC migration
To address how ECs behave in the presence of attractive and repulsive cues, we tracked the trajectories of human umbilical vein endothelial cells (HUVECs) when cultured under concentration gradients of VEGF and Sema3E proteins (Fig 1A and Appendix Fig S1A). The efficiency in directional cell migration was assessed by rose diagrams with the Rayleigh test (Fig 1B), in conjunction with the x‐ and y‐forward migration indexes (FMIs, Fig 1C and Appendix Fig S1B). In HUVECs transfected with control non‐targeting siRNA, VEGF promoted forward cell migration toward its higher concentrations. In contrast, Sema3E not only prevented the forward cell migration but also promoted reverse cell migration away from its source with an equivalent y‐FMI to that under VEGF gradients. Furthermore, co‐stimulation with inverse gradients of VEGF and Sema3E additively enhanced the efficiency in directional cell migration. These single and combined effects of VEGF and Sema3E were completely abolished by RhoJ knockdown, resulting in random cell migration similar to those without any ligand stimulation. Thus, RhoJ is required for synergistic integration of attractive and repulsive cues in directional EC migration.
Figure 1. RhoJ integrates chemotactic actions of VEGF and Sema3E on directional EC migration.
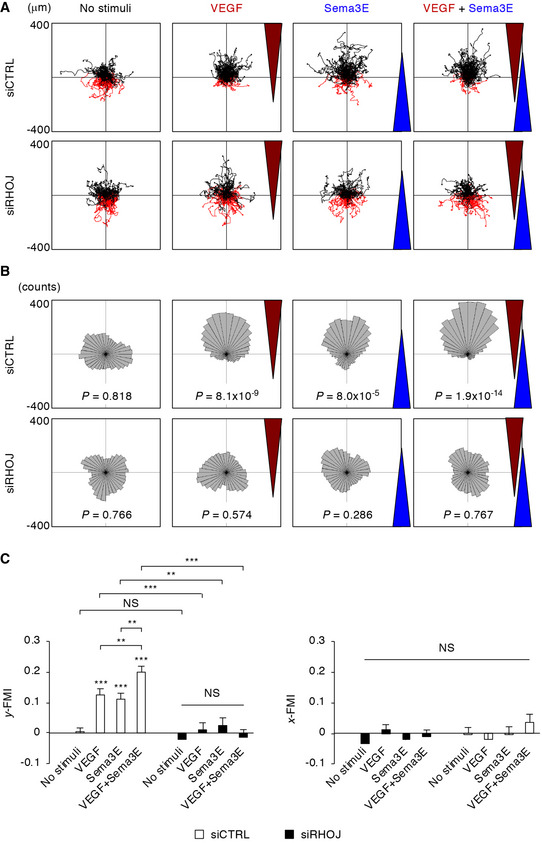
- Trajectory plots of siRNA‐transfected HUVECs under VEGF and/or Sema3E gradients for 12 h. Red trajectories indicate cells with displacement to the negative y‐axis at the end of analyses.
- Rose plots of cell trajectories in (A). Leaflets are grouped into 10° segments. The radius of each leaflet represents the cumulative number of cells migrating in the corresponding angular bin. P values for the Rayleigh test represent non‐random distributions of cell endpoints.
- Forward migration index (FMI) along the y‐ and x‐axes during cell migration. n = 90 cells per group. Data represent mean ± SEM. **P < 0.01; ***P < 0.001; NS, not significant, by the Tukey–Kramer test.
GTP‐bound RhoJ and GDP‐bound RhoJ differentially associate with PlexinD1 and VEGFR2
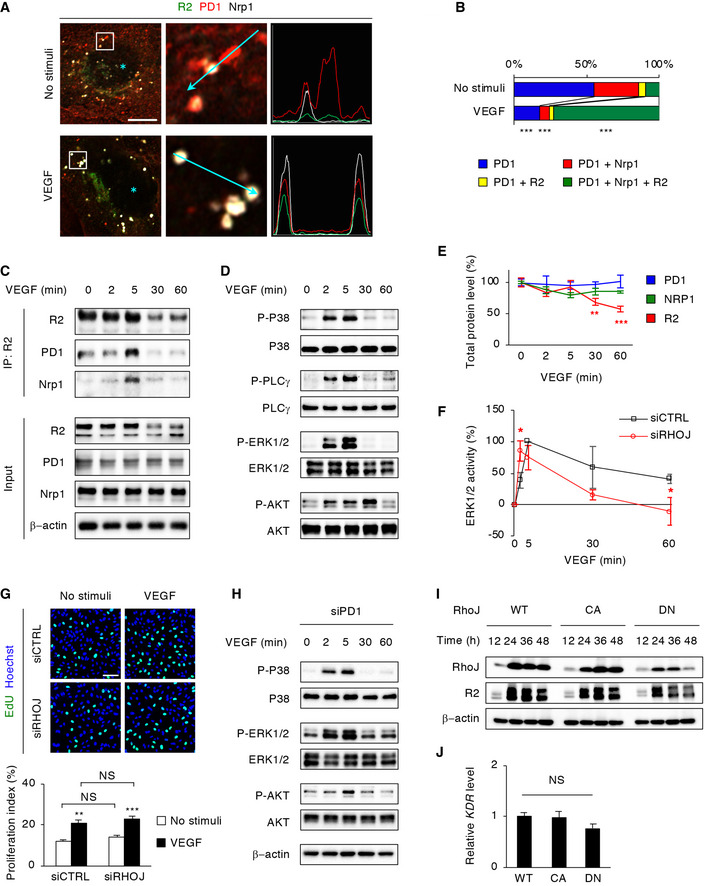
To uncover how RhoJ mediates VEGF and Sema3E signals, we next investigated the spatial associations of RhoJ with VEGFR2 and PlexinD1 in cultured HUVECs. RhoJ has triple palmitoylatable cysteine residues immediately upstream of the C‐terminal CAAX box (C, cysteine; A, aliphatic amino acid; X, any amino acid) that serve as membrane‐targeting sites (Vignal et al, 2000; Abe et al, 2003). Indeed, ectopically expressed RhoJ lacking the CAAX box distributed diffusely in the cytoplasm of cultured HUVECs (Fig EV1A). By contrast, both wild‐type (WT) and constitutively active (CA) RhoJ (Q79L) (Vignal et al, 2000) were distributed at plasma membranes and perinuclear endosomes (Figs 2A and EV1A), most of which colocalized with the early endosomal marker Rab5 (Fig EV1B). Dominant‐negative (DN) RhoJ (T35N) (Vignal et al, 2000; Abe et al, 2003) was absent from plasma membranes but localized in cytoplasmic and nuclear puncta (Figs 2A and EV1A), with frequent colocalization with the late endosomal marker Rab7 (Fig EV1B) and the proteasome marker Rpt5 (Fig EV1C). Moreover, ectopic DN‐RhoJ enlarged the size of and colocalized with promyelocytic leukemia (PML) nuclear bodies (Fig EV1C), which have been implicated in protein degradation (Lallemand‐Breitenbach & de The, 2010).
Figure EV1. Subcellular distribution of GTP‐RhoJ and GDP‐RhoJ in ECs.

- Subcellular distribution of ectopic RhoJ in HUVECs 12 h after transfection. WT‐RhoJ and CA‐RhoJ localized in plasma membranes and endosomal vesicles, whereas DN‐RhoJ localized in cytoplasmic and nuclear puncta. Note the diffuse cytoplasmic distribution of RhoJ lacking the CAAX motif (ΔCAAX). Scale bar, 10 μm.
- Super‐resolution images of fluorescence signals in HUVECs 12 h after co‐transfection of GFP‐RhoJ with RFP‐Rab5 or DsRed‐Rab7. Scale bar, 1 μm.
- Confocal images of immunostaining for ectopic RhoJ (green) and endogenous Rpt5 (red) or PML nuclear bodies (red) in HUVECs 12 h after transfection. Nuclei were labeled with Hoechst. Scale bar, 2 μm.
- Immunostaining for endogenous VEGFR2 and trans‐Golgi network protein p230 in HUVECs. Scale bar, 2 μm.
- Schematic representation of the PlexinD1 constructs. Numbers indicate amino acid positions. TM, transmembrane; C1 and C2, segmented GAP domains; RBD, Rho‐binding domain; Myr, Src myristoylated signal (MGSSKS) for membrane targeting.
- Fluorescence intensity of internalized PlexinD1 in Sema3E‐stimulated HUVECs. n = 10 cells per indicated time point.
- Fluorescence microscopy of COS7 cells 48 h after co‐transfection of vectors expressing RhoJ and EGFP. Scale bar, 50 μm.
- Proportion of collapsing cells in GFP‐positive COS7 cells (n > 100) in a fixed area (4.1 × 105 μm2) 48 h after co‐transfection. n = 3 areas per group.
Figure 2. GTP‐RhoJ and GDP‐RhoJ differentially associate with PlexinD1 and VEGFR2.

- Confocal (left three columns) and super‐resolution (right‐most columns) images of subcellular distributions of ectopic RhoJ (red), endogenous VEGFR2 (green), and PlexinD1 (left, white; right, blue) in cultured HUVECs 6 h after transfection with the indicated RhoJ constructs. Note the colocalization of WT‐RhoJ and CA‐RhoJ (Q79L) with PlexinD1 in perinuclear endosomes and DN‐RhoJ (T35N) with VEGFR2 in cytoplasmic and nuclear puncta. Light‐blue asterisks in the PlexinD1 panels indicate nuclei. See also Appendix Fig S2 for the full images of super‐resolution microscopy. Scale bar, 5 μm (left); 1 μm (right).
- Proportion of ectopic RhoJ that colocalized with endogenous VEGFR2 and/or PlexinD1. n = 10 cells per group. Data represent mean.
- Co‐immunoprecipitation (co‐IP) from 293T cells 48 h after transfection. WT‐RhoJ and CA‐RhoJ associated with PlexinD1, while DN‐RhoJ associated with VEGFR2.
Source data are available online for this figure.
Both ectopic WT‐RhoJ and CA‐RhoJ colocalized with endogenous PlexinD1 in perinuclear endosomes (Fig 2A and B, Appendix Fig S2), but not with VEGFR2 which predominantly accumulated in the trans‐Golgi network (Fig EV1D). By contrast, ectopic DN‐RhoJ colocalized with VEGFR2, but not PlexinD1, in cytoplasmic and nuclear puncta (Fig 2A and B, Appendix Fig S2). Similarly, in 293T cells that ectopically expressed RhoJ with PlexinD1 or VEGFR2, both WT‐RhoJ and CA‐RhoJ co‐immunoprecipitated (co‐IPed) with PlexinD1, whereas DN‐RhoJ co‐IPed with VEGFR2 (Fig 2C). These results collectively indicate that GTP‐bound RhoJ and GDP‐bound RhoJ differentially associate with PlexinD1 and VEGFR2, respectively, in distinct subcellular compartments.
Sema3E induces EC contraction by releasing RhoJ from PlexinD1
We next examined how RhoJ mediates the repulsive action of the Sema3E‐PlexinD1 signal in ECs. In the absence of Sema3E, the extracellular domain (ECD) of PlexinD1 is folded, thereby auto‐inhibiting signal transduction from its cytoplasmic domain (Kruger et al, 2005; Gay et al, 2011). Binding of Sema3E to the N‐terminal Sema domain of the PlexinD1‐ECD induces conformational changes in the cytoplasmic GTPase activating protein (GAP) domains (Kruger et al, 2005; Gay et al, 2011). Given the physical association of GTP‐RhoJ with PlexinD1, we speculated that RhoJ might bind to a Rho‐binding domain (RBD) that is flanked by the segmented GAP domains (Uesugi et al, 2009; Wang et al, 2011). In 293T cells, binding of ectopic RhoJ to full‐length PlexinD1 was disrupted by deleting the RBD (Figs 3A and B, and EV1E). Furthermore, RhoJ was incapable of binding to a mutated PlexinD1 lacking its ECD, which mimicked the removal of the auto‐inhibition of the cytoplasmic domain (Figs 3A and B, and EV1E). Similarly, Sema3E stimulation prevented RhoJ binding to the full‐length PlexinD1 (Fig 3B), which was corroborated by the spatial dissociation of RhoJ from internalized PlexinD1 in Sema3E‐stimulated HUVECs (Figs 3C and D, and EV1F). Together, these results indicate that Sema3E releases RhoJ from the PlexinD1‐RBD.
Figure 3. Sema3E induces EC contraction by releasing RhoJ from PlexinD1.

- Schematic representation of full‐length PlexinD1 (PD1) and its deletion mutants.
- Co‐IP from 293T cells 48 h after transfection. RhoJ‐PlexinD1 binding was disrupted by deleting the RBD or ECD of PlexinD1 and by 30‐min stimulation with Sema3E. Arrows indicate PD1 Full (upper, 250 kDa) and PD1▵RBD (lower, 1.7 kDa smaller than PD1 Full).
- PlexinD1 internalization in HUVECs 30 min after Sema3E stimulation. Scale bar, 10 μm.
- Super‐resolution images of subcellular distributions of ectopic WT‐RhoJ (red) and endogenous PlexinD1 (green) in HUVECs 24 h after transfection. RhoJ dissociated from internalized PlexinD1 30 min after Sema3E stimulation. Scale bar, 1 μm.
- Labeling of siRNA‐transfected HUVECs with phalloidin (white) 30 min after Sema3E stimulation. Scale bar, 50 μm.
- Quantification of areas of individual HUVECs. n > 100 cells per group. Data represent mean ± SEM. ***P < 0.001; NS, not significant, by the Tukey–Kramer test.
- A model of Sema3E‐induced cell contraction.
Source data are available online for this figure.
Overexpression of WT‐RhoJ and CA‐RhoJ, but not DN‐RhoJ, induced cell collapse, even in non‐endothelial COS7 cells (Fig EV1G and H), indicating the cell collapsing ability of GTP‐RhoJ independently of PlexinD1. To clarify how the Sema3E‐PlexinD1 signal regulates this activity of RhoJ in ECs, we quantitatively measured the cell surface areas of individual HUVECs after the transfection of siRNAs targeting RhoJ and/or PlexinD1 with Sema3E stimulation (Fig 3E and F). Consistent with our previous report (Fukushima et al, 2011), RhoJ knockdown alone had no effects on the cell surface area but abrogated Sema3E‐induced cell contraction. In contrast, PlexinD1 knockdown alone resulted in cell contraction even without Sema3E stimulation. This effect was alleviated by the double knockdown of PlexinD1 and RhoJ. Together, these results suggest that PlexinD1 prevents EC contraction by sequestering GTP‐RhoJ via its RBD, which is released upon Sema3E stimulation, thereby allowing RhoJ to stimulate cell contraction (Fig 3G).
RhoJ facilitates Sema3E‐induced reverse EC migration via activation of VEGFR2 and p38 MAPK
To further elucidate the mechanisms underlying the Sema3E‐induced reverse EC migration, we examined the spatial correlations of PlexinD1 with VEGFR2 and Nrp1 in perinuclear endosomes of cultured HUVECs. In the absence of Sema3E stimulation, over 50% of the PlexinD1‐positive vesicles were independent of Nrp1 or VEGFR2, and smaller proportions of PlexinD1 associated with Nrp1 and/or VEGFR2 (Fig EV2A and B). Notably, Nrp1 mostly associated with PlexinD1 but not VEGFR2 (Fig EV2A). However, Sema3E stimulation significantly increased the proportion of PlexinD1‐VEGFR2 complex (Fig EV2A and B). Indeed, co‐IP experiments demonstrated that Sema3E stimulation rapidly increased the association of PlexinD1 with VEGFR2, but not Nrp1 (Fig 4A). Moreover, Sema3E stimulation resulted in VEGFR2 phosphorylation at Y1214, but not Y1175, without affecting its total protein levels (Figs 4A, and EV2C and D). Furthermore, Sema3E activated p38 MAPK, but not PLCγ, Erk1/2, and Akt (Fig 4B), and this was abolished by VEGFR2 knockdown (Fig 4C). Consistent with these results, Sema3E‐induced VEGFR2 phosphorylation at Y1214 and p38 MAPK activation was abolished by knockdown of PlexinD1, but not Nrp1, and by treatment with the VEGFR2 tyrosine kinase inhibitor SU5416 (Fig EV2E and F). RhoJ knockdown prevented Sema3E‐induced PlexinD1‐VEGFR2 association (Fig 4D and E), VEGFR2 transphosphorylation at Y1214 (Fig EV2G), and p38 MAPK activation (Fig 4C). As the p38 MAPK inhibitor SB203580 abrogated the directional EC migration under Sema3E gradients (Fig 4F), these observations collectively indicate that RhoJ mediates Sema3E‐induced reverse EC migration by facilitating association of PlexinD1 with VEGFR2, transphosphorylation of VEGFR2 at Y1214, and activation of p38 MAPK (Fig 4G).
Figure EV2. Sema3E‐induced PlexinD1‐VEGFR2 association.

- Super‐resolution images of receptor colocalization in HUVECs 30 min after Sema3E stimulation. Left panels show immunolabeling of VEGFR2 (green), PlexinD1 (red), and Nrp1 (white), with light‐blue asterisks indicating nuclei. Middle panels show magnified views of white boxes in the left panels. Right panels represent fluorescence intensity profiles along 5‐μm arbitrary lines in the middle panels. Scale bar, 5 μm.
- Proportion of receptor complexes in the PlexinD1‐positive vesicles in HUVECs. n = 10 cells per group. Data represent mean. *P < 0.05, by Student's t‐test.
- Quantification of total VEGFR2 protein levels as a percentage of the values at 0 min. n = 3 per group. Data represent mean ± SEM.
- Immunoblots in HUVECs.
- Immunoblots in siRNA‐transfected HUVECs.
- Immunoblots in HUVECs treated with SU5416.
- Immunoblots in siRHOJ‐transfected HUVECs.
Source data are available online for this figure.
Figure 4. RhoJ facilitates Sema3E‐induced reverse EC migration via activation of VEGFR2 and p38 MAPK .

- Co‐IP from HUVECs. Blots for β‐actin shown in (A) were used to confirm equal loading in (B), as the same cell lysates were analyzed in both experiments.
- Immunoblots in HUVECs.
- Immunoblots in siRNA‐transfected HUVECs.
- Co‐IP from siRNA‐transfected HUVECs. The graph shows relative levels of PlexinD1‐VEGFR2 association, in which the values at 0 min are set to 1. n = 3 per group.
- Super‐resolution images of receptor colocalization in siRNA‐transfected HUVECs 30 min after Sema3E stimulation. Left panels show immunolabeling of VEGFR2 (green), PlexinD1 (red), and Nrp1 (white), with light‐blue asterisks indicating nuclei. Middle panels show magnified views of white boxes in the left panels. Right panels represent fluorescence intensity profiles along 5‐μm arbitrary lines in the middle panels. Scale bar, 10 μm. The graph shows the proportion of receptor complexes in PlexinD1‐positive vesicles. n = 10 cells per group.
- Trajectory plots (left) and rose plots (right) of HUVECs pre‐treated with the p38 MAPK inhibitor SB203580 under Sema3E gradient for 12 h. Red trajectories indicate cells with displacement to the negative y‐axis at the end of analyses. P values for the Rayleigh test represent non‐random distributions of cell endpoints. The graphs show FMI along the y‐ and x‐axes during cell migration. n = 90 cells per group.
- A model of Sema3E‐induced reverse cell migration.
RhoJ maintains VEGF signals by preventing degradation of internalized VEGFR2
In contrast to Sema3E, VEGF phosphorylated VEGFR2 at both Y1175 and Y1214 in cultured HUVECs (Fig EV2D), as previously recognized (Olsson et al, 2006; Koch & Claesson‐Welsh, 2012). In this process, VEGF stimulation induced formation of a trimeric holoreceptor comprising VEGFR2, PlexinD1, and Nrp1 that localized in perinuclear endosomes, with concomitant decrease in the presence of the monomeric form PlexinD1 and dimeric form of the PlexinD1‐Nrp1 receptors (Fig EV3A–C). VEGF stimulation activated p38 MAPK, PLCγ, Erk1/2, and Akt (Fig EV3D). Protein levels of VEGFR2, but not PlexinD1 and Nrp1, were significantly decreased at 30 min after VEGF stimulation (Fig EV3C and E), indicating that the fates of PlexinD1 and Nrp1 were independent of VEGFR2. RhoJ knockdown reduced the interaction of internalized VEGFR2 with PlexinD1 and Nrp1 even after VEGF stimulation (Fig 5A), which was further validated by co‐IP experiments (Fig 5B). RhoJ knockdown significantly decreased VEGFR2 levels within 5 min of VEGF stimulation (Fig 5C). Peak VEGF‐induced activation of PLCγ, Erk1/2, and Akt was earlier and more transient in RhoJ‐knockdown ECs than control cells (Figs 5D and EV3F). However, RhoJ depletion had negligible effects on EC proliferation under VEGF stimulation (Fig EV3G). By contrast, VEGF‐induced activation of p38 MAPK, which was dependent on the VEGFR2 kinase activity (Fig EV2F), was unaffected by RhoJ knockdown (Fig 5D). In line with these observations, PlexinD1 knockdown also resulted in VEGF‐induced internalization of VEGFR2 alone without Nrp1 association (Fig 5A), accelerated VEGFR2 degradation (Fig 5C), and earlier activation of Erk1/2 and Akt, but not p38 MAPK (Fig EV3H). Nrp1 knockdown had no effect on VEGFR2‐PlexinD1 association (Fig 5A), but delayed VEGFR2 degradation (Fig 5C). Together, these results indicate that upon VEGF stimulation, RhoJ initially facilitates VEGFR2‐PlexinD1 association, which is subsequently accompanied by the recruitment of Nrp1 pre‐coupled with PlexinD1. RhoJ‐bound PlexinD1 contributes to maintaining VEGFR2 signal transduction by preventing the degradation of internalized VEGFR2, which is inversely regulated by Nrp1.
Figure EV3. VEGF‐induced receptor complex formation and signal transduction.
- Super‐resolution images of receptor colocalization in HUVECs 30 min after VEGF stimulation. VEGFR2 (green), PlexinD1 (red), and Nrp1 (white) are shown as in Fig EV2A. Scale bar, 5 μm.
- Proportion of receptor complexes in the PlexinD1‐positive vesicles in HUVECs. Data of “no stimuli” are the same from Fig EV2B. n = 10 cells per group.
- Co‐IP from HUVECs. Blots for β‐actin shown in (C) were used to confirm equal loading in (D), as the same cell lysates were analyzed in these experiments.
- Immunoblots in HUVECs.
- Changes in total VEGFR2 protein levels as a percentage of the values at 0 min. n = 3 per group.
- Quantification of Erk1/2 activity in siRNA‐transfected HUVECs. Phosphorylated Erk1/2 levels normalized to total Erk1/2 levels are presented as a percentage of the value of Ctrl siRNA‐transfected HUVECs stimulated with VEGF for 10 min. n = 3 per group.
- Labeling for EdU (green) and Hoechst (blue) in siRNA‐transfected HUVECs 4 h after VEGF stimulation. HUVECs were incubated with EdU for 2 h prior to analysis. Scale bar, 50 μm. The graph shows the proliferation index of siRNA‐transfected HUVECs. n = 4 per group.
- Immunoblots in siRNA‐transfected HUVECs.
- Immunoblots in 293T cells after co‐transfection of vectors expressing RhoJ and VEGFR2.
- Relative expression levels of KDR/VEGFR2 mRNA from HUVECs 12 h after transfection of RhoJ‐expressing vectors. The values of WT‐RhoJ are set to 1. n = 3 per group.
Figure 5. RhoJ sustains VEGF signals by preventing degradation of internalized VEGFR2.
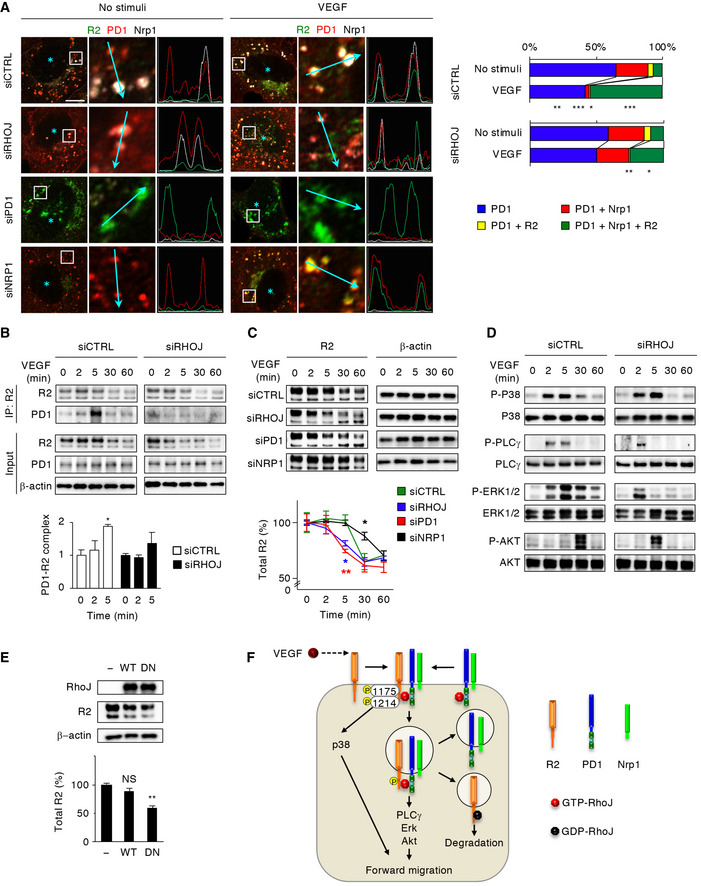
- Super‐resolution images of receptor colocalization in siRNA‐transfected HUVECs 30 min after VEGF stimulation. VEGFR2 (green), PlexinD1 (red), and Nrp1 (white) are shown as in Fig 4E. Scale bar, 10 μm. The graph shows the proportion of receptor complexes in the PlexinD1‐positive vesicles. Data of “no stimuli” are the same from Fig 4E. n = 10 cells per group.
- Co‐IP from siRNA‐transfected HUVECs. The graph shows relative levels of PlexinD1‐VEGFR2 association, in which the values at 0 min are set to 1. n = 3 per group.
- Immunoblots in siRNA‐transfected HUVECs. Blots for β‐actin shown in (C) were used to confirm equal loading in (D), as the same cell lysates were analyzed in these experiments. The graph shows changes in total VEGFR2 protein levels as a percentage of the values at 0 min. n = 3 per group.
- Immunoblots in siRNA‐transfected HUVECs. Note the early and transient activation of PLCγ, Erk1/2, and Akt, but not p38 MAPK, in RhoJ‐knockdown ECs.
- Immunoblots for ectopic RhoJ and endogenous VEGFR2 in HUVECs 12 h after transfection of RhoJ‐expressing vectors. The graph shows protein levels of VEGFR2 relative to those of the control set as 100%. n = 3 per group.
- A model of VEGF‐induced forward cell migration.
In line with its colocalization with protein degradation markers (Fig EV1B and C), protein levels of ectopic DN‐RhoJ decreased earlier than those of WT‐RhoJ and CA‐RhoJ in 293T cells (Fig EV3I). Moreover, ectopic DN‐RhoJ reduced the protein expression levels of ectopic VEGFR2 in 293T cells (Fig EV3I) and endogenous VEGFR2 in HUVECs (Fig 5E), without affecting its transcription levels (Fig EV3J). Considering that VEGF stimulation reduces GTP‐RhoJ in ECs (Fukushima et al, 2011), our results indicate that after a period of signal transduction from internalized VEGFR2, RhoJ is converted to the GDP‐bound state and shifts from PlexinD1 to VEGFR2. VEGFR2 is then sorted away from the PlexinD1‐Nrp1 complex and undergoes degradation (Fig 5F).
RhoJ promotes directional EC migration in sprouting vessels
To further dissect the role of RhoJ in VEGF‐dependent EC migration, we performed time‐lapse imaging of an ex vivo angiogenesis model using aortic rings derived from wild‐type (WT) and Rhoj knockout (KO) mice (Movie EV1). In this model, endogenous Sema3E was absent (Fig EV4A and B), and exogenous VEGF drives forward EC migration within sprouting vessels (Arima et al, 2011). In the Rhoj‐KO vessels, most stalk ECs failed to continuously migrate forward to the tips of vascular sprouts (Fig 6A). Indeed, the reverse, but not forward, migration of individual ECs in the Rhoj‐KO vessels was significantly increased compared with those in WT vessels (Fig 6B), which was responsible for the decreased vessel elongation (Fig 6C). These results indicate that RhoJ is required for VEGF‐dependent forward EC migration in sprouting vessels.
Figure EV4. Analyses of directional EC migration in sprouting vessels.

- RT–PCR using total RNAs from an aortic ring angiogenesis model. Total RNAs from 293T cells transfected with or without a Sema3E‐expressing vector were used as a positive or negative control, respectively.
- ELISA for mouse Sema3E proteins. Absorbance at 450 nm of the culture supernatants of the aortic ring model and the control Sema3E proteins was measured in triplicate. Data represent mean ± SEM.
- Labeling for YFP/GFP (green) and CD31 (red) in retinas of P5 CAG‐MerCreMer:R26R‐EYFP flox/WT and CAG‐MerCreMer:Rhoj floxflox mice after single i.p. injection of 10 μg of 4OHT at P1. Scale bar, 100 μm.
Source data are available online for this figure.
Figure 6. RhoJ promotes directional EC migration in sprouting vessels.
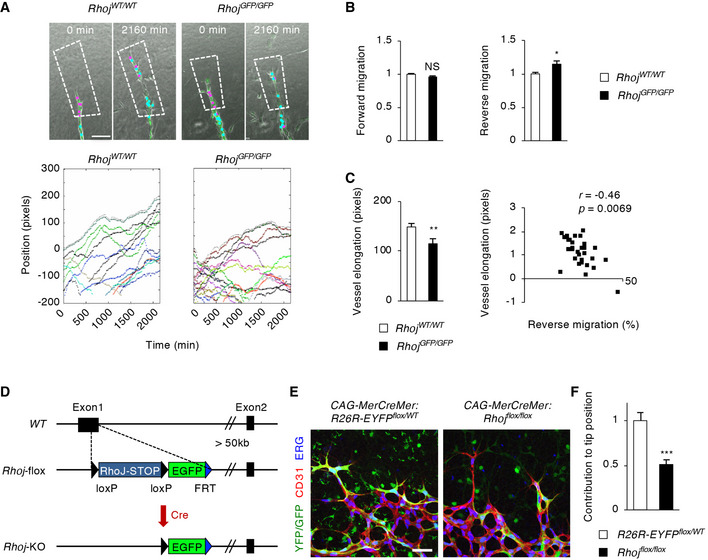
- Kinetic analyses of EC migration in an ex vivo aortic ring angiogenesis model. Upper panels are snapshots of time‐lapse imaging in which EC nuclei are labeled by SYTO dye (green). In analyzed regions framed by dotted lines, the centroids of the first five EC nuclei from the tip at 0 min are pseudocolored in magenta and those of the remaining EC nuclei are shown in cyan. Initial discrete labeling becomes intermixed over time (see also Movie EV1). Lower panels show trajectory analyses representing normalized positional changes in individual ECs during vessel elongation. Scale bar, 100 μm.
- Quantification of the kinetics of EC migration. Values of forward and reverse migration for Rhoj WT/WT are normalized to 1. n = 33 vessel branches per group.
- Quantification of vessel elongation (left graph). n = 33 vessel branches per group. Scatter plot shows relationship between “vessel elongation” and “reverse migration” in Rhoj GFP/GFP aortic rings as evaluated by a Pearson correlation coefficient.
- Schematic representation for construction of Rhoj mutant mice. Cre‐loxP‐mediated genetic recombination of the Rhoj‐flox allele generates the Rhoj‐KO allele by excising the RhoJ cDNA‐polyA cassette inserted into exon 1 of the Rhoj gene.
- Labeling for YFP/GFP (green), CD31 (red), and ETS transcription factor ERG (blue) in retinal vascular fronts of P5 CAG‐MerCreMer:R26R‐EYFP flox/WT and CAG‐MerCreMer:Rhoj floxflox mice after intraperitoneal (i.p.) injections of 10 μg of 4‐hydroxytamoxifen (4OHT) at P1. Scale bar, 50 μm.
- Relative contribution of YFP/GFP‐positive ECs to the tip positions. n = 16 per group. Values for CAG‐MerCreMer:R26R‐EYP flox/WT are normalized to 1.
To better understand the in vivo roles of RhoJ in directional EC migration, we tracked the positions of individual ECs during two‐dimensional extension of the vascular network in postnatal mouse retinas, in which ECs are exposed to VEGF derived from astrocytes in the same horizontal plane and Sema3E from underlying neurons in the ganglion cell layer (GCL) (Fukushima et al, 2011). We injected CAG‐MerCreMer mice (Egawa et al, 2009) carrying the R26R‐EYFP (Srinivas et al, 2001) or Rhoj‐flox (Kim et al, 2014) alleles intraperitoneally with a low dose of tamoxifen at postnatal day 1 (P1). This procedure allowed visualization of cells with inducible genetic recombination in a mosaic‐like manner by fluorescence reporter expression (Fig 6D). In P5 retinas, ECs were the major cell population expressing the fluorescence reporter in both of these mouse lines (Figs 6E and EV4C). However, scattered reporter expression was also found in other cell types (Figs 6E and EV4C), due to the global transgene expression under the CAG and Rosa26 promoters and endogenous RhoJ expression in non‐ECs as described below. Among the GFP‐positive retinal ECs labeled by CD31 and ETS transcription factor ERG, the proportions of tip ECs were reduced by about 50% in CAG‐MerCreMer:Rhoj flox/flox mice compared with those in the control CAG‐MerCreMer:R26R‐EYFP flox/WT mice (Fig 6F). These results indicate that RhoJ contributes to the adequate positioning of angiogenic ECs within the growing retinal vessels.
RhoJ deficiency delays vascular development and suppresses pathological angiogenesis
Despite the pivotal roles of RhoJ in directional EC migration, homozygous Rhoj‐KO mice were born at normal Mendelian ratios from the intercrossing of heterozygous males and females. However, the body weight of the Rhoj‐KO pups was significantly reduced compared with their WT littermates (Fig EV5A). Given the broad RhoJ expression in ECs of embryonic blood vessels (Fig EV5B), we suspected that RhoJ deficiency might cause non‐lethal alterations in vascular development. Indeed, the number of large vessels and their branches was significantly decreased in the head and somite of Rhoj‐KO mouse embryos (Fig EV5C), which prompted us to further evaluate the effects of RhoJ deficiency on the patterning of the retinal vasculature.
Figure EV5. Vascular development in Rhoj‐KO mice.

- Quantification of body weight of P4 Rhoj‐KO mice. n = 8 per group.
- Labeling for GFP in E12.5 Rhoj GFP/GFP mouse.
- Labeling for CD31 in head and somite of E10.5 Rhoj‐KO mice. The graphs show morphometric analyses of cranial and intersomitic vessels (ISVs). n = 8 per group.
- Labeling for GFP (green), α‐smooth muscle actin (αSMA, red), and CD31 (blue) in retinas of Rhoj GFP/WT mice. In arteries, RhoJ was downregulated in ECs and upregulated in αSMA‐positive vascular smooth muscle cells during vascular remodeling and maturation. RhoJ expression in ECs was maintained in veins and capillaries in adult retinas. Scale bar, 100 μm (top three rows); 20 μm (bottom two rows).
- Labeling for EdU (green), ERG (red), and CD31 (gray) in retinas of P4 Rhoj‐KO mice 2 h after i.p. EdU injections. Scale bar, 50 μm. The graph shows EC proliferation index in retinal veins. n = 9 per group.
- Labeling for CD31 in retinas of 6‐week‐old Rhoj‐KO mice. Scale bar, 200 μm (upper); 50 μm (lower). The graphs show quantification of each vascular layer. n = 3 per group.
In the developing retinal vessels, ubiquitous RhoJ expression in ECs was validated in the Rhoj‐KO mice (Figs 7A and EV5D). Although RhoJ deficiency did not affect the proliferation of retinal ECs (Figs 7B and EV5E), Rhoj‐KO mice demonstrated significant reductions in radial extension, branching, and density of the retinal vascular networks (Fig 7C and D). In addition to ECs, RhoJ was weakly expressed in a subpopulation of neurons in the GCL (Fig 7A). Moreover, in retinal arteries, RhoJ expression was downregulated in ECs but upregulated in vascular smooth muscle cells in accordance with the vascular remodeling and maturation (Fig EV5D). To exclude the possible effects of RhoJ deficiency in non‐ECs on the retinal vessels, we induced EC‐specific Rhoj‐KO (Rhoj iΔEC) by systemically injecting a high dose of tamoxifen to Pdgfb‐iCreERT2:Rhoj flox/flox mice (Claxton et al, 2008) once at P1. As similar results were observed in retinas of the Rhoj iΔEC mice and the Rhoj‐KO mice (Fig 7E and F), we speculated that defective motility of RhoJ‐deficient ECs was responsible for the retardation of retinal vascular development. However, the initial retardation of the retinal vascular formation was recovered in the Rhoj‐KO mice at later stages (Fig EV5F and Appendix Fig S3A), which may also be the case outside the retina.
Figure 7. RhoJ deficiency delays vascular development and suppresses pathological angiogenesis.

- Labeling for GFP (green) and CD31 (red) in retina of P4 Rhoj GFP/GFP mouse. Scale bar, 200 μm (left); 20 μm (right).
- Labeling for EdU (green), ERG (red), and CD31 (gray) in retinas of P4 Rhoj WT/WT and Rhoj GFP/GFP mice 2 h after i.p. EdU injections. Scale bar, 20 μm. The graph shows EC proliferation index in retinal capillaries behind the angiogenic fronts. n = 12 per group.
- Labeling for CD31 in retinas of P4 and P7 Rhoj WT/WT and Rhoj GFP/GFP mice. Scale bars, 500 μm.
- Morphometric analyses of retinal vessels in Rhoj WT/WT (P4, n = 18; P7, n = 8) and Rhoj GFP/GFP (P4, n = 16; P7, n = 11) mice.
- Labeling for CD31 in retinas of P4 Rhoj flox/flox and Pdgfb‐iCreERT2:Rhoj flox/flox (Rhoj iΔEC) mice after single i.p. injection of 100 μg of 4OHT at P1. Scale bar, 500 μm.
- Morphometric analyses of retinal vessels in P4 Rhoj flox/flox (n = 7) and Rhoj i∆EC (n = 6) mice.
- Relative expression levels of Vegfa and Sema3e mRNA from WT mouse retinas. n = 3 per group.
- Labeling for GFP (green) and CD31 (red) in retina of P18 OIR‐Rhoj GFP/WT mouse. Scale bar, 200 μm.
- Labeling for CD31 in retinas of P18 OIR‐Rhoj flox/flox and OIR‐Rhoj i∆EC mice after daily i.p. injection of 200 μg of 4OHT from P12. Scale bar, 500 μm.
- Quantification of areas of neovascular tufts in P18 OIR‐Rhoj flox/flox and OIR‐Rhoj i∆EC mice. n = 6 per group.
Finally, we assessed the role of RhoJ in pathological angiogenesis using a mouse model of oxygen‐induced retinopathy (OIR). In this model, preformed retinal capillaries regress under 5‐day exposure to hyperoxia (75% O2) from P7, followed by the formation of neovascular tufts protruding from the ischemic retinal surfaces after return to room air (Smith et al, 1994). In the ischemic retina, a subset of neurons in the GCL constantly expresses Sema3E, whereas VEGF is abundantly expressed in neurons, but not astrocytes, in the GCL and the inner nuclear layer (Fukushima et al, 2011). The retinal expression level of Vegfa, but not Sema3e, was upregulated (by 4.5‐fold) in the OIR model compared with that under physiological vascular development (Fig 7G). In this process, RhoJ was intensely expressed in ECs of the neovascular tufts (Fig 7H), and the OIR‐Rhoj iΔEC mice generated by daily tamoxifen injections to Pdgfb‐iCreERT2:Rhoj flox/flox mice from P12 showed an 80% reduction in the neovascular tufts (Fig 7I and J). As RhoJ deficiency did not affect EC proliferation in the neovascular tufts (Appendix Fig S3B), these results imply an enhanced anti‐migratory potential of RhoJ depletion under a high ratio of VEGF to Sema3E.
Discussion
As represented by their alternative nomenclature “collapsin”, the semaphorin family members were initially identified as ligands that induce collapse of the axonal growth cone (Semaphorin Nomenclature Committee, 1999). This effect has been largely attributed to the activation or inactivation of an array of small GTPases following conformational changes in Plexin receptors (Kruger et al, 2005). Among the secreted class III semaphorins (Sema3A to Sema3G), Sema3E is unique in its direct binding to PlexinD1, but not Nrp coreceptors (Gu et al, 2005). In ECs of growing retinal vessels, PlexinD1 is upregulated in response to VEGF, whereas binding of neuron‐derived Sema3E to PlexinD1 activates RhoJ and counteracts VEGF‐induced filopodia formation, thereby preventing disoriented angiogenesis (Fukushima et al, 2011; Kim et al, 2011). Given this repulsive action of Sem3E on tip ECs, we started our present study by evaluating its role in directional EC migration, which is an integral part of collective migration of tip and stalk ECs in sprouting blood vessels (Jakobsson et al, 2010; Arima et al, 2011).
In two‐dimensional culture conditions, Sema3E suppresses overall motility of ECs via actin depolymerization and cell‐matrix dissociations by inactivating R‐Ras, Rac1, and Cdc42 and activating Arf6 (Sakurai et al, 2010; Fukushima et al, 2011; Aghajanian et al, 2014; Tata et al, 2014). In contrast to this well‐recognized anti‐migratory activity, we have shown that Sema3E gradients promoted reverse EC migration away from its source with an equivalent efficiency to that of forward EC migration under VEGF gradients. Moreover, inverse gradients of VEGF and Sema3E synergistically enhanced directional EC migration. RhoJ was required for these individual and compound effects of VEGF and Sema3E. Thus, our present results indicate that RhoJ integrates the chemotactic cues from VEGF and Sema3E for migrating ECs.
Unlike the conventional mode of Rho GTPase activation through GDP‐GTP exchange by guanine nucleotide exchange factors (GEFs) (Heasman & Ridley, 2008), Sema3E enabled GTP‐RhoJ to induce cell contraction by release from the RBD of PlexinD1. This suggests that PlexinD1 prevents EC contraction by sequestering GTP‐RhoJ in the absence of Sema3E. In neurons, the repulsive actions of Sema3E require internalization and trafficking of PlexinD1 destined for recycling endosomes via interaction of its C‐terminal PDZ‐binding motifs with the adapter protein GIPC1 (Burk et al, 2017). This process may also occur in ECs, in which internalized PlexinD1 is ultimately recycled to the plasma membrane after Sema3E stimulation. Given that RhoJ regulates endocytic trafficking of cargo proteins such as transferrin receptor, podocalyxin, and α5β1 integrin (de Toledo et al, 2003; Richards et al, 2015; Sundararaman et al, 2020), it will be interesting to investigate how the binding and release of RhoJ influence the trafficking route of PlexinD1. Of note, Rnd2 also binds to the RBD of PlexinD1 and regulates its R‐Ras GAP activity (Uesugi et al, 2009). Thus, RhoJ and Rnd2 may interact to modulate the Sema3E signal, and this possibility requires further investigation.
Unexpectedly, we found that GTP‐RhoJ functioned as a hinge for the ligand‐induced association of PlexinD1 with VEGFR2, but not Nrp1. Accordingly, RhoJ facilitated Sema3E‐induced VEGFR2 transphosphorylation at Y1214 and p38 MAPK activation, which was required for its repulsive action. As the protein levels of both PlexinD1 and VEGFR2 were maintained after Sema3E stimulation, this signaling event may occur at the cell surface, consistently with the VEGF‐induced p38 MAPK activation (Chen et al, 2010). Nevertheless, Sema3E stimulation increased internalization of the PlexinD1‐VEGFR2 complex to perinuclear endosomes. Thus, these receptors may be recycled to the plasma membrane without undergoing degradation, although how the Sema3E‐induced VEGFR2 signal is terminated awaits further elucidation.
Based on the present results, we postulate that the repulsive action of Sema3E comprises its paradoxical cell‐contracting versus pro‐migratory effects in individual ECs, which may partly be explained by the front‐rear polarity in migrating cells (Friedl & Gilmour, 2009). In this scenario, RhoJ released from PlexinD1 mediates a typical repulsive action at the rear proximal to the source of Sema3E, while RhoJ facilitates PlexinD1‐VEGFR2 association, VEGFR2 transphosphorylation, and p38 MAPK activation at the front, which in concert promote reverse EC migration. This hypothesis should be validated by detecting polarized subcellular localization of VEGFR2, PlexinD1, RhoJ, and p38 MAPK in migrating ECs under Sema3E gradients.
In PlexinD1‐expressing neurons, Sema3E can be converted to an attractant in a VEGFR2‐dependent manner (Bellon et al, 2010). Unlike ECs, however, Sema3E transphosphorylates VEGFR2 at Y1175 and activates Akt in neurons (Bellon et al, 2010). This discrepancy is likely to be ascribed to the involvement of Nrp1 in the holoreceptor complex in neurons (Chauvet et al, 2007; Bellon et al, 2010), but not ECs after Sema3E stimulation. Considering the VEGF‐induced formation of the trimeric PlexinD1‐VEGFR2‐Nrp1 complex in ECs, further studies on the context‐dependent holoreceptor formation are required.
Upon VEGF stimulation, RhoJ facilitated PlexinD1‐VEGFR2 association in ECs, followed by the recruitment of Nrp1 pre‐coupled with PlexinD1, which may partly underlie the less requirement of direct binding of VEGF to Nrp1 for vascular development (Fantin et al, 2014; Gelfand et al, 2014). While VEGF stimulation increased internalization of the trimeric holoreceptor complex to perinuclear endosomes, protein levels of only VEGFR2, but not PlexinD1 and Nrp1, were decreased, indicating degradation of VEGFR2 and recycling of the PlexinD1‐Nrp1 complex after the termination of signal transduction. Notably, this VEGFR2 degradation was accelerated and delayed by siRNA knockdown of PlexinD1 and Nrp1, respectively, suggesting that the C‐terminal PDZ‐binding motifs of PlexinD1 and Nrp1 may inversely regulate the endocytic trafficking of VEGFR2 via GIPC1 and myosin VI (Simons et al, 2016; Burk et al, 2017; Carretero‐Ortega et al, 2019). In VEGF‐stimulated ECs, RhoJ‐mediated PlexinD1‐VEGFR2 association was required for sustained activation of PLCγ, Erk, and Akt, but not p38 MAPK, by preventing VEGFR2 degradation. These observations indicated that RhoJ facilitates forward EC migration by maximizing VEGFR2 activation. Activation of Arf6 is also implicated in Sema3E‐induced cell contraction as well as the VEGF‐induced VEGFR2‐Nrp1 association and VEGFR2 trafficking in ECs (Sakurai et al, 2010, 2011; Zhu et al, 2017). This similarity of RhoJ and Arf6 raises the possibility of their cooperative or compensatory functions downstream of Sema3E and VEGF signals.
In ECs, VEGF mediates the conversion of RhoJ to its GDP‐bound state partly via the Cdc42 GEF Arhgef15 (Fukushima et al, 2011; Kusuhara et al, 2012). In the present study, we used the T35N RhoJ mutant as a “GDP‐locked” DN form (Vignal et al, 2000; Abe et al, 2003), because the TN mutant of small GTPases abolishes the Mg2+ binding site, resulting in greatly reduced affinity for GTP, but not GDP, in in vitro culture conditions with low Mg2+ concentrations (Farnsworth & Feig, 1991). Intriguingly, GDP‐RhoJ was distinct from GTP‐RhoJ in terms of subcellular distributions and associations with VEGFR2 and PlexinD1 in cultured HUVECs. Ectopic GDP‐RhoJ mobilized VEGFR2 from its reservoir in the trans‐Golgi networks to cytoplasmic and nuclear puncta, which may be sites of protein degradation (Nethe & Hordijk, 2010). Consequently, ectopic GDP‐RhoJ decreased the protein levels of VEGFR2. In this respect, GDP‐RhoJ is not merely an “inactive” form, but instead it actively facilitates VEGFR2 degradation via proteasomal or lysosomal pathways. Taken together, we propose that RhoJ acts as a rate‐limiting system of VEGFR2 signal transduction by dual modes: the prevention of VEGFR2 degradation in its GTP‐bound form, and the promotion of VEGFR2 degradation in its GDP‐bound form. While degradation of GDP‐RhoJ may be regulated by its ubiquitylation (Nethe & Hordijk, 2010), the specific GAPs that mediate GTP hydrolysis of RhoJ downstream of VEGF and how GDP‐RhoJ associates with VEGFR2 should be clarified in further studies.
Considering that GDP‐RhoJ is prone to degradation without being recycled to the GTP‐bound status, RhoJ may exist as a GTP‐bound form by default. In this setting, the RBD of PlexinD1 may function as a reservoir of GTP‐RhoJ. As Sema3E inhibits VEGF‐induced reduction in GTP‐RhoJ (Fukushima et al, 2011), Sema3E‐mediated release of RhoJ from PlexinD1 may prevent consumption of the intracellular RhoJ pool by VEGF. This notion further indicates that the available amount of GTP‐RhoJ is pivotal for balancing the magnitude of VEGF and Sema3E signals in ECs. Nonetheless, retarded formation of retinal vasculature in RhoJ‐deficient mice was recovered at later stages, which may be explained by the compensatory roles of alternative small GTPases such as Arf6 or another Cdc42 subfamily member TC10 (RhoQ) (Heasman & Ridley, 2008). In the OIR model, however, endothelial RhoJ deficiency dramatically suppressed aberrant angiogenesis. This discrepancy may be, at least in part, ascribable to VEGF predomination over Sema3E in ischemic retina, which is also true in human eyes with diabetic retinopathy (Aiello et al, 1994; Kwon et al, 2015). Thus, RhoJ is a potential target for the efficient inhibition of pathological angiogenesis under excessive VEGF in neovascular eye diseases and cancer (Kim et al, 2014).
In addition to ECs, RhoJ was expressed in a subset of retinal neurons in the GCL. Although whether the RhoJ‐expressing neurons concurrently express PlexinD1 and VEGFR2 remains to be determined (Yang & Cepko, 1996; Fukushima et al, 2011), we envisage an involvement of RhoJ in the Sema3E and VEGF signals in the nervous system. Notably, RhoJ has been implicated in metastatic spreading of gastric cancer cells and melanoma cells (Kim et al, 2016; Ruiz et al, 2017). Furthermore, tumor invasiveness and metastatic spreading are promoted by binding of Sema3E to PlexinD1 and subsequent transactivation of the ErbB2 oncogenic kinase (Casazza et al, 2010). These notions suggest the possibility that RhoJ may facilitate Sema3E‐induced association of PlexinD1 with ErbB2 in metastatic tumor cells. RhoJ disruption may thus be effective for cancer therapy by suppressing not only angiogenesis (Kim et al, 2014) but also tumor cell migration. Alternatively, an agent targeting RhoJ‐PlexinD1 binding that interferes in the associations of PlexinD1 with VEGFR2 in ECs and ErbB2 in tumor cells may also be useful.
In conclusion, here we uncovered pleiotropic roles of RhoJ in the control of directional EC migration. First, GTP‐RhoJ mediated Sema3E‐induced cell contraction after its release from PlexinD1. Second, GTP‐RhoJ facilitated PlexinD1‐VEGFR2 association upon stimulation with Sema3E or VEGF, which subsequently activated distinct downstream signals. Third, GDP‐RhoJ terminated the signal transduction from internalized VEGFR2 by mobilizing it to the degradation pathway. Our present results suggest that RhoJ, or an alternative Rho GTPase, may act as a cell‐specific integrator of attractive and repulsive signals in directional cell migration and may thus be a therapeutic target in disease or tissue regeneration.
Materials and Methods
Cell culture
Primary HUVECs from pooled donors (Lonza) were seeded at a density of 1 × 104 cells/cm2 on 0.1% gelatin‐coated culture dishes (BD Falcon) and maintained at 37°C in endothelial growth medium EGM‐2 Bullet Kit (Lonza) before passage 7. HUVECs were serum‐starved with endothelial basal medium EBM‐2 (Lonza) containing 1% bovine serum albumin (BSA) 3–6 h prior to stimulation with 50 ng/ml human VEGF165 (HumanZyme) or 500 ng/ml human Sema3E (R&D) recombinant proteins. To inhibit VEGFR2 tyrosine kinase activity, HUVECs were incubated with 5 μM of SU5416 (Selleck) 2 h prior to stimulation with VEGF. COS‐7 and 293T cells were cultured at 37°C in Dulbecco's modified Eagle medium (DMEM, Sigma‐Aldrich) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin.
Migration assay
For the directional cell migration assay, HUVECs were seeded at a density of 1.5 × 106 cells/cm2 into a μ‐Slide Chemotaxis2D chamber (ibidi) according to the manufacturer's instructions. At the start of imaging, the upper and lower reservoirs of the chamber were filled with 50 ng/ml VEGF165 and 500 ng/ml Sema3E proteins, respectively. Chemotactic gradients were then generated by diffusion in the cell‐cultured area (Isfort et al, 2011). Images were acquired every 10 min for 12 h with a 5 × objective on an Axio Observer microscope (Zeiss) equipped with a CO2‐ and temperature‐controlled incubator (Incubation System XL, Zeiss). Individual cells were manually tracked using the MTrackJ plugin (Meijering et al, 2012) of ImageJ (version 1.48, NIH). Only viable and visible cells were tracked throughout sequential frames of a series of images. The datasets from each cell were analyzed to generate trajectory plots and rose diagrams to evaluate directional migration using the Chemotaxis and Migration Tool plugin (ibidi) of ImageJ. The efficiency of directional cell migration was evaluated by the x‐ and y‐forward migration index, which was defined as the ratio of displacement in the direction of the gradient (i.e., parallel to the x‐ or y‐axis) to the total path length (Foxman et al, 1999). The directional preference was evaluated by Rayleigh test. To inhibit p38 phosphorylation, HUVECs were incubated with 10 μM of SB203580 (Wako) 2 h prior to exposure to Sema3E gradients.
Gene silencing by siRNA
HUVECs were washed with Opti‐MEM (Life Technologies) and then transfected with 30 nM of pre‐designed Stealth RNAi siRNA duplexes (PlexinD1, HSS118224; VEGFR2, HSS105791; Nrp1, HSS113022; RhoJ, HSS126217; and HSS183814, Life Technologies) using Lipofectamine RNAiMAX (Life Technologies). Non‐targeting siRNA (Stealth RNAi Negative Control Medium GC Duplex, Life Technologies) was used as a negative control. The transfection medium was changed to complete culture medium after 4 h. Cells were re‐transfected with siRNAs 48 h after the initial transfection and cultured for another 48 h.
Transfection of plasmid vectors
A cDNA cassette encoding myc‐tagged mouse PlexinD1 (PD1), PD1ΔECD, or PD1ΔRBD was replaced with an EGFP cassette in a pEGFP‐N1 plasmid vector (Clontech). A cDNA cassette encoding EGFP, human VEGFR2, or human WT‐RhoJ, CA‐RhoJ (Q79L), DN‐RhoJ (T35N), or ΔCAAX‐RhoJ tagged with HA or GFP was subcloned into a pCAGGS plasmid vector (Kusuhara et al, 2012). The PD1ΔRBD and ΔCAAX‐RhoJ constructs were generated using the QuikChange II XL Site‐Directed Mutagenesis Kit (Agilent Technologies). The RFP‐tagged human Rab5 and DsRed‐tagged human Rab7 vectors were purchased from Addgene. Cultured cells were transfected with plasmid vectors using Lipofectamine 2000 (Life Technologies), in conjunction with PLUS Reagent (Life Technologies) for HUVECs. For co‐transfection experiments, equal amounts of each plasmid vector were used except for the DN‐RhoJ vector. Due to rapid DN‐RhoJ protein degradation, we used threefold and fivefold more of the DN‐RhoJ vector in microscopy and immunoblotting experiments, respectively.
Immunocytochemistry
HUVECs cultured in 35‐mm glass‐bottom dishes coated with type I collagen (Matsunami Glass) were fixed with 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS). Cells were then labeled with rabbit anti‐VEGFR2 (1:200, 55B11, Cell Signaling Technology), goat anti‐PlexinD1 (1:1,000, R&D), mouse anti‐Nrp1 (1:200, 446921, R&D), mouse anti‐PML (1:100, PG‐M3, Santa Cruz), mouse anti‐Rpt5 (1:200, TBP1‐19, Enzo Life Sciences), and mouse anti‐p230 trans‐Golgi (1:200, 611280, BD Biosciences) antibodies. Ectopically expressed HA‐tagged RhoJ was detected with a rat anti‐HA antibody (1:1,000, 3F10, Roche). The secondary antibodies were donkey IgGs conjugated with Alexa Fluor 488, Alexa Fluor 633 (Life Technologies), Cy3, and Cy5 (Jackson ImmunoResearch). Hoechst 33342 (Life Technologies) was used for nuclear staining. Images were taken with an LSM700 or LSM710 confocal microscope (Zeiss). Super‐resolution images were acquired using an LSM 880 with Airyscan confocal microscope (Zeiss). To monitor cell surface and internalized PlexinD1 proteins, serum‐starved HUVECs were incubated with 5 μg/ml goat anti‐PlexinD1 antibody (R&D) diluted in EBM‐2 containing 0.5% BSA for 30 min at 4°C. After washing with ice‐cold EBM‐2 to remove excess antibody, HUVECs were stimulated with 500 ng/ml Sema3E for 30 min at 37°C. Cells were then fixed with 4% PFA/PBS to detect surface PlexinD1, or washed with acid buffer (pH 2.2, 100 mM glycine, 20 mM magnesium acetate, and 50 mM KCl) for 1 min followed by washing with PBS and fixation with 4% PFA/PBS to detect internalized PlexinD1. The bound antibodies were detected with Cy3‐labeled donkey anti‐goat IgG. Images were taken with an Axio Observer microscope. For the Sema3E‐induced cell contraction assay, siRNA‐transfected HUVECs were stimulated with 500 ng/ml Sema3E for 30 min, fixed with 4% PFA/PBS, and labeled with Alexa Fluor 488‐conjugated phalloidin (Life Technologies). Fluorescence images were taken with an IX81 inverted microscope (Olympus). Areas of single HUVECs were measured using WinROOF software (version 6.5, Mitani Corp.). To analyze receptor colocalization, fluorescence intensity profiles across 5‐μm arbitrary lines in super‐resolution images acquired under a constant setting were measured using ZEN. The proportion of the protein colocalization was calculated by manually analyzing 30–100 vesicles positive for RhoJ (Fig 2B) or PlexnD1 (Figs 4E and 5A, and EV2B and 3B) per cell in confocal images. The proliferation index of cultured HUVECs was calculated as the proportion of EdU‐positive cells in > 400 Hoechst‐positive cells 4 h after stimulation with 50 ng/ml VEGF165. HUVECs incubated with 10 μM of EdU for 2 h were processed for Click Chemistry (Life Technologies) according to the manufacturer's instructions. Fluorescence images of COS‐7 cells co‐transfected with vectors expressing EGFP and RhoJ were taken with an IX81 inverted microscope.
Immunoprecipitation and immunoblotting
HUVECs were lysed with RIPA buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.5, 10 mM EDTA, and 1% NP40) containing Protease Inhibitor Cocktail (Nacalai Tesque), Phosphatase Inhibitor Cocktail (Nacalai Tesque), and 1 mM dithiothreitol (DTT, WAKO). After centrifugation at 13,000 × g for 10 min at 4°C, cell lysates for immunoprecipitation (IP) were incubated with Protein G Sepharose beads (GE Healthcare) and goat anti‐PlexinD1 (R&D) or rabbit anti‐VEGFR2 (55B11) antibodies overnight at 4°C. Lysis of 293T cells was performed with modified RIPA buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.5, 1 mM EGTA, 10 mM MgCl2, and 1% NP40) containing Protease Inhibitor Cocktail and 1 mM DTT. After centrifugation, cell lysates were incubated with Protein G Sepharose beads and goat anti‐GFP (GeneTex) or rabbit anti‐Myc (Abcam) antibodies. The immunoprecipitates and total cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to Immobilon‐P membranes (Millipore), and probed with the following antibodies: goat anti‐PlexinD1 (1:1,,000, R&D); rabbit anti‐VEGFR2 (1:2,000, 55B11); mouse anti‐Nrp1 (1:1,000, 446915, R&D); mouse anti‐phosphotyrosine (1:1,000, PY20, Enzo Life Sciences); rabbit anti‐phospho‐VEGFR2 Tyr1175 (1:1,000, 19A10, Cell Signaling Technology); rabbit anti‐phospho‐VEGFR2 Tyr1212 (1:1,000, 11A3, Cell Signaling Technology); rabbit anti‐phospho‐p38 (1:2,000, D3F9, Cell Signaling Technology); rabbit anti‐phospho‐PLCγ (1:2,000, Cell Signaling Technology); rabbit anti‐phospho‐Erk1/2 (1:2,000, D13.14.4E, Cell Signaling Technology); rabbit anti‐phospho‐Akt (1:2,000, 193H12, Cell Signaling Technology); rabbit anti‐p38 (1:2,000, D13E1, Cell Signaling Technology); rabbit anti‐PLCγ (1:2,000, D9H10, Cell Signaling Technology); rabbit anti‐Erk1/2 (1:2,000, Cell Signaling Technology); rabbit anti‐Akt (1:2,,000, Cell Signaling Technology); goat anti‐GFP (1:2,000, GeneTex); rabbit anti‐Myc (1:2,000, Abcam); rat anti‐HA (1:1,000, 3F10, Roche); and mouse anti‐β‐actin (1:2,000, AC‐15, Santa Cruz). HRP‐conjugated secondary antibodies (1:20,000, Wako) were used. Chemiluminescence signals were generated using ImmunoStar LD (Wako) or ImmunoStar Zeta (Wako) and detected with the Image Quant LAS 4000 (GE Healthcare) or ChemiDoc XR+ system (Bio‐Rad). The signal intensity was quantified using ImageJ. For quantifying the PlexinD1‐VEGFR2 complex, the ratio of co‐IPed VEGFR2 to IPed PlexinD1 (Fig 4D), and co‐IPed PlexinD1 to IPed VEGFR2 (Fig 5B) at each time point was measured and then normalized to the value at 0 min.
Animals
All animal research was conducted in compliance with the ARVO statement for the use of animals in ophthalmic and vision research and was approved by the Institutional Animal Care and Use Committee at Kobe University, Osaka University, the University of Tokyo, RIKEN CDB, and Nagoya City University. Rhoj GFP/GFP mice (Kim et al, 2014) were obtained by intercrosses or in vitro fertilization using heterozygous Rhoj GFP/WT males and females in a C57BL/6 background. Mosaic‐like Cre‐loxP‐mediated genetic recombination in postnatal retinas was induced by intraperitoneal (i.p.) injection of 10 μl of 1 mg/ml 4‐hydroxytamoxifen (4OHT, Sigma‐Aldrich) dissolved in 1:9 ethanol/sunflower oil (Sigma‐Aldrich) into CAG‐MerCreMer:R26R‐EYFP flox/WT or CAG‐MerCreMer:Rhoj flox/flox mice (Srinivas et al, 2001; Egawa et al, 2009; Kim et al, 2014) in a C57BL/6 background once at P1. After genetic recombination, the R26R‐EYFP allele expresses EYFP, while the Rhoj‐flox allele expresses EGFP with RhoJ loss of function. For tamoxifen‐inducible Rhoj deletion in ECs (Rhoj iΔEC), Pdgfb‐iCreERT2:Rhoj flox/flox mice (Claxton et al, 2008) in a C57BL/6 background were i.p. injected with 10 μl of 10 mg/ml 4OHT once at P1 or with 20 μl of 10 mg/ml 4OHT daily from P12 to P17 in the OIR model. In these experiments, Rhoj flox/flox mice without the Pdgfb‐iCreERT2 allele were used as controls. The OIR model was generated by maintaining mouse pups under 75% oxygen concentration from P7 to P12 using a ProOX oxygen controller (BioSpherix). Mice were not subjected to any prior procedures. All mice were fed with unlimited access to food and water and housed in specific pathogen‐free condition.
Ex vivo angiogenesis model
The ex vivo aortic ring assay was performed as previously described (Arima et al, 2011). Briefly, aortic rings were embedded in cell‐matrix type I collagen gel (Nitta Gelatin) on 8‐well Lab‐Tek II Chambered Coverglass (Nunc) and cultured in Medium‐199 containing 5% FBS, 100 U/ml penicillin, 10 μg/ml streptomycin, and 50 ng/ml human VEGF165 recombinant protein (R&D). ECs were visualized with FITC‐conjugated Isolectin B4 (5 μg/ml, Sigma‐Aldrich). Time‐lapse live imaging was started on the 5th or 6th day of the assay. For selective EC tracking, nuclei were labeled with the fluorescent probe SYTO‐16 (Life Technologies). Time‐lapse images were taken at 10‐μm intervals in the z‐axis every 15 min over 36 h with a 10 × objective on a FluoView FV10i confocal laser scanning microscope (Olympus).
In silico analysis of ex vivo angiogenesis
Cell tracking, data extraction, and data analysis were performed using ImageJ, MTrackJ plugin, and MATLAB (MathWorks) as previously described (Arima et al, 2011). Briefly, the nuclei of ECs stained with SYTO were manually selected and tracked in accordance with defined rules. The term “tip cells” refers to “cells at the tip of a certain time point”. The other non‐tip cells were termed “stalk cells”. The area of analysis was defined for use in the following kinetic assays by setting four coordinates that surround all nuclei in a branch. The axis and direction of elongation were determined by the coordinates of the tip at the first and last observed time points, and the coordinates of each cell were calculated by orthogonal projection to the axis of elongation. The normalized coordinates were plotted against time in the trajectory analysis, which enabled the visualization of single‐ and multi‐cellular movements in an elongating branch. “Vessel elongation” was determined as net elongation of a branch within an analyzed period. For cell kinetic analyses at stalks, the percentages of ECs migrating forward or backward were calculated as a ratio to the total number of cells throughout the observation period.
ELISA
Sema3E levels in the culture supernatants from the ex vivo angiogenesis model were assessed using an ELISA Kit for mouse Sema3E (USCN Life Science Inc.) according to the manufacturer's instructions. Serially diluted mouse Sema3E proteins (R&D) were used as controls. Absorbance at 450 nm was measured using a spectrophotometer (Ultrospec Visible Plate Reader II 96, GE Healthcare).
Immunohistochemistry
Whole‐mount immunohistochemistry of mouse embryos and retinas was performed as previously described (Fukushima et al, 2011). The primary antibodies were rat anti‐CD31 (1:500, Mec13.3, BD Biosciences), rabbit anti‐GFP (1:1,000, Life Technologies), goat anti‐GFP (1:1,000, GeneTex), rabbit anti‐ERG (1:1,000, EPR3864, Epitomics), and Cy3‐conjugated mouse anti‐α‐smooth muscle actin (1:1,000, 1A4, Sigma‐Aldrich). Signals were detected with donkey IgGs conjugated with Alexa Fluor 488, Alexa Fluor 647 (Life Technologies), Cy3, and Cy5. For the EdU incorporation assay, 100 μl (P4 pups) or 300 μl (P17 OIR) of 1 mg/ml EdU in PBS was i.p. injected 2 h before enucleation, and whole‐mount retinas were processed for Click Chemistry. Fluorescence images of retinas were taken with an LSM700 or LSM710 confocal microscope or an Axio Observer microscope, and images of embryos were acquired using an IX81 inverted microscope. For coloring reaction using diaminobenzidine (Dojindo), whole‐mount embryos were labeled with biotin‐conjugated rat anti‐CD31 antibody (1:500, Mec13.3), followed by incubation with avidin–biotin–peroxidase complex (Vector Laboratories). Images were taken with a Leica MZ10F stereomicroscope (Leica).
Morphometric analysis of retinal vasculature
Images of retinal quadrants were combined using the photomerge utility of Photoshop CS5 Extended software. The radius of the retinal vasculature was calculated by averaging the distances from the optic disk to the sprouting vascular fronts in four quadrants per retina. The number of vessel branch points was calculated in a fixed field (1.5 × 105 μm2) behind the sprouting vascular fronts in four quadrants and averaged per retina. The vascular density in postnatal mice was measured as a proportion of the CD31‐positive area in the total retinal area encircled by the growing vascular fronts using WinROOF software. The vascular areas in each retinal layer of 6‐week‐old mice were measured as the CD31‐positive areas in a fixed field (1.0 × 105 μm2) using Photoshop CS5 Extended software. Areas of neovascular tufts in the OIR model were measured using the Lasso tool of Photoshop software as previously described (Connor et al, 2009). The EC proliferation index was calculated as the proportions of ERG/EdU double‐positive ECs to total ERG‐positive ECs (> 20 ECs for capillaries and > 50 ECs for veins in P4 retina; > 40 ECs for neovascular tufts in P17 OIR retina). For each group, three to four quadrants per retina from three animals were analyzed. To assess the relative EC contribution to the tip positions after mosaic‐like genetic recombination, the proportions of GFP‐positive tip ECs to total GFP‐positive ECs were calculated in a fixed field (1.0 × 105 μm2) ranging from the angiogenic fronts to the second or third vascular loops in each quadrant per retina and corrected for those of GFP‐negative ECs in the same field. The values for CAG‐MerCreMer:R26R‐EYFP flox/WT were normalized to 1.
Reverse transcription PCR (RT–PCR)
From total RNAs prepared using Isogen (Nippon Gene) or an RNeasy Mini Kit (Qiagen), cDNAs were synthesized using the SuperScript III First‐Strand Synthesis System for RT–PCR (Life Technologies). Conventional RT–PCR (Fig EV4A) was performed with a HotStarTaq Master Mix Kit (Qiagen). Quantitative RT–PCR (Figs 7G and EV3J) was performed with SYBR Premix DimerEraser (Perfect Real Time, Takara Bio) using the ABI Prism 7500 Fast Sequence Detection System (Life Technologies). The mRNA values were normalized to actb or GAPDH mRNA levels. Primers were as follows: Vegfa, 5′‐GTACCTCCACCATGCCAAGT‐3′ and 5′‐GCATTCACATCTGCTGTGCT‐3′; Sema3e, 5′‐GGGGCAGATGTCCTTTTGA‐3′ and 5′‐AGTCCAGCAAACAGCTCATTC‐3′; Actb, 5′‐GGCTGTATTCCCCTCCATCG‐3′ and 5′‐CCAGTTGGTAACAATGCCATGT‐3′; KDR, 5′‐ACCCACGTTTTCAGAGTTGG‐3′ and 5′‐TCCAGAATCCTCTTCCATGC‐3′; and GAPDH, 5′‐ACCACAGTCCATGCCATCAC‐3′ and 5′‐TCCACCACCCTGTTGCTGTA‐3′.
Statistical analysis
Statistical analysis was performed with JMP Pro (version 11.2.0, SAS Institute Inc.). No sample size estimation, randomization, or stratification of the data was performed. For two‐group comparison, an unpaired two‐tailed Student's t‐test or Mann–Whitney U‐test was used. For multiple‐group comparison, ANOVA with post hoc Tukey–Kramer test or Kruskal–Wallis test with post hoc Steel–Dwass test was used. Error bars represent mean ± SEM. Statistical significance was set at P < 0.05.
Author contributions
YF performed the experiments and analyzed the data with help from KNishida. KNishiyama performed ex vivo angiogenesis assay and analyzed the data with help from HKu. HKa and S‐IN contributed to the generation of the Rhoj mutant mice. MF provided the Pdgfb‐iCreERT2 mice. SF and NM contributed to the design of the cell culture and biochemical experiments. AU conceived the study, designed the experiments, interpreted the data, and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank F. Costantini (Columbia University, USA) for the R26R‐EYFP mice; M. Shibuya (Jobu University, Japan), M. Negishi (Kyoto University, Japan), and I. Oinuma (University of Hyogo, Japan) for plasmid vectors; and H. Ishizaki and M. Kumai (KAN Research Institute, Inc., Japan) for in vitro fertilization of the Rhoj mutant mice. This work was supported by grants to AU from JSPS KAKENHI (25293078, 16H05155, 16K15737, and 19H03437), JST CREST “Spontaneous pattern formation ex vivo” (JPMJCR14W4), the Takeda Science Foundation, and the Eye Research Foundation for the Aged. We thank S.T. Fraser (University of Sydney, Australia) and Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
EMBO Journal (2020) 39: e102930
References
- Abe T, Kato M, Miki H, Takenawa T, Endo T (2003) Small GTPase Tc10 and its homologue RhoT induce N‐WASP‐mediated long process formation and neurite outgrowth. J Cell Sci 116: 155–168 [DOI] [PubMed] [Google Scholar]
- Aghajanian H, Choi C, Ho VC, Gupta M, Singh MK, Epstein JA (2014) Semaphorin 3d and semaphorin 3e direct endothelial motility through distinct molecular signaling pathways. J Biol Chem 289: 17971–17979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE et al (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 331: 1480–1487 [DOI] [PubMed] [Google Scholar]
- Arima S, Nishiyama K, Ko T, Arima Y, Hakozaki Y, Sugihara K, Koseki H, Uchijima Y, Kurihara Y, Kurihara H (2011) Angiogenic morphogenesis driven by dynamic and heterogeneous collective endothelial cell movement. Development 138: 4763–4776 [DOI] [PubMed] [Google Scholar]
- Bellon A, Luchino J, Haigh K, Rougon G, Haigh J, Chauvet S, Mann F (2010) VEGFR2 (KDR/Flk1) signaling mediates axon growth in response to semaphorin 3E in the developing brain. Neuron 66: 205–219 [DOI] [PubMed] [Google Scholar]
- Burk K, Mire E, Bellon A, Hocine M, Guillot J, Moraes F, Yoshida Y, Simons M, Chauvet S, Mann F (2017) Post‐endocytic sorting of Plexin‐D1 controls signal transduction and development of axonal and vascular circuits. Nat Commun 8: 14508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero‐Ortega J, Chhangawala Z, Hunt S, Narvaez C, Menendez‐Gonzalez J, Gay CM, Zygmunt T, Li X, Torres‐Vazquez J (2019) GIPC proteins negatively modulate Plexind1 signaling during vascular development. Elife 8: e30454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A, Finisguerra V, Capparuccia L, Camperi A, Swiercz JM, Rizzolio S, Rolny C, Christensen C, Bertotti A, Sarotto I et al (2010) Sema3E‐Plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J Clin Invest 120: 2684–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvet S, Cohen S, Yoshida Y, Fekrane L, Livet J, Gayet O, Segu L, Buhot MC, Jessell TM, Henderson CE et al (2007) Gating of Sema3E/PlexinD1 signaling by neuropilin‐1 switches axonal repulsion to attraction during brain development. Neuron 56: 807–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela‐Arispe ML (2010) Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol 188: 595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala‐Dilke K, Fruttiger M (2008) Efficient, inducible Cre‐recombinase activation in vascular endothelium. Genesis 46: 74–80 [DOI] [PubMed] [Google Scholar]
- Connor KM, Krah NM, Dennison RJ, Aderman CM, Chen J, Guerin KI, Sapieha P, Stahl A, Willett KL, Smith LE (2009) Quantification of oxygen‐induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc 4: 1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa G, Osawa M, Uemura A, Miyachi Y, Nishikawa S (2009) Transient expression of ephrin b2 in perinatal skin is required for maintenance of keratinocyte homeostasis. J Invest Dermatol 129: 2386–2395 [DOI] [PubMed] [Google Scholar]
- Fantin A, Herzog B, Mahmoud M, Yamaji M, Plein A, Denti L, Ruhrberg C, Zachary I (2014) Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF‐A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development 141: 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth CL, Feig LA (1991) Dominant inhibitory mutations in the Mg(2 + )‐binding site of RasH prevent its activation by GTP. Mol Cell Biol 11: 4822–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxman EF, Kunkel EJ, Butcher EC (1999) Integrating conflicting chemotactic signals. The role of memory in leukocyte navigation. J Cell Biol 147: 577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Gilmour D (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10: 445–457 [DOI] [PubMed] [Google Scholar]
- Fukushima Y, Okada M, Kataoka H, Hirashima M, Yoshida Y, Mann F, Gomi F, Nishida K, Nishikawa S, Uemura A (2011) Sema3E‐PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J Clin Invest 121: 1974–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay CM, Zygmunt T, Torres‐Vazquez J (2011) Diverse functions for the semaphorin receptor PlexinD1 in development and disease. Dev Biol 349: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand MV, Hagan N, Tata A, Oh WJ, Lacoste B, Kang KT, Kopycinska J, Bischoff J, Wang JH, Gu C (2014) Neuropilin‐1 functions as a VEGFR2 co‐receptor to guide developmental angiogenesis independent of ligand binding. Elife 3: e03720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D et al (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161: 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Lu MM, Epstein JA (2004) PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev Cell 7: 107–116 [DOI] [PubMed] [Google Scholar]
- Gu C, Yoshida Y, Livet J, Reimert DV, Mann F, Merte J, Henderson CE, Jessell TM, Kolodkin AL, Ginty DD (2005) Semaphorin 3E and plexin‐D1 control vascular pattern independently of neuropilins. Science 307: 265–268 [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701 [DOI] [PubMed] [Google Scholar]
- Isfort K, Ebert F, Bornhorst J, Sargin S, Kardakaris R, Pasparakis M, Bahler M, Schwerdtle T, Schwab A, Hanley PJ (2011) Real‐time imaging reveals that P2Y2 and P2Y12 receptor agonists are not chemoattractants and macrophage chemotaxis to complement C5a is phosphatidylinositol 3‐kinase (PI3K)‐ and p38 mitogen‐activated protein kinase (MAPK)‐independent. J Biol Chem 286: 44776–44787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12: 943–953 [DOI] [PubMed] [Google Scholar]
- Kaur S, Leszczynska K, Abraham S, Scarcia M, Hiltbrunner S, Marshall CJ, Mavria G, Bicknell R, Heath VL (2011) RhoJ/TCL regulates endothelial motility and tube formation and modulates actomyosin contractility and focal adhesion numbers. Arterioscler Thromb Vasc Biol 31: 657–664 [DOI] [PubMed] [Google Scholar]
- Kim J, Oh WJ, Gaiano N, Yoshida Y, Gu C (2011) Semaphorin 3E‐Plexin‐D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev 25: 1399–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Yang H, Fukushima Y, Saw PE, Lee J, Park JS, Park I, Jung J, Kataoka H, Lee D et al (2014) Vascular RhoJ is an effective and selective target for tumor angiogenesis and vascular disruption. Cancer Cell 25: 102–117 [DOI] [PubMed] [Google Scholar]
- Kim C, Yang H, Park I, Chon HJ, Kim JH, Kwon WS, Lee WS, Kim TS, Rha SY (2016) Rho GTPase RhoJ is associated with gastric cancer progression and metastasis. J Cancer 7: 1550–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Claesson‐Welsh L (2012) Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2: a006502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger RP, Aurandt J, Guan KL (2005) Semaphorins command cells to move. Nat Rev Mol Cell Biol 6: 789–800 [DOI] [PubMed] [Google Scholar]
- Kusuhara S, Fukushima Y, Fukuhara S, Jakt LM, Okada M, Shimizu Y, Hata M, Nishida K, Negi A, Hirashima M et al (2012) Arhgef15 promotes retinal angiogenesis by mediating VEGF‐induced Cdc42 activation and potentiating RhoJ inactivation in endothelial cells. PLoS ONE 7: e45858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SH, Shin JP, Kim IT, Park DH (2015) Aqueous levels of angiopoietin‐like 4 and semaphorin 3E correlate with nonperfusion area and macular volume in diabetic retinopathy. Ophthalmology 122: 968–975 [DOI] [PubMed] [Google Scholar]
- Lallemand‐Breitenbach V, de The H (2010) PML nuclear bodies. Cold Spring Harb Perspect Biol 2: a000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I (2012) Methods for cell and particle tracking. Methods Enzymol 504: 183–200 [DOI] [PubMed] [Google Scholar]
- Nethe M, Hordijk PL (2010) The role of ubiquitylation and degradation in RhoGTPase signalling. J Cell Sci 123: 4011–4018 [DOI] [PubMed] [Google Scholar]
- Olsson AK, Dimberg A, Kreuger J, Claesson‐Welsh L (2006) VEGF receptor signalling ‐ in control of vascular function. Nat Rev Mol Cell Biol 7: 359–371 [DOI] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146: 873–887 [DOI] [PubMed] [Google Scholar]
- Richards M, Hetheridge C, Mellor H (2015) The formin FMNL3 controls early apical specification in endothelial cells by regulating the polarized trafficking of podocalyxin. Curr Biol 25: 2325–2331 [DOI] [PubMed] [Google Scholar]
- Ruiz R, Jahid S, Harris M, Marzese DM, Espitia F, Vasudeva P, Chen CF, de Feraudy S, Wu J, Gillen DL et al (2017) The RhoJ‐BAD signaling network: an Achilles’ heel for BRAF mutant melanomas. PLoS Genet 13: e1006913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Gavard J, Annas‐Linhares Y, Basile JR, Amornphimoltham P, Palmby TR, Yagi H, Zhang F, Randazzo PA, Li X et al (2010) Semaphorin 3E initiates antiangiogenic signaling through plexin D1 by regulating Arf6 and R‐Ras. Mol Cell Biol 30: 3086–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Jian X, Lee CJ, Manavski Y, Chavakis E, Donaldson J, Randazzo PA, Gutkind JS (2011) Phosphatidylinositol‐4‐phosphate 5‐kinase and GEP100/Brag2 protein mediate antiangiogenic signaling by semaphorin 3E‐plexin‐D1 through Arf6 protein. J Biol Chem 286: 34335–34345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semaphorin Nomenclature Committee (1999) Unified nomenclature for the semaphorins/collapsins. Cell 97: 551–552 [DOI] [PubMed] [Google Scholar]
- Simons M, Gordon E, Claesson‐Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17: 611–625 [DOI] [PubMed] [Google Scholar]
- Smith LE, Wesolowski E, McLellan A, Kostyk SK, D'Amato R, Sullivan R, D'Amore PA (1994) Oxygen‐induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 35: 101–111 [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararaman A, Fukushima Y, Norman JC, Uemura A (2020) RhoJ regulates α5β1 integrin trafficking to control fibronectin remodeling during angiogenesis. Curr Biol 10.1016/j.cub.2020.03.042 [DOI] [PubMed] [Google Scholar]
- Takase H, Matsumoto K, Yamadera R, Kubota Y, Otsu A, Suzuki R, Ishitobi H, Mochizuki H, Kojima T, Takano S et al (2012) Genome‐wide identification of endothelial cell‐enriched genes in the mouse embryo. Blood 120: 914–923 [DOI] [PubMed] [Google Scholar]
- Tata A, Stoppel DC, Hong S, Ben‐Zvi A, Xie T, Gu C (2014) An image‐based RNAi screen identifies SH3BP1 as a key effector of Semaphorin 3E‐PlexinD1 signaling. J Cell Biol 205: 573–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Toledo M, Senic‐Matuglia F, Salamero J, Uze G, Comunale F, Fort P, Blangy A (2003) The GTP/GDP cycling of rho GTPase TCL is an essential regulator of the early endocytic pathway. Mol Biol Cell 14: 4846–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesugi K, Oinuma I, Katoh H, Negishi M (2009) Different requirement for Rnd GTPases of R‐Ras GAP activity of Plexin‐C1 and Plexin‐D1. J Biol Chem 284: 6743–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignal E, De Toledo M, Comunale F, Ladopoulou A, Gauthier‐Rouviere C, Blangy A, Fort P (2000) Characterization of TCL, a new GTPase of the rho family related to TC10 and Cdc42. J Biol Chem 275: 36457–36464 [DOI] [PubMed] [Google Scholar]
- Walchli T, Wacker A, Frei K, Regli L, Schwab ME, Hoerstrup SP, Gerhardt H, Engelhardt B (2015) Wiring the vascular network with neural cues: a CNS perspective. Neuron 87: 271–296 [DOI] [PubMed] [Google Scholar]
- Wang H, Hota PK, Tong Y, Li B, Shen L, Nedyalkova L, Borthakur S, Kim S, Tempel W, Buck M et al (2011) Structural basis of Rnd1 binding to plexin Rho GTPase binding domains (RBDs). J Biol Chem 286: 26093–26106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson E, Leszczynska K, Poulter NS, Edelmann F, Salisbury VA, Noy PJ, Bacon A, Rappoport JZ, Heath JK, Bicknell R et al (2014) RhoJ interacts with the GIT‐PIX complex and regulates focal adhesion disassembly. J Cell Sci 127: 3039–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Cepko CL (1996) Flk‐1, a receptor for vascular endothelial growth factor (VEGF), is expressed by retinal progenitor cells. J Neurosci 16: 6089–6099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Sacharidou A, Stratman AN, Le Bras A, Zwiers PJ, Spokes K, Bhasin M, Shih SC, Nagy JA, Molema G et al (2011) RhoJ is an endothelial cell‐restricted Rho GTPase that mediates vascular morphogenesis and is regulated by the transcription factor ERG. Blood 118: 1145–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Shi DS, Winter JM, Rich BE, Tong Z, Sorensen LK, Zhao H, Huang Y, Tai Z, Mleynek TM et al (2017) Small GTPase ARF6 controls VEGFR2 trafficking and signaling in diabetic retinopathy. J Clin Invest 4569–4582 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5