

Abstract
Recent years have seen a great expansion in our knowledge of the roles that metabolites play in cellular signaling. Structural data has provided crucial insights into mechanisms through which amino acids are sensed. New nutrient-coupled protein and RNA modifications have been identified and characterized. A growing list of functions have been ascribed to metabolic regulation of modifications such as acetylation, methylation, and glycosylation. A current challenge lies in developing an integrated understanding of the roles that metabolic signaling mechanisms play in physiology and disease, which will inform the design of strategies to target such mechanisms. In this brief article, we review recent advances in metabolic signaling through post-translational modification during cancer progression, in order to provide a framework for understanding signaling roles of metabolites in the context of cancer biology and illuminate areas for future investigation.
Introduction
Cellular metabolism functions to break down nutrients to generate smaller molecules and release energy (catabolism), as well as to synthesize larger molecules from smaller building blocks (anabolism). Certain metabolites generated during these processes can also exert signaling functions to modulate cellular activities in a manner responsive to nutritional status or metabolic shifts. In this brief article, our goal is to provide an update on advances from the last two years in our understanding of the roles of metabolites in signaling via post-translational modification, with a focus on cancer biology. Other diverse aspects of nutrient signaling have been covered in recent reviews [1–8]. Throughout the article, we highlight developing concepts that are reinforced by recent findings, including that: 1) nuclear metabolite production plays crucial roles in chromatin regulation; 2) systemic metabolism modulates tumor cell metabolic signaling; 3) acetyl-CoA and α-ketoglutarate (αKG) have emerged as important signaling molecules involved in multiple processes throughout tumorigenesis; and 4) metabolic signaling within the tumor microenvironment can impact therapeutic responses. We first examine recent findings in metabolic signaling during tumor initiation and primary tumor growth, and then in cancer progression and metastasis. We also discuss metabolic signaling in other cell types in the tumor microenvironment, and finally, the role of nuclear metabolite production in chromatin regulation. We conclude with perspectives on emerging concepts in metabolic signaling and how they can be explored in the context of cancer.
Metabolic Signaling Mechanisms in Tumor Initiation and Growth
The multistep process of cancer development begins with tumor initiation, in which cells acquire the requisite traits that allow them to form tumors, involving oncogene activation and tumor suppressor silencing. As cellular metabolism is remodeled during tumorigenesis to facilitate survival and anabolic growth [9,10], metabolites also exert signaling functions that contribute to tumor formation. A canonical example is seen in cancer-associated mutations in the genes encoding the metabolic enzymes isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2), which result in aberrant production of the oncometabolite, D-2-hydroxyglutarate (D-2-HG) [11,12]. D-2-HG acts as a competitive inhibitor of oxoglutarate-dependent dioxygenases [13], including epigenetic enzymes such as TET methylcytosine oxidases and JmjC histone demethylases. IDH mutations result in genome-wide epigenetic dysregulation, linked to malignant transformation [14]. Beyond D-2-HG, several other metabolites, including αKG, acetyl-CoA, and UDP-GlcNAc, have emerged as mediators of early tumorigenesis, at least in part via signaling mechanisms.
By way of its regulation of oxoglutarate-dependent dioxygenases, the ratio of αKG to succinate, a product and inhibitor of these enzymes, has emerged as a key node for modulating cell phenotypes, including stem cell pluripotency and macrophage polarization [14,15]. αKG has also been recently linked to p53-mediated tumor suppression. Using a mouse model of pancreatic cancer with a doxycycline-inducible shRNA targeting p53, p53 re-activation in tumors was found to induce differentiation and suppress tumor growth, at least in part via control of αKG accumulation [15]. Restoration of p53 function boosted the αKG:succinate ratio, and genetic and pharmacological augmentation of this ratio was sufficient to promote tumor differentiation. Conversely, high levels of succinate interfered with p53-dependent tumor suppression. Accumulation of 5-hydroxymethylcytosine (5hmc), indicative of elevated TET activity, correlated with the αKG:succinate ratio (Fig. 1). Though correlative, promotion of TET activity is a plausible mechanism of tumor suppression, since reduced 5hmc as a consequence of impaired TET function is frequently observed in cancers. TET enzymes are also thought to be a crucial target of both D-2-HG and its stereoisomer L-2-HG [13,16–18], as well as of succinate and fumarate, which accumulate in SDH and FH deficient tumors [1,19] (Fig. 1). Accumulation of L-2-HG in cancer cells can result from promiscuous activity of lactate dehydrogenase and malate dehydrogenase under hypoxia [20,21] or from loss of L-2-HG dehydrogenase function [22].
Figure 1. Metabolites regulate TET methylcytosine oxidases to impact tumor formation.
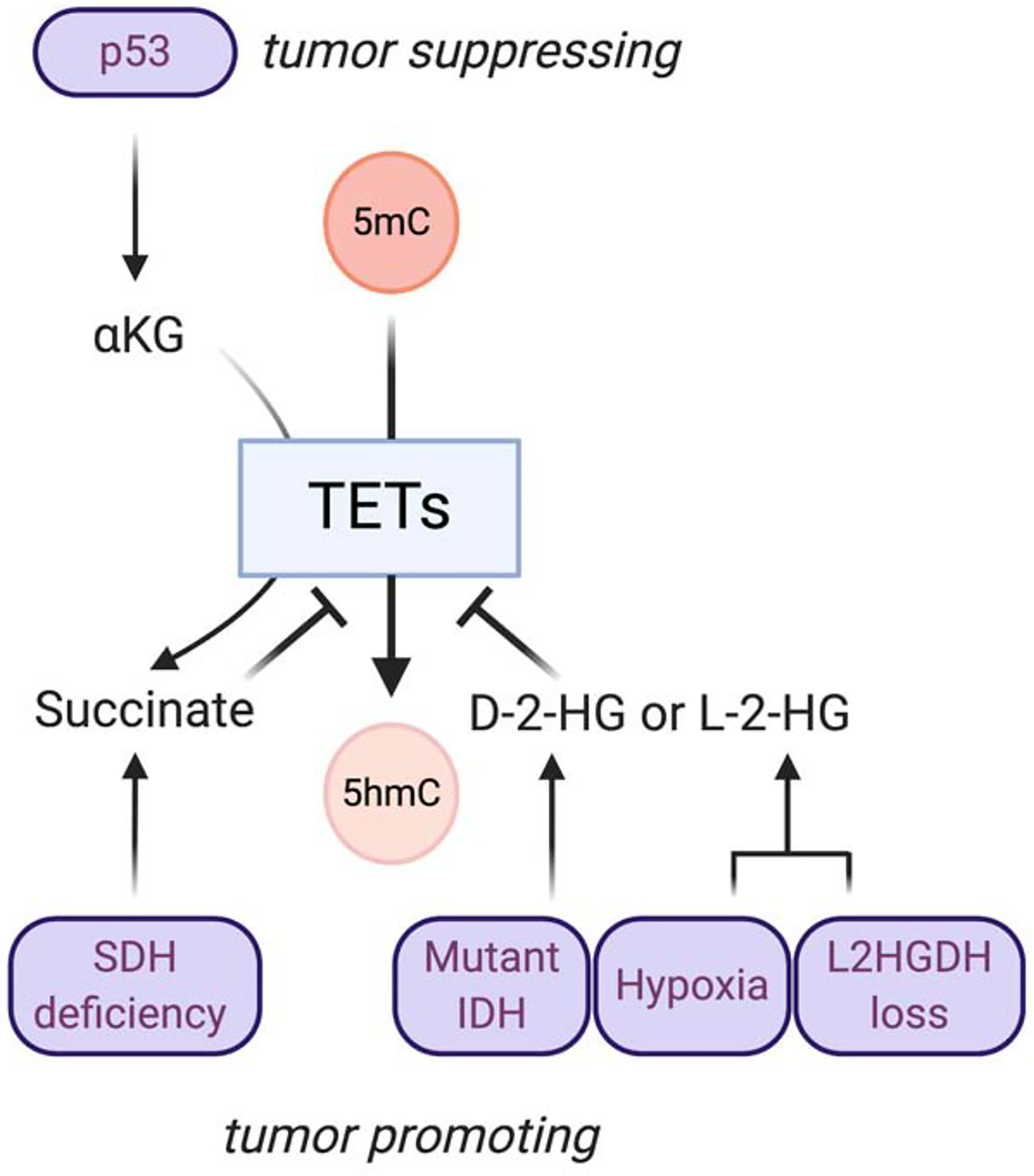
Metabolically-linked alterations in TET activity have been identified as consequences of both oncogene activation and tumor suppressor silencing. TET enzymes, which belong to the family of 2-oxoglutarate-dependent dioxygenases that rely on αKG as a cosubstrate, mediate the oxidation of 5-methylcytosine (5mC) to yield 5-hydroxymethylcytosine (5hmC) and subsequently 5-fC and 5-caC, to facilitate removal of DNA methylation. Oncogenic mutations in isocitrate dehydrogenase (IDH) lead to neomorphic activity and production of the oncometabolite D-2-hydroxyglutarate (D-2-HG). The stereoisomer L-2-HG can also be generated under hypoxia or accumulate upon loss of L-2-HG dehydrogenase activity. Succinate dehydrogenase (SDH) and fumarate hydratase (FH) loss of function drive accumulation of succinate and fumarate, respectively. 2-HG, succinate, and fumarate all inhibit the activity of TET enzymes. Conversely, increasing the αKG: succinate ratio, which has been identified as a component of the p53 tumor suppressive program[15], is associated with promotion of TET activity.
Other metabolites, including acetyl-CoA and pyruvate, have also been linked to tumor initiation. Acetyl-CoA was found to play a role in promoting pancreatic tumorigenesis [23]. Acetyl-CoA and global histone acetylation levels are elevated in murine pancreatic acinar cells harboring KRAS mutations, and genetic deletion of Acly suppressed acinar-to-ductal metaplasia in vitro and impaired tumor formation in vivo (Fig. 2). Pyruvate metabolism is intimately coupled to stem cell function [24,25], and inactivation of the mitochondrial pyruvate carrier was found to enhance intestinal tumor formation and promote gene expression signatures associated with stemness in adenomas[26]. Thus, metabolic reprogramming can facilitate tumor development, linked to regulation of chromatin modification and gene expression.
Figure 2. Acetyl-CoA mediates signaling functions throughout cancer progression.
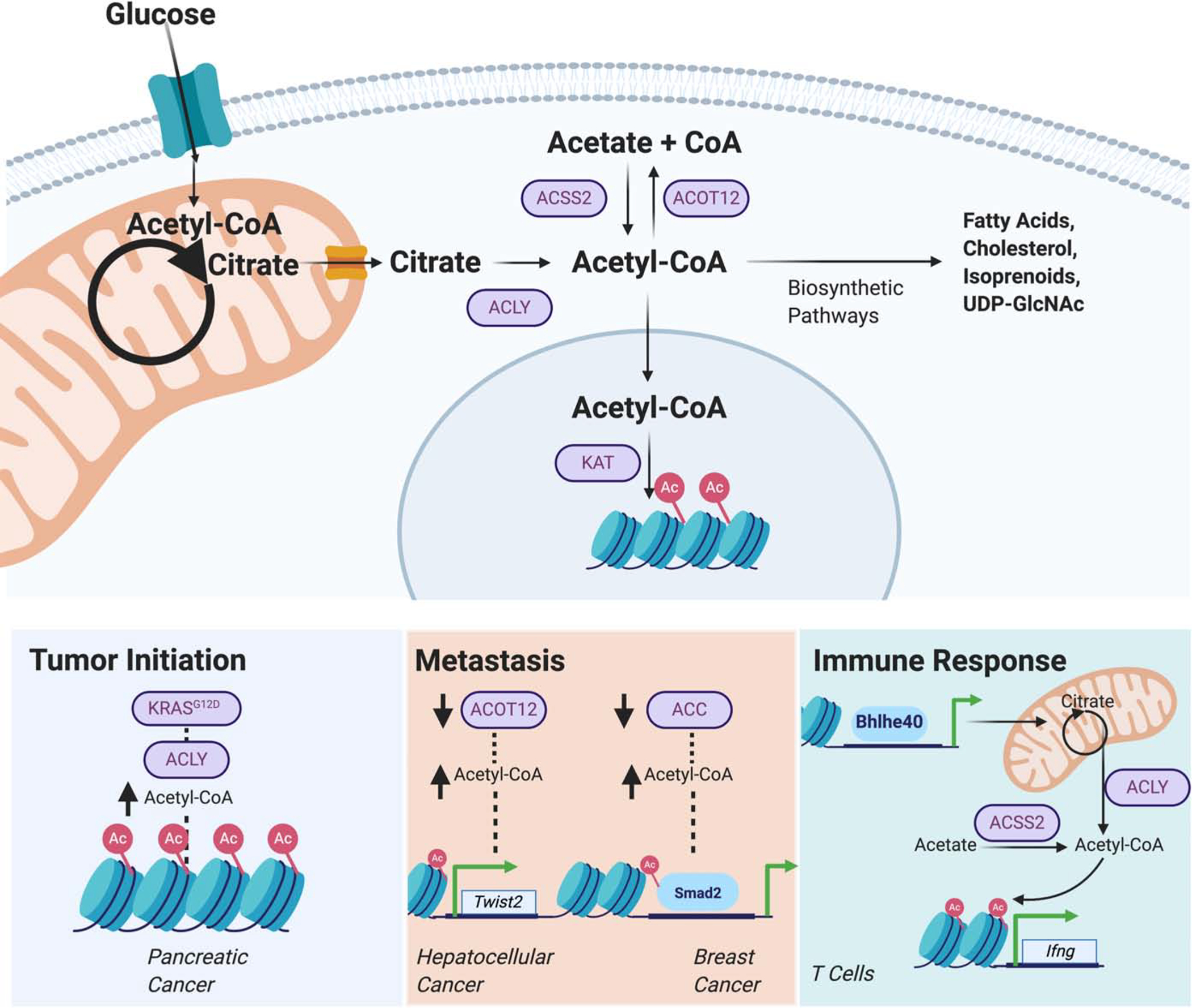
The signaling functions of metabolites have been implicated in multiple steps throughout tumorigenesis, including tumor initiation, metastasis, and in interaction with other cell types (immune, cancer-associated stroma). Here, acetyl-CoA is shown as a representative example. Nuclear-cytosolic acetyl-CoA is generated from mitochondria-derived citrate via ATP-citrate lyase (ACLY) or acetate via acyl-CoA synthetase short-chain family member 2 (ACSS2). This pool of acetyl-CoA feeds into multiple biosynthetic pathways and also serves as the sole substrate for acetylation reactions, mediated by lysine acetyl-transferases (KATs). ACLY-dependent elevations in acetyl-CoA and global histone acetylation were observed in KRAS mutant pancreatic acinar cells[23]. Accumulations in acetyl-CoA are also implicated in regulation of metastasis. In two models of cancer, inhibition of acetyl-CoA consuming enzymes acyl-CoA thioester 12 (ACOT12) and acetyl-CoA carboxylase (ACC) increased the expression or activity of EMT transcription factors Twist2 and Smad2 via histone or transcription factor acetylation, respectively[33,34]. Lastly, control of acetyl-CoA production in T cells, partly under the control of the transcription factor Bhlhe40, and histone acetylation are implicated in regulating IFNγ production and antitumor immunity[54,55,81].
Beyond the roles of cell autonomous metabolic alterations in cancer, epidemiological data identifies obesity as a risk factor for several cancers [27,28]. New evidence in mice suggests that hyperglycemia may increase DNA damage, resulting in KRAS mutations, in pancreatic cells [29]. This was attributed to a reduction in deoxyribonucleotides (dNTPs) due to glucose-sensitive O-GlcNAcylation and subsequent destabilization of the ribonucleotide reductase (RNR) complex, which generates dNTPs from their ribonucleotide counterparts. The O-GlcNAc modification depends on synthesis of UDP-N-acetyl-glucosamine (UDP-GlcNAc) through the hexosamine biosynthesis pathway and the subsequent transfer to the substrate protein by O-GlcNAc transferase (OGT) [30]. Although the determinants of OGT substrate specificity remain poorly understood, O-GlcNAcylation of specific enzymes has been proposed as a physiological mechanism to fine-tune metabolic regulatory pathways [31]. However, pathologic elevations of this modification, as observed under diabetic hyperglycemia, can have detrimental consequences, including the promotion of carcinogenesis. Interestingly, insulin stimulation of breast cancer cells was also associated with increased evidence of DNA damage [32]. Together, these emerging data suggest that increased mutational burden may be a contributing factor to the increased cancer incidence associated with hyperglycemia and hyperinsulinemia.
Metabolites Regulate Metastasis
Metabolites in the Transcriptional Regulation of EMT
Metastasis is a complex process that requires cells to be equipped with the ability to disseminate from primary tumors, survive in and extravasate from circulation, and ultimately colonize in distant sites. New studies demonstrate that the signaling roles of metabolites in metastasis may be multiple and context-dependent.
Two recent studies highlighted mechanistically distinct roles for acetyl-CoA in facilitating the transcriptional regulation of epithelial-mesenchymal transition (EMT) (Fig. 2). In investigating the adipokine leptin as a putative link between obesity and breast cancer pathogenesis, leptin-mediated activation of EMT was found to depend on AMPK-dependent phosphorylation of acetyl-CoA carboxylase (ACC), the rate limiting enzyme in fatty acid synthesis [33]. This inhibition of ACC led to an accumulation of acetyl-CoA and enhanced acetylation of the transcription factor Smad2, promoting invasiveness and activating EMT. A second mechanism through which acetyl-CoA promotes EMT was reported in hepatocellular carcinoma [34]. ACOT12, a predominantly cytosolic acetyl-CoA hydrolase which is expressed in healthy liver [35], is downregulated in tumors. Acetyl-CoA levels increased as a consequence of ACOT12 suppression, accompanied by elevated histone H3 acetylation at the Twist2 locus and expression of the encoded EMT transcription factor. The mechanisms by which such specificity in gene regulation is achieved remain to be further elucidated, although both of these studies align with a developing model that nuclear-cytosolic acetyl-CoA abundance modulates expression of specific sets of genes by controlling the levels or function of particular transcription factors [1,36,37].
Metabolites Define the Metastatic Niche
The final steps in the metastatic cascade involve colonization of cancer cells in a distant organ, which requires both remodeling of the extracellular matrix (ECM) and reprograming of other cell types within the local microenvironment [38]. Recent advances have revealed that nutrient availability and metabolic signaling may play a role in both of these processes, and thus help to define the metastatic niche.
In breast cancer cells, pyruvate uptake and metabolism can facilitate ECM remodeling at metastatic lung lesions by affecting collagen hydroxylation [39]. Stable collagen synthesis involves hydroxylation of proline residues on procollagen strands by the αKG-dependent collagen prolyl-4-hydroxylase (P4HA). Inhibition of pyruvate uptake in cancer cells reduced collagen hydroxylation, as well as lung metastases in vivo [39]. This effect was attributed to a decrease in production of αKG through alanine aminotransferase (ALT), which converts pyruvate and glutamate to αKG and alanine (Fig. 3). Remarkably, in vivo supplementation of αKG was sufficient to rescue collagen hydroxylation and metastatic burden in the lung. Notably, HIF1α, which is also prolyl hydroxylated via oxoglutarate-dependent enzymes to target HIF for degradation [40], transcriptionally regulates P4HA1 expression [41]. The activity of P4HA1 in consuming αKG for collagen hydroxylation has been proposed to limit HIF hydroxylation, resulting in its stabilization, and thereby promoting stemness and chemoresistance in breast cancer [42]. Thus, the availability of nutrients such as pyruvate at sites of metastasis may dictate regulation of and interplay between αKG-dependent enzymes, impacting ECM remodeling and cancer progression (Fig. 3). The notion that enzymes that consume αKG can restrict its availability to other enzymes is also supported by evidence in acute myeloid leukemia that overexpression of branched chain amino acid transaminase 1 (BCAT1), a reversible enzyme which transfers the α-amino group from BCAAs to αKG, depletes αKG pools, resulting in HIF1α stabilization and reduced TET activity [43]. As discussed, growing evidence supports the notion that metabolic signaling contributes to metastasis, though an outstanding question is whether distant signals from the primary tumor are involved in metabolically priming the metastatic niche. Although relatively little is known about this, glucose availability at sites of metastases has been proposed to be affected by secreted factors from the primary tumor [44].
Figure 3. Nutrient availability and αKG regulate aspects of metastatic biology in breast Cancer.
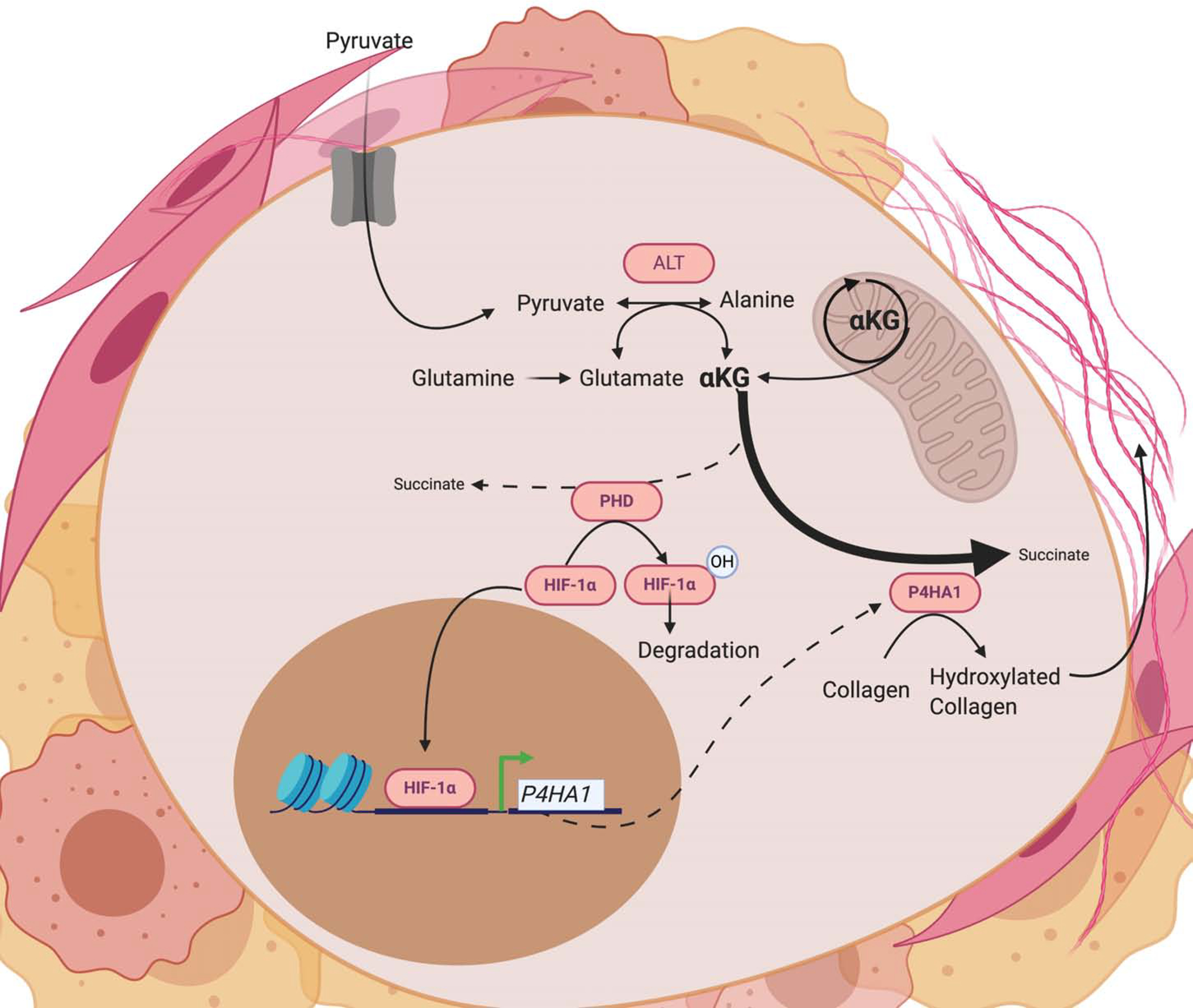
In metastatic breast cancer cells, αKG metabolism participates in extracellular matrix remodeling (ECM), stemness and chemoresistance. Pyruvate uptake promotes production of αKG through alanine aminotransferase (ALT). αKG is required by collagen prolyl-4-hydroxylase (P4HA) to hydroxylate procollagen for stable collagen synthesis in the process of ECM remodeling[39]. This shunting of αKG toward P4HA1 also limits HIF hydroxylation, resulting in stabilization and transcription of genes involved in chemoresistance and stemness[42].
Cancer-associated stromal cells also play a critical role in defining the metastatic niche. Proteomic analysis of matched primary vs metastatic ovarian cancer samples revealed a role for the metabolic enzyme nicotinamide N-methyltransferase (NNMT) in regulating gene expression in cancer associated fibroblasts (CAFs) [45]. NNMT, which is upregulated in the stroma of metastatic sites, depletes the universal methyl donor, S-adenosyl methionine (SAM), which it metabolizes to S-adenosyl homocysteine and a metabolically inactive product, 1-methylnicotinamide. High NNMT in cancer cells suppresses histone and DNA methylation, promoting altered gene expression and in glioblastoma, stem cell maintenance and tumor growth [46,47] Likewise, in CAFs, NNMT expression promoted hypomethylation of histones and DNA, as well as expression of cytokines and extracellular matrix components linked to metastasis [45]. Thus, metabolic signaling mechanisms can contribute to metastasis not only through direct roles in cancer cells but also within other cell types present in tumors.
Epigenetic Modifications Link Metabolites and Immune Function
Metabolic signals contribute to immune cell function and impact anticancer immunity, leading to efforts to target immune cell metabolism and to leverage cellular metabolism to optimize cancer immunotherapy [48]. As such, a critical issue in considering the roles of metabolic signals in cancer biology is how they play out within and between different cell types in the tumor microenvironment.
Acetyl-CoA metabolism and its links with histone acetylation represents a crucial node in T cell functioning. Building on prior work demonstrating roles for glucose and acetate metabolism and metabolic control of histone acetylation in regulating IFNγ production in T cells [49,50], the transcription factor Bhlhe40 was identified as a transcriptional regulator of mitochondrial metabolism in CD8+ T cells. Bhlhe40 was found to promote histone acetylation and IFNγ production, presumably via mitochondrial citrate export and ACLY-dependent acetyl-CoA production. Consistently, in a separate study, both the malate-aspartate shuttle and citrate export for acetyl-CoA synthesis were found to promote IFNγ production in CD4+ TH1 cells [51]. Notably, acetate, which can bypass the requirement for mitochondrial metabolism for acetyl-CoA production [52,53], could augment the production of IFNγ [54–56] in a manner that was further enhanced by histone deacetylase inhibition [55]. Conversely, silencing of ACSS2 in T cells suppressed IFNγ levels and impaired tumor-clearing [54]. ACSS2 has been proposed as a cancer therapeutic target [57], and its role in promoting anti-cancer immunity may be a caveat to targeting this enzyme. Interestingly, these pathways also appear to be relevant for maintenance of T cell stemness. High extracellular potassium, which is often present in tumors due to release of necrotic cell contents, impairs nutrient transport into T cells and results in a state of functional starvation that includes low acetyl-CoA and histone acetylation levels. Although these conditions limited expression of effector genes such as IFNγ, they also preserved T cell stemness and capacity for anti-tumor response [56], highlighting a nuanced role for nutrient regulation of T cell function in cancer.
Metabolic regulation of αKG-dependent enzymes is also an important determinant of T cell function. Glutaminase inhibition suppresses αKG levels in T cells, increasing global histone methylation and altering gene expression to promote TH1 specification [58]. TH17 differentiation was simultaneously impeded [58]. Analogously, complex III inhibition raised levels of succinate and 2-HG, promoting hypermethylation and interfering with the suppressive function of regulatory T cells [59]. Encouragingly, in vivo, inhibition of glutamine metabolism was found to impair cancer cell metabolic programs that promote an immunosuppressive environment, while also enhancing antitumor immune responses [60]. Glutamine metabolic blockade was linked to an upregulation of pyruvate carboxylase-dependent anaplerosis and acetate utilization in CD8+ T cells but not in cancer cells [60], pointing to the potential to optimize metabolic therapeutic strategies taking into account effects on both cancer cells and tumor-associated immune cells.
Compartmentalized Metabolism and Chromatin Modifications in Cancer
As links between metabolism and chromatin modification continue to emerge in the context of cancer progression, a central question has been the role of metabolic compartmentalization [61]. The role of compartmentalization is an obvious consideration for TCA cycle metabolites with nuclear signaling functions, such as αKG and succinate. Evidence has also emerged that nucleus and cytosol may be at least partially distinct metabolic compartments [62], even though small molecules can diffuse through nuclear pores. Empirical evidence for the nucleus as metabolically distinct from cytosol came from study of NAD+-dependent poly ADP-ribosylation (PARylation) during adipocyte differentiation. While whole cell NAD+ levels remained unchanged, a reduction in nuclear NAD+ was necessary for induction of an adipogenic transcription program [63]. Compartmentalized metabolite measurements were achieved using fluorescent biosensors, allowing monitoring of NAD+ levels in living cells [63].
Prior work has established the nuclear localization of multiple metabolic enzymes that produce acetyl-CoA (ACLY, ACSS2, PDC) [64–66] or are components of methionine or folate cycles, involved in SAM production (MAT2A, MTHFD1, SESAME complex) [67–69]. Recent studies have provided new insight into the potential pathways that may facilitate acetyl-CoA production within the nucleus. One study which investigated how histone acetyltransferase 1 (HAT1) coordinates growth-factor and nutrient-stimulated histone production proposed that the deacetylation of newly synthesized H4 histones after their nuclear translocation provides a local source of acetate for acetylation of promoter regions, including those encoding histone H4 genes [70] (Fig. 4). Another study investigating the functional significance of nuclear glycogen, which had been observed histologically in lung cancer cells, proposed that nuclear glycogen metabolism represents a nuclear source of acetyl-CoA [71]. Leveraging an isolated nuclei system, glycogen synthesis from glucose-6-phosphate and breakdown to pyruvate were found to occur within nuclei, with nuclear glycogenolysis contributing acetyl groups for histone acetylation (Fig. 4). Although this study did not document the mechanism of acetyl-CoA production from pyruvate, presumably this would require either pyruvate conversion to acetyl-CoA by a nuclear PDC [72] or generation of acetate from pyruvate [73] and subsequent acetyl-CoA production by ACSS2. The advantages of nuclear glycogen synthesis and breakdown to supply acetyl-CoA for histone acetylation versus more straightforward synthesis from citrate or acetate are not yet clear, though could potentially serve to maintain histone acetylation in the face of fluctuating nutrient availability. Nuclear glycogen accumulation may also provide other unknown benefits to the cancer cell.
Figure 4. Novel mechanisms toward the production of nuclear acetyl-CoA.
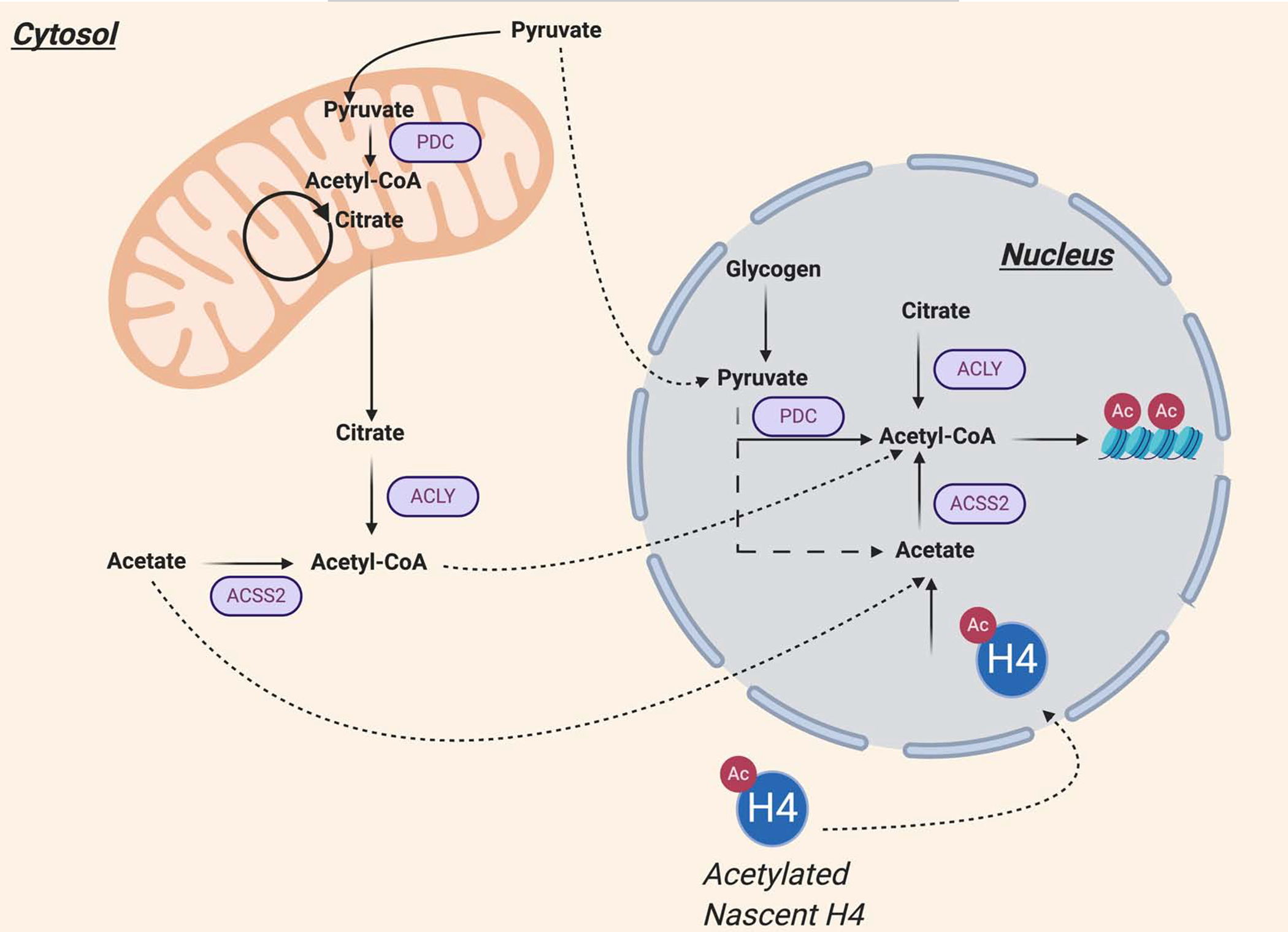
ACLY, ACSS2, and PDC have all been found within the nucleus for local and context-specific generation of acetyl-CoA. Breakdown of nuclear glycogen into pyruvate has been identified as a novel source of acetyl-CoA for histone acetylation [71]. Nuclear-generated pyruvate could in principle contribute to histone acetylation through PDC-dependent production of acetyl-CoA [72] or through non-enzymatic conversion of pyruvate to acetate and subsequent acetyl-CoA generation by ACSS2[73]. Nuclear transport and deacetylation of newly synthesized H4 histones has also been proposed as a source of acetate for acetylation of histones at promoters of histone H4 genes [70].
Conclusions and Emerging Questions
Within the past two years, several intriguing themes have emerged from the advances surrounding the signaling function of metabolites in cancer, as we discuss above, and the findings have equally raised many questions moving forward. One key question surrounds sources of particular metabolites in different contexts, both at the cellular and systemic level. For example, recent evidence points to ethanol metabolism as a source of acetate in the regulation of brain histone acetylation [74]. In addition to metabolites in circulation and produced intracellularly, metabolic crosstalk between cells within the tumor microenvironment likely participates in metabolic signaling between cancer cells, immune cells, and fibroblasts. A second important question surrounds the roles of the ever-growing list of acylations and other post-translational modifications that are metabolically sensitive [75]. For example, lysine lactylation, derived from lactate, was recently reported and may be mediated either enzymatically, by one or more acyltransferase, or non-enzymatically from lactoyl-glutathione [76,77]. Since lactate is produced abundantly and can be both exported and imported by cells, the modification offers a potential mechanism of crosstalk between cells in the tumor microenvironment. Additionally, newly identified cysteine modifications that are sensitive to the abundance of certain electrophilic metabolites have been described as metabolic signaling mediators of anti-oxidant and anti-inflammatory responses [78–80]. Ongoing research into the roles of metabolites as signaling molecules will continue to enhance our molecular understanding of how metabolites interface with signaling pathways and with chromatin to tune cellular responses in accordance with metabolic state.
Acknowledgement
We thank members of the KEW lab for helpful discussions. We apologize to authors whose work was not cited owing to space constraints. The lab of KEW is supported by R01CA174761, R01CA228339, and R01DK116005.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: none
References
- 1.Campbell SL, Wellen KE: Metabolic Signaling to the Nucleus in Cancer. Molecular Cell 2018, 71:398–408. [DOI] [PubMed] [Google Scholar]
- 2.Chantranupong L, Wolfson RL, Sabatini DM: Nutrient-Sensing Mechanisms across Evolution. Cell 2015, 161:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence RE, Zoncu R: The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 2019, 21:133–142. [DOI] [PubMed] [Google Scholar]
- 4.Wolfson RL, Sabatini DM: The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metabolism 2017, 26:301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandel NS: Evolution of Mitochondria as Signaling Organelles. Cell Metabolism 2015, 22:204–206. [DOI] [PubMed] [Google Scholar]
- 6.Garcia D, Shaw RJ: AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Molecular Cell 2017, 66:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reid MA, Dai Z, Locasale JW: The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol 2017, 19:1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, Mills EL: Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nature Metabolism 2018, 1:16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantor JR, Sabatini DM: Cancer Cell Metabolism: One Hallmark, Many Faces. Cancer Discovery 2012, 2:881–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeBerardinis RJ, Chandel NS: Fundamentals of cancer metabolism. Sci. Adv 2016, 2:e1600200–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. : The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. : Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S- H, Ito S, Yang C, Wang P, Xiao M- T, et al. : Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, Chi JT, Bigner DD, Vertino PM, Yan H: A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Research 2012, 22:2339–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **15.Morris JP, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen C- C, Baslan T, Marinkovic ZS, Sánchez-Rivera FJ, Leach SD, et al. : α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573:595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that reactivation of p53 in a murine model of pancreatic ductal adenocarcinoma drives an increase in the αKG:succinate ratio and that this change is necessary and sufficient for p53-mediated differentiation and tumor suppression, therefore identifying metabolic regulation as a key component of the tumor suppressive function of p53 in pancreatic cancer.
- 16.Kudo Y, Tateishi K, Yamamoto K, Yamamoto S, Asaoka Y, Ijichi H, Nagae G, Yoshida H, Aburatani H, Koike K: Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci 2012, 103:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cimmino L, Aifantis I: Alternative roles for oxidized mCs and TETs. Current Opinion in Genetics & Development 2017, 42:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. : Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yong C, Stewart GD, Frezza C: Oncometabolites in renal cancer. Nature Reviews Nephrology 2019, doi: 10.1038/s41581-019-0210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Intlekofer AM, Dematteo RG, Venneti S, Finley LWS, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, et al. : Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metabolism 2015, 22:304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oldham WM, Clish CB, Yang Y, Loscalzo J: Hypoxia-Mediated Increases in l-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metabolism 2015, 22:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shenoy N, Bhagat TD, Cheville J, Lohse C, Bhattacharyya S, Tischer A, Machha V, Gordon-Mitchell S, Choudhary G, Wong L- F, et al. : Ascorbic acid–induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J. Clin. Invest 2019, 129:1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrer A, Trefely S, Zhao S, Campbell SL, Norgard RJ, Schultz KC, Sidoli S, Parris JLD, Affronti HC, Sivanand S, et al. : Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discovery 2019, 9:416–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, Flores A, Mohlman J, Sorensen LK, Earl CS, et al. : Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat Cell Biol 2017, 19:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flores A, Schell J, Krall AS, Jelinek D, Miranda M, Grigorian M, Braas D, White AC, Zhou JL, Graham NA, et al. : Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat Cell Biol 2017, 19:1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bensard CL, Wisidigama DR, Olson KA, Berg JA, Krah NM, Schell JC, Nowinski SM, Fogarty S, Bott AJ, Wei P, et al. : Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metabolism 2019, doi: 10.1016/j.cmet.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calle EE, Kaaks R: Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 2004, 4:579–591. [DOI] [PubMed] [Google Scholar]
- 28.Sung H, L Siegel R, Rosenberg PS, Jemal A: Articles Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. The Lancet Public Health 2019, 4:e137–e147. [DOI] [PubMed] [Google Scholar]
- *29.Hu C-M, Tien S-C, Hsieh P-K, Jeng Y-M, Chang M-C, Chang Y-T, Chen Y-J, Chen Y-J, Lee EYHP, Lee W-H: High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metabolism 2019, 29:1334–1349.e10. [DOI] [PubMed] [Google Scholar]; This study shows that high concentrations of glucose promote O-GlcNAcylation of the enzyme ribonucleotide reductase, leading to an increase in genomic alterations including KRAS mutations, preferentially in pancreatic ductal cells. These findings provide one possible mechanism by which systemic metabolic dysregulation could contribute to increased cancer incidence.
- 30.Akella NM, Ciraku L, Reginato MJ: Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biology 2019, 17:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruan H- B, Singh JP, Li M- D, Wu J, Yang X: Cracking the O-GlcNAc code in metabolism. Trends in Endocrinology & Metabolism 2013, 24:301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Senapati P, Kato H, Lee M, Leung A, Thai C, Sanchez A, Gallagher EJ, LeRoith D, Seewaldt VL, Ann DK, et al. : Hyperinsulinemia promotes aberrant histone acetylation in triple-negative breast cancer. Epigenetics & Chromatin 2019, 12:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia MR, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A, Christian S, Hess-Stumpp H, Augustin HG, Müller-Decker K, et al. : Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metabolism 2017, 26:842–855.e5. [DOI] [PubMed] [Google Scholar]
- 34.Lu M, Zhu W-W, Wang X, Tang J-J, Zhang K-L, Yu G-Y, Shao W-Q, Lin Z-F, Wang S-H, Lu L, et al. : ACOT12-Dependent Alteration of Acetyl-CoA Drives Hepatocellular Carcinoma Metastasis by Epigenetic Induction of Epithelial-Mesenchymal Transition. Cell Metabolism 2019, 29:886–900.e5. [DOI] [PubMed] [Google Scholar]
- 35.Hunt MC, Siponen MI, Alexson SEH: The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism. BBA - Molecular Basis of Disease 2012, 1822:1397–1410. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez S, Viola JM, Torres A, Wallace M, Trefely S, Zhao S, Affronti HC, Gengatharan JM, Guertin DA, Snyder NW, et al. : Adipocyte ACLY Facilitates Dietary Carbohydrate Handling to Maintain Metabolic Homeostasis in Females. Cell Reports 2019, 27:2772–2784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JV, Berry CT, Kim K, Sen P, Kim T, Carrer A, Trefely S, Zhao S, Fernandez S, Barney LE, et al. : Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca 2+–NFAT signaling. Genes & Development 2018, 32:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massagué J, Obenauf AC: Metastatic colonization by circulating tumour cells. Nature 2016, 529:298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **39.Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, Boon R, Escalona-Noguero C, Torrekens S, Verfaillie C, et al. : Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that uptake of pyruvate by breast cancer cells leads to increased αKG levels, which subsequently promotes hydroxylation of procollagen by collagen prolyl-4-hydroxylase; inhibition of pyruvate uptake reduced collagen hydroxylation and metastatic burden in the lung, which could be rescued by supplementation of αKG, thereby linking nutrient availability with ECM remodeling in metastasis.
- 40.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr: HIF Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O. Science 2001, 292:464–468. [DOI] [PubMed] [Google Scholar]
- 41.Gilkes DM, Semenza GL, Wirtz D: Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer 2014, 14:430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, O’Connor K, Lane AN, Xu R: Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nature Communications 2018, 9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, et al. : BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2018, 551:384–388. [DOI] [PubMed] [Google Scholar]
- 44.Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O’Connor STF, Li S, Chin AR, et al. : Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nature Cell Biology 2015, 17:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **45.Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, Tienda SM, Nahotko DA, Li G, Blaženović I, et al. : Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569:723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]; By performing proteomic analysis of matched primary vs metastatic ovarian cancer samples, this study identified the metabolic enzyme NNMT as a major regulator of reprogrammed gene expression in ovarian cancer associated fibroblasts by its effect on substrate availability for chromatin methylation, and that NNMT was necessary and sufficient for acquisition of the CAF phenotype.
- 46.Jung J, Kim LJY, Wang X, Wu Q, Sanvoranart T, Hubert CG, Prager BC, Wallace LC, Jin X, Mack SC, et al. : Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2017, 2:766–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ulanovskaya OA, Zuhl AM, Cravatt BF: NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nature Chemical Biology 2019, 9:300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Sullivan D, Sanin DE, Pearce EJ, Pearce EL: Metabolic interventions in the immune response to cancer. Nature Reviews Immunology 2019, 19:324–335. [DOI] [PubMed] [Google Scholar]
- 49.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO: Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, et al. : Memory CD8+ T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 2016, 44:1312–1324. [DOI] [PubMed] [Google Scholar]
- 51.Bailis W, Shyer JA, Zhao J, Canaveras JCG, Khazal Al FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, et al. : Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, doi: 10.1038/s41586-019-1311-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo Y- M, et al. : ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Reports 2016, 17:1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. : Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu J, Villa M, Sanin DE, Buck MD, O’Sullivan D, Ching R, Matsushita M, Grzes KM, Winkler F, Chang C- H, et al. : Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Reports 2019, 27:2063–2074.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, et al. : The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8+ T Cell Fitness and Functionality. Immunity 2019, 51:491–507.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *56.Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha N- H, Lee P- H, Shin M, Patel SJ, Yu Z, et al. : T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363:eaau0135. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study documents that high extracellular potassium, a frequent feature of the tumor microenvironment, impairs nutrient transport into T cells, resulting in functional starvation. This is associated with reduced acetyl-CoA and histone acetylation levels and impaired expression of effector molecules, but also enhanced T cell stemness.
- 57.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. : Acetate Dependence of Tumors. Cell 2014, 159:1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. : Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175:1780–1795.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, et al. : Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, doi: 10.1038/s41586-018-0846-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *60.Leone RD, Zhao L, Englert JM, Sun I- M, Oh M- H, Sun I- H, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. : Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that global inhibition of glutamine metabolism differentially affected cancer cells vs. CD8+ T cells: while treatment with a glutamine antagonist compromised oxidative and glycolytic metabolism in cancer cells, effector T cells were able to remain highly activated and proliferative, which was linked to upregulation of compensatory acetate metabolism and pyruvate carboxylase-dependent anaplerosis.
- 61.Wellen KE, Snyder NW: Should we consider subcellular compartmentalization of metabolites, and if so, how do we measure them? Current Opinion in Clinical Nutrition and Metabolic Care 2019, 22:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sivanand S, Viney I, Wellen KE: Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends in Biochemical Sciences 2018, 43:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, Kraus WL: Metabolic regulation of transcription through compartmentalized NAD +biosynthesis. Science 2018, 360:eaan5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB: ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sivanand S, Rhoades S, Jiang Q, Lee JV, Benci J, Zhang J, Yuan S, Viney I, Zhao S, Carrer A, et al. : Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Molecular Cell 2017, 67:252–265.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C, Dhayade S, Schug ZT, Voorde JV, Blyth K, et al. : Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Reports 2017, 18:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, Igarashi K: Methionine Adenosyltransferase II Serves as a Transcriptional Corepressor of Maf Oncoprotein. Molecular Cell 2011, 41:554–566. [DOI] [PubMed] [Google Scholar]
- 68.Sdelci S, Rendeiro AF, Rathert P, You W, Lin J- MG, Ringler A, Hofstätter G, Moll HP, Gürtl B, Farlik M, et al. : MTHFD1 interaction with BRD4 links folate metabolism to transcriptional regulation. Nature Genetics 2019, 51:990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li S, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL, Suganuma T: Serine and SAM Responsive Complex SESAME Regulates Histone Modification Crosstalk by Sensing Cellular Metabolism. Molecular Cell 2015, 60:408–421. [DOI] [PubMed] [Google Scholar]
- 70.Gruber JJ, Geller B, Lipchik AM, Chen J, Salahudeen AA, Ram AN, Ford JM, Kuo CJ, Snyder MP: HAT1 Coordinates Histone Production and Acetylation via H4 Promoter Binding. Molecular Cell 2019, 75:711–724.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *71.Sun RC, Dukhande VV, Zhou Z, Young LEA, Emanuelle S, Brainson CF, Gentry MS: Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metabolism 2019, 30:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study introduces the novel concept that glycogen is synthesized and broken down in the nucleus and could serve as a source of acetyl groups on histones. It implicates abnormal accumulation of this nuclear glycogen as important to non-small cell lung cancer progression.
- 72.Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED: A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell 2014, 158:84–97. [DOI] [PubMed] [Google Scholar]
- 73.Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA, Liu J, Lund PJ, Lopes M, Garcia BA, et al. : Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 2018, 175:502–513.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mews P, Donahue G, Drake AM, Luczak V, Abel T, Berger SL: Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sabari BR, Di Zhang, Allis CD, Zhao Y: Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 2017, 18:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *76.Zhang Di, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. : Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses protein mass spectrometry and stable isotope tracing to identify histone lactylation, a previously unknown modification and show that it is sensitive to lactate levels in cells.
- 77.Gaffney DO, Jennings EQ, Anderson CC, Marentette JO, Shi T, Oxvig A- MS, Streeter MD, Johannsen M, Spiegel DA, Chapman E, et al. : Non-enzymatic Lysine Lactoylation of Glycolytic Enzymes. Cell Chemical Biology 2019, 27:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, Chin EN, Ahmad I, Chatterjee AK, Lairson LL, et al. : A metabolite-derived protein modification integrates glycolysis with KEAP1–NRF2 signalling. Nature 2018, 562:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. : Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang L- H, et al. : Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 2018, 556:501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bailis W, Shyer JA, Zhao J, Canaveras JCG, Khazal Al FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, et al. : Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, 571:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]