

Abstract
SARS-CoV-2 is causative agent of COVID-19, which is responsible for severe social and economic disruption globally. Lack of vaccine or antiviral drug with clinical efficacy suggested that drug repurposing approach may provide a quick therapeutic solution to COVID-19. Nonstructural protein-15 (NSP15) encodes for an uridylate-specific endoribonuclease (EndoU) enzyme, essential for virus life cycle and an attractive target for drug development. We have performed in silico based virtual screening of FDA approved compounds targeting EndoU in search of COVID-19 drugs from commercially available approved molecules. Two drugs Glisoxepide and Idarubicin used for treatment for diabetes and leukemia, respectively, were selected as stronger binder of EndoU. Both the drugs bound to the active site of the viral endonuclease by forming attractive intermolecular interactions with catalytically essential amino acid residues, His235, His250, and Lys290. Molecular dynamics simulation studies showed stable conformation dynamics upon drugs binding to endoU. The binding free energies for Glisoxepide and Idarubicin were calculated to be –141 ± 11 and –136 ± 16 kJ/mol, respectively. The IC50 were predicted to be 9.2 µM and 30 µM for Glisoxepide and Idarubicin, respectively. Comparative structural analysis showed the stronger binding of EndoU to Glisoxepide and Idarubicin than to uridine monophosphate (UMP). Surface area calculations showed buried are of 361.8Å2 by Glisoxepide which is almost double of the area occupied by UMP suggesting stronger binding of the drug than the ribonucleotide. However, further studies on these drugs for evaluation of their clinical efficacy and dose formulations may be required, which may provide a quick therapeutic option to treat COVID-19.
Communicated by Ramaswamy H. Sarma
Keywords: SARS-CoV-2, endonuclease, drug repurposing, molecular dynamic simulation, binding affinity, IC50
1. Introduction
Novel coronavirus (nCoV) originated from Wuhan, China, is a spherical or pleomorphic shaped, single-stranded RNA virus with characteristic crown-shaped glycoproteins on its surface (Graham Carlos et al., 2020). nCOV causes coronavirus disease-2019 (COVID-19) characterized by pneumonia and high fever (Corkery et al., 2015; Majid Rezaei, 2020). The nCOV is also named as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) upon the similarity of its symptoms to those induced by the severe acute respiratory syndrome (SARS). The first case of COVID-19 was reported in late December 2019 and was declared pandemic on March 11, 2020. As of June 3, 2020, more than 6.28 million cases of SARS-CoV-2 infection have been confirmed with 379,941 global fatalities (https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/). The genomic sequences of SARS-CoV-2 viruses isolated from a number of patients share sequence identity higher than 99.9%, suggesting a very recent host shift into humans (Kim, Chung, Jo, et al., 2020).
SARS-CoV-2 belongs to the beta CoVs category and has a diameter of approximately 60–140 nm. Like other CoVs, it is sensitive to UV rays and higher temperature (Cascella et al., 2020). Diagnosis of COVID-19 is based on the real-time polymerase chain reaction (RT-PCR) technique of isolated RNA from the patients. The SARS-CoV-2 genome consists of a long replicase gene encoding nonstructural proteins (NSPs), along with structural and accessory genes (Vijgen et al., 2005). The replicase gene encodes for two ORFs named as rep1a and rep1b by ribosomal frameshifting, which are translated into pp1a and pp1ab polyproteins (Cui et al., 2019). These two large polyproteins are processed by two viral proteases known as 3C-like protease (3CLpro) and papain-like protease (PLP) that are encoded by NSP5 and NSP3, respectively. Sixteen viral NSPs are produced by cleavage (Baez-Santos et al., 2015), which gets assembled into a membrane-bound large replicase complex having several functions. NSPs have been exhibiting many enzymatic activities along with RNA replication and modification of subgenomic RNAs. One of these proteins, NSP15 a nidoviral RNA uridylate-specific endoribonuclease (NendoU) containing carboxy-terminal catalytic domain linked to the EndoU family (Deng & Baker, 2018). Previous study has suggested that Nsp15 has crucial role in evasion of host defense mechanism and loss of EndoU function results in protective immune response (Deng & Baker, 2018; Liu et al., 2019). Therefore, Nsp15 is considered critical in coronavirus biology. Detection technique(s) also remains a critical factor for treatment and may still need improvements (Babadaei, Hasan, Bloukh, et al., 2020; Elasnaoui & Chawki, 2020). There is no existing vaccine or proven drug for this disease, but various treatment options such as utilizing medicines effective in other viral ailments, are being attempted against SARS-COV2 (Babadaei, Hasan, Vahdani, et al., 2020; Elmezayen et al., 2020; Khan, Jha, Amera, et al., 2020; Mahanta et al., 2020; Muralidharan et al., 2020). The use of de-risked compounds, with potentially lower overall development costs and shorter development timelines is being widely accepted against drug targets (Adeoye et al., 2020; Lobo-Galo et al., 2020). Novel and natural compounds as inhibitors of SARS-CoV-2 are also being explored using regress computational approaches (Abdelli et al., 2020; Bhardwaj et al., 2020; Elfiky, 2020a; Enmozhi et al., 2020; Islam et al., 2020; Kumar et al., 2020; Umesh et al., 2020; Wahedi et al., 2020).
In order to explore anitviral compounds several reports of using drug repurposing methods are also recently been made available (Aanouz et al., 2020; Al-Khafaji et al., 2020; Chetri et al., 2019; Elfiky, 2020b; Gupta et al., 2020; Gyebi et al., 2020; Joshi et al., 2020; Khan et al., 2020; Mittal et al., 2020; Sarma et al., 2020; Sinha et al., 2020). In the present study, in silico methods have been used to identify potential drugs which may act as specific inhibitor for EndoU enzyme of SARS-CoV-2. Food and drug administration (FDA) approved drug database has been used for structure based virtual screening (SBVS) and molecular docking. Top compounds were then re-docked and the protein complexes were analyzed. To remove false-positive hits further validation was performed using molecular dynamics (MD) simulation studies. Furthermore, the molecular mechanics-Poisson-Boltzmann surface area (MM/PBSA) based binding free energies between the EndoU-ligand complexes were calculated. Glisoxepide, sulfonylurea agent used for the treatment of type 2 diabetes mellitus and Idarubicin, anticancer drug were found most potent inhibitors of SARS-COV2 EndoU.
2. Material and methods
2.1. Selection of the drug target protein and ligand molecules
Crystal structure of the EndoU of SARS-Cov-2 was downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank. The crystal structure (PDB ID: 6W01) has the highest resolution among all EndoU crystal structures and hence was used in the study. 6W01 is a complex of EndoU with citrate ion and was solved at 1.9 Å resolution (Kim et al., 2020). The bound citrate ion was used as a control for docking and simulation studies. For ligand retrieval, the FDA approved drug database was downloaded from the ZINC12 database (Irwin & Shoichet, 2005; Irwin et al., 2012) in mol2 file format.
2.2. Preparations of protein and ligand molecules
Protein and ligand were prepared for virtual screening. For protein preparation, only single chain 3D structure of EndoU was retained from the PDB and all other heteroatoms were removed. The coordinates were then energy minimized to avoid close contact within atoms using Chimera (Pettersen et al., 2004). Amber ff99SB force field was employed for the minimization process with 100 steepest descent steps. Then, polar hydrogen along with Kollman charges were added to the protein structure. Finally, AD4 atom type was assigned and structure was saved in pdbqt (Protein Data Bank, Partial Charge (Q), & Atom Type (T)) format using Autodock Tools (ADT)) (Steffen et al., 2010). For ligand preparation, prepareligand4.py script provided by Autodock developers was used. First, the non-polar hydrogens were added and merged, then the atoms types were set, and the Gasteiger charges were added. Finally, each ligand from the FDA drug database (n = 2895) was converted to required pdbqt format.
2.3. Structure based virtual screening
SBVS is a technique used in the drug discovery and development methods to explore compound library in search of novel bioactive molecules against a target structure (Shukla & Singh, 2020). SBVS was performed by using Autodock Vina (Trott & Olson, 2009) and then redocked with Idock (Li et al., 2012) and Smina (Koes et al., 2013). The previously prepared protein coordinates were used to prepare the grid. The position of co-crystallized compound citrate was used as a center of grid. The active site of EndoU is comprised of His235, His250, and Lys290 residues. The citrate bound with the His235, Gln245, His250, Lys290, Val292, Thr341 and Tyr343 in the co-crystallized structure. The parameters for the grid were Center_X = −63.98, Center_Y = 72.101, Center_Z = 28.841 and Size_X = 40 Å, Size_Y = 40 Å and Size_Z = 40 Å. The number of binding mode and energy range were set to maximum with exhaustiveness at 50. The prepared ligands were screened against the EndoU enzyme. To avoid any false-positive identification, we further performed SBVS with Smina tool. Same parameter and ligands were used in the process. Top five drug molecules (Dihydroergotamine, Glisoxepide, Idarubicin, Ergotamine and Tasosartan) with good binding affinity to NSP15 were selected for further analysis.
2.4. IC50 prediction
In order to calculate binding affinity of the drugs Dihydroergotamine, Glisoxepide, Idarubicin, Ergotamine and Tasosartan, the half-maximal inhibitory concentration (IC50) against EndoU was predicted using Autodocktools package version (Steffen et al., 2010). The grid box spacing was 0.375 Å with Center_X = −63.98, Center_Y = 72.10, Center_Z = 28.84 and Size_X = 40 npts, Size_Y = 40 npts and Size_Z = 40 npts. Number of individuals in population and maximum number of energy evaluations in each run was kept 150 and 25,000,000, respectively to determine best docking conformer and other docking parameters were set to default. Overall, 100 conformation of each compound was generated with IC50 values. Finally, Glisoxepide, Idarubicin and Tasosartan complexes with EndoU were used for MD simulation.
2.5. Molecular dynamics simulation system preparation
To understand the conformational dynamics upon drugs binding to EndoU, MD simulation studies were performed. All atom simulation method was used to gain insights by solving Newton’s equation of motion. MD simulations of the EndoU-drugs complexes were performed with the GROMACS 4.6.5 package using gromos53a6 force field (Pronk et al., 2013; Van Der Spoel et al., 2005). The topology of EndoU was generated using pdb2gmx modules of GROMACS. The model was placed in a dodecahedron box using the editconf module keeping protein EndoU at the center and with 10 Å distance from the edges. The system was then solvated with simple point charge water model (SPC216) to attain real dynamics. Solvated systems were then energy minimized using the Steepest Descent algorithms with 5000 steps cutoff. Further, individual systems were slowly warmed up using a V-rescale thermostat with a coupling constant to reach 310 K to perform equilibration in NVT (number of atoms, volume of the system, and temperature of the system) ensemble. Later, the solvent density was maintained using a Parrinello-Rahman barostat with a coupling constant at 1 bar and 310 K to perform equilibration in NPT (number of atoms in the system, the pressure of the system and temperature of the system) ensemble. The systems were equilibrated for about 100 ps under both ensemble processes (NVT and NPT) with position restrained dynamics. The LINCS algorithm was used for constraining all the bonds. Finally the systems were submitted to molecular dynamics simulation for 100 ns to observe stability of each EndoU-ligand complex (Khan et al., 2020; Shukla et al., 2018; Sk et al., 2020).
2.6. MD simulations analysis
The resultant trajectories were sampled using GROMACS for analysis (Van Der Spoel et al., 2005). Protein dynamics and ligand interaction were analyzed for the whole trajectories. Respective frames of the trajectories were visualized and analyzed using Chimera (Pettersen et al., 2004). Initially, aligning and rotational fitting of the trajectory was performed to exclude simulation artifacts if any. The root mean square deviation (RMSD) of the Cα backbone atoms of the protein was calculated for each system. The root mean square fluctuation (RMSF) values of protein Cα atoms were also measured to identify the fluctuating residues during simulation. To evaluate the compactness of the protein structure upon ligand binding, radius of gyration (Rg) was measured. The correlated motion of protein was also measured using principal component analysis (PCA). In PCA, covariance matrix is generated and studied by diagonalizing all the motions of protein in form of eigenvector and eigenvalues. The principal component PC1 versus PC2 was plotted to examine the cluster in phase space. Finally, the binding free energy was calculated for each protein ligand complex. In addition, Poisson-Boltzmann surface area continuum solvation (MM/PBSA) (Fu et al., 2018; Hu et al., 2017; Kumari et al., 2014; Moesgaard et al., 2020; Xue et al., 2018) was also performed to calculate the free binding energy of the ligand. Distance of 3.2 Å was kept as cutoff for bond length for salt bridge and hydrogen bond interactions (Mohammad et al., 2020; Shukla et al., 2020). All graphical presentation was prepared using Origin 6.0.
3. Results and discussion
3.1. Virtual screening
The structure-based virtual screening was carried out for the identification of inhibitors against EndoU using Autodock vina, Idock and Smina tools (Koes et al., 2013; Li et al., 2012; Trott & Olson, 2009). Each molecule was scored on the basis of binding affinity. To validate screening and redocking protocol, bound citrate molecule was used as control. The binding affinity of resulted compounds ranged from –5.1 to –9.8 kcal/mol and –5.2 to –10.23 kcal/mol and –5.2 to –10.5 kcal/mol from Autodock vina, Idock and Smina, respectively (Table 1). Idarubicin was found to be the lowest energy molecule with binding affinity –10.5 kcal/mol and Tasosartan was found having lowest binding affinity (Table 1). All the compounds showed a favorable binding affinity towards EndoU indicating their competency of inhibiting enzyme (Table 1). To avoid toxicity, selected compounds were filtered by removing the drugs targeting human kinase receptors in apoptosis. Finally, Dihydroergotamine, Glisoxepide, Idarubicin, Ergotamine and Tasosartan that showed good binding affinity for EndoU were selected (Table 2).
Table 1.
List of top five commercially available drugs with their respective binding scores in kcal/mol calculated from different docking tools.
| ZINC ID | Name | Vina | Idock | Smina | IC50 (mM) |
|---|---|---|---|---|---|
| ZINC00537804 | Glisoxepide | –9.4 | –9.59 | –9.5 | 0.009 |
| ZINC03830924 | Idarubicin | –9.4 | –9.5 | –10.5 | 0.03 |
| ZINC03978005 | Dihydroergotamine | –9.8 | –10.23 | –10 | 0.52 |
| ZINC13444037 | Tasosartan | –9.3 | –9.28 | –9.2 | 0.09 |
| ZINC52955754 | Ergotamine | –9.4 | –9.59 | –9.6 | 0.19 |
| ZINC00895081 | Citrate | –5.1 | –5.63 | –5.8 | – |
Binding scores for citrate is also shown as docking control molecule.
Table 2.
Hydrogen bonded interaction between selected drugs and EndoU.
| Drug | Molecular Weight | Chemical Structure | Interacting residues | Distance [Å] |
|---|---|---|---|---|
| Glisoxepide | 449.533 | 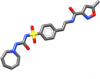 |
His235 Gly248 His250 Asn278 Lys290 Ser294 Thr341 Leu346 |
3.3 3.1 3.0 3.2 3.1 3.2 2.8 2.8 |
| Idarubicin | 497.5 | 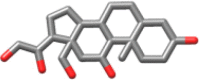 |
Gln245 Gly248 Val292 Val292 Lys290 Ser294 Tyr343 |
2.8 2.9 3.1 3.4 2.9 3.2 3.1 |
| Dihydroergotamine | 583.689 | 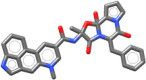 |
Gln245 Lys290 | 3.11 3.10 |
| Tasosartan | 411.469 | 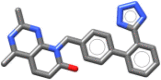 |
Thr341 | 3.00 |
| Ergotamine | 581.673 |  |
Gln245 | 3.11 |
| Citrate | 192.123 |  |
Gln245 Gly248 His250 Lys290 |
2.97 2.88 2.90 2.91 |
| Uridine Mono Phosphate (UMP) (Kim et al,. 2020) | 324.18 |  |
His250 Ser294 Lys290 | 3.20 2.70 2.80 |
Citrate ion bound to EndoU in PDB: 6W01 (Kim et al., 2020) was used as control.
3.2. IC50 prediction
The IC50 values of selected molecules were observed in the range from 0.0092 mM to 0.52 mM. The IC50 of Dihydroergotamine, Glisoxepide, Idarubicin, Ergotamine and Tasosartan were predicted to be 0.52 mM, 0.0092 mM, 0.039 mM, 0.19 mM and 0.09 mM, respectively (Table 1). From the runs of prediction by Autodock, Glisoxepide was observed to be the best possible inhibitory molecule against EndoU of SARS-Cov-2 with the IC50 value of 9.22 µM.
3.3. Molecular dynamics of apo EndoU and EndoU-complexes
In order to provide dynamics information, all the four models including citrate as control (EndoU-Idarubicin, EndoU-Glisoxepide, and EndoU-Tasosartan) were subjected to MD simulations for 100 ns each. Further, to determine stability of all the systems, RMSD values of backbone atoms were analyzed. During 100 ns simulations, all the systems achieved stability after 10 ns and average RMSD values were found to be 0.372 nm, 0.393 nm, 0.385 nm and 0.4 nm, respectively (Figure 1(a)). The RMSD profiles indicated that the binding of the all selected compounds significantly stabilized the EndoU structure. The RMSD value for individual drugs was also calculated. Average RMSD value of Citrate, Glisoxepide, Idarubicin and Tasosartan was 0.126 nm, 0.203 nm, 0.106 nm and 0.174 nm, respectively. Average RMSD value of Idarubicin was found to be lowest whereas rest of the ligand showed comparable RMSD values (Figure 1(b)). RMSD of EndoU and its all complexes got stabilized at 50 ns of simulation. Further analysis of complex was carried over for after 50 ns (Table 3).
Figure 1.

Molecular dynamics simulations of EndoU-drug complexes. (a) RMSD of the Cα backbone of EndoU complexes over the 100 ns, (b) RMSD of each ligand over the 100 ns, (c) RMSF of Cα atoms of EndoU complexes over the 100 ns, (d) number of hydrogen bond interactions for during simulation between EndoU and compounds. The EndoU complexes with Citrate (black), Idarubicin (red), Glisoxepide (green), and Tasosartan (blue) are represented in different color schemes.
Table 3.
Average values of systematic and energetic parameters indicating structural stability of EndoU upon ligand binding during 100 ns simulation.
| Citrate | Idarubicin | Glisoxepide | Tasosartan | |
|---|---|---|---|---|
| RMSF | 0.224 | 0.228 | 0.19 | 0.212 |
| RMSD | 0.372 | 0.393 | 0.385 | 0.4 |
| Rg | 2.35 | 2.41 | 2.3 | 2.98 |
| Hydrogen bonds | 4 | 4 | 5 | 4 |
To provide more detailed information of motions in EndoU upon binding of selected compounds, RMSF as function of the residue number for the last 50 ns trajectories was plotted (Figure 1(c)). The RMSF values of EndoU-Citrate, EndoU-Idarubicin, EndoU-Glisoxepide, and EndoU-Tasosartan were observed to be 0.224, 0.228, 0.19 and 0.212 nm, respectively. As shown in RMSF plot several residual fluctuations were observed in the RMSF profile of each complex. Upon the binding of Glisoxepide to EndoU, the RMSF of binding site was significantly reduced suggesting gain in thermodynamic stability of the enzyme.
Intramolecular hydrogen bonds in a protein are considered to play vital roles in molecular recognition, stability and overall conformation. The number of hydrogen bonds was analyzed to get insight into the protein-ligand interaction and stability. The possible number of hydrogen bonds in the complexes of EndoU with Citrate, Idarubicin, Glisoxepide, and Tasosartan were observed to 4, 7, 8 and 1, respectively (Figure 1(d)). Drugs also gained more van der Waals interactions with EndoU in comparison with the control citrate compound, which has provided base for stronger binding of drugs to the enzyme.
Furthermore, the Rg is defined as overall conformational shape of a protein and utilized to know about protein compactness. We observed the compactness of EndoU-complexes by plotting their Rg (Figure 2(a)). The Rg values of EndoU-Citrate, EndoU-Idarubicin, EndoU-Glisoxepide, and EndoU-Tasosartan were found to be 2.35, 2.41, 2.3, and 2.98 nm, respectively. The average Rg values of each compound were comparable while the binding of Glisoxepide showed the decrease in Rg values. The solvent accessible area was also calculated for each complex. The average solvent accessible area was 196, 199, 198 and 198 nm2 for Idarubicin, Glisoxepide, Tasosartan and Citrate complexes, respectively (Figure 2(b)).
Figure 2.

Stability of EndoU-drug complexes. (a) Number of hydrogen bond interactions between EndoU-complexes during simulation are shown. (b) Total solvent accessible area with respect to time is shown. The EndoU complexes with Citrate (black), Idarubicin (red), Glisoxepide (green), and Tasosartan (blue) are represented in different color schemes.
3.4. Principal component analysis
PCA investigate the total expansion of protein during the MD simulation. The PCA was performed for EndoU-Citrate, EndoU-Idarubicin, EndoU-Glisoxepide, and EndoU-Tasosartan via determining their collective motions and dynamics were illustrated utilizing gmx covar and gmx anaeig modules with reference to the backbone. PCA reveals the average mobility of a protein. The first ten eigenvectors for EndoU complexes with Citrate, Idarubicin, Glisoxepide, and Tasosartan accounted for 81%, 80%, 70% and 77% of the motions observed for the last 50 ns, respectively (Figure 3(a)). The results show that EndoU-Tasosartan showed fewer motions as compared to other predicted hits and the reference compound, whereas reference compound citrate showed higher motions among all ligands. EndoU-Idarubicin, EndoU-Glisoxepide, and EndoU-Tasosartan showed fewer motions during ligand binding, indicating their stabilizing effects on the enzyme. Since the first few eigenvectors play an important role in overall motions, we considered only the first two eigenvectors from each EndoU complex for a clear depiction of the results (Figure 3(b)). The PC1 versus PC2 principal component graph showed that EndoU-Tasosartan showed a much defined and stable cluster as compared to all other selected ligands, while EndoU-Citrate did not show a stable cluster in phase space. So, the two-dimensional PCA result analysis suggested that the predicted drugs are better than the reference citrate ion for recognizing endonuclease enzyme.
Figure 3.

Principal component analysis of EndoU complexes. (a) Plot of eigenvalues versus eigenvector index is shown. Only first 0.05% of eigenvectors are considered for representation. (b) Projection of the motion of the protein in phase space along the PC1 and PC2 is drawn. The EndoU complexes with Citrate (black), Idarubicin (red), Glisoxepide (green), and Tasosartan (blue) are represented in different color schemes.
3.5. Secondary structure analysis
Conformational behavior and degree of folding of a protein depends on its secondary structure. We have calculated the secondary structure elements of EndoU during the course of simulation to identify the stability of EndoU upon ligand binding. α-helix, β-sheet, bridge and turn of EndoU were analyzed considering individual residues per step, and were plotted as a function of time with the total secondary structure with respect to time. The total structural elements of all EndoU complexes remain stable, whereas, increment was observed in the secondary structure contents of selected ligands (Figure 4). This increase in secondary structure is due to the conversion of coils into β-sheet as a result of structural stabilization of enzyme due to drugs binding. Overall, no reduction in secondary elements was observed after drugs binding to EndoU, suggesting the strong stability of the complexes.
Figure 4.

Secondary structure analysis of EndoU-drug complexes after complex formation. The secondary structure component of EndoU was calculated from (a) EndoU-Citrate, (b) EndoU-Idarubicin, (c) EndoU-Glisoxepide, and (d) EndoU-Tasosartan complexes. Secondary structures (black), α-helices (yellow), β-sheet (green), 310-helices (navy blue), 5-helices (olive), coil (red), β-bridge (blue), bend (cyan) and turn (pink) are drawn.
3.6. Binding affinity calculations
The MM/PBSA tool is used to evaluate the protein and ligand binding energy during MD simulation. The free binding energy was determined using polar and apolar solvation energy. The free binding energy was investigated as electrostatic energy, polar solvation energy, van der Waals energy, SASA energy and average binding energy (Table 4). MM/PBSA based free binding energy of all EndoU-ligand complexes was calculated for the last 50 ns trajectories. The reference compound citrate showed free binding energy of –54 kJ/mol whereas Glisoxepide, Idarubicin and Tasosartan showed binding energy of –141, –134 and –103 kJ/mol suggesting Glisoxepide having highest affinity for EndoU.
Table 4.
Van der Waals, electrostatic, polar salvation, SASA and binding energy in kJ/mol for each EndoU-drug complex.
| ZINC ID | EndoU complex | Van der Waals energy | Electrostatic energy | Polar salvation energy | SASA energy | Binding energy |
|---|---|---|---|---|---|---|
| ZINC00537804 | Glisoxepide | –174 ± 50 | –11 ± 7 | 61 ± 27 | −16 ± 4 | −141 ± 11 |
| ZINC03830924 | Idarubicin | –194 ± 10 | −35 ± 18 | 113 ± 20 | –17 ± 1 | −134 ± 16 |
| ZINC13444037 | Tasosartan | –136 ± 16 | −2 ± 8 | 48 ± 10 | –13 ± 2 | −103 ± 18 |
| ZINC00895081 | Citrate | −81 ± 15 | −43 ± 31 | 79 ± 33 | −9 ± 1 | −54 ± 13 |
In order to elucidate the energy contribution prediction of potentially important residues of EndoU participating in ligand binding during the course of simulation, the energy decomposition plot was calculated. The plot showed that Gly240, Gly241, Gly247, Gly248, His250, Asn278, Ser294, Trp333, Thr341, Tyr343, Pro344, Lys345 and Leu346 were consistently involved in stabilization of the EndoU complexes during simulation (Figure 5).
Figure 5.

The binding free energy decomposition per-residue during complex formation. The energy contribution by interacted residues in each EndoU-Drug complex is shown. The energy change values upon EndoU complex formation with reference compound Citrate (black), Idarubicin (red), Glisoxepide (green), and Tasosartan (blue) are depicted.
3.7. Binding of idarubicin and glisoxepide to EndoU
NSP15 is shown to have endoribonuclease activity by hydrolyzing RNA with specificity towards uridylate (Bhardwaj et al., 2004, 2006, 2008; Kim et al., 2020). The catalytic site of NSP15 is located at the C-terminal NendoU domain in three-dimensional crystal structure (Kim et al., 2020). The active site is formed by a shallow groove between the two β-sheets, with six conserved key residues His235, His250, Ser294, Lys290, Thr341, and Tyr343 (Figure 6(a)) (Kim et al., 2020). Based on sequence similarities His235, His250, Lys290 have been known as catalytic triad of EndoU (Ricagno et al., 2006).
Figure 6.

Interactions between EndoU and the ligands. (a) Ribbon diagram showing major residues involved in catalysis and ligand binding in the structure of NSP15. (b) Interaction of Glisoxipide with NSP15 is shown. Hydrogen bonds are shown as dotted lines. (c) Interactions between Idarubicin and NSP15 are drawn. (d) Binding of UMP to NSP15 is shown. Interacting residues of NSP15 are shown in light yellow and ligand molecules are colored brown. Some of the interacting residues have been omitted for the sake of clarity. The figure was drawn using Chimera (Pettersen et al., 2004).
As Idarubicin and Glisoxepide are shown to have maximum affinity for EndoU based on binding energies and IC50 predictions, the intermolecular interactions between EndoU and both the drug molecules were analyzed. The drug complexes were stabilized by noncovalent interactions including hydrogen bonds and van der Waals interactions between drug molecules and EndoU. Both the drug molecules occupied active site of the enzyme. In EndoU-citrate complex (PDB: 6W01), citrate formed four hydrogen bond with the side chains of Gln245, Gly248, His250 and Lys290 amino acid residues of the active site. Idarubicin and Glisoxepide occupied same place as citrate ion in the active site of EndoU. Glisoxepide formed maximum eight number of hydrogen bonded interactions with Gly248 (NH), His235 (Nε2), His250 (Nε2), Asn278 (NH2), Lys290 (Nζ), Ser294 (Oγ), Thr341 (Oλ) and Leu346 (NH), whereas Val292, Trp333, Tyr343 and Glu340 of EndoU were involved in vander Waals interaction with the Glisoxepide (Figure 6(b)). Idarubicin bound EndoU less strongly than Glisoxepide with seven number of hydrogen bonded interactions. It formed hydrogen bonds with Gln245 (NH2), Gly248 (NH), Val292 (O) and Ser294 (NH, Oγ), Lys290 (Nζ), Tyr343 (OH) and also made van der Waals contacts with the side chains of Val292, Trp333, Thr341, Tyr343, and Lys345 residues of the enzyme (Figure 6(c)).
3.8. Comparison of the binding of drugs and ribonucleotide to EndoU
Recently, crystal structure of SARS-COV2 EndoU complex with uridine mono phosphate (UMP) has been solved (6WLC.pdb) (Kim et al., 2020). Idarubicin and Glisoxepide both share same place with UMP in the active site of EndoU. UMP formed only four hydrogen bonded interaction with His250 (Nε2), Ser294 (Oγ, NH) and Lys290 (Nζ) (Figure 6(d)). UMP also formed van der Waal contacts with Tyr343 and Val292 residues of EndoU. Comparatively, analysis showed that both Glisoxepide and Idarubicin formed more number of attractive interactions with EndoU and hence bound more strongly than UMP. Buried surface area calculations of EndoU showed 361.8 Å2, 310.5 Å2 and 186.6 Å2 area occupied by Glixoxepide, Idarubicin and UMP, respectively further indicating more contact area for the drugs than the UMP.
4. Conclusions
We aim to contribute to tackle the therapeutic crisis arisen due to SARS-COV-2, by using commercially available FDA approved drugs for the development of novel class of antivirals. As EndoU is found to be involved in host envision mechanism and thus can be utilized as an attractive drug target for the development of anti-COVID-19 therapeutics. We have utilized in-silico based approach by using structure based virtual screening of the FDA approved database against EndoU to identify its potent inhibitors. Two drugs Idarubicin and Glisoxepide, were selected out of 2895 compounds based on their binding affinity towards EndoU. Idarubicin inserts itself into DNA and prevents DNA unwinding by interfering with the enzyme topoisomerase II in order to perform its function as antileukemic drug (Fukushima et al., 1993). Glisoxepide is an anti-diabetic drug belongs to second-generation sulfonylureas (Seino, 2012).
Both the drugs recognized active site of EndoU suggesting them to be competitive inhibitors of the enzyme. MD simulation studies confirmed that selected drugs efficiently bind to EndoU and formed stable complexes with least structural perturbations. Glisoxepide has shown to be most potent inhibitors of EndoU with predicted IC50 of 9.2 µM whereas Idarubicin binds enzyme with IC50 of 30 µM. A comparative analysis also showed stronger binding of drugs than UMP and citrate ion to EndoU. We assume that Glisoxepide and Idarubicin both may have a good potential of controlling SARS-COV2 infection and are needed to be further validated by in vitro and in vivo studies. Furthermore, these compounds may also act synergistically to develop novel class of inhibitor against SARS-CoV2.
Glossary
Abbreviations
- COVID-19
coronavirus disease 2019
- PCA
principal component analysis
- MD
molecular dynamics
- MM/PBSA
molecular mechanics Poisson-Boltzmann surface area
- nCoV
novel coronavirus
- PME
particle-mesh Ewald
- RMSD
root mean square deviations
- RMSF
root mean square fluctuations
- SARS-CoV-2
severe acute respiratory syndrome-coronavirus-2
- SASA
solvent accessible surface area
- SPC
simple point charge
Funding Statement
We are thankful to the Indian Council of Medical Research (ICMR), Ministry of Health and Family Welfare, Govt. of India [BIC/12(31)/2012] and Council of Scientific and Industrial Research (CSIR), Human Resource Development Group (HRDG), India [37(1668)-16-EMR-II].
Acknowledgements
We are thankful to Bioinformatics Resources and Applications Facility (BRAF), CDAC, Pune, for providing access to the computational facility.
Disclosure statement
Disclosure Statement.
No potential conflict of interest was reported by the authors.
References
- Aanouz I., Belhassan A., El Khatabi K., Lakhlifi T., El Idrissi M., & Bouachrine M. (2020). Moroccan Medicinal plants as inhibitors of COVID-19: Computational investigations. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1758790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelli I., Hassani F., Brikci S. B., & Ghalem S. (2020). In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1763199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeoye A. O., Oso B. J., Olaoye I. F., Tijjani H., & Adebayo A. I. (2020). Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1765876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khafaji K., Al-DuhaidahawiL D., & Tok T. T. (2020). Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1764392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babadaei M. M. N., Hasan A., Bloukh S. H., Edis Z., Sharifi M., Kachooei E., & Falahati M. (2020). The expression level of angiotensin-converting enzyme 2 determine the severity of COVID-19: Lung and heart tissue as targets. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1767211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babadaei M. M. N., Hasan A., Vahdani Y., Bloukh S. H., Sharifi M., Kachooei E., & Falahati M. (2020). Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1767210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y. M., St. John S. E., & Mesecar A. D. (2015). The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Research, 115, 21–38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj K., Guarino L., & Kao C. C. (2004). The severe acute respiratory syndrome coronavirus Nsp15 protein is an endoribonuclease that prefers manganese as a cofactor. Journal of Virology, 78(22), 12218–12224. 10.1128/JVI.78.22.12218-12224.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj K., Palaninathan S., Alcantara J. M. O., Yi L. L., Guarino L., Sacchettini J. C., & Kao C. C. (2008). Structural and functional analyses of the severe acute respiratory syndrome coronavirus endoribonuclease Nsp15. The Journal of Biological Chemistry, 283(6), 3655–3664. 10.1074/jbc.M708375200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj K., Sun J., Holzenburg A., Guarino L. A., & Kao C. C. (2006). RNA recognition and cleavage by the SARS coronavirus endoribonuclease. Journal of Molecular Biology, 361(2), 243–256. 10.1016/j.jmb.2006.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj V. K., Singh R., Sharma J., Rajendran V., Purohit R., & Kumar S. (2020). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2020.1766572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella M., Rajnik M., Cuomo A., Dulebohn S. C., & Di Napoli R. (2020). Features, evaluation and treatment coronavirus (COVID-19). StatPearls [PubMed] [Google Scholar]
- Chetri P. B., Shukla R., & Tripathi T. (2019). Identification and characterization of glyceraldehyde 3-phosphate dehydrogenase from Fasciola gigantica. Parasitology Research, 118(3), 861–872. 10.1007/s00436-019-06225-w [DOI] [PubMed] [Google Scholar]
- Corkery D. P., Holly A. C., Lahsaee S., & Dellaire G. (2015). Connecting the speckles: Splicing kinases and their role in tumorigenesis and treatment response. Nucleus (Austin, TX), 6(4), 279–288. 10.1080/19491034.2015.1062194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., & Shi Z. L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17(3), 181–192. 10.1038/s41579-018-0118-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., & Baker S. C. (2018). An “Old” protein with a new story: Coronavirus endoribonuclease is important for evading host antiviral defenses. Virology, 517, 157–163. https://doi.org/https://doi.org/ 10.1016/j.virol.2017.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elasnaoui K., & Chawki Y. (2020). Using X-ray images and deep learning for automated detection of coronavirus disease. Journal of Biomolecular Structure and Dynamics, 1–22. 10.1080/07391102.2020.1767212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. a). Natural products may interfere with SARS-CoV-2 attachment to the host cell. Journal of Biomolecular Structure & Dynamics. 10.1080/07391102.2020.1761881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. (2020. b). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure & Dynamics. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen A. D., Al-Obaidi A., Şahin A. T., & Yelekçi K. (2020). Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1758791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi S. K., Raja K., Sebastine I., & Joseph J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1760136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu T., Zheng G., Tu G., Yang F., Chen Y., Yao X., Li X., Xue W., & Zhu F. (2018). Exploring the binding mechanism of metabotropic glutamate receptor 5 negative allosteric modulators in clinical trials by molecular dynamics simulations. ACS Chemical Neuroscience, 9(6), 1492–1502. 10.1021/acschemneuro.8b00059 [DOI] [PubMed] [Google Scholar]
- Fukushima T., Ueda T., Uchida M., & Nakamura T. (1993). Action mechanism of idarubicin (4-demethoxydaunorubicin) as compared with daunorubicin in leukemic cells. International Journal of Hematology, 57(2), 121–130. [PubMed] [Google Scholar]
- Graham Carlos W., Dela Cruz C. S., Cao B., Pasnick S., & Jamil S. (2020). Novel Wuhan (2019-NCoV) coronavirus. American Journal of Respiratory and Critical Care Medicine, 201(4), P7–P8. 10.1164/rccm.2014P7 [DOI] [PubMed] [Google Scholar]
- Gupta M. K., Vemula S., Donde R., Gouda G., Behera L., & Vadde R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1751300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi G. A., Ogunro O. B., Adegunloye A. P., Ogunyemi O. M., & Afolabi S. O. (2020). Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. Journal of Biomolecular Structure and Dynamics, 1–19. 10.1080/07391102.2020.1764868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Xie J., Hu S., Zhang L., & Dong Y. (2017). Exploration of the binding affinities between ecdysone agonists and EcR/USP by docking and MM-PB/GBSA approaches. Journal of Molecular Modeling, 23(5), 1–11. 10.1007/s00894-017-3329-5 [DOI] [PubMed] [Google Scholar]
- Irwin J. J., & Shoichet B. K. (2005). ZINC - A free database of commercially available compounds for virtual screening. Journal of Chemical Information and Modeling, 45(1), 177–182. 10.1021/ci049714+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J., Sterling T., Mysinger M. M., Bolstad E. S., & Coleman R. G. (2012). ZINC: A free tool to discover chemistry for biology. Journal of Chemical Information and Modeling, 52(7), 1757–1768. 10.1021/ci3001277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam R., Parves R., Paul A. S., Uddin N., Rahman M. S., Mamun A. A., Hossain M. N., Ali A. A., & Halim M. A. (2020). A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure & Dynamics. 10.1080/07391102.2020.1761883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R. S., Jagdale S. S., Bansode S. B., Shankar S. S., Tellis M. B., Pandya V. K., Chugh A., Giri A. P., & Kulkarni M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1760137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. T., Ali A., Wang Q., Irfan M., Khan A., Zeb M. T., & Wei D.-Q. (2020). Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2. A molecular dynamic study. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1769733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R. J., Jha R. K., Amera G. M., Jain M., Singh E., Pathak A., & Singh A. K. (2020). Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2’-O-ribosemethyltransferase. Journal of Biomolecular Structure & Dynamics. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S. A., Zia K., Ashraf S., Uddin R., & Ul-Haq Z. (2020). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1751298 [DOI] [PubMed] [Google Scholar]
- Kim Y., Maltseva N., Jedrzejczak R., Endres M., Godzik A., Michalska K., Joachimiak A., Center for Structural Genomics of Infectious Diseases (CSGID). (2020). RCSB PDB - 6WLC: Crystal structure of NSP15 endoribonuclease from SARS CoV-2 in the complex with uridine-5’-monophosphate. Retrieved May 7, 2020, from https://www.rcsb.org/structure/6WLC
- Kim J.-M., Chung Y.-S., Jo H. J., Lee N.-J., Kim M. S., Woo S. H., Park S., Kim J. W., Kim H. M., & Han M.-G. (2020). Identification of coronavirus isolated from a patient in Korea with covid-19. Osong Public Health and Research Perspectives, 11(1), 3–7. 10.24171/j.phrp.2020.11.1.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Jedrzejczak R., Maltseva N. I., Wilamowski M., Endres M., Godzik A., Michalska K., & Joachimiak A. (2020). Crystal structure of Nsp15 endoribonuclease NendoU from SARS‐CoV‐2. Protein Science. 10.1002/pro.3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes D. R., Baumgartner M. P., & Camacho C. J. (2013). Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. Journal of Chemical Information and Modeling, 53(8), 1893–1904. 10.1021/ci300604z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D., Kumari K., Jayaraj A., Kumar V., Kumar R. V., Dass S. K., & Singh P. (2020). Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1752310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Consortium, & Lynn A., Open Source Drug Discovery Consortium. (2014). g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962. 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Li H., Leung K. S., & Wong M. H. (2012). Idock: A multithreaded virtual screening tool for flexible ligand docking [Paper presentation]. 2012 IEEE Symposium on Computational Intelligence and Computational Biology, CIBCB 2012, San Diego, CA. 10.1109/CIBCB.2012.6217214 [DOI]
- Liu X., Fang P., Fang L., Hong Y., Zhu X., Wang D., Peng G., & Xiao S. (2019). Porcine deltacoronavirus nsp15 antagonizes interferon-β production independently of its endoribonuclease activity. Molecular Immunology, 114, 100–107. 10.1016/j.molimm.2019.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Galo N., Terrazas-López M., Martínez-Martínez A., & Díaz-Sánchez Á. G. (2020). FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1764393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta S., Chowdhury P., Gogoi N., Goswami N., Borah D., Kumar R., Chetia D., Borah P., Buragohain A. K., & Gogoi B. (2020). Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: An in silico based approach. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1768902 [DOI] [PubMed] [Google Scholar]
- Majid Rezaei B. (2020). Theory about treatments and morbidity prevention of corona virus disease (Covid-19). Journal of Pharmacy and Pharmacology. 10.17265/2328-2150/2020.03.004 [DOI] [Google Scholar]
- Mittal L., Kumari A., Srivastava M., Singh M., & Asthana S. (2020). Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. Journal of Biomolecular Structure and Dynamics, 1–26. 10.1080/07391102.2020.1768151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moesgaard L., Reinholdt P., Wüstner D., & Kongsted J. (2020). Modeling the sterol-binding domain of aster-A provides insight into its multiligand specificity. Journal of Chemical Information and Modeling, 60(4), 2268–2281. 10.1021/acs.jcim.0c00086 [DOI] [PubMed] [Google Scholar]
- Mohammad T., Siddiqui S., Shamsi A., Alajmi M. F., Hussain A., Islam A., Ahmad F., & Hassan M. I. (2020). Virtual screening approach to identify high-affinity inhibitors of serum and glucocorticoid-regulated kinase 1 among bioactive natural products: Combined molecular docking and simulation studies. Molecules, 25(4), 823 10.3390/molecules25040823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., & Gromiha M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1752802 [DOI] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., & Ferrin T. E. (2004). UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., & Lindahl E. (2013). GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics (Oxford, England), 29(7), 845–854. 10.1093/bioinformatics/btt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricagno S., Egloff M.-P., Ulferts R., Coutard B., Nurizzo D., Campanacci V., Cambillau C., Ziebuhr J., & Canard B. (2006). Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proceedings of the National Academy of Sciences of the United States of America, 103(32), 11892–11897. 10.1073/pnas.0601708103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma P., Sekhar N., Prajapat M., Avti P., Kaur H., Kumar S., Singh S., Kumar H., Prakash A., Dhibar D. P., & Medhi B. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure & Dynamics. 10.1080/07391102.2020.1753580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S. (2012). Cell signalling in insulin secretion: The molecular targets of ATP, cAMP and sulfonylurea. Diabetologia, 55(8), 2096–2108. 10.1007/s00125-012-2562-9 [DOI] [PubMed] [Google Scholar]
- Shukla R., Shukla H., & Tripathi T. (2018). Activity loss by H46A mutation in Mycobacterium tuberculosis isocitrate lyase is due to decrease in structural plasticity and collective motions of the active site. Tuberculosis (Edinburgh, Scotland), 108, 143–150. 10.1016/j.tube.2017.11.013 [DOI] [PubMed] [Google Scholar]
- Shukla R., Shukla H., & Tripathi T. (2020). Structure-based discovery of phenyl-diketo acids derivatives as Mycobacterium tuberculosis malate synthase inhibitors. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1758787 [DOI] [PubMed] [Google Scholar]
- Shukla R., & Singh T. R. (2020). Virtual screening, pharmacokinetics, molecular dynamics and binding free energy analysis for small natural molecules against cyclin-dependent kinase 5 for Alzheimer’s disease. Journal of Biomolecular Structure and Dynamics, 38(1), 248–262. 10.1080/07391102.2019.1571947 [DOI] [PubMed] [Google Scholar]
- Sinha S. K., Shakya A., Prasad S. K., Singh S., Gurav N. S., Prasad R. S., & Gurav S. S. (2020). An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.2020.1762741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sk M. F., Roy R., Jonniya N. A., Poddar S., & Kar P. (2020). Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations. Journal of Biomolecular Structure and Dynamics, 1–21. 10.1080/07391102.2020.1768149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen C., Thomas K., Huniar U., Hellweg A., Rubner O., & Schroer A. (2010). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–91. 10.1002/jcc.21576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., & Olson A. J. (2009). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesh K., D., Selvaraj C., Singh S. K., & Dubey V. K. (2020). Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. Journal of Biomolecular Structure and Dynamics, 1–7. 10.1080/07391102.2020.1763202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., & Berendsen H. J. C. (2005). GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 26(16), 1701–1718. 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- Vijgen L., Keyaerts E., Moës E., Thoelen I., Wollants E., Lemey P., Vandamme A.-M., & Van Ranst M. (2005). Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. Journal of Virology, 79(3), 1595–1604. 10.1128/JVI.79.3.1595-1604.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahedi H. M., Ahmad S., & Abbasi S. W. (2020). Stilbene-based natural compounds as promising drug candidates against COVID-19. Journal of Biomolecular Structure & Dynamics. 10.1080/07391102.2020.1762743 [DOI] [PubMed] [Google Scholar]
- Xue W., Yang F., Wang P., Zheng G., Chen Y., Yao X., & Zhu F. (2018). What contributes to serotonin-norepinephrine reuptake inhibitors’ dual-targeting mechanism? The key role of transmembrane domain 6 in human serotonin and norepinephrine transporters revealed by molecular dynamics simulation. ACS Chemical Neuroscience, 9(5), 1128–1140. 10.1021/acschemneuro.7b00490 [DOI] [PubMed] [Google Scholar]