

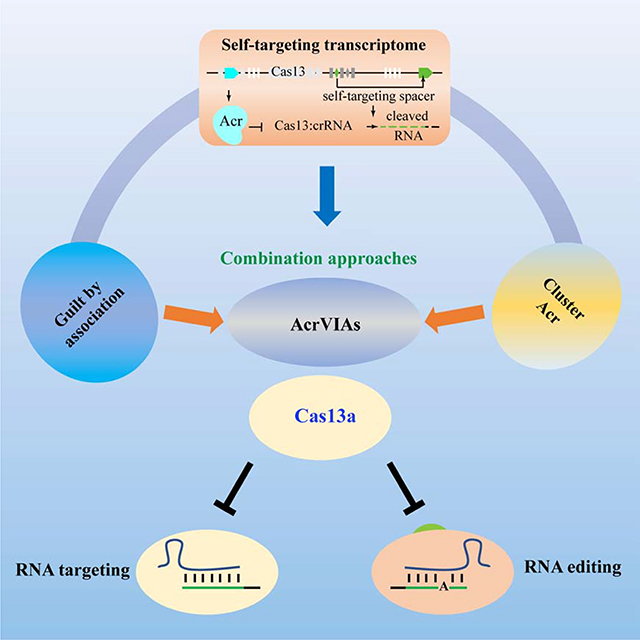
Summary
Cas13 has demonstrated unique and broad utility in RNA editing, nucleic acid detection, and disease diagnosis; however, a constantly active Cas enzyme may induce unwanted effects. Bacteriophage- or prophage regions-encoded anti-CRISPR (acr) molecules provide the potential to control targeting specificity and potency to allow for optimal RNA editing and nucleic acid detection by spatiotemporally modulating endonuclease activities. Using integrated approaches to screen acrVI candidates and evaluate their effects on Cas13 function, we discovered a series of acrVIA1–7 genes that block the activities of Cas13a. These VI-A CRISPR inhibitors substantially attenuate RNA targeting and editing by Cas13a in human cells. Strikingly, AcrVIAs also significantly muffle the single nucleic acid editing ability of the dCas13a RNA editing system. Mechanistically, AcrVIA1, 4, 5 and 6 bind LwaCas13a while AcrVIA2 and 3 can only bind LwaCas13-crRNA complex. These identified anti-CRISPR molecules may enable precise RNA-editing in Cas13-based application and studying phage-bacterium interaction.
eTOC Blurb
Lin et al. reveal inhibitors of CRISPR-Cas13a can block RNA targeting and editing in bacteria and human cells, providing a means to modulate Cas13a activity.
Graphical Abstract
Introduction
CRISPR-Cas systems provide microbes with RNA-guided adaptive immunity against bacteriophages through sequence-specific destruction of invading nucleic acids by crRNA-Cas effector complexes (Barrangou et al., 2007; Marraffini and Sontheimer, 2008). These systems have recently been developed into powerful, versatile tools for genome editing, agricultural engineering, and biotechnology (Knott and Doudna, 2018). Six distinct types of CRISPR-Cas systems (I to VI) are currently described and are categorized into two classes (Koonin et al., 2017). Type I, II, III and V CRISPR-Cas systems target DNA, while type VI is thought to specifically target RNA (Abudayyeh et al., 2016; Shmakov et al., 2015). Type VI systems were initially repurposed for transcript knockdown or RNA editing in eukaryotic organisms (Abudayyeh et al., 2017; Cox et al., 2017) and subsequently modified as programmable tools to extend our capacity for genetic mutation correction, disease diagnosis, and targeted killing of RNA viruses
Cas13 (Type VI) is the only member of CRISPR-Cas systems that specifically targets and cleaves RNA (Shmakov et al., 2015). Cas13 nucleases contain a separate RNase activity used for processing precursor crRNA into mature crRNA. The conserved higher eukaryotes prokaryotes nucleus-binding (HEPN) domains of Cas13 form a composite RNase active center responsible for catalyzing target RNA cleavage (East-Seletsky et al., 2017; Liu et al., 2017a; Liu et al., 2017b; Shmakov et al., 2015). At present, four distinct subtypes VI-A, B, C, and D have been identified based on Cas13 effector and additional Cas genes (Smargon et al., 2017; Yan et al., 2018). Though the utility of Cas 13 RNA targeting as tools is widely acknowledged (Abudayyeh et al., 2016; Cox et al., 2017; Gootenberg et al., 2018; Gootenberg et al., 2017; Konermann et al., 2018; Myhrvold et al., 2018; Terns, 2018), running a constantly active Cas13 imposes a risk to control its ribonuclease activity, causing safety concerns. However, currently no proteins are identified to show the potential to off-switch RNA-targeting CRISPR-Cas13 system.
The fierce arms race between bacteria and phages has led to the emergence of phage-generated anti-CRISPR (Acr) proteins that inhibit CRISPR-Cas-mediated immunity, allowing phages to successfully invade or lyse bacteria (Koonin and Makarova, 2018; Pawluk et al., 2018). AcrIFs were first discovered from Pseudomonas aeruginosa type I-F CRISPR-Cas system (Bondy-Denomy et al., 2013; Pawluk et al., 2014). AcrIIAs and AcrIICs were shown to inhibit Cas9 and modulate its genome-editing potency (Harrington et al., 2017; Pawluk et al., 2016a; Rauch et al., 2017). More recently, AcrVAs have been shown to inhibit Cas12a, another strong tool for DNA editing (Marino et al., 2018; Watters et al., 2018). In addition, the discovery of AcrIIIB from archaeal virus inhibits the type III-B CRISPR-Cas system by interacting with Cmr effector complexes to hamper Csx1 RNase-mediated function (Bhoobalan-Chitty et al., 2019). Discovery of Acrs may enable better control of Cas activity to improve specificity of targeting. However, Acrs are highly heterogeneous in nature, with very few conserved sequences or structures, making the discovery of Acrs a challenge. To date, Acr molecules have been identified in type I, II, III, and V CRISPR-Cas system (Athukoralage et al., 2020; Bhoobalan-Chitty et al., 2019; Harrington et al., 2017; Marino et al., 2018; Pawluk et al., 2016a; Pawluk et al., 2016b; Rauch et al., 2017; Watters et al., 2018), but no Acr proteins have been reported for VI CRISPR-Cas systems.
In type VI CRISPR-Cas13 systems, the activated Cas13-crRNA effector complexes that are triggered by target RNA binding cleave both the crRNA-bound target RNA (cis-cleavage) and non-target RNAs (trans-cleavage) (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Liu et al., 2017b). Therefore, self-targeting spacers in type VI CRISPR arrays might be lethal to host cells through mediation of targeted RNA cleavage and undesired degradation of microbial transcriptomes (Abudayyeh et al., 2016). The trans--cleavage of transcripts by activated Cas13-crRNA effector complexes, in particular, could induce cellular dormancy to halt host propagation (Meeske et al., 2019). Since self-targeting spacers act as a genomic marker for frequently co-existing with inhibitors of CRISPR-Cas immune systems in the same host strain (Marino et al., 2018; Rauch et al., 2017; Watters et al., 2018), we reasoned that these type VI strains encode unrecognized inhibitors of CRISPR-Cas13 that allow for co-existence of a spacer-protospacer pair (Figure 1A). Using an integrated approach as detailed in Figure 1B, we report the discovery of a series of acrVIA genes directly interact with LwaCas13 or LwaCas13-crRNA binary complex that not only block RNA targeting activities of Cas13a in bacteria and human cells but also inhibit dCas13a-mediated RNA editing.
Figure 1. An Integrated Approach for Screening Acr Candidate Genes of Type VI CRISPR-Cas Systems.

(A) Schematic of anti-CRISPRs in type VI strains enabling survival of cells containing self-targeting CRISPR arrays.
(B) An integrated workflow for identifying the self-targeting spacers in type VI CRISPR strains and discovering Acr candidate genes flanked by more highly conserved genes (Aca, anti-CRISPR-associated) that are located in prophage regions and often contain a helix-turn-helix (HTH) DNA-binding domain.
(C) Numbers of strains containing type VI CRISPR-Cas systems with self-targeting spacers (ST).
(D) Leptotrichia wadei strain F0279 possessing self-targeting spacers that contain predicted Acr candidate genes in the prophage regions.
Results
Developing an Integrated Approach to Screen Acr Candidates for CRISPR-Cas13 System
To discover the CRISPR-Cas13 inhibitors, we used an integrated approach with detailed procedure in Figure 1B. We applied a bioinformatic pipeline, the Self-Targeting Spacer Searcher platform (STSS) developed by the Doudna lab with modification (Watters et al., 2018), to identify the strains containing Cas13 self-targeting spacers from type VI-A (19 strains) (East-Seletsky et al., 2017), VI-B (74 strains) (Smargon et al., 2017), and VI-D (13 strains) systems (Konermann et al., 2018; Yan et al., 2018) (Step 1, Figure 1B). We determined the number of reference RNA sequences that had at least one self-targeting spacer. Interestingly, we found self-targeting spacers in 1 strain for type VI-A, 10 strains for type VI-B, and none for type VI-D strains (Figure 1C and Table S1). These strains each contain perfectly matched self-targeting spacers by CRISPR-Cas13 systems that should lead to lethal self-targeting in the absence of Acrs. Specifically, three self-target protospacers for type VI-A CRISPR were found in Leptotrichia wadei F0279 (Figure 1D and Table S1), raising the possibility of the existence of potential inhibitors of type VI-A CRISPR-Cas systems in this strain.
To reveal potential Acrs in the L. wadei F0279 genome, we refer to a highly conserved Aca protein containing HTH domain along with the “guilt by association” approach to predict Acrs (Step 2–3, Figure 1B) (Koonin and Makarova, 2018; Marino et al., 2018; Pawluk et al., 2016a; Pawluk et al., 2016b). First, using the PHAge Search Tool (PHAST), we identified 3 prophage regions encoding 117 open reading frames (ORFs) in L. wadei F0279 (Table S2). Next, using the protein family’s database (Pfam), the three distinct families of genes encoding proteins containing HTH as Aca candidates in the predicted prophage regions were identified (Figure 1D and Table S2). Using our approach 1 (Step 1–3, Figure 1B), we identified eleven previously uncharacterized proteins adjacent to Aca candidates as potential Acr candidates as they are flanked by the same strand of Aca (Figure 1D). Herein, we predicted that these Acr candidates encoding inhibitors of CRISPR-Cas13 may allow for co-occurrence of Cas13-crRNA effector complexes interacting with the protospacer transcripts.
Identification of Acrs that Inactivate CRISPR-Cas13a
To functionally characterize the identified putative Acrs, we used a cell-free transcription-translation (TXTL) system (Sun et al., 2013) to test the inhibitory activity of these candidate Acr-encoding genes. Plasmids encoding LwaCas13a and deGFP-crRNA were added to the TXTL mix with a deGFP reporter construct in the presence or absence of candidate Acr plasmids we constructed (Figure 2A). Of the eleven candidates tested for inhibition activity for type VI-A CRISPR-Cas, five proteins were shown to increase the expression of GFP reporters (Figures 2B and 2C). Moreover, the various rates of GFP expression pattern were reproducible and consistently observed during the entire experimental period (Figure 2B), which actually measured the cleaved mRNA of target gene (GFP), indicating their blockade roles in Cas13a-mediated RNA targeting. In addition, these Acr candidates exhibited different inhibitory effects on LwaCas13a enzymatic activity (61.2% for AcrVIA1, 98.5% for AcrVIA2, 71.1% for AcrVIA3, 88.7% for AcrVIA4, and 109% for AcrVIA5), with AcrVIA5 showing the highest inhibition at endpoint GFP expression (the entire 20 h observation being recorded) (Figure 2D). Our data indicate that these Acr proteins effectively inhibited LwaCas13a-mediated deGFP mRNA cleavage. Therefore, these results demonstrate that our AcrVI candidates successfully blocked RNA targeting activities of Cas13a protein.
Figure 2. Identification and Validation of Type VI-A Inhibitors.

(A) Overview of the transcription-translation (TXTL) reaction. DNA expressing Cas13a, deGFP fluorescent reporter plasmid, and GFP crRNA are mixed with or without plasmid potentially containing Acr candidate genes. Schematic of GFP reporter plasmid that contains a protospacer matching crRNA spacer. PFS: protospacer flanking site.
(B) GFP fluorescence measured over the course of 16 hours, normalized to the maximum level of expression in the TXTL reaction with non-target crRNA plasmid. Each acr genomic fragment from prophage region 1, prophage region 2, or prophage region 3 in L. wadei F0279 presented in Figure 1D. GFP reporters are only highly expressed when orf1, orf2, orf5, orf7, or orf11 are present. GFP target or Non-target (NT) indicating whether or not crRNA targeted GFP mRNA or not in the TXTL reaction. The line showed the mean values of three biological replicates; **P<0.01, *P<0.05 (one-way ANOVA plus Tukey test).
(C) GFP image assessed the blockade of LwaCas13 activity with various Acr candidate genes.
(D) Testing the individual Acr candidate genes from the prophage regions resulted in percent inhibition of CRISPR-Cas13a activity, which represents the level of GFP reporter expression observed relative to the non-targeting at 20 h of the TXTL reaction. Box plots: center line represents median, min to max (whiskers) and dots represent six independent relicates, and **P<0.01, *P<0.05 (one-way ANOVA plus Tukey test).
To investigate whether Acrs inhibit diverse Cas13 orthologs, we assayed CRISPR-Cas13 interference against the lytic RNA bacteriophage MS2 in the presence or absence of AcrVIA1 to AcrVIA5, respectively. We performed a plaquing assay with serial dilutions of MS2 phage using Cas13a with a spacer targeting MS2 genome in rep gene (non-targeting spacer as controls) in the presence or absence of respective AcrVIA. We observed that AcrVIA5 strongly impeded RNA interference with target spacers of all three Cas13a strains by forming apparent plaques (Figure 3A). AcrVIA1, AcrVIA2, AcrVIA3, and AcrVIA4 inhibited the RNA interference of LwaCas13a and LbuCas13a, but not LshCas13a (Figure 3A). In contrast, the remaining six candidate genes (orf3, 4, 6 and orf8–10) had no inhibitory activity (Figure S1A and S1B). To determine the degree of RNA interference in vivo, we established a plasmid interference assay with a protospacer placed either in-frame at the 5’-end of the GmR gentamycin-resistance gene (transcribed target) or on the opposite strand (non-transcribed target) (Figure 3B). For bacteria co-transformed with the transcribed target, the colony-forming unit (CFU) under antibiotic selection was increased compared to the non-transcribed target in the presence of AcrVIA1, AcrVIA2, AcrVIA3, and AcrVIA4 for LwaCas13a and LbuCas13a bacterial strains, but not LshCas13a (Figures 3B and S1C–S1E). Strikingly, expression of the AcrVIA5 resulted in equal transformation frequencies for LshCas13a with targeted or untargeted GmR gentamycin-resistance genes (Figures 3B and S1C–S1E), reflecting a blockade of CRISPR interference in all three bacterial strains (LwaCas13a, LshCas13a and LbuCas13a) and confirming that AcrVIA5 has the most widespread inhibitory activity of Cas13a amongst these AcrVIA inhibitors. These data implied that diverse type VI-A CRISPR-Cas systems were indeed inhibited by AcrVIA1 to 5. Conversely, none of the AcrVIAs inhibited type VI-B and type VI-D systems (Figures 3C and 3D), indicating that the inhibitory effects of these proteins on type VI-A enzymes are highly specific.
Figure 3. AcrVIA Inhibitors Block Cas13a but not Cas13b/Cas13d.

(A) Phage plaque assays with 10-fold serial dilutions of MS2 RNA phage to assess inhibition of the type VI-A CRISPR-Cas system.
(B) RNA interference assay schematic (right) and results (left). A target sequence is placed in-frame at the start of the transcribed gene containing plasmid or empty vectors conferring GenR resistance and plated on selection antibiotic plates.
(C) Phage plaque assays with 10-fold serial dilutions of MS2 RNA phage to assess inhibition of CRISPR-Cas type VI-B and VI-D.
(D) RNA interference assay of type VI-B and VI-D CRISPR-Cas systems by AcrVIA1-VIA5.
Results for dots represents three biological replicates and error bars indicate SEM between three replicates; **P<0.01, n.s., non-significant (one-way ANOVA plus Tukey test).
To explore the existence of AcrVIs homologs, we used the protein sequences of AcrVIA3 and AcrVIA5 to perform blastp in other type VI-A strains and identified one homolog family (AcrVIA3Ere) in Eubacterium rectate T1–815 (Figure S1F) and two homolog families (AcrVIA5Lwa, and AcrVIA5Lsp) in Leptotrichia wadei and Leptotrichia sp. oral taxon 879 (Figure S1G). By testing the potential inhibition of these three Acr homolog candidates in LwaCas13a nucleases, we discovered that three homologs of each protein showed strong inhibition of LwaCas13a activities (Figure S1F and S1G).
Additional Acrs are Found in Rhodobacter and Leptotrichia
Because acr genes are often located within close proximity to each other (Marino et al., 2018), we used approach 2 (based on cluster together with known acr genes) to screen additional Acr molecules (Figure 1B and 4A). We found that both of type VI-A Rhodobacter capsulat R121 and Leptotrichia buccalis DSM 1135 strains possess self-targets of type I-C and I-B CRISPR-Cas systems, respectively (Figure 4A). We identified an acrICl homolog in R. capsulat R121 prophage region, which was identified as type I-C Acr protein in the Gram-negative bovine pathogen Moraxella bovoculi (Marino et al., 2018). Next, we selected three genes adjacent to acrIC1Rca as candidate acr genes for discovering additional AcrVIA against type VI-A CRISPR-Cas systems (Figure 4B, upper). We measured the ability of the candidates to inhibit Cas13a activity and found that orf1Rca (named AcrVIA6), but neither orf2Rca, nor orf3Rca, significantly inhibited LwaCas13a RNA-targeting, as demonstrated by CRISPR-sensitive phage plaque formation resulting from expression of these proteins in bacteria (Figure 4C, left) and increased expression levels of GFP reporters in vitro (Figures 4D and 4E). These data identify AcrVIA6 as a Acr member for Cas13a.
Figure 4. Type VI-A Acr Proteins are also Identified in R. capsulatus and L. buccalis.

(A) Schematic of R. capsulatus R121 and L. buccalis DSM1135 intragenomic self-targeting spacers encoded by type I-C and I-B CRISPR-Cas, respectively.
(B) Schematic showing type VI-A (acrVIA6, acrVIA7), type I-C (acrIC1Rca), and type I-B (acrIB) inhibitors in R. capsulatus and L. buccalis. orf represents genome sequences with unknown function. Vertical arrows indicate the percent protein sequence identity to known Acr proteins.
(C) Phage plaque assays with 10-fold serial dilutions of the MS2 RNA phage to assess inhibition of type VI-A CRISPR-Cas.
(D) Digital imaging of the extent of GFP fluorescence to assess the inhibition of type VI-A CRISPR-Cas observed relative to identification of AcrVIA6 and AcrVIA7.
(E-F) Kinetic TXTL data for acr genes measured over the course of 16 h of gene expression in R. capsulatus R121 (E) and L. buccalis DSM1135 (F). The line showed the mean values of three biological replication, and **P<0.01, *P<0.05 (one-way ANOVA plus Tukey test).
Similar to R. capsulatus, the homologs of acrIIA1 (Rauch et al., 2017) and acrIIC4 (Lee et al., 2018), which we called acrIIA1Lbu, and acrIIC4Lbu (Figure 4B, lower), were also found in L. buccalis DSM 1135 by BLAST searches. The inhibition activities of Cas13a for the 5 nearby genes around the acrIIA1Lbu, and acrIIC4Lbu were investigated and revealed that orf3Lbu (named AcrVIA7) was capable of inhibiting LwaCas13a RNA-targeting both in vivo (Figure 4C, right) and in vitro (Figures 4D and 4F). AcrIB1Lbu also inhibited L. buccalis type I-B CRISPR-Cas (Figures S2A–S2C), explaining the tolerance of self-targeting for type I-B CRISPR-Cas systems in L. buccalis DSM 1135 strains. It remains unknown why a different targeting CRISPR-Cas system can be targeted by the Acrs originating from different species. We speculate that these Acrs may interfere with the early stage of nucleic acid targeting, recognition or crRNA genesis. In addition, our results revealed that none of AcrVIA molecules had inhibitory activity for type VI-B and VI-D systems (Figures S2D and S2E). Critically, these anti-CRISPRs exhibited no influence on phage growth independent of Cas13a (Figure S2F) and may have potential for modulating RNA targeting ability of Cas13a in mammals.
Type VI-A Acr Proteins Interact with LwaCas13a or LwaCas13a-crRNA complex
To determine whether type VI-A anti-CRISPRs inhibit LwaCas13a activity by interacting with LwaCas13a, we mixed purified anti-CRISPR proteins with LwaCas-msfGFP protein and conducted Co-Immunoprecipitation (Co-IP) by anti-GFP antibody to assess whether the anti-CRISPRs bind LwaCas13 in vitro. We found that AcrVIA1, AcrVIA4, AcrVIA5, or AcrVIA6 were recruited to LwaCas13a, reflecting the association of the indicated AcrVIAs with LwaCas13a (Figure S3A). Co-IP assays also showed that AcrVIA2 and AcrVIA3 interacted with crRNA-bound Cas13a but not the free form of LwaCas13a (Figure S3B), reminiscent of the anti-CRISPR AcrIIA4 targeting only crRNA-bound SpCas9 (Dong et al., 2017; Yang and Patel, 2017) and AcrVA4 interacting with crRNA-bound crMbCas12a (Dong et al., 2019; Knott et al., 2019; Zhang et al., 2019).
Next, we used purified, 6x His-tagged anti-CRISPR proteins as bait protein to capture LwaCas13a from mammalian cell lysate using the Pierce™ His Protein Interaction Pull-Down Kit. Nickel affinity chromatography analysis showed that AcrVIA1, AcrVIA4, AcrVIA5, or AcrVIA6 bound to LwaCas13a in the absence of crRNA (Figure S3C), whereas AcrVIA2 and AcrVIA3 only interacted with crRNA-bound LwaCas13a (Figure S3D). Using fluorescent microscopy, we not only observed co-localization of AcrVIA1, AcrVIA4, AcrVIA5, or AcrVIA6 and LwaCas13a (Figure S3E), but also observed co-localization of AcrVIA2 or AcrVIA3 with LwaCas13a-crRNA complex (Figure S3F). These data illustrate that AcrVIs specifically bound to LwaCas13a or LwaCas13a-crRNA complex.
Acr Genes are Widespread in Leptotrichia and Fusobacterium
To explore the homology landscape of anti-CRISPRs activity on type VI-A CRISPR-Cas systems, we attempted to create phylogenetic trees for AcrVIA1, AcrVIA2, AcrVIA3, AcrVIA5, AcrVIA6 and AcrVIA7. A comprehensive phylogenetic analysis of AcrVIA 1 revealed that homologs were conserved widely across Leptotrichia (Figure S4A). Two distinct sequence families of AcrVIA2 and AcrVIA3 were mostly restricted in Leptotricha and Fusobacterium (Figures S4B and S4C), while there were no homologs for AcrVIA4. AcrVIA5 families were observed in the genomes of Leptotrichia (Figure S4D). Three distinct sequence families of AcrVIA6 were identified in many genera including Rhodobacteraceae, Sulfitibacte, Rhodobacter, Defluviimonas, Leisingera, Epibacterium, Aliiroseovarius, Citreimonas, Salmonella, etc. (Figure S4E). Lastly, AcrVIA7 homologs were widely distributed in Campylobacter, Taylorella, Helicobacter, Leptotrichia, Acaryochloris, and Streptobacillus (Figure S4F). Now that anti-CRISPRs inhibiting CRISPR-Cas systems, which target DNA and RNA, have been described, it is speculated that anti-CRISPRs are able to inhibit all types and subtypes of CRISPR-Cas system may exist.
AcrVIAs Inhibit LwaCas13a RNA Targeting and Reduce the Collateral Activity in Human Cells
Given the widespread inhibitory effects of AcrVIA on Cas13a activity in bacteria, we imagine that these anti-CRISPRs might be used as off-switches for CRISPR-Cas13 RNA targeting in mammalian cells. To this end, we developed a dual-luciferase reporter system that expressed both Gaussia luciferase and red firefly luciferase under different promoters, allowing one transcript to serve as the Cas13 target and the other as a dosing control. We then designed guide RNAs against Gaussia transcript and cloned them into a U6 promoter-driven guide expression vector. We cloned each of AcrVIA candidates, including negative controls, into a mammalian expression vector and co-transfected with the LwaCas13a, PspCas13b, or CasRX (Cas13d) expression vector, guide vector, and dual-luciferase construct into human HEK293T cells to quantify RNA targeting efficiency by measuring luciferase activity after 48 h transfection (Figure 5A). Anti-LwaCas13a molecules resulted in Gaussia transcript knockdown (50.0% for AcrVIA 1, 35.0% for AcrVIA2, 43.0% for AcrVIA3, 23.1% for AcrVIA4, 1.3% for AcrVIA5, 44.7% for AcrVIA6 and 44.5% for AcrVIA7) in human cells compared to no Acr controls (91.1%) (Figure 5B). Titration of the AcrVIA plasmids relative to LwaCas13a expression plasmid revealed comparable dose-dependent responses to inhibition of LwaCas13a by all seven AcrVIAs (Figure S5A). Consistent with the findings in bacteria, these anti-CRISPRs exhibited no effects on RNA targeting mediated by type VI-B and VI-D CRISPR-Cas systems (Figures S5B and S5C). Lastly, these AcrVIA proteins have no effect on cell proliferation or toxicity to human cells (Figures S5D and S5E).
Figure 5. AcrVIAs Specifically Inhibit RNA Targeting by CRISPR-Cas13a Nucleases in Mammalian Cells.

(A) Schematic of mammalian luciferase reporter system for evaluating effects of AcrVIAs on RNA targeting of Cas13 systems.
(B) Quantification of RNA targeting of Gaussia luciferase using the Cas13a in human cells HEK293T expressing the indicated AcrVIA proteins or control (empty vector: no Acr).
(C) Inhibition assay for knockdown of three different endogenous transcripts with Cas13a in the presence of AcrVIAs by qRT-PCR.
Results were repeated at least three biological replicates (n=6 for B and n=3 for C), and error bars indicate mean SEM between replicates; **P<0.01, *P<0.05 (one-way ANOVA plus Tukey test).
We next tested whether these Cas13a inhibitors affected the disruption of endogenous genes (KRAS, CXCR4, and PPIB) by Cas13a in human embryonic kidney cells (HEK293T). Of all Acrs tested, AcrVIA5 showed the strongest inhibition (Figure 5C). These results confirm that AcrVIAs might be useful for regulating LwaCas13 RNA targeting in mammalian cells.
Despite becoming the most useful tools for gene editing or regulation, the currently available Cas systems are imperfect due to the potential off-target effects, immunogenicity and cytotoxicity (Fu et al., 2013). To evaluate whether AcrVIAs can inhibit collateral (unintended RNA targeting to influence mRNA expression) activity, we performed transcriptome-wide mRNA sequencing for LwaCas13a knockdown experiments with or without AcrVIAs. In the Gaussia transcript assay, LwaCas13a induced significant knockdown of the target transcript (Figure S6A), whereas AcrVIA2 and AcrVIA5 inhibited this transcript knockdown (Figure S6B and S6C). Moreover, we found that guide-independent RNA cleavage activities of LwaCas13a (Figure S6A) were alleviated by AcrVIA2 (Figure S6B) and AcrVIA5 (Figure S6C), respectively. Differential expression analysis indicated 546 significant off-targets in LwaCas13a knockdown but only 111 and 101 when adding AcrVI2 or AcrVIA5 (Figure S6D), suggesting a reduction in guide-independent RNA degradation by the Cas13a inhibitors.
AcrVIA5 Inhibits dCas13a-mediated RNA Editing in Human Diseases
Given that AcrVIA5 achieved the most robust inhibition of Cas13a activity (Figures 2D, 5B, and 5C), we speculate that AcrVIA5 may have potential for precise control of activity of Cas13a-mediated RNA editing. We tested whether AcrVIA5 can repress the programmable RNA-editing function of nuclease-deficient/deactivated LwaCas13a (dLwaCas13a)-mediated RNA-targeting tools, such as the human ADAR (adenosine deaminase acting on RNA enzymes). To determine whether AcrVIA5 prevents stable RNA binding by dLwaCas13a, we used a previously developed system in which dLwaCas13a-msfGFP are simultaneously colocalized to endogenous mRNA loci by cognate crRNA upon co-transfection of their expression plasmid in HEK293T cells (Abudayyeh et al., 2017) (Figure 6A). We readily observed co-localizing dLwaCas13a-msfGFP foci as long as endogenous mRNA-directed crRNA were included for dLwaCas13 (Figures 6B and 6C), as reported previously (Abudayyeh et al., 2017). We then repeated the experiment with co-transfected, mTagBFP2-marked plasmid (Figure 6A) also carrying an AcrVIA5 expression cassette. Co-expression of AcrVIA5 prevented the co-localization of dLwaCas13a-msfGFP with endogenous mRNA (Figure 6D), suggesting that the function of AcrVIA5 may involve blocking target RNA recognition by Cas13a. Furthermore, dLwaCas13a-msfGFP foci were observed in 94.7 % of cells in the absence of AcrVIA5 protein. By contrast, 8.06% of cells exhibited dLwaCas13a-msfGFP foci when AcrVIA5 was applied, a more than 11.7-fold blockage (Figure 6E). These results confirm the robust inhibitory effect of AcrVIA5 on stable, crRNA-programmed RNA binding in mammalian cells by dLwaCas13a.
Figure 6. AcrVIA5 Prevents dCas13a-Mediated RNA Editing Programmable A to I Replacement.

(A) Schematic representation of plasmids used for dLwaCas13a-msfGFP with crRNA and AcrVAIA5 protein. The plasmid encoding the AcrVIA5 inhibitor is also marked with the blue fluorescent protein mT agBFP2.
(B-D) Fluorescence images of HEK293 cells transiently transfected with plasmids depicted in (A). Each plasmid set (in the absence or presence of crRNAs, with or without AcrVIA5) is given on the right of each row: No crRNA for dLwaCas13a-msfGFP (B), No Acr protein (C), with AcrVIA5 (D). Scale, 5 μm.
(E) Quantitation of dLwaCas13a-msfGFP foci in cells that express no Acr or AcrVIA5.
(F) Overview of AcrVIAs on RNA editing by dLwaCas13a-ADAR2DD fusion proteins and diagram of Gaussia luciferase 140G>A target and targeting guide design. Deamination of target A restored the stop codon to amino acid.
(G) The ratio of dLwaCas13a-ADAR2DD for Guassia luciferase restoration by AcrVIA5 through the use of target guides normalized by a nontargeting guide.
(H) TA cloning sequencing for quantification of RNA editing ratio of Gaussia luciferase 140G>A target with or without AcrVIA5.
(I) Quantification of RNA editing ratio of endogenous genes (MECP2, SMN1, CFTR) linking with three different disease-relevant G>A mutation selected from ClinVar by dLwaCas13a-ADAR2DD with or without AcrVIA5.
Results for dots represents at least three biological replicates and error bars indicate SEM between samples; **P<0.01, *P<0.05 (one-way ANOVA plus Tukey test).
To investigate this mammalian application, the engineered dLwaCas13a-ADAR2DD fusion system was constructed via dLwaCas13a fused with the deaminase domain of human ADAR with E488Q/T375G mutation to enhance the catalytic activity of RNA editing for programmable A to I replacement (Cox et al., 2017). To test the inhibition activity of dLwaCas13a-ADAR2DD in the presence of AcrVIA5, we generated an RNA-editing reporter on Gaussia by introducing a nonsense mutation (UG140G>UA140G) and showed that the repair ability of the wild-type code through A>I editing was completely abolished by AcrVIA5 (Figure 6F) as reflected by restoration of Gaussia (140G>A) luminescence. We found that dLwaCas13a-ADAR2DD could effectively alter the codon UAG to UGG through A>I and then be detected as a restoration of Gaussia luciferase, which was inhibited by adding AcrVIA5 (Figure 6G). To further characterize the inhibitory effects and potential application of AcrVIA5 in effective repression of RNA editing, we measured dLwaCas13a-ADAR2DD RNA editing of Gaussia (140G>A) transcripts via reverse transcription and TA cloning. The data from TA cloning demonstrate that dLwaCas13a-ADAR2DD achieved excellent editing rates for A to I replacement (29.2% for Guide 1, 23.3% for Guide 2), whereas AcrVIA5 addition drastically reduced propensity for this editing (3.9% for Guide 1, 6.1% for Guide 2) (Figure 6H). These results suggest that AcrVIA5 may be used to reduce the target RNA editing by adjusting CRISPR-Cas13 activities.
Realizing that clinical or embryonic gene-editing requires developing efficient methods of controlling the Cas toolkit, we then examined the ability of AcrVIA5 to affect the correction of gene mutations in human disease by dLwaCas13a-ADAR2DD. We designed gRNAs targeting the gene mutations associated with important human diseases such as MECP2 (311G>A, Trp104Ter) in Rett syndrome, SMN1 (305G>A, Trp102Ter) in spinal muscular atrophy, and CFTR (3846G>A, Trp1282Ter) in cystic fibrosis. We were able to achieve 21.9% editing rate of MECP2, 18.8% editing rate of SMN1, and 12.5% editing rate of CFTR, whereas AcrVIA5 substantially blocked propensity for this editing (3.1% for MECP2, 3.1% for SMN1, 0% for CFTR) (Figure 6I). Given that AcrVIA5 can be used to inhibit RNA editing technologies, it may have potential for optimally modulating activities of Cas13a-based RNA editing tools.
Discussion
Nuclear acids of bacteriophages and other MGEs are natural targets for bacterial CRISPR-Cas systems, and the latter also encodes Acr proteins to counteract CRISPR-Cas function under the selective pressures in bacterial war on phage infection. Using a combination of advanced searches and comprehensive analytical approaches, we report the systematic discovery of anti-CRISPR molecules hidden within type VI CRISPR strains. Importantly, the AcrVIAs, particularly AcrVIA5, potently inhibit Cas13a in both bacteria and human cells and may be useful for regulating RNA targeting and editing. Although most gene-editing studies are focused on Cas9 or Cas12, which target DNA, manipulation of RNA is highly desired since it does not interfere with the mammalian or plant genome and can be broadly used to alleviate and/or cure of genetic and acquired diseases. Cas13a has recently shown broad utility in RNA targeting and editing, regulation of transcription, and biotechnological engineering (Abudayyeh et al., 2016; Cox et al., 2017; Gootenberg et al., 2018; Gootenberg et al., 2017; Konermann et al., 2018; Myhrvold et al., 2018; Terns, 2018). We showed that AcrVIAs function as an off-switch to improve the strategy to control the effective Cas13a activity.
Excessive, prolonged Cas13 nuclease exposure may exacerbate undesirable RNA mutations and induce significant off-target effects, thus limiting CRISPR-based applicability and precision targeting addressing important safety concerns (Abudayyeh et al., 2017; Cullot et al., 2019; Montiel-Gonzalez et al., 2018; Zuo et al., 2019). Hence, designing and testing Cas13 off-switches will undoubtedly pave the way for reducing the non-specific targeting or toxicity and improving gene editing for eventual clinical applications. The natural AcrVIAs from different bacterial species that we characterized with type VI-A CRISPR-Cas systems showed the potential as off-switches for LwaCas13 RNA editing in human cells, resulting into reduced guide-independent RNA degradation for the off-target events. Conceptually, these Acrs may serve as a means of controlling Cas13a activities after completion of the intended editing.
To date, multiple strategies have been used to discover Acrs, and the first example is rationalized by the existence of CRISPR-resistant phages that replicate in bacteria (Bondy-Denomy et al., 2013). Another strategy for discovery of Acrs involves bioinformatics analysis using self-targeting CRISPR sequences on the same genome, which requires inactivation of the CRISPR-Cas systems, and Acrs are the most likely solution (Rauch et al., 2017; Watters et al., 2018). Furthermore, both the clustering with known acr genes (Marino et al., 2018) and proximity to aca genes (Pawluk et al., 2016a) enable the use of methods to predict Acr candidates. Nevertheless, the discovery of Acrs has been painstakingly slow due to limited high throughput and systemic screening.
Encouraged by the recent development of an STSS system that could systemically find self-targets in bacteria, we have integrated this approach with a bioinformatics-based strategy that uses crRNAs targeting reference RNA sequences in the host strains and refers to HTH motif in prophage regions along with guilt of association to identify Acr candidates. Through integration of these aforementioned strategies, we were able to discover AcrVIA1–7. We anticipate that this integrated approach will provide a consistent and relatively easy method for identifying Acrs in other CRISPR-Cas systems, especially by using a similar mechanism for type VI-B and -D systems (Konermann et al., 2018; Smargon et al., 2017; Yan et al., 2018; Zhang et al., 2018) and a distinct mechanism for type III systems (Goldberg et al., 2014; Jia et al., 2019; Samai et al., 2015; Wang et al., 2019).
Some mechanisms by which Acrs inactivate CRISPR-Cas systems have been proposed, such as by inhibiting the effector binding or directly disrupting nuclease activity (Bondy-Denomy et al., 2015). We demonstrate that AcrVIAs directly interact with LwaCas13 or LwaCas13-crRNA complex and repress RNA cleavage. Multiple sequence alignment and phylogenetic analyses have been conducted, but no significantly homologous sequences of these AcrVIAs were identified, indicating that they may abrogate CRISPR-Cas13 activity through different mechanisms. Strikingly, AcrVIA5 prevents dCas13-ADAR2DD-mediated RNA editing programmable A to I replacement in mammalian cells that has been tested for accurate nucleic base modification. Although the detailed mechanisms of CRISPR inhibition by AcrVIAs need to be further investigated, it is possible that AcrVIA5 binding to Cas13a may hinder the subsequent RNA targeting or directly prevent catalytic activation (Bondy-Denomy et al., 2015; Wang et al., 2016). Identification and mechanistic dissection of Acr molecules will expand our understanding of CRISPR-Cas biology and phage-host interactions.
Given the rapid evolutionary arms race between phage and bacteria, Acr proteins are probably much more widespread than that are currently reported. The identification of AcrVIAs will facilitate anti-CRISPR discovery by providing important tools for revealing other Acr systems, such as type VI-B, -C, and -D CRISPR-Cas systems. Continued discovery of Acrs as well as analysis of their functional mechanisms will provide unique opportunities to regulate and/or turn off CRISPR systems while illuminating the biological significance of Acrs in CRISPR diversity. Finally, AcrVIAs may be used to enhance the CRISPR-Cas13 RNA editing and detection toolkit and study phage-bacterium interactions.
STAR ★ Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to Lead Contact Min Wu (min.wu@und.edu).
The materials generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbes
E. coli DH5a, BL21, C-3000 and its-derived mutation (table S6) strains were routinely grown in LB or LB-agar media and, when required, supplemented with ampicillin (100 μg/mL, Fisher Scientific), chloramphenicol (12.5 μg/mL, Sigma), kanamycin (100 μg/mL, Fisher Scientific), and gentamicin (75 μg/mL, ACROS).
Cell lines
Human Embryonic Kidney 293 plus T cell antigen (HEK293T cells, CRL-3216, ATCC) cells were cultured in RMPI 1640 (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (VWR) and 1 X penicillin-streptomycin (Thermo Fisher Scientific).
METHOD DETAILS
Bioinformatic Pipeline for approach 1 to identify self-targeting type V CRISPR-Cas system strains and anti-CRISPR
The methods for identification of type VI-A, VI-B, and VI-D CRISPR-Cas strains were described previously (East-Seletsky et al., 2017; Konermann et al., 2018; Smargon et al., 2017; Yan et al., 2018),. Spacer sequences from CRISPR arrays were extracted and used for analyzing the RNA target according to the bioinformatic pipeline of Self-Targeting Spacer Searcher platform (STSS) (Watters et al., 2018) with modification for targeting transcripts. The type VI strains containing self-targeting spacer were used to identify the presence of prophage regions through the PHAge Search Tool (PHAST) (Zhou et al., 2011). Protein families database (Pfam) (El-Gebali et al., 2018) was used to identify the helix-turn-helix (HTH) domain as bona fide Acr-association (Aca) gene candidates in the predicted prophage regions, which act as the mark to screen the uncharacterized open reading frames (ORFs) as anti-CRISPR (Acr) candidates in the prophage regions. To determine the type VI strains with no self-targeting spacers contained an Acr gene, all known Acr genes (He et al., 2018; Hynes et al., 2018; Rauch et al., 2017) were used to perform a BLASTP against the ORFs in the prophage regions.
Plasmids construction
Plasmids used in this study are listed in Table S5, which purchased from Addgene or were synthesized by Gene Universal or were performed for molecular cloning steps using E. coli DH5a when required, supplemented with ampicillin (Fisher Scientific), chloramphenicol (Sigma), kanamycin (Fisher Scientific), and gentamicin (ACROS).
Transcription-translation (TXTL) reactions in vitro
The TXTL reaction contained up to five DNA plasmids: the P70a-deGFP reporter plasmid (Arbor Bioscience), a Cas13a genomic amplicon or plasmid (Addgene #91865), a GFP-crRNA plasmid, a pTXTL-P70a-T7rnap HP (Arbor Bioscience), and an optional acr candidate plasmid. The crRNA plasmid was synthesized (Gene Universal) and cloned into the pCDFDuet-1 vector. Individual acr candidate genes were synthesized (Gene Universal) and cloned into a custom expression system derived from the pET-16b.
To prepare all plasmids for TXTL reaction, the plasmids were isolated using the QIAGEN Plasmid Mini Kit (QIAGEN) to elute DNA plasmid with 200 μL nuclease-free H2O. Using the 200 μL of AMPure XP beads (Beckman Coulter) we purified the plasmid product according to the handbook and by adding 20 μL of nuclease-free H2O to elute the plasmids.
The TXTL Sigma 70 master mix was purchased from Arbor Biosciences and the reactions were set up in a total of 24 μL each according to the manufacturer’s instructions. Each reaction contained 18 μL of TXTL master mix, 0.1 nM of pTXTL-P70a-T7rnap HP, 0.125 nM of GFP reporter plasmid, 1 nM of Cas13a plasmid, 2 nM of GFP-crRNA plasmid, 2nM of Acr candidate plasmid, and 5 μM of β-D-1-thiogalactopyranoside (IPTG). The reactions were incubated to measure GFP fluorescence kinetics through BioTeK Synergy HT Multi-Mode Microplate Reader with excitation 485 nm, emission 528 nm at 29 °C for up to 16 or 20 h.
Inhibition activity for TXTL reactions was calculated:
GFP20h is the GFP fluorescence level measured at the 20 h, GFPmin is the minimum fluorescence measured, GFPev,20h is the GFP level of no Acr positive control, GFPNT,20h is the GFP level of non-targeting negative control, GFPNT, min is the minimum GFP level for non-targeting negative control.
GFPTP is the GFP fluorescence level measured at each time-course during the 16 h, GFPNT, max is the maximum GFP level for non-targeting negative control.
GFP fluorescence images were photographed at the fluorescence mode with excitation filter 465 and emission filter GFO by IVIS XRII system (PerkinElmer).
Type VI-A, VI-B, and VI-D expression in E. coli C-3000
The Cas13a, Cas13b, or Cas13d plasmids were purchased from Addgene. The pre-crRNA plasmids for transcribed target or non-transcribed target for MS2 RNA phage or gentamicin gene in pRU 1103 were synthesized by Gene Universal or through the annealed oligos ligating to BbsI digested Cas13 background vector. The E. coli C-3000 strains were electroporated with both Cas13 and pre-crRNA plasmid to construct the E. coli C-3000 derived strains containing CRISPR-Cas13 systems. The targeting was confirmed in the derived strains containing CRISPR-Cas system using phage challenge assay and RNA interference assays as described below.
Approach 2 to expand Acr discovery
To identify additional AcrVI, we use approach 2 (based on cluster with known Acr) to amplify type I-B CRISPR-Cas system was amplified from the Leptotrichia buccalis DSM 1135 (ATCC 14201) and ligated into the pMQ70 to generate pMQ70-IB plasmid. CRISPR array for targeting or non-targeting gentamicin was synthesized (Gene Universal) and cloned into pgRNA vector. Both plasmids were transformed into E. coli BL21 per the manufacturer’s instructions. The E. coli strain with I-B CRISPR-Cas system was confirmed through transformation efficiency assay via pRU1103 plasmid, as described in the “bacterial transformation” section.
Bacteriophage MS2 drop plaque assays
The bacteriophage MS2 RNA phage was purchased from the ATCC (ATCC 15597-B1). Individual spacers and repeat sequences for MS2 RNA phage interference were synthesized (Gene Universal) into the pCDFDuet-1 vector corresponding to Cas13a, Cas13b, or Cas13d. Plaque assay on bacterial lawns of E. coli C-3000 or derived strains was performed at 37 °C on LB agar (1.5%) plates with a lower percentage of LBTop agar (0.3%) that were supplemented with 1 mM isopropyl IPTG and/or arabinose. Bacterial cells were mixed with LBTop agar and poured onto an LB agar plate as an even layer. The plates were spot with 3 μL of each phage lysate on the lawn and grown overnight. Anti-CRISPR activity was assessed by measuring replication of MS2 phages on bacterial lawns relative to the vector control.
Bacterial transformation assays
Gene Universal The pRU1103 plasmid with gentamicin resistance (Addgene #14467) was used for transformation of efficiency assay for type I-B in E. coli BL21 and derived mutation strains with or without an optional Acr candidate plasmid according to a previously published protocol (Lin et al., 2019a; Lin et al., 2019b).
RNA interference assays
To investigate the anti-CRISPR activity in VI CRISPR-Cas system, the pRU1103 plasmid was added to 50 μL of E. coli C-3000 strain containing Cas13a, crRNA targeted gentamicin RNA, and Acr candidate or empty vector. The strains were incubated for 30 min on ice and heat shock at 42 °C for 60 seconds. Then, 500 μL of SOC was added to strains and incubated with 220 rpm/min at 37 °C for 60 min. Next, we performed a plating of SOC medium on the plates containing gentamicin (75 μg/mL, ACROS).
Phylogenetic reconstructions
Homologous protein sequences of AcrVIAs were acquired through BLASTp searching for the non-redundant protein sequence database. The sequences of high homology (E value < 1e-03, >70% coverage) were downloaded and aligned by ClustalW in MEGA7(Kumar et al., 2016). Reconstruction of phylogenetic trees with each AcrVIA protein family was performed in MEGA7 using the minimum evolution method (Desper and Gascuel, 2004) with the Poisson model for amino acid substitution model, uniform rates among sites and pairwise deletion of gaps/missing data treatment. The resulting phylogeny images were then edited for clarity using Illustrator (Adobe).
Cloning of constructs for human cell expression
Human cells LwaCas13a, PspCas13b, and CasRx expression plasmids were acquired from Addgene. Human cell U6 promoter expression plasmids for crRNA targeted Gaussia mRNA were generated by annealing and ligating the oligonucleotide duplexes into BbsI-digested LwaCas13a guide expression backbone, PspCas13b crRNA backbone, and CasRx pre-gRNA backbone vector, respectively. And the crRNA targeted endogenous genes KRAS, CXCR4, and PPIB were cloned into LwaCas13a guide expression backbone. The Gaussia and Firefly luciferase reporter plasmids were purchased from ThermoFisher Scientific. Human codon optimized AcrVIA sequences with a C-terminal nuclear localization signal (SV40) were cloned into pcDNA3.1+ via Gibson cloning.
Designing and cloning of human cell constructs for RNA editing
dLwaCas13a-msfGFP and dPspCas13b-GS-ADAR2DD(E488Q/T375G) (REPAIRv2 Plasmid 10387) were obtained from Addgene. The EcoRI-digested dLwaCas13a-msfGFP plasmid were added at the XbaI and AscI sites in the C-terminus. The deaminase domains of human ADAR2dd were amplified from the dPspCas13b-GS-ADAR2DD(E488Q/T375G) and cloned into dLwCas13a plasmid via XbaI and AscI double digestion. The recombinant dLwaCas13a-ADAR2DD plasmid was used to RNA editing.
The Gaussia luciferase mutation reporter for measuring anti-CRISPR-mediated inhibition of dCas13-ADAR2dd RNA - editing activity was generated by creating a mutation at 140 site (TGG>TAG) in the Gaussia reporter plasmid. The red firefly luciferase reporter acts as a normalization control, but a defective Gaussia due to the addition of the TGG>TAG pre-termination site by dCas13-ADAR2DD in the presence or absence of anti-CRISPR proteins.
For guide cloning for RNA editing, the oligonucleotide duplexes for targeting RNA editing sites were heated to 95 °C for 5 min and then annealed by cooling to 4 °C. The annealed oligos were ligated to BbsI-digested LwaCas13a guide expression backbone under U6 expression,
Human cell transfection
Human cells were cultured and LipofectAmine 3000 (Thermo Fisher Scientific) transfection was performed with 300 ng of Cas13 nuclease plasmid, 300 ng of crRNA plasmid, and 750 ng of anti-CRISPR protein plasmid that isolated using the QIAGEN Plasmid Mini Kit (QIAGEN); and experiments testing knockdown of reporter plasmids were supplemented with 50 ng Gaussia luciferase and red firefly luciferase construct per well. Twelve hours before transfection, cells were plated in 96-well at approximately 10,000 cells per well. For each well, plasmids were combined with Opti-MEM I Reduced Serum Medium (Thermo Fisher Scientific) and P3000 reagent added to a total of 10 μL (separately 0.5 μL of Lipofectamine 3000 was mixed with 9.5 μL of Opti-MEM). Then we added diluted plasmids to each tube of diluted LipofectAmine solutions to incubate for 15 min and incubated cells for 48 h at 37 °C in 5% CO2 in a cell culture incubator. For endogenous genes test, all experiments were performed in 6-well plates.
Purification of His anti-CRISPR proteins
6xHis-AcrVIA proteins were purified from pET-28a vector expressed in E. coli BL21. Cells were cultured in LB medium at 37°C until OD600=0.6. Protein expression was induced by adding 1 mM IPTG for overnight at 18°C. Cells were lysed by sonication with buffer 1 (50 mM Tris-HCl PH=7.0, 500 mM NaCl, 1 mM TCEP, and 5 % glycerol with protease inhibitor cocktail (Thermo Fisher Scientific) and centrifuged with 16000 rpm/min. The supernatant lysates were incubated with HisPurTM Ni-NTA Spin Columns (Thermo Fisher Scientific) and eluted with 200 mM imidazole. The purified proteins were dialyzed into buffer 2 (50 mM Tris-HCl PH=7.5, 150 mM NaCl, 1mM EDTA, 0.05 % NP-40) for protein interaction experiments.
Co-Immunoprecipitation (Co-IP)
Fifty μL of Dynabeads™ Sheep anti-Mouse IgG were washed by PBS containing 0.1% BSA and 2 mM EDTA. Four μg of mouse monoclonal GFP antibody (Biolegend) were bound to the beads for 60 min at room temperature. After washing, we added 200 μL of HEK293T cell lysates containing LwaCas13a-GFP proteins for rotating the tubes for 2 h incubation at 4 °C. The beads were washed five times with 50 mM Tris-HCl PH=7.5, 150 mM NaCl, 1mM EDTA, 0.05 % NP-40 and then incubated with different His-AcrVIA proteins for overnight. After washing, the eluted proteins were analyzed by SDS-PAGE and western blot with anti-GFP antibody (IP) or anti-his tag antibody (Co-IP).
His pull-down
His pull-down was preformed using the PierceTM His Protein Interaction Pull-Down Kit (The. Briefly, the purified anti-CRISPR proteins were dialysis against Thermo Scientific BupH Tris Buffered Saline (Thermo Fisher Scientific). After washing with wash solution (1:1 wash solution of TBS:Pierce Lysis Buffer with 10 mM imidazole), the HisPur Cobalt Resin was incubated with AcrVIA proteins for 1 h with gentle rocking at 4 °C. After washing with five times, add up to 800 uL of HEK293T cell lysates containing LwaCas13a-GFP proteins and incubated at 4 °C for overnight with gentle rocking. The beads were collected by centrifugation at 1250 g/min for 1 min after washing five times. Bound proteins were eluted with 290 mM imidazole in wash solution. The eluted proteins were analyzed by western blot with anti-GFP and anti-his tag antibody.
Measurement of luciferase activity
Cells containing luciferase were harvested 48 h after transfection with washing PBS buffer. Cells were disrupted with 100 μL/well of 1 X Cell Lysis buffer and shaken for 15 min. Luciferase activity was measured using Pierce™ Gaussia-Firefly Luciferase Dual Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Fluorescence microscopy
pC035-dLwaCas13a-msfGFP plasmid was purchased from Addgene (Addgene #91925) and used directly for no-crRNA control experiments. We also modified dLwaCas13a-msfGFP into an all-in-one plasmid that also included an crRNA-expressing cassette cloning from LwaCas13a guide expression backbone with U6 promoter, with crRNAs targeted to endogenous gene via Gibson assembly to generate dLwaCas13a-msfGFP-crRNA plasmid. To make the AcrVIA plasmids, we amplified an AcrVIA5 cassette and incorporated in pEJS481-pCDest2-AcrE2-mTagBFP2-IRES (Addgene #85746) to replace the AcrE2 by Gibson assembly. HEK293T cells were plated in 24-well tissue culture plates on poly-d-lysine coverslip (Corning) and transfected by LipofectAmine 3000 (Thermo Fisher Scientific). After 48 h incubation, cells were washed with PBS, and fixed in 4% paraformaldehyde for 15 min. Images were observed under a Zeiss LSM 510 Meta Confocal Microscope. Only cells that exhibited mTagBFP2 and msfGFP fluorescence were assessed for the presence or absence of co-localizing dLwaCas13a-msfGFP foci, and such imaged cells were included in the quantifications.
RNA isolation and qRT-PCR
Total RNA was isolated using the Direct-zol™ RNA MiniPrep kit according to the manufacturer’s protocol with DNase I treatment. 2 μg total DNA-free RNA was reverse transcribed to cDNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qPCR was performed using CFX Connect™ Real-Time System (Bio-Rad), Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific).
RNA editing in human cells
To assess the inhibition activity of dLwaCas13a-ADAR2DD by anti-CRISPR proteins, 300 ng of dLwaCas13a-ADAR2DD plasmid, 300 ng of guide expression plasmid, 759 ng of AcVIA5 were used to test knockdown of reporter plasmids with 50 ng Gaussia luciferase mutation and red firefly luciferase plasmid per well through LipofectAmine 3000 kit. After 48 h transfection, Luciferase activity was measured using Pierce™ Gaussia-Firefly Luciferase Dual Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
For cloning disease-relevant mutations (pathogenic G>A mutation) to test AcrVIA5 inhibition activity, three G>A mutations related to disease pathogenesis as defined in ClinVar were selected and 200 bp regions surrounding these mutations were synthesized and Gibson cloned under expression of CMV.
For counting RNA editing rates, RNA from cells was harvested and reverse transcribed using the above described method with a gene specific reverse transcription primer. The cDNA was used to PCR amplification and the cloned into T-Vector pMD19 (Simple) (TaKaRa) for sequencing. RNA editing rates were calculated at 140 site (TGG>TAG) within the sequencing window.
RNA sequencing and analysis
For specificity analysis of AcrVIAs-mediated reduction of the collateral activity of LwaCas13a knockdown, RNA-seq for the express package was performed on HiSeq4000 system from LwaCas13a knockdown in the presence or absence of AcrVIA2 and AcrVIA5. Total RNA was isolated using the Direct-zolTM RNA MiniPrep kit according to the manufacturer’s protocol with DNase I treatment. RNA libraries were prepared using NEBNext Ultra RNA Library Prep Kit for Illumina. Sequencing was done by Hiseq4000 systems for 400 million reads per library. Fragments per kilo base per million mapped reads (fpkm) values were used for expression counts and were transformed to log-space by taking the log2(fpkm+1). For differentially expressed genes, only genes with a fold change ≥2 or less than 0.5 in at least two of the three replicates. The variation of control vs., LwaCas13a, LwaCas13a with AcrVIA2 or AcrVIA5 was analyzed by considering the distribution of standard deviations for gene expression across three replicates and represented as violin plots.
Cell viability assay and cell cytotoxicity assay
Mammalian cells were transfected with anti-CRISPR protein plasmid. Twenty-four hours after transfection, cell viability was measured using the CellTiter 96® AQueous One Solution Cell Proliferation assay (Promega) according to manufacturer’s instructions. Cell cytotoxicity was measured through LDH activity by Pierce LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific).
QUANTIFICATION AND STATISTICAL ANALYSIS
Please refer to the Figure Legends for description of sample size and statistical analyses. One-way ANOVA plus Tukey’s post hoc tests were used for comparisons with GraphPad (GraphPad Software, La Jolla, CA). P>0.05 was considered no significant difference (NS), and *P<0.05 or **P<0.01 were considered statistically significant.
DATA AND CODE AVAILABILTY
The published article includes all datasets generated during this study. Additional amino acid sequences of AcrVIAs and Aca are reported in Table S3
Supplementary Material
Table S1. Bioinformational Analysis for Screening Type VI Strains Containing Self-targets, Related to Figure 1.
Table S2. Domain Architectures Identified for Prophage Regions in L. wadei F0279 with Type VI-A CRISPR-Cas Strain Containing Self-targets, Related to Figure 1.
Table S5. Plasmids Used for Expression in Bacteria and Human Cells, Related to STAR Methods.
Table S6. Bacterial Strains and Phage Used in this Study, Related to STAR Methods.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Oligonucleotides | ||
| crRNAs | This study | Table S4 |
| Recombinant DNA | ||
| Plasmids | This study | Table S5 |
| Antibodies | ||
| mouse monoclonal GFP antibody | Biolegend | Cat# 668206 |
| anti-his tag anti-body | Thermo Fisher Scientific | Cat# TA150087 |
| Bacterial and Virus Strains | ||
| E. coli c-3000 | ATCC | Cat# 15597 |
| NEB 5-alpha Competent E. coli | New England BioLabs | Cat# C2987H |
| BL21 Competent E. coli | New England BioLabs | Cat# C2530H |
| E. coli bacteriophage MS2 | ATCC | Cat# 15597-B1 |
| Leptotrichia buccalis DSM 1135 | ATCC | Cat# 14201 |
| E. coli c3000-derived strains | This study | Table S6 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LB Broth Base | Thermo Fisher Scientific | Cat# 12780029 |
| LB Agar | Fisher Scientific | Cat# BP1425-500 |
| SOC medium | New England BioLabs | Cat# B9020S |
| ampicillin | Fisher Scientific | Cat#11593027 |
| chloramphenicol | Sigma | Cat# C0378 |
| kanamycin | Fisher Scientific | Cat# 25389-94-0 |
| gentamicin | ACROS | Cat# 1405-41-0 |
| IPTG | Invitrogen | Cat# 15529019 |
| penicillin-streptomycin | Thermo Fisher Scientific | Cat# 10378016 |
| fetal bovine serum | VWR Seradigm | Cat# 97068-085 |
| LipofectAmine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| Opti-MEM I Reduced Serum Medium | Thermo Fisher Scientific | Cat# 11058021 |
| RMPI 1640 Medium | Thermo Fisher Scientific | Cat# 11875119 |
| protease inhibitor cockt | Thermo Fisher Scientific | Cat# 78425 |
| Critical Commercial Assays | ||
| QIAGEN Plasmid Mini Kit | QIAGEN | Cat# 12125 |
| myTXTL Sigma 70 Master Mix Kit | Arbor Bioscience | Cat# 507024 |
| AMPure XP beads | Beckman Coulter | Cat# A63882 |
| Dynabeads™ Sheep anti-Mouse IgG | Invitrogen | Cat# 11201D |
| Pierce™ His Protein Interaction Pull-Down Kit | Thermo Fisher Scientific | Cat# 21277 |
| Pierce™ Gaussia-Firefly Luciferase Dual Assay Kit | Thermo Fisher Scientific | Cat# 16181 |
| High Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4368814 |
| Maxima SYBR Green qPCR Master Mix | Thermo Fisher Scientific | Cat# K0252 |
| Direct-zol™ RNA MiniPrep kit | Zymo Reseach | Cat# R2052 |
| the CellTiter 96 AQueous One Solution Cell Proliferation assay | Promega | Cat# G3580 |
| Pierce LDH Cytotoxicity Assay Kit | Thermo Fisher Scientific | Cat# C20300 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216 |
Highlights.
An integrated approach is designed to screen anti-CRISPRs (AcrVIA) candidates
A series of prophages are discovered in regions encoding AcrVIAs
AcrVIAs inhibit Cas13a RNA targeting in bacteria and human cells
AcrVIAs impede programmable RNA base editing by blocking dCas13a-based toolkit
Acknowledgments
This work was supported by National Institutes of Health Grants R01 AI109317-01A1, R01 AI138203, P20 GM113123 and AI097532-01A1 to MW and UND Post-Doc Pilot Grant; this work was also supported by the Key Program of National Nature Science Foundation of China (NSFC, 81530063) to Jianxin Jiang. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Dr, Bony De Kumar, Danielle Perley, and Hannah Huffman in Genomics Core of the University of North Dakota for RNA-seq and bioinformatics analysis, as well as Dr. Sergei Nechaev, Dr. Archana Dhasarathy, and Dr. Beiswenger Kristina of the University of North Dakota for critical reading of the manuscript.
Footnotes
Declaration of interests
A provisional patent application pertaining to AcrVIA genes for CRISPR-related technologies and their applications has been filed by the University of North Dakota, Daping Hospital, and Army Medical University in China.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DB, Kellner MJ, and Regev A (2017). RNA targeting with CRISPR–Cas13. Nature 550, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, and Minakhin L (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athukoralage JS, McMahon SA, Zhang C, Grüschow S, Graham S, Krupovic M, Whitaker RJ, Gloster TM, and White MF (2020). An anti-CRISPR viral ring nuclease subverts type III CRISPR immunity. Nature 577, 572–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bhoobalan-Chitty Y, Johansen TB, Di Cianni N, and Peng X (2019). Inhibition of Type III CRISPR-Cas Immunity by an Archaeal Virus-Encoded Anti-CRISPR Protein. Cell 179, 448–458. [DOI] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, and Davidson AR (2015). Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, and Davidson AR (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, and Zhang F (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, Teichmann M, Rousseau E, Lamrissi-Garcia I, and Guyonnet-Duperat V (2019). CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 10, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desper R, and Gascuel O (2004). Theoretical foundation of the balanced minimum evolution method of phylogenetic inference and its relationship to weighted least-squares tree fitting. Mol Biol Evol 21, 587–598. [DOI] [PubMed] [Google Scholar]
- Dong, Guo M, Wang S, Zhu Y, Wang S, Xiong Z, Yang J, Xu Z, and Huang Z (2017). Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–439. [DOI] [PubMed] [Google Scholar]
- Dong L, Guan X, Li N, Zhang F, Zhu Y, Ren K, Yu L, Zhou F, Han Z, Gao N, et al. (2019). An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol. 26, 308–314. [DOI] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Burstein D, Knott GJ, and Doudna JA (2017). RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol. Cell 66, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, and Doudna JA (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. (2018). The Pfam protein families database in 2019. Nucleic Acids Res 8, 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, and Sander JD (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. biotechnol. 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GW, Jiang W, Bikard D, and Marraffini LA (2014). Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature 514, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, and Zhang F (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu JJ, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. (2017). A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 170, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Bhoobalan-Chitty Y, Van LB, Kjeldsen AL, Dedola M, Makarova KS, Koonin EV, Brodersen DE, and Peng X (2018). Anti-CRISPR proteins encoded by archaeal lytic viruses inhibit subtype I-D immunity. Nat Microbiol 3, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Agudelo D, Goulet A, Amigues B, Loehr J, Romero DA, Fremaux C, Horvath P, Doyon Y, et al. (2018). Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat Commun 9, 2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia N, Mo CY, Wang C, Eng ET, Marraffini LA, and Patel DJ (2019). Type III-A CRISPR-Cas Csm Complexes: Assembly, Periodic RNA Cleavage, DNase Activity Regulation, and Autoimmunity. Mol. Cell 73, 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, and Doudna JA (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Thornton BW, Lobba MJ, Liu JJ, Al-Shayeb B, Watters KE, and Doudna JA (2019). Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat. Struct. Mol. Biol. 26, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, and Hsu PD (2018). Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, and Makarova KS (2018). Anti-CRISPRs on the march. Science 362, 156–157. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, and Tamura K (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Mir A, Edraki A, Garcia B, Amrani N, Lou HE, Gainetdinov I, Pawluk A, Ibraheim R, Gao XD, et al. (2018). Potent Cas9 Inhibition in Bacterial and Human Cells by AcrIIC4 and AcrIIC5 Anti-CRISPR Proteins. MBio 9, e02321–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Pu Q, Shen G, Li R, Guo K, Zhou C, Liang H, Jiang J, and Wu M (2019a). CdpR inhibits CRISPR-Cas adaptive immunity to lower anti-viral defense while avoiding self-reactivity. iScience 29, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Pu Q, Wu Q, Zhou C, Wang B, Schettler J, Wang Z, Qin S, Gao P, Li R, et al. (2019b). High-throughput screen reveals sRNAs regulating crRNA biogenesis by targeting CRISPR leader to repress Rho termination. Nat. Commun. 10, 3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li X, Ma J, Li Z, You L, Wang J, Wang M, Zhang X, and Wang Y (2017a). The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 170, 714–726. [DOI] [PubMed] [Google Scholar]
- Liu L, Li X, Wang J, Wang M, Chen P, Yin M, Li J, Sheng G, and Wang Y (2017b). Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell 168, 121–134. [DOI] [PubMed] [Google Scholar]
- Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, Rauch BJ, Walton RT, Berry JD, Joung JK, Kleinstiver BP, et al. (2018). Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 362, 240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske AJ, Nakandakari-Higa S, and Marraffini LA (2019). Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature 570, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Gonzalez MF, Quiroz JFD, and Rosenthal JJ (2018). Current strategies for Site-Directed RNA Editing using ADARs. Methods 156, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrvold C, Freije CA, Gootenberg JS, Abudayyeh OO, Metsky HC, Durbin AF, Kellner MJ, Tan AL, Paul LM, and Parham LA (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, et al. (2016a). Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 167, 1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Bondy-Denomy J, Cheung VH, Maxwell KL, and Davidson AR (2014). A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio 5, e00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Davidson AR, and Maxwell KL (2018). Anti-CRISPR: discovery, mechanism and function. Nat Rev Microbiol 16, 12–17. [DOI] [PubMed] [Google Scholar]
- Pawluk A, Staals RH, Taylor C, Watson BN, Saha S, Fineran PC, Maxwell KL, and Davidson AR (2016b). Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol 1, 16085. [DOI] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, and Bondy-Denomy J (2017). Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 168, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, and Marraffini LA (2015). Co-transcriptional DNA and RNA cleavage during type III CRISPR-Cas immunity. Cell 161, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, and Severinov K (2015). Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol. Cell 60, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargon AA, Cox DBT, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, Abudayyeh OA, Essletzbichler P, Shmakov S, Makarova KS, et al. (2017). Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol. Cell 65, 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, and Noireaux V (2013). Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J Vis Exp. 16, e50762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terns MP (2018). CRISPR-Based Technologies: Impact of RNA-Targeting Systems. Mol. Cell 72, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mo CY, Wasserman MR, Rostøl JT, Marraffini LA, and Liu S (2019). Dynamics of Cas10 govern discrimination between self and Non-self in type III CRISPR-Cas immunity. Mol. Cell 73, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yao D, Xu JG, Li AR, Xu J, Fu P, Zhou Y, and Zhu Y (2016). Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat. Struct. Mol. Biol. 23, 868–870. [DOI] [PubMed] [Google Scholar]
- Watters KE, Fellmann C, Bai HB, Ren SM, and Doudna JA (2018). Systematic discovery of natural CRISPR-Cas12a inhibitors. Science 362, 236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, and Scott DA (2018). Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol. Cell 70, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, and Patel DJ (2017). Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol. Cell 67, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Konermann S, Brideau NJ, Lotfy P, Wu X, Novick SJ, Strutzenberg T, Griffin PR, Hsu PD, and Lyumkis D (2018). Structural basis for the RNA-guided ribonuclease activity of CRISPR-Cas13d. Cell 175, 212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Z, Daczkowski CM, Gabel C, Mesecar AD, and Chang L (2019). Structural Basis for the Inhibition of CRISPR-Cas12a by Anti-CRISPR Proteins. Cell Host Microbe 25, 815–826. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, and Wishart DS (2011). PHAST: a fast phage search tool. Nucleic Acids Res 39, W347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo E, Sun Y, Wei W, Yuan T, Ying W, Sun H, Yuan L, Steinmetz LM, Li Y, and Yang H (2019). Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science, 364, 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Bioinformational Analysis for Screening Type VI Strains Containing Self-targets, Related to Figure 1.
Table S2. Domain Architectures Identified for Prophage Regions in L. wadei F0279 with Type VI-A CRISPR-Cas Strain Containing Self-targets, Related to Figure 1.
Table S5. Plasmids Used for Expression in Bacteria and Human Cells, Related to STAR Methods.
Table S6. Bacterial Strains and Phage Used in this Study, Related to STAR Methods.