

Abstract
Photoreceptor cyclic nucleotide-gated (CNG) channels regulate Ca2+ influx in rod and cone photoreceptors. Mutations in cone CNG channel subunits CNGA3 and CNGB3 are associated with achromatopsia and cone dystrophies. Mice lacking functional cone CNG channel show endoplasmic reticulum (ER) stress-associated cone degeneration. The elevated cyclic guanosine monophosphate (cGMP)/cGMP-dependent protein kinase (PKG) signaling and upregulation of the ER Ca2+ channel ryanodine receptor 2 (RyR2) have been implicated in cone degeneration. This work investigates the potential contribution of RyR2 to cGMP/PKG signaling-induced ER stress and cone degeneration. We demonstrated that the expression and activity of RyR2 were highly regulated by cGMP/PKG signaling. Depletion of cGMP by deleting retinal guanylate cyclase 1 or inhibition of PKG using chemical inhibitors suppressed upregulation of RyR2 in CNG channel deficiency. Depletion of cGMP or deletion of Ryr2 equivalently inhibited unfolded protein response/ER stress, activation of the CCAAT-enhancer-binding protein homologous protein, and activation of the cyclic adenosine monophosphate response element binding protein, leading to early-onset cone protection. In addition, treatment with cGMP significantly enhanced Ryr2 expression in cultured photoreceptor-derived Weri-Rb1 cells. Findings from this work demonstrate the regulation of cGMP/PKG signaling on RyR2 in the retina and support a role of RyR2 upregulation in cGMP/PKG signaling-induced ER stress and photoreceptor degeneration.
Keywords: CNG channel, cone photoreceptors, retinal degeneration, ryanodine receptor, cGMP/PKG signaling
Introduction
Photoreceptor cyclic nucleotide-gated (CNG) channels play a pivotal role in rod and cone phototransduction. In darkness or dim light, the CNG channel is kept open by binding of cyclic guanosine monophosphate (cGMP), maintaining a steady Ca2+ and Na+ influx. Light induces hydrolysis of cGMP by photoreceptor phosphodiesterase 6, resulting in closure of the channel and hyperpolarization of the cell (1, 2). The cone photoreceptor CNG channel is composed of two structurally related subunits, CNGA3 and CNGB3. CNGA3 is an ion-conducting subunit, whereas CNGB3 functions as a modulator (2). Mutations in CNGA3 and CNGB3 genes are associated with achromatopsia, progressive cone dystrophies, and early-onset macular degeneration (3–5).
Cone loss in patients with achromatopsia and cone dystrophies associated with CNG channel mutations has been well documented by optical coherence tomography (6–12). Impaired cone function and endoplasmic reticulum (ER) stress-associated cone apoptosis/progressive cone degeneration have also been characterized in Cnga3−/− and Cngb3−/− mice (13–16) and in Cnga3−/−/Nrl−/− mice with CNG channel deficiency on a cone-dominant Nrl−/− background (17, 18). One typical cellular alteration in CNG channel deficiency is the remarkable elevation of cellular cGMP level and increased activity of the cGMP-dependent protein kinase (protein kinase G, PKG) (17, 19, 20). Retinal cGMP levels in Cnga3−/−/Nrl−/− mice sharply increased at postnatal day 8 (P8), peaked around P10-15, remained high through P30-60, and returned to near control levels at P90 (19). The cGMP elevation pattern correlated with apoptotic cone death (13, 17, 19). The contribution of elevated cGMP/PKG signaling to ER stress/cone death was demonstrated by genetic deletion of retinal guanylate cyclase 1 (retGC1), an enzyme responsible for biosynthesis of cGMP in photoreceptors, and inhibition of PKG using chemical inhibitors (19, 21). How cGMP/PKG signaling induces ER stress and cone death, however, remains unidentified.
Along with elevated cGMP/PKG signaling, CNG channel-deficient cones show cellular calcium/ER calcium dysregulation. Cellular calcium/ER calcium homeostasis is essential to normal cellular signaling, including photoreceptor light adaptation and protein folding. Perturbation of cellular calcium has been associated with cellular dysfunction, ER stress, and cell death (22–26). As non-selective cation channels, CNG channels play a pivotal role in cellular calcium homeostasis/ER calcium homeostasis. Measurements of intracellular Ca2+ levels showed reduced cytosolic Ca2+ level and altered cellular Ca2+ dynamics in CNG channel-deficient cones (27). ER calcium homeostasis is primarily regulated by two ER calcium channels, inositol-1,4,5-trisphosphate receptor (IP3R) (28) and ryanodine receptor (RyR) (29), which efflux calcium out of the ER, and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) (30), which influxes calcium into the ER. Three RyR isoforms have been identified; RyR1, RyR2, and RyR3. All three isoforms of RyR are expressed in photoreceptors, and RyR2 has been identified as the major form in photoreceptors and is localized in the inner segments and outer nuclear layer (27, 31–33). CNG channel-deficient retinas show elevated RyR2 expression/activity and treatment with RyR inhibitor or deletion of Ryr2 reduced ER stress/cone death and improved cone protein outer segment localization (27, 34).
In the current study, we examined the regulation of cGMP/PKG signaling on RyR2 and investigated whether RyR2 contributes to cGMP/PKG signaling-induced ER stress/cone death. We demonstrated that the expression and activity of RyR2 were highly regulated by cGMP/PKG signaling. Depletion of cGMP or inhibition of PKG abolished upregulation of RyR2 in CNG channel-deficient retinas. Depletion of cGMP or deletion of Ryr2 equivalently inhibited unfolded protein response (UPR)/ER stress, activation of the CCAAT-enhancer-binding protein homologous protein (CHOP), and activation of the cyclic adenosine monophosphate response element binding protein (CREB), leading to early-onset cone protection. In addition, treatment with cGMP significantly enhanced Ryr2 expression in cultured photoreceptor-derived Weri-Rb1 cells. Our findings demonstrate a tight regulation of cGMP/PKG signaling on RyR2 in the retina, and support a role of RyR2 upregulation in cGMP/PKG signaling-induced ER stress and cone degeneration. This work provides insight into the mechanism of photoreceptor degeneration in CNG channel deficiency and provides a better understanding of how cGMP/PKG signaling induces photoreceptor cell death.
Materials and Methods
Mice, antibodies, and other reagents
The Cnga3−/− (14), Nrl−/− (35), Gucy2e−/− (36), and Hrgpcre (transgenic mice expressing Cre recombinase directed by the human red/green pigment [HRGP] gene promoter) (37) mouse lines were generated as described previously. The Ryr2flox/flox mouse line (38) was obtained from Jackson Laboratory (Bar Harbor, ME). The Cnga3−/−/Nrl−/− (17), Cnga3−/−/Nrl−/−/Gucy2e−/− (21), and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre (34) mouse lines were generated by cross-breeding. Lack of expression of CNGA3 in the retina was shown by immunofluorescence labeling using anti-CNGA3 antibody (Supplementary Fig. 1). Genotyping was conducted to confirm the knockout/expression of the specific genes, including Nrl, Gucy2e, Ryr2fl/fl, and Hrgpcre (Supplementary Fig. 2). NRL is a basic-motif leucine zipper transcription factor essential for the normal development of rods. Mice lacking the Nrl gene have no rods, but have increased numbers of cones, functionally manifested as a loss of rod function coupled with super-normal cone function (35). Morphologically, Nrl−/− photoreceptors have a cone-like nucleus, short and disorganized outer segments, and frequently form rosette-like structures in the retina (35). Electrophysiological studies demonstrate the expression of functional S- and M-opsins (39, 40), whereas gene expression studies confirm the transformation of all rods into cones in Nrl−/− retinas (41). As a valuable mammalian cone-dominant model (cones comprise only 2-3% of the total photoreceptor population in the wild-type mouse retina), Nrl−/− mouse line has been commonly used for studies of biochemical alterations in cones (17, 42, 43). The present study used Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice to explore the regulation of cGMP/PKG signaling on RyR2 in cones lacking CNG channel. It should be noted that Cnga3−/−/Nrl−/− mice display the phenotype of loss of cone function, cone degeneration, and apoptotic cone death, similar to that in Cnga3−/− mice (17, 18). All mice were maintained under cyclic light (12 h light-dark) conditions. In the light cycle, cage illumination was 7 foot-candles. All animal maintenance and experiments were approved by the local Institutional Animal Care and Use Committee (University of Oklahoma Health Sciences Center, Oklahoma City, OK) and conformed to the guidelines on the care and use of animals adopted by the Society for Neuroscience and the Association for Research in Vision and Ophthalmology (Rockville, MD).
The primary antibodies used are listed in Table 1. Horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse secondary antibodies were purchased from Kirkegaard & Perry Laboratories, Inc. (Gaithersburg, MD). All other reagents were purchased from Sigma Aldrich (St Louis, MO), Bio-Rad (Hercules, CA), and Thermo Fisher Scientific (Waltham, MA).
Table 1.
List of primary antibodies
| Antibody | Provider/Reference | Catalog No. | Dilutions used in IB and IF |
|---|---|---|---|
| M-opsin | EMD Millipore, Billerica, MA | AB5405 | 1: 500 (IB) |
| Cone arrestin (CAR) | EMD Millipore, Billerica, MA | AB15282 | 1: 500 (IB) 1:200 (IF) |
| p-eIF2α | Cell signaling, Danvers, MA | 3398 | 1: 500 (IB) |
| p-Ire1α | Abcam, Cambridge, MA | ab48187 | 1: 500 (IB) |
| p-CREB | Cell signaling, Danvers, MA | 91985 | 1: 500 (IB) |
| CREB | Cell signaling, Danvers, MA | 9197 | 1: 500 (IB) |
| p-RyR2 | Abcam, Cambridge, MA | ab59225 | 1: 500 (IB) |
| p-CaMKII | Cell signaling, Danvers, MA | 12716 | 1: 500 (IB) |
| GFAP | Dako | Z0334 | 1: 500 (IB) |
| Derl-1 | Abcam, Cambridge, MA | ab176732 | 1: 500 (IB) |
| Creb3l3 | Novus, Centennial, CO | NBP2-16008 | 1: 500 (IB) |
| Uggt2 | Thermo Fisher Scientific, Waltham, MA | PA5-27960 | 1: 500 (IB) |
| ATF6 | Active Motif, Carlsbad, CA | 40962 | 1: 500 (IB) |
| CHOP | Cell signaling, Danvers, MA | 2985 | 1: 500 (IB) |
| TBP | Thermo Fisher Scientific, Waltham, MA | MA1-10883 | 1: 1000 (IB) |
| Iba1 | FUJIFILM Wako Chemicals, Richmond, VA | 019-19741 | 1:200 (IF) |
| CNGA3 | Matveev et al., (43) | - | 1:200 (IF) |
| β-actin | Abcam, Cambridge, MA | ab6276 | 1: 2000 |
PKG inhibitor treatment
Two PKG inhibitors, KT5823 (Sigma Aldrich, St Louis, MO) and (Rp)-8-Br-cGMPS (Santa Cruz Biotechnology, Dallas, TX), were used. (Rp)-8-Br-cGMPS is a cyclic nucleotide analog (an Rp-diastereomer of cGMP), whereas KT5823 is a K-series inhibitor that inactivates the ATP-binding site through competition with ATP (44). These two compounds have been shown to reduce PKG activity in CNG channel-deficient mice and reduce ER stress/cone death (21). Starting at postnatal day 7 (P7), Cnga3−/−/Nrl−/− mice received intraperitoneal injection of KT5823 (1.0 μmol/kg body weight/day), (Rp)-8-Br-cGMPS (5.0 μmol/kg body weight/day), or vehicle for 9 days, as described previously (21). At the end of the treatment, retinas were collected for biochemical evaluations.
Retinal cross-section preparation
Mouse eyes were enucleated (the superior portion of the cornea was marked for orientation before enucleation) and fixed in Prefer (Anatech Ltd., Battle Creek, MI) for 25 min at room temperature. Fixed eyes were then stored in 70% ethanol until processing for sections, as described previously (27). Paraffin-embedded sections (5-μm thickness) passing vertically through the retina (along the vertical meridian passing through the optic nerve head, to allow an examination of the retina in the dorsal and ventral hemisphere) were prepared with a microtome (Leica, Bannockburn, IL).
TUNEL assay
The terminal deoxynucleotidyltransferase dUTP nick end-labeling (TUNEL) was carried out to evaluate photoreceptor apoptotic death, as described previously (45). Paraffin-embedded retinal cross sections and the In Situ Cell Death Fluorescein Detection kit (Roche Diagnostics, Mannheim, Germany) were used. The images of the immunohistochemistry were taken with the FV1000 confocal laser scanning microscope (Olympus, Center Valley, PA). The total TUNEL-positive cells in the outer nuclear layer were counted and averaged from at least three sections/eye.
PCR array and quantitative RT-PCR
Total RNA preparation and reverse transcription were performed as described previously (18). The Mouse Unfolded Protein Response RT² Profiler™ PCR Array (Qiagen, Hiden, Germany), which profiles the expression of 84 genes involved in unfolded protein binding, protein folding, endoplasmic-reticulum-associated protein degradation, and heat shock proteins, was used under the manufacturer’s instructions. The quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays were performed with a real-time PCR detection system (iCycler; Bio-Rad). The primers used are listed in Table 2. The relative gene expression value was calculated based on the ∆∆Ct method (18).
Table 2.
List of primers used for qRT-PCR
| Gene symbols | Species | Forward primer | Reverse primer |
|---|---|---|---|
| Hprt1 | mouse | GCAAACTTTGCTTTCCCTGGTT | CAAGGGCATATCCAACAACA |
| Ryr2 | mouse | CTACCCGAACCTCCAGCGATACT | GCAAAAGAAGGAGATGATGGTGTG |
| Hprt1 | human | GCTATAAATTCTTTGCTGACCTGCTG | AATTACTTTTATGTCCCCTGTTGACTGG |
| Itpr1 | human | ACGCTGACATCCTGATTGAG | ATTCCCTCACACTCTTGCTG |
| Ryr2 | human | TGGAGAAATCAGAAGGGCAAG | CTTATCAGTTGAAGACCGGGAG |
| Serca2 | human | ATCTTTGAATCCCCATACCCG | CGACATAGAGGATCAGGAAGTG |
| Calr | human | CCTATGATAACTTTGGCGTGC | TTGTCCTTCATTTGTTTCTCTGC |
Retinal protein preparation, SDS-PAGE, and western blot analysis
Retinal protein preparation, SDS-PAGE, and western blot analysis were performed as described previously (21). Briefly, retinas were homogenized in homogenization buffer A (0.32 M sucrose, 20 mM HEPES, pH 7.4, and 3 mM EDTA containing protease and phosphatase inhibitors) (Sigma Aldrich), and homogenates were centrifuged at 3000 rpm for 10 min at 4 °C. The resulting supernatant was then centrifuged at 13,000 rpm for 35 min at 4 °C to separate cytosolic (supernatant) and membrane (pellet) fractions. The resulting pellet in the first step and the membrane pellet in the second step were resuspended in homogenization buffer B (0.32 M sucrose, 20 mM HEPES, pH 7.4, 3 mM EDTA, and 0.1% Triton X-100 containing protease and phosphatase inhibitors, as described above), sonicated twice for 15 s on ice at medium speed using a Masonix XL2000 ultrasonic cell disruptor with a 30-s recovery between disruptions, and incubated for 1 h at 4 °C with gentle agitation. After incubation, the homogenate was centrifuged at 13,000 rpm for 35 min at 4 °C. The resulting supernatant was used as the nuclear and membrane fraction. All protein concentrations were determined using a protein-assay kit from Bio-Rad. Retinal protein samples were then subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes, which were subsequently blocked in 5% nonfat milk for 1 h at room temperature. Immunoblots were incubated with primary antibody overnight at 4 °C (see Table 1 for antibody information). After washing in Tris-buffered saline with 0.1% Tween 20, the immunoblots were incubated with HRP-conjugated secondary antibody (1:20,000) for 1 h at room temperature. Chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA) was used to detect primary antibodies binding to respective cognate antigens. A Li-Cor Odyssey machine and Li-Cor software (Li-Cor Biosciences, Lincoln, NE) were used for detection and densitometric analysis.
Weri-Rb1 cell culture and cGMP treatment
The Weri-Rb1 (ATCC, Manassas, VA) retinoblastoma cell line was cultured in RPMI-1640 medium (ATCC) with 15% FBS, 2 mM L-glutamine (Mediatech, Inc., Herndon, VA), 0.1% Fungizone (Invivogen, San Diego, CA), 5 μM plasmocin (Invivogen), 55 μM β-mercaptoethanol (Invitrogen), and 10 μg/ml insulin (I5500, Sigma-Aldrich, St. Louis, MO) at 37 °C in a humidified incubator with 5% CO2, as described (46). For cGMP treatment, cells cultured in RPMI-1640 medium were treated with cGMP (Santa Cruz, Dallas, TX) at various concentrations for 24 hours. At the end of the experiments, cells were harvested for evaluation of gene expression.
Statistical analysis
One-way analysis of variance and unpaired Student’s t-test were used to evaluate significant differences between multiple groups and two groups, respectively. Results are expressed as means ± SEM of number of observations. Differences were considered statistically significant when p < 0.05. Data were analyzed and graphed using GraphPad Prism® software (GraphPad Software).
Results
Depletion of cGMP suppressed upregulation of RyR2 activity and expression in Cnga3−/−/Nrl−/− retinas
To explore the potential regulation of cGMP/PKG signaling and determine whether RyR2 upregulation is a consequence of cGMP/PKG signaling, we analyzed the activity and expression of RyR2 in CNG channel-deficient retinas after cGMP depletion. Genetic deletion of retGC1 (encoded by Gucy2e) was used to deplete cGMP in Cnga3−/−/Nrl−/− mice (Cnga3−/−/Nrl−/−/Gucy2e−/−). This approach has been shown to abolish the elevation of cGMP level and PKG activity in Cnga3−/−/Nrl−/− retina and improve cone survival (19, 21). Retinas of Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Nrl−/− mice at P15, P30 and P60 were analyzed for RyR2 activity and expression. The activity of the channel was evaluated by examining the level of phosphorylated protein using immunoblotting (47). Expression was evaluated by examining the mRNA level of the channel using qRT-PCR. We found that depletion of cGMP nearly completely abolished the elevation of phospho-RyR2 in CNG channel-deficient mice. As shown in Fig. 1A, phospho-RyR2 level was doubled in Cnga3−/−/Nrl−/− mice, compared with Nrl−/− mice, and was reduced by about 50% in Cnga3−/−/Nrl−/−/Gucy2e−/− mice. Similarly, RyR2 mRNA level was decreased by about 50% in Cnga3−/−/Nrl−/−/Gucy2e−/− retinas, compared with age-matched Cnga3−/−/Nrl−/− controls (Fig. 1B).
Figure 1. Depletion of cGMP suppressed upregulation of RyR2 in Cnga3−/−/Nrl−/− retinas.
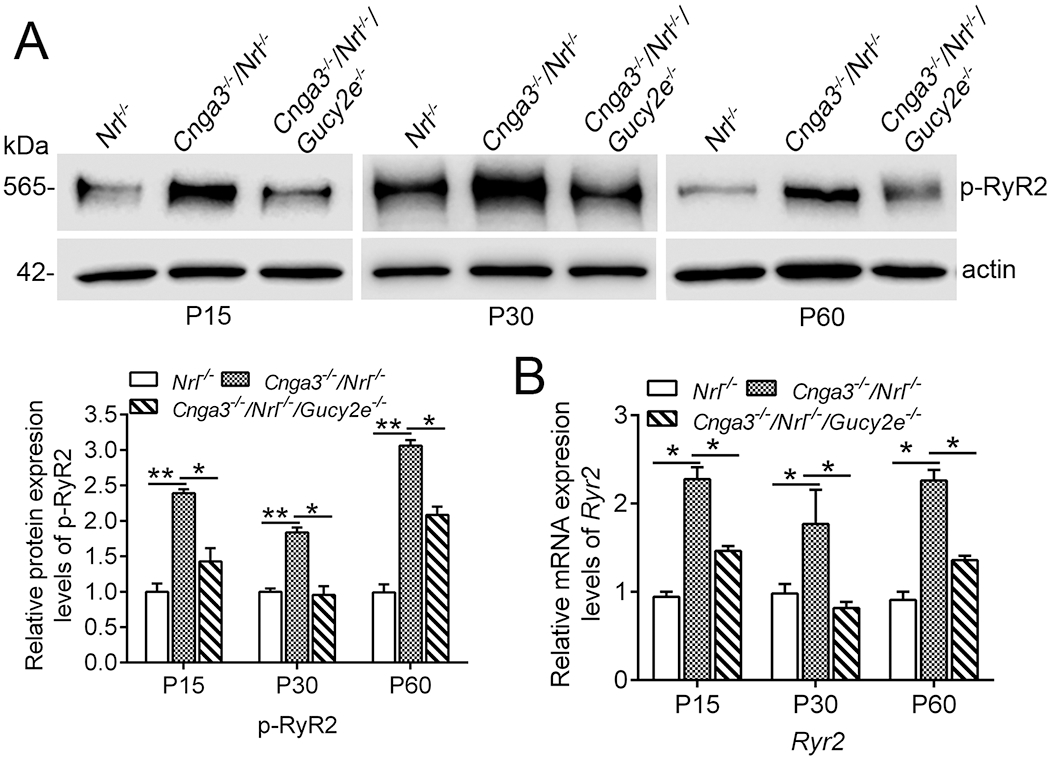
Expression of phospho-RyR2 and Ryr2 mRNA levels in retinas of Nrl−/−, Cnga3−/−/Nrl−/−, and Cnga3−/−/Nrl−/−/Gucy2e−/− mice at P15, P30, and P60 were analyzed by immunoblotting and qRT-PCR. (A) Increased phospho-RyR2 level in Cnga3−/−/Nrl−/− retinas and deletion of retGC1 suppressed the upregulation. Shown are representative immunoblotting images of phospho-RyR2 and corresponding quantitative analysis. (B) qRT-PCR results showing increased mRNA level of RyR2 in Cnga3−/−/Nrl−/− retinas. Deletion of retGC1 suppressed the upregulation. Data are represented as mean ± SEM 3-4 assays using retinas prepared from 8-10 mice in each group (*P < 0.05, **P < 0.01).
Treatment with PKG inhibitors suppressed upregulation of RyR2 activity and expression in Cnga3−/−/Nrl−/− retinas
In addition to genetic manipulation, we used PKG inhibitors to explore the contribution of PKG signaling. Starting at P7, Cnga3−/−/Nrl−/− mice received intraperitoneal injection of PKG inhibitor KT5823 (1.0 μmol/kg body weight/day), (Rp)-8-Br-cGMPS (5.0 μmol/kg body weight/day), or vehicle for 9 days (21). At the end of the treatment, retinas were collected for evaluation of phospho-RyR2 and RyR2 mRNA levels. We found that the treatment completely abolished the elevation of phospho-RyR2 in CNG channel-deficient mice. The phospho-RyR2 level in Cnga3−/−/Nrl−/− mice was reduced by about 65% and 80%, respectively, after KT5823 or (Rp)-8-Br-cGMPS treatment, compared with vehicle-treated controls (Fig. 2A). qRT-PCR results showed that the RyR2 mRNA level was decreased by about 30% after treatment with (Rp)-8-Br-cGMPS, but not KT5823 (Fig. 2B). Taken together, the results from genetic deletion and pharmacological inhibition experiments showed that cGMP/PKG signaling increased RyR2 activity and expression in Cnga3−/−/Nrl−/− retinas.
Figure 2. Treatment with PKG inhibitor suppressed upregulation of RyR2 in Cnga3−/−/Nrl−/− retinas.

Cnga3−/−/Nrl−/− mice received intraperitoneal injection of KT5823 (1.0 μmol/kg body weight/day), (Rp)-8-Br-cGMPS (5.0 μmol/kg body weight/day), or vehicle for 9 days, starting at P7, and were then analyzed for phospho-RyR2 and Ryr2 mRNA levels in the retina by immunoblotting and qRT-PCR. (A) Increased phospho-RyR2 level in Cnga3−/−/Nrl−/− retinas and reduced level after treatment with PKG inhibitors. Shown are representative immunoblotting images of phospho-RyR2 and the corresponding quantitative analysis. (B) qRT-PCR results showing an increased mRNA level of RyR2 in Cnga3−/−/Nrl−/− retinas and decreased expression after treatment with PKG inhibitors. Data are represented as mean ± SEM 3-4 assays using retinas prepared from 6-8 mice in each group (*P < 0.05, **P < 0.01).
CaMKII activity was not altered in Cnga3−/−/Nrl−/− retinas
Several kinases, including PKG, PKA (protein kinase A, cAMP-dependent protein kinase), and calmodulin-dependent protein kinase II (CaMKII), have been shown to phosphorylate RyR2 in vitro and in vivo (48–52). We previously showed that the cellular cAMP level was not elevated in CNG channel-deficient mice (19), suggesting that PKA is less likely responsible for the elevated RyR2 activity. In the present study, we examined CaMKII activity in CNG channel-deficient retinas to determine whether this kinase plays a role in RyR2 activation. Retinas of Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Nrl−/− mice at P15, P30, and P60 were analyzed for levels of phospho-CaMKII α and β isoforms by immunoblotting. The analysis showed that the levels of phospho-CaMKII were not different between Cnga3−/−/Nrl−/− and age-matched Nrl−/− controls (Fig. 3), suggesting that this kinase is less likely to be responsible for the upregulation of RyR2. Of note, the levels of phospho-CaMKII were significantly reduced in Cnga3−/−/Nrl−/−/Gucy2e−/− retinas, compared with Cnga3−/−/Nrl−/− and Nrl−/− retinas, particularly at P15 and P60 (Fig. 3), suggesting a regulation of cGMP signaling on CaMKII.
Figure 3. Phospho-CaMKII level was not altered in Cnga3−/−/Nrl−/− retinas.
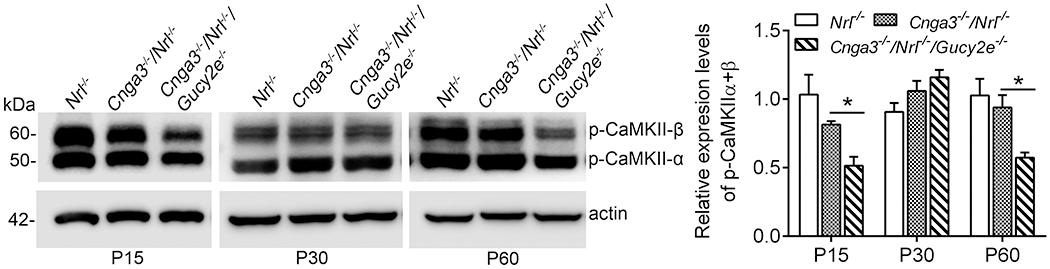
Expression levels of phospho-CaMKII in retinas of Nrl−/−, Cnga3−/−/Nrl−/−, and Cnga3−/−/Nrl−/−/Gucy2e−/− mice at P15, P30, and P60 were analyzed by immunoblotting. Shown are representative immunoblotting images and corresponding quantitative analysis. Data are represented as mean ± SEM 3-4 assays using retinas prepared from 8-10 mice in each group (*P < 0.05).
Depletion of cGMP and deletion of Ryr2 equivalently suppressed UPR/ER stress in Cnga3−/−/Nrl−/− retinas
CNG channel-deficient retinas show ER stress (17, 21) and cone protein mislocalization (13, 53, 54), suggesting dysregulation of ER homeostasis and protein folding. Inhibition of cGMP/PKG signaling (21) or RyR2 function (27, 34) suppressed ER stress/improved cone outer segment protein localization. In the present study, we examined the effects of cGMP depletion and Ryr2 deletion on UPR to determine whether these manipulations exert similar actions on ER homeostasis. Retinas of Cnga3−/−/Nrl−/−, Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice at P15 were analyzed for expression of UPR genes using the Mouse Unfolded Protein Response RT2 Profiler™ PCR Array. This array profiles the expression of 84 genes involved in unfolded protein binding, protein folding, ER-associated protein degradation, heat shock proteins, and enzymatic regulation of unfolded glycoproteins. We found that the expression of fourteen genes was significantly increased in Cnga3−/−/Nrl−/− mice (Fig. 4A–B). These fourteen genes were Atf6b (encoding activating transcription factor 6 beta), Bax (encoding Bcl2-associated X protein), Calr (encoding calreticulin), Cebpb (encoding CCAAT/enhancer binding protein beta), Creb3l3 (encoding cAMP responsive element binding protein 3-like 3), Derl1 (encoding degradation in ER protein 1), Dnajc3 (encoding DnaJ [Hsp40] homolog, subfamily C, member 3), Edem1 (encoding ER degradation enhancer, mannosidase alpha-like 1), Ern2 (encoding ER to nucleus signaling 2), Ero1lb (encoding ER oxidoreductase 1 like protein beta), Ganc (encoding glucosidase, alpha; neutral C), Hspb9 (encoding heat shock protein, alpha-crystallin-related, B9), Srebf2 (encoding sterol regulatory element binding factor 2), and Uggt2 (encoding UDP-glucose glycoprotein glucosyltransferase 2), with Ern2 most significantly elevated. These genes are primarily involved in UPR (Atf6b, Calr, Hspb9, and Dnajc3), ER-associated protein degradation (Derl1 and Edem1), ER stress-associated cell death (Cebpb, Ern2, and Creb3l3), and reglucosylation of unfolded glycoproteins (Uggt2), supporting UPR at the gene expression level. Depletion of cGMP abolished elevation of ten genes among the list (Fig. 4A), while deletion of Ryr2 abolished elevation of the thirteen genes (Fig. 4B). The expression of Derl-1, Creb3l3, and Uggt2, encoded by Derl1, Creb3l3, and Uggt2, respectively, were further evaluated using immunoblotting. Similar to findings at mRNA levels, the expression levels of Derl-1 and Creb3l3 were increased in CNGA3-deficient retina, and depletion of cGMP or deletion of RyR2 reduced the protein expression levels. However, we did not detect elevation of Uggt2 in CNGA3-deficient retina (Figure 4C). Thus, the data from the gene expression and protein expression levels suggest that depletion of cGMP and deletion of Ryr2 equivalently suppressed UPR in Cnga3−/−/Nrl−/− retinas.
Figure 4. Depletion of cGMP and deletion of Ryr2 suppressed upregulation of UPR genes in Cnga3−/−/Nrl−/− retinas.

Expression of UPR genes were evaluated in retinas of Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice by PCR array and immunoblotting. (A-B) Depletion of cGMP (A) or deletion of Ryr2 (B) suppressed upregulation of UPR genes in Cnga3−/−/Nrl−/− retinas. Shown are results of PCR array obtained from retinas of P15 Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice. (C) Immunoblotting evaluation of expression levels of Derl-1, Creb313, and Uggt2 in the retinas of P15 Nrl−/−, Cnga3−/−/Nrl−/−, and Cnga3−/−/Nrl−/−/Gucy2e−/− mice. Shown are representative immunoblotting images and the corresponding quantitative analysis. Data are represented as mean ± SEM 3-4 assays using retinas prepared from 6-10 mice in each group (*P < 0.05, **P < 0.01, ***P < 0.001).
We also evaluated the effects of cGMP depletion and Ryr2 deletion on the ER stress pathway activities. The three hallmarks of ER stress, eIF2α (eukaryotic translation initiation factor 2 alpha), Ire1α (serine/threonine-protein kinase/endoribonuclease), and ATF6 (activating transcription factor 6), were examined. At P15, the phospho-Ire1α and phospho-eIF2α levels were already increased in Cnga3−/−/Nrl−/− mice, compared with Nrl−/− controls, and the elevations remained at P30 and P60 (Fig. 5A). Depletion of cGMP appeared to differentially affect the pathway activities. The levels of phospho-Ire1α were reduced in Cnga3−/−/Nrl−/−/Gucy2e−/− mice at P15 and P30, but not P60, compared with age-matched Cnga3−/−/Nrl−/− controls, whereas the levels of phospho-eIF2α were reduced at P30 and P60, but not at P15 (Fig. 5A). Upon ER stress, ATF6 is cleaved. The cleaved form translocates to the nucleus, where it regulates transcription of ER stress-related genes and subsequent ER stress-related cellular responses (55). The ATF6 cleavage and nuclear localization was significantly increased in Cnga3−/−/Nrl−/− retinas, and depletion of cGMP or deletion of Ryr2 effectively inhibited activation of ATF6 (Fig. 5B). The effects of cGMP depletion and RyR2 deficiency were further examined by evaluating the expression of CHOP, a member of the C/EBP (CCAAT enhancer-binding protein) transcription factor family induced in ER stress through the Grp78/Bip-phospho-eIF2α pathway (56). The expression level of CHOP was significantly increased in Cnga3−/−/Nrl−/− mice. Depletion of cGMP or deletion of Ryr2 effectively suppressed expression of CHOP (Fig. 5C).
Figure 5. Depletion of cGMP and deletion of Ryr2 suppressed ER stress and CHOP activation in Cnga3−/−/Nrl−/− retinas.
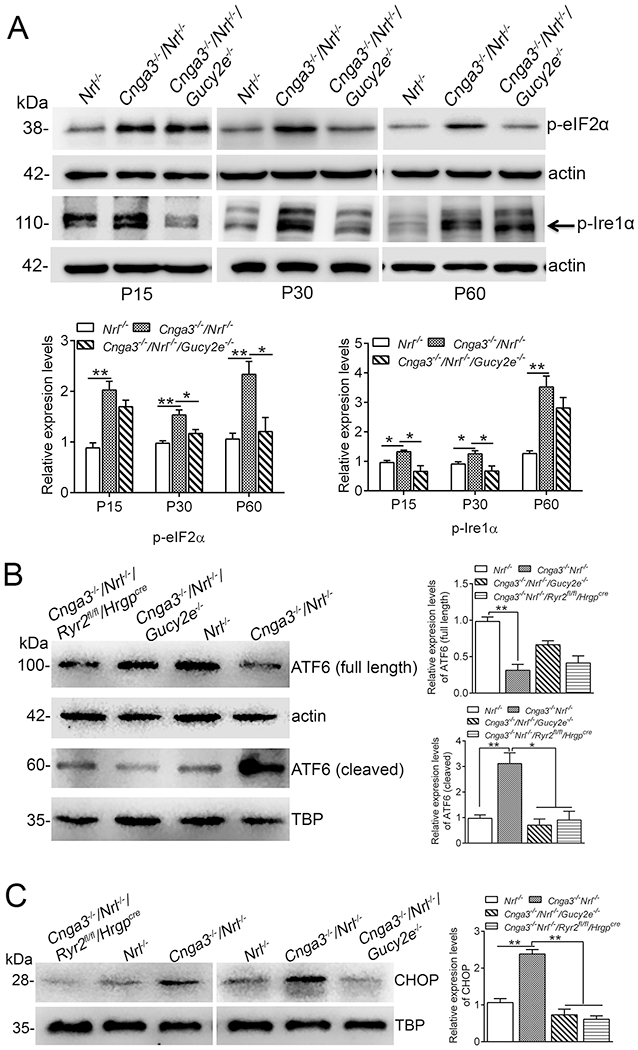
Expression of ER stress markers and CHOP were evaluated in retinas of Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice by immunoblotting. (A) Levels of phospho-eIF2α and phospho-Ire1α were evaluated in retinas of Nrl−/−, Cnga3−/−/Nrl−/−, and Cnga3−/−/Nrl−/−/Gucy2e−/− mice at P15, P30, and P60. Shown are representative immunoblotting images and the corresponding quantitative analysis. (B) ATF6 cleavage and nuclear translocation were evaluated in retinas of P15 Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice. Shown are representative immunoblotting images and the corresponding quantitative analysis. (C) The expression levels of CHOP were evaluated in retinas of P15 Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice. Shown are representative immunoblotting images and corresponding quantitative analysis. Data are represented as mean ± SEM 3-4 assays using retinas prepared from 6-10 mice in each group (*P < 0.05, **P < 0.01).
Depletion of cGMP and deletion of Ryr2 reduced CREB activity in Cnga3−/−/Nrl−/− retinas
CREB, a 43kDa basic/leucine zipper transcription factor, is activated/phosphorylated by various kinases, including PKG (57). We evaluated the effects of cGMP depletion on CREB activity in CNG channel deficiency. Retinas of Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Nrl−/− mice at P15, P30, and P60 were analyzed for levels of phospho-CREB by immunoblotting. The analysis showed that the phospho-CREB level was increased by one- to two-fold in Cnga3−/−/Nrl−/− mice, compared with Nrl−/− retinas (Fig. 6A). Depletion of cGMP abolished the activation of CREB at P15 and P30, and significantly reduced it at P60 (Fig. 6A), supporting a cGMP-dependent activation of CREB in CNG channel deficiency. CREB activity was also analyzed in Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre retinas. The analysis showed that deletion of Ryr2 abolished the activation of CREB at P30 (Fig. 6B). However, CREB activity was increased in Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre retinas at P15, compared with age-matched Cnga3−/−/Nrl−/− controls (Fig. 6B).
Figure 6. Depletion of cGMP and deletion of Ryr2 suppressed upregulation of CREB activity in Cnga3−/−/Nrl−/− retinas.
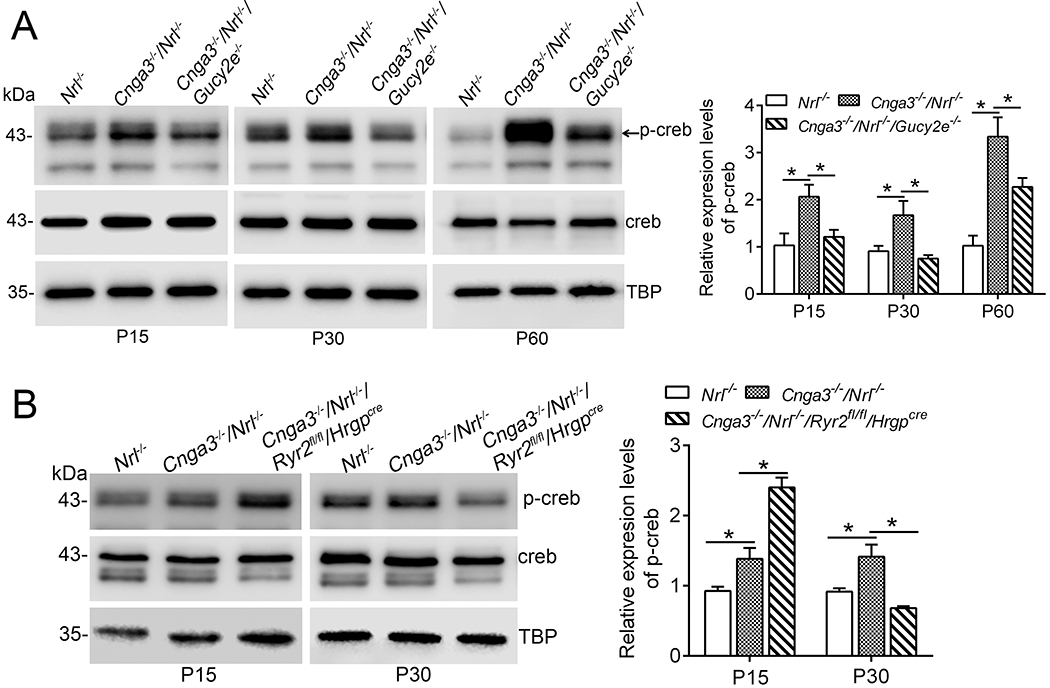
Expression of phospho-CREB and total CREB were analyzed in retinas of Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice at P15, P30, and P60 by immunoblotting. (A) Depletion of cGMP suppressed upregulation of phospho-CREB in Cnga3−/−/Nrl−/− retinas. Shown are representative immunoblotting images of phospho-CREB and the corresponding quantitative analysis. (B) Deletion of Ryr2 increased the phospho-CREB level in Cnga3−/−/Nrl−/− mice at P15, but reduced it at P30. Shown are representative immunoblotting images of phospho-CREB and the corresponding quantitative analysis. Data are represented as mean ± SEM 3-4 assays using retinas prepared from 8-10 mice in each group (*P < 0.05).
Depletion of cGMP and deletion of Ryr2 led to early-onset cone protection in Cnga3−/−/Nrl−/− mice
CNG channel-deficient mice display early-onset, progressive cone degeneration (14, 16). In the present study, we evaluated early age cone survival/protection in Cnga3−/−/Nrl−/− mice after cGMP depletion or Ryr2 deletion. The expression levels of cone proteins, including cone arrestin (CAR) and M-opsin, and photoreceptor apoptosis were analyzed in mice at P15 and P30. We found that CAR level was increased by about two- and five-fold in Cnga3−/−/Nrl−/−/Gucy2e−/− mice, compared with age-matched Cnga3−/−/Nrl−/− controls, and M-opsin level was increased by about one-fold (Fig. 7A). Similar findings were obtained in mice with Ryr2 deletion. M-opsin and CAR levels were increased by about one- to two-fold in Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice, compared with age-matched Cnga3−/−/Nrl−/− controls (Fig. 7B). The increased expression of CAR after depletion of cGMP or deletion of Ryr2 was also shown by immunofluorescence labeling (Fig. 7C, the abnormal retinal morphology reflected rosette-like structures formed in mice lacking Nrl). Photoreceptor apoptosis/death was evaluated by TUNEL. The number of TUNEL-positive cells detected in the outer nuclear layer (ONL) of Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre retinas was significantly reduced, compared with age-matched Cnga3−/−/Nrl−/− controls (Fig. 7D). To determine whether microglia cells contribute to the TUNEL-positive labeling, we examined location/activity of these cells in Cnga3−/−/Nrl−/− and Nrl−/− retinas by immunofluorescence labeling using an antibody against the microglia cell marker ionized calcium binding adaptor molecule 1 (Iba1) (58). The assay showed that the Iba1 labeling was mainly observed in the outer plexiform layers (OPL) and the inner plexiform layers (IPL), but not in the ONL, and there was no increased labeling intensity in Cnga3−/−/Nrl−/− retinas, compared with that in Nrl−/− retinas (see Supplementary Fig. 3). These results do not support the contribution of microglia cells to the TUNEL-positive labeling detected in the ONL of Cnga3−/−/Nrl−/− retinas. Thus, depletion of cGMP or deletion of Ryr2 effectively reduced early-onset cone degeneration/death in Cnga3−/−/Nrl−/− mice. Müller glia activate in response to retinal stress with increased expression of glial fibrillary acidic protein (GFAP) in intermediate filaments, and upregulation of GFAP has been previously shown in CNG channel deficient-retinas (13, 21). The present work examined GFAP expression in Cnga3−/−/Nrl−/− and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice at P30 by immunoblotting. As shown in Fig. 7E, GFAP level was increased by about one-fold in Cnga3−/−/Nrl−/− mice, compared with controls. This elevation was completely abolished by deletion of Ryr2. This observation was in line with the reduced photoreceptor death after Ryr2 deletion.
Figure 7. Depletion of cGMP and deletion of Ryr2 led to early-onset cone protection in Cnga3−/−/Nrl−/− mice.

Cone survival/death and retinal stress were analyzed in Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice at P15 and P30. (A) Depletion of cGMP increased expression levels of cone proteins in Cnga3−/−/Nrl−/− mice. Shown are representative immunoblotting images of CAR and M-opsin and the corresponding quantitative analysis. (B) Deletion of Ryr2 increased expression levels of cone proteins in Cnga3−/−/Nrl−/− mice. Shown are representative immunoblotting images of CAR and M-opsin and the corresponding quantitative analysis. (C) Shown are representative confocal images of immunofluorescence labeling of CAR (Arr3) in Nrl−/−, Cnga3−/−/Nrl−/−, Cnga3−/−/Nrl−/−/Gucy2e−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice at P30. (D) Deletion of Ryr2 reduced cone apoptosis in Cnga3−/−/Nrl−/− mice. Cone apoptosis was evaluated by TUNEL labeling on retinal cross sections of Nrl−/−, Cnga3−/−/Nrl−/−, and Cnga3−/−/Nrl−/−/Ryr2fl/fl/Hrgpcre mice at P30. Shown are representative confocal images of TUNEL labeling and the corresponding quantitative analysis. ONL, outer nuclear layer; INL, inner nuclear layer. (E) Deletion of Ryr2 reduced activation of Müller cells in Cnga3−/−/Nrl−/− mice. Shown are representative immunoblotting images of GFAP and corresponding quantitative analysis in mice at P30. Data are represented as means ± SEM 3-4 assays using retinas prepared from 8-11 mice in each group (*P < 0.05, **P < 0.01, ***P < 0.001).
Treatment with cGMP increased Ryr2 expression in cultured photoreceptor-derived Weri-Rb1 cells
The regulation of cGMP signaling on RyR2 was also evaluated in an in vitro cell culture system using the photoreceptor-derived Weri-Rb1 cells (45, 59). This approach examines the regulation in a system in which cGMP signaling is artificially elevated. Cells were treated with various concentrations of cGMP for 24 hours and were harvested at the end of treatment for evaluation of Ryr2 expression by qRT-PCR. We found that treatment with cGMP increased RyR2 mRNA level by 10-to-15-fold (Fig. 8). This treatment did not alter calreticulin (an ER chaperone protein) mRNA level, increased IP3R1 mRNA level, and decreased SERCA2 mRNA level (Fig. 8).
Figure 8. Treatment with cGMP increased expression of RyR2 mRNA in cultured photoreceptor-derived Weri-Rb1 cells.
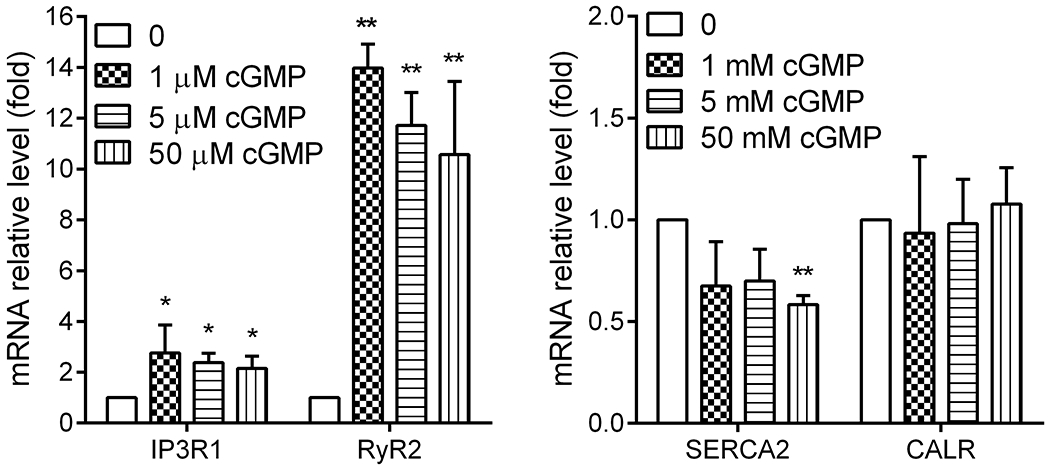
Cells were treated with various concentrations of cGMP for 24 hours and were harvested at the end of treatment for gene expression evaluation by qRT-PCR. Shown are mRNA expression levels of RyR2, IP3R1, SERCA2, and calreticulin. Data are represented as mean ± SEM of three assays from two independent experiments (*P < 0.05, **P < 0.01, compared with untreated controls).
Discussion
We and others previously demonstrated that CNG channel-deficient retinas undergo remarkable elevation of cellular cGMP and increased PKG activity (17, 19, 20), and further provided evidence that cGMP/PKG signaling contributes to ER stress and cone degeneration (19, 21). We also documented the upregulation of RyR2 in CNG channel deficiency, and its contribution to ER stress/cone degeneration and protein mislocalization (27, 34). In the current work, we tested our hypothesis that upregulation of RyR2 mediates cGMP/PKG signaling-induced ER stress and cone death. We examined the activity and expression of RyR2 in CNG channel-deficient retinas after cGMP/PKG inhibition. We also examined the effects of cGMP depletion and Ryr2 deletion on UPR/ER stress pathway activity, downstream signaling activity, and cone survival/death. We demonstrated that RyR2 activity and expression are highly regulated by cGMP/PKG signaling, and depletion of cGMP and deletion of Ryr2 equivalently suppressed UPR/ER stress and activation of CHOP, resulting in early-onset cone protection. These findings support the view that RyR2/ER calcium regulation is a significant downstream effector of cGMP/PKG signaling, and upregulation of RyR2 contributes to cGMP/PKG signaling-induced UPR/ER stress. Although regulation of RyR by cGMP/PKG signaling has been investigated in other tissues (51, 60, 61), no study investigating this phenomenon in retina/photoreceptors has been reported. This is an important area to explore because perturbation of cGMP/PKG signaling and cellular calcium/ER calcium dysregulation represent significant cellular defects in many distinct types of retinal degenerations (62, 63). Our work for the first time demonstrates the regulation of cGMP/PKG signaling on RyR2 in the retina, and its significance in retinal degeneration.
The regulation of cGMP/PKG on RyR2 activity and expression was demonstrated by cGMP depletion and PKG inhibition. The regulation of RyR2 gene expression by cGMP signaling was also shown in cultured photoreceptor-derived cells. Two interesting findings were obtained. First, the cGMP/PKG signaling regulation of RyR2 was significant and powerful. RyRs have been shown to be primarily regulated by cytosolic Ca2+ and protein kinases, including PKG (60, 61, 64, 65). Depletion of cGMP or inhibition of PKG nearly completely abolished the channel upregulation in CNG channel deficiency, although the cellular calcium homeostasis perturbation was still present. Second, cGMP/PKG signaling regulated both activity and expression of the channel. We observed that the two PKG inhibitors had more significant effects on the channel’s activity than on gene expression, and Rp-8-Br-cGMPS, but not KT5823, suppressed the gene expression. How KT5823 affected the channel expression differently from (Rp)-8-Br-cGMPS is not known at this time, but the two compounds exert their inhibition of PKG activity through different mechanisms (44). How cGMP/PKG signaling regulates the channel at a gene expression level remains unknown and merits further investigation. It should be pointed out that cGMP/PKG signaling also regulates activity of IP3R1, the other ER calcium efflux channel, in CNG channel deficiency. Unlike RyR2, IP3R1 phosphorylation decreases the channel’s activity and reduces Ca2+ release from the ER, though the reported data were controversial (66–71). Our previous report demonstrated that depletion of cGMP or inhibition of PKG increased phosphorylation of IP3R1 (21), thereby reducing the channel activity. The mechanisms underlying how cGMP/PKG signaling increases phosphorylation of RyR2 and decreases phosphorylation of IP3R1 remain to be determined; the former likely results from direct phosphorylation by PKG, and the latter might involve a more complex mechanism, involving a number of regulatory enzymes that directly interact with IP3R1. The protein kinases PKG, PKA, and CaMKII have been shown to phosphorylate RyRs (48–52). Our previous studies showed no significant elevation of cAMP in CNG channel-deficient retina (19), suggesting a less important role of PKA. In this study, we examined the level of CaMKII in the retinas. We found that the level of phospho-CaMKII was not different between Cnga3−/−/Nrl−/− and Nrl−/− mice, suggesting that the increased level of phospho-RyR2 is less likely to result from CaMKII signaling. However, depletion of cGMP was shown to lower the level of phospho-CaMKII in the retina, supporting a regulation of cGMP signaling on CaMKII. Nevertheless, data from this study favor the view that CaMKII is less likely to be responsible for RyR2 upregulation in CNG channel deficiency.
Depletion of cGMP and deletion of Ryr2 suppressed CREB signaling. CREB is a 43kDa basic leucine zipper transcription factor that is expressed in most tissues, and regulates cell proliferation, differentiation, and survival. It is activated by phosphorylation from various kinases, including PKA, PKG, mitogen-activated protein kinase signaling pathway, and CaMKII (57). In particular, PKG was found to phosphorylate CREB in neuronal cells, including in the hippocampus (72) and cerebellar neurons (73). Our data showing reduced CREB activity/phospho-CREB levels after depletion of cGMP support the cGMP/PKG regulation of CREB in the retina. In addition to protein kinases, CREB has been shown to be regulated by other cellular events, including ER stress and calcium signaling/ER calcium signaling (74, 75). Data from the present study showed reduced CREB activity after Ryr2 deletion, supporting a regulation of RyR2/ER calcium homeostasis on CREB. Thus, the upregulation of CREB in CNG channel deficiency is likely a consequence of both elevated cGMP/PKG signaling and enhanced RyR2 function/ER calcium dysregulation. CREB signaling has been shown to have pro-survival effects (76–78) and anti-survival effects (79–81). Alterations of CREB activity have been implicated in human neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease (82), and in animal models of retinal degeneration (83–85). Upregulation of CREB was observed in canine models of retinitis pigmentosa and an N-methyl-D-aspartate-induced rat model of retinal degeneration (83, 84). However, downregulation of CREB has been reported in retinal degeneration 10 (rd10) mice (85). In the present work, we showed that CREB activity was significantly elevated in CNG channel-deficient retinas, and depletion of cGMP or deletion of Ryr2 abolished upregulation of CREB activation. The significance of CREB upregulation in CNG channel deficiency and its role in photoreceptor survival/degeneration are not clear. Because the suppression of CREB upregulation by depletion of cGMP or deletion of Ryr2 was accompanied by improved cone survival/reduced cone death, one may presume that upregulation of CREB in CNG channel deficiency is likely anti-survival. However, experiments using genetic or chemical approaches to manipulate CREB activity would provide more definitive information.
In summary, we demonstrate the regulation of cGMP/PKG signaling on RyR2 in CNG channel-deficiency. Our findings support the view that upregulation of RyR2 contributes, at least in part, to cGMP/signaling-induced ER stress and photoreceptor degeneration. This work provides insight into how elevation of cGMP/PKG signaling induces ER stress and photoreceptor degeneration in CNG channel deficiency. Because perturbation of cGMP/PKG signaling and cellular calcium dysregulation are common throughout multiple neurodegenerative diseases, including many distinct types of photoreceptor degenerations, it is critical to understand how these dynamic signaling pathways interact and direct cell fate and determine the pathogenesis components.
Supplementary Material
Acknowledgments
We thank Drs. Martin Biel, Anand Swaroop, Wolfgang Baehr, and Yun-Zheng Le for providing Cnga3−/−, Nrl−/−, Gucy2e−/−, and Hrgpcre mouse lines. We thank the Imaging Core Facility and the Histology Core Facility of the Department of Cell Biology at OUHSC for technical assistance.
This work was supported by grants from the National Eye Institute (R01EY027754 and P30EY021725) and the Oklahoma Center for the Advancement of Science and Technology.
Abbreviations used:
- ATF6
activating transcription factor 6
- CaMKII
calmodulin-dependent protein kinase II
- cAMP
cyclic adenosine monophosphate
- CAR
cone arrestin
- cGMP
cyclic guanosine monophosphate
- CHOP
CCAAT-enhancer-binding protein homologous protein
- CNG
cyclic nucleotide-gated
- CREB
cyclic adenosine monophosphate response element binding protein
- ER
endoplasmic reticulum
- eIF2α
eukaryotic translation initiation factor 2 alpha
- GFAP
glial fibrillary acidic protein
- HRGP
human red/green pigment
- Iba1
ionized calcium binding adaptor molecule 1
- IP3R
inositol-1,4,5-trisphosphate receptor
- Ire1α
serine/threonine-protein kinase/endoribonuclease 1α
- PKA
cAMP-dependent protein kinase
- PKG
cGMP-dependent protein kinase
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- RetGC1
retinal guanylate cyclase 1
- RyR2
ryanodine receptor 2
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- TUNEL
terminal deoxynucleotidyltransferase dUTP nick end-labeling
- UPR
unfolded protein response
Footnotes
All authors declare that they have no conflict of interest.
References
- 1.Kaupp UB, and Seifert R (2002) Cyclic nucleotide-gated ion channels. Physiol Rev 82, 769–824 [DOI] [PubMed] [Google Scholar]
- 2.Michalakis S, Becirovic E, and Biel M (2018) Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int J Mol Sci, 7;19(3). pii: E749. doi: 10.3390/ijms19030749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohl S, Baumann B, Broghammer M, Jagle H, Sieving P, Kellner U, Spegal R, Anastasi M, Zrenner E, Sharpe LT, and Wissinger B (2000) Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 9, 2107–2116 [DOI] [PubMed] [Google Scholar]
- 4.Kohl S, Marx T, Giddings I, Jagle H, Jacobson SG, Apfelstedt-Sylla E, Zrenner E, Sharpe LT, and Wissinger B (1998) Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nature Genetics 19, 257–259 [DOI] [PubMed] [Google Scholar]
- 5.Nishiguchi KM, Sandberg MA, Gorji N, Berson EL, and Dryja TP (2005) Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum Mutat 25, 248–258 [DOI] [PubMed] [Google Scholar]
- 6.Varsanyi B, Somfai GM, Lesch B, Vamos R, and Farkas A (2007) Optical coherence tomography of the macula in congenital achromatopsia. Invest Ophthalmol Vis Sci 48, 2249–2253 [DOI] [PubMed] [Google Scholar]
- 7.Thiadens AA, Somervuo V, van den Born LI, Roosing S, van Schooneveld MJ, Kuijpers RW, van Moll-Ramirez N, Cremers FP, Hoyng CB, and Klaver C (2010) Progressive Loss of Cones in Achromatopsia. An Imaging Study using Spectral-Domain Optical Coherence Tomography. Invest Ophthalmol Vis Sci 51, 5952–5957 [DOI] [PubMed] [Google Scholar]
- 8.Genead MA, Fishman GA, Rha J, Dubis AM, Bonci DM, Dubra A, Stone EM, Neitz M, and Carroll J (2011) Photoreceptor structure and function in patients with congenital achromatopsia. Invest Ophthalmol Vis Sci 52, 7298–7308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fahim AT, Khan NW, Zahid S, Schachar IH, Branham K, Kohl S, Wissinger B, Elner VM, Heckenlively JR, and Jayasundera T (2013) Diagnostic fundus autofluorescence patterns in achromatopsia. Am J Ophthalmol 156, 1211–1219 e1212 [DOI] [PubMed] [Google Scholar]
- 10.Thiadens AA, Slingerland NW, Roosing S, van Schooneveld MJ, van Lith-Verhoeven JJ, van Moll-Ramirez N, van den Born LI, Hoyng CB, Cremers FP, and Klaver CC (2009) Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology 116, 1984–1989 e1981 [DOI] [PubMed] [Google Scholar]
- 11.Liang X, Dong F, Li H, Yang L, and Sui R (2015) Novel CNGA3 mutations in Chinese patients with achromatopsia. Br J Ophthalmol 99, 571–576 [DOI] [PubMed] [Google Scholar]
- 12.McClintock M, Peden MC, and Kay CN (2014) Spectral domain optical coherence tomography findings in CNGB3-associated achromatopsia and therapeutic implications. Adv Exp Med Biol 801, 551–557 [DOI] [PubMed] [Google Scholar]
- 13.Michalakis S, Geiger H, Haverkamp S, Hofmann F, Gerstner A, and Biel M (2005) Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Invest Ophthalmol Vis Sci 46, 1516–1524 [DOI] [PubMed] [Google Scholar]
- 14.Biel M, Seeliger M, Pfeifer A, Kohler K, Gerstner A, Ludwig A, Jaissle G, Fauser S, Zrenner E, and Hofmann F (1999) Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc Natl Acad Sci U S A 96, 7553–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Morris L, Fliesler SJ, Sherry DM, and Ding X-Q (2011) Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Invest Ophthalmol Vis Sci 52, 3557–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding XQ, Harry CS, Umino Y, Matveev AV, Fliesler SJ, and Barlow RB (2009) Impaired cone function and cone degeneration resulting from CNGB3 deficiency: down-regulation of CNGA3 biosynthesis as a potential mechanism. Hum Mol Genet 18, 4770–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thapa A, Morris L, Xu J, Ma H, Michalakis S, Biel M, and Ding XQ (2012) Endoplasmic reticulum stress-associated cone photoreceptor degeneration in cyclic nucleotide-gated channel deficiency. J Biol Chem 287, 18018–18029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma H, Thapa A, Morris LM, Michalakis S, Biel M, Frank MB, Bebak M, and Ding XQ (2013) Loss of cone cyclic nucleotide-gated channel leads to alterations in light response modulating system and cellular stress response pathways: a gene expression profiling study. Hum Mol Genet 22, 3906–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu J, Morris L, Thapa A, Ma H, Michalakis S, Biel M, Baehr W, Peshenko IV, Dizhoor AM, and Ding XQ (2013) cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J Neurosci 33, 14939–14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michalakis S, Muhlfriedel R, Tanimoto N, Krishnamoorthy V, Koch S, Fischer MD, Becirovic E, Bai L, Huber G, Beck SC, Fahl E, Buning H, Paquet-Durand F, Zong X, Gollisch T, Biel M, and Seeliger MW (2010) Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol Ther 18, 2057–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma H, Butler MR, Thapa A, Belcher J, Yang F, Baehr W, Biel M, Michalakis S, and Ding XQ (2015) cGMP/Protein Kinase G Signaling Suppresses Inositol 1,4,5-Trisphosphate Receptor Phosphorylation and Promotes Endoplasmic Reticulum Stress in Photoreceptors of Cyclic Nucleotide-gated Channel-deficient Mice. J Biol Chem 290, 20880–20892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, Jeffrey KD, Sampaio AV, Underhill TM, and Johnson JD (2009) Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 58, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, and Paterlini-Brechot P (2005) Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24, 4921–4933 [DOI] [PubMed] [Google Scholar]
- 24.Duncan RS, Goad DL, Grillo MA, Kaja S, Payne AJ, and Koulen P (2010) Control of intracellular calcium signaling as a neuroprotective strategy. Molecules 15, 1168–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chao CC, Huang CC, Lu DY, Wong KL, Chen YR, Cheng TH, and Leung YM (2012) Ca2+ store depletion and endoplasmic reticulum stress are involved in P2X7 receptor-mediated neurotoxicity in differentiated NG108–15 cells (vol 113, pg 1377, 2012). J Cell Biochem 113, 2178–2178 [DOI] [PubMed] [Google Scholar]
- 26.Mekahli D, Bultynck G, Parys JB, De Smedt H, and Missiaen L (2011) Endoplasmic-Reticulum Calcium Depletion and Disease. Csh Perspect Biol 3, a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butler MR, Ma H, Yang F, Belcher J, Le YZ, Mikoshiba K, Biel M, Michalakis S, Iuso A, Krizaj D, and Ding XQ (2017) Endoplasmic reticulum (ER) Ca(2+)-channel activity contributes to ER stress and cone death in cyclic nucleotide-gated channel deficiency. J Biol Chem 292, 11189–11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor CW, and Tovey SC (2010) IP3 Receptors: Toward Understanding Their Activation. Csh Perspect Biol 2, a004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fill M, and Copello JA (2002) Ryanodine receptor calcium release channels. Physiol Rev 82, 893–922 [DOI] [PubMed] [Google Scholar]
- 30.Periasamy M, and Kalyanasundaram A (2007) SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 35, 430–442 [DOI] [PubMed] [Google Scholar]
- 31.Shoshan-Barmatz V, Zakar M, Shmuelivich F, Nahon E, and Vardi N (2007) Retina expresses a novel variant of the ryanodine receptor. Eur J Neurosci 26, 3113–3125 [DOI] [PubMed] [Google Scholar]
- 32.Krizaj D, Lai FA, and Copenhagen DR (2003) Ryanodine stores and calcium regulation in the inner segments of salamander rods and cones. J Physiol 547, 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W, Xing W, Ryskamp DA, Punzo C, and Krizaj D (2011) Localization and phenotype-specific expression of ryanodine calcium release channels in C57BL6 and DBA/2J mouse strains. Exp Eye Res 93, 700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma H, Yang F, Butler MR, Rapp J, Le YZ, and Ding XQ (2019) Ryanodine Receptor 2 Contributes to Impaired Protein Localization in Cyclic Nucleotide-Gated Channel Deficiency. eNeuro 6 (3), 10.1523/ENEURO.0119-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, and Swaroop A (2001) Nrl is required for rod photoreceptor development. Nat Genet 29, 447–452 [DOI] [PubMed] [Google Scholar]
- 36.Yang RB, Robinson SW, Xiong WH, Yau KW, Birch DG, and Garbers DL (1999) Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J Neurosci 19, 5889–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le YZ, Ash JD, Al-Ubaidi MR, Chen Y, Ma JX, and Anderson RE (2004) Targeted expression of Cre recombinase to cone photoreceptors in transgenic mice. Mol Vis 10, 1011–1018 [PubMed] [Google Scholar]
- 38.Bround MJ, Asghari P, Wambolt RB, Bohunek L, Smits C, Philit M, Kieffer TJ, Lakatta EG, Boheler KR, Moore ED, Allard MF, and Johnson JD (2012) Cardiac ryanodine receptors control heart rate and rhythmicity in adult mice. Cardiovasc Res 96, 372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikonov SS, Daniele LL, Zhu X, Craft CM, Swaroop A, and Pugh EN Jr. (2005) Photoreceptors of Nrl −/− mice coexpress functional S- and M-cone opsins having distinct inactivation mechanisms. J Gen Physiol 125, 287–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daniele LL, Lillo C, Lyubarsky AL, Nikonov SS, Philp N, Mears AJ, Swaroop A, Williams DS, and Pugh EN Jr. (2005) Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Invest Ophthalmol Vis Sci 46, 2156–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshida S (2004) Expression profiling of the developing and mature Nrl−/− mouse retina: identification of retinal disease candidates and transcriptional regulatory targets of Nrl. Hum. Mol. Genet. 13, 1487–1503 [DOI] [PubMed] [Google Scholar]
- 42.Farjo R, Skaggs JS, Nagel BA, Quiambao AB, Nash ZA, Fliesler SJ, and Naash MI (2006) Retention of function without normal disc morphogenesis occurs in cone but not rod photoreceptors. J Cell Biol 173, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matveev AV, Quiambao AB, Browning Fitzgerald J, and Ding XQ (2008) Native cone photoreceptor cyclic nucleotide-gated channel is a heterotetrameric complex comprising both CNGA3 and CNGB3: a study using the cone-dominant retina of Nrl−/− mice. J Neurochem 106, 2042–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolfertstetter S, Huettner JP, and Schlossmann J (2013) cGMP-Dependent Protein Kinase Inhibitors in Health and Disease. Pharmaceuticals 6, 269–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang F, Ma H, Belcher J, Butler MR, Redmond TM, Boye SL, Hauswirth WW, and Ding XQ (2016) Targeting iodothyronine deiodinases locally in the retina is a therapeutic strategy for retinal degeneration. FASEB J 30, 4313–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, Liu A, Jhanwar SC, Abramson DH, and Cobrinik D (2009) Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell 137, 1018–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozawa T (2010) Modulation of ryanodine receptor Ca2+ channels (Review). Mol Med Rep 3, 199–204 [DOI] [PubMed] [Google Scholar]
- 48.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, and Valdivia HH (2007) Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase a phosphorylation site in the cardiac ryanodine receptor. Circ Res 101, 819–829 [DOI] [PubMed] [Google Scholar]
- 49.Witcher DR, Kovacs RJ, Schulman H, Cefali DC, and Jones LR (1991) Unique Phosphorylation Site on the Cardiac Ryanodine Receptor Regulates Calcium-Channel Activity. J Biol Chem 266, 11144–11152 [PubMed] [Google Scholar]
- 50.Takasago T, Imagawa T, and Shigekawa M (1989) Phosphorylation of the Cardiac Ryanodine Receptor by Camp-Dependent Protein-Kinase. J Biochem-Tokyo 106, 872–877 [DOI] [PubMed] [Google Scholar]
- 51.Takasago T, Imagawa T, Furukawa K, Ogurusu T, and Shigekawa M (1991) Regulation of the Cardiac Ryanodine Receptor by Protein Kinase-Dependent Phosphorylation. J Biochem-Tokyo 109, 163–170 [DOI] [PubMed] [Google Scholar]
- 52.Dixit SS, Wang T, Manzano EJ, Yoo S, Lee J, Chiang DY, Ryan N, Respress JL, Yechoor VK, and Wehrens XH (2013) Effects of CaMKII-mediated phosphorylation of ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PloS one 8, e58655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu J, Morris L, Fliesler SJ, Sherry DM, and Ding XQ (2011) Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Invest Ophthalmol Vis Sci 52, 3557–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carvalho LS, Xu J, Pearson RA, Smith AJ, Bainbridge JW, Morris LM, Fliesler SJ, Ding XQ, and Ali RR (2011) Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet 20, 3161–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong M, Luo S, Baumeister P, Huang JM, Gogia RK, Li M, and Lee AS (2004) Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J Biol Chem 279, 11354–11363 [DOI] [PubMed] [Google Scholar]
- 56.Li J, Lee B, and Lee AS (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 281, 7260–7270 [DOI] [PubMed] [Google Scholar]
- 57.Johannessen M, Delghandi MP, and Moens U (2004) What turns CREB on? Cell Signal 16, 1211–1227 [DOI] [PubMed] [Google Scholar]
- 58.Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, Lopez-Cuenca I, Rojas P, Trivino A, and Ramirez JM (2017) The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci 9, 10.3389/fnagi.2017.00214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Fu L, Chen DG, and Deeb SS (2007) Identification of novel retinal target genes of thyroid hormone in the human WERI cells by expression microarray analysis. Vision Res 47, 2314–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santulli G, and Marks AR (2015) Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol 8, 206–222 [DOI] [PubMed] [Google Scholar]
- 61.Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D’Ascia SL, Cannone M, Marziliano N, Trimarco B, Guise TA, Lacampagne A, and Marks AR (2015) Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Invest 125, 1968–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bowes C, Li TS, Danciger M, Baxter LC, Applebury ML, and Farber DB (1990) Retinal Degeneration in the Rd Mouse Is Caused by a Defect in the Beta-Subunit of Rod Cgmp-Phosphodiesterase. Nature 347, 677–680 [DOI] [PubMed] [Google Scholar]
- 63.Woodruff ML, Olshevskaya EV, Savchenko AB, Peshenko IV, Barrett R, Bush RA, Sieving PA, Fain GL, and Dizhoor AM (2007) Constitutive excitation by Gly90Asp rhodopsin rescues rods from degeneration caused by elevated production of cGMP in the dark. J Neurosci 27, 8805–8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamilton SL (2005) Ryanodine receptors. Cell Calcium 38, 253–260 [DOI] [PubMed] [Google Scholar]
- 65.Verkhratsky A (2004) Endoplasmic reticulum calcium signaling in nerve cells. Biol Res 37, 693–699 [DOI] [PubMed] [Google Scholar]
- 66.Murthy KS, and Zhou H (2003) Selective phosphorylation of the IP3R-I in vivo by cGMP-dependent protein kinase in smooth muscle. Am J Physiol Gastrointest Liver Physiol 284, G221–230 [DOI] [PubMed] [Google Scholar]
- 67.Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Soderberg O, Bootman MD, and Roderick HL (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A 105, 2427–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arguin G, Regimbald-Dumas Y, Fregeau MO, Caron AZ, and Guillemette G (2007) Protein kinase C phosphorylates the inositol 1,4,5-trisphosphate receptor type 2 and decreases the mobilization of Ca2+in pancreatoma AR4–2J cells. J Endocrinol 192, 659–668 [DOI] [PubMed] [Google Scholar]
- 69.Caron AZ, Chaloux B, Arguin G, and Guillemette G (2007) Protein kinase C decreases the apparent affinity of the inositol 1,4,5-trisphosphate receptor type 3 in RINm5F cells. Cell Calcium 42, 323–331 [DOI] [PubMed] [Google Scholar]
- 70.Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, and Pinton P (2012) Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis 3, e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bai GR, Yang LH, Huang XY, and Sun FZ (2006) Inositol 1,4,5-trisphosphate receptor type 1 phosphorylation and regulation by extracellular signal-regulated kinase. Biochem Biophys Res Commun 348, 1319–1327 [DOI] [PubMed] [Google Scholar]
- 72.Lu YF, Kandel ER, and Hawkins RD (1999) Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci 19, 10250–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ciani E, Guidi S, Bartesaghi R, and Contestabile A (2002) Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J Neurochem 82, 1282–1289 [DOI] [PubMed] [Google Scholar]
- 74.Kikuchi D, Tanimoto K, and Nakayama K (2016) CREB is activated by ER stress and modulates the unfolded protein response by regulating the expression of IRE1alpha and PERK. Biochem Biophys Res Commun 469, 243–250 [DOI] [PubMed] [Google Scholar]
- 75.Lin L, Cao J, Yang SS, Fu ZQ, Zeng P, Chu J, Ning LN, Zhang T, Shi Y, Tian Q, Zhou XW, and Wang JZ (2018) Endoplasmic reticulum stress induces spatial memory deficits by activating GSK-3. J Cell Mol Med 22, 3489–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balogh A, Nemeth M, Koloszar I, Marko L, Przybyl L, Jinno K, Szigeti C, Heffer M, Gebhardt M, Szeberenyi J, Muller DN, Setalo G Jr., and Pap M (2014) Overexpression of CREB protein protects from tunicamycin-induced apoptosis in various rat cell types. Apoptosis 19, 1080–1098 [DOI] [PubMed] [Google Scholar]
- 77.Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, and Montminy M (2003) cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes & development 17, 1575–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reusch JE, and Klemm DJ (2002) Inhibition of cAMP-response element-binding protein activity decreases protein kinase B/Akt expression in 3T3-L1 adipocytes and induces apoptosis. J Biol Chem 277, 1426–1432 [DOI] [PubMed] [Google Scholar]
- 79.Francois F, Godinho MJ, and Grimes ML (2000) CREB is cleaved by caspases during neural cell apoptosis. FEBS letters 486, 281–284 [DOI] [PubMed] [Google Scholar]
- 80.Lee YY, Moujalled D, Doerflinger M, Gangoda L, Weston R, Rahimi A, de Alboran I, Herold M, Bouillet P, Xu Q, Gao X, Du XJ, and Puthalakath H (2013) CREB-binding protein (CBP) regulates beta-adrenoceptor (beta-AR)-mediated apoptosis. Cell Death Differ 20, 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang Y, Ma S, Wei F, Liang G, Yang X, Huang Y, Wang J, and Zou Y (2019) Pivotal role of cAMP-PKA-CREB signaling pathway in manganese-induced neurotoxicity in PC12 cells. Environ Toxicol. 34,1052–1062 [DOI] [PubMed] [Google Scholar]
- 82.Sen N (2018) ER Stress, CREB, and Memory: A Tangled Emerging Link in Disease. Neuroscientist, 25(5):420–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Isenoumi K, Kumai T, Kitaoka Y, Motoki M, Kitaoka Y, Kuribayashi K, Munemasa Y, Kogo J, Kobayashi S, and Ueno S (2004) N-methyl-D-aspartate induces phosphorylation of cAMP response element (CRE)-binding protein and increases DNA-binding activity of CRE in rat retina. J Pharmacol Sci 95, 108–114 [DOI] [PubMed] [Google Scholar]
- 84.Beltran WA, Allore HG, Johnson E, Towle V, Tao W, Acland GM, Aguirre GD, and Zeiss CJ (2009) CREB1/ATF1 activation in photoreceptor degeneration and protection. Invest Ophthalmol Vis Sci 50, 5355–5363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong E, Bachleda A, Xiong Y, Osawa S, and Weiss ER (2017) Reduced phosphoCREB in Muller glia during retinal degeneration in rd10 mice. Mol Vis 23, 90–102 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.