

Abstract
Mitigation of total-body irradiation (TBI) in C57BL/6 mice by two drugs, which target apoptosis and necroptosis respectively, increases survival compared to one drug alone. Here we investigated whether the biomarker (signature)-directed addition of a third anti-ferroptosis drug further mitigated TBI effects. C57BL/6NTac female mice (30–33 g) received 9.25 Gy TBI, and 24 h or later received JP4–039 (20 mg/kg), necrostatin-1 (1.65 mg/kg) and/or lipoxygenase-15 inhibitor (baicalein) (50 mg/kg) in single-, dual- or three-drug regimens. Some animals were sacrificed at days 0, 1, 2, 3, 4 or 7 postirradiation, while the majority in each group were maintained beyond 30 days. For those mice sacrificed at the early time points, femur bone marrow, intestine (ileum), lung and blood plasma were collected and analyzed for radiation-induced and mitigator-modified levels of 33 pro-inflammatory and stress response proteins. Each single mitigator administered [JP4–039 (24 h), necrostatin-1 (48 h) or baicalein (24 h)] improved survival at day 30 after TBI to 25% (P = 0.0432, 0.2816 or 0.1120, respectively) compared to 5% survival of 9.25 Gy TBI controls. Mice were administered the drug individually based on weight (mg/kg). Drug vehicles comprised 30% cyclodextrin for JP4–039 and baicalein, and 10% Cremphor-EL/10% ethanol/80% water for necrostatin-1; thus, dual-vehicle controls were also tested. The dual-drug combinations further enhanced survival: necrostatin-1 (delayed to 72 h) with baicalein 40% (P = 0.0359); JP4–039 with necrostatin-1 50% (P = 0.0062); and JP4–039 with baicalein 60% (P = 0.0064). The three-drug regimen, timed to signature directed evidence of onset after TBI of each death pathway in marrow and intestine, further increased the 30-day survival to 75% (P = 0.0002), and there was optimal normalization to preirradiation levels of inflammatory cytokine and stress response protein levels in plasma, intestine and marrow. In contrast, lung protein levels were minimally altered by 9.25 Gy TBI or mitigators over 7 days. Significantly, elevated intestinal proteins at day 7 after TBI were reduced by necrostatin-1-containing regimens; however, normalization of plasma protein levels at day 7 required the addition of JP4–039 and baicalein. These findings indicate that mitigator targeting to three distinct cell death pathways increases survival after TBI.
INTRODUCTION
The notion that mitigation of the effects of total-body irradiation (TBI) can be optimally achieved by administration of a single drug that resolves all effects of the multiple mechanisms of radiation-induced networks, including inflammation and cell death, is overly optimistic. It has been shown that mitigation of TBI effects in C57BL/6 mice by two drugs, which target apoptosis and necroptosis respectively, increases survival compared to one drug alone (1). There are several programs of regulated cell death involved in the response to radiation. Switches between these programs (when one fails or is suppressed by a radiomitigator) are followed by takeover of the response by another pathway, which continues the death process (1). Therefore, it is logical to conclude that optimal and durable radiation mitigation will only be achieved by administration of multiple agents that target multiple injury response pathways.
Radiation-induced cell death signaling pathways are distinct, yet overlapping with respect to the control of their biochemical mechanisms (1–12). Radiation-induced apoptosis is initiated within minutes after nuclear DNA double strand breaks and communication of signals from nucleus to mitochondria (13, 14), which lead to mitochondrial cardiolipin disassociation from cytochrome-C, oxidation of cardiolipin, its transport to the outer mitochondrial membrane, cytoplasmic leakage of cytochrome C, activation of caspase-3 and cleavage of nuclear poly-ADP-ribosyl polymerase causing nuclear DNA fragmentation, and cell death (2, 6, 13–15). A distinct necroptosis death pathway (1, 11) is delayed to 48 h or longer postirradiation and follows the elevation of inflammatory cytokines including tumor necrosis factor-α (TNF-α) that bind to both local, and distant (via the circulation) cell membranes, activate the TNF-α receptor, phosphorylation of RIP1 and RIP3 kinase, cell membrane permeability and cell death (11). Properly timed delivery of two mitigators that block apoptosis (JP4–039) and necroptosis (necrostatin-1) after TBI results in improved survival compared to the delivery of a single mitigator (1).
A third distinct radiation-induced cell death pathway is ferroptosis (16–25), which is initiated by free radicals and oxygenation products, such as peroxides and hydroperoxides, and involves mitochondrial glutathione peroxidase 4 (GPX4) (21, 22). Depletion of GPX4 leads to, among other events, the separation of phosphatidyl ethanolamine from its binding protein in the endoplasmic reticulum, activation of 15-lipoxygenase (18–20), additional lipid oxidations, cell membrane permeability and cell death (9, 16, 17).
We investigated whether targeting ferroptosis by a third mitigator increased survival after TBI. We tested three known ferroptosis inhibitors, ferrostatin (24), liproxstatin (23) (15-LOX) and baicalein (25), as radiation mitigators using cell lines in vitro, and in vivo in TBI mice. We determined the effect of the best ferroptosis inhibitor delivered alone or in combination with anti-apoptosis drug JP4–039 (26) and/or anti-necroptosis drug necrostatin-1 on survival after TBI and the effect on biomarkers of radiation-induced damage to bone marrow, small intestine and lung, as well as reflected in plasma. The results demonstrate superior TBI mitigation by delivery of a three-drug regimen including the ferroptosis inhibitor baicalein. The data extend the concept of signature-directed sequential delivery of TBI mitigators to include targeting the ferroptosis cell death pathway.
MATERIALS AND METHODS
Animals and Animal Care
C57BL/6NTac adult (30–33 g) female mice received 9.25 Gy TBI (LD50/30) using a Mark I Gamma Cell Irradiator (JL Shepherd & Associates, San Fernando, CA) with filter removed. This action resulted in a dose rate of 343 cGy per min. The dosimetry of this irradiator and orthovoltage below spectrum have been reported (1). To investigate the change in the energy spectrum of the incident radiation field due to the removal of the filter, percentage-depth-dose (PDD) profiles were measured using Gafchromice™ EBT3 radiochromic film (Ashlande™, Covington, KY). Measurements of the PDD profiles both with and without the filter in place were completed with the films aligned parallel to the beam axis between two 14 × 14 × 0.2-cm3 pieces of polymethyl methacrylate (PMMA). The films were centered within the cabinet irradiator for measurements (1).
The measured PDD profiles have been published elsewhere (1). The profiles were normalized to a depth of 5 cm. The PDD profile measured without the filter decreased more rapidly at shallow depths than did the profile measured with the filter, indicating a greater proportion of low-energy photons detected in the absence of the filter. Beyond a depth of 50 mm, the two PDD profiles were similar, indicating that the high-energy region of the energy spectrum was minimally effected by the presence of the filter. Therefore, removal of the filter lowered the average energy of the incident radiation, but was not expected to significantly alter the radiation dose received by the animal placed in a Lucite® holding chamber during exposure (1).
Cell Lines, Tissue Culture and In Vitro Radiation Survival Curves
Bone marrow stromal cell lines and hematopoietic progenitor cell lines were derived from C57BL/6NTac (10) and 129/Sv (5) mouse long-term marrow cultures. Clonogenic radiation survival curves were performed using methods described elsewhere (5).
In vitro radiation survival curves.
Radiation survival curves were performed in vitro, as described elsewhere (3, 5). Briefly, radiation mitigators JP4–039, baicalein, necrostatin-1, ferrostatin and liproxstatin were dissolved in DMSO at a concentration of 10 mM. Radiation survival curves were performed by resuspending C57BL/6 bone marrow cells at 1 × 106 cells per ml of tissue culture media. The cells were exposed to doses of 0 to 8 Gy using a Mark 1, Model 68A cesium irradiator (JL Shepherd & Associates). Radiation mitigators were added to the cells (1 μl/ml tissue culture media) for a final concentration of 10 μM. The cells were plated in 50-mm petri dishes at 500, 1,000 or 2,500 cells per petri dish. The cells were plated in a CO2 incubator at 37°C for 7 days. The cells were stained with crystal violet and colonies of greater than 50 cells were counted. The data were analyzed using linear-quadratic or single-hit, multiple-target models.
Preparation and Delivery of Radiation Mitigator Drugs
The synthesis of JP4–039, a peptide isostere conjugate of 4-amino-TEMPO (4-AT) nitroxide, was performed as described elsewhere (4). The drug was dissolved in 30% 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich® LLC, St. Louis, MO) at 8 mg/ml. During the preparation, the active ingredient (JP4–039) is added to the 30% cyclodextrin solution and incubated for 2 h at 50°C on a magnetic spin plate. JP4–039 was administered intramuscularly (I.M.) in 50 ll containing 400 μg of drug. In the current experiments, JP4–039 was delivered 24 h after TBI (1).
Necrostatin-1 was administered as described elsewhere (1). Briefly, the drug was dissolved in 10% ethanol, 10% Cremophor-EL®, and 80% deionized water, to obtain a concentration of 0.33 mg/ml. Mice were intravenously (I.V.) injected with 100 μl containing 33 μg of drug at several time points after TBI. The drug dose was thus 1.65 mg/kg. In some TBI experiments, necrostatin-1 was delivered after previous administration of JP4–039. Anti-ferroptosis drugs tested were dissolved in 30% 2-hydroxypropyl-β-cyclodextrin by heating to 50°C on a magnetic stir plate. The drugs were I.M. injected in 50 μl at 10 mg/kg for ferrostatin (23), 50 mg/kg for liproxstatin (24) and 50 mg/kg for baicalein (25). For baicalein, drug was delivered at 24 h, 48 h or 72 h postirradiation.
Delivery of Single or Multiple Mitigators to TBI Mice
A total of 120 C57BL/6NTac female mice received 9.25 Gy TBI (1). The mice were divided into eight treatment groups of 15 mice, as follows: 9.25 Gy only; JP4–039 at 24 h after TBI: baicalein at 24 h after TBI; JP4–039 and baicalein at 24 h after TBI; necrostatin-1 at 48 h after TBI; JP4–039 at 24 h after TBI and necrostatin-1 at 72 h after TBI; baicalein at 24 h after TBI and necrostatin-1 at 72 h after TBI; and JP4–039 and baicalein at 24 h after TBI with necrostatin-1 at 72 h after TBI. The mice were followed for development of hematopoietic syndrome at which time they were sacrificed. Vehicles for JP4–039 and baicalein were 30% 2-hydroxypropyl-β-cyclodextrin, and necrostatin-1 was delivered in 10% Cremophor-EL, 10% ethanol and 80% water.
Protein Level Measurements in Plasma and Tissues after Irradiation and Administration of Radiation Mitigators
A total of 135 mice received 9.25 Gy TBI and were assigned to one of eight different treatment groups, as described below.
30 mice received TBI only; 5 mice per day were sacrificed on day 0, 1, 2, 3, 4 or 7.
20 mice injected with JP4–039 at 24 h; 5 mice per day sacrificed on day 2, 3, 4 or 7.
20 mice injected with baicalein at 24 h; 5 mice per day sacrificed on day 2, 3, 4 or 7.
20 mice injected with both JP4–039 and baicalein; 5 mice per day sacrificed on day 2, 3, 4 or 7.
15 mice injected with necrostatin-1 on day 2 after TBI; 5 mice per day sacrificed on day 3, 4 or 7.
10 mice injected with JP4–039 at 24 h and necrostatin-1 at 72 h; 5 mice per day sacrificed on day 4 or 7.
10 mice injected with baicalein at 24 h and necrostatin-1 at 72 h; 5 mice per day sacrificed on day 4 or 7.
10 mice injected with JP4–039 at 24 h, baicalein at 24 h and necrostatin-1 at 72 h; 5 mice per day sacrificed on day 4 or 7.
At the time of sacrifice, blood, bone marrow (femur and tibia), small intestine (ileum) and lungs were isolated from each individual animal and placed on ice. The bone marrow, ileum and lungs were homogenized in 1 ml of 0.1% Tween® 80 in phosphate buffered saline (PBS) to prevent clumping, and centrifuged at 1,000 rpm for 10 min at 4–68°C. Supernatant was removed, protein determined using a Bio-Rad® Protein Assay Kit (Hercules, CA) and supernatant frozen at −80°C. The blood was collected in EDTA tubes, centrifuged at 1,000 rpm for 10 min with the plasma removed and frozen at −80°C. The protein was used in a Luminex® assay (Austin, TX) to determine protein level.
Standardization of Plasma, Lung, Intestine and Bone Marrow Protein Levels for Luminex Assays
A list of the 33 proteins assayed and the known primary function of each is provided elsewhere (1). Selection of these 33 proteins was based on the pathways involved in the known inflammatory and radiation-induced damage responses (1). A Bio-Rad protein assay was run for each tissue type (plasma, lung, intestine, bone marrow) and experimental group [9.25 Gy irradiation only (control); 9.25 Gy with JP4–039; 9.25 Gy with necrostatin-1; 9.25 Gy with JP4–039 at 24 h and necrostatin-1 at 72 h after TBI; baicalein at 24 h after TBI; baicalein and JP4–039 at 24 h after TBI; baicalein at 24 h and necrostatin-1 at 72 h after TBI; and JP4–039 and baicalein at 24 h with necrostatin-1 at 72 h after TBI]. We utilized the TGF-β1 Single Plex Magnetic Bead Kit, as well as the 32-Multiplex Mouse Cytokine/Chemokine Magnetic Bead Panel (EMD Millipore, Billerica, MA), which tested protein concentrations for eotaxin, G-CSF, GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, IP-10, KC, LIF, LIX, MCP-1, M-CSF, MIG, MIP-1α, MIP-1β, MIP-2, regulated upon activation normal T cell expressed and secreted (RANTES), TNF-α and VEGF.
Each kit provided reagents for use in the Luminex protein immunoassay, which included one vial of mouse cytokine/chemokine quality controls, and one vial serum matrix, 96-well plate, 30 ml assay buffer, 60 ml 10× wash buffer, one 3.2-ml bottle of mouse cytokine detection antibodies and one 3.2-ml bottle of streptavidin-phycoerythrin. The 32-Multiplex kit provided one 3.5-ml of pre-mixed 32-plex beads. The TGF-β1 kit provided the above reagents, as well as 5 ml of sample diluent, 1.0 ml of 1.0 N HCl, 1.0 ml of 1 N NaOH and 3.5 ml of anti-TGF-β1 beads.
Preparation of Reagents for Immunoassay Beads, Quality Controls, Wash Buffer and Serum Matrix
Antibody-immobilization beads were sonicated for 30 s and then vortexed for 1 min prior to use. For quality control, the tubes were handled as a 1:10 dilution with 250 μl of assay buffer. The 60 ml of 10× wash buffer were diluted 1:10 with 540 ml of deionized water. Using the Luminex method, 32 proteins were assayed. TGF-β1 (the 33rd protein) was assayed using a different immunoassay technique.
For the 32-multiplex assay, serum matrix for plasma samples was reconstituted in 2.0 ml of assay buffer, and allowed to sit at room temperature for 10 min.
Preparation of Standards for Protein Luminex Assays
Mouse cytokine standards were reconstituted in 250 μl of deionized water, inverted to mix, vortexed and then transferred to a polypropylene microfuge tube and labeled as “standard 6.” Serial dilutions were performed, by which 50 ll of standards 6, 5, 4, 3 and 2 were micropipetted to a microfuge tube containing either 200 μl of assay buffer (for 32-multiplex) or 150 μl of assay buffer (for TGF-β1), until standard 1 was obtained.
Determination of Protein Concentration
Lungs, intestine and bone marrow homogenates were tested for protein concentration using the Bio-Rad protein detection assay by diluting the detection solution 1:5 in deionized water. Each sample and standard (2 mg/ml, 1 mg/ml, 0.75 mg/ml, 0.5 mg/ml, 0.25 mg/ml, 0.125 mg/ml and 0.0 mg/ml) was plated in triplicate on a 96-flat bottom-well plate with 5 μl of sample/standard combined. The 1:5 diluted protein detection solution (250 μl) was added to each well, mixed and run for analysis on spectrophotometer at a wavelength of 595 nm.
The average optical density value for each sample and standard was calculated, and a standard curve was formulated based on the averages of the standards. The standard curve was used to obtain the actual protein concentrations of each sample, which was used to standardize the Luminex assay. Immunoassay results for plasma were converted from “μg/ml” to “μg/mg protein” for comparison to intestine and bone marrow.
Dilution of Samples
The 32-multiplex plasma samples were diluted 1:2 by combining 25 μl of plasma with 25 μl of assay buffer. TGF-β1 plasma samples were diluted 1:5 by combining 12.5 μl of plasma with 50 μl sample diluent, 25 μl of each 1:5-diluted sample was plated on a 96-well plate and treated with 2.0 μl of 1.0 N HCl, then tested to ensure pH was below 3.0. The plate was sealed and covered with aluminum foil, then shaken at room temperature for 15 min. The acid-treated plasma samples were then further diluted at 1:6 by combining 10 μl of acid-treated plasma with 50 μl of assay buffer, resulting in a final dilution of 1:30.
Procedures for the 32-Multiplex Luminex Immunoassay
All plasma, lung, intestine and bone marrow samples were assayed in 96-well plates, which were conditioned first by adding 200 μl of wash buffer to each well. Plates were then sealed and covered on a plate shaker for 10 min at room temperature. Wash buffer was then removed via suction.
Prepared standards and controls (25 μl of each) were added to the appropriate well, followed by 25 μl of assay buffer to the background sample wells. Either serum matrix (for plasma; 25 μl) or 0.1% Tween 80 in PBS (for intestine and bone marrow; 25 μl) was added to each background, standard and control well. Then, 25 μl of sample was added to each sample well. The bottle of pre-mixed beads was vortexed and then 25 μl was added to each well. The plates were then sealed, covered and incubated overnight at 2–8°C.
Between 16–18 h later, the plate was placed on a magnet, the well contents were removed via suction, and then the plates were washed twice with 200 μl of wash buffer, as described in the first step. Detection antibodies (25 μl) were then added to each well, and the plate was sealed, covered and incubated for 1 h on a plate shaker at room temperature. After incubation, 25 μl of streptavidin-phycoerythrin was added to each well, and the plate was again sealed, covered and incubated for 30 min on a plate shaker at room temperature. After incubation, well contents were removed via suction, and the plates were washed twice with 200 μl of wash buffer, as described above. After the two washes, 150 μl of wash buffer was added to all wells, the plate was placed on a plate shaker for 5 min, and then read using the Luminex 100/200™.
Data Analysis and Standardization of Intestine, Lung and Bone Marrow Results
We reported the protein concentrations by Luminex assay in lg/ml. A protein concentration assay was run on each sample. For each intestine, plasma and bone marrow sample, the data from the Luminex assay for that sample was divided by the determined protein concentration to give standardized concentrations of pg/mg protein.
TGF-beta-1 Immunoassay
TGF-β was not measured by Luminex, as it requires a different method for tissue preparation. Intestine, lung and bone marrow samples for the TGF-β1 immunoassay required an acidification step. Tissue samples (25 μl of each) were plated onto a 96-well plate, treated with 2.0 μl of 1.0 N HCl and tested to ensure pH was below 3.0. The plate was sealed and covered with aluminum foil, then shaken at room temperature for 15 min. The acid-treated samples were then neutralized with 2 μl of 1.0 N NaOH prior to adding to the sample well.
For the TGF-β1 assay of plasma, a serum matrix was reconstituted in 1.0 ml of deionized water and 4.0 ml of assay buffer, and was allowed to sit for 10 min at room temperature. Then, 0.1 ml of the reconstituted serum matrix was micropipetted and diluted in 0.5 ml of assay buffer, for a final dilution of 1:30. The next procedures with the 96-well plate assay were then performed as described above for the 32-multiplex Luminex immunoassay.
Western Blot Analysis of Three Proteins and Measurement of Phosphorylation of p-RIP3 Kinase, Acyl-CoA-Synthase and GPX4
Three proteins were measured using an alternative method to the Luminex assay to confirm the results. We used Western blot analysis rather than ELISA assay for consistency with our previously published work (1). Using a Western blot analysis, we measured receptor-interacting serine/threonine-protein kinase (RIP3) phosphorylation as a marker for necroptosis, or glutathione peroxidase 4 (FPX4) and Acyl-CoA-synthase (ALSC) as indicators of ferroptosis (1, 11). C57BL/6NTac female mice received 9.25 Gy TBI. The bone marrow and ileum were collected from 5 mice per time point, preirradiation, and daily for 7 days postirradiation (eight time points). The bone marrow was centrifuged at 1,500 rpm for 5 min, and the pellet resuspended in ACK Lysing Buffer (Quality Biological Inc., Gaithersburg, MD) for 10 min on ice to lyse the red blood cells. The cells were centrifuged at 1,500 rpm for 5 min with the pellet resuspended in PBS and transferred to 1.5 ml Eppendorf tubes, and microcentrifuged at 2,500 rpm for 5 min. The pellet was resuspended in 1 ml of cell lysis buffer (1% Nonidet™ P-40, 20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml of leupeptin and 10 μg/ml of aprotinin), placed on a rotator, rocked gently at 4°C for 30 min and then centrifuged in a microcentrifuge at 13,000 rpm and 4°C for 10 min. The supernatant was removed and placed in a new Eppendorf tube and stored on ice.
The ileum and lung were excised from each mouse. A 100-mg piece of each tissue was placed in a round-bottom tube, 1 ml of cell lysis buffer added and placed on ice. Each sample was homogenized using a Polytron® PT3000 (Brinkman Inc., Riverview, FL), placed on ice for 2 h, transferred to a 1.5-ml Eppendorf tube, centrifuged in a microcentrifuge for 15 min at 13,000 rpm and 4°C, and the supernatant was then transferred to a new Eppendorf tube and stored on ice. Protein concentration was determined using Bio-Rad Protein Assay Dye Reagent Concentrate. The protein samples were added to 4× Loading Buffer (Bio-Rad) with 10% 2-mercaptoethanol, vortexed and heated at 95–100°C for 5 min.
The samples were loaded on 4–12% Bis-Tris Criterion XT Precast Gels (Bio-Rad) and electrophoresized at 145 V for 70 mins using XT MES as the running buffer (Bio-Rad). The proteins were transferred to PVDF membranes (Bio-Rad) at 0.45A for 90 min using Tris/Glycine buffer (Bio-Rad) with an ice block. Nonspecific binding was blocked by incubating the membrane in 5% nonfat milk for 30 mins. As the loading control, we used an anti-GADPH mouse monoclonal antibody (Cell Signaling Technology® Inc., Danvers, MA). The Western blots were left in 5% nonfat milk overnight at 4°C and then washed three times with TBS (Bio-Rad) for 5 mins. The secondary antibody (either goat anti-rabbit IgG-HRP, goat anti-mouse IgG-HRP or donkey anti-goat IgG-HRP; all from Santa Cruz Biotechnologyt Inc., Dallas, TX) was diluted 1:10,000 in 5% nonfat milk, incubated with the membrane for 1 h at room temperature, and washed three times with TBS for 5 min. The proteins were detected using SuperSignale West Pico PLUS Chemiluminescent Substrate (Thermo Fisher, Pittsburgh, PA). Western blot quantification was performed using LabWorks™ Management System (Lablogics Inc., Mission Viejo, CA).
For Western blot measuring RIP3 kinase phosphorylation in intestine (ileum) or lung, the membrane was incubated with the primary antibody (anti-phospho RIP3 Antibody; Abcam®, Cambridge, MA), Acyl-CoA-synthase (anti-mouse Acyl-CoA-Synthase monoclonal antibody; Santa Cruz Biotechnology) and GPX4 (anti-mouse antibody; Santa Cruz Biotechnology).
Graphical and Statistical Analysis
Student’s unpaired t test was used to compare the time points of each group that received mitigating drugs to the corresponding time points in the control group, as well as to compare all days in each experimental group (including the control) to day 0 (nonirradiated baseline) of the control group.
Use of Standard Deviation or Standard Error of the Mean
We summarized the estimates of D0 and n from multiple experiments with mean ± standard error of the mean (SEM; Fig. 1). We used SEM to indicate the precision of the mean values as estimates for the parameters. We used mean and SEM to make the bar graphs with errors indicated (Figs. 6–9). As is usually presented for this type of plot, the SEMs indicate the uncertainty around the estimate of the mean measurement. In Supplementary Tables S1–8 (https://doi.org/10.1667/RR15486.1.S1) we used mean ± standard deviations (SD) to summarize the data for protein levels in each group. We used SD to estimate the variability of the population from which the sample was drawn, because it is estimated that approximately 95% of observations of any distribution usually fell within the two standard deviation limits of the mean. The mean and SEM for each time point were determined and graphed using GraphPad Prism software (LaJolla, CA) to compare each experimental group for each of the 33 proteins tested in plasma, bone marrow, intestine (ileum) and lung.
FIG. 1.

Radiation mitigation of 129/Sv mouse bone marrow stromal cell lines by anti-ferroptosis drugs: ferrostatin, liproxstatin and baicalein. Cell lines were irradiated and plated in clonogenic survival curves, as described in Materials and Methods. We compared the anti-ferroptosis drugs with the anti-apoptosis mitigator, JP4–039, as a control. Each mitigator was dissolved in DMSO at a concentration of 10 mM and used at a concentration of 10 μM. The survival curve is for one of three separate experiments, each with triplicate cultures scored for each point; however, the results shown for D0 and n are means and standard error of all three separate experiments.
FIG. 6.
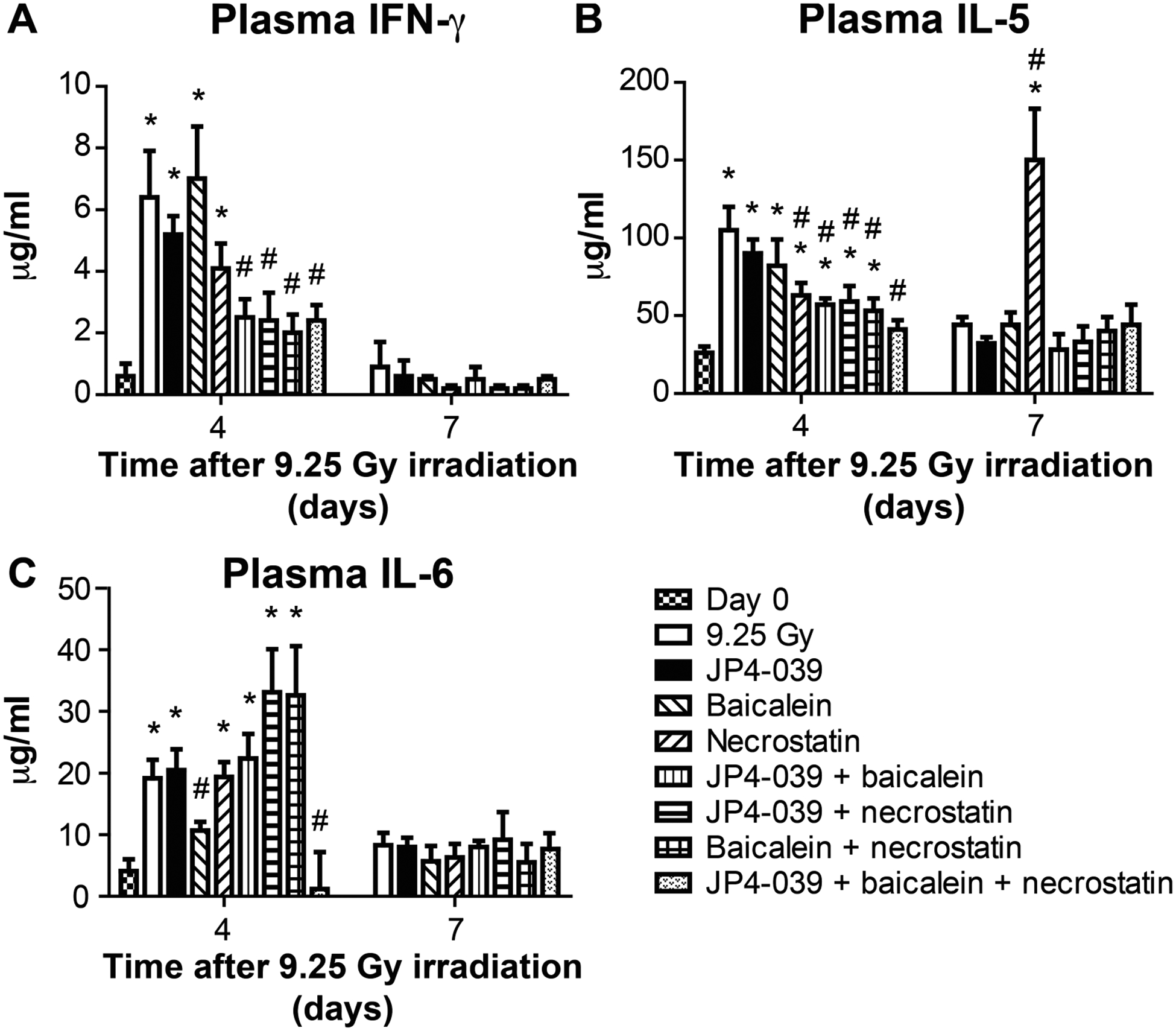
Effect in C57BL/6NTac mice of 9.25 Gy TBI and mitigator combinations on protein levels in plasma at days 4 and 7 postirradiation. Subgroups of mice were injected at 24 h with JP4–039 (20 mg/kg), baicalein (50 mg/kg), or both, at 48 h with necrostatin-1 (1.65 mg/kg) or JP4–039 (20 mg/kg), and baicalein (50 mg/kg) at 24 h followed by necrostatin-1 (1.65 mg/kg) at 72 h. On days 4 and 7, plasma was isolated and Luminex assay was performed for the expression of inflammatory proteins. Panels A–C: Expressions are shown for IFN-γ, IL-5 and IL-6 (n = 5 mice per time point), respectively. Results are mean ± SEM. *P < 0.05 compared to day 0; #P < 0.05 compared to 9.25 Gy irradiation on same day.
FIG. 9.
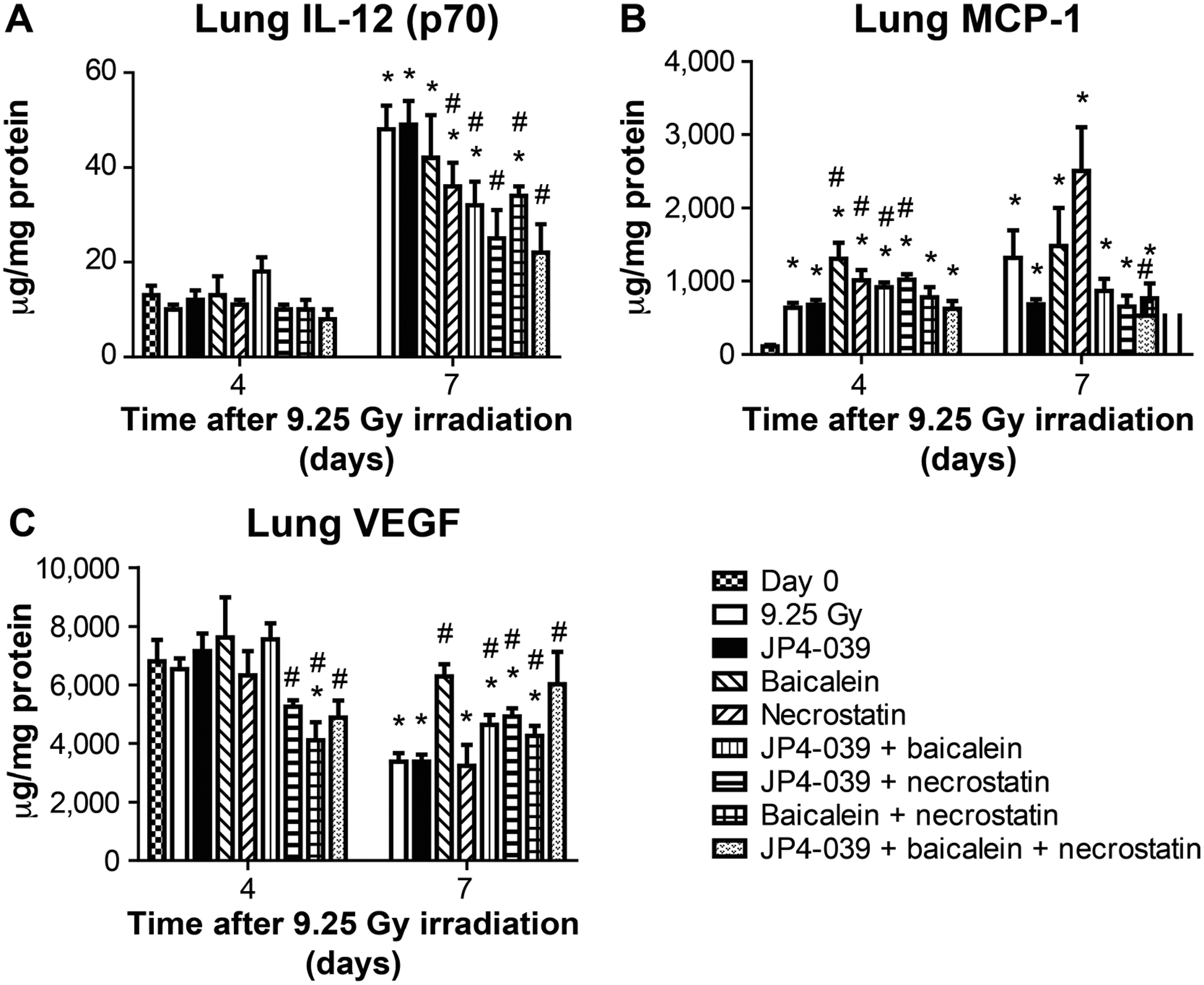
Effect of 9.25 Gy TBI and mitigator combinations on protein levels in lung at days 4 and 7. C57BL/6NTac female mice received 9.25 Gy TBI. Subgroups were injected at 24 h postirradiation with JP4–039 (20 mg/ kg), baicalein (50 mg/kg) or both JP4–039 (20 mg/kg) and baicalein (50 mg/kg). At 48 h after TBI a subgroup was injected with necrostatin-1 (1.65 mg/kg). Another subgroup was injected at 24 h after TBI with JP4–039 (20 mg/kg) and baicalein (50 mg/kg) followed at 72 h after TBI by an injection of necrostatin-1 (1.65 mg.kg). On day 4 and 7 the lungs were analyzed using Luminex assay for expression of inflammatory proteins. Panels A–C: Results are shown for IL-12 (p70), MCP-1 and VEGF (n = 5 mice per time point), respectively. Results are mean ± SEM. *P < 0.05 compared to day 0; #P < 0.05 compared to 9.25 Gy irradiation on same day.
Power Calculation
The sample size estimate of n = 15 per group was based on a one-sided log-rank test with 78% power to detect (at a 0.05 significance level) an estimated difference of 50% 30-day survival in the radiation-only control group versus 90% 30-day survival in a treated group.
For presentation of absolute and relative levels of proteins, we used two formats. The mean and SEM for each time point was determined and graphed, using GraphPad Prism software (LaJolla, CA) to compare each experimental group for each of the 33 proteins. Student’s unpaired t test was used to compare the time points of each group that received each single-, dual- or three-mitigator-drug regimen to the corresponding time point in the control radiation-alone group, as well as compared to each day in each experimental group (including the irradiated control) with the baseline day 0 (nonirradiated baseline) of the control group.
RESULTS
Small Molecule Inhibitors of Ferroptosis are Radiation Mitigators for Cells in Culture
To determine the effect of an anti-ferroptosis drug on radiation survival, we first tested the effect on cell lines in vitro in clonogenic survival assay of each of three drugs compared to JP4–039 and necrostatin-1. Mouse marrow stromal cell lines from the C57BL/6 and 129/Sv strains (5, 10) were irradiated in vitro to doses between 0 and 8 Gy, and plated in clonogenic radiation survival assays according to methods published elsewhere (5). Each of three drugs, previously reported to inhibit ferroptosis, was added at 1, 10 or 100 μM 24 h postirradiation and cells were trypsinized and plated for clonogenic survival assays. The anti-ferroptosis drugs tested were liproxstatin, ferrostatin and baicalein (Fig. 1). The optimal dose of each anti-ferroptosis drug was 10 μM for all three agents. Since the results were similar for each of the three anti-ferroptosis agents in vitro, we next tested each of the three in vivo.
Baicalein is an Effective Mitigator at 24, 48 or 72 h after TBI of C57BL/6NTac Mice
Based on a comparison of the effects of the three anti-ferroptosis drugs in vivo when delivered at 24 h after TBI, baicalein was determined to be superior to ferrostatin or liproxstatin (Fig. 2A). We next tested the effect of baicalein as a radiation mitigator added 24 h compared to 48 or 72 h after the LD50/30 dose of 9.25 Gy. Baicalein significantly increased survival of TBI mice at all times, 24, 48 and 72 h, but was most effective at 24 h (Fig. 2B). Based on these results, baicalein was chosen for use in the other experiments due to its relatively high solubility. The results for the radiation-only (control) treatment group (Fig. 2A) with 25% survival at 30 days differed from those shown in Fig. 2B, where there was 0% survival at 30 days. The difference in the control groups’ survival reflects the dose-drift in animal survival after TBI over the several months required for repeat experiments. The difference in survival of irradiated control mice between experiments could not be attributed to differences in animal age, weight, gender or radiation dose rate. The dose drift over time in the survival after a single dose of TBI has been attributed to differences in supply of animal feed in the diet (vendors are required to provide nutritional value, but not components, e.g., addition of seasonal additives such as alfalfa) and/or possible differences in veterinary personnel in the animal facility (27). These differences may contribute to changes in the intestinal microbiome, which can influence survival after TBI. Further studies will be required to explain differences in the radiation-only survival groups.
FIG. 2.
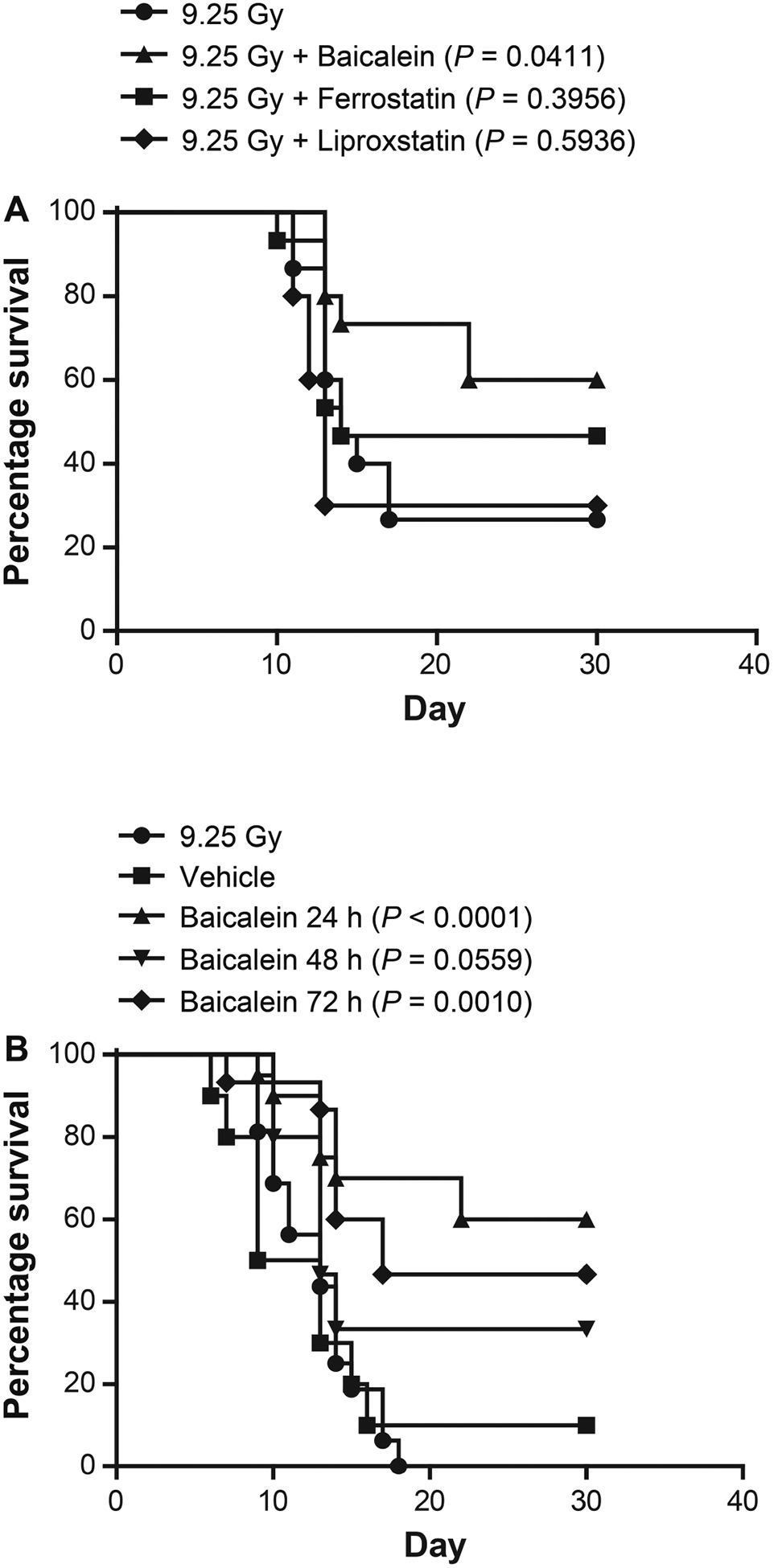
Improved survival by baicalein administered at 24 h after 9.25 Gy TBI in C57BL/6NTac mice. Panel A: At 24 h after TBI, subgroups of 15 mice were I.M. injected with either ferrostatin (10 mg/kg) or baicalein (50 mg/kg) dissolved in 30% cyclodextrin. Panel B: Other subgroups were I.M. injected with baicalein (50 mg/kg in 30% cyclodextrin) at 24, 48 or 72 h after TBI (n = 16).
Optimization of Timing for Integrated Delivery of Baicalein along with JP4–039 and Necrostatin-1 after TBI
Previously published studies have demonstrated that addition of the anti-necroptosis drug, necrostatin-1, is effective as a radiation mitigator, when the pathway for necroptosis (TNF-α binding to its cell surface receptor) has been activated. This process is detectable at 48 h after TBI, making the administration of necrostatin-1 suboptimal if delivered before 48 h. Previously reported studies have also demonstrated that administration of the anti-apoptosis drug, JP4–039, at 24 h after TBI, delayed by 24 h the plasma and tissue elevation of TNF-α as an indication of onset of necroptosis, thus, delaying the appropriate time for delivery of the anti-necroptosis drug to 72 h (12).
We evaluated the time of detection of biomarkers (signatures) for the onset of ferroptosis in plasma and tissues of TBI mice. As shown in Fig. 3, a significant decrease in levels of glutathione peroxidase, and increase in the ferroptosis-initiating enzyme Acyl-CoA-synthase, is detectable at 24 h postirradiation in both lung and intestine. Furthermore, in mice administered JP4–039 at 24 h, there was no significant further delay or change in time of appearance of the two signatures indicating the onset of ferroptosis. The current data establish that no significant alteration in the onset of ferroptosis was detected if an anti-apoptosis drug was administered at 24 h after TBI.
FIG. 3.
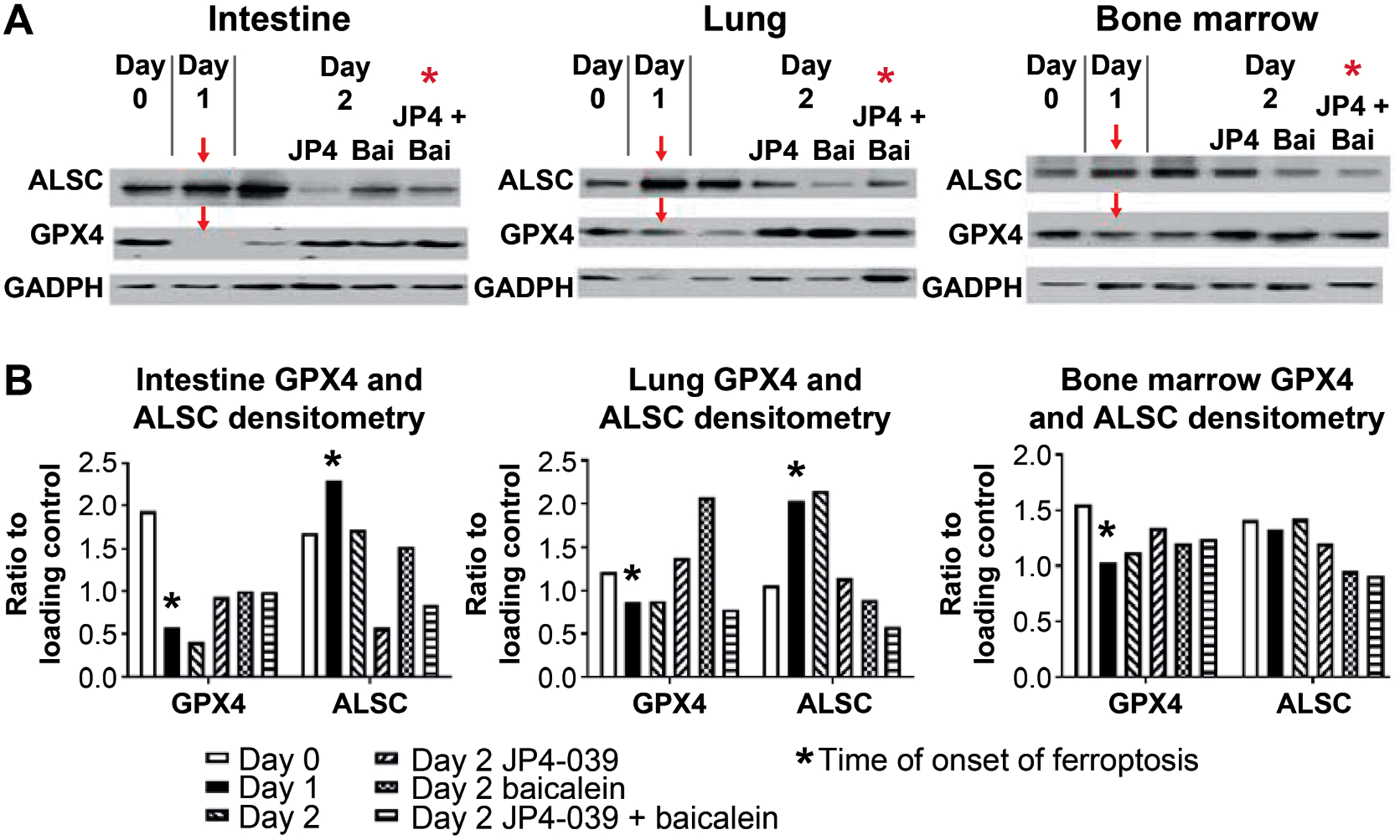
Signature-directed optimal time of baicalein administration to C57BL/6 mice at 24 h after TBI. To determine when radiation-induced ferroptosis begins as an indicator of when to administer baicalein, C57BL/6NTac mice received 9.25 Gy TBI. At 24 h postirradiation, mouse subgroups were I.M. injected with JP4–039 (20 mg/kg), baicalein (50 mg/kg) or both JP4–039 and baicalein. On days 0, 1 and 2, mice were sacrificed and bone marrow, plasma and intestine isolated. Panel A: Western blot analysis was performed for the expression of Acyl-CoA-synthase (ALSC) and glutathione peroxidase (GPX4) in intestine, lung and bone marrow. Red arrows indicate the onset of ferroptosis, as demonstrated by increased ALSC and decreased GPX4. Panel B: Densitometric normalization of ALSC and GPX4 gene expression was by comparison to GADPH expression. *Onset of ferroptosis. P < 0.05 compared to 9.25 Gy TBI.
We next investigated whether administration of baicalein along with JP4–039 at 24 h after TBI affected the delayed onset of necroptosis, using assay for RIP3 kinase phosphorylation (1). To determine whether the onset of necroptosis was further delayed past 72 h by administration of JP4–039 and baicalein at 24 h, we performed Western blot for the signatures of onset of necroptosis (RIP3 kinase phosphorylation) (Fig. 4). These results are shown in Western blots for intestine, lung and bone marrow (Fig. 4). There was no change in the delay of p-RIP3 in marrow or intestine to 72 h by adding the third mitigator, baicalein, at 24 h (Fig. 4).
FIG. 4.
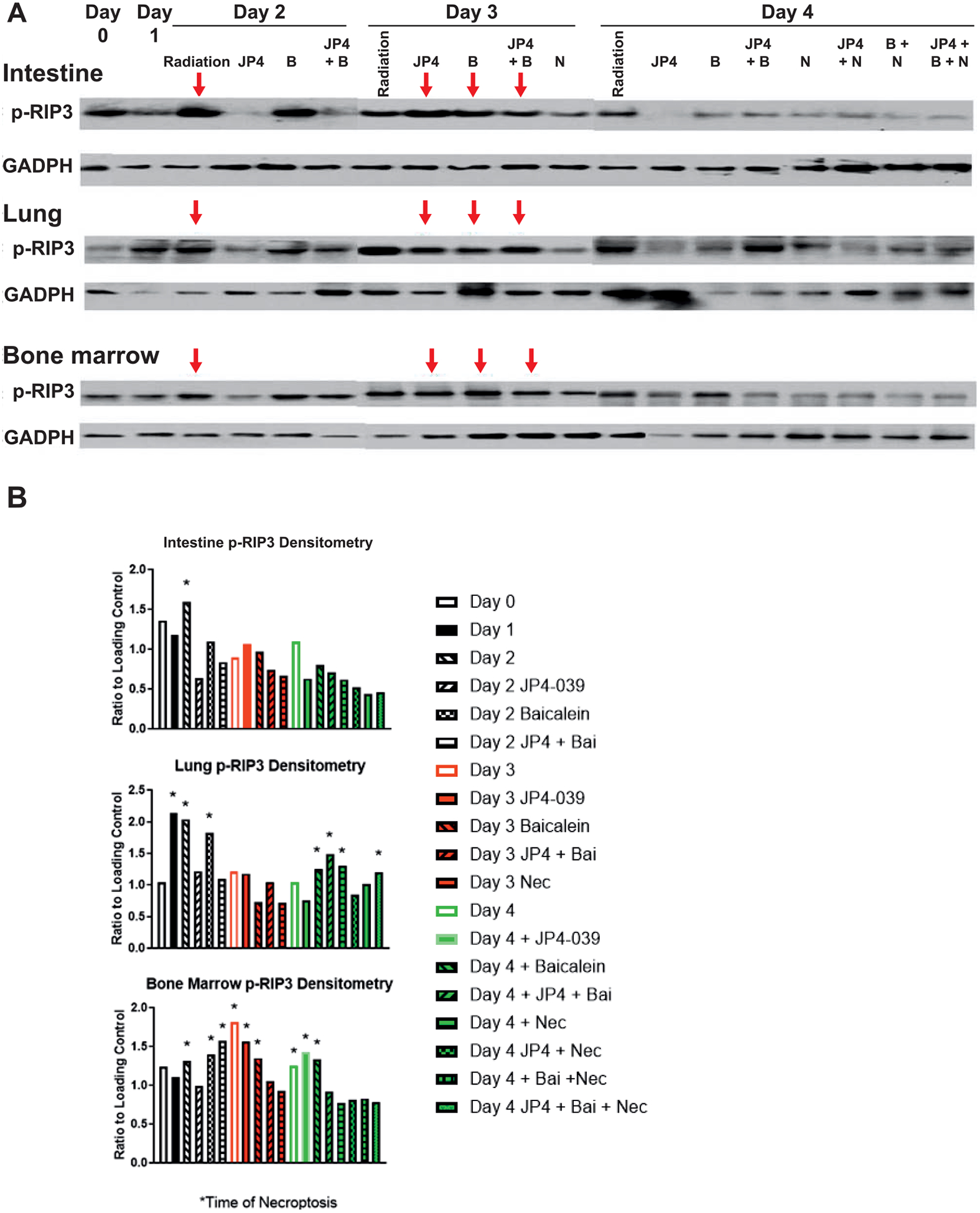
Administration of baicalein at 24 h along with JP4–039 delays signature-directed RIP3 phosphorylation to 72 h after TBI in intestine, lung and bone marrow. C57BL/6NTac mice received 9.25 Gy TBI, then injected 24 h later with JP4–039 (20 mg/kg), baicalein (50 mg/kg) or both JP4–039 (20 mg/kg) and baicalein (50 mg/kg). Necrostatin (1.65 mg/kg) was injected at 48 h if injected alone or 72 h if mice were previously administered JP4–039 or baicalein. Mice were sacrificed on days 0, 1, 2, 3 or 4 followed by isolation of bone marrow, intestine and plasma. Panel A: Western Blot analysis for increased p-RIP3 as an indicator of necroptosis (red arrow) was performed. Panel B: Densitometric normalization of p-RIP3 expression by comparison with GADPH was performed. *Onset of necroptosis.
Superior Mitigation of TBI Effects by Addition of Baicalein to JP4–039 and Necrostatin-1
We next tested the effect of the addition of baicalein to the established dual-drug regimen of JP4–039 at 24 h and necrostatin-1 at 72 h (1). Single-, dual and three-drug regimen results were then compared.
As shown in Fig. 5, there was superior survival at day 30 after TBI in mice administered JP4–039 and baicalein at 24 h and necrostatin-1 at 72 h. In these experiments, mice were maintained for 30 days to establish the LD50/30. In other experiments, mice surviving to day 30 were maintained, and all survivors at day 30 were still alive at day 100 or longer. We did not maintain animals beyond the 100-day time point due to limitations in the animal facility, including cage space.
FIG. 5.
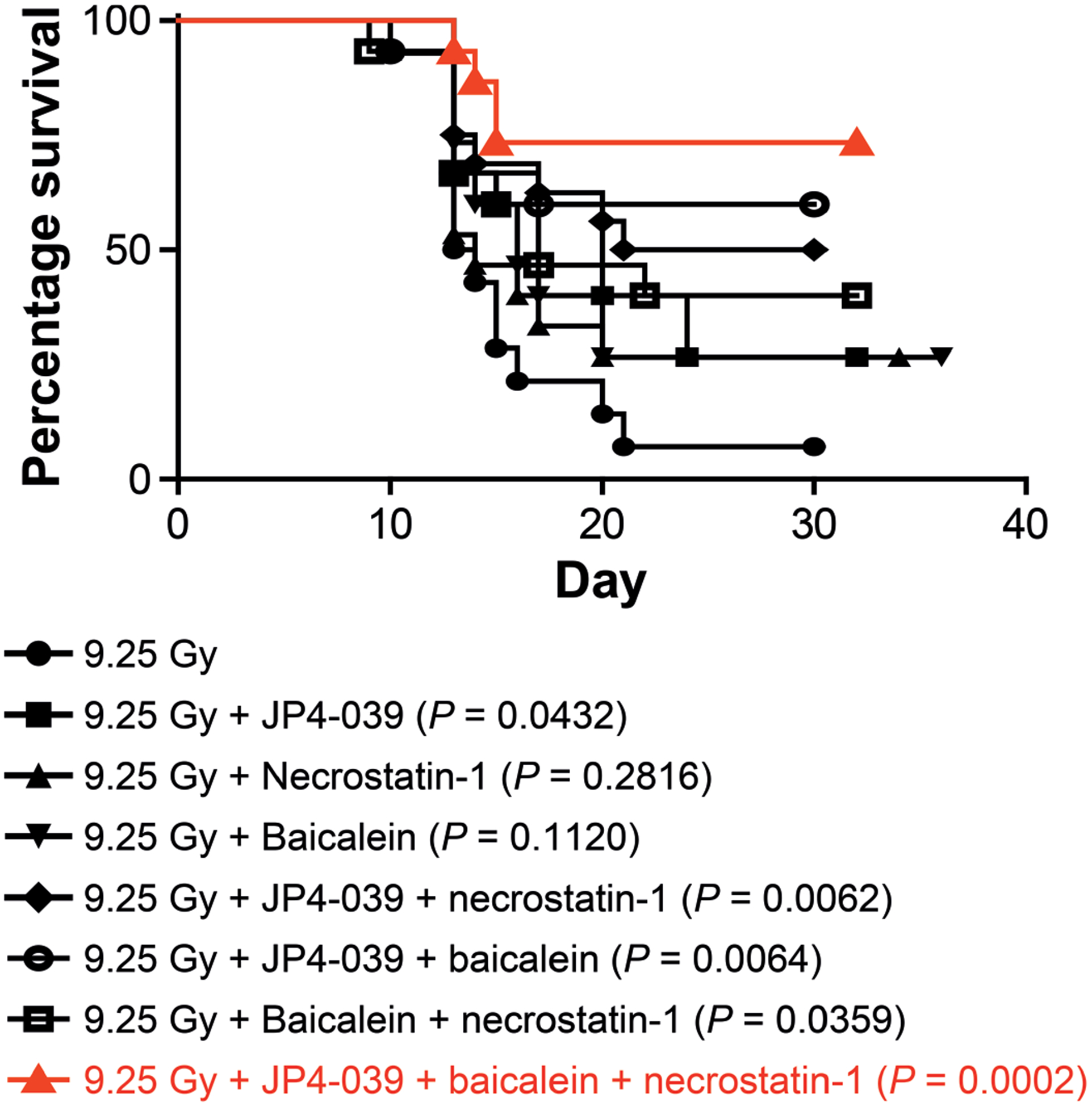
Superior mitigation by three-mitigator regimen of baicalein and JP4–039 at 24 h and necrostatin-1 at 72 h after 9.25 Gy TBI. C57BL/6NTac mice received 9.25 Gy TBI. At 24 h postirradiation subgroups of 15 mice were injected with JP4–039 (20 mg/kg), baicalein (50 mg/kg) or both. At 48 h one subgroup was injected with necrostatin-1 (1.65 mg/kg). Necrostatin-1 was given at 72 h in another subgroup if mice received JP4–039 (20 mg/kg) and baicalein (50 mg/kg) at 24 h. Mice in each group (n = 15) were monitored for development of hematopoietic syndrome, at which time the mice were sacrificed.
Tissue-Specific Differences in Radiation Mitigator-Mediated Modulation of TBI-Altered Levels of Inflammatory Cytokines and Stress Response Gene Proteins
We removed tissues from TBI mice that had been administered a single mitigator, combination of two mitigators or the superior three-drug combination. We tested tissues removed at days 1, 2, 3, 5 or 7 after TBI, and compared their levels to preirradiation levels. There were clear differences in the kinetics of increased expression of specific inflammatory cytokines and stress response gene products between tissues. The three-mitigator-drug combination had a dramatic effect on normalizing radiation-induced changes at day 7. The results for representative plasma, bone marrow, ileum and lung are shown in Figs. 6–9. Normalization by mitigators of radiation-induced elevation or suppression of inflammatory cytokine and stress response proteins was prominent in marrow and intestine, while less apparent in plasma and lung (Fig. 10). The three-mitigator-drug combination had a dramatic effect on normalizing radiation-induced damage on day 7. The presence of necrostatin-1 had the greatest consistency of effect; however, there were clearly instances where the three-drug regimen was more effective.
FIG. 10.

Relative effects of single-, dual- and three-mitigator-drug regimens on levels of 33 stress response and inflammatory cytokine proteins over 7 days after TBI. Panels A–D: Protein levels for plasma, bone marrow, intestine and lung are shown, respectively. C57BL/6NTac mice received 9.25 Gy TBI. Subgroups were injected at 24 h postirradiation with JP4–039 (20 mg/kg), baicalein (50 mg/kg) or both JP4–039 (20 mg/kg) and baicalein (50 mg/kg), at 48 h with necrostatin-1 (1.65 Gy), or JP4–039 (20 mg/kg) plus baicalein (50 mg/kg) at 24 h plus necrostatin-1 (1.65 mg/kg) at 72 h. Mice were sacrificed on days 0, 1, 2, 3, 4 or 7 (n = 5 mice/time point). From each mouse, plasma, bone marrow, intestine and lungs (panels A–D) were isolated and Luminex assay performed for inflammatory proteins. (n = 5 per time point). Ir = irradiation alone; J = JP4–039 at h; B = baicalein at 24 h; JB and JP4–039 baicalein at 24 h; N = necrostatin 1 at 48 h; JN = JP4–039 at 24 h and necrostatin-1 at 72 h; BN = baicalein at 24 h and necrostatin-1 at 72 h; JBN JP4–039 and baicalein at 24 h and necrostatin-1 at 72 h. Significant increases in gene expression compared to day 0 are indicated in red; significant decreases in gene expression compared to day 0 are indicated in green. Data for protein expression in plasma, lung, intestine and bone marrow are summarized with the mean and standard deviation for each treatment at each time point. The two-sided two-sample t test was used to compare all days of each experimental group (including the control) to day 0 (nonirradiated baseline) of the control group.
Effects of Mitigators on TBI-Induced Alterations in Plasma Proteins
The effect of single-, dual- or three-drug combinations on mitigation of radiation effects with respect to plasma proteins is shown in Figs. 6 and 10A. Representative levels of the proteins IL-3, GM-CSF and exotaxin are shown in absolute numbers as μg/ml in Fig. 6, while Fig. 10A shows relative changes. The daily changes in each correlation, and the absolute protein levels, are shown in Supplementary Figs. S1–4 and Tables S1–8, respectively (https://doi.org/10.1667/RR15486.1.S1). The effects of radiation on plasma proteins over 7 days were similar to those in the previously reported experiments (1). Radiation-induced-detectable increases in G-CSF, IL-6, KC, MCP-1, while inducing a decrease in RANTES. There was normalization by day 7, which was optimal in mice that received the three-drug regimen.
In contrast to previously published results (1), a lack of decrease in IL-13 was noted here. The effect of JP4–039 with respect to plasma levels of protein signatures was different from that of the prior study (1) (Fig. 6A).
While there was a clear radiation-induced increase in G-CSF in the current studies, there was less of an increase in exotaxin and less of an increase in KC and MIP-1 than the levels observed in previously reported studies (1). There was no detectable radiation-induced decrease in VEGF or MIP-1 in the current studies. These data may reflect genetic drift of the Taconic C57BL/6 mice after two years, and/or seasonal or diet differences in response to radiation exposure.
The results with necrostatin-1 alone with respect to plasma were also different from those previously published (1) with respect to the decrease, overall, in expression of many cytokine genes. In contrast to previously reported results (1), there were no significant decreases in exotaxin, GM-CSF, IFN-γ, IL-10, IL-12 (p40), IL-12 (p70), IL-2, IL-7, MCSF, MIP-1α, MIP-2 or TGF-β. There was a decrease in RANTES (Fig. 10A).
The results with the administration after TBI of the combination of JP4–039 and necrostatin-1 with respect to protein levels in plasma are shown in Fig. 10A. As noted elsewhere (1), necrostatin-1 alone lessened the magnitude of TBI-induced increases and decreases in proteins detected in the plasma at days 1–7. There was a significant effect of necrostatin-1 alone at day 7. The three-drug regimen returned all protein levels to near preirradiation levels, as did JP4–039 alone. Normalization of protein levels was not observed in plasma of mice treated with necrostatin-1 alone at 48 h after TBI (Fig. 10A). With baicalein alone, there were still increases in levels of G-CSF, IP-10 and KC at day 3; however, by day 7, in all proteins these had returned to preirradiation levels with the exception of a decrease in IL-2 and IL-4, and a modest remaining increase in MIP-1α. The effect of combined JP4–039 and baicalein was similar to that of JP4–039 alone with respect to normalizing protein levels by day 7.
The combination of baicalein with necrostatin-1 produced results similar to that observed with baicalein alone (Fig. 10A). Three drugs (JP4–039, baicalein, and necrostatin-1) produced a near-complete normalization of plasma protein to preirradiation levels by day 7 with the exception of a modest, continued increase in G-CSF and decrease in IL-4 (Fig. 10A).
Effect of TBI on Bone Marrow Stress Response and Inflammatory Cytokine Protein Levels, and Amelioration by Single-, Dual- or Three-Mitigator Combinations
TBI-induced elevation or decrease in levels of multiple proteins in the bone marrow compared to levels in plasma. Figure 7 shows absolute levels of representative bone marrow proteins at day 4 and 7, while Fig. 10B shows relative levels over 7 days (see also Supplementary Figs. S2 and Tables S2 and S6; https://doi.org/10.1667/RR15486.1.S1). There were significant radiation-induced changes in levels at day 4 and 7 of M-CSF, G-CSF and IL-3 (Fig. 7).
FIG. 7.
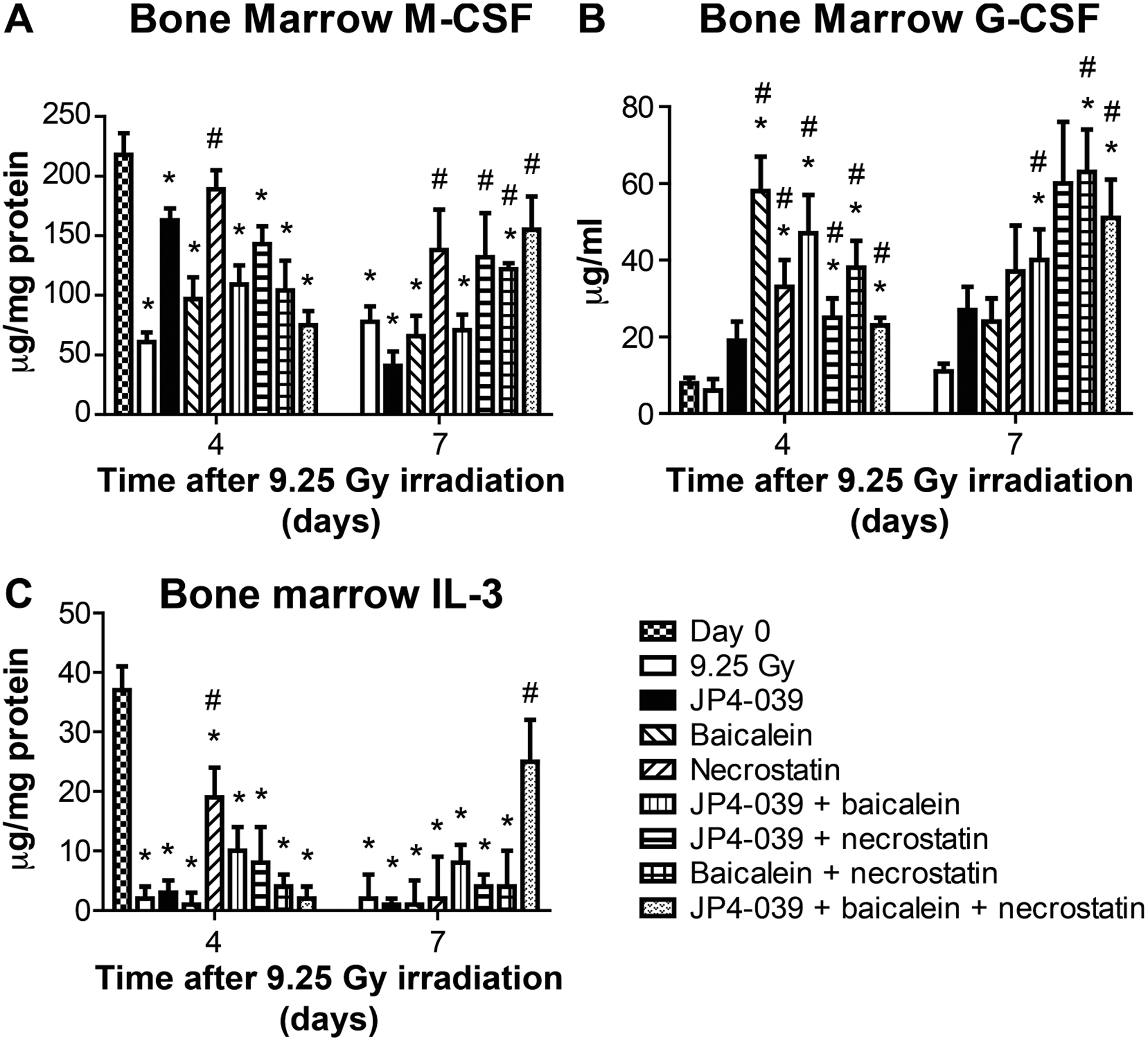
Effect of 9.25 Gy TBI and mitigator combinations on protein levels in bone marrow at days 4 and 7. Luminex assay for expression of inflammatory proteins was performed on bone marrow isolated from mice that received 9.25 Gy TBI, then injected at 24 h after TBI with JP4–039 (20 mg/kg), baicalein (50 mg/kg) or both JP4–039 and baicalein (75 mg/kg), at 48 h with necrostatin-1 (1.65 mg/kg) or JP4–039 (20 mg/kg) and baicalein (50 mg/kg) at 24 h with necrostatin-1 (1.65 mg/kg) at 72 h. Panels A–C: Protein expressions are shown for M-CSF, G-CSF and IL-3 (n = 5 mice per time point), respectively. Results are mean ± SEM. *P < 0.05 compared to day 0; #P < 0.05 compared to 9.25 Gy irradiation on same day.
The effects of mitigators with respect to modulation of bone marrow protein responses to radiation were most dramatic at day 7. The three-drug regimen was best at restoring preirradiation protein levels at day 7 with M-CSF (Fig. 7A) and IL-3 (Fig. 7C), as well as other proteins (Fig. 10B). JP4–039 suppressed radiation-induced levels of several cytokines compared to radiation alone, prominently, in MCP-1, MIG, MIP-1α, MIP-1β and RANTES (Fig. 10B). The effect of necrostatin-1 alone was different in marrow than in plasma at day 7. In marrow, levels were low, while still elevated in plasma. The combination of JP4–039 with necrostatin-1 showed that bone marrow protein levels at day 7 were restored to preirradiation levels, particularly the radiation-induced elevation in IL-10 (Fig. 10B).
With baicalein alone, bone marrow protein levels were normalized by day 7 after TBI; however, this was not as complete as that observed in mice that received the three-drug regimen.
The effects of JP4–039 and baicalein on bone marrow were similar to those of baicalein alone by day 7; however, IL-10 remained elevated. Baicalein combined with necrostatin-1 also normalized protein levels at day 7. Bone marrow results at day 7, with the three-drug regimen (JP4–039, baicalein and necrostatin-1) revealed significant normalization of protein levels, including reduction of some cytokine and stress response protein levels; however, there was still persistent elevation of some proteins, in particular IL-4, and decrease in others, particularly IL-12 (p40) (Fig. 10B).
Effect of TBI on Intestine (Ileum) Levels of Stress Response and Inflammatory Proteins and Amelioration by Single-, Dual- or Three-Mitigator Regimens
The results of the effects of mitigators on radiation-induced changes in protein levels in intestine were very different from those observed with plasma or bone marrow. Figure 8 shows absolute levels of representative proteins TNF-α, IP-10 and IFN-γ, and Fig. 10C shows relative levels (see also Supplementary Fig. S3 and Tables S3 and 7; https://doi.org/10.1667/RR15486.1.S1). A comparison of the current results of TBI alone with those in prior experiments (1) confirmed the difference between protein levels after TBI alone in intestine and those in bone marrow and plasma. Representative levels of the proteins, TNF-α, IP-10 and IFN-γ, showed that the three-drug regimen effectively reduced the persistent TBI-induced elevations in these proteins at day 7 (Fig. 10C).
FIG. 8.
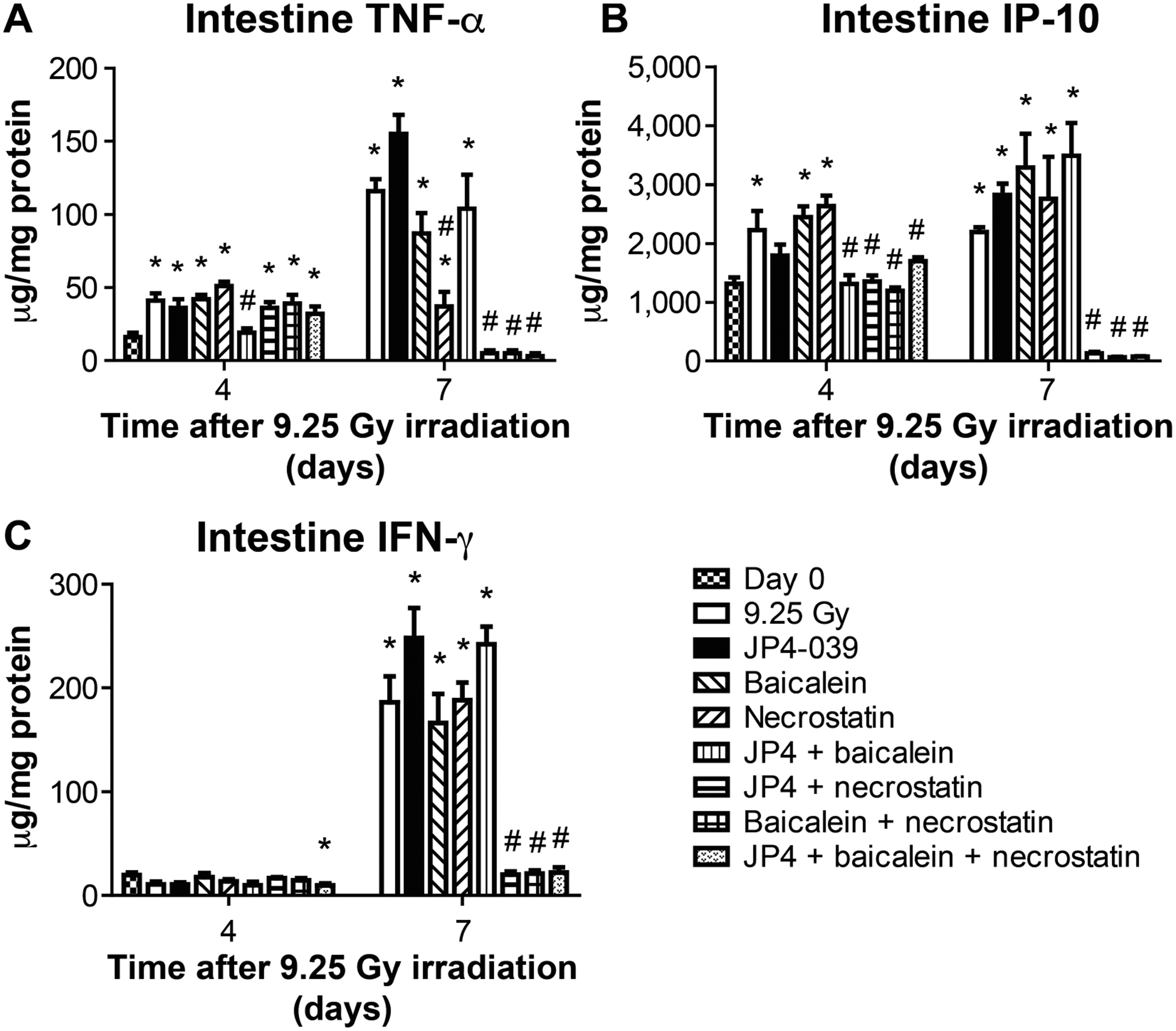
Effect of 9.25 Gy TBI and mitigator combinations on protein levels in intestine (ileum) on days 4 and 7. C57BL/6NTac mice received 9.25 Gy TBI with subgroups injected at 24 h with JP4–039 (20 mg/kg), baicalein (50 mg/kg) or both, at 48 h with necrostatin-1 (1.65 mg/kg) only, or JP4–039 (20 mg/kg), baicalein (50 mg/kg) at 24 h and necrostatin-1 at 72 h. The ileum was isolated from mice sacrificed on day 4 or 7. Luminex assay for inflammatory proteins was performed. Panels A–C: Expressions are shown for TNF-α, IP-10 and IFN-γ (n = 5 mice per time point), respectively. Results are mean ± SEM. *P < 0.05 compared to day 0; #P < 0.05 compared to 9.25 Gy irradiation on same day.
Radiation alone induced a significant increase in multiple proteins in the intestine by day 7, including: GM-CSF, IL-15, IL-3, IL-5, IL-7, IL-9, LIX, LIF, M-CSF and MIP-1β. Results at days 1–4 were similar to those reported elsewhere (12); however, in the current study upregulation of protein levels was detected early at day 4 and was persistent at day 7 in intestine. After administration of JP4–039 alone, intestinal elevation of all proteins remained high at day 7.
In contrast, necrostatin-1 administered alone at 48 h after TBI, normalized the significant elevation in multiple proteins in the intestine on day 4 with further reduction by day 7 (Fig. 10C). When JP4–039 and necrostatin-1 were given together, the results were similar to that obtained with necrostatin-1 alone.
Baicalein administered alone at 24 h after TBI showed less of an effect on intestinal protein levels at day 7 with elevation of GM-CSF, IL-12 (p40), IL-15, IL-6, IL-7, M-CSF and MIP-1β. JP4–039, baicalein alone or with JP4–039 was less effective at normalizing protein levels in the intestine compared to those mitigator regimens that included necrostatin-1 (Fig. 10C). Baicalein with either necrostatin-1 or JP4–039 was quite effective at normalizing or reducing protein levels in the intestine at day 7 compared to preirradiation levels. The three-drug combination (JP4–039, baicalein and necrostatin-1) (Fig. 10C) was also very effective at reducing the elevations of radiation-induced protein responses, and doing so rapidly at day 4.
Modest Effects of TBI on Lung Protein Levels Over 7 Days After TBI with Normalization by Mitigators
Radiation effects on the lung over 7 days after TBI included few elevations and greater suppression of protein levels than those observed with plasma, marrow or intestine. Figure 9 shows absolute levels of three representative proteins, IL-12 (p70), MCP-1 and VEGF, and Fig. 10D shows relative levels (see also Supplementary Fig. S4 and Tables S4 and S8; https://doi.org/10.1667/RR15486.1.S1). The data are consistent with the fact that the lung displays late effects rather than acute effects at 9.25 Gy TBI. The results are in contrast to the observed acute effects of radiation to the C57BL/6 mouse lung after 20 Gy subtotal-body thoracic cavity irradiation (3). In lung specimens from days 1, 2, 3, 4 and 7 after 9.25 Gy TBI, there were modest increases in G-CSF, MCP+1, KC and LIF, and decreases in MIP-1α, MIP-1β, RANTES and TGF-β.
The effect of JP4–039 delivered at 24 h after TBI on lung protein levels was modest compared to effects on bone marrow or intestine. When necrostatin-1 was delivered either alone or with JP4–039, there were also modest changes in levels of radiation-induced stress response and inflammatory cytokine proteins. Baicalein alone or in combination with JP4–039 or necrostatin-1 showed modest effects on radiation-induced protein level changes in lung. There was complete normalization by the three-drug regimen at day 7 after TBI with the exception of a modest increase in IL-5 and decrease in MIP-1b. The current results are consistent with the lung as a late responding organ to 9.25 Gy TBI, with fewer radiation-induced changes in the levels of the 33 proteins studied compared to the changes observed in bone marrow and intestine (ileum) in the same animals. The current data are in contrast to the significant acute effects of a higher 20 Gy thoracic radiation dose in the same mouse strain (2).
The possible intersections of the known signaling pathways for apoptosis, necroptosis and ferroptosis are shown in Fig. 11.
FIG. 11.
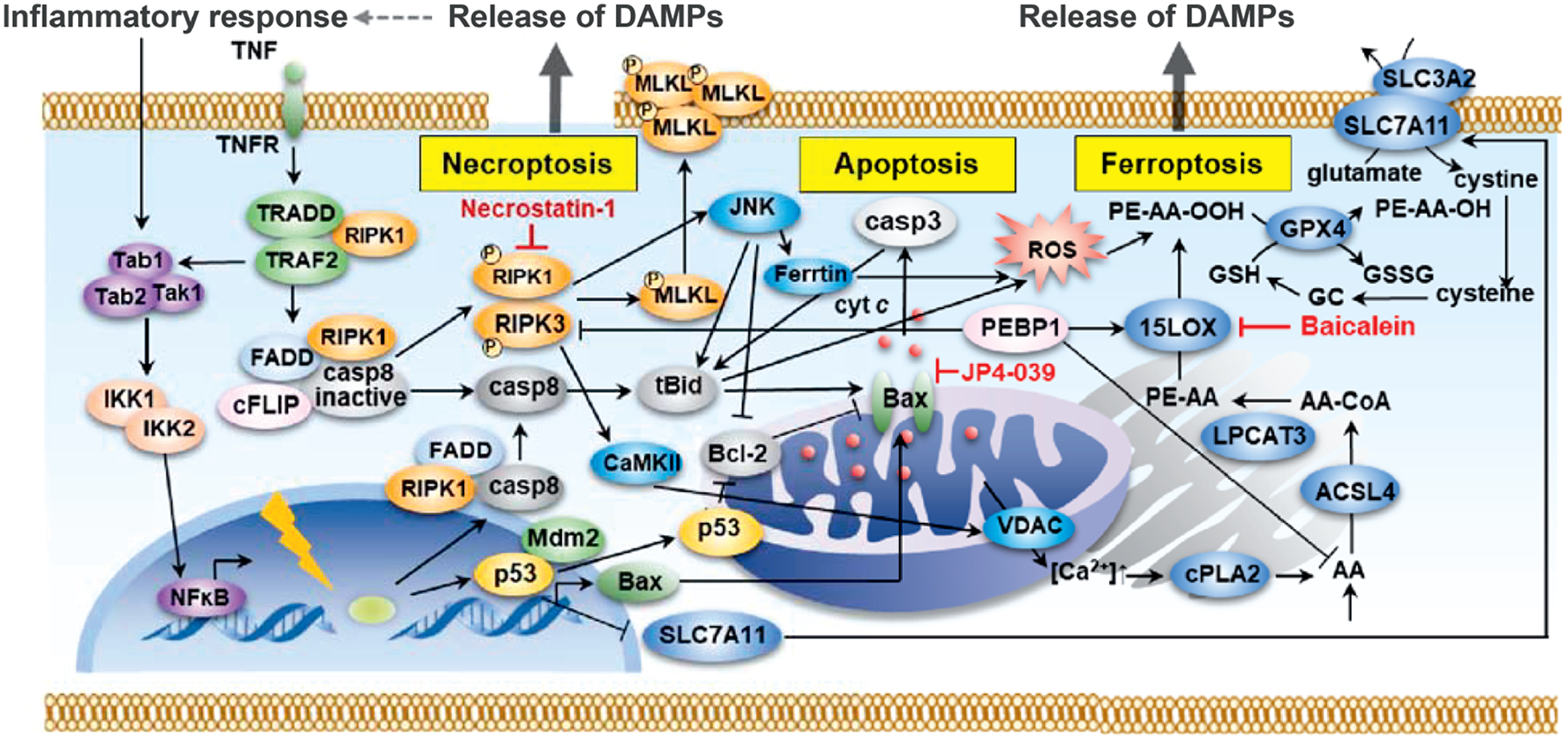
Schematic of the signaling pathway interactions in apoptosis, necroptosis and ferroptosis. Programmed cell death pathways are not independent pathways but are connected at many levels. This scheme demonstrates how the pathways are related and why blocking only one pathway may not be adequate to mitigate radiation damage.
DISCUSSION
Radiation-induced death pathways are multiple and inter related, and switch depending on failure or mitigation of the biochemical steps in one followed by takeover by another. The effect of one radiomitigator that blocks one pathway may initiate or delay the switch to another death pathway (1). Among those pathways known to be switched by the response to TBI, is that involving the phosphatidylethanolamine binding protein-1 (PEBP1), which is a regulator switching from necroptosis to ferroptosis (17).2 We reasoned that an Understanding of the mechanisms of these responses to TBI would be necessary to derive the best regimen of radio mitigators. The current results demonstrate improvement in mitigation of TBI effects by the addition of a third anti-ferroptosis drug to a regimen of two other radiation mitigators, i.e., an anti-apoptosis drug (GS-nitroxide, JP4–039), and an anti-necroptosis drug (necrostatin-1). Ferroptosis is initiated by radiation-induced depletion of glutathione peroxidase-4 (21, 22), and elevation of the enzyme acyl-coA-synthase, both of which are biomarkers (signatures) for the onset of oxidative lipid changes in the endoplasmic reticulum specific to ferroptosis (18–20). We evaluated three anti-ferroptosis drugs, ferrostatin (23), liproxstatin (24) and baicalein (25), for inclusion in a regimen of radiation mitigators. Baicalein was the most effective anti-ferroptosis mitigator when delivered after irradiation to cell lines in vitro or at 24 h after TBI in vivo. While baicalein was also effective in vivo at 48 or 72 h after TBI, the optimal effectiveness was observed at 24 h after the LD50/30 dose of 9.25 Gy to C57BL/6NTac mice.
For optimal sequencing of delivery of the three-mitigator regimen, baicalein was administered at its optimum time of 24 h after TBI along with JP4–039, which is also most effective at 24 h (1). Administration of baicalein and JP4–039 together at 24 h did not alter the delay in onset of necroptosis to 72 h after TBI, measured as p-RIP3 in lung, intestine and bone marrow. The regimen of JP4–039 and baicalein at 24 h, followed by delayed administration of necrostatin-1 at 72 h after TBI, provided improvement in survival compared to single or dual mitigators.
If different programs of cell death were always activated sequentially, and each was dependent upon the prior one, then delivery of the first mitigator, JP4–039, which suppresses apoptosis, should have been sufficient to provide the best increase in survival after TBI. If necroptosis were the second pathway to be initiated after TBI, then necrostatin-1 alone should have been sufficient to provide optimal survival if delivered at its optimal time (1). The current data reveal that TBI-induced death pathways do not work this way in sequence. Rather, we discovered that blocking one cell death program likely prompted a switch to the next pathway, as was the case with apoptosis, necroptosis and ferroptosis. An alternative explanation is that the drugs could influence PK and PD effects, i.e., the relative concentrations of each drug, and thus, efficacy would be dependent on each other’s presence in a pathway independent fashion. Since a single inhibitor of a specific death program was not sufficient, our paradigm-shifting concept further justifies the need for a time-optimized delivery of multiple mitigators of TBI.
There were three major observations regarding the effect of our optimal three-mitigator-drug regimen on inflammatory and stress response protein levels after TBI: 1. Significant increases in multiple proteins in the intestine on day 7 after TBI were greater than those observed in bone marrow, lung or plasma. 2. Administration of necrostatin-1 alone, in combination with baicalein or JP4–039, or in the three-drug combination, significantly ameliorated TBI-induced increases in intestinal proteins at day 7; in contrast, necrostatin-1 modulation of TBI effects was not observed in bone marrow, lung or plasma; and 3. Despite the dramatic normalizing effect of necrostatin-1 at day 7 after TBI in intestine, normalization of plasma protein elevations required addition of the other mitigators. Thus, radiation induced protein levels in plasma likely reflect TBI effects not restricted to bone marrow, intestine and lung, and may reflect radiation effects on other organs not analyzed in the current studies, including liver, muscle and brain.
There were modest changes in lung protein levels over 7 days after TBI, consistent with late-radiation effects detected in that organ after the 9.25 Gy TBI dose used in the current experiments. These data are in contrast to other experiments showing that a higher dose of 20 Gy to lung did induce acute effects in the same C57BL/6 mouse strain (3).
The kinetics of change observed in 33 protein levels in plasma did not correlate with biomarkers for drug effects over 7 days in intestine or marrow. Plasma protein levels did confirm optimal effectiveness of the three-drug regimen with respect to survival after TBI. Other plasma proteins, or other categories of biomarkers, including oxidized phospholipids, microRNAs, or mitochondrial or cellular DNA, may prove to be better biomarkers (signatures) of the effects of the three mitigators studied on TBI.
There were differences in TBI-induced levels of the 33 proteins studied in plasma, marrow and intestine compared to those reported elsewhere (1). The age and gender of mice (7–9 weeks, female) was the same, as was the animal supplier (Taconic Farms) and the time of day (9:00–11:00 a.m.) of irradiation. However, the current TBI experiments were performed during the winter into early spring, while prior (1) studies were performed in the autumn. Seasonal differences in TBI response may prove to be important relative to diet and the microbiome. While animal feed suppliers are required to disclose nutrient levels, the ingredients may vary over the seasons depending on grain harvests, and alter the intestinal microbiome. We are investigating these variables in an attempt to explain seasonal dose drift in the TBI LD50/30 (27), which may explain the changes in biomarker levels among experiments.
The complexity of the multiple ionizing radiation-induced cell death pathways has led to a requirement to understand the molecular biologic, as well as, cellular defenses that can modulate each cell death pathway. This expanding knowledge has led to the discovery of three small molecule mitigators targeted to each of three pathways. Further studies are needed to determine whether enhanced mitigation of TBI can be achieved by adding mitigators that target yet other cell death pathways, including parthanatos and pyroptosis (28–32).2
Supplementary Material
Table S1. Analysis of data at day 4: plasma
Table S2. Analysis of data at day 4: bone marrow.
Table S3. Analysis of data at day 4: intestine.
Table S4. Analysis of data at day 4: lung.
Fig. S1. Effect of single-, dual or three-mitigator combination on 9.25 Gy TBI-induced plasma signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.
Fig. S2. Effect of single-, dual- or three-mitigator combination on 9.25 Gy TBI-induced bone marrow signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.
Fig. S3. Effect of single-, dual- or three-mitigator combination on 9.25 Gy TBI-induced intestine (ileum) signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.
Fig. S4. Effect of single-, dual- or three-mitigator combination on 9.25 Gy TBI-induced lung signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (NIH/NIAID grant no. U19A168021) and the UPCI-Hillman Animal Research Core Facility award, no. P30CA047904.
Footnotes
Lamade AM, Huang Z, Mao G, Shrivastava I, Epperly MW, St. Croix CM, et al. Deadly duality of PEBP1: shutting off necroptosis, turning on ferroptosis. (Manuscript submitted for publication).
REFERENCES
- 1.Steinman J, Epperly M, Hou W, Willis J, Wang H, Fisher R, et al. Improves total-body irradiation survival by delivery of two radiation mitigators that target distinct cell death pathways. Radiat Res 2018; 189:68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rwigema J-CM, Beck B, Wang W, Doemling A, Epperly MW, Shields D, et al. Two strategies for the development of mitochondrial-targeted small molecule radiation damage mitigators. Int J Radiat Oncol Biol Phys 2011; 80:860–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalash R, Epperly MW, Goff J, Dixon T, Sprachman MM, Zhang X, et al. Amelioration of irradiation pulmonary fibrosis by a water soluble bi-functional sulfoxide radiation mitigator (MMS350). Radiat Res 2013; 180:474–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frantz M-C, Skoda EM, Sacher JR, Epperly MW, Goff JP, Greenberger JS, et al. Synthesis of analogs of the radiation mitigator JP4–039 and visualization of BODIPY derivatives in mitochondria. Org Biomol Chem 2013; 11: 4147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berhane H, Epperly MW, Goff J, Kalash R, Cao S, Franicola D, et al. Radiobiologic differences between bone marrow stromal and hematopoietic progenitor cell lines from Fanconi Anemia (Fancd2–/–) mice. Radiat Res 2014; 181:76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthus TS, et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem 2014; 6:542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Epperly MW, Goff JP, Franicola D, Wang H, Wipf P, Li S, et al. Esophageal radioprotection in thoracic irradiated mice with transgenic lung tumors by swallowed JP4–039/F15. In Vivo 2014; 28:435–40. [PMC free article] [PubMed] [Google Scholar]
- 8.Berhane H, Shinde A, Kalash R, Xu K, Epperly MW, Goff J, et al. Amelioration of irradiation induced oral cavity mucositis and distant bone marrow suppression in Fancd2–/– (FVB/N) mice by intraoral JP4–039/F14. Radiat Res 2014; 182:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, St. Croix C, et al. Oxidized arachidonic and adrenic Pes navigate cells to ferroptosis. Nat Chem Biol 2016; 13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinde A, Berhane H, Rhieu BH, Kalash R, Xu K, Goff J, et al. Intraoral mitochondrial-targeted GS-Nitroxide, JP4–039, radio protects normal tissue in tumor-bearing radiosensitive Fancd2–/– (C57Bl/6) mice. Radiat Res 2016; 185:134–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Z, Epperly M, Watkins SC, Greenberger JS, Kagan VE, Bayir H. Necrostatin-1 rescues mice from lethal irradiation. Biochimica at Biophysica Acta 2016; 1862:850–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christner S, Guo J, Parise RA, Ringeval M, Hoye AT, Wipf P, et al. Liquid chromatography-tandem mass spectrometric assay for the quantitation of the novel radiation protective agent and radiation mitigator JP4–039 in murine plasma. J Pharmaceut Biomed Anal 2017; S0731–7085:32605–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinman J, Epperly M, Hou W, Willis J, Wang H, Fisher R, et al. Improves total-body irradiation survival by delivery of two radiation mitigators that target distinct cell death pathways. Radiat Res 2018; 189:68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Epperly MW, Sikora C, Defilippi S, Gretton J, Zhan Q, Kufe DW, et al. Manganese superoxide dismutase (SOD2) inhibits radiation induced apoptosis by stabilization of the mitochondrial membrane. Radiat Res 2002; 157:568–77. [DOI] [PubMed] [Google Scholar]
- 15.Epperly MW, Gretton JE, Bernarding M, Nie S, Rasul G, Greenberger JS. Mitochondrial localization of copper/zinc superoxide dismutase (Cu/ZnSOD) confers radioprotective functions in vitro and in vivo. Radiat Res 2003; 160:568–78. [DOI] [PubMed] [Google Scholar]
- 16.Tyurina YY, St. Croix C, Watkins S, Epperly M, Anthonymuthu T, Kisin E, et al. Redox (phospho)lipidomics of signaling in inflammation and programmed cell death. J Leukocyte Biol 2019; 106:57–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 2016; 113:E4966–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 2017; 171:628–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuch EM, Vellaramkalayil R, Zhang I, Lehnen D, Brugger B, Sreemmel W, et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta 2014; 1841:227–39. [DOI] [PubMed] [Google Scholar]
- 20.Li LO, Klett EL, Coleman RA. Acyl-CoA synthesis, lipid metabolism, and lipotoxicity. Biochim Biophys Acta 2010; 1801:246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas JP, Maiorino M, Ursini F, Firotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J Biol Chem 1990; 265:454–61. [PubMed] [Google Scholar]
- 22.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156:317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 2018; 14:507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 2014; 136:4551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zilka O, Shah R, Li B, Friedmann AJP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci 2017; 3:232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Y, Song X, Sun X, Huang J, Zhong M, Lotze MT, et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun 2016; 473:775–80. [DOI] [PubMed] [Google Scholar]
- 27.Brand R, Epperly MW, Stottlemyer JM, Skoda E, Gao X, Li S, et al. A topical mitochondria-targeted redox cycling nitroxide mitigates oxidative stress induced skin damage. J Investigative Dermatology 2017; 137:576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Epperly MW, Wipf P, Fisher R, Franicola D, Beumer J, Li S, et al. Evaluations of different formulations and routes for the delivery of the ionizing radiation mitigator GS-nitroxide (JP4–039). In Vivo 2018; 32:1009–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dar HH, Tyurina YY, Mikulska KA, Shrivastava I, Tyurin VA, Krieger J, et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest 2018; 128:4639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sivananthan A, Shields D, Fisher R, Hou W, Zhang X, Franicola D, et al. Continuous 1 year oral administration of the radiation mitigator, MMS350, after total body irradiation restores bone marrow stromal cell proliferative capacity and reduces age-related senescence in Fanconi anemia (Fanca–/–) mice. Radiat Res 2019; 191:139–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krainz T, Du L, Lewis T, Lamade AM, Calderon M, Watkins SC, et al. Synthesis and evaluation of a mitochondria-targeting Poly(ADP-ribose) Polymerase-1 inhibitor. ACS Chem Biol 2018; 13:2868–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014; 15:135–47. [DOI] [PubMed] [Google Scholar]
- 33.Dunnett CW. A multiple comparison procedure for comparing several teatments with a control. J Am Stat Assoc 1955; 50:1096–121. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Analysis of data at day 4: plasma
Table S2. Analysis of data at day 4: bone marrow.
Table S3. Analysis of data at day 4: intestine.
Table S4. Analysis of data at day 4: lung.
Fig. S1. Effect of single-, dual or three-mitigator combination on 9.25 Gy TBI-induced plasma signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.
Fig. S2. Effect of single-, dual- or three-mitigator combination on 9.25 Gy TBI-induced bone marrow signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.
Fig. S3. Effect of single-, dual- or three-mitigator combination on 9.25 Gy TBI-induced intestine (ileum) signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.
Fig. S4. Effect of single-, dual- or three-mitigator combination on 9.25 Gy TBI-induced lung signatures of radiation damage after (panels A–H): irradiation alone; JP4–039; necrostatin-1; baicalein; JP4–039 plus necrostatin-1; baicalein plus necrostatin-1; JP4–039 plus baicalein; and three-drug regimen, respectively.