

Abstract
Harnessing metal-free photoinduced reversible-deactivation radical polymerization (photo-RDRP) in organic and aqueous phases, we report a synthetic approach to enzyme-responsive and pro-apoptotic peptide brush polymers. Thermolysin-responsive peptide based polymeric amphiphiles assembled into spherical micellar nanoparticles that undergo a morphology transition to worm-like micelles upon enzyme-triggered cleavage of coronal peptide sidechains. Moreover, pro-apoptotic polypeptide brushes show enhanced cell uptake over individual peptide chains of the same sequence, resulting in a significant increase in cytotoxicity to cancer cells. Critically, increased grafting density of pro-apoptotic peptides on brush polymers correlates with increased uptake efficiency and concurrently, cytotoxicity. The mild synthetic conditions afforded by photo-RDRP, make it possible to access well-defined peptide-based polymer bioconjugate structures with tunable bioactivity.
Keywords: Peptide Brush Polymer, Photo-RDRP, Metal-Free, Biomaterials, Nanomaterials
Graphical Abstract
Empowered by light: Photo-induced reversible deactivation radical polymerization enabled the access to a new class of bioactive peptide brush polymers. Those peptide-based brush polymers exhibited well-defined structures and tunable bioactivities, highlighting their immense potential in biomedical applications.
Introduction
The convergence of photochemistry and controlled polymerization techniques has led to the development of new living polymerization methodologies, post-polymerization modification strategies, and to the production of advanced materials.[1] In comparison with common triggers for polymerization, light has the unique advantage of providing mild reaction conditions, without the need for adding additional reactive molecules, and providing spatiotemporal control over reactions.[2–6] The toolbox of photo-induced controlled polymerization techniques is expanding, giving rise to photo-induced reversible deactivation radical polymerization (photo-RDRP),[1–6] photo-induced ring-opening metathesis polymerization (photo-ROMP),[7] photo-controlled cationic polymerization,[8] and photo-triggered ring opening polymerization (photo-ROP) of cyclic esters or N-carboxyanhydride.[9] Among these photo-induced polymerization methods, photo-RDRP techniques have received the most interest due to their broad vinyl monomer scope and relatively mild reaction conditions conducted at room temperature, in metal-free systems, and with high tolerance to oxygen and water.[10–11] We reasoned that these mild conditions should provide a route for the incorporation of peptide-modified vinyl monomers into bioactive, highly functionalized polymers, and polymeric materials. The mild conditions would minimize side reactions and thus retain the integrity of the biomolecules during polymerization and provide clean materials following polymerization. [6, 12–13]
Photo-electron transfer reversible addition-fragmentation transfer polymerization (PET-RAFT) represents a powerful tool in the arsenal of photo-RDRP approaches.[5] This technique can be performed under visible blue or green light in the presence of a biocompatible organo-photocatalyst such as eosin Y.[2] More generally, RAFT polymerization has demonstrated tolerance towards many functional groups pendent on monomers.[14] Therefore, we postulated that PET-RAFT could serve as an ideal photo-RDRP approach to explore photo polymerization of peptide-modified vinyl monomers.
The multi-valent display of peptides as side chains in brush polymers can lead to materials with enhanced biological activities such as higher binding affinities to targets and increased cell-penetration.[15–17] Examples include peptide brush polymers prepared via ROMP and atom transfer radical polymerization which involve the use of ruthenium and copper-based catalysts.[18–20] The possibility of residual heavy metals remaining after synthesis raises concerns in biomedical applications. Herein we demonstrate a metal-free photo-RDRP approach to peptide brush polymers (Figure 1). Two bioacitive peptide vinyl monomers featuring enzyme-responsive and pro-apoptotic amino acid sequences were successfully copolymerized with dimethylacrylamide (DMA) via PET-RAFT protocol in both organic and aqueous solutions. Trithiocarbornate based RAFT agents were used because the resultant polymers with terminal trithiocarbonate moeity have been demonstrated non-toxic in vitro and can be easily removed upon the polymerization.[21] Incorporation of the DMA comonomer not only lessened the steric hindrance from the peptide macromonomer, but also facillitated the preparation of peptide brush polymers with different grafting densities. Furthermore, the robust nature of PET-RAFT allowed access to various architectures including brush and linear-brush diblock copolymers consisting of enzyme-responsive peptide side chains. Linear-brush diblock copolymers self-assembled into micelles, capable of further morphing into worm-like structure upon treatment with thermolysin. By variation of the grafting density of pro-apoptotic peptide, the cellular uptake efficiency and cytotoxicity of peptide brush polymers can be controlled, revealing the crucial role of architecture (i.e., grafting density) in governing the bioactivity of polypeptide brushes. These results highlight the potential of photo-RDRP for the preparation of peptide brush polymer materials with well-defined structures and highly tunable properties in biomedical applications.
Figure 1.

Synthesis of peptide brush polymers via photo-RDRP. Two bioactive peptide vinyl monomers were designed. A thermolysin-responsive amino acid sequence GPLGLAGG, and pro-apoptotic peptide KLAKLAKKLAKLAK.
Results and Discussion
Peptide monomers containing acrylamide moieties, serving as the polymerizable group for radical polymerization were synthesized by addition of acrylic acid to an amino-hexanoic spacer unit on the N-terminus of the peptide chain (Figure 1). Two amino acid sequences were chosen to prepare two proof-of-concept systems. The first sequence GPLGLAGG, is a known substrate for various proteolytic enzymes including thermolysin.[19] The other is a sequence KLAKLAKKLAKLAK, which, when internalized, triggers apoptosis of cells by mitochondrial membrane disruption.[22] The chemical structure and purity of the peptide monomers (PepAm and KLAAm in Figure 1) were verified by high performance liquid chromatography (HPLC), 1H NMR spectroscopy, and electrospray ionization mass spectrometry (ESI-MS) (Figures S1-S5).
Due to the steric bulk of the peptide macromonomers, we reasoned that random copolymerization with a spacer monomer would enhance overall monomer conversions as well as improve control over the course of photo-RDRP. To examine this, the homopolymerization of PepAm in DMSO was first conducted (Table S1, Entry 1). According to the kinetics, no polymerization was observed after 18 hours, suggesting that steric hindrance stemming from the peptide side chains significantly hampered the photo-RDRP process. In view of this, a comonomer, dimethyl acrylamide was employed in the preparation of enzyme-responsive polypeptide brushes (Table S1, Entries 2–5). DMA was chosen due to its similar vinyl substructure (i.e., acrylamide) in comparison with the peptide monomer (Figure 2).
Figure 2.

(a) Photo-RDRP of PepAm and DMA in DMSO; (b) Kinetic plot of monomer concentrations versus time for PepAm and DMA over the course of photo-RDRP; (c) GPC traces of enzyme-responsive peptide brush polymers (P1-P4, table S1) with different grafting densities. P1-P4 represent poly(PepAm6-co-DMA7), poly(PepAm15-co-DMA45), poly(PepAm34-co-DMA117), and poly(PepAm21-co-DMA71), respectively.
To understand the composition and distribution of PepAm and DMA in the polymers, 1H NMR spectroscopy was used to study the rate of polymerization and conversion of the two monomers in photo-RDRP under blue LED (Table S1, entry 4, Figure S6). According to 1H NMR analysis (Figure 2b and Figure S7), both the peptide monomer and DMA comonomer had similar propagation rates, indicative of a statistical distribution of peptide monomers along the polymer backbone. Gel permeation chromatography (GPC) analysis showed a narrow molecular weight distribution for all polypeptide brushes with different grafting densities (Figure 2c, table S1). Moreover, theoretical molecular weights of all polymers are on par with those from GPC results, suggesting good control over the photo polymerizations. While full monomer conversions were not achieved, residual PepAm and DMA monomers were effectively removed by dialysis of the crude polymer mixtures in water, as confirmed by its disappearance in the GPC trace (Figure S8). We note, natural sun-light was also effective in triggering the photo-polymerization of PepAm, leading to well-defined polypeptide brushes (Figures S9 and S10).
To examine the bio-activity of polypeptide brushes, polymer poly(PepAm21-co-DMA71) (P4) was treated with thermolysin, an enzyme that can selectively cleave the amide bond between glycine (G) and leucine (L) (Figure 3). HPLC was employed to monitor the cleavage reaction, showing that the polypeptide brushes were rapidly cleaved within 1 hour under the investigated conditions (Figures S11). The cleaved peptide fragment was further analyzed by ESI-MS and shown to have an identical mass to that of the genuine synthetic cleavage fragment LAGG (Figure S12). These results clearly indicate that the side-chain peptides remain accessible and reactive towards enzyme cleavage following polymerization. This is counter to our previous observations of highly dense peptide brushes generated using ring-opening metathesis polymerization, where peptides can be made entirely resistant to aggressive proteolytic treatments.[15] The different activity of peptide brush polymers to enzyme digestion likely stems from the structural variation in polymer backbones prepared by photo-RDRP and ROMP. In comparison with rigid polynorbornene backbone containing sp2 hybridized carbon-carbon double bonds, vinyl polymer backbone is more flexible and hence increases the accessibility of side chain peptides to surrounding enzymes. Moreover, the radical approach (i.e., photo-RDRP) to peptide brush polymers was achieved via copolymerization of peptide vinyl monomers and spacer monomers, resulting in random copolymers with a lower grafting density than that of polynorbornene-type polymers, which were synthesized by homopolymerization of norbornene modified peptide monomers.
Figure 3.

(a) Schematic of thermolysin-promoted cleavage of poly(PepAm21-co-DMA71) (P4); (b) TEM micrograph of PMMA90-b-poly(PepAm21-co-DMA63) based micelles (P8) before treatment with thermolysin; (c) TEM micrograph of PMMA90-b-poly(PepAm21-co-DMA63) based micelles (P8) after treatment with thermolysin; (d) DLS traces of P8 based nano-objects before and after thermolysin-induced cleavage.
To capitalize on this accessibility to substrate, amphiphilic block copolymers were prepared (P7-P9, Table S2) by chain extension of poly(methyl methacrylate) or poly(n-butyl acrylate) based macro chain transfer agents with PepAm and DMA (Scheme S1, Figures S13 –S18). The resulting amphiphilic diblock copolymers assembled into micelles in water. For example, PMMA90-b-poly(PepAm21-co-DMA63) (P8, table S2) were spherical micelles, 24 nm in diameter as characterized by transmission electron microscope (TEM), in good agreement with the hydrodynamic diameter (28 nm) determined by dynamic light scattering (DLS) (Figures 3b and 3d).
The critical packing parameter (CPP) dictates the thermodynamic morphology of amphiphilic block copolymers. In principle, a higher CPP (> 1/3) can lead to the formation of higher order morphologies such as cylinders and bilayer vesicles.[23] Since polypeptide brush polymer P4 showed rapid cleavage in the presence of thermolysin (vide supra), we expected that the micelles formed from polypeptide containing diblock polymers could respond to thermolysin, resulting in truncation of the hydrophilic polypeptide corona and thus a reduction in interfacial curvature (i.e., increment in CPP), leading to a change in morphology. Indeed, TEM and DLS showed that the spherical micellar structure of P8 underwent a phase transition into a worm-like phase upon treatment with thermolysin (Figures 3c and 3d). By contrast, no change in diameter was observed for particles treated under the same cleavage conditions using deactivated thermolysin which had been pretreated with ethylenediaminetetraacetic acid (Figure 3d). In addition, similar morphological transformations were observed in other block copolymer micelles including PMMA90-b-poly(PepAm9-co-DMA30) (P7) and PnBA200-b-poly(PepAm36-co-DMA123) (P9), demonstrating the versatility of this approach to enzyme-responsive shape-shifting nanoparticles (Figures S19–21).
The ability to conduct polymerizations directly in water is of significant interest to the field of biomedical polymer materials, as it not only avoids the use of toxic organic solvents but also eliminates the time-consuming step of transferring the polymeric materials from the organic to aqueous phase. To explore the feasibility of aqueous photo-RDRP of peptide monomers, we examined the photo-RDRP of both enzyme-responsive peptide acrylamide (PepAm) and pro-apoptotic KLA peptide acrylamide (KLAAm) in water. Table S3 summarizes the polymerization results for PepAm and DMA, indicating dramatically higher monomer conversions in water compared to those obtained by photo-polymerizations in DMSO (Table S1). We hypothesize that this is due to the hydrogen bonding between the amide carbonyl groups with water molecules, leading to enhanced solubility of PepAm in aqueous solution.[24] Polymers (P10-P12) were analyzed by GPC and NMR (Figures S22-S24) and showed molecular weights that were in good agreement with theoretical values, confirming that photo-RDRP of PepAm was unaffected under aqueous conditions.
For the KLAAm monomer, which contains abundant amine groups, acetate buffer (pH 5) was utilized to fully protonate the amine groups, reducing their nucleophilicity (pKa = 9) and thus precluding undesired aminolysis of the chain transfer agents (Figure 4). By adjusting the feed ratio of DMA to KLAAm, polypeptide brushes with various grafting densities of KLA side chains were prepared (Table S4, P13-P16). Based on NMR analysis, monomer conversions were quantitative for all random copolymerizations of DMA and KLAAm (Figures S25 and S26). However, the homopolymerization of KLAAm led to a modest monomer conversion (40%) possibly due to the steric bulk of the KLA peptide macromonomer. The narrow and symmetric GPC traces of KLA based polypeptide brushes indicates good control over the aqueous photo-RDRP of KLAAm (Figure 4b, Figures S27–28). Furthermore, the secondary structure of KLA peptides and brush polymers were assessed by circular dichroism (CD) spectroscopy, which showed a mixture of α-helix and random coil. The CD spectra of the KLA monomer and resulting polymers are identical, suggesting the polymerization process does not alter the secondary structure of the peptide (Figure 4c).
Figure 4.

(a) Aqueous photo-RDRP of KLAAm and DMA in acidic buffer (pH 5); (b) GPC traces of KLA based peptide brush polymers with different grafting densities (P13-P16, table S4); (C) Circular dichroism spectra of KLA peptide, KLAAm, and representative polymer: poly(KLAAm25-co-DMA25).
The KLA peptide sequence used in these studies is a known pro-apoptotic peptide which is capable of inducing cell apoptosis via disruption of mitochondrial membranes.[25] It is typically fused with a cell-penetrating peptide because of its otherwise poor cellular uptake.[26] The KLA based brush polymers, while lacking a cell-penetrating peptide moiety, collectively possess a number of cationic charges when polymerized, which could enhance the affinity to the negatively charged cell membrane and consequently promote delivery of the KLA based polymer brushes into cells.[16] To elucidate the role of KLA grafting densities on the cellular uptake and cytotoxicity of KLA brush polymers, we conducted in vitro cell studies of different brush polymers (P13-P16, table S4) in HeLa cells (Figures 5–7). Flow cytometry was used to quantify the cell uptake efficiency of brush polymers. All KLA peptide brush polymers show significantly more cellular uptake compared to the free peptide (Figure 5). This observation is consistent with the multivalency of KLA peptides organized as polypeptide brushes, which enhanced the affinity and cell uptake of the KLA containing materials. In addition, cell internalization of densely grafted polypeptides including poly(KLAAm25-co-DMA25) and poly(KLAAm10) clearly outperformed more sparsely grafted polypeptide brushes such as poly(KLAAm25-co-DMA75) (Figures 5 and S29).
Figure 5.

Flow cytometry analysis (λex/em = 548/570 nm) of HeLa cells incubated with rhodamine B-labeled KLA peptide (a), poly(KLAAM25-co-DMA75) (b), poly(KLAAM25-co-DMA25) (c), and poly(KLAAM10) (d) at a concentration of 0.25 μM with respect to the dye. Chemical structures of each dye-labeled materials are shown adjacent to the corresponding histogram. KLA based peptide brush polymers possessed markedly higher cell uptake ability than that of KLA peptide.
Figure 7.
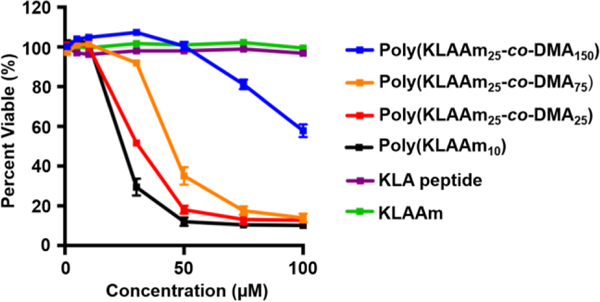
Cell viability assay of KLA peptide, KLAAm, and a library of KLA based peptide brush polymers with different grafting densities. Hella cells were treated with peptide based materials and incubated for 72 hours at 37°C (CellTilter-Blue assay, n = 3 independent experiments with three independent samples in each). KLA peptide and KLAAm did not exhibit cytotoxicity to Hela Cells even at a concentration of 100 μM. The IC50 value of peptide brush polymers decreased as the grafting density of peptide brush polymer increased, indicating a higher cytotoxicity of KLA peptide brush polymers with a more compact brush architecture.
The cellular uptake behavior of KLA peptide and polymer brushes were further studied by confocal laser scanning microscopy (CLSM). Cells treated with rhodamine labeled KLA peptide showed no uptake even at a high concentration (50 μM with respect to peptide). On the other hand, the cellular uptake of all KLA peptide brush polymers was clearly visible at the same peptide concentration, as evidenced by the increase in rhodamine fluorescence in HeLa cells (Figures 6 and S30). Finally, cytotoxicity assays demonstrated that KLA based polymer brushes had significantly higher cytotoxicity than either the free KLA peptide or the KLAAm monomer (Figure 7). Notably, the cytotoxicity of polypeptide brushes was dependent on the grafting density of KLA peptides. As the grafting density of KLA peptide increased and the polymer brushes became more compact, the half-maximum inhibitory concentration IC50 values decreased. These cytotoxicity results are consistent with the observed cellular uptake behavior of KLA brush polymers, further demonstrating the role of grafting density on the material properties.
Figure 6.

Confocal laser scanning microscopy images of Hela cells treated with rhodamine-labeled peptide based materials at a concentration of 0.25 μM with respect to rhodamine B (λex/em = 548/570 nm). From top to bottom: KLA peptide (a-c), poly(KLAAm25-co-DMA75) (d-f), poly(KLAAm25-co-DMA25) (g-i), and poly(KLAAm10) (j-l). Cell nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI, λex/em = 360/460 nm). Cell membrane was stained with wheat germ agglutinin, Alexa Fluor 488 conjugate (WGA 488, λex/em = 495/519 nm). Scale bar: 20 μm, inset scale bar 10 μm.
Conclusion
In summary, we present examples of photo-RDRP of peptide acrylamide monomers. This is a robust synthetic approach to prepare bioactive polypeptide brushes under mild conditions using visible light, in aqueous solution, and at room temperature. We envision that a wide variety of other functional peptide monomers such as therapeutic and cell-penetrating peptides will be compatible with this technique. Moreover, we demonstrated the important role that the architecture (i.e., grafting density) of peptide brush polymers has on function such as cell penetration and cytotoxicity towards cancer cells. Given the widespread interest in peptides as therapeutics and targeting moieties in biomedicine, we envision these mild synthetic procedures will open the door to entirely new peptide brush polymer biomaterials.
Experimental Section
Preparation of Peptide Vinyl Monomers via Solid-Phase Peptide Synthesis (SPPS):
peptides were synthesized on Rink resins (0.67 mmol/g) using standard FMOC SPPS procedures on an AAPPTec Focus XC automated synthesizer. A typical SPPS procedure included deprotection of the N-terminal Fmoc group with 20 % 4-methyl-piperidine in DMF (1 × 20 min, followed by 1 × 5 min), and 30 min amide couplings (twice) using 3.0 equiv. of the Fmoc-protected amino acid, 2.9 equiv. of HBTU and 6.0 equiv. of DIPEA. After that, peptide acrylamide monomers were prepared by amide coupling to Fmoc-6-aminohexanoic acid, followed by Fmoc deprotection and final amidation with acrylic acid (3 equiv.) in the presence of HBTU (2.9 equiv.), and DIPEA (6.0 equiv.). The crude peptide monomers were obtained by cleavage from the resins and further purified by preparative HPLC.
Aqueous photo-RDRP of peptide acrylamide monomers:
In a typical aqueous photoinduced polymerization (P14), KLA peptide (KLAKLAKKLAKLAK) acrylamide monomer (30 mg, 25 equiv.) and DMA (1.8 mg, 25 equiv.) were dissolved in 150 μL of acetate buffer (0.1 M, pH 5). Then 10 μL (1.0 equiv.) of water-soluble RAFT agent stock solution (2.2 mg in 100 μL of acetate buffer) was added into the reaction mixture. Following that, 10 μL (0.05 equiv.) of eosin Y disodium salt stock solution (2.5 mg in 1 mL of acetate buffer) and PMDETA (0.12 mg, 1.0 equiv.) were added. The solution was degassed by N2 flow for 30 min and then placed into the photo-reactor (450 nm, 2.8 mW/cm2) for 24 h. Upon the polymerization, the polymer product was purified by dialysis into DIW, followed by lyophilization.
Supplementary Material
Acknowledgements
The authors are grateful for the support of an NIH Common Fund, the NHLBI (R01HL139001), and a National Science Foundation fund (DMR-1710105). C.B. acknowledges the Swiss National Science Foundation (SNSF) for a postdoctoral fellowship.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Corrigan N, Yeow J, Judzewitsch P, Xu JT, Boyer C, Angew. Chem. Int. Ed. 2019, 58, 5170–5189; Angew. Chem. 2019, 131, 5224–5243; [DOI] [PubMed] [Google Scholar]; b) Xu JT, Jung K, Atme A, Shanmugam S, Boyer C, J. Am. Chem. Soc. 2014, 136, 5508–5519; [DOI] [PubMed] [Google Scholar]; c) Chen M, Zhong MJ, Johnson JA, Chem. Rev. 2016, 116, 10167–10211; [DOI] [PubMed] [Google Scholar]; d) Sun H, Kabb CP, Sims MB, Sumerlin BS, Prog. Polym. Sci. 2019, 89, 61–75; [Google Scholar]; e) Molla MR, Rangadurai P, Antony L, Swaminathan S, de Pablo JJ, Thayumanavan S, Nat. Chem. 2018, 10, 659–666; [DOI] [PubMed] [Google Scholar]; f) Yang L, Sun H, Liu Y, Hou WJ, Yang Y, Cai R, Cui C, Zhang PH, Pan XS, Li XW, Li L, Sumerlin BS, Tan WH, Angew. Chem. Int. Ed. 2018, 57, 17048–17052; Angew. Chem. 2018, 130, 17294–17298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Niu J, Lunn DJ, Pusuluri A, Yoo JI, O’Malley MA, Mitragotri S, Soh HT, Hawker CJ, Nat. Chem. 2017, 9, 537–545. [DOI] [PubMed] [Google Scholar]
- [3].a) Pan XC, Lathwal S, Mack S, Yan JJ, Das SR, Matyjaszewski K, Angew. Chem. Int. Ed. 2017, 56, 2740–2743; Angew. Chem. 2017, 129, 2784–2787; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pan XC, Fang C, Fantin M, Malhotra N, So WY, Peteanu LA, Isse AA, Gennaro A, Liu P, Matyjaszewskit K, J. Am. Chem. Soc. 2016, 138, 2411–2425. [DOI] [PubMed] [Google Scholar]
- [4].Carmean RN, Becker TE, Sims MB, Sumerlin BS, Chem 2017, 2, 93–101. [Google Scholar]
- [5].Xu JT, Shanmugam S, Fu CK, Aguey-Zinsou KF, Boyer C, J. Am. Chem. Soc. 2016, 138, 3094–3106. [DOI] [PubMed] [Google Scholar]
- [6].Tan JB, Sun H, Yu MG, Sumerlin BS, Zhang L, ACS Macro Lett. 2015, 4, 1249–1253. [DOI] [PubMed] [Google Scholar]
- [7].Ogawa KA, Goetz AE, Boydston AJ, J. Am. Chem. Soc. 2015, 137, 1400–1403. [DOI] [PubMed] [Google Scholar]
- [8].a) Kottisch V, Supej MJ, Fors BP, Angew. Chem. Int. Ed. 2018, 57, 8260–8264; Angew. Chem. 2018, 130, 8392–8396; [DOI] [PubMed] [Google Scholar]; b) Michaudel Q, Kottisch V, Fors BP, Angew. Chem. Int. Ed. 2017, 56, 9670–9679; Angew. Chem. 2017, 129, 9798–9808; [DOI] [PubMed] [Google Scholar]; c) Kottisch V, Michaudel Q, Fors BP, J. Am. Chem. Soc. 2016, 138, 15535–15538. [DOI] [PubMed] [Google Scholar]
- [9].a) Eisenreich F, Kat han M, Dallmann A, Ihrig SP, Schwaar T, Schmidt BM, Hecht S, Nat. Catal. 2018, 1, 516–522; [Google Scholar]; b) Fu CK, Xu JT, Boyer C, Chem. Commun. 2016, 52, 7126–7129. [DOI] [PubMed] [Google Scholar]
- [10].Ng G, Yeow J, Chapman R, Isahak N, Wolyetang E, Cooper-White JJ, Boyer C, Macromolecules 2018, 51, 7600–7607. [Google Scholar]
- [11].Yang QZ, Lalevee J, Poly J, Macromolecules 2016, 49, 7653–7666. [Google Scholar]
- [12].Varlas S, Blackman LD, Findlay HE, Reading E, Booth PJ, Gibson MI, O’Reilly RK, Macromolecules 2018, 51, 6190–6201. [Google Scholar]
- [13].Blackman LD, Varlas S, Arno MC, Houston ZH, Fletcher NL, Thurecht KJ, Hasan M, Gibson MI, O’Reilly RK, ACS Central Sci. 2018, 4, 718–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hill MR, Carmean RN, Sumerlin BS, Macromolecules 2015, 48, 5459–5469. [Google Scholar]
- [15].Blum AP, Kammeyer JK, Yin J, Crystal DT, Rush AM, Gilson MK, Gianneschi NC, J. Am. Chem. Soc. 2014, 136, 15422–15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Blum AP, Kammeyer JK, Gianneschi NC, Chem. Sci. 2016, 7, 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Maynard HD, Okada SY, Grubbs RH, J. Am. Chem. Soc. 2001, 123, 1275–1279. [DOI] [PubMed] [Google Scholar]
- [18].Ayres L, Vos MRJ, Adams PJHM, Shklyarevskiy IO, van Hest JCM, Macromolecules 2003, 36, 5967–5973. [Google Scholar]
- [19].Adamiak L, Touve MA, LeGuyader CLM, Gianneschi NC, ACS Nano 2017, 11, 9877–9888. [DOI] [PubMed] [Google Scholar]
- [20].Conrad RM, Grubbs RH, Angew. Chem. Int. Ed. 2009, 48, 8328–8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fairbanks BD, Gunatillake PA, Meagher L, Adv. Drug Deliver. Rev. 2015, 91, 141–152. [DOI] [PubMed] [Google Scholar]
- [22].Cieslewicz M, Tang JJ, Yu JL, Cao H, Zavaljevski M, Motoyama K, Lieber A, Raines EW, Pun SH, Proc. Nat.l. Acad. Sci. USA 2013, 110, 15919–15924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Doncom KEB, Blackman LD, Wright DB, Gibson MI, O’Reilly RK, Chem. Soc. Rev. 2017, 46, 4119–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Valdebenito A, Encinas MV, Polym. Int. 2010, 59, 1246–1251. [Google Scholar]
- [25].Agemy L, Friedmann-Morvinski D, Kotamraju VR, Roth L, Sugahara KN, Girard OM, Mattrey RF, Verma IM, Ruoslahti E, Proc. Natl. Acad. Sci. USA 2011, 108, 17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fantin VR, Berardi MJ, Babbe H, Michelman MV, Manning CM, Leder P, Cancer Res. 2005, 65, 6891–6900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.