

Abstract
The involvement of complement in B2 cell responses has been regarded as occurring strictly via complement components in plasma. Herein, we show that antibody (Ab) production and class switch recombination (CSR) depend on autocrine C3a and C5a receptor (C3ar1/C5ar1) signaling in B2 cells. CD40 upregulation, IL-6 production, growth in response to BAFF or APRIL, and AID/Bcl-6 expression, as well as follicular CD4+ cell CD21 production all depended on this signal transduction. Ova immunization of C3ar1−/−C5ar1−/− mice elicited IgM Ab but no other isotypes, whereas decay accelerating factor (Daf1)−/− mice, elicited more robust IgM Ab production and CSR than WTs. Comparable differences occurred in ova immunized muMT recipients of WT, C3ar1−/−C5ar1−/−, and Daf1−/− B2 cells, and in HEL immunized muMT recipients of MD4 B2 cells on each genetic background. B2 cells produced factor-I and C3, and auto-phosphorylated CD19, and immunized C3−/−C5−/− recipients of WT MD4 BM efficiently produced Ab. Thus, B2 cell produced complement participates in B2 cell activation.
INTRODUCTION
While complement participates in B2 cell responses, current concepts are that it does so via plasma derived complement proteins that are activated after antigen (Ag) engagement of the B cell receptor (BCR). Complement initially was regarded solely as serving as an effector system for IgM and complement fixing IgG isotypes (Owen et al., 2012). A large body of data over many years however has shown that it functions on B2 cells themselves (Fearon and Carroll, 2000). This work has shown that it is integrally involved in B2 cell costimulation as well as in class switch recombination (CSR) after IgM antibody (Ab) is produced.
The classical view of complement’s role in the B2 cell response is as follows: B2 cell costimulation occurs as a result of ligation of B2 cell expressed CD21 [complement receptor 2 (CR2) which induces phosphorylation of closely associated CD19. C3dg is the ligand for CD21. It is generated from C3b that covalently associates with IgM Ab-antigen complexes (Ag-Ab) comprised of the BCR and the cognate Ag that triggers its activation. C3b covalently incorporates into both the Ab and Ag (Takahashi et al., 1977; Takahashi et al., 1978). Current concepts (Fearon and Carroll, 2000) are that the incorporated C3b derives from plasma C3 and that its uptake in the Ag-Ab occurs close to the B2 cell surface or after release of the Ag-Ab from the B2 cells. C3b in the Ag-Ab-C3b complex is cleaved to C3dg by the enzyme factor I yielding Ag-Ab-C3dg. This cleavage is thought to be mediated by factor I which circulates in plasma. For Ag-Ab C3b near the B2 cell surface, CD35 [complement receptor 1 (CR1)] expressed on B2 cells themselves can serve as the obligate cofactor for the factor I conversion of C3b to C3dg (Iida and Nussenzweig, 1983; Medof et al., 1982a). For Ag-Ab-C3b released from the B2 cells or that form remotely and enter the circulatory system, CD35 on erythrocytes (E) can serve as the obligate cofactor (Medof et al., 1982a; Medof and Nussenzweig, 1984; Medof et al., 1982b). CD19 phosphorylation that is evoked by C3dg ligation of CD21 enables the binding activation of PI-3Kα on CD19. The activated PI-3Kα then coordinately signals together with downstream signaling intermediates of the activated BCR to promote B2 cell activation and Ab secretion. Co-ligation of the BCR and CD21 increases B2 cell activation 10–1000-fold (Fearon and Carroll, 2000). Ag and C3dg in the same Ag-Ab-C3dg complexes can simultaneously ligate the BCR and CD21 respectively, to augment Ab production (Carter and Fearon, 1992) and promote CSR (Owen et al., 2012). While these findings implicate complement quantitatively as well as qualitatively in the Ab response, both have been presumed to derive from liver-produced complement proteins in plasma. Complement has not been directly implicated in B2 cell processes that precede IgM Ab secretion and CSR.
B2 cell activation that primes Ab production against most polypeptide antigens requires CD4+ cell help (Owen et al., 2012). The coupling of activation-induced CD40 ligand (CD40L) on cognate CD4+ cells to B2 cell expressed CD40 is an essential process in this help. This engagement in conjunction with Ag specific BCR stimulation induces B2 cell proliferation. It also induces expression of activation-induced cytidine deaminase (AID) and B-cell lymphoma 6 (Bcl-6), two proteins that enable CSR and affinity maturation (AFM) of the Ab variable sequence. Much literature (Hoshino et al., 2000; Litinskiy et al., 2002; Snapper et al., 1992) has implicated CD4+ cell produced cytokines in regulating CSR. Among these cytokines are IL-21, IFN-ɣ, IL-4, IL-5, and TGF-β (Owen et al., 2012). Past work has implicated Bcl-6 in directing B2 cells into splenic and lymph node (LN) germinal centers where they can undergo AFM rather than differentiate to plasma cell commitment (Owen et al., 2012).
CD4+ cell expression of CD40L that confers B2 cell help is activation induced (Owen et al., 2012). Our prior studies of CD4+ cell activation (Liu et al., 2008; Strainic et al., 2008), found that CD40L upregulation depends on endogenous C3a and C5a generation and autocrine C3a and C5a receptor (C3ar1/C5ar1) signaling in CD4+ cells during their activation by primed antigen presenting cells [APCs, e.g. dendritic cells (DCs)] (Lalli et al., 2008; Strainic et al., 2008). That work showed that the intensity of this G protein coupled receptor (GPCR) signaling is governed by the cell-associated regulator, decay accelerating factor (DAF or CD55) which controls the generation of C3a/C5a from locally produced C3/C5. C3ar1/C5ar1 signaling operates tonically in CD4+ cells in low levels to sustain viability. Amplified C3ar1/C5ar1 signaling occurring in concert with lifted DAF restraint on C3ar1/C5ar1 signaling potentiates T effector cell (Teff) responses, whereas repressed C3ar1/C5ar1 signaling occurring in concert with upregulated DAF expression suppresses Teff responses. Recent studies by others documented the critical role of immune cell produced complement in shaping the Teff response and extended the above findings by showing that C3 cleavage and signaling operates intracellularly in human CD4+ cells (Arbore et al., 2016; Liszewski et al., 2013).
In this study, we investigated whether autocrine C3ar1/C5ar1 signaling functions endogenously in B2 cells and whether this signaling is involved in B2 cell processes upstream of Ab production and is needed for CSR. Our experiments found that C3ar1/C5ar1 signaling is required for B2 cell homeostasis as well as for multiple steps in B2 cell activation, i.e. CD40 upregulation, initial B2 cell proliferation, plus AID and Bcl-6 expression needed for CSR and AFM. Importantly, we further found that autocrine C3ar1/C5ar1 signaling in B2 cells leads to B2 cell synthesis of C3 and factor I, as well as endogenous phosphorylation of CD19 that provides B2 cell costimulation (Owen et al., 2012). The data thus argue that endogenously produced complement by B2 cells participates in many upstream steps in B2 cell activation including CD21 dependent amplification of Ab production and CSR. The findings thus provide important new insights regarding the interconnection of complement with critical processes in B2 cells not previously recognized.
RESULTS
Ab production and CSR depend on C3ar1/C5ar1 signaling
As an initial test of whether autocrine C3ar1/C5ar1 signaling is involved in B2 cell responses, we immunized C3ar1−/−C5ar1−/− and WT C57BL/6 mice with ovalbumin (ova) in IFA and compared the Ab responses. We drew blood samples on d 8, 13, and 21 post-immunization, boosted the mice on d 23, and drew samples 7 d post boost (d 30) (diagrammed in Fig 1A). By ELISA, overall ova-specific Ig production was ~80% attenuated in C3ar1−/−C5ar1−/− mice compared to that in WTs (Fig 1B). Moreover, C3ar1−/−C5ar1−/− mice produced only ova-specific IgM Ab (Fig 1C) which did not convert into other isotypes (IgG1 and IgG2b in C3ar1−/−C5ar1−/− vs WT = ___and __) irrespective of longer times (data not shown). Identical ova immunization of Daf1−/− mice, in which autocrine C3ar1/C5ar1 signaling is potentiated, conversely showed heightened anti-ova total IgG production (Fig 1D). Notably, the immunized Daf1−/− mice showed a more rapid (>5-fold at d 7) increase in overall Ig as well as increased class switching (70–100%) to IgG1 and to complement fixing IgG2b and presumably IgG2c, the C57BL/6 allelic variant of IgG2a cross reactive with anti-IgG2a Ab (Fig 1E). A repeat experiment (5 mice each group) gave comparable results. These findings pointed to C3ar1/C5ar1 signaling participating in both initial B2 cell activation and CSR.
Figure 1. Ab production by mice deficient in C3ar1/C5ar1 is impaired.

WT, C3ar1−/−C5ar1−/−, and Daf1−/− mice were immunized subcutaneously with ova/IFA (n=5). A) Flow diagram of experimental design. Blood was drawn at five time points: 1 d pre-immunization and 8, 13, 21, and 30 d post-immunization with ova. Mice were boosted with ova/IFA 1 wk prior to 30 d bleed. (AU = relative absorbance units). B) Plasma from WT and C3ar1−/−C5ar1−/− mice 21 d post-immunization assayed for ova-specific total Ig. C) 21 d plasmas from WT and C3ar1−/−C5ar1−/− mice assayed for ova-specific anti-IgM, -IgG1, -IgG2a, -IgG2b, IgG2c, IgG3, -IgA, -Ig-κ, and -Ig-λ Abs. Inset: ratios of IgM and IgG isotypes in the three genotypes. D) Plasmas from WT and Daf1−/−7 d post-immunization assayed for ova-specific Ig. E) 7 d WT and Daf1−/− plasmas assayed for anti-ova-Ig isotypes as in C) above.
Upon activation, B2 cells produce complement components
The reduced and heightened in vivo Ab responses in C3ar1−/−C5ar1−/− and Daf1−/− mice could reflect the dependence of B2 cell help on CD4+ cell CD40L which our previous studies showed is regulated by C3ar1/C5ar1 signaling (Strainic et al., 2008). Therefore, we next examined purified B2 cells in vitro for C3a/C5a production and C3ar1/C5ar1 expression. A FACS analysis of the isolated B2 cells is shown in Fig S1. Anti-IgM F(ab’)2/anti-CD40 stimulated splenic B2 cells showed increases in C3/C5/C3ar1/C5ar1 mRNA expression levels at 24 h (Fig 2A). B2 cells stimulated with F(ab)2 alone did not upregulate complement (not shown). LPS plus IL-4 stimulation, however, evoked increases in C3/C5/C3ar1/C5ar1 mRNAs comparable to those following anti-IgM F(ab’)2/anti-CD40 stimulation (Fig S2A). Serum free supernatants from anti-IgM F(ab’)2/anti-CD40 stimulated B2 cell cultures showed increases in C3a and C5a (Fig 2B). Relevant to this, flow cytometric analyses of B2 cells stained intracellularly revealed preexisting pools of intracellular C3a/C5a and C3ar1/C5ar1 in unstimulated cells with marked intracellular increases of both the ligands and receptors after stimulation, arguing that the extracellular C3a/C5a derived from their intracellular generation (Fig 2C). These data argued that autocrine C3ar1/C5ar1 signaling operates in B2 cells as in CD4+ T cells and suggested that abrogated vs heightened C3ar1/C5ar1 signaling in B2 cells as well as CD4+ T cells is involved in the decreased vs increased in vivo Ab response in C3ar1−/−C5ar1−/− vs Daf1−/− mice.
Figure 2. B2 cells generate C3a/C5a from endogenously produced complement and autocrine C3ar1/C5ar1 signaling in B2 cells is needed for Ab production.
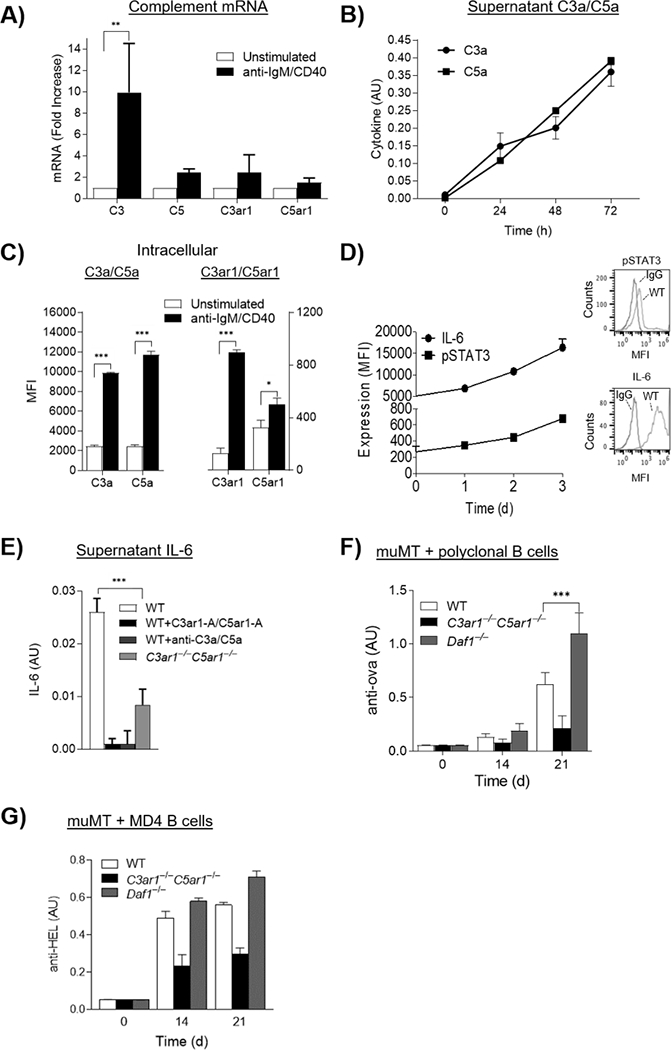
A) WT splenic B2 cells were incubated with anti-IgM F(ab’)2/anti-CD40 (3 μg/ml each) for 24 h at 37°C after which C3/C3ar1/C5/C5ar1 mRNA expression were quantified by qPCR. (n=3) p<05 B) Culture supernatants of the cells in A) were assayed for C3a and C5a by ELISA over time. N=3, p<.05 C) Unstimulated and anti-IgM (Fab’)2 stimulated WT B2 cells were assayed for intracellular C3a, C3ar1, C5a, C5ar1 by flow cytometry of permeabilized cells. n=3, p<.05. D) WT B2 cells were stimulated as in A) and intracellular IL-6 and p-STAT3 were assayed by FACS of permeabilized cells. n=3, p<.05 E) Anti-IgM (Fab’)2/CD40 stimulated WT B2 cells were incubated with media alone, C3ar1-A/C5ar1-A (30 ng/ml each), or anti-C3a/C5a mAbs (3 ug/ml each) and identically stimulated C3ar1−/−C5ar1−/− B2 cells were incubated with media alone after which supernatants were assayed for IL-6 by ELISA. AU =Absorbance units, n=3, p,.05. F) WT, C3ar1−/−C5ar1−/−, or Daf1−/− B2 cells (3×106) were adoptively transferred into muMT recipients (n=5 respectively). Recipients were then immunized with ova/CFA and assayed at 0, 14, and 21 d for anti-ova Ab by ELISA. The anti-ova Ab is total Ig. G) MD4 B2 cells on the WT, C3ar1−/−C5ar1−/−, or Daf1−/− backgrounds (n=5 each) were transferred to another set of muMT recipients after which the animals were immunized with HEL/CFA. Anti-HEL IgM Ab levels were assayed at 0, 14, and 21 d by ELISA.
IL-6 production by B2 cells accompanies endogenous C3a/C5a production
B2 cell activation is connected with the production of IL-6 (Hobbs et al., 1991; Yoshizaki et al., 2018) which not only confers proinflammatory effects on other cells but feeds back to promote B2 cell proliferation and Ab production (Taher et al., 2017). Adding exogenous C5a to B2 cells has been reported to augment B2 cell IL-6 production (Bröker et al., 2018; Gasque, 2004; Kupp et al., 1991). To determine if IL-6 production by B2 cells is dependent on autocrine C3ar1/C5ar1 signaling in the B2 cells, we assayed B2 cells and their supernatants for IL-6 following anti-IgM F(ab’)2/anti-CD40 activation in the absence or presence of C3ar1/C5ar1 pharmaceutical antagonists (C3ar1-A/C5ar1-A). These assays showed increases in IL-6 and activated STAT3 (Fig 2D) paralleling those of C3a/C5a (Fig 2B) in the B2 cells, both of which were abolished by the C3ar1/C5ar1 antagonists (Fig 2E). The increases in B2 cell C3a/C5a and IL-6 production were dependent on B2 cell CD40 signaling as they did not occur when the B2 cells were stimulated with anti-IgM F(ab’)2 alone (Fig S2B). Substituting C3ar1−/−C5ar1−/− B2 cells for WT B2 cells markedly decreased both C3a/C5a and IL-6 production (Fig 2E). These data indicated that autocrine C3ar1/C5ar1 signaling drives IL-6 production by B2 cells.
To test whether downregulated and upregulated anti-ova responses in C3ar1−/−C5ar1−/− and Daf1−/− mice resulted from altered C3ar1/C5ar1 signaling in vivo in B2 cells rather than only in CD4+ cells, we adoptively transferred purified WT, C3ar1−/−C5ar1−/−, or Daf1−/− B2 cells to muMT recipients that have normal CD4+ T cells and DCs but lack B2 cells. We then immunized the three sets of recipient mice with ova and assayed anti-ova Ig levels at 14 and 21 d. Consistent with autocrine C3ar1/C5ar1 signaling in B2 cells being integral to Ab production, the overall anti-ova Ab response was reduced 66% in recipients of C3ar1−/−C5ar1−/− B2 cells, whereas it was increased 76% in recipients of Daf1−/− B2 cells (Fig 2F). Comparable results were obtained for hen egg lysozyme (HEL) immunization of muMT recipients of HEL specific MD4 B2 cells on each genetic background (Fig 2G). These findings documented that in vivo Ab production is dependent on concomitant C3ar1/C5ar1 signaling in B2 cells as well as in CD4+ cells.
BAFF and APRIL function in B2 cells depend on autocrine C3ar1/C5ar1 signaling
The B cell growth factors BAFF [aka (Blys) B cell Activator of the TNF family and APRIL (A Proliferation-Inducing Ligand) provide viability and growth signals to B2 cells. To determine whether C3ar1/C5ar1 signaling is involved in their growth inductive functions, we cultured WT and C3ar1−/−C5ar1−/− splenic B2 cells with anti-IgM F(ab’)2 plus BAFF or APRIL and quantified cell growth at 24 h. Both growth factors increased the number of WT B2 cells 25–40%, but neither induced the growth of C3ar1−/−C5ar1−/− B2 cells over that of unstimulated cells (Fig 3A right). Consistent with this difference, adding C3ar1-A/C5ar1-A to the cultures of WT B2 cells abolished both BAFF or APRIL induced growth (Fig 3A left). Luminex assays showed that disabled C3ar1/C5ar1 signaling in B2 cells abolished AKT S473, Src and ERK phosphorylation (Fig 3B).
Figure 3. B2 cell growth responses to BAFF and APRIL depend on C3ar1/C5ar1 signaling in B2 cells.

A) Left: WT B2 cells were incubated with anti-IgM F(ab’)2 (3 μg/ml) with either BAFF or APRIL (100 ng/ml each) in the absence or presence of C3ar1/C5ar1 pharmaceutical antagonists (RA) (100 ng/ml) for 24 h at 37°C and growth quantitated. Right: WT and C3ar1−/−C5ar1−/− B2 cells were incubated with anti-IgM F(ab’)2 (3 μg/ml) with either BAFF or APRIL (100 ng/ml each) for 24 h at 37°C and B2 cell growth compared. B) WT B2 cells were incubated at 37°C for 3, 9, and 27 min with anti-IgM (Fab’2)/anti-CD40 (3 ug/mL each) in the absence or presence of RA (100 ng/ml) and p-Src, p-AKT and p-Erk assayed by Luminex assay. C) WT B2 cells were incubated with either anti-IgM F(ab’)2/anti-CD40 (3 μg/ml each) ± RA (100 ng/ml) (left), LPS (1 μg/ml) and IL-4 (10 ng/ml) ± RA (100 ng/ml) (middle) or anti-IgM F(ab’)2 (3 μg/ml) ± C5a (300 ng/mL) (right), all for 48 h at 37°C and assayed for BAFF-R and TACI expression by FACS. D) WT DCs were incubated at 37°C for 72 h with LPS (1 μg/ml) ± RA (100 ng/ml) or with C5a (300 ng/ml) ± RA (100 ng/ml), after which intracellular BAFF was assayed by FACS.
The functions of BAFF and APRIL are transmitted via their B2 cell receptors BAFF-R (BAFF receptor), and TACI (transmembrane activator of cyclophilin ligand interactor). Culturing WT splenic B2 cells with anti-IgM F(ab’)2/anti-CD40 upregulated the expression of BAFF and TACI ~70% and >100 % respectively (Fig 3C left). Adding C3ar1-A/C5ar1-A to the cultures virtually reversed the upregulations of both. We observed corresponding suppressive effects of C3ar1/C5ar1 antagonism following stimulation of B2 cells with LPS and IL-4 in the presence of the C3ar1-A/C5ar1-A (Fig 3C middle). Conversely, adding C5a in the presence of anti-IgM F(ab’)2 but absence of anti-CD40 stimulation up-regulated TACI ~70% and BAFF-R ~45% (Fig 3C right). In follicles, the source of BAFF is follicular DCs and BAFF production can be induced by LPS (Huard et al., 2004). Adding LPS to cultures of WT DCs increased the production of BAFF by 6-fold (Fig 3D, left). Adding C5a had the same effect, whereas including C3ar1-A/C5ar1-A in the LPS or C5a (as a control) containing cultures totally abolished the increase in BAFF production in response to both stimulants. (Fig 3D right). These data further supported the proposition that C3ar1/C5ar1 signaling is integral to the viability and growth functions of the TACI and BAFF-R signaling systems.
Autocrine C3ar1/C5ar1 signaling in B2 cells is required for proliferation and induction of AID and Bcl-6
The failed B2 cell responses to BAFF and APRIL prompted examination of BCR and CD40 dependent B2 cell proliferation. Adding C3ar1-A/C5ar1-A to cultures of anti-IgM F(ab’)2/anti-CD40 stimulated WT B2 cells virtually abolished B2 cell proliferation as assessed by CFSE (WT MFI =1400+ vs C3ar1−/−C5ar1−/− MFI= 20,000, unstimulated 20,000) (Fig 4A). We observed similar growth inhibition with C3ar1−/−C5ar1−/− B2 cells (Fig 4B), the two complementary experiments indicating that B2 cell proliferation depends on autocrine C3ar1/C5ar1 signaling in B2 cells.
Figure 4. C3ar1/C5ar1 signaling is needed for B2 cell expression of CD40, AID and Bcl-6 and CD4+ cell production of IL-21.

A) WT B2 cells prelabeled with CFSE were incubated at 37°C for 5 d with anti-IgM F(ab’)2/ anti-CD40 (3 μg/ml each) in the absence or presence of C3ar1/C5ar1 pharmaceutical antagonists (RA) (100 ng/ml) and proliferation assessed by CFSE dilution. Cells treated with RA overlapped the shaded area corresponding to unstimulated cells B) C3ar1−/−C5ar1−/− B2 cells prelabeled with CFSE were incubated as in A) and proliferation assessed by CFSE dilution. C3ar1−/−C5ar1−/− B2 cells overlapped the shaded area corresponding to unstimulated cells. C) WT and C3ar1−/−C5ar1−/− B2 cells were incubated at 37°C for 48 h with anti-IgM F(ab’)2/CD40L (3 μg/ml each) ± RA (100 ng/ml) (left) or C5a (300 ng/ml) ± RA (100 ng/ml) (right), after which CD40 expression was quantified by flow cytometry. D) WT B2 cells were incubated at 37°C for 72 h with LPS (1 μg/mL) and IL-4 (10 ng/mL) ± RA (300 ng/mL) after which AID and Bcl-6 mRNA expression were quantified by qPCR. E) WT CD4+ and C3ar1−/−C5ar1−/− CD4+ cells were incubated at 37°C for 48 h with anti-CD3/anti-CD28 Dynabeads (1:1 bead-to-cell ratio) in the absence or presence of RA (300 ng/mL) after which IL-21 was assayed by ELISA. N=3, p< .001
Our previous studies of DC-CD4+ cell interactions (Liu et al., 2008; Strainic et al., 2008) showed that DC up-regulation of CD40 depends on autocrine C3ar1/C5ar1 signaling in DCs. In accordance with B2 cell CD40 expression being similarly dependent on this signaling, stimulated WT showed 400% CD40 upregulation that was markedly attenuated in the presence of C3ar1-A/C5ar1-A (Fig 4C left). In accordance with this diminution by C3ar1-A/C5ar1-A in WT B2 cells, stimulated C3ar1−/−C5ar1−/− B2 cells, showed markedly blunted CD40 upregulation (Fig 4C left). Conversely, adding C5a to WT B2 cells increased CD40 by >100% partially simulating the effect of anti-IgM F(ab’)2/anti-CD40 stimulation (Fig 4C right).
Because our ova immunizations of C3ar1−/−C5ar1−/− mice showed IgM but no increases in other isotypes (Fig 1C), we next examined the role of C3ar1/C5ar1 signaling in induction of both AID and Bcl-6 needed for CSR. LPS/IL-4 stimulated WT B2 cells upregulated AID mRNA expression by >1500%. C3ar1/C5ar1 antagonism abolished the upregulation by >90% (Fig 4D left) explaining the disabled isotype switching in our in vivo studies (Fig 1C). C3ar1/C5ar1 antagonism likewise markedly blunted the induction of Bcl-6 mRNA expression (Fig 4D right) needed for germinal center function (Dent et al., 1997). Also important for CSR, activated WT CD4+ cells upregulated IL-21 production by ~150%. In contrast, pharmacological disruption of C3ar1/C5ar1 signaling reduced the IL-21 upregulation by >85%. Substitution of C3ar1−/−C5ar1−/− CD4+ cells had a similar dampening effect (Fig 4E). The lesser diminution of IL-21 in C3ar1−/−C5ar1−/− CD4+ cells as compared to C3ar1/C5ar1 antagonized WT CD4+ cells is due to compensations in other signaling pathways (Huang M, Strainic MG and Medof ME JOCS in press).
B2 cells produce factor I together with C3 and endogenously phosphorylate CD19
CD21 (aka CR2) signaling lowers the threshold for B2 cell activation (Carter and Fearon, 1992) and markedly augments Ab production. The ligand for CD21 is C3dg which is generated by factor I cleavage of C3b. As discussed (Introduction), the C3dg generation is thought to occur in after released Ab complexes with its specific antigen (Ag), and the resulting Ag-Ab complexes incorporate C3b. Our experiments above (Figs 2AB) showed that B2 cells endogenously synthesize C3 and locally generate C3a. Since the C3a generation is accompanied by concomitant generation of C3b, we hypothesized that B2 cells might synthesize factor I in conjunction with C3 and that factor I cleavage of the endogenously produced C3b could enable autocrine C3dg ligation of CD21 and consequent autocrine phosphorylation of CD19. B2 cell expressed CR1 (CD35) could serve as the obligate cofactor for this cleavage (Iida and Nussenzweig, 1983; Medof et al., 1982a).
To test this idea, we first cultured B2 cells from WT and C3−/− mice and assayed each for the presence of C3 protein by flow cytometry. We detected C3 antigen on WT but not on the C3−/− B2 cells confirming specificity (Fig 5A). Stimulation of WT B2 cells with anti-IgM F(ab’)2/anti-CD40 upregulated C3 mRNA production >800% (Fig 5B). The stimulation concomitantly up-regulated factor I mRNA levels by >650% (Fig 5C). These data indicated that activated B2 cells in and of themselves possess the machinery i.e. C3b, factor I, and CR1, needed to generate C3dg and consequently induce CD21 activation.
Figure 5. B2 cells endogenously phosphorylate CD19 by virtue of locally producing C3 and factor I.

A) WT B cells were adoptively transferred into C3−/− mice (n=5) after which C3 expression on B cells was assayed by flow cytometry. B) WT splenic B2 cells were incubated at 37°C for 72 h with anti-IgM F(ab’)2/anti-CD40, after which C3 mRNA expression was quantified by qPCR C) WT splenic B2 cells were incubated as in B) after which factor I mRNA levels were quantitated by qPCR. D) WT B2 cells in the absence or presence of RA (100 ng/mL each) or C3ar1−/−C5ar1−/− B2 cells were incubated at 37°C with anti-IgM F(ab’)2/anti-CD40 (3 μg/mL) and CD19 phosphorylation assayed on immunoblots as a function of time. E) MD4 BM was transplanted into irradiated C3−/−C5nul recipients. Following immunization of the chimeras and the untreated C3−/−C5nul mice with HEL in CFA, blood samples were drawn at 1, 3, 5, 7 and 10 d and assayed for by ELISAs for anti HEL IgM (left) and IgG (right) Abs. As controls, blood samples were assayed from identically immunized WT mice and from unimmunized C3−/−C5nul-MD4 chimeras.
C3dg ligation of CD21 induces phosphorylation of CD19 enabling the association and activation of PI-3Kα (Owen et al., 2012). To test whether this process operates endogenously in activated B2 cells, we activated WT B2 cells and assayed extracts of the cells for p-CD19 (Y531) as a function of time. An increase in amounts of p-CD19 (Y531) was detectable 1 min post ex vivo stimulation with anti-IgM F(ab’)2)/anti-CD40 with levels gradually declining through 60 min (Fig 5D), consistent with amplification of CD19 phosphorylation occurring endogenously in the absence of systemic C3 or factor I. The process was dependent on autocrine C3ar1/C5ar1 signaling in the B2 cells as it was attenuated when C3ar1-A/C5ar1-A were added to activated WT B2 cells or if activated C3ar1−/−C5ar1−/− B2 cells were used (Fig 5D). These data indicated that autocrine CD19 signaling in activated B2 cells is dependent on B2 cell complement production and C3ar1/C5ar1 signaling. They argue that activated B2 cells can trigger CD19 signaling in and of themselves and raise the possibility that the uptake of C3b into Ab-Ag and the conversion of C3b into C3dg could occur in the B2 cells themselves.
B2 cells in mice devoid of plasma C3 produce Ab in response to immunization
Taken together, the above studies centrally implicated B2 cell produced complement in B2 cell activation and Ab production but did not exclude the requirement during in vivo activation of systemic complement. To address this issue, we transplanted C3−/−C5def mice with WT MD4 BM and immunized the chimeras and the untreated recipient C3−/−C5def mice with HEL in CFA. We then assayed blood samples drawn at progressively increasing times for anti-HEL Ab. The chimeric mice but not the C3−/−C5def mice produced anti-HEL IgM Ab which increased in amounts with time (Fig 5E). Characteristic of the MD4 cell response in which CSR does not occur, no anti-HEL IgG Ab was generated. These data together with the data in Fig 5D that CD19 phosphorylation depends on endogenous complement production in B2 cells indicated that local complement production in the MD4 cells was sufficient for the B2 cell anti-HEL Ab response.
DISCUSSION
The experiments in this study provide unexpected insights into the involvement of complement in key processes involved in B2 cell activation. They show that B2 cells locally generate C3a/C5a that enter into autocrine C3ar1/C5ar1 signaling loops. These loops operate tonically in B2 cells and amplify with B2 cell stimulation. The studies show that this GPCR signaling is required for BAFF and APRIL growth and viability signaling. They further demonstrate that B2 cell upregulation of CD40 required for Ab production and AID/Bcl-6 expression needed for CSR/AFM depend on C3ar1/C5ar1 signaling. Importantly, the experiments show that in addition to producing C3 protein that leads to C3a (and C3b) generation, B2 cells endogenously produce factor I. This concomitance of factor I upregulation with B2 cell activation indicates that B2 cells possess the capability to endogenously generate C3dg and thereby ligate CD21 which induces costimulatory signaling by associated CD19. Analyses in vitro of CD19 phosphorylation in purified B2 cells following their activation confirmed that this process is dependent on autocrine C3ar1/C5ar1 signaling. The finding that immunization of chimeric C3−/−C5def mice possessing WT MD4 BM taken together with the multiple sets of in vitro and in vivo findings that autocrine C3ar1/C5ar1 signaling is required for B2 activation demonstrated that local complement is sufficient for the initial B2 cell Ab. The studies thus show that B2 cell viability, B2 cell proliferation, B2 cell activation of AID/Bcl-6 all interconnect with autocrine C3ar1/C5ar1 signaling. As with the above steps in B2 cell activation, CD19 phosphorylation was dependent on autocrine C3ar1/C5ar1 signaling. Collectively, the above findings thus argue that endogenous complement production in B2 cells is necessary and sufficient at least for initial Ab production in vivo in the absence of systemic C3 and C5. While our findings that CD40 upregulation is defective in C3ar1−/−C5ar1−/− B2 cells suggest an upstream involvement, our findings that C3ar1−/−C5ar1−/− B2 cells are defective in BAFF and APRIL signaling and CD40 upregulation, but that all three genotypes showed the same IgM response suggest both upstream and early IgM Ab involvement, but more studies will be needed to confirm this.
The findings that autocrine C3ar1/C5ar1 signaling operates both tonically and during activation in B2 cells parallels our findings for CD4+ T cells and DCs (Lalli et al., 2008; Strainic et al., 2008). Those studies showed that the underlying mechanism is that C3ar1/C5ar1 signaling activates phosphatidylinositol-3 kinase-ɣ (PI-3Kγ) which promotes inner leaflet phosphatidylinositol 3,4,5 trisphosphate [PtdIns (3,4,5)P3] assembly needed for AKT phosphorylation and its downstream signaling to mTOR. Multiple lines of in vitro and in vivo evidence (Lalli et al., 2008; Strainic et al., 2008) documented that C3ar1/C5ar1 signals are needed for CD4+ cell viability and proliferation. The studies herein show that autocrine C3ar1/C5ar1 signaling in B2 cells likewise is needed for AKT as well as Src and ERK phosphorylation. While others have found that B2 cell activation is associated with the production of IL-6 (Kawano et al., 1988; Muraguchi et al., 1988), the experiments herein show that IL-6 and C5a generation in B2 cells are mechanistically interdependent. In support of this, IL-6 signaling promotes B2 cell C3a/C5a generation and inhibiting C3a/C5a generation suppresses IL-6 production. The former finding is consistent with prior unexplained findings that C5a induces IL-6 in retinal pigment epithelial cells (Fukuoka et al., 2003; Montz et al., 1991) and IL-6 conversely promotes C5a production in CD4+ cells (Strainic et al., 2013).
In follicles, BAFF secretion by follicular DCs maintains B2 cell viability (Thompson et al., 2001). In the bone marrow, B2 cell viability is supported by the combination of BAFF-R and BCR signaling (Dallos et al., 2009). The linkage of BAFF-R signaling with viability has been mechanistically connected with several processes. Among these processes is PI-3K dependent generation of PtdIns (3,4,5)P3 that enables AKT phosphorylation (Patke et al., 2006). Our prior findings in CD4+ cells (Strainic et al., 2008) that C3ar1/C5ar1 induction of PI-3Kɣ activation promotes PtdIns (3,4,5)P3 assembly and AKT phosphorylation connects C3ar1/C5ar1 signaling with this process. Observations by others (Russkamp et al., 2015) that C5ar1 signaling can activate ERK provide further support for this linkage. Others have found that BCR activation of splenic B2 cells in the absence of BAFF-R signaling leads to B2 cell apoptosis (Rauch et al., 2009). This finding parallels our previous findings that disabled C3ar1/C5ar1 signaling in anti-CD3 activated CD4+ cells similarly eventuates in Teff cell apoptosis (Strainic et al., 2008).
In B2 cells, while CD19 signaling is principally connected with costimulatory signaling needed to drive B2 cell proliferation, it also has been implicated in operating tonically to sustain viability (Depoil et al., 2008; Fearon and Carroll, 2000). Unlike C3a that is generated as a soluble polypeptide that functions as the ligand for C3ar1, nascent C3b binds covalently to targets. Our findings that CD19 phosphorylation is dependent on B2 cell produced C3a imply the concomitant B2 cell generation of C3b. Our finding of B2 cell production of factor I opens up the mechanistically attractive possibility that factor I and B2 cell expressed CR1 could convert the C3b to C3dg which in turn could ligate CD21 and activate CD19. Further experiments will be needed to determine if the Ag-Ab-C3dg complex initially produced by the B2 cell links the BCR to CD21 or whether C3dg bound to another protein endogenously ligates CD21. Since the experiments herein showed that autocrine C3ar1/C5ar1 signaling in B2 cells is connected with viability signaling as well as with BAFF/APRIL function, the previously reported connections of CD19 signaling with viability, in principle, could be mechanistically connected with autocrine C3ar1 signaling, since endogenous C3b production (that leads to C3dg production) is accompanied by C3a generation.
Like costimulation conferred by CD28 signaling in CD4+ cells in which CD28 phosphorylation triggers recruitment and activation of PI-3Kα (promoting PtdIns (3,4,5)P3 dependent phosphorylation of AKT), CD19 phosphorylation in B2 cells triggers the same events. Relevant to this process, the experiments in this study show that B2 cell proliferation is linked to up-regulated production of C3/C5 and C3ar1/C5ar1 which would evoke C3ar1/C5ar1 signaling (Strainic et al., 2008) and, in principle, concomitantly evoke C3dg-(CD21 induced) CD19 signaling. Consequently, both autocrine C3a/C5a-C3ar1/C5ar1 signaling and autocrine C3dg-CD21 signaling could be involved in maintaining viability. Our previous findings that C3ar1/C5ar1 signaling drives CD4+ cell upregulation of CD40L (Strainic et al., 2008) together with the findings herein that C3ar1/C5ar1 signaling drives B2 cell upregulation of CD40 would support the concept that the CD4+ cell and B2 cell produced complement that upregulates costimulatory molecules on both partners functions to augment CD4+ cell coupling to B2 cells. Based on the above described interconnections of C3ar1/C5ar1 signaling with BAFF and APRIL function, this signaling consequently could not only underlie CD4+ cell help but also participate in maintaining the viability of both CD4+ cell and B2 cell partners.
Past studies (Fearon and Carroll, 2000) have shown that CD19 signaling together with BCR signaling markedly lowers the amount of Ag needed for B2 cell activation. This work found that the combined signaling lowers the amount of antigen needed by as much as 10,000% and promotes CSR (Carter and Fearon, 1992). The CD19 signaling occurs as a result of C3dg ligation of CD21 evoking p-SYK induced phosphorylation of the CD19 Sh2 domain, thereby enabling the binding and activation of PI-3Kα (Fearon and Carroll, 2000). While current concepts are that the PI-3Kα activation generates sufficient amounts of PtdIns (3,4,5)P3 to enable phosphorylation of AKT, the data herein argue that PI-3Kɣ activation induced by C3ar1/C5ar1 co-signaling also is required. The findings that B2 cell AKT, Src, and ERK phosphorylation are disabled by blockade of C3ar1/C5ar1 signaling which activates PI-3Kɣ (Strainic et al., 2008) support this concept.
The above findings that B2 cells themselves produce C3 raise the question of whether the B2 cell produced C3 is functionally relevant in the secondary Ab response. The classical view is that secreted IgM Ab binds to Ag and that the resulting soluble Ab-Ag complex covalently incorporates C3b. The Ag-Ab-C3b complex then binds to CR1 on erythrocytes or B2 cells, after which the incorporated C3b is converted to C3dg by plasma factor I (Medof et al., 1982a; Medof et al., 1983; Medof et al., 1982b). The Ag-Ab-C3dg complex then returns to B2 cells by virtue of the Ag binding to the BCR and C3dg binding to CD21. This model thus envisions the process occurring outside of the parental B2 cell followed by return of the complex to the B2 cell surface. The joint ligation of C3dg and CD21 amplifies the Ab response (Carter and Fearon, 1992) as well as promotes CSR (Carter and Fearon, 1992). The data herein raise the possibility that these processes can occur by B2 cell produced C3 and factor I as well as by systemic C3 and factor I and in and on the B2 cell. In this regard, it will be important to distinguish the respective role of plasma and B2 cell produced C3 and factor I in generating Ab-Ag-C3dg.
Taken together, the findings in this study are relevant to several past observations. They potentially also have important clinical implications. Among past observations are that impaired B2 cell development in C3−/− mice (Jacobson et al., 2009) has been attributed to loss of plasma C3 function (Owen et al., 2012). It will be informative to re-examine these findings to distinguish the respective roles of exogenous vs B2 cell produced C3. Potentially relevant to the data herein is the finding (Liszewski et al., 2013) that individuals deficient in plasma C3 can locally generate C3a in human CD4+ cells and still produce autoantibody. Among potential clinical implications is that DAF blockade which amplifies Ab production by potentiating B2 cell C3ar1/C5ar1 signaling could be exploited to develop more potent vaccines. Conversely, interference with B2 cell C3a/C5a production or C3ar1/C5ar1 signaling could be leveraged to suppress auto-Ab production. Such interference would concomitantly suppress B2 cell proinflammatory cytokine production. The abrogation of CD40 upregulation could suppress antigen presentation in autoimmune diseases such as rheumatoid arthritis or other disorders driven by heightened B2 cell activation.
MATERIALS AND METHODS
Reagents and antibodies
Murine C5a was from Cell Sciences (Canton, ME). Anti-mouse C3a and C5a mAbs were from BD Biosciences (San Jose, CA). C3ar1-A and C5ar1-A were purchased from Calbiochem (EMB Biochemicals). CFSE was used according to the manufacturer’s instructions (Invitrogen). Anti-mouse IgM F(ab’)2 was purchased from Jackson Labs (Bar Harbor, ME). FITC anti-mouse CD19, anti-mouse CD40, PE anti-mouse CD40, anti-mouse IL-6, biotin-anti-mouse IL-6, anti-mouse C5a, biotin anti-mouse C5a, BAFF, APRIL, PE anti-mouse TACI, and APC anti-mouse BAFF-R were purchased from BD Biosciences (San Jose, CA). HRP anti-mouse IgG was purchased from Cell Signal Technology (Danvers, MA) Recombinant mouse IL-4 was from Miltenyi Biotech, (San Diego, CA). and LPS (Escherichia coli O26:B6) from Sigma-Aldrich (St. Louis, MO)
Animals
C57BL/6, MD4, and muMT were from Jackson labs (Bar Harbor, ME). C3−/− mice were received from Dr. Feng Lin (Cleveland Clinic, Cleveland, OH). C3ar1−/− and C5ar1−/− mice were gifts from Dr. Michael Carroll and Dr. Craig Gerard (Harvard Medical School and Children’s Hospital, Boston, MA). All studies were approved by the Case Western Reserve University Institutional Animal Care and Use Center.
Ova immunizations
WT, C3ar1−/−C5ar1−/−, and Daf1−/− mice (n=5 each) (C57BL/6) were immunized s.c. with 100 μg of ova in IFA (Hooke). Mice were tail vein bled one day prior to immunization and 8, 13, and 21 d post-immunization. All mice were boosted with 100 μg of ova in IFA 23 d post-immunization. Mice were tail vein bled once more 7 d post-boost. All blood samples were heparinized, plasma separated, and stored at −70°C.
Anti-ova-ELISAs
Ninety-six-well plates were coated overnight at 4°C with 200 μg/mL ovalbumin/PBS. Plates were blocked for 1 h at 20o C with 1% BSA/PBS. Plasma samples were diluted and incubated for 6 h at RT. HRP-labeled horse anti-mouse IgG mAb (Cell Signalling Technology, catalogue number 70765) was incubated for 1 h at 20o C to assay total anti-ova IgG levels. For isotype specific ELISA assays, plasma samples were diluted 1:200 and incubated for 6 h at 20o C followed by rat anti-mouse IgM, IgG1, IgG2a, IgG2b, IgG3, or IgA or Ig-κ, -λ heavy chains from Mouse Immunoglobulin Isotyping ELISA Kit (BD Pharmingen, catalogue number: 550487). Plates were then incubated with HRP-labeled goat anti-rat Ig.
B cell, T cell and DC isolations
Mice were euthanized for 5 min in CO2 and spleens were collected in FACS buffer. Spleens were crushed into single cell suspensions. B2 cell CD4+ T and CD11c negative selection isolation kits (Miltenyi, San Diego, CA) and AutoMACS were used to sort B cells, CD4+ T cells and DCs respectively. Purity of B cells following isolation was validated by FACS. In many cases, analyses were done gating on CD19. In all cases this confirmed purity.
Cell culture and activation assays
Splenic B2 cells, CD4+ and DCs were cultured in RPMI 1640 media supplemented with 0.5% fetal bovine serum, 1% penicillin/streptomycin, 1% Glutamax (Invitrogen), and 1% sodium pyruvate (Invitrogen) in round-bottom 96-well plates, 2×105 cells per well. B2 cells were stimulated by incubating them with either 3 ug/mL anti-IgM F(ab’)2, 3 ug/mL anti-CD40, 1 μg/mL LPS, 10 ng/mL IL-4 or 5 ug/mL C5a. B2 cells were labeled with 2 uM CFSE for 7 min. CD4+ cells were stimulated using Dynabeads Mouse T-Activator CD3/CD28 for T-Cell Expansion and Activation (Gibco, MD) at a 1:1 bead-to-cell ratio. DCs were stimulated by incubating with 1 μg/mL LPS or 300 ng/mL C5a To inhibit proliferation, 100 ng/mL C3ar1/C5ar1 pharmaceutical antagonists were used.
Flow cytometry
Cells were Fc receptor blocked for 10 min at 4°C with anti-mouse CD16/CD32. Cells were incubated for 15 min with respective fluorescent antibodies at 4°C. Analysis was performed on BD FACSAria or BD LSR II in Case Western Reserve University’s Comprehensive Cancer Center Flow Cytometry Core.
RNA Purification, cDNA synthesis, and qPCR
Cells were extracted for 5 min at 20°C with TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. When C3, C5, fB, fD, and AID mRNAs were analyzed, preparations were treated with DNAse I (standard protocol) for removal of genomic DNA. We synthesized cDNAs by incubating 20 uL of mRNAs in Sprint PowerScript Single Shots (Clontech, Mountain View, CA). A total of 10 uL of diluted cDNA was mixed with 2 uL of primer and 10 uL SYBR green master mix (Applied Biosystems, Foster City, CA) and assayed in triplicate on an ABI prism 7000 cycler. In all assays, fold increases are relative to each basal level and standardized to actin.
MD4 adoptive transfers and HEL immunization
3×106 MD4 C3ar1−/−C5ar1−/− or MD4 Daf1−/− B cells were injected via tail vein into muMT recipient mice. Recipient mice were administered s.c. 100 ug HEL (Sigma) in CFA 1 wk after adoptive transfers and bled weekly. Plasma anti-HEL Ab levels were assayed as in anti-ova detection ELISAs with 200 ug/mL HEL/PBS coating instead of ova.
AKT, Src, and ERK phosphorylation studies
B2 cells were stimulated with anti-IgM F(ab’)2 and anti-CD40 (5 ug/mL) at 37 C for 3, 9, and 27 min after which they were assayed for p-Src, t-Src, p-AKT, t-AKT, p-Erk, t-Erk, using Millipore’s (Burlington, MA) Luminex assay according to the manufacturer’s instructions.
Immunoprecipitations
Cells were washed twice with PBS and extracted on ice for 10 min with 1% NP-40, 150 mM NaCl, 1 mM PMSF, 0.4 mM EDTA, and a protease-inhibitor cocktail (Complete Mini, Roche, Mannheim, Germany). After centrifugation of extracts for 10 min at 13,000 x g, supernatants were incubated for 1 hr at 4°C with appropriate antibody, after which Sepharose A beads were added and the mixture incubated overnight at 4°C. Centrifuged pellets were washed 5 x, SDS sample buffer was added, and boiled samples were loaded onto SDS-PAGE gels.
Immunoblotting
All blots were performed by standard procedures as described (Lin et al., 2001) with HRP-conjugated secondary antibody and an ECL enhancer (GE Healthcare, Buckinghamshire, UK).
Supplementary Material
Figure S1. Isolation and purity of B cells. Gating strategy: cells in the B cell location on forward scatter and side scatter were depleted of doublets and then stained for CD19.
Figure S2. LPS + IL-4 evokes C3a/C5a generation by B2 cell similarly to anti-IgM (Fab2’)/anti-CD40 stimulation and this synthesis requires CD40 A) WT B2 cells were incubated with LPS (1 μg/mL) and IL-4 (10 ng/mL) for 24 h at 37°C and C3/C3ar1/C5/C5ar1 mRNA expression levels assayed by qPCR. B) Unstimulated and anti-IgM (Fab’)2 stimulated WT B2 cells were assayed for extracellular C3a, C3ar1, C5a, C5ar1 by flow cytometry of cells. n=3. C) WT B2 cells were incubated at 37°C for 72 h with anti-IgM F(ab’)2 (3 μg/mL) alone after which C3a, C5a and IL-6 released into culture supernatants were assayed by ELISAs.
References
- Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, Dutow P, Woodruff TM, Yu ZX, O’Neill LA, et al. (2016). T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science 352, aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröker K, Figge J, Magnusen AF, Manz RA, Köhl J, and Karsten CM (2018). A Novel Role for C5a in B-1 Cell Homeostasis. Frontiers in Immunology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RH, and Fearon DT (1992). CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science 256, 105–107. [DOI] [PubMed] [Google Scholar]
- Dallos T, Krivosikova M, Chorazy-Massalska M, Warnawin E, Zanova E, Rudnicka W, Radzikowska A, and Maslinski W (2009). BAFF from bone marrow-derived mesenchymal stromal cells of rheumatoid arthritis patients improves their B-cell viability-supporting properties. Folia biologica 55, 166–176. [DOI] [PubMed] [Google Scholar]
- Dent AL, Shaffer AL, Yu X, Allman D, and Staudt LM (1997). Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 276, 589–592. [DOI] [PubMed] [Google Scholar]
- Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VL, and Batista FD (2008). CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nature immunology 9, 63–72. [DOI] [PubMed] [Google Scholar]
- Fearon DT, and Carroll MC (2000). Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annual review of immunology 18, 393–422. [DOI] [PubMed] [Google Scholar]
- Fukuoka Y, Strainic M, and Medof ME (2003). Differential cytokine expression of human retinal pigment epithelial cells in response to stimulation by C5a. Clinical and experimental immunology 131, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasque P (2004). Complement: a unique innate immune sensor for danger signals. Molecular immunology 41, 1089–1098. [DOI] [PubMed] [Google Scholar]
- Hobbs MV, McEvilly RJ, Koch RJ, Cardenas GJ, and Noonan DJ (1991). Interleukin-6 production by murine B cells and B cell lines. Cellular immunology 132, 442–450. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Yagita H, Ortaldo JR, Wiltrout RH, and Young HA (2000). In vivo administration of IL-18 can induce IgE production through Th2 cytokine induction and up-regulation of CD40 ligand (CD154) expression on CD4+ T cells. European journal of immunology 30, 1998–2006. [DOI] [PubMed] [Google Scholar]
- Huard B, Arlettaz L, Ambrose C, Kindler V, Mauri D, Roosnek E, Tschopp J, Schneider P, and French LE (2004). BAFF production by antigen-presenting cells provides T cell co-stimulation. International immunology 16, 467–475. [DOI] [PubMed] [Google Scholar]
- Iida K, and Nussenzweig V (1983). Functional properties of membrane-associated complement receptor CR1. Journal of immunology 130, 1876–1880. [PubMed] [Google Scholar]
- Jacobson AC, Roundy KM, Weis JJ, and Weis JH (2009). Regulation of murine splenic B cell CR3 expression by complement component 3. Journal of immunology 183, 3963–3970. [DOI] [PubMed] [Google Scholar]
- Kawano M, Hirano T, Matsuda T, Taga T, Horii Y, Iwato K, Asaoku H, Tang B, Tanabe O, Tanaka H, and et al. (1988). Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature 332, 83–85. [DOI] [PubMed] [Google Scholar]
- Kupp LI, Kosco MH, Schenkein HA, and Tew JG (1991). Chemotaxis of germinal center B cells in response to C5a. European journal of immunology 21, 2697–2701. [DOI] [PubMed] [Google Scholar]
- Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, and Heeger PS (2008). Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 112, 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, et al. (2013). Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39, 1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, and Cerutti A (2002). DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nature immunology 3, 822–829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liu J, Lin F, Strainic MG, An F, Miller RH, Altuntas CZ, Heeger PS, Tuohy VK, and Medof ME (2008). IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol 180, 5882–5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Iida K, Mold C, and Nussenzweig V (1982a). Unique role of the complement receptor CR1 in the degradation of C3b associated with immune complexes. The Journal of experimental medicine 156, 1739–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Lam T, Prince GM, and Mold C (1983). Requirement for human red blood cells in inactivation of C3b in immune complexes and enhancement of binding to spleen cells. J Immunol 130, 1336–1340. [PubMed] [Google Scholar]
- Medof ME, and Nussenzweig V (1984). Control of the function of substrate-bound C4b-C3b by the complement receptor Cr1. The Journal of experimental medicine 159, 1669–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Prince GM, and Mold C (1982b). Release of soluble immune complexes from immune adherence receptors on human erythrocytes is mediated by C3b inactivator independently of Beta 1H and is accompanied by generation of C3c. Proceedings of the National Academy of Sciences of the United States of America 79, 5047–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montz H, Koch KC, Zierz R, and Gotze O (1991). The role of C5a in interleukin-6 production induced by lipopolysaccharide or interleukin-1. Immunology 74, 373–379. [PMC free article] [PubMed] [Google Scholar]
- Muraguchi A, Hirano T, Tang B, Matsuda T, Horii Y, Nakajima K, and Kishimoto T (1988). The essential role of B cell stimulatory factor 2 (BSF-2/IL-6) for the terminal differentiation of B cells. The Journal of experimental medicine 167, 332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JA, Punt J, Stranford SA, Jones PP, and Kuby J (2012). Kuby immunology, Seventh Edition. edn.
- Patke A, Mecklenbrauker I, Erdjument-Bromage H, Tempst P, and Tarakhovsky A (2006). BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. The Journal of experimental medicine 203, 2551–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch M, Tussiwand R, Bosco N, and Rolink AG (2009). Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PloS one 4, e5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russkamp NF, Ruemmler R, Roewe J, Moore BB, Ward PA, and Bosmann M (2015). Experimental design of complement component 5a-induced acute lung injury (C5a-ALI): a role of CC-chemokine receptor type 5 during immune activation by anaphylatoxin. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 29, 3762–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper CM, McIntyre TM, Mandler R, Pecanha LM, Finkelman FD, Lees A, and Mond JJ (1992). Induction of IgG3 secretion by interferon gamma: a model for T cell-independent class switching in response to T cell-independent type 2 antigens. The Journal of experimental medicine 175, 1367–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, and Medof ME (2008). Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strainic MG, Shevach EM, An F, Lin F, and Medof ME (2013). Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nature immunology 14, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taher TE, Ong VH, Bystrom J, Hillion S, Simon Q, Denton CP, Pers JO, Abraham DJ, and Mageed RA (2017). Association of Defective Regulation of Autoreactive Interleukin-6-Producing Transitional B Lymphocytes With Disease in Patients With Systemic Sclerosis. Arthritis & rheumatology. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Tack BF, and Nussenzweig V (1977). Requirements for the solubilization of immune aggregates by complement: assembly of a factor B-dependent C3-convertase on the immune complexes. The Journal of experimental medicine 145, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Takahashi S, Brade V, and Nussenzweig V (1978). Requirements for the solubilization of immune aggregates by complement. The role of the classical pathway. The Journal of clinical investigation 62, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, et al. (2001). BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 293, 2108–2111. [DOI] [PubMed] [Google Scholar]
- Yoshizaki K, Murayama S, Ito H, and Koga T (2018). The Role of Interleukin-6 in Castleman Disease. Hematology/oncology clinics of North America 32, 23–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Isolation and purity of B cells. Gating strategy: cells in the B cell location on forward scatter and side scatter were depleted of doublets and then stained for CD19.
Figure S2. LPS + IL-4 evokes C3a/C5a generation by B2 cell similarly to anti-IgM (Fab2’)/anti-CD40 stimulation and this synthesis requires CD40 A) WT B2 cells were incubated with LPS (1 μg/mL) and IL-4 (10 ng/mL) for 24 h at 37°C and C3/C3ar1/C5/C5ar1 mRNA expression levels assayed by qPCR. B) Unstimulated and anti-IgM (Fab’)2 stimulated WT B2 cells were assayed for extracellular C3a, C3ar1, C5a, C5ar1 by flow cytometry of cells. n=3. C) WT B2 cells were incubated at 37°C for 72 h with anti-IgM F(ab’)2 (3 μg/mL) alone after which C3a, C5a and IL-6 released into culture supernatants were assayed by ELISAs.