

Summary
Bone marrow (BM) mesenchymal stem and progenitor cells (MSPCs) are a critical constituent of the hematopoietic stem cell (HSC) niche. Previous studies have suggested that the zinc-finger epithelial-mesenchymal transition transcription factor Snai2 (also known as Slug) regulated HSCs autonomously. Here, we show that Snai2 expression in the BM is restricted to the BM stromal compartment where it regulates the HSC niche. Germline or MSPC-selective Snai2 deletion reduces the functional MSPC pool, their mesenchymal lineage output, and impairs HSC niche function during homeostasis and after stress. RNA-sequencing analysis revealed that Spp1 (osteopontin) expression is markedly upregulated in Snai2-deficient MSPCs. Genetic deletion of Spp1 in Snai2-deficient mice, rescues MSPCs’ functions. Thus, SNAI2 is a critical regulator of the transcriptional network maintaining MSPCs by the suppression of osteopontin expression.
Keywords: Bone marrow, mesenchymal stem and progenitor cells, stromal cells, Snai2/Slug, osteopontin (OPN)/Spp1, self-renewal, hematopoietic stem cell niche, transcriptional regulation
eTOC Blurb
Wei et al found that Snai2 is preferentially expressed and required in bone marrow mesenchymal stem and progenitor cells (MSPC) for their maintenance and regeneration. The authors show that Snai2 promotes self-renewal by suppressing Spp1 expression in MSPCs.
Introduction
Bone marrow (BM) mesenchymal stem cells (MSPCs) comprise self-renewing and multipotent precursor cells that can give rise to bone, cartilage, fat and hematopoiesis-supporting marrow stroma (Bianco et al., 2013; Frenette et al., 2013). Their critical roles in supporting hematopoiesis and, in particular, hematopoietic stem cells (HSCs) have increasingly been recognized over the past two decades. During development, MSPCs were reported to associate with hematopoietic sites (Mendes et al., 2005). In adult animals, MSPCs are a major provider of the essential niche factors (e.g. SCF, CXCL12, Pleiotrophin and IL-7) for HSC maintenance and progenitor differentiation (Cordeiro Gomes et al., 2016; Ding and Morrison, 2013; Ding et al., 2012; Greenbaum et al., 2013; Himburg et al., 2018; Mendez-Ferrer et al., 2010). However, despite these critical functions, the mechanisms regulating MSPC maintenance in the bone marrow remain largely unknown.
Transcription factors are critical cell fate determinants. A few transcription factors have been suggested to regulate BM development or HSC niche (Kieslinger et al., 2010; Liu et al., 2018; Omatsu et al., 2014; Seike et al., 2018), yet the network of factors that specify MSPC and maintain BM homeostasis have not been elucidated. The challenge is in part due to the poorly defined organization of the heterogeneity and differentiation hierarchy within the mesenchymal lineage (Baryawno et al., 2019; Severe et al., 2019; Tikhonova et al., 2019). A transcription regulatory network specific to MSPCs and their progeny will be needed to understand the BM niche maintenance and function.
Our transcriptomic analyses of MSPCs have revealed that Snai2 (also known as Slug), an epithelial-mesenchymal transition (EMT) transcription factor, was highly expressed in BM Nes-GFP+ niche cells (Kunisaki et al., 2013; Nakahara et al., 2019). EMT transcription factors have been associated with mesenchymal traits and stem cell characteristics during healthy developmental and pathogenic malignant programs (Guo et al., 2012; Mani et al., 2008; Medici et al., 2010), raising the possibility that Snai2 may regulate MSPCs in the BM niche.
Results
Snai2 is selectively expressed in BM MSPCs
Our transcriptome analysis of Nes-GFP+ BM MSPCs (Kunisaki et al., 2013) revealed high expression levels of EMT transcription factors, among which, Snai2, a Snail family zinc finger protein, showed the highest transcript levels (Figure 1A). Two independent transcriptome profilings (Cabezas-Wallscheid et al., 2014; Maryanovich et al., 2018) found negligible Snai2 expression in HSCs. To map precisely Snai2 expression within the BM, we crossed Snai2YFP reporter mice, in which the yellow fluorescent protein (YFP) driven by an internal ribosome entry site (IRES) was targeted into the 3’UTR of the Snai2 locus (Guo et al., 2012), with Ng2-Cre; R26-tdTomato double transgenic mice in which the Tomato expression labels Nes-GFP+ niche cells (Asada et al., 2017). Both imaging and flow cytometry analyses revealed that Snai2YFP expression labeled virtually all (~97%) the NG2-marked BM mesenchymal cells, including the peri-arteriolar, sinusoidal and interstitial stromal cells (Figure 1B and C), while rare single positive cells may reflect heterogeneity of the BM stroma or the dynamic expression of the reporter transgenes. Similar results were obtained in the Snai2YFP; Lepr-Cre; R26-tdTomato mice consistent with the largely overlapping expression of these two Cre lines in the non-arteriolar stromal cells (Asada et al., 2017; Ding et al., 2012) (Figure S1A and B). Furthermore, Snai2YFP expression was absent in endosteal osteoblasts (Figure 1B and Figure S1A and C), suggesting that Snai2 was specifically expressed in the mesenchymal stem and progenitor fraction but not in their differentiated progeny. Additionally, our analysis of the Snai2YFP reporter and endogenous Snai2 transcript in sorted BM populations only detected Snai2 expression in the mesenchymal cell fractions but not in hematopoietic cells, including purified HSCs and progenitors (Figure 1D–G). Snai2YFP+ cells were enriched in niche factor expression and fibroblastic colony forming activity (CFU-F) in BM, further confirming Snai2 expression in the MSPC compartment (Figure 1H and I).
Figure 1. Snai2 is specifically expressed in bone marrow MSPCs.
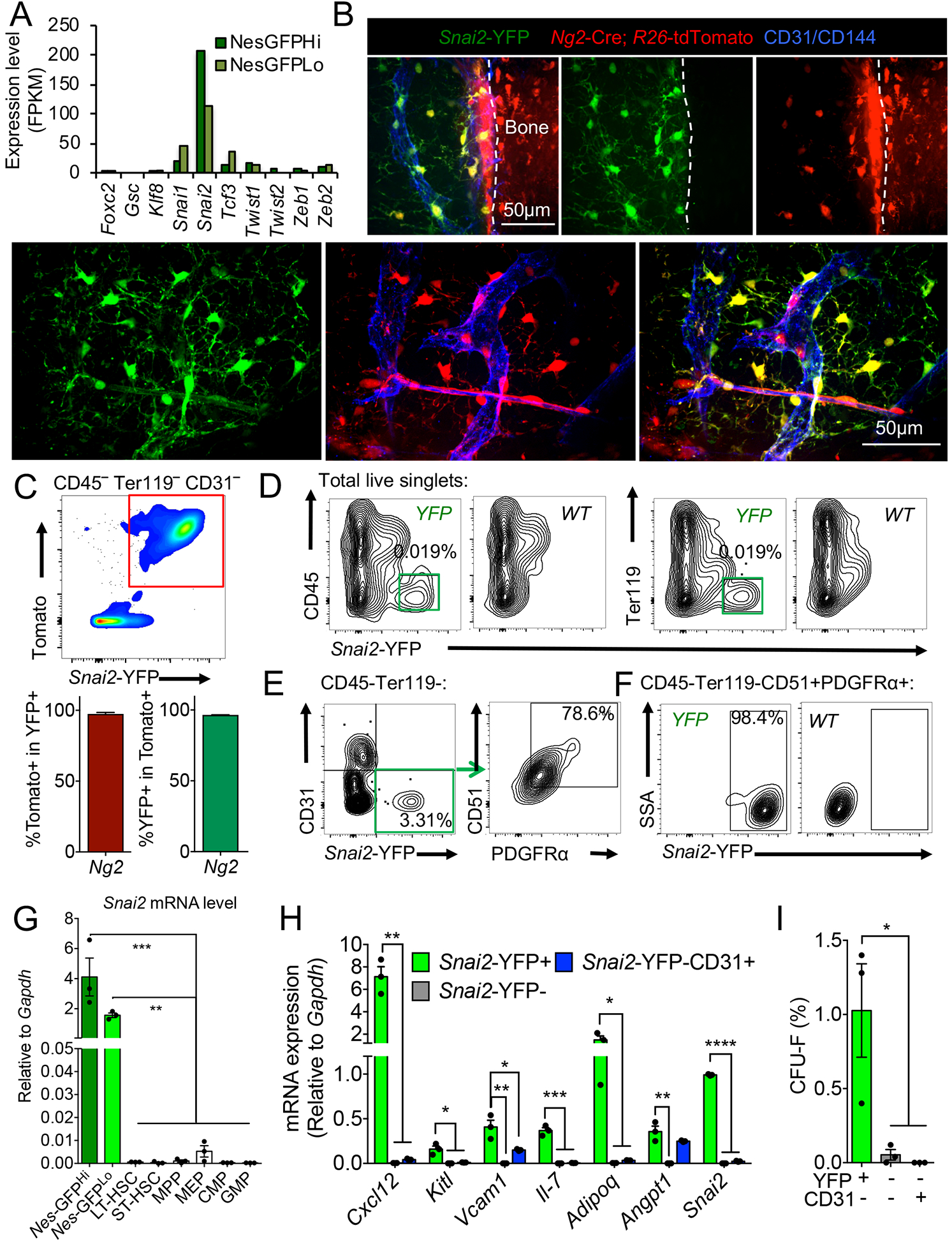
(A) Expression of EMT factors in Nes-GFPHi and Nes-GFPLo cells from previous RNA-seq analysis (Kunisaki et al., 2013). (B) Immunofluorescence images of whole-mount sterna from Snai2-YFP; Ng2-Cre; R26-tdTomato mice. A647-conjugated anti-CD31/CD144 antibodies were injected intravenously to label functional blood vessels. Scale bar=50μm. (C) FACS plot of CD45− Ter119− CD31− BM cells and quantifications showing nearly complete overlap between Snai2-YFP and Ng2-Cre; R26-tdTomato expression (n=3). (D) FACS plot showing restricted Snai2-YFP expression in the CD45− Ter119− fraction of the BM nucleated cells (BMNCs). (E) FACS plot showing Snai2-YFP labels mostly CD45− Ter119− CD31− CD51+PDGFRα+ MSPCs. (F) FACS plot showing that most CD45− Ter119− CD31− CD51+PDGFRα+ MSPCs are Snai2-YFP+. (G) qRT-PCR analysis of Snai2 mRNA expression in sorted MSPC and hematopoietic stem and progenitor populations (Nes-GFPhi and GFPlo: CD45− Ter119− CD31−Nes-GFPhigh and GFPlow respectively. LT-HSC: long term HSCs, ST-HSC: short term HSC, MPP: multipotent progenitors, MEP: megakaryocyte-erythrocyte progenitors, CMP: common myeloid progenitors, GMP: granulocyte-macrophage progenitors). (H) Niche factor mRNA levels in sorted CD45− Ter119− CD31− stromal populations from Snai2-YFP BM. (I) Colony forming unit-fibroblast (CFU-F) activity of sorted CD45− Ter119− CD31− stromal populations from Snai2-YFP BM.
Snai2 deletion reduces bone marrow MSPC numbers
To investigate Snai2’s function, we first analyzed BM MSPCs in previously described Snai2-deficient (Snai2LacZ, hence as Snai2−/−) mice (Jiang et al., 1998), bred with Nes-GFP transgenic mice to label MSPCs (Mendez-Ferrer et al., 2010). As these mice do not survive into adulthood on a pure C57/BL6 background, we intercrossed Snai2+/− with 129Sv wild-type mice to obtain 129Sv/C57BL/6 F1 generation and hence Snai2−/− and littermate control animals. At 8 weeks of age, Snai2−/− BM showed a mild reduction of cellularity compared to wild-type littermates (Figure 2A), and a reduced frequency and absolute number of CD45− Ter119− CD31− CD51+ PDGFRα+ MSPCs (Pinho et al., 2013) or Nes-GFP+ cells (Figure 2B–D). Furthermore, sorted Snai2−/− MSPCs formed significantly fewer and smaller mesenspheres (Figure 2E–G) and CFU-F colonies (Figure S1D and E), which indicate an impaired self-renewal capacity. Immunofluorescence examination of Snai2−/− bones revealed a reduced lining of the trabeculae by mature osteoblasts marked by osteocalcin, a noncollagenous secreted protein produced exclusively by osteoblasts (Figure 2H). We also observed an absence of adipocytes marked by BODIPY, a lipid droplet tracing dye, and Perilipin, the major lipid droplet coating protein (Figure 2I), consistent with the reported absence of white adipose tissues in Snai2−/− mice (Perez-Mancera et al., 2007). To investigate further if the reduced adipocytes originated from an MSPC defect, we challenged control and Snai2−/− mice with 6 Gy irradiation, an injury shown to induce adipogenesis from BM MSPCs in vivo (Mizoguchi et al., 2014). We found that Snai2−/− MSPCs differentiation into Perilipin+ adipocyte was markedly reduced (Figure S1F). In addition, micro-CT analysis of Snai2−/− femurs revealed that the bone formation was impaired as shown by significantly reduced mineralized trabecular and cortical bone volumes (Figure 2J, K). These data suggest that Snai2 deletion impairs the self-renewal capacity and multi-lineage output of BM MSPCs.
Figure 2. Snai2 deletion impairs BM MSPC pool size and activity.

(A) Quantification of total BMNCs from femurs of wild type (+/+) and Snai2-deficient (−/−) mice. (B-D) FACS plot of CD45− Ter119− CD31− BM cells and quantifications showing reduced CD51+PDGFR + (B) and Nes-GFP+ MSPCs (C) in the BM of Snai2− /− mice. EC, endothelial cells, defined as CD45− Ter119−CD31+. (E) Mesensphere-forming capacity of Snai2+/+ and Snai2− /− BM Nes-GFP+ MSPCs. (F) Sphere-forming units from femurs of Snai2+/+ and Snai2− /− mice. (G) Bright-field images of mesenspheres formed by Snai2+/+ and Snai2− /− BM Nes-GFP+ MSPCs. (H and I) immunofluorescence images from frozen sections and quantifications of Osteocalcin+ (Ocn+) osteoblasts (H) and BODIPY+ Perilipin+ adipocytes (I) in control and Snai2−/− femurs. Scale bars =100μm. (J) Micro-CT 3D reconstruction of femurs from control and Snai2−/− mice. (K) Micro-CT analysis of femurs from control and Snai2− /− mice (n=4 mice). BS, bone surface. BV, bone volume. TV, total (tissue) volume.
Snai2 deletion alters the HSC niche both at steady state and after stress
We next asked if the MSPC defects produced by Snai2 deletion resulted in alterations of hematopoiesis. Snai2−/− mice presented a mild anemia and a slight increase in white blood cell counts and platelet numbers in blood (Figure S1G). The frequency and absolute number of Lineage− Sca1+ ckit+ CD150+ CD48− HSCs (Figure 3A) and CD45− CD11b− CD71+ Ter119+ erythroblasts (Figure S2A) were significantly reduced in Snai2−/− BM compared to controls. Cell cycle analysis indicated that higher proportion of HSCs resided in the G0 phase in Snai2−/− BM compared to controls (Figure 3B), suggesting increased quiescence.
Figure 3. Snai2 deletion impairs BM HSC niche at steady state and after stress.
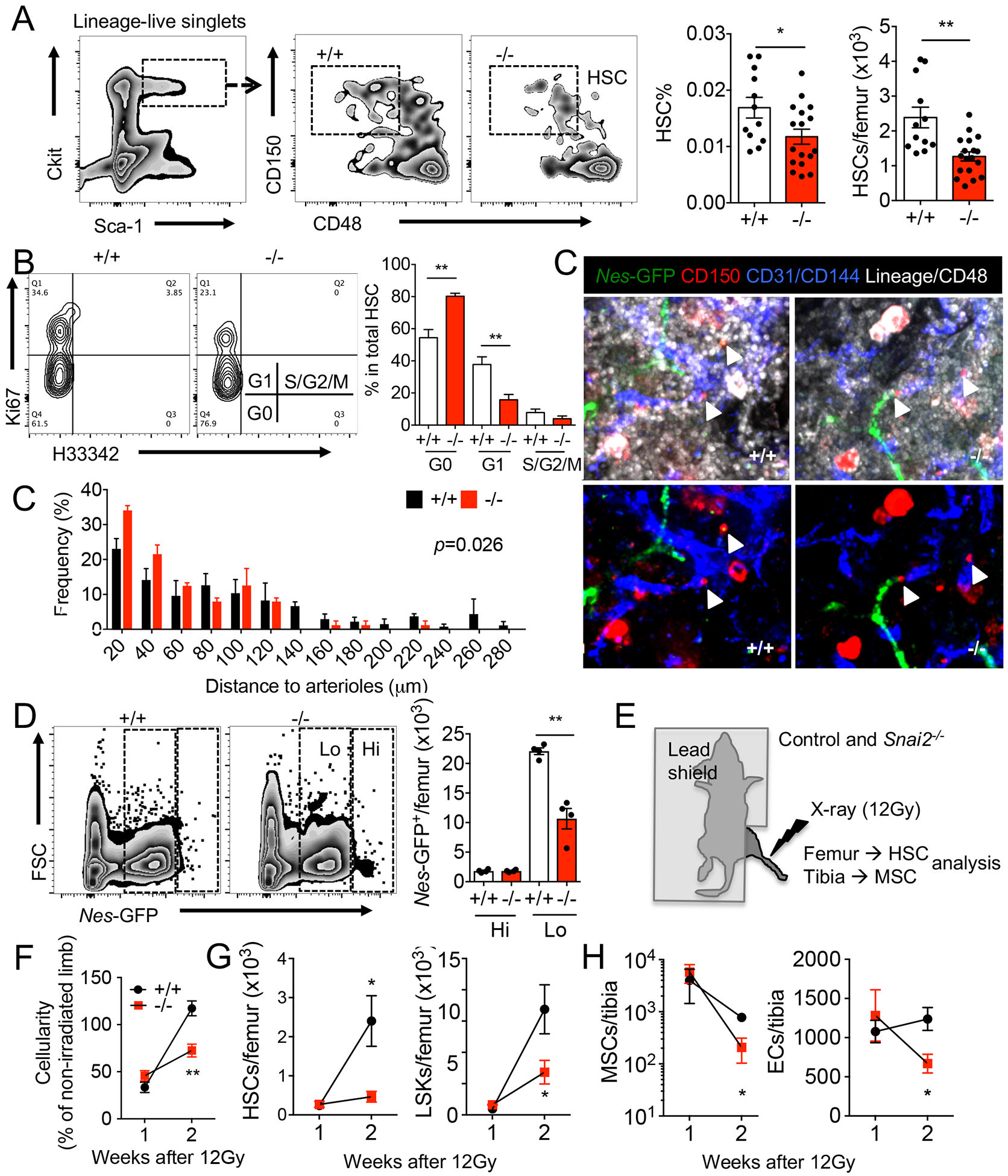
(A) FACS plots and quantification of lineage−ckit+ Sca1+ CD150+ CD48− HSCs in steady state BM of Snai2+/+ and Snai2− /− mice. (B) Cell cycle profiling of control and Snai2− /− BM HSCs by Ki-67 and H33342 staining. n=7 biological samples per group. (C) Whole mount immunofluorescence images of control and Snai2− /− mouse sternum and localization of HSCs (Lineage− CD48− CD150+) observed in situ relative to Nes-GFPHi arterioles. p value was determined by two-sample Kolmogorov-Smirnov test. (D) FACS plots and quantification of the absolute numbers of Nes-GFPHi and Nes-GFPLo cells in control and Snai2− /− mouse bone marrow. (E) Experimental design of targeted limb irradiation of control and Snai2−/− mice. (F-H) BM cellularity (F), HSC and LSK (lineage−Sca-1+Ckit+) (G) and MSPC and EC numbers (H) in irradiated legs of control and Snai2− /− mice. n=3–5 biological samples per group.
Previous studies have reported that Snai2-null mice die from total body irradiation at a dose that wild-type control mice could survive and attributed it to hematopoietic cell-intrinsic defects (Inoue et al., 2002; Perez-Losada et al., 2003; Wu et al., 2005). By contrast, we have found that wild-type BM donor cells could not rescue lethally irradiated Snai2−/− recipients and anemia was observed in the dying Snai2−/− recipient animals (Figure S2C–E), suggesting a defective BM microenvironment during recovery. This defect was not due to impaired homing of donor cells into the Snai2−/− BM as the number of migrated hematopoietic cells in lethally irradiated mutant and wild-type mice was similar (Figure S2F). Similarly, most Snai2−/− mice died after a single dose of 250 mg/kg 5-flurouracil (5FU) challenge while all wild-type littermates survived (Figure S2G). We found no significant difference in rates of MSPC apoptosis (Figure S2H) one day after 5FU. Moreover, in radiation chimeras in which the recipients are wild type, Snai2−/− BM cells did not show any deficit in recovery after 5FU (Figure S2I). These functional results, combined with the expression data, clearly indicate that SNAI2 is acting on the non-hematopoietic compartment and is critical in the recovery after genotoxic insults.
We then surmised that the impaired MSPC niche in Snai2−/− BM led to reduced HSC self-renewal and retarded BM recovery after myeloablation. We evaluated HSC distributions relative to various quiescence-regulating niches in the Snai2−/− BM by confocal immunofluorescence imaging (Bruns et al., 2014; Kunisaki et al., 2013; Pinho et al., 2018; Zhao et al., 2014; Zhao et al., 2019), and found significantly closer association of HSCs with Nes-GFPhigh MSPC-associated arterioles, but not with megakaryocytes or bones (Figure 3C and S2J and K). Interestingly, we observed a disproportional loss of Nes-GFPlow sinusoidal and interstitial MSPCs but not the Nes-GFPhi MSPCs in the Snai2−/− BM (Figure 3D and S2L). This also suggests that Snai2 is required in Nes-GFPlow mesenchymal niche cells but not in the hematopoietic or bone-associated stromal populations (Figure 1B and Figure S1A and C). To further exclude the possibility that hematopoiesis might be affected indirectly by Snai2 expression outside of BM, we carried out targeted irradiation studies in which we irradiated one limb of the control or Snai2−/− mice using CT-guided X-ray such that only a small localized fraction of the total bone marrow was ablated (Figure 3E). One week after targeted irradiation, control or Snai2−/− mice showed similar numbers of live hematopoietic and stromal cells. However, at two weeks post-irradiation, the irradiated limbs from wild-type mice showed full recovery in cellularity (~120% of the non-irradiated limb) whereas Snai2−/− limbs exhibited significantly lower cellularity (~75% of the non-irradiated side; Figure 3F). The HSC and LSK numbers of the control limbs also recovered to non-irradiated levels at two weeks while their recovery in Snai2−/− limbs remained significantly impaired (Figure 3G). This was associated with a severe reduction of the MSPCs and endothelial cells in the irradiated Snai2−/− limbs (Figure 3H). Therefore, our results indicate that the recovery defects of Snai2−/− animals are not due to the impaired cell survival after stress, but rather to a compromised recovery of niche cells.
Gene expression changes in Snai2-deficient MSPCs
To gain mechanistic insights on Snai2-mediated regulation of BM MSPC activity, we conducted a transcriptome analysis on sorted control and Snai2−/− BM MSPCs. We detected 154 significantly up-regulated and 85 down-regulated (p value<0.05, counts per million>20, fold change>2) genes in Snai2−/− MSPCs compared with controls. Ingenuity Canonical Pathway analysis revealed that the most differentially expressed genes were enriched in extracellular matrix (ECM) signaling and remodeling, fibrosis and integrin signaling (Table S1–S2). An unbiased Gene Set Enrichment Analysis (GSEA) also identified a significant up-regulation of genes in ECM-receptor interaction in Snai2−/− MSPCs (Figure S3A). GSEA further revealed that genes involved in osteogenesis and chondrogenesis were highly up-regulated (Figure S3B) while adipogenesis was not significantly changed (Figure S3C), suggesting an altered mesenchymal differentiation program. By contrast, genes encoding HSC niche factors, such as Cxcl12, Kitl, Angpt1, Vcam1, and Il7,, were minimally affected by Snai2 deletion with the exception of Spp1(osteopontin) which was highly up-regulated and confirmed by RT-PCR (Figure 4A and B and S3D). Snai2 was reported to antagonize the p53-mediated apoptosis pathway in other cell types (Kurrey et al., 2009; Wu et al., 2005). Although we detected a significant up-regulation of p53 targets (Kannan et al., 2001) in Snai2−/− MSPCs, a reported SNAI2 targets involved in DNA damage response, Puma (encoded by Bbc3) (Wu et al., 2005), was unchanged (Figure S3E and F). Cdkn1a, encoding the cyclin-dependent kinase inhibitor p21Waf1/Cip1 and a known SNAI2 target (Chen and Gridley, 2013), was significantly up-regulated (Figure S3F) while the expression of Foxc1, a critical transcription regulator of MSPC differentiation (Omatsu et al., 2014), was unchanged (Figure S3D).
Figure 4. Snai2 regulates adult BM MSPC via suppression of Spp1.
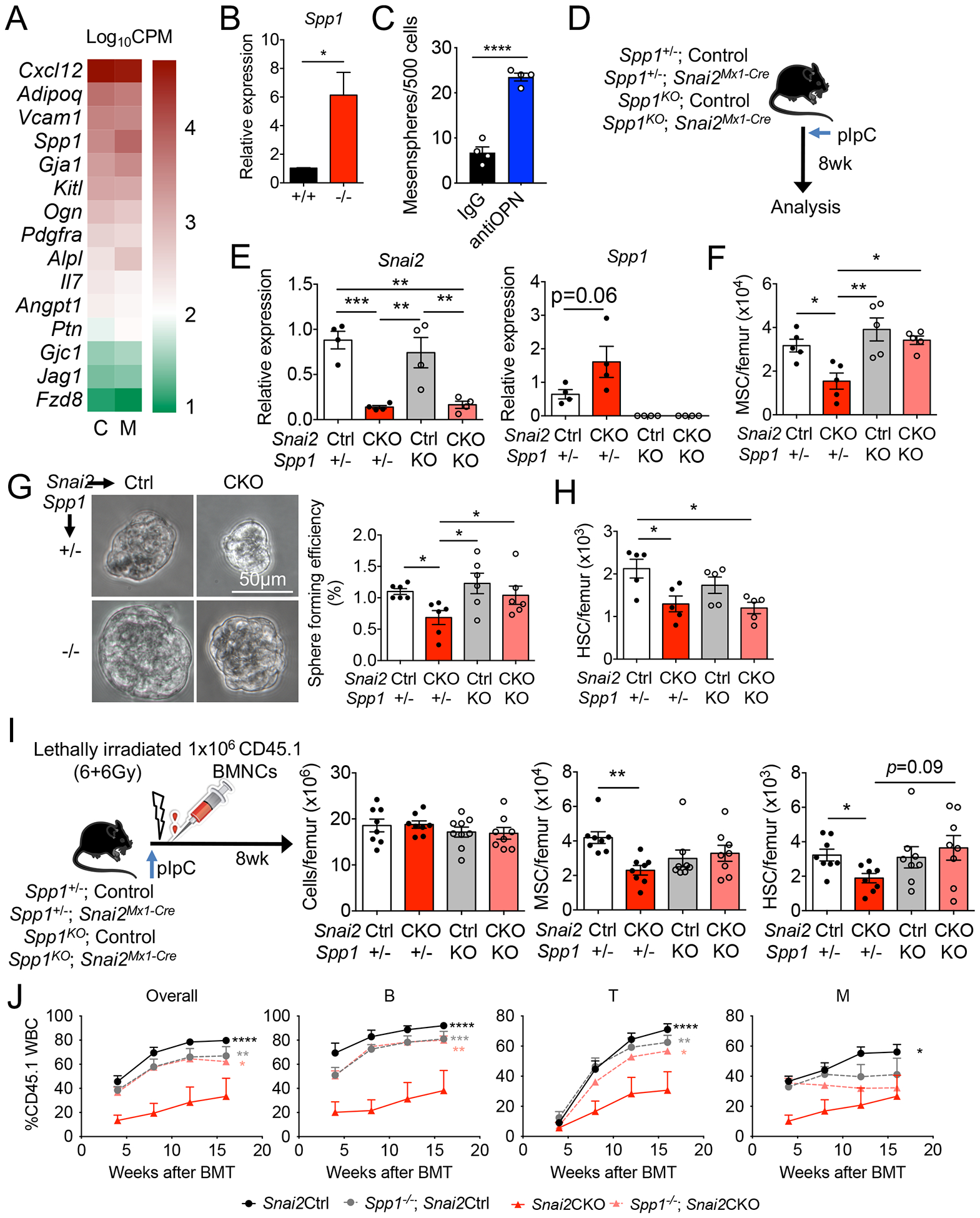
(A) RNA-seq analysis of niche factor gene expression in control (C) and Snai2− /− (M) Nes-GFP+ MSPCs. n=3 per group. Gene expression levels are shown as Log10CPM (counts per million). (B) qRT-PCR analysis of Spp1 mRNA in sorted Nes-GFP+ MSPC from control and Snai2−/− femurs. n=4 per group. (C) Mesensphere formation assay using sorted wild-type BM Nes-GFP+ MSPCs in the presence of 10μg/ml anti-OPN or goat IgG. n=4 biological samples per group from two independent experiments. (D) Experimental scheme of inducible deletion of Snai2 in Snai2Mx1-Cre; Spp1− /− compound knockout mice. (E) qRT-PCR analysis of Snai2 and Spp1 mRNA in sorted CD45− Ter119− CD31− CD51+PDGFRα+ MSPC from Snai2Mx1-Cre; Spp1− /− knockout femurs. (F) MSPC numbers in Snai2Mx1-Cre; Spp1−/− compound knockout mice. (G) Bright field images and quantification of mesensphere forming efficiency of sorted BM MSPCs from Snai2Mx1-Cre; Spp1−/− compound knockout mice. (H) Quantification of HSC numbers in Snai2Mx1-Cre; Spp1− /− mice. (I) Experimental design and analysis of lethally irradiated Snai2Mx1-Cre; Spp1−/− mice reciprocally transplanted with wild-type (CD45.1) BM donor cells. (J) Peripheral blood analysis of donor-derived leukocytes (CD45.1) in lethally irradiated recipients transplanted with equal volume of BM cells from (I) (CD45.1) and WT competitors (CD45.2). n=4 mice per group. Statistical analysis was done using multiple comparisons Two-way ANOVA and significance was shown comparing Snai2CKO to all the other groups.
Snai2 regulates adult BM MSPCs via suppression of osteopontin
Based on these results, we focused on Spp1 (osteopontin or OPN) as a candidate downstream effector of Snai2 in BM niche regulation. Previous studies have suggested that Snai2 directly binds to the Spp1 promoter (Kurrey et al., 2009). OPN is an extracellular matrix glycoprotein previously suggested to be a negative regulator of BM HSC number and ageing (Guidi et al., 2017; Nilsson et al., 2005; Stier et al., 2005). Although it is highly enriched in the endosteum (McKee et al., 1993), Spp1 transcript is mostly expressed by subsets of mesenchymal stromal cells that are found throughout the BM (Tikhonova et al., 2019). In addition, MSPCs express receptors (integrins and CD44) for OPN (Denhardt et al., 2001) (Figure S3G), enabling a potential autocrine signaling. Yet, the role of OPN in MSPC activity is unclear. We thus reasoned Snai2 might regulate MSPC self-renewal and HSC number via Spp1 suppression. To test this hypothesis, we cultured wild-type BM MSPCs at clonal density in the presence of an OPN-blocking antibody (Kiefer et al., 2010). We found that OPN blockade markedly increased the ability to form mesenspheres compared to the IgG control group (Figure 4C), suggesting that extracellular OPN may play an autocrine inhibitory role in MSPC self-renewal.
To overcome the lethality of Snai2 deletion in the germline, we generated a floxed allele of Snai2 (Snai2floxed) using CRISPR/Cas9 technology (Figure S3H) and crossed it with Mx1-Cre (Snai2floxed/floxed; Mx1-Cre, hereafter as Snai2Mx1-Cre) to knockout Snai2 postnatally (Figure 4D). Polyinosine-polycytidylic acid (pIpC) injections induced efficient deletion of Snai2 in adult BM MSPCs of Snai2Mx1-Cre mice as previously described (Figure 4E) (Park et al., 2012). Consistent with Snai2−/− mice, postnatal Snai2 deletion resulted in significant reductions of BM MSPC numbers, mesensphere-forming capacity, up-regulation of Spp1 mRNA and reduction in BM HSC numbers (Figure 4E–H), while BM cellularity and MSPC niche factor expression were not affected (Figure S4A–C). These results confirmed that Snai2 is required for not just the development of BM MSPCs but also their maintenance during homeostasis. We then crossed Snai2Mx1-Cre mice with Spp1 knockout (Spp1−/−) animals to evaluate the impact of OPN on the phenotype of Snai2-deficient mice. We found that in the absence of OPN, MSPC numbers and sphere-forming capacity were rescued and Cdkn1a upregulation after Mx1-Cre-induced Snai2 deletion was also reversed (Figure 4E–G and S4D), clearly indicating that Snai2 suppression of Spp1 expression is critical for BM MSPC self-renewal and maintenance. Interestingly, BM HSC numbers were reduced in Snai2 conditional knockout mice (CKO) regardless of Spp1 expression (Figure 4H), suggesting that other factors might contribute to the reduced HSC niche function of Snai2-deficient MSPCs.
To further examine the role of Snai2 and Spp1 in BM regeneration and exclude the potential contribution of Snai2 expression in hematopoietic cells, we subjected the pIpC-induced Snai2Mx1-Cre; Spp1−/− mice to lethal irradiation and reciprocal BM transplantation using CD45.1 wild-type donor cells (Figure 4I). Snai2Mx1-Cre CKO mice were viable after reciprocal BM transplant and their BM cellularity were able to recover to near the wild-type levels (Figure S4E–G). However, the BM MSPC numbers were significantly reduced in Snai2 CKO (Figure S4G), despite a complete replacement of their BM by wild-type hematopoietic cells. Reciprocally transplanted Snai2 CKO mice also displayed reduced HSC numbers (CD45.1) in their BM (Figure S4H), which was confirmed by reduced repopulation in secondary competitive BM transplantation (Figure S4I). Analysis of the CKO MSPCs revealed unchanged niche factor but up-regulated Spp1 and Cdkn1a mRNA expression (Figure S4J), consistent with our observations in Snai2−/− mice. Importantly, the reduction in HSC repopulation activity was due to Snai2 deletion in niche cells—not HSCs— because when HSCs were sorted from Snai2CKO BM, they performed slightly better than those from control mice (Figure S4K), consistent with their more quiescent cell cycle profile (Figure 3B). Intriguingly, Snai2Mx1-Cre; Spp1−/− mice displayed wild-type levels of BM MSPC (Figure 4I), indicating that Snai2-mediated Spp1 suppression in MSPCs is critical for BM recovery. In addition, reduced HSC number in Snai2Mx1-Cre mice was also restored upon Spp1 deletion, as shown by both phenotypic FACS analysis (Figure 4I), secondary competitive BM transplantation (Figure 4J), and normalized repopulation activity from isolated HSCs (Figure S4K). Altogether, our results indicate that Snai2 regulates BM MSPC and HSC activities via suppression of Spp1 in BM MSPCs.
Discussion
The molecular basis of BM MSPCs’ maintenance and multi-lineage potency remains poorly understood. MSPCs originate at least in part from the neural crest (Isern et al., 2014; Morikawa et al., 2009), a developmental cell population that gives rise to diverse lineages including peripheral neurons, muscle and bone. The formation of neural crest involves the EMT process (Kalcheim, 2015), and studies in lower vertebrates have implicated a critical role of the EMT transcription factor Snai2 in the formation of these neural-crest derivatives (Mancilla and Mayor, 1996; Nieto et al., 1994). Although Snai2 is also expressed in migratory neural crest cells of mouse embryos, it is not required for the formation of these cells (Jiang et al., 1998), indicating an evolutionarily distinct functions. Here, our results indicate that Snai2, is essential for maintaining the functional pool of MSPCs in adult BM. Deletion of Snai2 results in reduced MSPC numbers and impaired MSPC self-renewal and lineage output. This is associated with an altered lineage differentiation gene signature, up-regulation of p53 target genes and fibrosis-related genes. Thus, Snai2 represents a master regulator of MSPC’s multipotent state in part by suppressing aberrant differentiation or senescence program.
MSPCs can gain the capacity to induce a hematopoiesis-supporting marrow and maintain definitive HSCs. The present data raise the possibility that Snai2 may take part of the regulatory network ensuring HSC niche function and this activity is not mediated via the modulation of the niche factor CXCL12 or SCF. It is notable that Snai2 deletion resulted in a preferential loss of Nes-GFPLo, ubiquitously distributed, peri-sinusoidal MSPCs, but relative enrichment of Nes-GFPHi peri-arteriolar niche cells. This may explain the alterations in HSC quiescence status since the peri-arteriolar niche contribute to maintaining HSC quiescence (Asada et al., 2017; Kunisaki et al., 2013; Pinho et al., 2018).
Our results have uncovered an autocrine function of OPN in MSPC regulation and Snai2 regulates the BM MSPC pool size at least in part via suppression of OPN. OPN is one of the most abundantly deposited glycoprotein in the BM extracellular matrix and a negative regulator of BM HSPC pool size via microenvironment-mediated mechanisms that have not been clearly defined (Nilsson et al., 2005; Stier et al., 2005). It is intriguing that MSPCs are a major producer of OPN in the BM based on mRNA abundance and they express receptors for OPN as well. Our results suggest that, in addition to a direct restrictive role on HSC expansion, OPN might also restrict HSC numbers by restricting BM MSPC numbers. Our findings also indicate that OPN’s autocrine inhibitory role on MSPC self-renewal may be a potential mechanism by which BM MSPCs sense the need from the BM microenvironment to regenerate. In summary, our study identifies a Snai2-Spp1 axis regulating BM MSPC self-renewal and HSC niche function.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by: Paul S. Frenette (paul.frenette@einsteinmed.org).
Materials Availability
This study has generated a Snai2floxed mouse line, which can be made available upon request and requires a Material Transfer Agreement (MTA).
Data and Code Availability
Raw and processed reads data from the RNA-seq have been deposited in the Gene Expression Omnibus under accession number GSE142705.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Snai2−/−, previously as SlugLacZ, (Jiang et al., 1998) and Snai2YFP knockin (Guo et al., 2012) mice are previously described. Snai2floxed mice were generated at the Gene Modification Facility of Albert Einstein College of Medicine as described below. C57BL/6 (CD45.2) and Bl6-Ly5.1 (CD45.1) mice were purchased from Charles River Laboratories (Frederick Cancer Research Center, Frederick, MD)/NCI or the Jackson Laboratories (B6.SJL-Ptprca Pepcb/BoyJ). Ng2-Cre (B6; FVB-Tg(Cspg4-cre)1Akik/J), Lepr-Cre (B6.129-Leprtm2(cre)Rck/J), R26-tdTomato (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J), Mx1-Cre (B6.Cg-Tg(Mx1-cre)1Cgn/J) and Spp1− /− (Spp1Tm1Blh/J) mice were from Jackson Laboratories. All animals were housed in specific pathogen-free barrier facility and all experimental procedures were approved by the Animal Care and Use Committee of Albert Einstein College of Medicine. All experiments or start of treatments were performed on mice of both genders with littermate controls from the same colony between 8–16 weeks of age.
Generation of Snai2floxed mice by CRISPR/Cas9
Two loxp sites were inserted into the endogenous Snai2 locus sequentially by CRISPR/Cas9 technology. gRNAs targeting to intron 1 (gRNA1: ggagattgctgctcaggcga ggg) and Exon 3 un-translated region (gRNA2: ttactgacagctagattgaa agg) of mouse Snai2 gene respectively were designed by an online tool (http://crispr.mit.edu/) and generated by in vitro transcription (Yang et al., 2013). Cas9 mRNA was purchased from SBI. The single-stranded Snai2 conditional knockout homologous recombination donor (HRDs) carrying 60nt homologous arms at each side and surrounded loxp sites (HRD1: ttctctgagcctgtgtgtacgcaaaaggagtgcacggacgtgatcgcacccttcccctcg TCTGATAACTTCGTATAATGTATGCTATACGAAGTTATtgagcagcaatctcctgaagccaagcacttgggaaagcagctggattcgtgcatgccatt, HRD2: gtacttaaagttaattcgttctatgtgaagtttaaaattatatttactgacagctagattATAACTTCGTATAATGTATGCTATACGAAGTTATcgataccgtcgacctcggaaaggataaagataagaat ctttctctttaaagatgaagtgaaaagcacattgcatct) were synthesized from IDT. C57BL6 female mice (3–4 weeks old) were super-ovulated by intra-peritoneal PMS (5 IU/mouse day1) and HCG (5 IU/mouse day 3) injections and mated to C57BL6 males. Fertilized embryos were collected from oviducts. The gRNAs, Cas9 mRNA and Snai2 conditional knockout HRDs were mixed and microinjected into the cytoplasm of fertilized eggs. The injected zygotes were transferred into pseudopregnant CD1 females, and the resulting pups were obtained and genotyped by sequencing PCR clones from the targeted region. Mice successfully inserted 3’ loxp site were obtained first and expanded as donors for 5’ loxp insertion. Mice successfully incorporated both loxp sites were bred to C57BL6 mice to remove potential off-targets and establish the Snai2floxed colony. The top ten predicted off-target sites were examined by targeted sequencing. A null allele of Snai2 was also generated as a by-product of the gRNA injections. Once established, the Snai2floxed allele was genotyped by ear clip genomic DNA PCR using primers: Snai2 5’GTF: tgagatctttgctgacaaggaa and Snai2 5’GTR: ctagctgtaccgtgcctgtg (WT: 196bp, Snai2floxed: 233bp). Snai2 Rev_3: tgatgacaaccaggcatcat and Snai2 FOR_6: tgacaaatgaaagtccaaagaca (WT: 500bp, Snai2floxed: 550bp).
Primary cell culture
For CFU-F assays, 1–3×103 sorted Nes-GFP+ or CD45− Ter119− CD31− CD51+PDGFRα+ MSPCs were seeded per well in a 6-well adherent tissue culture plate using phenol-red freeα-MEM (Gibco) supplemented with 20% FBS (Hyclone), 10% MesenCult stimulatory supplement (Stem Cell Technologies) and 0.5% penicillin-streptomycin. One-half of the media was replaced after 7 days and at day 14 colonies were stained with Giemsa staining solution (EMD Chemicals) and numerated under a bright field microscope. For mesensphere assays, MSPCs were plated in sphere media (15% chicken embryo extract, 0.1mM 2-Mercaptoethanol, 1% non-essential amino acids, 1% Pen-Strep, 1% N2 and 2% B27 supplements (Gibco) in 1:2 DMEM/F12: human endothelial-SFM, filter-sterilized and supplemented with 20 ng/ml fibroblast growth factor (FGF)-basic, insulin-like growth factor-1 (IGF-1), epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and oncostatin M (OSM) (Peprotech) at clonal density (<500 cells/cm2) in ultralow adherent-culture dishes (Stem Cell Technology) as previously described (Pinho et al., 2013). Once plated, cells were kept at 37°C 5% CO2 in a water jacketed incubator untouched for 1 week to prevent cell aggregation. Half media change was performed at 1 week and spheres with >4 cells were numerated at day 9. For testing the relevance of OPN in mesensphere formation, MSPCs were sorted at 500 /well into 96-well ultralow attachment plates (Corning #3474) in sphere media supplemented with 10 μg/ml anti-OPN (R&D#: AF808) or goat IgG.
METHOD DETAILS
Flow cytometry and cell sorting
Bone marrow stromal cells were isolated as described previously with minor modifications (Kunisaki et al., 2013). Briefly, bone marrow plugs were flushed out by inserting a 21G needle attached to a 1 ml syringe filled with digestion buffer (1 mg/ml Collagenase IV, 2 mg/ml Dispase and 1 mg/ml DNase I in PBS) and then incubated for ~30 min in the digestion buffer with gentle rocking at 37°C. Single cell suspension was then generated by gently inverting the tubes. When stromal components are not required for analysis, femurs were flushed gently with 1 ml of ice-cold PEB (PBS/2 mM EDTA / 0.5% BSA) buffer using a 1 ml syringe (BD) into single cell suspension by passing through a 21G needle (BD) into FACS tubes. The single cell suspension was then pelleted, and RBCs lysed by an ammonium chloride solution. The resulted BM nucleated cells (BMNCs) were then identified and counted as trypan blue excluding cells under the microscope. When analyzing erythroblasts, the BM was processed as described before (Wei et al., 2019). About 2×106 BMNCs were then processed for each FACS staining. Stained sample suspensions were acquired on an LSR II flow cytometer. DAPI-negative singlets were analyzed for all live samples unless otherwise specified. For sorting, samples were processed under sterile conditions and sorted on a FACSAria II (BD). Data were analyzed using FACS Diva (BD) or FlowJo (Tree Star) software.
Complete blood count
Mice were bled ~25 μl into an Eppendorf tube containing 2 μl of 0.5 M EDTA (Life Technologies) using heparinised micro-haematocrit capillary tubes (Fisherbrand) under isoflurane anaesthesia. Blood was diluted 1:20 in PBS and analysed on an Advia counter (Siemens).
Immunofluorescence imaging
Whole-mount sternum HSC immunofluorescence staining and imaging analysis were performed as previously described (Bruns et al., 2014; Kunisaki et al., 2013; Pinho et al., 2018). Briefly, Alexa Fluor 647-anti-VE-Cadherin (BV13) (Biolegend) and APC-anti-CD31/PECAM-1 (MEC13.3) were infused 10 min before sacrificing the mice to label vasculature in vivo. Sternum segments were then harvested, cleaned and bisected sagittally with a surgical blade into individual halves to expose the marrow cavity. Fragments were re-fixed with 4% PFA; blocked and permeabilized in PBS with 10% normal goat serum and 0.5% Triton X-100 and stained with primary antibodies (biotin anti-Lineage panel cocktail, biotin anti-CD48 and PE anti-CD150) for 3 days. After three PBS washes, the tissues were then incubated with streptavidin eFluor 450 for 2 h. Images were acquired on a ZEISS Axio examiner D1 microscope with a confocal scanner unit, CSUX1CU (Yokogawa) and analyzed using Slide Book software. After image acquisition, the Euclidean distance of each CD150+ CD48− Lineage− HSC to the closest arteriole/megakaryocyte/bone was measured in Slide Book software to generate distribution maps. For cryosections, tissues were fixed with 4% PFA via perfusion, and femoral or tibial bone tissues were further fixed with 4% PFA after dissection for 30 min at 4°C. Fixed bones were then incubated in 10%, 20%, and 30% sucrose each for 1 h at 4°C for cryoprotection and embedded in 5% carboxymethyl cellulose (SECTION-LAB). Sections, 20–30 μm thick, were prepared using Kawamoto’s film method (Kawamoto and Shimizu, 2000). Alexa Fluor 488-anti-GFP (Molecular Probes) was used for enhancement of the Snai2-YFP signal. Anti-mouse osteocalcin antibody (R21C-01A, Takara) was used to stain osteoblasts and anti-perilipin antibody (D1D8, Cell Signaling) and BODIPY 493/503 (Molecular Probes) for adipocytes.
Micro-CT analysis
Micro-CT analysis of mouse femurs was done as previously described with minor modifications (Hanoun et al., 2014). Bones were fixed in 4%PFA for 48 h and then kept in PBS at 4°C until scan. The bone samples were scanned using a high-resolution SkyScan micro-CT system following a standard protocol. Images were acquired using a 10 MP digital detector, 10W power energy (100 kV and 100 μA) and a 0.5 mm aluminium filter. X-ray projections were generated from the sample obtaining both transverse and coronal slices with a 6.6 μm image pixel size. Trabecular and cortical regions were defined as positions along the long axis of the femur relative to the growth plate reference (trabecular ROI: 0.215–1.94 mm and cortical ROI: 2.15–2.58 mm from growth plate). Morphometric analysis and 3D reconstruction were done using the SkyScan CT-analyzer software.
In vivo treatments and bone marrow transplantation
For 5-FU challenge, a single dose (250 mg/kg body weight) of freshly made solution (12.5 mg/ml in sterile PBS) was given i.v. to each mouse under isoflurane anaesthesia. PolyI:C (Invivogen) was administered intra-peritoneally (i.p.) every other day at 5 mg/kg for 5 doses for Mx1-Cre induction. For reciprocal BM transplantation, control and Snai2CKO mice (CD45.2) were lethally irradiated (600+600 cGy, at least 3 h apart) in a Shepherd Mark-1 137Cs irradiator and retro-orbitally injected with 1 × 106 RBC-lysed WT bone marrow nucleated cells (CD45.1) under isoflurane anaesthesia. Similarly, irradiated C57Bl/6 mice were transplanted with 1 × 106 C57Bl/6 BM cells (CD45.2) at the same time to provide competitor BM cells during secondary competitive bone marrow transplant (BMT) assays. For competitive BMT assays, 1 × 106 BM cells (CD45.2) from aforementioned competitor mice and equal volume of test bone marrow cells (CD45.1) were mixed before injection. For HSC transplant assays, 200 sorted HSCs (CD45.1) from reciprocally transplanted control and Snai2CKO mice were mixed with 0.4 × 106 BM cells (CD45.2) from aforementioned competitor mice before injection into lethally irradiated WT recipient mice (CD45.2).
Homing assay
Bone marrow nucleated cells (5 × 106 cells) from wild type mice (CD45.1/CD45.2) were transplanted into lethally irradiated Snai2+/+ and Snai2−/− recipient mice. Recipients were sacrificed at 3 h post transplantation and femurs and spleens were processed for FACS analysis.
Targeted limb irradiation
Animals were anesthetized by an intraperitoneal injection of ketamine/xylazine mix (80–100 mg/10 mg/kg) prior to irradiation using the Small Animal Radiation Research Platform, SARRP (XStrahl, Surrey, UK). The orthovoltage x-ray unit operates at 220 kVp and 13 mA. Prior to irradiation, a cone-beam computed tomography (CBCT) or static x-ray scan was acquired using 50 kVp and 0.7 mA tube current with Al filtration. Mice were maintained in a circular lucite jig at a source-surface distance (SSD) of 65cm for 771 seconds with whole body lead shielding (to protect the individualized compartments from unwanted irradiation) and a 7.5 mm port through which secured right limbs protruded and were irradiated to 12 Gy in a single fraction.
RNA isolation and quantitative real time PCR
Sorted cells were directly collected in 100 μl of lysis buffer and RNA isolation was performed using the Dynabeads® mRNA DIRECT™ Micro Kit following the manufacturer’s instructions (Invitrogen). Reverse transcription was performed using the RNA to cDNA EcoDry™ Premix system (Clontech). Quantitative real-time PCR was performed as previously described using SYBR Green (Roche) on a QuantStudio 6 Flex Real-time PCR System (Applied Biosystems). The relative mRNA abundance was calculated using the ΔCt method using the expression of Gapdh as the internal control. Primer sequences can be found in the Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD51 PE | eBioscience | Cat# 12-0512-83 |
| anti-mouse CD140a APC | eBioscience | Cat# 17-1401-81 |
| anti-mouse Ly-6A/E (Sca1) APC | eBioscience | Cat# 17-5981-83 |
| anti-mouse Ly-6A/E (Sca1) PE-Cy7 | eBioscience | Cat# 25-5981-82 |
| anti-mouse Ly-6A/E (Sca1) Alexa Fluor 700 | eBioscience | Cat# 56-5981-82 |
| anti-mouse CD117 (c-Kit) BV 421 | Biolegend | Cat#105828 |
| anti-mouse CD117 (c-Kit) PE-Cy7 | Biolegend | Cat#105814 |
| anti-mouse CD48 biotin | eBioscience | Cat#13-0481-85 |
| anti-mouse CD48 PerCP-eFluor 710 | eBioscience | Cat#46-0481-82 |
| anti-mouse CD48 FITC | eBioscience | Cat#11-0481-82 |
| anti-mouse CD41 APC | eBioscience | Cat#17-0411-82 |
| anti-mouse CD41 FITC | eBioscience | Cat#11-0411-81 |
| anti-mouse CD45.2 PE | eBioscience | Cat#12-0454-82 |
| anti-mouse CD45.2 PerCP-Cy5.5 | eBioscience | Cat#45-0454-82 |
| anti-mouse CD45.1 FITC | eBioscience | Cat#11-0453-85 |
| anti-mouse CD45.1 APC eFluor 780 | eBioscience | Cat#47-0453-82 |
| anti-mouse CD4 PE-Cy7 | eBioscience | Cat# 25-0041-82 |
| anti-mouse CD8a PE-Cy7 | eBioscience | Cat# 25-0081-82 |
| anti-mouse B220 APC eFluor 780 | eBioscience | Cat# 47-0452-82 |
| anti-mouse CD11b Alexa Fluor 647 | Biolegend | Cat# 101218 |
| anti-mouse CD31 Alexa Fluor 647 | Biolegend | Cat# 102516 |
| anti-mouse CD144 Alexa Fluor 647 | Biolegend | Cat# 138006 |
| anti-mouse Ki-67 eFluor 660 | eBioscience | Cat# 50-5698-82 |
| anti-mouse CD34 PE | Biolegend | Cat# 119308 |
| anti-mouse FcRII/III PerCP-eFluor 710 | eBioscience | Cat# 46-0161-82 |
| anti-mouse CD150 PE | Biolegend | Cat# 115904 |
| anti-mouse Ter119 PerCP-Cy5.5 | eBioscience | Cat# 45-5921-82 |
| anti-mouse Lineage panel cocktail biotin | BD Biosciences | Cat# 559971 |
| Streptavidin APC eFluor 780 | eBioscience | Cat# 47-4317-82 |
| Streptavidin eFluor 450 | eBioscience | Cat# 48-4317-82 |
| Streptavidin BV 570 | Biolegend | Cat# 405227 |
| anti-mouse osteocalcin | Takara | Cat# R21C-01A |
| anti-perilipin | Cell Signaling | Cat# D1D8 |
| Mouse Osteopontin (OPN) Affinity Purified Polyclonal Ab | R&D Systems | Cat# AF808 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BODIPY 493/503 | Molecular Probes | Cat# D3922 |
| 5-Fluorouracil (5-FU) | Sigma-Aldrich | Cat# F6627 |
| PolyI:C | Invitrogen | Cat# tlrl-pic |
| DAPI (4’,6-diamino-2-phenylindole) | Sigma-Aldrich | D9542 |
| Hoechst 33342 | Sigma-Aldrich | B2261 |
| Collagenase IV | Gibco | Cat# 17104-019 |
| Dispase | Gibco | Cat# 17105-041 |
| DNase I | Sigma-Aldrich | Cat# DN25 |
| MesenCult stimulatory supplement | StemCell Technologies | 05502 |
| Recombinant murine EGF | PeproTech | AF-315-09 |
| Recombinant murine Oncostatin M (OSM) | R&D Systems | 495-MO-025 |
| Recombinant human IGF-1 | R&D Systems | 291-G1-200 |
| Recombinant murine PDGF-AA | PeproTech | 315-17 |
| Recombinant human FGF-basic | R&D Systems | 233-FB-025 |
| Chick Embryo Extract, Ultrafiltrate (CEE) 20ml | USBiological | C3999 |
| Human Endothelial-SFM | Gibco | 11111-044 |
| DMEM/F12, GlutaMAX | Gibco | 10565-018 |
| Critical Commercial Assays | ||
| RNA to cDNA EcoDry™ Premix system | Clontech | 639546 |
| Dynabeads® mRNA DIRECT™ Micro Kit | Invitrogen | 61012 |
| Deposited Data | ||
| RNA-seq | GSE142705 | N/A |
| Oligonucleotides used for qRT-PCR | ||
| Gapdh-F: TGTGTCCGTCGTGGATCTGA | Eurofins Genomics | N/A |
| Gapdh-R: CCTGCTTCACCACCTTCTTGA | Eurofins Genomics | N/A |
| Snai2-F: TGGTCAAGAAACATTTCAACGCC | Eurofins Genomics | N/A |
| Snai2-R: GGTGAGGATCTCTGGTTTTGGTA | Eurofins Genomics | N/A |
| Cxcl12-F: TGCATCAGTGACGGTAAACCA | Eurofins Genomics | N/A |
| Cxcl12-R: CACAGTTTGGAGTGTTGAGGAT | Eurofins Genomics | N/A |
| Kitl-F: GAATCTCCGAAGAGGCCAGAA | Eurofins Genomics | N/A |
| Kitl-R: GCTGCAACAGGGGGTAACAT | Eurofins Genomics | N/A |
| Spp1-F: TCCCTCGATGTCATCCCTGTTG | Eurofins Genomics | N/A |
| Spp1-R: GGCACTCTCCTGGCTCTCTTTG | Eurofins Genomics | N/A |
| Angpt1-F: CTCGTCAGACATTCATCATCCAG | Eurofins Genomics | N/A |
| Angpt1-R: CACCTTCTTTAGTGCAAAGGCT | Eurofins Genomics | N/A |
| Il7-F: TTCCTCCACTGATCCTTGTTCT | Eurofins Genomics | N/A |
| Il7-R: AGCAGCTTCCTTTGTATCATCAC | Eurofins Genomics | N/A |
| Vcam1-F: GACCTGTTCCAGCGAGGGTCTA | Eurofins Genomics | N/A |
| Vcam1-R: CTTCCATCCTCATAGCAATTAAGGTG | Eurofins Genomics | N/A |
| Runx2-F: TTACCTACACCCCGCCAGTC | Eurofins Genomics | N/A |
| Runx2-R: TGCTGGTCTGGAAGGGTCC | Eurofins Genomics | N/A |
| Cdkn1a-F: CGAGAACGGTGGAACTTTGAC | Eurofins Genomics | N/A |
| Cdkn1a-R: CAGGGCTCAGGTAGACCTTG | Eurofins Genomics | N/A |
| Bbc3-F: CTACCTCTGCGCCCCCAC | Eurofins Genomics | N/A |
| Bbc3-R: CGACTCTAAGTGCTGCTGGG | Eurofins Genomics | N/A |
| Trp53-F: GCGTAAACGCTTCGAGATGTT | Eurofins Genomics | N/A |
| Trp53-R: TTTTTATGGCGGGAAGTAGACTG | Eurofins Genomics | N/A |
| Foxc1-F: CACTCGGTGCGGGAAATGT | Eurofins Genomics | N/A |
| Foxc1-R: GGTACAGAGACTGACTGGCA | Eurofins Genomics | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J | The Jackson Laboratory | RRID:IMSR_JAX:007900 |
| B6; FVB-Tg(Cspg4-cre)1Akik/J | The Jackson Laboratory | RRID:IMSR_JAX:008538 |
| B6.129-Leprtm2(cre)Rck/J | The Jackson Laboratory | RRID:MGI: 3776164 |
| B6.Cg-Tg(Mx1-cre)1Cgn/J | The Jackson Laboratory | RRID:IMSR_JAX:003556 |
| Spp1Tm1Blh/J | The Jackson Laboratory | JAX: 004936 |
| C57BL/6 (CD45.2) | National Cancer Institute | |
| Bl6-Ly5.1 (CD45.1) | The Jackson Laboratory | JAX: 002014 |
| Software and Algorithms | ||
| SlideBook | Intelligent Imaging Innovations | RRID:SCR_014300 |
| FlowJo | Tree Star | RRID:SCR_008520 |
| FACS Diva 6.1 | BD Biosciences | RRID:SCR_001456 |
| Gene Set Enrichment Analysis | Broad Institute | RRID:SCR_003199 |
| Ingenuity Pathway Analysis | QIAGEN | RRID:SCR_008653 |
| GraphPad Prism | GraphPad Software | RRID:SCR_002798 |
RNA-seq analysis
Total RNA from 2,000-sorted CD45− Ter119− CD31− Nes-GFP+ BM MSPCs was extracted using the RNAeasy Plus Micro kit (Qiagen), and assessed for integrity and purity using an Aligent 2100 Bioanalyzer (Agilent Technologies). Complementary DNA libraries were then generated using the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Clontech Laboratories) and the Nextera XT DNA Sample preparation Kit (Illumina). The libraries were then submitted for Illumina HiSeq 2500 sequencing (150bp single ended) at the Single Cell Genomics and Epigenomics Core facility at Albert Einstein College of Medicine according to the standard procedures. RNA-Seq data were then processed using a publicly available pipeline. Briefly, single-ended sequencing reads were aligned to the mouse genome (mm10) and quantified using htseq-count. Next, we normalized for RNA composition by calculating scaling factors for each sample using calcNormFactors in edgeR (version 3.12.0). Nominal p-values were corrected using the Benjamini–Hochberg procedure to adjust for multiple hypothesis testing. Differentially expressed gene sets were then identified using Ingenuity Pathways Analysis (GIAGEN Bioinformatics) and Gene Set Enrichment Analysis (GSEA) software (Broad Institute). Raw and processed reads data from the RNA-seq have been deposited in the Gene Expression Omnibus under accession number GSE142705.
QUANTIFICATION AND STATISTICAL ANALYSIS
In each experiment, each mouse was analyzed as a biological replicate. Data in all figures were obtained in at least two independent experiments. No statistical method was used to predetermine the sample size. The experiments were not randomized and the investigators were not blinded during the experiments and outcome analyses. Data visualization and statistical analysis were performed using Graphpad Prism 7. All data are shown as mean ± s.e.m. *p< 0.05, **p< 0.01, ***p< 0.001, ****p<0.0001 by unpaired Student’s t test unless otherwise indicated.
Supplementary Material
Highlights.
Snai2 is required for bone marrow MSPC maintenance and regeneration.
Snai2 deletion compromises the HSC niche during homeostasis and after stress.
Snai2 suppresses osteopontin (Spp1) expression in MSPCs.
Compound Snai2 and Spp1 knockouts rescue MSPC function.
Acknowledgements
We thank the National Institutes of Health (DK056638, HL069438, HL116340), the Leukemia and Lymphoma Society (LLS-TRP 6475-15) and the New York State Department of Health (NYSTEM IIRP C029570 and C029154) for their support of our laboratory. We also thank Y. Zhang of the Albert Einstein College of Medicine Gene Targeting and Transgenic Core, L. Tesfa of the Flow Cytometry Sorting Facility, R. Sellers of the Histology and Comparative Pathology Facility for technical assistance and guidance, and Dr. Luis Cardoso at City College of New York for micro-CT analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
P.S.F. has served as consultant for Pfizer, received research funding from Ironwood Pharmaceuticals and owns stock options of Cygnal Therapeutics.
References
- Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, Ma’ayan A, and Frenette PS (2017). Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol 19, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, Kokkaliaris KD, Mercier F, Tabaka M, Hofree M, et al. (2019). A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 177, 1915–1932 e1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, and Wang CY (2013). The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med 19, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. (2014). Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. [DOI] [PubMed] [Google Scholar]
- Chen Y, and Gridley T (2013). Compensatory regulation of the Snai1 and Snai2 genes during chondrogenesis. J Bone Miner Res 28, 1412–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, Tani-Ichi S, Schlenner S, Richie E, Rodewald HR, et al. (2016). Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 45, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denhardt DT, Noda M, O’Regan AW, Pavlin D, and Berman JS (2001). Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest 107, 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, and Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G, and Morrison SJ (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenette PS, Pinho S, Lucas D, and Scheiermann C (2013). Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu Rev Immunol 31, 285–316. [DOI] [PubMed] [Google Scholar]
- Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, and Link DC (2013). CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi N, Sacma M, Standker L, Soller K, Marka G, Eiwen K, Weiss JM, Kirchhoff F, Weil T, Cancelas JA, et al. (2017). Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J 36, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Duhrsen U, and Frenette PS (2014). Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 15, 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himburg HA, Termini CM, Schlussel L, Kan J, Li M, Zhao L, Fang T, Sasine JP, Chang VY, and Chute JP (2018). Distinct Bone Marrow Sources of Pleiotrophin Control Hematopoietic Stem Cell Maintenance and Regeneration. Cell Stem Cell 23, 370–381 e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Seidel MG, Wu W, Kamizono S, Ferrando AA, Bronson RT, Iwasaki H, Akashi K, Morimoto A, Hitzler JK, et al. (2002). Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2, 279–288. [DOI] [PubMed] [Google Scholar]
- Isern J, Garcia-Garcia A, Martin AM, Arranz L, Martin-Perez D, Torroja C, Sanchez-Cabo F, and Mendez-Ferrer S (2014). The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. Elife 3, e03696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Lan Y, Norton CR, Sundberg JP, and Gridley T (1998). The Slug gene is not essential for mesoderm or neural crest development in mice. Dev Biol 198, 277–285. [PubMed] [Google Scholar]
- Kalcheim C (2015). Epithelial-Mesenchymal Transitions during Neural Crest and Somite Development. J Clin Med 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Amariglio N, Rechavi G, Jakob-Hirsch J, Kela I, Kaminski N, Getz G, Domany E, and Givol D (2001). DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20, 2225–2234. [DOI] [PubMed] [Google Scholar]
- Kawamoto T, and Shimizu M (2000). A method for preparing 2- to 50-micron-thick fresh-frozen sections of large samples and undecalcified hard tissues. Histochem Cell Biol 113, 331–339. [DOI] [PubMed] [Google Scholar]
- Kiefer FW, Zeyda M, Gollinger K, Pfau B, Neuhofer A, Weichhart T, Saemann MD, Geyeregger R, Schlederer M, Kenner L, et al. (2010). Neutralization of osteopontin inhibits obesity-induced inflammation and insulin resistance. Diabetes 59, 935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieslinger M, Hiechinger S, Dobreva G, Consalez GG, and Grosschedl R (2010). Early B cell factor 2 regulates hematopoietic stem cell homeostasis in a cell-nonautonomous manner. Cell Stem Cell 7, 496–507. [DOI] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, and Bapat SA (2009). Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 27, 2059–2068. [DOI] [PubMed] [Google Scholar]
- Liu X, Ma Y, Li R, Guo D, Wang N, Zhao Y, Yin J, Ren Q, Lin Y, and Ma X (2018). Niche TWIST1 is critical for maintaining normal hematopoiesis and impeding leukemia progression. Haematologica 103, 1969–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancilla A, and Mayor R (1996). Neural crest formation in Xenopus laevis: mechanisms of Xslug induction. Dev Biol 177, 580–589. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, Wei Q, Wang X, Ciero P, Xu J, et al. (2018). Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat Med 24, 782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee MD, Farach-Carson MC, Butler WT, Hauschka PV, and Nanci A (1993). Ultrastructural immunolocalization of noncollagenous (osteopontin and osteocalcin) and plasma (albumin and alpha 2HS-glycoprotein) proteins in rat bone. J Bone Miner Res 8, 485–496. [DOI] [PubMed] [Google Scholar]
- Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, and Olsen BR (2010). Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16, 1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes SC, Robin C, and Dzierzak E (2005). Mesenchymal progenitor cells localize within hematopoietic sites throughout ontogeny. Development 132, 1127–1136. [DOI] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, and Frenette PS (2010). Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, Ono N, Kronenberg HM, and Frenette PS (2014). Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev Cell 29, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa S, Mabuchi Y, Niibe K, Suzuki S, Nagoshi N, Sunabori T, Shimmura S, Nagai Y, Nakagawa T, Okano H, et al. (2009). Development of mesenchymal stem cells partially originate from the neural crest. Biochem Biophys Res Commun 379, 1114–1119. [DOI] [PubMed] [Google Scholar]
- Nakahara F, Borger DK, Wei Q, Pinho S, Maryanovich M, Zahalka AH, Suzuki M, Cruz CD, Wang Z, Xu C, et al. (2019). Engineering a haematopoietic stem cell niche by revitalizing mesenchymal stromal cells. Nat Cell Biol 21, 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Sargent MG, Wilkinson DG, and Cooke J (1994). Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 264, 835–839. [DOI] [PubMed] [Google Scholar]
- Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, Bertoncello I, Bendall LJ, Simmons PJ, and Haylock DN (2005). Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 106, 1232–1239. [DOI] [PubMed] [Google Scholar]
- Omatsu Y, Seike M, Sugiyama T, Kume T, and Nagasawa T (2014). Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature 508, 536–540. [DOI] [PubMed] [Google Scholar]
- Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, and Scadden DT (2012). Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Losada J, Sanchez-Martin M, Perez-Caro M, Perez-Mancera PA, and Sanchez-Garcia I (2003). The radioresistance biological function of the SCF/kit signaling pathway is mediated by the zinc-finger transcription factor Slug. Oncogene 22, 4205–4211. [DOI] [PubMed] [Google Scholar]
- Perez-Mancera PA, Bermejo-Rodriguez C, Gonzalez-Herrero I, Herranz M, Flores T, Jimenez R, and Sanchez-Garcia I (2007). Adipose tissue mass is modulated by SLUG (SNAI2). Hum Mol Genet 16, 2972–2986. [DOI] [PubMed] [Google Scholar]
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, and Frenette PS (2013). PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med 210, 1351–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Marchand T, Yang E, Wei Q, Nerlov C, and Frenette PS (2018). Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev Cell 44, 634–641 e634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seike M, Omatsu Y, Watanabe H, Kondoh G, and Nagasawa T (2018). Stem cell niche-specific Ebf3 maintains the bone marrow cavity. Genes Dev 32, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, Kokkaliaris KD, Rhee C, Lee D, Scadden EW, et al. (2019). Stress-Induced Changes in Bone Marrow Stromal Cell Populations Revealed through Single-Cell Protein Expression Mapping. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, Cheng T, Dombkowski D, Calvi LM, Rittling SR, et al. (2005). Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 201, 1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, Pinho S, Akhmetzyanova I, Gao J, Witkowski M, et al. (2019). The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Boulais PE, Zhang D, Pinho S, Tanaka M, and Frenette PS (2019). Maea expressed by macrophages, but not erythroblasts, maintains postnatal murine bone marrow erythroblastic islands. Blood 133, 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, and Look AT (2005). Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 123, 641–653. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, and Jaenisch R (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, Ahamed J, and Li L (2014). Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med 20, 1321–1326. [DOI] [PubMed] [Google Scholar]
- Zhao M, Tao F, Venkatraman A, Li Z, Smith SE, Unruh J, Chen S, Ward C, Qian P, Perry JM, et al. (2019). N-Cadherin-Expressing Bone and Marrow Stromal Progenitor Cells Maintain Reserve Hematopoietic Stem Cells. Cell Rep 26, 652–669 e656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed reads data from the RNA-seq have been deposited in the Gene Expression Omnibus under accession number GSE142705.