

Abstract
A double functionalization of vicinal sp3 C-H bonds has been developed, wherein a β amine and γ iodide are incorporated onto an aliphatic alcohol in a single operation. This approach is enabled by an imidate radical chaperone, which selectively affords a transient β alkene that is amino-iodinated in situ. Overall, the radical-polar-crossover cascade entails the following key steps: (i) β C-H iodination via 1,5-hydrogen atom transfer (HAT), (ii) desaturation via I2 complexation, and (iii) vicinal amino-iodination of an in situ generated allyl imidate. The synthetic utility of this double C-H functionalization is illustrated by conversion of aliphatic alcohols to a diverse collection of α,β,γ substituted products bearing heteroatoms on three adjacent carbons. The radical-polar crossover mechanism is supported by various experimental probes, including isotopic labeling, intermediate validation, and kinetic studies.
Graphical Abstract

Introduction
In nature, form and function are often intertwined. Likewise, increased molecular complexity may increase utility – affording, for instance, more potent and selective medicines.1 Illustrating this link between value and complexity, Figure 1a includes a series of molecules, each containing a propanol backbone with varying heteroatom substitution. As compared to 1-propanol (< $0.1/mL), its N-containing analogs [serinol ($40/mL), 2,3-diaminopropan-1-ol ($1000/mL), 2-aziridinemethanol (>$10,000/mL)] are costlier and more challenging to synthesize.2 Yet, the latter, N-rich analogs are more likely to be found in medicines,3 and thus methods for synthesizing such densely oxidized scaffolds remain valuable.1 Typically, vicinal amino alcohols may be synthesized by alkene difunctionalization of allylic alcohols (Figure 1b).4 As a complementary route to access products with α,β,γ heteroatom substitution, we sought to directly employ aliphatic alcohols, without pre-installation of a reactive alkene – via double C-H oxidation.
Figure 1.

Radical-polar crossover enables synthetically valuable, double C-H functionalization.
To access products with three contiguous oxidized carbons, we proposed a radical-polar cascade strategy may address this challenge. Yet, whereas radical-polar crossover mechanisms5 are typically initiated by radical addition to alkenes,6 we were interested in pursuing a reversed strategy, entailing H• abstraction to first generate an alkene and then harness its polar reactivity. We were inspired by pioneering studies of Barluenga et al 7 converting alkane solvents to iodo-acetates under highly oxidizing conditions via O-radicals, as well as an N3I-mediated iodo-lactamization by J-Q Yu et al.8 To extend this approach to enable selective C-H difunctionalization of synthetically useful alcohols, we envisioned a radical chaperone strategy, wherein an alcohol is converted to an imidate N-radical, may allow in situ alkene generation via desaturation (Figure 1c). Amino-halogenation of this transient intermediate would afford an iodo-oxazoline, which may then be readily converted to a family of α,β,γ substituted amino alcohols.
Building on our recent work in the area of directed C-H functionalization by 1,5-hydrogen atom transfer (HAT),9–10 we were cognizant of the unique ability of imidate radicals to selectively convert alcohols to their β amines11–12 or β halides.13 As shown in Figure 2, this radical chaperone strategy, entails temporary conversion of an alcohol A to an imidate by coupling with a nitrile. Subjecting this imidate radical precursor to AcOI (prepared in situ from NaI and PhI(OAc)2) then affords a transient N-iodo-imidate B, whose weak N-I bond is readily homolyzed with visible light. The resulting N-centered radical C is then well-suited to undergo regioselective 1,5-HAT to afford β C-centered radical D. Upon recombination with the caged iodine radical, the key intermediate, β iodo imidate E, is formed. In our previous studies, a polar solvent (e.g. MeCN) facilitates in situ cyclization to yield oxazoline F, which can be hydrolyzed by acidic work-up to afford β amino alcohol G.11a,c In non-polar solvents (e.g. PhMe), 4-aryl oxazolines are further oxidized to heretoaromatic azoles.11d Alternatively, β iodo imidate E may undergo a second, iterative 1,5-HAT to selectively abstract the remaining β C-H bond, which is weaker by ~2 kcal/mol due to C-X polarization, to afford β di-iodide H. Upon aminolysis, geminal β di-iodide I is obtained – complementing Pd-catalyzed β C-H iodination by J-Q Yu et al, which forms distal di-iodides.14 We have found the key to this divergent reactivity rests in the judicious choice of solvents. For example, whereas MeCN yields cyclization to oxazoline F, a MeCN:CH2Cl2 mixture affords iterative HAT (to β di-iodide I). We anticipate this less polar solvent mixture allows a second N-oxidation to outcompete cyclization. Furthering this hypothesis, we envisioned a solvent that increases NaI (and thus AcOI) concentration without increasing polarity might enable I-oxidation – facilitating an alternate radical-polar crossover mechanism.
Figure 2.

Several single and double C-H functionalization mechanisms enabled by imidate radical chaperone strategy.
In this third mechanistic possibility, iodine-centered oxidation of β iodo imidate E may afford β hypervalent iodane J. This alkyl λ3-iodane is a hypernucleofuge, which is 106 times faster of a leaving group than triflate.15 For this reason, we anticipated its rapid β elimination would afford allyl imidate K. The regioselectivity of this elimination – away from the imidate – was expected based on imidate polarization. Again this reactivity would complement Pd-catalyzed mechanisms, including 1,5-HAT pathways recently developed by Gevorgyan and coworkers to access enols, enamines, and terminal alkenes via desaturation.16 Finally, we hoped allyl imidate K would directly undergo AcOI-mediated halo-cyclization in a radical-polar crossover cascade to afford γ iodo oxazoline L. We expected this intermediate could also be hydrolyzed to β amino alcohols – with an additional γ iodo functional handle to enable further nucleophilic substitution. With this added versatility, a family of α,β,γ substituted amino alcohols would be rapidly accessible from aliphatic alcohols by a desaturation-mediated cascade.
Results and Discussion
To test our radical-polar crossover cascade hypothesis, we subjected imidate 1, whose tertiary β iodide intermediate we expected to be prone to oxidative elimination, to excess AcOI (in situ combination of NaI and PhI(OAc)2) in MeCN with visible light irradiation (26W compact fluorescent light) (Figure 3). We were pleased to find the cascade was indeed feasible, affording iodo oxazoline 2 (43% yield) along with des-iodo oxazoline 3 (18% yield). Moreover, substituting a less polar solvent (CH2Cl2) slows cyclization and affords more 2 (63% yield), along with β iodide 4 (18% yield) and oxazoline 3 (12% yield). Extending this trend, non-polar PhCF3 solvent arrests cyclization and exclusively affords β iodide 4 (60% yield). Alternatively, a more polar 3:1 HFIP:CH2Cl2 solvent mixture yields oxazoline 3 (83% yield) selectively. Building on these observations, we tested tBuOH, a moderately polar, protic solvent, and were delighted to observe selective vicinal C-H difunctionalization to 2 (92% yield). Notably, the proposed intermediate alkene 5 was not recovered from any of these experiments, and tBuOH affords < 5% of side-products 3 or 4. It is worth highlighting the strong solvent effect observed, wherein three divergent transformations are controlled by solvent choice. For example, imidate 1 can be tuned to selectively afford: β C-H iodination (4) in PhCF3, β C-H amination (3) in HFIP:CH2Cl2, or vicinal C-H β-amino-γ-iodination (2) in tBuOH.
Figure 3.
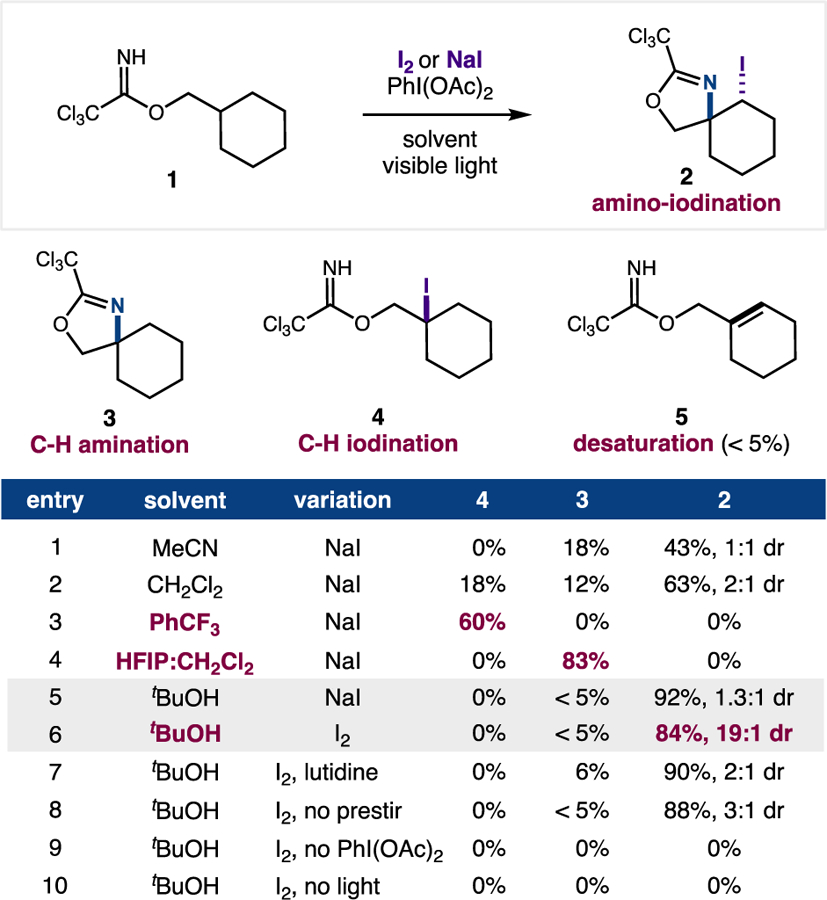
Development of vicinal, double C-H functionalization. Conditions: 0.2 mmol imidate, I2 or NaI (3 equiv), PhI(OAc)2 (3 equiv), tBuOH [0.3 M], 3 min stir before visible light irradiation for 1 h. Yields and dr determined by 1H NMR vs internal standard.
In further probing our hypothesis that tBuOH is optimal because it best solubilizes AcOI precursors and increases oxidant concentration, we switched the iodine reagent from NaI to I2. Although we previously found I2 to work well in some cases,11c its photolytic initiation often affords significant side-product formation and poor desired reactivity.10a Thus, we were pleasantly surprised to find that in tBuOH, I2 forms iodo oxazoline 2 efficiently (84% yield) and with high diastereoselectivity (19:1 dr). As reaction controls, we probed the effects of added base (2,6-lutidine) and immediate irradiation (without a 3-minute pre-stir to ensure I2 solubility before irradiation). Both changes resulted in significantly lower diastereoselectivity with similar efficiency. Lastly, as expected, absence of PhI(OAc)2 (to generate AcOI), I2 (to facilitate iodide elimination), or light (to initiate N-I homolysis) affords no reactivity.
Synthetic Scope
Having developed a regio- and diastereo- selective vicinal, double C-H amino-iodination of alcohols via an imidate-radical-polar crossover mechanism, we sought to investigate the generality and utility of this cascade reaction (Figure 4). To this end, we found a range of cyclic alcohols are efficiently amino-iodinated to afford spirocyclic oxazolines fused to 4–8 membered carbocycles (2, 6–9). Notably, even cyclobutane (a common motif in medicines and natural products)17 is amino-iodinated (9), likely through a strained cyclobutene intermediate, showcasing the unique utility of this radical-mediated strategy to doubly modify small rings. A variety of functional groups are tolerated under these reaction conditions, including ethers, amides, esters, and nitriles, affording spiro-fused bis-heterocycles (10–13, 19). In addition to primary alcohols, this cascade is also amenable to secondary alcohols, which selectively undergo radical-polar crossover reactivity to afford spirocyclic oxazolines (14–15) rather than distal, iterative HAT products.13 Additionally, γ acyclic alcohols afford γ iodo oxazolines as well. In this case, both symmetric (di-Me, di-nBu) or asymmetric (Me, nBu) substituents are tolerated – with the latter affording 8:1 to 20:1 regioselectivity for the more substituted γ iodide (16–19).
Figure 4.

Synthetic utility of vicinal C-H amino-iodination of alcohols. Conditions: 0.2 mmol imidate, I2 (3 equiv), PhI(OAc)2 (3 equiv), tBuOH [0.3 M], 3 min stir before visible light irradiation. aNaI (3 equiv). bI2 (2 equiv). cPhI(OAc)2 (5 equiv). d NMR yield. ePhI(OAc)2 (4 equiv). Isolated yields indicated, along with diastereoselectivity (dr) or regioselectivity (rr), as determined by NMR of the crude reaction mixture.
We have shown the cyclization of intermediate β iodo imidates is challenging in the absence of benzylic activation, and thus, more nucleophilic benzimidates (vs trichloroimidates) are necessary to afford amination in these cases.11a Therefore, to test the limits of this new radical-polar crossover, we investigated such amination-prone imidates. Unexpectedly, a wide range of benzimidates (accessed by addition of alcohols to benzonitriles) are amenable to this cascade. For example, electronically diverse, para-aryl substituents, ranging from -CH3 to -CF3 afford iodo oxazolines (20–24) with excellent efficiency and diastereoselectivity (>70% yield, >20:1 dr). Additionally, meta- and bis-halide substitution are tolerated, as well as oxidatively sensitive naphthalenes (25–28). Lastly, and perhaps, most surprisingly, alkyl nitrile-derived imidates also efficiently afford spirocyclic oxazolines (29–30) despite increased nucleophilicity of these imidates, which may otherwise cyclize to afford mono-amination.
To further probe the functional group tolerance of this radical-polar cascade, a robustness screen was performed.18 In this investigation of 1 to 2, we observed medicinally relevant, five- and six-membered N-containing heterocycles (e.g. imidazole, pyridine) are well-tolerated. Interestingly, we observed a slight decrease in diastereoselectivity in the presence of these bases. We attribute this effect to I2-base complexation,19 which effectively decreases the concentration of I2 and rate of the resulting polar amino-iodination pathway (see SI for more details). Next, we were pleased to find alkyl chlorides, which are prone to displacement by I− (generated upon alkyl-iodide elimination), are also tolerated. Additionally, alcohols, aldehydes, and amides are preserved, despite the possibility of their consumption under these oxidative conditions (see SI for an extended table of functional group tolerance investigations).
Mechanistic Investigations
A detailed description of our proposed mechanism is shown in Figure 5a. First, in situ generation of AcOI occurs by combination of PhI(OAc)2 and I2 via a ligand exchange mechanism.20 Next, an alcohol-derived imidate I undergoes N-iodination by displacement of AcOI, which is electrophilic at iodine.21 The resulting N-iodo imidate II contains a weak N-I bond that is homolyzed under visible light irradiation. The electrophilic, N-centered radical III may then undergo a thermodynamically and kinetically driven 1,5-HAT to generate nucleophilic, C-centered radical IV. Upon radical recombination with I•, β alkyl iodide V is formed – terminating the radical component of the radical-polar crossover mechanism. To promote polar elimination, Lewis acidic complexation of I2 to iodide V would form the alkyl triiodide nucleofuge VI.22 Upon net elimination of HI and I2, a resulting allyl imidate VII is generated and amino-iodinated under these oxidative conditions. This halo-cyclization may occur by either a polar, iodonium (VIII) or radical, π-addition (5-exo-trig cyclization of IX) mechanism. The high diastereoselectivity observed for trans-iodo-oxazoline X suggests a polar mechanism is operative – entailing intramolecular cyclization of iodonium VIII by the tethered imidate. Nucleophile-induced pre-polarization via this pathway may also account for the observed stereoselectivity.23
Figure 5.
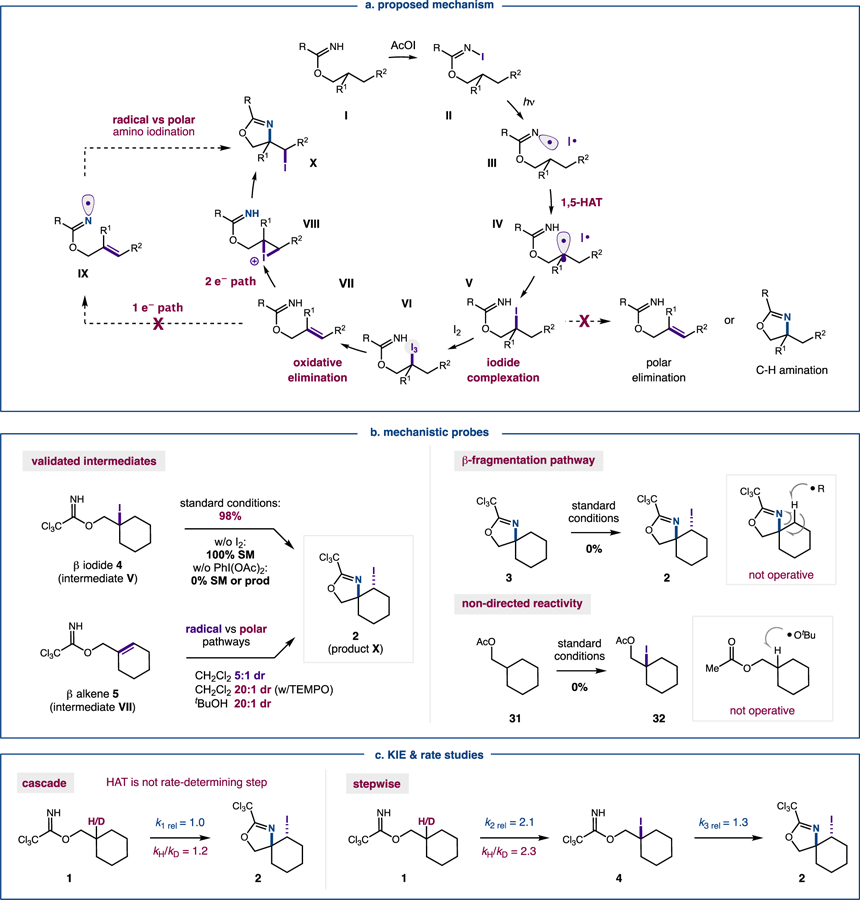
Vicinal C-H aminoiodination via imidate radical chaperone desaturation-addition cascade: (a) Proposed mechanism. (b) Mechanistic probes for various key steps. (c) KIE & rate studies.
To better understand this radical-polar crossover mechanism, we conducted a series of experiments probing the elementary steps of this cascade. First, two of the key proposed intermediates, β alkyl iodide 4 and β alkene 5 (representing V and VII, respectively), were independently synthesized and subjected to the reaction (Figure 5b). Both of these imidates afford iodo oxazoline 2 (up to 98%) – validating their likely intermediacy in the cascade mechanism. As further support, iodide 4 was observed by 1H NMR during the course of the reaction (alkene 5 is also observed when the reaction is performed in toluene; see SI for details).
We then interrogated the mechanism of β alkyl iodide elimination – an important question, given its similarity to the stable γ iodo product. Since a variety of relevant additives (e.g. NaOAc, HOAc; see SI for full list) do not promote elimination of intermediate 4 (or V), we hypothesized the iodide may be first oxidized to a λ3-iodane, which is an elimination-prone hypernucleofuge.15 Upon subjecting 4 to each reaction component (individually or as combinations), the oxidation/elimination/cyclization sequence to form iodo oxazoline 2 was only observed in the presence of AcOI (98% yield with I2, PhI(OAc)2; 36% yield with I2, NaOAc). Interestingly, PhI(OAc)2 alone does not oxidize the alkyl iodide, unlike what has been observed in other systems.24 Another notable observation is that I2 alone provides full consumption of 4, albeit without formation of 2. Further discussion of the nature of this β iodide-selective oxidation/elimination is provided in subsequent sections.
As our next line of enquiry, we focused on the proposed β alkene 5 and its ability to afford iodo oxazoline 2 via either a radical or polar pathway. Interestingly, when CH2Cl2 is used as solvent (vs tBuOH), only 5:1 dr is observed (vs 20:1 dr in tBuOH). Suspecting the lower stereoselectivity might be a result of a radical pathway (via IX), we added TEMPO to the CH2Cl2 reaction and observed a recovery of high stereoselectivity (20:1 dr). Based on this data, we reasoned the stereoselective tBuOH conditions suggest a polar mechanism for the amino-iodination of alkene 5 (via trans intramolecular imidate addition to iodonium VIII).
Next, we sought to probe if alternate mechanisms, such as β fragmentation or non-directed functionalization, are operative (Figure 5b). In the first case, the C-H amination side-product (oxazoline 3) could afford amino-iodinated product (iodo oxazoline 2) by β fragmentation of a γ C-radical (inset). While there is no obvious driving force for γ selective C-H abstraction (or regeneration of an N-centered radical via this pathway), a related mechanism was identified by J-Q Yu and coworkers in their iodo-lactamization.8 Nevertheless, when resubjected to reaction conditions, oxazoline 3 remains intact and does not afford iodo oxazoline 2. Similarly, we sought to investigate if a non-directed pathway, as described by Barluenga et al,7 is operative. Noting that AcOI (I2, PhI(OAc)2) in tBuOH may form tBuO-I, whose homolysis would generate a reactive HAT reagent (tBuO•),25 we replaced the imidate directing group with a similarly polar ester. In this case, subjecting acetate 31 to reaction conditions does not form β iodide 32 or any other relevant products. This observation supports our hypothesis that the regioselective cascade is mediated by imidate radical 1,5-HAT rather than non-directed HAT (by tBuO•; see inset).
Finally, we investigated the role of HAT in the double C-H functionalization by measuring relative reaction rates of individual steps, while also considering the effects of heavy atom labels on these rates (Figure 5c). In the overall reaction, a negligible, primary kinetic isotope effect (KIE) was determined by measuring parallel rates of reactivity of 1 vs β deutero 1 (kH/kD=1.2). Interestingly, when rates of formation of intermediate 4 are measured (vs cascade product 1), a slightly larger primary KIE is observed (kH/kD=2.3). Together, this data suggests that while HAT is rate-determining for intermediate C-H iodination, it is not the rate-determining step of the overall amino-iodination cascade. Similarly, comparison of the relative rates of the overall reaction (double C-H amino-iodination; k1 rel = 1.0) to stepwise formation of β iodide 4 (mono C-H iodination; k2 rel = 2.1) and its subsequent conversion to product (elimination/cyclization; k3 rel = 1.3) illustrates the bottleneck in this cascade. Specifically, radical-mediated C-H iodination was found to be 1.6-times faster than subsequent conversion to the iodo-oxazoline, which supports our earlier observation that HAT precedes the rate-limiting step.
Strategy for Secondary β C-H Bonds
Building on this mechanistic understanding that conversion of the β alkyl iodide to product represents the slowest step(s) and biggest challenge, we sought to expand the scope of this cascade beyond its synthetic limitation of tertiary (3°) β C-H bonds. This goal is further complicated by the observation that secondary (2°) γ iodide products are stable, while tertiary β iodide intermediates are not. Our rationale for this observed selectivity is that Lewis acidic I2 may reversibly bind to alkyl halides, especially in non-polar solvents (Figure 6).22 Notably, this complexation is substrate-controlled, with highly substituted alkyl iodides binding more strongly than less substituted variants (binding constants, K: tertiary, 1.3 >> secondary, 0.4). Thus, we propose that preferential formation of the tertiary triiodide allows selective dissociation to I3– and a 3° cation – enabling elimination and subsequent cyclization. Based on our experimental data, it appears this equilibrium-dependent heterolysis is driven in the forward direction by AcOI-mediated oxidation of I3–. This mechanistic pathway provides an explanation for the observed selectivity for oxidation of tertiary β iodide intermediate, but not 2° γ iodide product. Nevertheless, this proposal suggests an inherent synthetic limitation that we wished to address.
Figure 6.

Selective oxidation of tertiary over secondary alkyl iodide.
To overcome this β methylene challenge, we designed a suite of strategies to access amino-iodination of 2° C-H bonds – even in cases where oxidation of, or I2 complexation to, a 2° β iodide intermediate is disfavored (Figure 7). First, incorporating electron-withdrawing groups (e.g. ketone, ester) at the γ position sufficiently enhances acidity of the γ proton and facilitates β iodide elimination – affording cascade products with four consecutively oxidized carbons (33–36). A second strategy entails use of a γ halide leaving group to promote iodonium formation from the vicinal dihalide intermediate (37–38). A third approach employs the β-silicon effect – enhancing the leaving group ability of a β iodide by incorporating a γ silyl group (39–40). Finally, if no additional functionality is desired, simply incorporating a 3° C-H bond at the γ (vs β) position also facilitates the cascade (41–42). In this case, since 1,5-HAT is favored over 1,6-HAT,11c we anticipate the sec-isopentyl β iodide is sufficiently strained to behave more like the tertiary β iodides (undergoing I2 complexation and subsequent I3– oxidative elimination).
Figure 7.
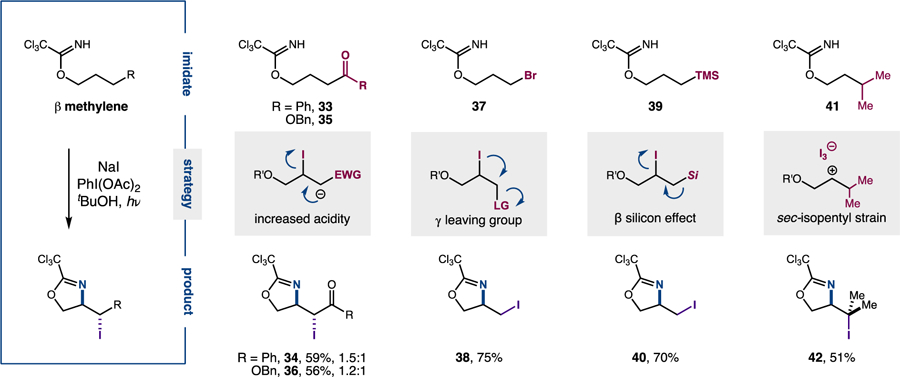
Double C-H functionalization of imidates with β methylenes.
Single vs Double C-H Functionalizations
To illustrate the utility of this new double C-H functionalization strategy for accessing divergent β,γ amino-iodination reactivity (versus our previous studies on single β C-H amination),11 we compared the fate of several alcohols under either reaction condition (Figure 8). In each case, selective formation of complementary product classes was observed. For example, imidates containing β methines exclusively afforded oxazolines previously, whereas we have now illustrated two examples (17–18) of a radical-polar crossover mechanism providing access to γ-iodo oxazolines. Similarly, while a γ ester-substituted imidate previously yielded single β C-H amination efficiently, β-amino-γ-iodide 36 is exclusively obtained by the new strategy described herein. Finally, whereas an imidate bearing a γ tertiary C-H solely afforded C-H amination before (with >20:1 β regioselectivity), we now show alternate access to a more densely functionalized, β-amino-γ-iodide (42) is also possible.
Figure 8.

Single vs double C-H functionalizations.
Diversification of Vicinal Products
Having demonstrated the broad scope of this double C-H functionalization, we next sought to showcase the synthetic utility of these γ iodo-oxazolines (Figure 9). Notably, we envisioned both the γ C-I bond and the oxazoline C-O bond could be orthogonally diversified to access a range of densely functionalized products. First, the γ iodide was displaced with a variety of nucleophiles to access α,β,γ heteroatom substitution. For example, phthalimide and azide afford γ amination (43–44), while phenoxide and acetates afford γ oxygenation, including with biotin (45–47). Next, the oxazoline was hydrolyzed by either HCl (aq) to access the free γ-iodo-β-amino alcohol (48) or with TsOH to afford the N-protected variant (49). Interestingly, in the absence of water, HCl causes a C-O to C-Cl displacement to afford α-chloro-β-amino-γ-iodide (50). Alternatively TMS-Br mediates C-O to C-Br conversion to afford another nonsymmetric dihalide: α-bromo-β-amino-γ-iodide (51).
Figure 9.

Post-synthetic diversification of iodo-oxazolines by C-I, C-O, and multi-site functionalization.
Finally, multi-site reactivity of γ iodo-oxazoline was observed in the presence of some nucleophiles. For example, EtSH displaces the C-I to form a thioether while also incorporating another thioether onto the oxazoline through chloride displacement/reduction of the -CCl3 group (52).26 On the other hand, the “hard” nucleophile, cyanide, selectively adds to the imidate carbon, allowing subsequent N-anion attack of the adjacent C-I to form bicyclic aziridine 53. Finally, through a one-pot protocol, the oxazoline is converted to β hydroxy aziridine 54 by acidic hydrolysis, base-mediated ring closure, and then Ts-protection.
To illustrate the value of this radical chaperone-mediated strategy for the rapid diversification of alcohols, we also developed a streamlined, one-pot protocol for the sequence, including: (i) chaperone addition, (ii) vicinal C-H amino-iodination, and (iii) chaperone removal (Figure 10). In the case of cyclohexyl methanol, either the protected, or free, γ-iodo-β-amino alcohol can be obtained, depending on whether TsOH or HCl is used to facilitate the final hydrolysis (55 or 56, respectively). In both cases, a single diastereomer is easily isolated by column chromatography. Additionally, the free amino alcohol 56 obtained by the latter protocol afforded single crystals, enabling X-ray structure determination to confirm the trans relationship between the iodide and amine.
Figure 10.
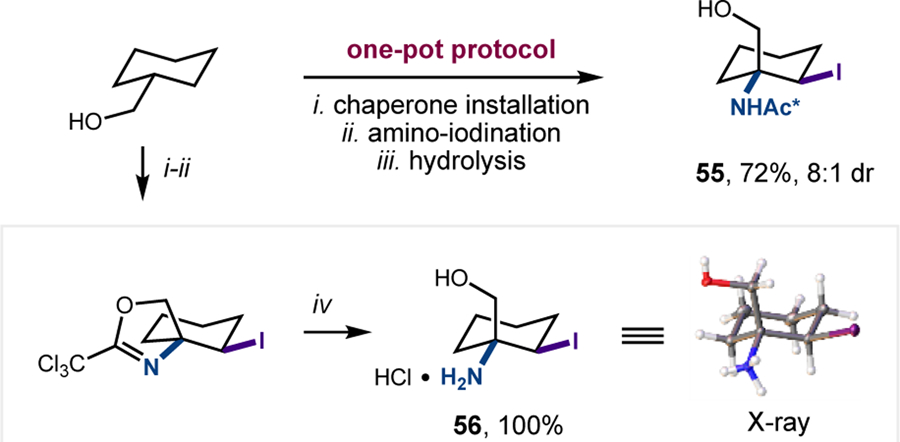
One-pot protocol and structure validation. Conditions: (i) Cl3CCN, DBU, DCM; (ii) I2, PhI(OAc)2, tBuOH; (iii) TsOH, MeOH, H2O; (iv) HCl (aq)
One-Step Strategy
With a one-pot protocol in hand to convert alcohols to their β-amino-γ-iodo analogs, we questioned if this strategy may be further streamlined into a single step. Although we showed in situ conversion of an alcohol to its imidate and subsequent, double C-H functionalization without purification is possible (see Figure 10), initial combination of all reagents (alcohol, nitrile, and oxidants) appeared an insurmountable challenge. In this case, an alcohol must couple the nitrile without first reacting with the potent oxidants. Yet, given the 68% recovery of octanol in our functional group tolerance studies (see Figure 4), we posited that the desired competition might be possible. To test this hypothesis, all reagents (alcohol, nitrile, base, and oxidants) were combined at once and subjected to visible light irradiation in tBuOH (Figure 11). Remarkably, this one-step procedure affords 20% amino-iodinated product 2 directly from alcohol. To further improve this protocol, we investigated the rate of intermediate formation. In the absence of oxidants, we observed imidate synthesis is slow in tBuOH (only 15% 1 after 15 minutes). Moreover, a parallel reaction including oxidants yields 0% 1 after 60 mins – albeit with full alcohol consumption, suggesting that competitive oxidation of the alcohol (to aldehydes, acids, etc.) is operative.
Figure 11.

Challenge of single-step strategy: competitive oxidation.
With the hopes of outcompeting oxidative degradation of the alcohol by desired imidate formation, we conducted a survey of the competitive rates of these processes in various solvents. Interestingly, we found non-polar solvents, such as PhCF3, facilitate more rapid imidate synthesis (vs oxidative degradation). For example, in the absence of oxidants, 100% imidate 1 is formed within 15 mins (vs 15% in tBuOH). Upon inclusion of oxidants, 78% imidate 1 is still formed. Notably, this rapid formation of imidate (vs oxidation) allows ensuing double C-H functionalization to occur efficiently – yielding 75% product 2. An explanation for this improved overall reactivity in PhCF3 (75% vs 20% in tBuOH) entails slower oxidation in the non-polar solvent (24 h needed vs 1 h, typically; see also Figure 3, entry 3) coupled with accelerated imidate formation.
To probe the generality of this single-step strategy, a range of alcohols was subjected to this streamlined protocol that combines both imidate synthesis and the amino-iodination cascade in a single operation (Figure 12). To our delight, combining all reagents in PhCF3 and irradiating with visible light for 24 hours directly yields β-amino-γ-iodo heterocycles from several alcohols. For example, both cyclic and acyclic alcohols with varied branching, ring size, and substituents are amino-iodinated efficiently (55–59, 58–75% yields by NMR). Subsequent hydrolysis with TsOH affords the free γ-iodo-β-amino alcohol in each case (51–70% isolated yields).
Figure 12.
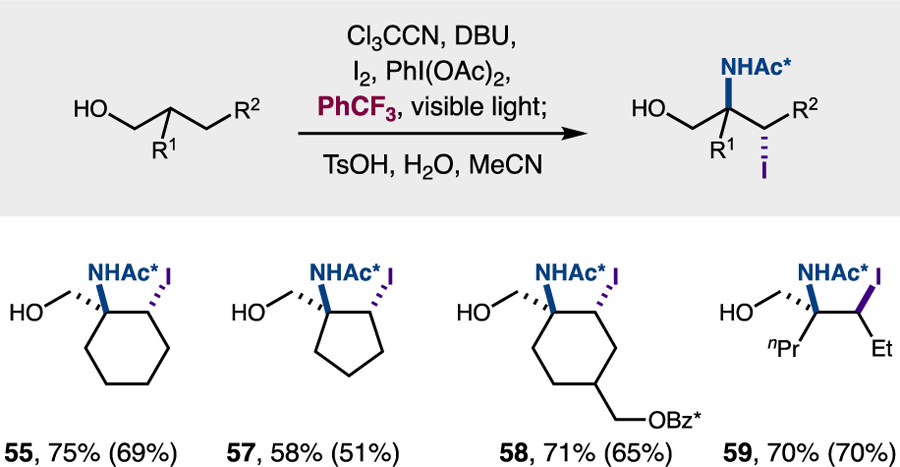
Direct, double C-H functionalization of alcohols in one step.
Conclusions
In summary, we have developed a radical-mediated cascade that enables vicinal C-H di-functionalization of alcohols by regioselective desaturation and in situ amino-iodination of their imidates. Our current understanding of strong solvent effects observed in imidate radical-mediated HAT now enables selective access to any one of three classes of divergent reactivity: (i) mono C-H amination, (ii) geminal C-H di-halogenation, or (iii) vicinal C-H amino-iodination. Mechanistic studies of the vicinal C-H di-functionalization, reported herein, are consistent with a radical-polar crossover cascade that entails (a) directed HAT by imidate radicals, (b) regioselective desaturation by I2 complexation, and (c) diastereoselective iodo-cyclization. Additionally, we have shown the γ iodo-oxazolines are easily manipulated to enable access to a family of densely functionalized molecules bearing three-consecutively oxidized carbons. We expect this directed radical-polar crossover strategy for rapidly upgrading the molecular complexity of alcohols is applicable to other valuable classes of vicinal C-H di-functionalizations.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (R35 GM119812), National Science Foundation (CAREER 1654656), and Sloan Foundation for financial support. RKT is supported by an NSF graduate fellowship. Structural data was collected by Dr Nicholas Settineri at the UC Berkeley X-ray Crystallography Facility (supported by NIH S10-RR027172).
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Experimental procedures and characterization for all new compounds (PDF)
1H and 13C NMR spectral data (PDF)
X-ray crystallographic data (CIF) can also be found at The Cambridge Crystallographic Data Centre (CCDC 1967638).
The authors declare no competing financial interest.
References
- (1).(a) Campos KR; Coleman PJ; Alvarez JC; Dreher SD; Garbaccio RM; Terrett NK; Tillyer RD; Truppo MD; Parmee ER The Importance of Synthetic Chemistry in the Pharmaceutical Industry. Science 2019, 363 (6424), eaat0805; [DOI] [PubMed] [Google Scholar]; (b) Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem 2018, 10 (4), 383–394.; [DOI] [PubMed] [Google Scholar]; (c) Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-like Molecules. Chem. Soc. Rev 2016, 45 (3), 546–576. [DOI] [PubMed] [Google Scholar]
- (2). Prices from following vendors: 1-propanol (Sigma-Aldrich, 1L); serinol (Sigma-Aldrich, 1 mL); 2,3-diaminopropan-1-ol (Enamine, 1g), 2-aziridinemethanol (Chemieliva, 1g).
- (3).Smith BR; Eastman CM; Njardarson JT Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem 2014, 57 (23), 9764–9773. [DOI] [PubMed] [Google Scholar]
- (4).(a) Müller TE; Beller M Metal-Initiated Amination of Alkenes and Alkynes. Chem. Rev 1998, 98 (2), 675–703; [DOI] [PubMed] [Google Scholar]; (b) Jensen KH; Sigman MS Mechanistic Approaches to Palladium-Catalyzed Alkene Difunctionalization Reactions. Org. Biomol. Chem 2008, 6 (22), 4083–4088; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McDonald RI; Liu G; Stahl SS Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev 2011, 111 (4), 2981–3019; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chemler SR; Bovino MT Catalytic Aminohalogenation of Alkenes and Alkynes. ACS Catal 2013, 3 (6), 1076–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Murphy JA The Radical–Polar Crossover Reaction In Radicals in Organic Synthesis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001; pp 298–314; [Google Scholar]; (b) Pitzer L; Schwarz JL; Glorius F Reductive Radical-Polar Crossover: Traditional Electrophiles in Modern Radical Reactions. Chem. Sci 2019, 10 (36), 8285–8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Kischkewitz M; Okamoto K; Mück-Lichtenfeld C; Studer A Radical-Polar Crossover Reactions of Vinylboron Ate Complexes. Science 2017, 355 (6328), 936–938; [DOI] [PubMed] [Google Scholar]; (b) Silvi M; Sandford C; Aggarwal VK Merging Photoredox with 1,2-Metallate Rearrangements: The Photochemical Alkylation of Vinyl Boronate Complexes. J. Am. Chem. Soc 2017, 139 (16), 5736–5739; [DOI] [PubMed] [Google Scholar]; (c) Phelan JP; Lang SB; Compton JS; Kelly CB; Dykstra R; Gutierrez O; Molander GA Redox-Neutral Photocatalytic Cyclopropanation via Radical/Polar Crossover. J. Am. Chem. Soc 2018, 140 (25), 8037–8047.; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Touney EE; Foy NJ; Pronin SV Catalytic Radical-Polar Crossover Reactions of Allylic Alcohols. J. Am. Chem. Soc 2018, 140 (49), 16982–16987. [DOI] [PubMed] [Google Scholar]
- (7).(a) Barluenga J; González-Bobes F; González JM Activation of Alkanes upon Reaction with PhI(OAc)2-I2. Angew. Chem. Int. Ed 2002, 41 (14), 2556–2558; [DOI] [PubMed] [Google Scholar]; (b) Chouthaiwale PV; Suryavanshi G; Sudalai A NaIO4-KI-NaN3 as a New Reagent System for C-H Functionalization in Hydrocarbons. Tetrahedron Lett 2008, 49 (45), 6401–6403; [Google Scholar]; (c) Bering L; Antonchick AP Selective Transition-Metal-Free Vicinal Cis-Dihydroxylation of Saturated Hydrocarbons. Chem. Sci 2017, 8, 452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu T; Mei T-S; Yu J-Q γ,δ,ε-C(Sp(3))-H Functionalization through Directed Radical H-Abstraction. J. Am. Chem. Soc 2015, 137 (18), 5871–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Review: Stateman LM; Nakafuku KM; Nagib DA Remote C-H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50 (8), 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) δ C-H functionalization via 1,5-HAT: Wappes EA; Fosu SC; Chopko TC; Nagib DA Triiodide-Mediated δ-Amination of Secondary C-H Bonds. Angew. Chem. Int. Ed 2016, 55 (34), 9974–9978; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Z; Stateman LM; Nagib DA δ C-H (Hetero)Arylation via Cu-Catalyzed Radical Relay. Chem. Sci 2019, 10 (4), 1207–1211; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang Z; Zhang X; Nagib DA Chiral Piperidines from Acyclic Amines via Enantioselective, Radical-Mediated δ C–H Cyanation. Chem 2019, 5, 3127–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Wappes EA; Nakafuku KM; Nagib DA Directed β C–H Amination of Alcohols via Radical Relay Chaperones. J. Am. Chem. Soc 2017, 139, 10204–10207; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nakafuku KM; Fosu SC; Nagib DA Catalytic Alkene Difunctionalization via Imidate Radicals. J. Am. Chem. Soc 2018, 140, 11202–11205; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stateman LM; Wappes EA; Nakafuku KM; Edwards KM; Nagib DA Catalytic β C–H Amination via an Imidate Radical Relay. Chem. Sci 2019, 10, 2693–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen AD; Herbort JH; Wappes EA; Nakafuku KM; Mustafa DN; Nagib DA Radical Cascade Synthesis of Azoles via Tandem Hydrogen Atom Transfer. Chem. Sci 2020, 10.1039/c9sc06239d. [DOI] [PMC free article] [PubMed]
- (12).(a) Mou XQ; Chen XY; Chen G; He G Radical-Mediated Intramolecular β-C(sp3)-H Amidation of Alkylimidates: Facile Synthesis of 1,2-Amino Alcohols. Chem. Commun 2018, 54, 515–518; [DOI] [PubMed] [Google Scholar]; (b) Kumar Y; Jaiswal Y; Kumar A Visible-Light-Mediated Remote γ-C(Sp3)-H Functionalization of Alkylimidates: Synthesis of 4-Iodo-3,4-Dihydropyrrole Derivatives. Org. Lett 2018, 20 (16), 4964–4969; [DOI] [PubMed] [Google Scholar]; (c) Shaw M; Kumar A Visible-Light-Mediated β-C(Sp3)-H Amination of Glycosylimidates: En Route to Oxazoline-Fused/Spiro Nonclassical Bicyclic Sugars. Org. Lett 2019, 21 (9), 3108–3113; [DOI] [PubMed] [Google Scholar]; (d) Wang F; Stahl SS Merging Photochemistry with Electrochemistry: Functional-Group Tolerant Electrochemical Amination of C(sp3)–H Bonds. Angew. Chem. Int. Ed 2019, 58 (19), 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wappes EA; Vanitcha A; Nagib DA β C-H Di-Halogenation via Iterative Hydrogen Atom Transfer. Chem. Sci 2018, 9 (19), 4500–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Giri R; Chen X; Yu JQ Palladium-Catalyzed Asymmetric Iodination of Unactivated C-H Bonds under Mild Conditions. Angew. Chem. Int. Ed 2005, 44 (14), 2112–2115; [DOI] [PubMed] [Google Scholar]; (b) Giri R; Wasa M; Breazzano SP; Yu JQ Converting Gem-Dimethyl Groups into Cyclopropanes via Pd-Catalyzed Sequential C-H Activation and Radical Cyclization. Org. Lett 2006, 8 (25), 5685–5688. [DOI] [PubMed] [Google Scholar]
- (15).Okuyama T; Takino T; Sueda T; Ochiai M Solvolysis of Cyclohexenyliodonium Salt, a New Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for Internal Return from an Intimate Ion—Molecule Pair. J. Am. Chem. Soc 1995, 117 (12), 3360–3367. [Google Scholar]
- (16).(a) Parasram M; Chuentragool P; Sarkar D; Gevorgyan V Photoinduced Formation of Hybrid Aryl Pd-Radical Species Capable of 1,5-HAT: Selective Catalytic Oxidation of Silyl Ethers into Silyl Enol Ethers. J. Am. Chem. Soc 2016, 138 (20), 6340–6343; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Parasram M; Chuentragool P; Wang Y; Shi Y; Gevorgyan V General, Auxiliary-Enabled Photoinduced Pd-Catalyzed Remote Desaturation of Aliphatic Alcohols. J. Am. Chem. Soc 2017, 139 (42), 14857–14860; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chuentragool P; Parasram M; Shi Y; Gevorgyan V General, Mild, and Selective Method for Desaturation of Aliphatic Amines. J. Am. Chem. Soc 2018, 140 (7), 2465–2468; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chuentragool P; Yadagiri D; Morita T; Sarkar S; Parasram M; Wang Y; Gevorgyan V Aliphatic Radical Relay Heck Reaction at Unactivated C(sp3)–H Sites of Alcohols. Angew. Chem. Int. Ed 2019, 58 (6), 1794–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Xu Y; Conner ML; Brown MK Cyclobutane and Cyclobutene Synthesis: Catalytic Enantioselective [2+2] Cycloadditions. Angew. Chem. Int. Ed 2015, 54 (41), 11918–11928; [DOI] [PubMed] [Google Scholar]; (b) Poplata S; Tröster A; Zou YQ; Bach T Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 +2] Photocycloaddition Reactions. Chem. Rev 2016, 116 (17), 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Collins KD; Glorius F A Robustness Screen for the Rapid Assessment of Chemical Reactions. Nat. Chem 2013, 5 (7), 597–601; [DOI] [PubMed] [Google Scholar]; (b) Collins KD; Glorius F Intermolecular Reaction Screening as a Tool for Reaction Evaluation. Acc. Chem. Res 2015, 48 (3), 619–627. [DOI] [PubMed] [Google Scholar]
- (19).Aloisi GG; Beggiato G; Mazzucato U Charge Transfer Complexes between Halogens and Pyridines. Part 4. - Effect of the Acid Strength of the Acceptors. Trans. Faraday Soc 1970, 66 (0), 3075–3080. [Google Scholar]
- (20).Yoshimura A; Zhdankin VV Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev 2016, 116 (5), 3328–3435. [DOI] [PubMed] [Google Scholar]
- (21).Chen EM; Keefer RM; Andrews LJ Acetyl Hypoiodite as an Aromatic Iodinating Agent. J. Am. Chem. Soc 1967, 89 (2), 428–430. [Google Scholar]
- (22).Keefer RM; Andrews LJ The Interaction of Iodine and Bromine with Organic Halides. J. Am. Chem. Soc 1952, 74 (8), 1891–1893. [Google Scholar]
- (23).Ashtekar KD; Vetticatt M; Yousefi R; Jackson JE; Borhan B Nucleophile-Assisted Alkene Activation: Olefins Alone Are Often Incompetent. J. Am. Chem. Soc 2016, 138 (26), 8114–8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Martínez C; Muñiz K An Iodine-Catalyzed Hofmann-Löffler Reaction. Angew. Chem. Int. Ed 2015, 54 (28), 8287–8291. [DOI] [PubMed] [Google Scholar]
- (25).Courtneidge JL; Lusztyk J; Pagé D Alkoxyl Radicals from Alcohols. Spectroscopic Detection of Intermediate Alkyl and Acyl Hypoiodites in the Suárez and Beebe Reactions. Tetrahedron Lett 1994, 35 (7), 1003–1006. [Google Scholar]
- (26).Caballero-García G; Romero-Ortega M; Barroso-Flores J Reactivity of Electrophilic Chlorine Atoms Due to σ-Holes: A Mechanistic Assessment of the Chemical Reduction of a Trichloromethyl Group by Sulfur Nucleophiles. Phys. Chem. Chem. Phys 2016, 18 (39), 27300–27307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.