

Abstract
Age-related macular degeneration (AMD) is one of the most common causes of vision loss. Advanced forms of AMD are seen in primarily 2 types—neovascular AMD (nAMD) with the presence of choroid neovascularization and nonneovascular AMD (nnAMD) with geographic atrophy. Although there are 4 anti-vascular endothelial growth factor drugs either widely used or approved for the former, there are no current treatments for the latter. This review will highlight upcoming treatments for AMD currently in clinical trials. For nAMD: Abicipar pegol, an intravitreal anti-vascular endothelial growth factor based on designed ankyrin repeat proteins (DARP) in protein, is currently pending approval. Conbercept and Faricimab, 2 intravitreal anti-growth factors, are currently in phase 3. Nine other upcoming agents have at least produced results in the 2A phase including intravitreal injections (KSI-301, OPT-302, RGX-314, ICON-1, and DE-122), depot (GB-102), drug reservoir (PDS), topical drops (PAN-90806), and oral formulations (AKST4290). We summarize all the newer molecules.
Keywords: emerging treatments, neovascular AMD, wet AMD
Age-related macular degeneration (AMD) is a disease that is characterized by an acquired degeneration of the retina that causes visual deficits primarily in the central vision.1 The key feature of AMD is the presence of drusen within the macula. Modification of the Bruch membrane/choroid complex and the retinal pigment epithelium and photoreceptor cells leads to degeneration of the retina. The prevalence of advanced AMD has been estimated to be 1.6% with exudative form in at least 1 eye being 1.2% and Geographic atrophy in 1 eye being 0.6%.2 Another study showed prevalence of exudative AMD in the population older than 52 years to be 1.5%.3 The number of patients globally with AMD is estimated to be 196 million in 2020 and 288 million in 2040.4
There is wet or neovascular AMD (nAMD), which is defined by choroidal neovascularization (CNV). CNV affects only 10% to 15% of patients with AMD but accounts for 90% of severe vision loss caused by AMD.5 The mainstay of modern nAMD treatment has been targeted anti-vascular endothelial growth factor (VEGF) therapy. VEGF is an important cytokine in angiogenesis.6 In particular vascular endothelial growth factor-A (VEGF-A) is a current target for retinal diseases. It binds to the extracellular ligand-binding domains of 2 tyrosine kinase receptors (VEGFR-1 and VEGFR-2), which leads to activation of an internal signaling pathway that alters the genes for angiogenesis and vascular permeability. Elevated levels of VEGF-A are found in the vitreous of patients with nAMD, diabetic retinopathy, and macular edema.7 As a result of the increased VEGF, there can be leakage of fluid and bleeding underneath the retina causing disruption and eventually resulting in the loss of vision.6
Bevacuzimab (Avastin), manufactured by Genetech/Roche, is a recombinant humanized monoclonal IgG1antibody that binds to VEGF.8 It binds to all forms of VEGF-A preventing it from binding to endothelial cell receptors. The off-label intravitreal formulation of bevacizumab has been used since 2005. Although it was never submitted to the US Food and Drug Administration (FDA) for AMD use, it is widely used “off label” and has now a long track record of safety and efficacy.9 Ranibizumab (Lucentis), manufactured by Genentech, is a monoclonal, humanized antibody fragment that blocks VEGF-A. FDA-approved intravitreal Ranibizumab for nAMD treatment in 2006 is on the basis of the improvement in visual acuity (VA) for nAMD in the MARINA (Minimally Classic/Occult Trial of the Anti-VEGF Antibody Ranibizumab in the Treatment of Neovascular AMD) and ANCHOR (Anti-VEGF Antibody for the Treatment of Predominantly Classic Choroidal Neovascularization in AMD) trials.10–14 Aflibercept, manufactured by Regeneron Pharmaceuticals, is a receptor decoy that binds to VEGF. Fc portion of human IgG1 is fused to the binding domains of VEGF receptor 1 and 2 to make this compound.15 Aflibercept was approved by the FDA in 2011 for nAMD based on the data from VIEW 1 and 2 (VEGF Trap-Eye: Investigation of Efficacy and Safety in Wet AMD) trials, which showed that the agent could be dosed every 2 months, after a loading dose.16
Bevacizumab (Avastin), ranibizumab (Lucentis), and Aflibercept (Eylea) are 3 drugs used in nAMD treatment. A fourth recently approved drug for intravitreal injection, Brolucizumab (Beovu), manufactured by Novartis, was approved by the FDA in the last quarter of 2019.17 Brolucizumab is a single-chain antibody fragment. Based on the data from the HAWK and HARRIER studies where brolucizumab was noninferior to aflibercept based on mean change in best-corrected visual acuity (BCVA) in naïve patients with nAMD. Also, >50% of the patients who received brolucizumab were maintained on Q12 weeks interval.18 This is the largest dosing schedule allowed by any of the drugs on the market. About 30% of the patients gained 15 letters in BCVA compared with baseline by year 1. Overall, the drug arms showed noninferiority in BCVA overall compared the controls (P < 0.001).
There are many other emerging drugs for the treatment of nAMD on the horizon. This review has 3 goals: first to provide a comprehensive list of drugs in development for AMD; second to provide updated results from existing trials; and third to gather expert correspondence and opinions about each of these results. Trials highlighted here have completed phase 2A, have future plans, and have yet to receive FDA approval.
nAMD STUDIES
Abicipar Pegol
Abicipar pegol (AGN-150998), developed by Allergan plc (Coolock, Dublin) and Molecular Partners AG (Zurich, Switzerland), is an anti-VEGF molecule based on ankyrin repeat proteins (DARPin) family that binds to VEGF-A. It has a longer intraocular half-life compared with ranibizumab.19 This drug is currently filing Biologics License Application with the FDA.20
Two Phase 3, randomized, double-blinded, multicentered, parallel, noninferiority trials titled CEDAR (NCT02462928)21 and SEQUOIA (NCT02462486)22 tested the effectiveness of abicipar pegol versus ranibizumab on treatment-naïve nAMD patients. In both trials, the first arm received 2 mg of intravitreal injections of the agent monthly for the first 3 months and then every 8 weeks. The second arm received 2 mg of intravitreal injections of the agent monthly for the first 2 months, then an injection 8 weeks after last treatment, and then every 12 weeks. The third arm was a control that received intravitreal injections of 0.5 mg of ranibizumab every 4 weeks.
The primary endpoint of both trials was percentage of patients with stable BCVA (loss of <15 letters) from baseline at 52 weeks. CEDAR enrolled 939 patients. Results showed 91.7% Q8w abicipar and 91.2% Q12w abicipar were stable compared with 94.5% of the ranibizumab Q4w arm.20 SEQUOIA enrolled 946 patients. Results showed 94.8% of Q8w abicipar arm and 91.3% of Q12w abicipar had stable vision compared with 96% of Q4w ranibizumab arm.20 Abicipar is the only anti-VEGF that has maintained stable vision in >91% of patients on a fixed 12-week regimen.
A major side-effect of the agent was intraocular inflammation. In CEDAR, it occurred on 15.1% on the Q8w agent arm and 15.4% in the Q12w agent arms, compared with 0% in the ranibizumab treated group. In SEQUOIA it occurred on 15.7% and 15.3% of Q8w and Q12w arms, compared with 0.6% of the control arm.
As such impurities in formulations were identified including potentially fragments of Escherichia coli, an integral part of manufacturing of DARPin.23 A new Phase 2 trial, MAPLE (NCT03539549), was designed with a new reformulated agent.24 A prospective open-labeled, multicentered, one-armed study with this new formulation in treatment-naïve or previous anti-VEGF nAMD patients (n = 124). Patients received loading dose of 2 mg of agent monthly for 3 months and then again after 8 weeks. The primary endpoint was changed from baseline in BCVA in the study eye at 28 weeks. Intraocular inflammation occurred at 8.9% with 1.8% being severe inflammation.25
The drug is currently being reviewed for regulatory application by the FDA with expected action coming in mid-2020.25
The rate on intraocular inflammation in MAPLE was similar to phase 2 trials with the old formulation. Phase 2 BAMBOO (n = 25) and CYPRESS (n = 25) trials had 7.5% intraocular inflammation in 16 weeks. Phase 2 REACH trial with the “old formulation” had 2 arms of abicipar 1 mg (n = 25) and abicipar 2 mg (n = 23), with an intraocular inflammation adverse events of 5 of 48 (10.4%) in abicipar-treated patients at 20 weeks. The effectiveness of the new formulation will be presented soon.26 In the MAPLE trial there was an exclusion criterion for active periocular, ocular, or intraocular infection at baseline, which was not present in the CEDAR and SEQUOIA trials. It is still possible that having a baseline inflammation can be worsened by the drug.
KSI-301
KSI-301, developed by KODIAK sciences, Palo Alto, CA, is an antibody biopolymer conjugate compromised of humanized anti-VEGF monoclonal antibody and phosphorylcholine-based polymer to augment the stability and increase residence time in the eye. An increased formulation strength delivers higher molecular doses of anti-VEGF. Design features include improvement in ocular pharmacokinetic parameter, greater long-term concentrations without same or increased bioactivity as compared with standard of care. Phase 1 in 9 patients showed primary safety and tolerability endpoint with no serious adverse events.
NCT03790852 is a phase 1b, randomized, open-label study comparing 2 doses of KSI-301 in patients with nAMD (n = 35), diabetic macular edema (n = 35), and retinal vein occlusion (n = 35).27 Patients were randomized in a 1:3 ratio to receive either 2.5 mg or 5 mg of KSI-301. Both arms had monthly doses for the first 3 months, followed by subsequent doses per retreatment criteria in the protocol. In the nAMD arm, 55% (16/29) have achieved a 6-month interval before first retreatment.28 Results showed in a pooled analysis at 24 weeks a mean gain of 5.9 early treatment diabetic retinopathy study (ETDRS) letters and a reduction of central subfield thickness of 58 μm in this group. This trial is currently ongoing with primary endpoint at 36 weeks.
DAZZLE (NCT04049266) is a Phase 2, randomized, double-blinded, noninferiority multicentered, trials on KSI-301 in treatment-naïve nAMD patients (n = 368).29 The first arm will receive 5 mg of intravitreal injections at either 12-, 16-, and 20-week intervals after 3 monthly loading doses. Patients in the second control arm will receive 2 mg of aflibercept intravitreal injections monthly for the first 3 months and then every 8 weeks. The primary outcome is the mean change in BCVA from baseline at 52 weeks. After year 1, patients in the aflibercept treatment arm will be re-randomized 1:1 into KSI-301 5 mg and aflibercept 2 mg arms and re-analyzed at 96 weeks. Pivotal studies looking at diabetic macular edema and retinal vein occlusion are currently being planned.
OPT-302
OPT-302, developed by OPTHEA Limited, inhibits VEGF C/D activity. Animal studies have shown benefits to combined inhibition of OPT-302 along with aflibercept (VEGF-A inhibitor) than the latter alone.
NCT03345082 is a Phase 2b, randomized, double-masked, prospective, multicentered, controlled study on intravitreal OPT-302 in patient (n = 366 patients) with nAMD.30 Patients were randomized to receive 1 of 3 arms in a 1:1:1 ratio. Patients received either 2 mg of OPT-302 along with 0.5 mg of ranibizumab (Group A), 0.5 mg of the agent OPT-302 with 0.5 mg of ranibizumab (Group B), or 0.5 mg ranibizumab alone (Group C). Patients received monthly injections in all 3 groups. The primary outcome of mean change from baseline in BCVA at 24 weeks was 14.22 letters for Group A, 9.44 letters for Group B, and 10.84 letters for Group C.31 Group A showed a superiority with a gain of +3.4 letters (P = 0.0107) compared with Group C. Secondary endpoints were also met at 24 weeks. Group A had more patients gaining ≥15 or more ETDRS letters and less patients losing ≥15 or more ETDRS letters Group C at 24 weeks. Additionally, at week 24 there was superiority in number of patients with sub-retinal (18.5% in Group A versus 29.3% in Group C) and intra-retinal cysts (16.8% versus 21.6%). The change in the total lesion area from baseline was −4.33 mm2 for Group A compared with −3.11 mm2 for Group C (P = 0.0137). Similarly, change in the CNV area from baseline was −4.97 mm2 for Group A compared with −3.59 mm2 for group C (P = 0.0052) at 24 weeks. The safety profile was comparable between Group A [69.4% treatment emergent adverse event (AE) and 8.3% serious AE] and Group C (75.0% treatment emergent AE and 5.6% serious AE).
Conbercept
Conbercept (KH902), developed by Chengdu Kanghong Pharmaceuticals Group Co., Ltd. (Chengdu, China), is a molecule that combines second Ig domain of VEGFR1 and the third and fourth Ig domain of VEGFR2 of Fc human IgG1.
PHOENIX (NCT01436864) was a phase 3, prospective, double-masked, multicentered, sham-controlled, trial on intravitreal Conbercept in patients with nAMD (n = 124) conducted in China.32 Patients were randomized into 2:1 “Conbercept” versus “sham” arms. The first Conbercept arm received 0.5 mg of the agent monthly for the first 3 months, as a loading dose, and then 0.5 mg every 3 months afterwards. The second sham arm received sham injections monthly for the first 3 months, then 0.5 mg monthly for 3 months and lastly 0.5 mg every 3 months afterwards. The primary endpoint showed +9.20 letter improvement in Conbercept group versus +2.02 letters in sham group at 3 months (P < 0.001). Further analysis at 12 months showed no statistical difference in the BCVA improvement between the 2 groups, with +9.98 BCVA letter improvement in the Conbercept arm versus +8.81 letters in sham arm, (P = 0.64). Following this trial, Conbercept was approved in China.
Ethical concerns have also been raised about the design of the trial. Per the authors rescue therapy was not offered in this study because there were no anti-VEGF therapies approved in China when the trial began in 2011. Ranibizumab was not approved in China till 2012. Additionally if a patient was elected to receive another AMD therapy, he or she was withdrawn from the study.32 However, Grzybowski and Kanclerz note that anti-VEGF agents were already being used off-label for intravitreal injections in China by the time the study began. Additionally, because photodynamic therapy was an established treatment for nAMD in Asia, a noninferiority study comparing Conbercept with another established therapy was certainly possible.33
PANDA-1 (NCT03577899) and PANDA-2 (NCT03630952) are phase 3, randomized, quadruple-blinded, multicentered, dose-ranging trial on intravitreal Conbercept on patients with nAMD.34 Patients will be randomized 1:1:1 to 3 arms and receive 0.5 mg of Conbercept, 1.0 mg of Conbercept, or 2.0 mg aflibercept, respectively. All 3 arms will receive monthly doses for the first 2 months, followed by doses every 2 months for the remainder of the trial. The primary endpoint is the mean change from BCVA at 36 weeks. Secondary outcomes include proportion of subjects gaining ≥15 ETDRS BCVA letters, mean changes in BCVA letters, mean changes from baseline in central retinal thickness (CRT) at 36 weeks, number of significant adverse effect (SAE), and blood concentration of Conbercept at 96 weeks. The trials are currently recruiting.
There is controversy regarding the lack of screening for polypoidal choroidal vasculopathy (PCV), a type of CNV seen in AMD patients.35 According to Călugă and Călugăru, since proportion of PCV in Asian patients with nAMD is between 20% and 60%, not stratifying for them via indocyanine green angiography (ICG) limits the results from this trial, especially since aflibercept has been shown to have better response against certain types of PCVs.36,37 In response Liu and Xu stated that the use of fundus fluorescein angiography was consistent with previous landmark anti-VEGF trials. Additionally, because of a shortage of ICG supply in China during the trial, it was impossible to perform ICG examinations for all study subjects.38 Additionally, there was no comparison done between the baselines of the 2 groups of patients. Călugă and Călugăru et al noted differences between the 2 groups in age of patients, previous treatment applied, and central retinal thickness among other factors.35
Faricimab
Faricimab (formerly known as RG7716), developed by Roche/Genentech, is the first bispecific antibody that blocks VEGF-A and angiopoietin-2. It also has a modified Fc portion to reduce systemic absorption and potential of intraocular inflammation. Ang-2 levels are elevated in patients with AMD and blocking Ang-2 reduces inflammation and leakage. Phase I study results confirmed Faricimab was safe and well-tolerated with improvements in BCVA and anatomic parameters for patients with difficult-to-treat neovascular AMD.39
AVENUE (NCT02484690) was a Phase II, multiple-center, randomized, double-masked, active comparator-controlled trial of intravitreal faricimab on treatment-naïve nAMD patients (n = 273).40 Patients were randomized in 3:2:2:2:3 fashion and received 0.5 mg of ranibizumab Q4, 1.5 mg of faricimab Q4, 6 mg of faricimab Q4, 6 mg of faricimab Q4 × 4 followed by Q8, 0.5 mg of ranibizumab Q4 weeks x 3 doses followed by faricimab 6 mg Q4 weeks, respectively. 1.5 mg of faricimab arm Q4 had best gains of +9.1 letters at 36 weeks. Although all arms showed significant decreases in central subfield thickness (CST), the combination therapy arm demonstrated the largest reduction of −185 μm. AVENUE concluded efficacy and safety of Faricimab Q4w and Q8w compared with monthly doses of ranibizumab.
STAIRWAY (NCT03038880) was another Phase II, randomized, double masked study evaluating the durability of trial of Faricimab in treatment-naïve nAMD patients.41 Patients were randomized in 2:2:1 fashion and received faricimab 6 mg Q4w × 4 doses then Q12w, faricimab 6 mg Q4w × 4 then Q16w, ranibizumab 0.5 mg Q4w, respectively. At the primary endpoint at week 52, visual acuity was fully maintained in patients treated with Q16W faricimab and Q12W faricimab compared with Q4W ranibizumab. Patients in the 12-week group gained 10.08 letters, those in the 16-week group gained 11.42 letters, and the ranibizumab group had a mean gain of 9.59 letters. 65% of patients treated with Faricimab had no protocol-defined disease activity 12 weeks after their last injection.
LUCERNE (NCT03823300) and TENAYA (NCT03823287) are a Phase III, randomized, triple masked, parallel assignment trial on intravitreal Faricimab in patients (n = 640) with nAMD. Patients will be randomized into either the Faricimab or Aflibercept arm. Patients in the first arm will be given Faricimab based on the study protocol. Patient in the second arm will be given aflibercept monthly for 3 months, followed by once every 2 months. The primary outcome will be changed in BCVA at 48 weeks. Secondary outpoints include change in BCVA, percentage in patients gaining or losing certain number of letters, change from baseline in CST thickness, percentage of patients without intraretinal fluid, patients without subretinal fluid and changes in the CNV area and leakage at 112 weeks.
RGX-314
RGX-314, manufactured by Regen BioPharma (La Mesa, CA), uses a novel AAV8 vector to deliver a genome that induces production of an anti-VEGF Fab, similar to ranibizumab, delivered via subretinal injection during vitrectomy. Per the company, their gene delivery method yields higher levels of anti-VEGF expression than earlier AAV vectors. In animal models, maximal expression of therapeutic protein in the anterior chamber with the RGX-314 was 4992 ng/mL, versus 528 ng/mL and 0.217 ng/mL for the previous Genzyme and Avalanche vectors, respectively.42
NCT03066258 was an phase I/IIa, open-labeled, nonrandomized, sequential assignment, dose escalation, multicentered trial on previously treated nAMD patients (n = 42) that required ≥4 anti-VEGF injections in the 8 months before trial entry.43 This was a 5-armed study with first 2 arms (A and B) of 6 patients and 3 arms (C, D, E) of 12 patients. All cohorts received anti-VEGF injection of ranibizumab to evaluate a response before enrolling in the study. Two weeks after the ranibizumab injection, each cohort received RGX-314. Cohort A, B, C, D, and E received 3 × 109 genome copies (GC)/eye, 1 × 1010 GC/eye, 6 × 1010 GC/eye, 1.6 × 1011 GC/eye, and 2.5 x 1011 GC/eye, respectively. Anti-VEGF was given 4 weeks later post-treatment and as needed every 4 weeks thereafter, per investigator discretion. Reasons for anti-VEGF intervention included if CNV increased, there was new or persistent fluid, there was vision loss of ≥5 letters associated with fluid or there was new retinal hemorrhage. The primary endpoint to determine the safety and tolerability of RGX-314 in subjects with nAMD through 26 weeks has been met. Cohorts ranged between 45.9 and 71.7 weeks since first anti-VEGF injection. No drug-related SAE was observed.44
Secondary endpoints include changes in BCVA, CRT, rescue injections, and mean change in area of CNV at 106 weeks. Interim data presented show the requirement injection (annualized) for cohort A, B, and C was 10.5, 9.0, and 2.6, respectively. The requirement injections (per 6 months) for cohort D and E were 4.4 and 1.6, respectively. In cohort 3, 3 of 6 patients did not require injections over 1.5 years. In cohort D and E, there were 5 of 12 and 9 of 12 patients who did not need injections in a 5- to 6-month period. All groups had an improvement in BCVA. Cohort C, D, and E had a BCVA improvement of +9, +2, and +4 letters and a reduction of CRT of −40, −42, and −68 μm, respectively. This trial is ongoing till 106 weeks per secondary endpoints.
GB-102
GB-102, developed by GrayBug Vision, is an injectable form of sunitinib maleate, a tyrosine kinase inhibitor that targets both VEGF-A and platelet-derived growth factor (PDGF). When injected it forms a depot in the inferior vitreous, which gradually biodegrades over time and functions as a durable method of antiangiogenesis. GB-102 treatment can last up to 6 months with comparable visual acuity and central subfield thickness outcomes before another dose is required.
ADAGIO (NCT03249740) was a Phase 1/2a, open-labeled, single-dose, multicentered study on GB-102 in nAMD patients (n = 32). Patients who showed positive response to any anti-VEGF agents after 3 previous intravitreal injections were enrolled in the study if they showed disease activity before study treatment. Four escalating dose cohorts of 8 patients each received a single dose of either 0.25, 0.5, 1, or 2 mg of GB-102. The primary endpoints of safety and tolerability were successfully met. Secondary endpoints evaluated stability of visual acuity and subretinal thickness. Eighty-eight percent of the patients at 3 months and 68% of the patients at 6 months were maintained on a single dose of GB-102. Some patients maintained positive outcomes as far as 8 months.45 optical coherence tomography measurements showed reduction of CST at all months compared with pre-treatment (P < 0.05). However, the 2-mg arm had microparticle dispersion to the anterior chamber, causing a decrease in visual acuity.46 A new manufacturing process was developed to eliminate the microparticle dispersion and incomplete aggregation, which was used for future trials.
ALTISSIMO is a Phase 2b (NCT03953079), randomized, single-masked, multicentered, controlled study on GB-102 for CNV lesions secondary in previously treated nAMD patients (n = 160). Patients will be put into 3 arms and will be given either 1 mg of GB-102, 2 mg of GB-102, or 2 mg of aflibercept at baseline. The GB-102 arms will then receive their same initial dose every 6 months, whereas the latter control group will continue to receive aflibercept 2 mg every 2 months. The primary outcome is the proportion of treated subjects remaining rescue-free through month 10. Secondary outcome includes BCVA from baseline at months 10 and through month 12, mean changes to CST at months 9 of 10 and through 12 months, mean number of intravitreal injections, and absence of exudation at month 9 and 12 of treatment.
One advantage of this device is that it bridges the gap between current intravitreal injection treatments and real-life solution for wet AMD. Of the emerging therapies, GB-102 has by far the longest time needed between treatments, and thus can have a significant impact on the patient's life.46
PAN-90806
PAN-90806, from PanOptica (Mount Arlington, NJ), is a tyrosine kinase inhibitor of VEGF-A and PDGF. The agent is a topical drop that was hypothesized to reach the target tissues in the retina using the trans-scleral vascular route. Previous phase 1/2 confirmed role as monotherapy in nAMD patients over 8 weeks of treatment (n = 20) and as a maintenance therapy in nAMD following singe injection of ranibizumab for 12 weeks (n = 10). However, a side-effect of these studies was punctate keratopathy due to off target inhibition of corneal epithelial epidermal growth factor receptor. A new formula was made for future studies.
NCT03479372 was a Phase 1/2, randomized, double-masked, uncontrolled, multicenter study in treatment naive nAMD patients, using PAN-90806 topical drops.47 Patients were randomized 1:1:1 and treated with either 2, 6, or 10 mg/mL drops daily for 12 weeks and were evaluated every week with rescue injections starting week 2. Patients were screened every week with rescue injections starting week 2. The primary endpoint of safety and tolerability was met with “no major or serious untoward (unfavorable and unintended) safety issues or trends.”48 The secondary endpoints evaluated the anti-VEGF biological response. Through the 1-month post-treatment visit, 51% of patients never needed rescue therapy and completed the study on the agent alone. Twenty-three (88%) of the 26 nonrescued patients showed clinical improvement or stability. Subjects requiring at least 1 ranibizumab rescue injections were 7 of 17 in the 2 mg/mL arm, 9 of 18 in the 6 mg/mL arm, and 9 of 16 in the 10 mg/mL arm. Still, at least 79% of scheduled possible rescue injections were avoided by patients, supporting the topical agent as not only a maintenance therapy couple with ranibizumab, but potentially a monotherapy. Masked review showed that patients with visual acuity (<20/50) and baseline severity (CST >400 μm) predisposed patients to early rescue.
PAN-90806 shows potential as a monotherapy treatment for nAMD, with possibilities for prophylaxis or chronic maintenance. This is the first time a topical anti-VEGF eyedrop has demonstrated safety and biological response as monotherapy. However, it may be only appropriate for certain patients, as seen in the results of the masked review and further studies are needed to confirm these findings.
Port Delivery System
The port delivery system (PDS), manufactured by Genentech (South San Francisco, CA), is a drug delivery system that is a permanent, refillable device surgically inserted into the patient's eye.49 The patient must undergo surgery to insert the self-sealing implant in the sclera and the pars plana. The reservoir in the implant allows for drug replenishment in clinic without the need to remove it from the eye. Ranibizumab in the PDS moves by passive diffusion down a concentration gradient from the implant into the vitreous.
LADDER (NCT02510794) was a Phase 2, multicenter, randomized, active treatment-controlled study that compared PDS with ranibizumab with intravitreal ranibizumab in nAMD patients (n = 220).49 Patients had to have received ≥2 previous anti-VEGF injections and been responsive to treatment to be qualified. Participants were randomized into 3:3:3:2 arms comparing PDS with ranibizumab 10 mg/mL, 40 mg/mL, 100 mg/mL, or monthly intravitreal ranibizumab 0.5 mg injections, respectively. The primary endpoint was time to first implant refill and the results showed that the patients went 8.7, 13.0, and 15.0 months in the PDS 10 mg/mL, 40 mg/mL, and 100 mg/mL arms, respectively.49 In the latter arm, 79.8% went ≥6 months without needing a refill. At 9 months, BCVA change from baseline was –3.2, –0.5, +5.0, and +3.9 letters in the PDS 10 mg/mL, PDS 40 mg/mL, PDS 100 mg/mL, and monthly intravitreal ranibizumab 0.5-mg arms, respectively. Vision outcomes for the PDS 100 mg/mL arm seemed to be comparable with vision outcomes for the monthly intravitreal injections, in post hoc analysis. The CFT change from baseline was similar in PDS 100 mg/mL and monthly control arm at 9 months. There was initially increased vitreous hemorrhage as high as 50% during initial surgical procedures. A modified procedure was developed which included scleral dissection to the pars plana followed by cauterization of the choroid with laser coagulation. With this new surgical procedure optimization, postoperative vitreous hemorrhage was 4.5% (7/157), including 1 serious event.
ARCHWAY is a Phase 3 multicenter, randomized, active comparator-controlled trial that is evaluating patients with nAMD (n = 418) with fixed interval dosing. Patients in the PDS arm will receive the surgical implant with refills of 100 mg/mL ranibizumab at a fixed interval of every 24 weeks. The control arm patients will receive 10 mg/mL ranibizumab on a monthly basis. The primary outcome for this study is the change in BCVA from baseline to week 40, whereas secondary outcomes include changes in BCVA for a variety of time frames, changes in CST at week 36, and participants in the PDS arm that require supplemental treatments before their scheduled refill.
The sustained delivery device allows for patients to avoid monthly injections, lowering their treatment burden for the patients and may possibly improve real world outcomes. Potential drawbacks to this device include the fact that an additional surgery is required for the insertion of this device, which has its own complication. However, with wider spread usage, surgical precision should also improve.46 During the LADDER study, refinements were made to decrease hemorrhage during the procedure which included ablation of the pars plana at the incision site.50 Additionally, further studies need to be done to analyze patients on anti-thrombotic therapy who will undergo the implant. Lastly, longer-term studies are needed to reveal local effect and durability of the permanent implant in the eye.
ICON-1
ICON-1, a product of Iconic Therapeutics (South San Francisco, CA), is a recombinant modified factor VIIIa protein linked with the Fc portion of a human immunoglobulin G1. It binds to tissue factor, which is overexpressed in choroidal neovasculature of AMD, but does not interfere with normal blood coagulation. The Fc portion of this molecule can bind to the Fc receptor of cells like Natural Killer cells and prompt antibody-dependent cellular toxicity to reduce CNV. There is potential for ICON-1 to be combined with existing anti-VEGF medication.
NCT03452527 was a Phase 1, open-label, dose-escalation, nonrandomized, multicentered study on intravitreal injection of ICON-1 in patients (n = 18) with CNV due to nAMD.51 The primary endpoint of safety and tolerability was met, as no patients had any SAEs caused by the study drug.
EMERGE is a phase 2, randomized, double-masked study on intravitreal intravitreal in patients (n = 88) with CNV secondary to nAMD. Patients were randomized 1:1:1 to receive a combination of 0.5 mg ranibizumab and 0.3 mg of ICON-1, 0.5 mg of ranibizumab only, and 0.3 mg ICON-1 only.52 All arms received monthly injections for 3 months. After this point, patients could receive further treatments if necessary. The primary endpoints were BCVA, CRT, and safety. ICON-1 monotherapy group only had mild reduction in CRT and stable BCVA. CNV decreased by 40% in the combination arm, 14.6% in the ranibizumab only arm, and 17.2% in the ICON-1 only arm at 6 months.52 In the combination and ranibizumab arm, improvement in BCVA (+8.4 letters in the vs +8.3 letters) and decrease in CRT (−83.9 vs −91.4 μm) were equivalent. There was no SAE related to the drug. The average number of treatments needed after month 3 was 1 for the combination arm and 1.4 for the ranibizumab only arm.
DECO (NCT03452527) is another Phase 2 randomized, open-label, parallel, multicentered study in patients (n = 15) with CNV secondary to nAMD.53 All patients will receive initial treatment with aflibercept and will then receive maintenance therapy with 0.6 mg of intravitreal injection of ICON-1 or 2 mg of intravitreal injection of aflibercept. The primary outcome of this study is the main change in the CNV area from baseline at 9 months’ period. The secondary outcomes are changes in BCVA from baseline at 9 months and duration of treatment-free periods over time.
Currently an anti-tissue factor monoclonal antibody ICON-4 is being evaluated as a potential therapy for AMD with clinical trials starting in 2020.54
AKST4290
AKST4290 (formerly ALK4290), from Alkahest, (San Carlos, CA) is an oral treatment that targets eotaxin, an immunomodulatory chemokine highly expressed in CNVs, choroidal endothelial cells, and circulation in AMD. The agent is an inhibitor against CCR3, which is the natural receptor for eotaxin. Increase of eotaxin and C-C chemokine receptor type 3 increases membrane permeability and degradation, immune cell recruitment, and disturbs regulation. CCR3 and its ligand CCL11 are expressed in neovascular lesions, and CCL11 levels are increased in choroidal endothelial cells and systemic circulations in patients with AMD.55 AKST4290 decreases inflammatory cytokines in preclinical models.
AKST4290–201 (NCT03558061) and AKST4290–202 (NCT03558074) were both phase 2a, single-arm, open-label studies of oral 400 mg bis in die monotherapy administrate of AKST4290 in nAMD patients. AKST4290–201 included naive patients (n = 29). AKST4290–202 included patients (n = 26) no longer responding to anti-VEGF therapy, which was defined as persistent fluid and no BCVA improvement after at least 3 monthly injections.56 The primary endpoint for both trials was mean change in BCVA. Secondary endpoints for both included safety, exploratory morphological endpoints at biomarkers at 6 weeks, CRT, intraretinal fluid, subretinal fluid, and changes to pigment epithelial detachments. AKST4290–201 shows that 24 patients (83%) had stable or better BCVA change from baseline at 6 weeks. The mean improvement in BCVA was +7 letters. AKST4290–202 showed 72% of patients had stable or improved BCVA.57 Mean improvement in BCVA was 2 letters with 8% gaining ≥10 letters. CST did not change during 6-week treatment. However, there was improvement in the CST in the 4-week follow-up after treatment was completed. Other secondary points including retinal pigment epithelium detachment maximum height, CNV size, and CNV leakage size did not show significant change from baseline.55 Safety profile revealed no SAEs in either trials. Currently randomized, placebo-controlled studies are planned in the future.
DE-122
DE-122 (Carotuximab), from Santen (Osaka, Japan) and TRACON Pharmaceuticals (San Diego, CA), is an antibody to endoglin, a protein that plays a critical role in angiogenesis.58 Endoglin is a growth factor that is expressed in the endothelium. Endoglin has been shown to release angiogenic factors from inflammatory cells in vivo. Endoglin has also been shown to be in high amounts in endothelial cells contained within choroidal neovascular membranes. Mice models have shown benefit of combined targeting of endoglin and anti-VEGF signal pathway.59 This drug is an ophthalmic reformulation of TRC 105, an anti-cancer drug made by TRACON.
PAVE (NCT02555306) was a Phase 1/2, open-labeled, dose-escalating, sequential cohort, multicentered study of intravitreal DE-122 on patients (n = 12) with nAMD refractory to VEGF inhibitors. Patients had to have received previous treatment in the study eye in intravitreal anti-VEGF. Patients were separated into 4 arms and received either 0.5 mg, 1.0 mg, 2.0 mg, or 4.0 mg via a single intravitreal injection of D-122. The primary outcome, the change in BCVA, was −1.3 (2.1 SD) for the 0.5-mg arm, 2.7 (5.0 SD) for the 1.0-mg arm, 2.0 (7.9SD) for the 2.0 mg arm, and 3.3 (4.5 SD) for the 4.0-mg arm at 90 days. A secondary outcome, changes in the CST, was −116.3 (194.9 SD) for the 0.5-mg arm, 62.7 (68.1 SD) for 1.0-mg arm, −36.0 (42.6 SD) for 2.0-mg arm, and −111.3 (171.2 SD) for the 4.0-mg arm.60 There was no medication-related SAE at 90 days.
AVANTE (NCT03211234) is a Phase 2, multicenter, randomized, double-masked, active control study of intravitreal injections DE-122 on nAMD patients (n = 76). Patients were randomized 1:1:1 arm that received either DE-122 with Lucentis, high-dose DE-122 with Lucentis, or Lucentis alone. The primary outcome was the mean change from baseline in BCVA at 24 weeks. This study recently completed enrollment.61
Below in Table 1 we have summarized the results of recent nAMD trials.
TABLE 1.
Ongoing nAMD Trials
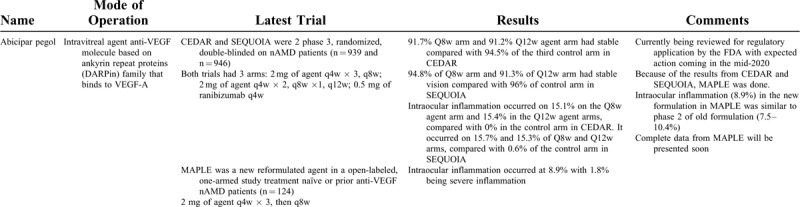
EMERGING AND HALTED TRIALS
Recent nAMD trials that have been terminated or not moving forward are listed in Table 2.
TABLE 1 (Continued).
Ongoing nAMD Trials
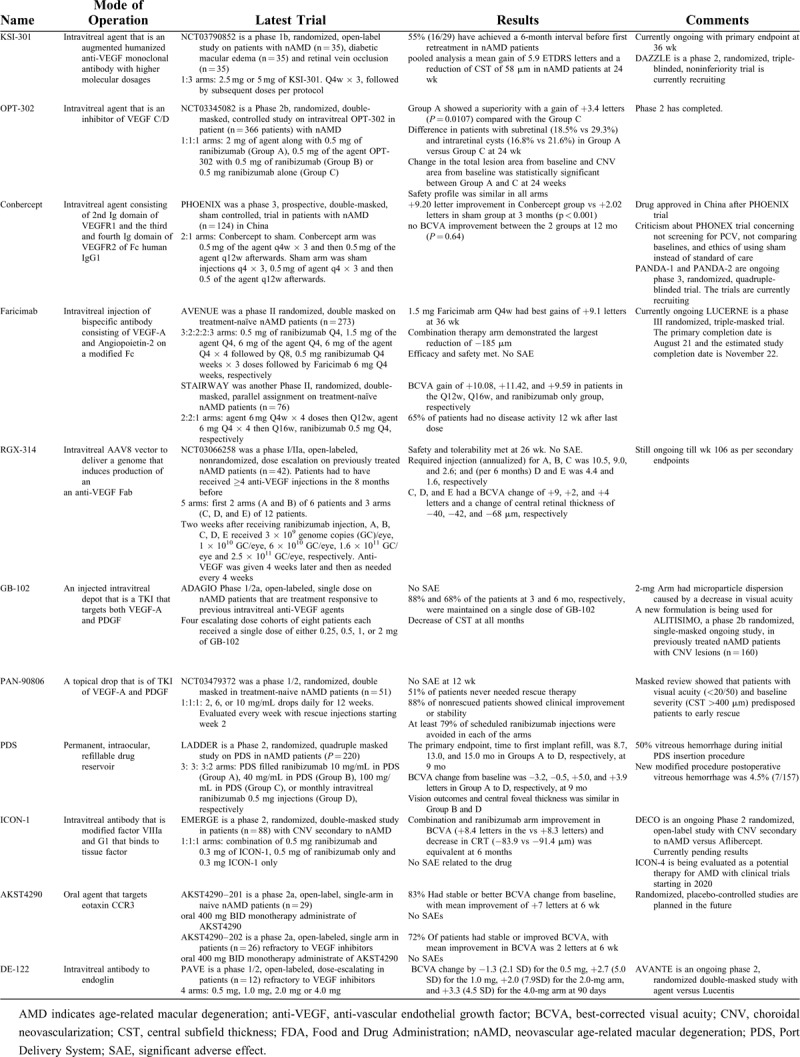
nAMD trials that are currently in earlier phases have been listed in Table 3.
TABLE 2.
nAMD Trials Terminated or Not Moving Forward
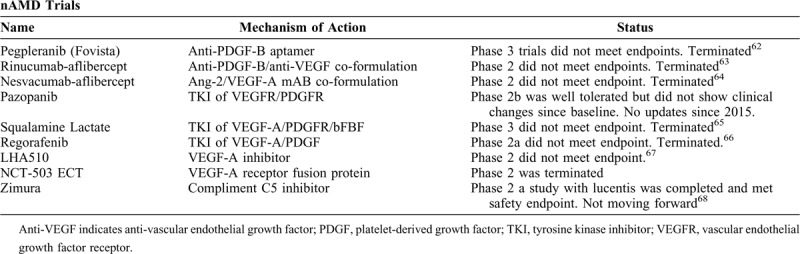
TABLE 3.
nAMD Emerging Trials

CONCLUSIONS
There are exciting newer therapies emerging for AMD. On the nAMD front, abicipar pegol is already seeking FDA approval, whereas Faricimab and Conbercept are already in phase 3. There are also alternatives to intravitreal agents that show promise. PAN-90906 as a topical drop, oral AKST4290, and PDS as a depot offer alternative solutions to patients with compliancy issues with intravitreal agents. For the year 2020, when it comes to AMD, there are exciting treatments on the horizon.
Footnotes
The authors have no conflict of interest to declare.
A.K.: Consultant—Adverum, Alcon, Allegro, Allergan, Bausch and Lomb, Chengdu Kanghong Biotechnology Co., Genentech, Gemini, Gyroscope, Inc., Eyepoint, Kodiak Sciences Inc., Novartis, Opthea, Oxurion, PolyPhotonix, Recens, Regenxbio, Roche.
Research support—Adverum, Allergan, Chengdu Kanghong Biotechnology Co., Gemini, Genentech, Inc., Roche, Novartis, Gyroscope, Kodiak Sciences Inc., Novartis, Opthea, Ophthotech, Oxurion, Regenxbio.
Lecture fees — Allergan, Genentech, Inc., Novartis.
AAA: None
J.C.: Novartis; Allergan; OD-OS.
REFERENCES
- 1. Elshatory Y, Feldman B, Tripathy K, Kim L, Shah V. Age-related macular degeneration - EyeWiki Aaoorg. 2019. https://eyewiki.aao.org/Agerelated_macular_degeneration. [Google Scholar]
- 2.Klein R, Klein BEK, Linton KLP. Prevalence of age-related maculopathy: The Beaver Dam Eye Study. Ophthalmology 1992; 99:933–943. [DOI] [PubMed] [Google Scholar]
- 3.Leibowitz HM, Krueger DE, Maunder LR, et al. The Framingham Eye Study monograph: An ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973-1975. Surv Ophthalmol 1980; 24: suppl: 335–610. [PubMed] [Google Scholar]
- 4.Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health 2014; 2:e106–e116. [DOI] [PubMed] [Google Scholar]
- 5.Bressler NM, Bressler SB, Congdon NG, et al. Potential public health impact of age-related eye disease study results: AREDS Report No. 11. Arch Ophthalmol 2003; 121:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lien S, Lowman HB. Therapeutic anti-VEGF antibodies. Handb Exp Pharmacol 2008; 131–150. [DOI] [PubMed] [Google Scholar]
- 7.Chong V. Ranibizumab for the treatment of wet AMD: a summary of real-world studies. Eye (Lond) 2016; 30:270–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldman B, Lim J, Tripathy K, Kim L, Karth P, Shah V. Bevacizumab - EyeWiki Aao org 2019; https://eyewiki.aao.org/Bevacizumab [Google Scholar]
- 9.Lynch SS, Cheng CM. Bevacizumab for neovascular ocular diseases. Ann Pharmacother 2007; 41:614–625. [DOI] [PubMed] [Google Scholar]
- 10.Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006; 355:1419–1431. [DOI] [PubMed] [Google Scholar]
- 11.Chang TS, Bressler NM, Fine JT, Dolan CM, Ward J, Klesert TR. Improved vision-related function after ranibizumab treatment of neovascular age-related macular degeneration: results of a randomized clinical trial. Arch Ophthalmol 2007; 125:1460–1469. [DOI] [PubMed] [Google Scholar]
- 12.Kaiser PK, Blodi BA, Shapiro H, Acharya NR. Angiographic and optical coherence tomographic results of the MARINA Study of ranibizumab in neovascular age-related macular degeneration. Ophthalmology 2007; 114:1868–1875. [DOI] [PubMed] [Google Scholar]
- 13.Brown DM, Michels M, Kaiser PK, Heier JS, Sy JP, Ianchulev T. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR Study. Ophthalmology 2009; 116:57–65. [DOI] [PubMed] [Google Scholar]
- 14.Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med 2006; 355:1432–1444. [DOI] [PubMed] [Google Scholar]
- 15.Semeraro F, Morescalchi F, Duse S, Parmeggiani F, Gambicorti E, Costagliola C. Aflibercept in wet AMD: specific role and optimal use. Drug Des Devel Ther 2013; 7:711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trichonas G, Kaiser PK. Aflibercept for the treatment of age-related macular degeneration. Ophthalmol Ther 2013; 2:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delfaro A. Brolucizumab wins FDA approval, introducing extended dosing for wet AMD. Am Acad Ophthalmol 2019; https://www.aao.org/headline/brolucizumab-wins-fda-approval-introducing-extendi [Google Scholar]
- 18.Dugel PU, Koh A, Ogura Y, et al. HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology 2020; 127:72–84. [DOI] [PubMed] [Google Scholar]
- 19.Kunimoto D, Ohji M, Maturi RK, et al. Evaluation of abicipar pegol (an anti-VEGF DARPIN therapeutic) in patients with neovascular age-related macular degeneration: Studies in Japan and the United States. Ophthalmic Surg Lasers Imaging Retina 2019; 50:e10–e22. [DOI] [PubMed] [Google Scholar]
- 20. Khurana R. Safety and Efficacy of Abicipar in Patients with Neovascular Age-related Macular Degeneration. In: American Academy of Ophthamology, 10/26/2019, San Francisco, CA; 2019. Available at: https://www.molecularpartners.com/allergan-and-molecular-partners-announce-topline-safety-results-from-maple-study-of-abicipar-pegol/. Accessed December 12, 2019. [Google Scholar]
- 21. A Safety and Efficacy Study of Abicipar Pegol in Patients With Neovascular Age-related Macular Degeneration (CDER). Available at: https://clinicaltrials.gov/ct2/show/NCT02462928. Published 2015. [Google Scholar]
- 22.2015; Safety and efficacy of abicipar pegol in patients with neovascular age-related macular degeneration. https://clinicaltrials.gov/ct2/show/NCT02462486 [Google Scholar]
- 23.Sharma A, Kumar N, Kuppermann BD, Bandello F. Abicipar pegol: the non-monoclonal antibody anti-VEGF. Eye (Lond) 2020; 34:797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evaluating abicipar for safety and treatment effect in patients with neovascular age-related macular degeneration (AMD) 2018; https://clinicaltrials.gov/ct2/show/NCT03539549 [Google Scholar]
- 25.Allergan and molecular partners present late-breaking data from phase 3 studies of investigational abicipar pegol in neovascular wet age-related macular degeneration – molecular partners. Molecularpartnerscom 2019; https://www.molecularpartners.com/allergan-and-molecular-partners-present-late-breaking-data-from-phase-3-studies-of-investigational-abicipar-pegol-in-neovascular-wet-age-related-macular-degeneration/ [Google Scholar]
- 26. Allergan and Molecular Partners Announce Topline Safety Results from MAPLE study of Abicipar pegol. Available at: https://www.molecularpartners.com/allergan-and-molecular-partners-announce-topline-safety-results-from-maple-study-of-abicipar-pegol/. Published 2019. Accessed December 12, 2019. [Google Scholar]
- 27.Exploratory study to investigate the bioactivity, ocular and systemic safety, tolerability, and pharmacokinetics following single and multiple intravitreal administrations of KSI-301 in subjects with wAMD, DME and RVO - full text view – ClinicalTrialsgov clinicaltrialsgov 2020; https://clinicaltrials.gov/ct2/show/NCT03790852?term=ksi-301&draw=2&rank=1 [Google Scholar]
- 28. Do DV. Update on phase 1b and phase 2 studies of KSI-301: a novel anti-VEGF antibody biopolymer conjugate with potential for extended durability in wet AMD. In: Angiogenesis, Exudation and Degeneration 2020, February 8, 2020, Miami, FL. [Google Scholar]
- 29. A Study to Evaluate the Efficacy and Safety of KSI-301, an Anti-VEGF Antibody Biopolymer Conjugate, Versus Aflibercept in Patients With Neovascular (Wet) Age-Related Macular Degeneration. (DAZZLE). Available at; https://clinicaltrials.gov/ct2/show/study/NCT04049266#eligibility. Published 2019. [Google Scholar]
- 30. A dose-ranging study of intravitreal OPT-302 in combination with ranibizumab, compared with ranibizumab alone, in participants with neovascular age-related macular degeneration (wet AMD). Available at: https://clinicaltrials.gov/ct2/show/NCT03345082. Published 2017. [Google Scholar]
- 31. Baldwin M. Phase 2b Clinical Results of OPT-302 (VEGF-C/D ‘Trap’) Combination Treatment in nAMD. In: October 10, 2019, Ophthalmology Innovation Summit @ American Academy of Ophthalmlogy, San Francisco, CA. San Francisco. [Google Scholar]
- 32.Liu K, Song Y, Xu G, et al. Conbercept for treatment of neovascular age-related macular degeneration: results of the randomized phase 3 PHOENIX Study. Am J Ophthalmol 2019; 197:156–167. [DOI] [PubMed] [Google Scholar]
- 33.Grzybowski A, Kanclerz P. Comment on: conbercept for treatment of neovascular age-related macular degeneration: results of the randomized Phase 3 PHOENIX Study. Am J Ophthalmol 2020; 209:216. [DOI] [PubMed] [Google Scholar]
- 34.Efficacy and safety trial of conbercept intravitreal injection for neovascular age-related macular degeneration (PANDA-1) - full text view - ClinicalTrials.gov. Clinicaltrialsgov 2020; https://clinicaltrials.gov/ct2/show/NCT03577899 [Google Scholar]
- 35.Călugă D, Călugăru M. Conbercept for treatment of neovascular age-related macular degeneration: results of the randomized phase 3 Phoenix Study. Am J Ophthalmol 2019; 198:262–263. [DOI] [PubMed] [Google Scholar]
- 36.Jeong S, Sagong M. Short-term efficacy of intravitreal aflibercept depending on angiographic classification of polypoidal choroidal vasculopathy. Br J Ophthalmol 2017; 101:758–763. [DOI] [PubMed] [Google Scholar]
- 37.Cheung CMG, Lai TYY, Ruamviboonsuk P, et al. Polypoidal choroidal vasculopathy: definition, pathogenesis, diagnosis, and management. Ophthalmology 2018; 125:708–724. [DOI] [PubMed] [Google Scholar]
- 38.Liu K, Xu X. Reply Am J Ophthalmol 2019; 198:263.doi:10.1016/j.ajo.2018.09.029. [DOI] [PubMed] [Google Scholar]
- 39.Chakravarthy U, Bailey C, Brown D, et al. Phase I trial of anti–vascular endothelial growth factor/anti-angiopoietin 2 bispecific antibody RG7716 for neovascular age-related macular degeneration. Ophthalmol Retin 2017; 1:475–485. [DOI] [PubMed] [Google Scholar]
- 40.2020; A proof-of-concept study of Faricimab (RO6867461) in participants with choroidal neovascularization (CNV) secondary to age-related macular degeneration (AMD) - FULL TEXT VIEW - ClinicalTrials.gov. Clinicaltrials.gov. https://clinicaltrials.gov/ct2/show/NCT02484690 [Google Scholar]
- 41.Study to evaluate faricimab (RO6867461; RG7716) for extended durability in the treatment of neovascular age related macular degeneration (nAMD) - full text view - ClinicalTrials.gov. Clinicaltrialsgov 2020; https://clinicaltrials.gov/ct2/show/NCT03038880 [Google Scholar]
- 42. Regenxbio announces additional positive interim phase I/IIA trial update for RGX-314 for the treatment of wet amd at the American Academy of Ophthalmology 2019 Annual Meeting. Available at: https://regenxbio.gcs-web.com/news-releases/news-release-details/regenxbio-announces-additional-positive-interim-phase-iiia-trial. Published 2019. [Google Scholar]
- 43. RGX-314 gene therapy for neovascular AMD trial. Available at: https://clinicaltrials.gov/ct2/show/NCT03066258. Published 2017. [Google Scholar]
- 44.Heier J. Key takeaways from the RGX-314 phase I/IIa clinical trial for wet AMD (Cohorts 1-5). In: American Academy of Ophthamology, 10/21/2019, San Francisco, CA ; 2019; https://www.regenxbio.com/wp-content/uploads/2019/10/Key-Takeaways-From-The-RGX-314-Phase-I-IIa-Clinical-Trial-For-Wet-AMD-Cohorts-1-5.pdf [Google Scholar]
- 45. Boyer D. New developments in drug therapy for retinal disorders. In: Hawaiian Eye & Retina Annual Meeting, January 21, 2019, Kona, Hawaii; 2019. [Google Scholar]
- 46. Baker-Schena L. Drug delivery for the posterior segment. American Academy of Ophthamology. Available at: https://www.aao.org/eyenet/article/drug-delivery-for-the-posterior-segment. Published 2019. [Google Scholar]
- 47.Study of PAN-90806 eye drops, suspension for neovascular AMD - full text view - ClinicalTrials.gov. Clinicaltrialsgov 2020; https://clinicaltrials.gov/ct2/show/NCT03479372 [Google Scholar]
- 48. Chaney P. PAN-90806: Once-daily topical anti-VEGF eye drop for wet AMD and other neovascular eye disease. In: Ophthamology Innovation Summit, October 10, 2019, San Francisco, CA; 2019. Available at: https://www.panopticapharma.com/wp-content/uploads/2019/10/PAN-90806-Data-at-OIS@AAO.pdf.. [Google Scholar]
- 49.Campochiaro PA, Marcus DM, Awh CC, et al. The port delivery system with ranibizumab for neovascular age-related macular degeneration: results from the randomized phase 2 ladder clinical trial. Ophthalmology 2019; 126:1141–1154. [DOI] [PubMed] [Google Scholar]
- 50.Sharma A, Kumar N, Kuppermann BD, Francesco B. Re: Campochiaro et al.: The Port Delivery System with ranibizumab for neovascular age-related macular degeneration: results from the randomized phase 2 Ladder clinical trial (Ophthalmology 2019; 126: 1141-1154). Ophthalmology 2019; 126:e87–e88. [DOI] [PubMed] [Google Scholar]
- 51.Wells JA, Gonzales CR, Berger BB, Gonzalez VH, Sippy BD, Burian G. A phase 1, open-label, dose-escalation trial to investigate safety and tolerability of single intravitreous injections of ICON-1 targeting tissue factor in wet AMD. Ophthalmic Surg Lasers Imaging Retin 2018; 49:336–345. [DOI] [PubMed] [Google Scholar]
- 52.Gonzales CR, Burian G. A phase 2 study (EMERGE) evaluating repeated intravitreal administration of ICON-1 in patients with choroidal neovascularization (CNV) secondary to age-related macular degeneration (AMD). Invest Ophthalmol Vis Sci 2017; 58:3766. [Google Scholar]
- 53. Open-label study of intravitreal ICON-1 in patients with choroidal neovascularization secondary to age-related macular degeneration (AMD) - full text view - ClinicalTrials.gov. Clinicaltrials.gov. 2020. Available at: https://clinicaltrials.gov/ct2/show/NCT03452527. [Google Scholar]
- 54.2020; Annes S. Iconic therapeutics signs ophthalmology option agreement. http://iconictherapeutics.com/wp-content/uploads/2019/08/press-release-novartis-ophthalmology-option-agreement-08222019.pdf [Google Scholar]
- 55.Krader CG. Green light given in early trial for agent targeting neovascular AMD. Ophthamology Times 2019; 50.https://www.ophthalmologytimes.com/sites/default/files/legacy/mm/digital/media/OT091519_ezine.pdf [Google Scholar]
- 56. Jeffords E. AKST4290: An Oral Small Molecule CCR3 Antagonist. In: Ophthamology Innovation Summit, July 25, 2019, Chicago, IL. Available at: https://ois.net/wp-content/uploads/2019/07/Alkahest-FINAL-7-22-19.pdf. [Google Scholar]
- 57.Abbey AM. Downside of finger flicks, upside of abicipar changes. Retin Spec 2019; 38.https://www.retina-specialist.com/CMSDocuments/2019/10/rs0919i.pdf [Google Scholar]
- 58.Grisanti S, Canbek S, Kaiserling E, et al. Expression of endoglin in choroidal neovascularization. Exp Eye Res 2004; 78:207–213. [DOI] [PubMed] [Google Scholar]
- 59.Shen W, Lee SR, Yam M, et al. A combination therapy targeting endoglin and VEGF-A prevents subretinal fibro-neovascularization caused by induced müller cell disruption. Invest Ophthalmol Vis Sci 2018; 59:6075–6088. [DOI] [PubMed] [Google Scholar]
- 60. Inc S. Santen presents phase I/II data on DE-122 (Carotuximab) in patients with refractory wet age-related macular degeneration. Prnewswire.com. 2018. Available at: https://www.prnewswire.com/news-releases/santen-presents-phase-iii-data-on-de-122-carotuximab-in-patients-with-refractory-wet-age-related-macular-degeneration-300597011.html. [Google Scholar]
- 61. TRACON pharmaceuticals I. TRACON pharmaceuticals reports second quarter 2019 financial results and provides corporate update. GlobeNewswire News Room. 2019. Available at: https://www.globenewswire.com/news-release/2019/08/07/1898645/0/en/TRACON-Pharmaceuticals-Reports-Second-Quarter-2019-Financial-Results-and-Provides-Corporate-Update.html. [Google Scholar]
- 62. A Phase 3 Safety and Efficacy Study of Fovista® (E10030) Intravitreous Administration in Combination With Lucentis® Compared to Lucentis® Monotherapy. Available at: https://clinicaltrials.gov/ct2/show/NCT01944839. Published 2013. [Google Scholar]
- 63. Regeneron Pharmaceuticals I. Regeneron announces phase 2 study of aflibercept co-formulated with Rinucumab (anti-PDGFR-beta) shows no benefit over aflibercept alone in neovascular age-related macular degeneration. Prnewswire.com. 2016. Available at: https://www.prnewswire.com/news-releases/regeneron-announces-phase-2-study-of-aflibercept-co-formulated-with-rinucumab-anti-pdgfr-beta-shows-no-benefit-over-aflibercept-alone-in-neovascular-age-related-macular-degeneration-300337055.html. [Google Scholar]
- 64. Regeneron provides update on EYLEA (AFLIBERCEPT) injection and NESVACUMAB (ANG2 Antibody) Combination Program. Available at: https://newsroom.regeneron.com/news-releases/news-release-details/regeneron-provides-update-eylear-aflibercept-injection-and?ReleaseID=1049746. Published 2017. [Google Scholar]
- 65. A study of MSI-1256F (Squalamine Lactate) to treat “wet” age-related macular degeneration - full text view - ClinicalTrials.gov. Clinicaltrials.gov. 2020. Available at: https://clinicaltrials.gov/ct2/show/NCT00333476. [Google Scholar]
- 66.Joussen AM, Wolf S, Kaiser PK, et al. The Developing Regorafenib Eye drops for neovascular Age-related Macular degeneration (DREAM) study: an open-label phase II trial. Br J Clin Pharmacol 2019; 85:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poor SH, Adams CM, Ferriere M, Weichselberger A, Grosskreutz CL, Weissgerber G. Topical VEGF receptor inhibitor, LHA510, did not demonstrate efficacy in a Proof-of-Concept study in patients with neovascular age-related macular degeneration (nv AMD). Invest Ophthalmol Vis Sci 2018; 59:2394. [Google Scholar]
- 68. Gingerich CP. Zimura/Lucentis Combo Safe, But Company Decides Not to Move Forward. MD Mag. 2018. Available at: https://www.mdmag.com/medical-news/zimura-lucentis-combo-safe-but-company-decides-not-to-move-forward. [Google Scholar]