

Abstract
One of the challenges of the post genomic era is to provide a more realistic representation of cellular processes by combining a systems biology description of functional networks with information on their interacting components. Here we carried out a systematic large-scale computational study on a structural protein-protein interaction network dataset in order to dissect thermodynamic characteristics of binding determining the interplay between protein affinity and specificity. As expected, interactions involving specific binding sites display higher affinities than those of promiscuous binding sites. Next, in order to investigate a possible role of modular distribution of hot spots in binding specificity, we divided binding sites into modules previously shown to be energetically independent. In general, hot spots that interact with different partners are located in different modules. We further observed that common hot spots tend to interact with partners exhibiting common binding motifs, whereas different hot spots tend to interact with partners with different motifs. Thus, energetic properties of binding sites provide insights into the way proteins modulate interactions with different partners. Knowledge of those factors playing a role in protein specificity is important for understanding how proteins acquire additional partners during evolution. It should also be useful in drug design.
Keywords: affinity, protein hubs, protein networks, protein-protein interactions, specificity
1. Introduction
Protein-protein interactions play an important role in all biological processes, including gene expression control, signal transduction and immune response. Networks of protein-protein interactions are increasingly being studied to provide a systems-level description of the cell [1,2,3,4]. Yet, most of these studies examine global aspects of network topology, ignoring the molecular nature of the interactions. A thermodynamic and structural characterization of protein interactions is essential for the understanding of biological mechanisms and complements the systems biology perspective [5,6,7]. One of the interesting problems which require studies from a systems and molecular points of view is the interplay between protein binding affinity and specificity. Protein-protein interaction networks may impose evolutionary constraints on protein interfaces to maintain favorable and avoid unfavorable interactions, and to preserve the optimal binding affinity for the biological process [8]. Proteins are able to bind multiple partners with a wide range of affinities, either simultaneously or individually. Lower affinities are often associated with highly transient regulated interactions, crucial for the multiple-partners-binding function of hubs.
A number of experimental studies have focused on the molecular basis of protein-protein interactions which leads to such diversity in binding affinity and specificity [9,10,11]. Computational analyses of the architecture [12] and physico-chemical characteristics [13,14,15,16] of binding sites have provided insights into important features of protein interfaces (recently reviewed in [17]). Furthermore, sequence and structural plasticity of binding sites could facilitate interaction with several protein partners [18]. The nature and distribution of binding hot spots also play an important role in protein associations. Alanine scanning analysis has revealed that some binding hot spots are conserved within protein families [19], whereas others are specific to each family member [20,21,22]. Experiments have emphasized the modular design of binding sites, with energetic cooperative contribution of single residues within the module; and additive between modules [23,24,25]. Computational protein design methods have also been used to elucidate the tradeoff between stability and specificity for the optimization of biological function [26,8]. However, most of these studies have been carried out on a limited set of protein examples, while the general principles governing protein-protein binding remain elusive (reviewed in [17]).
A characteristic of protein interactions is that some binding sites are able to interact with multiple partners, whereas others are specific to a single interaction. Here we study the linkage between protein binding specificity and affinity in protein interaction networks. We compiled a non-redundant structural protein-protein interaction network dataset of protein hubs from the yeast interactome [27]. We estimated the binding free energy of each interaction. As expected, our results show that interactions occurring through specific binding sites usually display higher affinities than those taking place through promiscuous binding sites, indicating that the ability of binding sites to interact with multiple partners is achieved at the cost of binding affinity. This is expected since multiple-partner binding through the same binding site should be transient, readily responding to the changing conditions in the cell. We further observed a tendency of binding sites to interact with partners exhibiting similar degree of specificity/promiscuity and these interactions to occur with similar affinities. This could indicate multiple paths in the network toward the same aim; that is, system robustness.
In an attempt to explore the linkage between specificity/promiscuity with respect to the affinity of the interaction, we carried out a comprehensive in silico alanine-scanning to determine binding hot spots. Our analysis revealed that most hot spots in promiscuous binding sites are involved in individual interactions whereas only a few hot spots participate in more than one interaction (common hot spots). Binding sites comprising these common hot spots usually interact with partners that contain the same binding motifs; whereas hot spots that interact with different partners and are located in different modules, tend to interact with partners with different binding motifs.
We have previously shown that the modular organization of protein structures divides the binding site into energetically independent modules which contain highly cooperative hot spot residues [28]. Here, we study a potential role of the modular distribution of hot spot residues in binding site specificity. Interestingly, the number of binding site modules containing hot spots increases with promiscuity. Hence, hot spots in promiscuous binding sites tend to be more distributed over different modules, whereas specific binding sites generally contain hot spots within one module. This situation is nicely illustrated with promiscuous binding sites in ubiquitin and a small rho-like GTPase; and with specific binding sites in cytochrome b and calmodulin-dependent protein kinase.
Despite the fact that hot spot modular distribution plays an important role in modulating protein specificity, the mechanism by which binding sites are capable of interacting with many binding partners varies considerably. For example, not surprisingly most structurally disordered binding sites are promiscuous, suggesting multiple states to accommodate different binding partners. Similarly, binding sites containing a large fraction of hydrophobic patches are highly promiscuous.
To our knowledge, our study is the first which systematically estimates protein binding free energies for all interactions in a large-scale experimental protein network in order to address the relationship between binding specificity/promiscuity and affinity. We observed that the modular distribution of binding hot spots is an important factor in determining binding specificity. Our approach addresses properties of large-scale protein interaction networks and provides useful information for binding site design. We note that since this study is carried out on a large number of proteins, it does not account for protein dynamics and structural changes are not considered.
2. Materials and Methods
2.1. Structural interactome
We compiled the yeast interactome from several experimental sources [27,29], eliminating redundancy by selecting the set of non redundant partners (sequence similarity < 80%) for each interacting protein. We next searched for representative PDB structures of the complexes, with the additional restriction of non-redundancy of pairs of interacting chains on each node. We obtained an initial subset of 259 hubs (proteins with more than 5 partners with representative structure) participating in 877 different interactions (see Table S1). Each hub was associated with a representative template structure.
2.2. Mapping of binding sites
Binding sites were mapped onto the surfaces of each hub template structure by means of a multiple alignment, and clustered into mutually exclusive interfaces using an agglomerative hierarchical algorithm [30], following several steps:
Starting from N binding sites, compute the N x N matrix of binary distances between pairs of binding sites, where a value is set to 1 if a pair overlaps in at least one residue and 0 otherwise.
Compute the k-means clustering of the distance matrixes for k = 1…N clusters, re-computing at each step distances between the new merged clusters.
Stop step 2) at the first k clusters where all binding sites within clusters overlap.
Using this method we identified a total of 539 interfaces involved in different interactions (1 to 5 interactions).
2.3. In silico energetic analysis
Binding energies and alanine scanning for each interacting complex were computed by using FoldX [31]. A residue was considered a hot spot in one interaction if the free binding energy change upon mutation to alanine was equal or higher than 2 kcal/mol. The accuracy of the energetic predictions for these point mutations was estimated in [32] to be within a standard deviation of 0.5 kcal/mol.
2.4. Interacting motifs
Sequence motifs for each ORF in yeast were downloaded from the PEP database [33], and mapped into the binding site of the interacting partners for each complex structure.
2.5. Modular decomposition of protein structures
We modeled template structures of hubs as graphs, with residues corresponding to vertices, and their contact to edges. Two residues were considered to be in contact if their distance was less than or equal to 5 Å. These networks were subsequently decomposed into modules by means of the edge-betweenness algorithm [34], based on the iterative removal of edges with the highest number of paths running through it.
2.6. Model evaluation
For those interactions in our analysis where no complex structures from yeast were available, we used the closest complex homologues from other organisms. In order to estimate the accuracy of our predictions in these cases, we focused our analysis on the subset of protein-protein interactions with existing yeast structures (experimental structures). For each protein-protein interaction of this subset, we selected the corresponding closest homologue in another organism (modeled structure), (see Table S4).
We carried out a comparison between the experimental and modeled structures for each step in our analysis:
Complex structural alignment. A comparison between the experimental and the modeled structures of the interacting complexes was performed by using the structural alignment program mammoth [35]. An average root mean square deviation (RMSD) of 2.2 Å was obtained, which can be considered a robust performance based on standard docking evaluations [36].
Modular decomposition. Further, we compared the modular decomposition of both protein structures, and we found that on average almost 71.9% of residues within a module in the experimental structure overlap with the corresponding aligned residues contained within a module in the modeled structure.
Modular distribution of hot spots. We further compared modules containing predicted hot spots in both protein structures, finding that 72.2% of modules that contain hot spots in the experimental structure match modules that also contain hot spots in the modeled structure.
Energetic analysis. Finally, we computed the binding free energy per residue for both complexes. The averaged difference between these energies was found to be close to zero, whereas the estimated standard deviation of the averaged differences for our dataset was 0.01 (kcal/mol)/residue, a value that supports the robustness of our main result in Figure 1A.s
Figure 1.
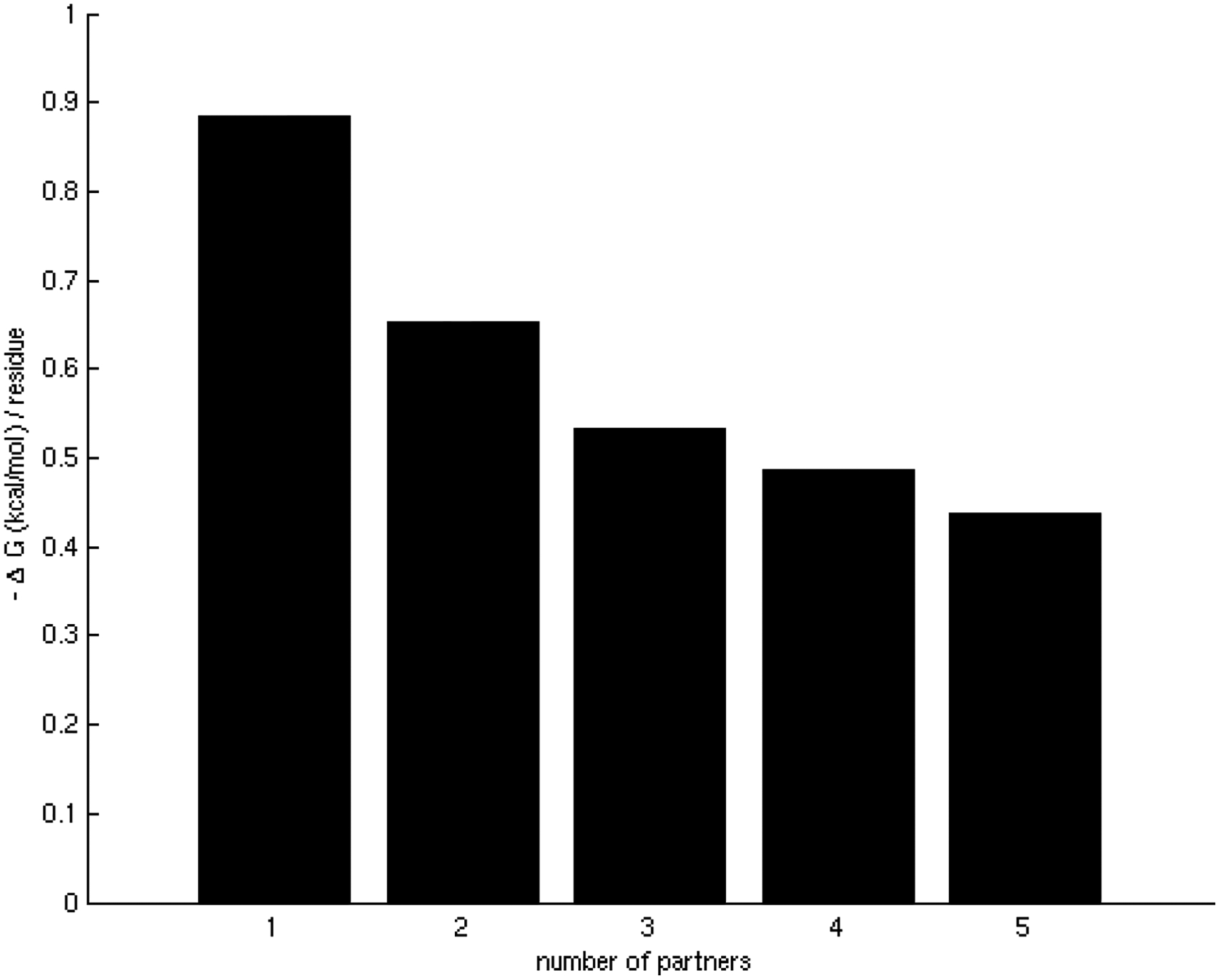
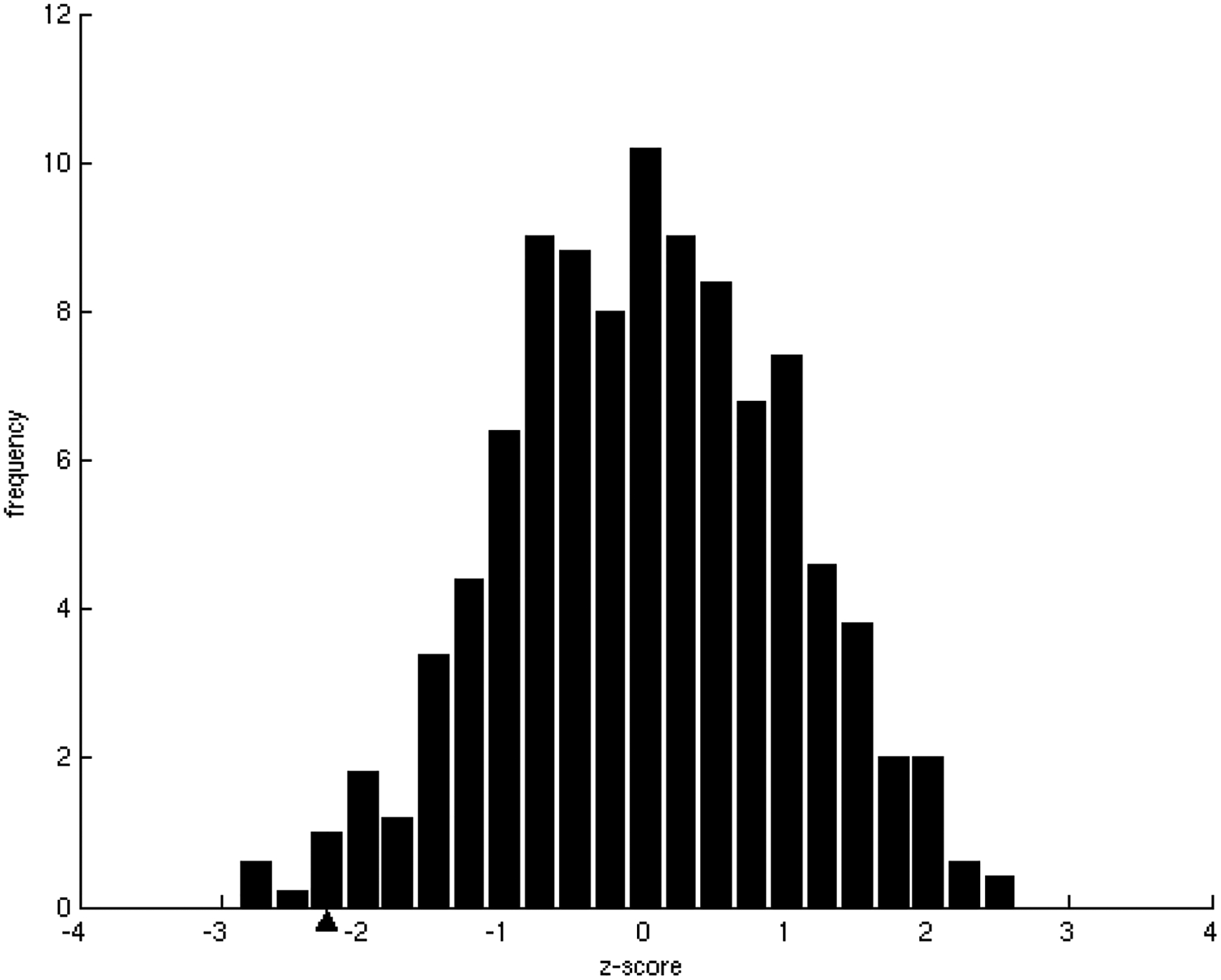
Relationship between the number of interacting partners (specificity/promiscuity) and binding energy per residue (affinity).
A) Binding energy per residue in binding sites averaged for different number of partners in binding sites. There is a clear tendency for the binding affinities to become weaker as the number of interacting partners of binding sites increases.
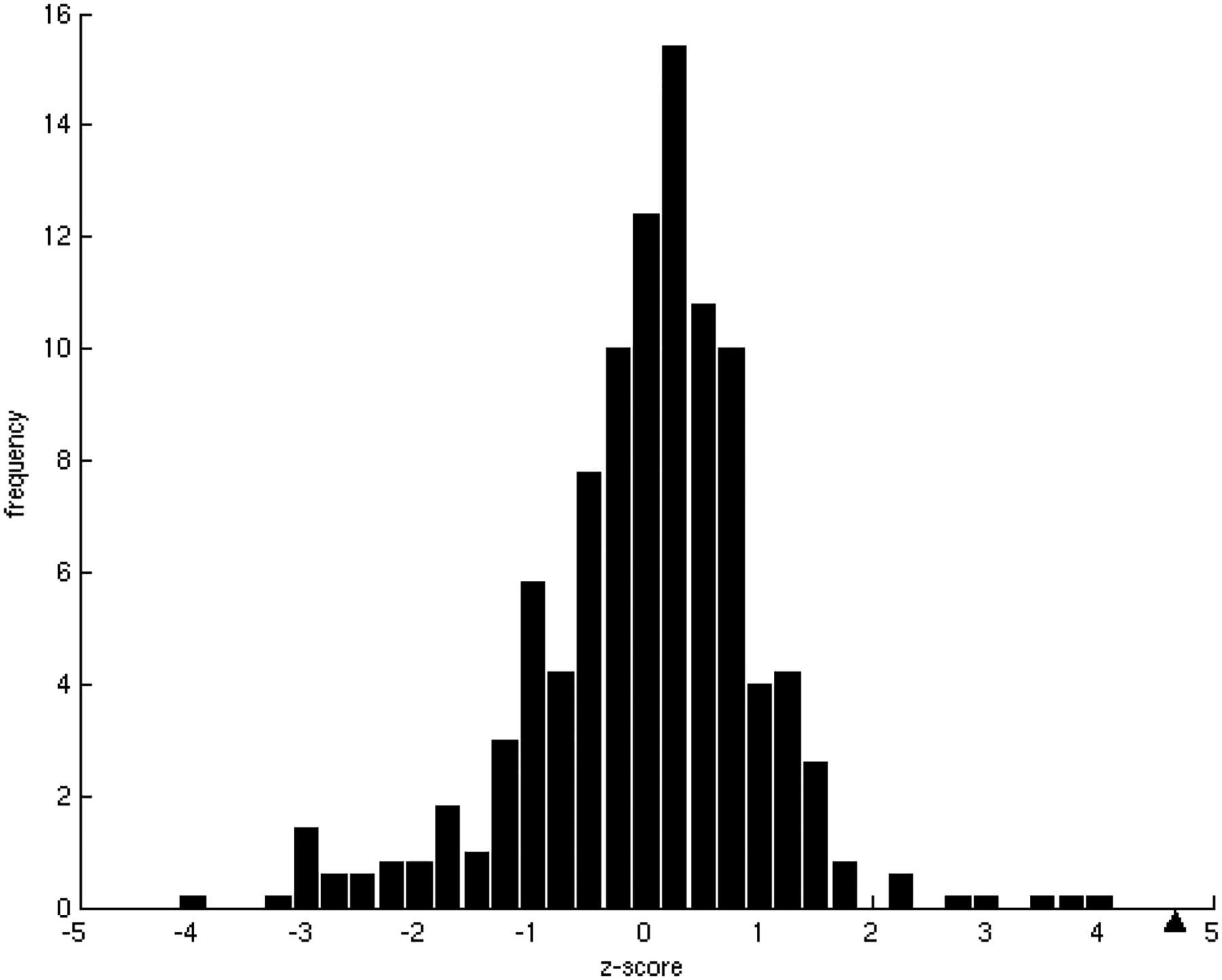
B) Z-score frequency distribution of the correlation coefficients of 500 randomizations of the binding energies. In our dataset, the observed tendency, marked with a black triangle, has a statistically significant z-score = −2.17(p-value = 1.50 × 10−2).
2.7. Random tests
The random tests were performed for 500 randomizations of the dataset that were generated by the following protocols: a) randomization of interacting affinities by a random shuffle of the interacting energies; b) random interactions between hubs by random shuffle of the partners of each hub; c) randomization of hot spots by randomly rearranging hot spots on each binding site. The statistical significance of the Pearson correlation coefficients r was computed by means of the Fisher transformation [37]:
| (1) |
2.8. Binding site disorder and hydrophobic patches
Disordered regions on the binding sites were calculated by using DisEMBL [38]. Hydrophobic patches on the binding sites were calculated as in [13].
3. Results and Discussion
3.1. Affinity and specificity of interactions
In order to investigate the linkage between affinity and binding site specificity in protein-protein interactions, we compiled a non-redundant structural protein-protein interaction network dataset of protein hubs from the yeast interactome [27,29]. Based on the structures of protein complexes, we mapped binding sites on the surface of each hub (see Materials and Methods), and annotated the numbers of their interacting partners. Hence, the hub interactome was represented as an interactome of hub interfaces, with the specificity/promiscuity determined by the numbers of binding partners. A binding site with only one partner was classified as specific; sites with multiple partners as promiscuous.
Interaction affinities, estimated as the per residue binding free energy on the interface [31], were calculated for all interactions of each binding site. As expected, we observed that specific binding sites tend to bind their partners with higher affinity than promiscuous sites. As the number of different partners increases, the interactions become weaker, ranging, on average, from approximately 0.9 kcal/mol per residue in binding sites with only one partner to less than 0.5 kcal/mol per residue in binding sites with 5 partners (Figure 1A), indicating a tradeoff between binding affinity and specificity. These results were shown to be statistically significant (z-score = −2.17, p-value = 1.50 × 10−2) by the generation of a random test based on the reshuffling of binding energies (Figure 1B). We further considered binding site specificities at both sides of the interacting pairs, observing that specific binding sites interact with other specific binding sites with higher affinity than with promiscuous ones. In contrast, interactions between promiscuous binding sites tend to be weaker (Table 1). The frequencies of specific-specific and promiscuous-promiscuous interactions between hubs is high as compared to frequencies from a random rewiring of the interacting hubs, with z-score values of 2.8 (p-value = 2.56 × 10−3) and 2.2 (p-value = 1.39 × 10−2) respectively (Figure 2A,B). In general, binding sites interact with their partners with similar affinities (Figure 3A) as compared to the random case (Figure 3B). Differences between binding affinities corresponding to interactions occurring through the same binding site were observed to be very small (close to 0), whereas energy differences between interactions occurring through distinct binding sites were around 0.5 kcal/mol per residue.
Table 1.
Averaged affinities for interactions between different types of binding sites: specific-specific, promiscuous-promiscuous, and specific-promiscuous.
| Interaction type | -ΔG [(kcal/mol)/residue] |
|---|---|
| specific-specific | 0.93 |
| specific-promiscuous | 0.85 |
| promiscuous-promiscuous | 0.50 |
Figure 2.
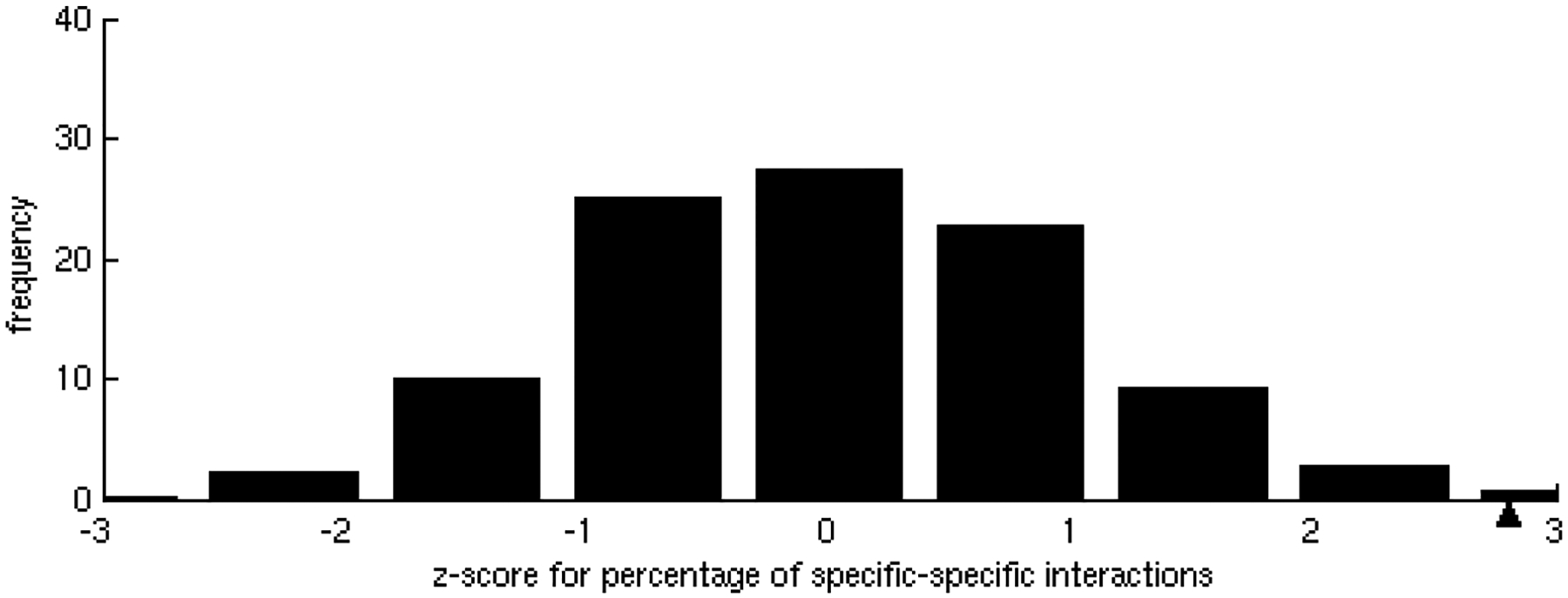
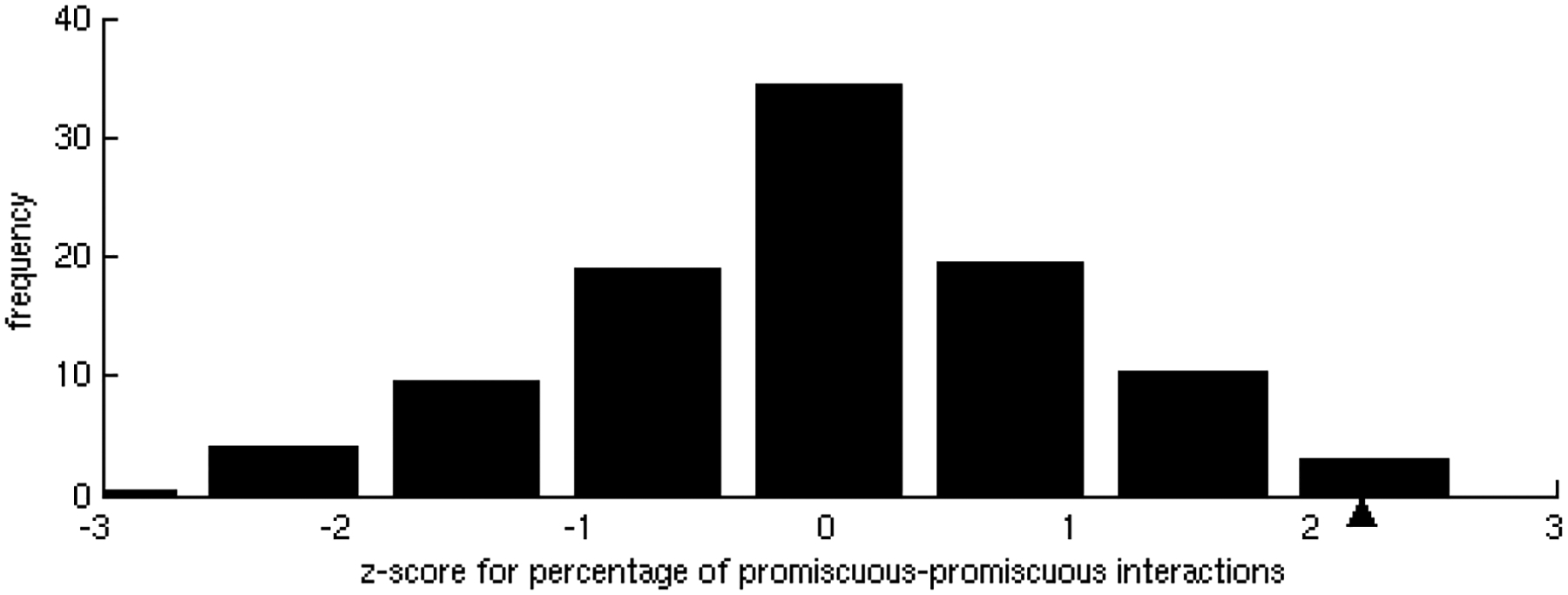
Homogeneity in specificities of interactions.
A) Z-score distribution of the percentage of specific-specific interactions found in 500 randomizations of the interacting partners. In the dataset, the observed percentage, marked with a black triangle, of specific-specific interactions is statistically significant (z-score = 2.8, p-value = 2.56 × 10−3).
B) Z-score distribution of the percentage of promiscuous-promiscuous interactions found in 500 randomizations of the interacting partners. In the dataset, the observed percentage, marked with a black triangle, of promiscuous-promiscuous interactions is statistically significant (z-score = 2.2, p-value = 1.39 × 10−2).
Figure 3.
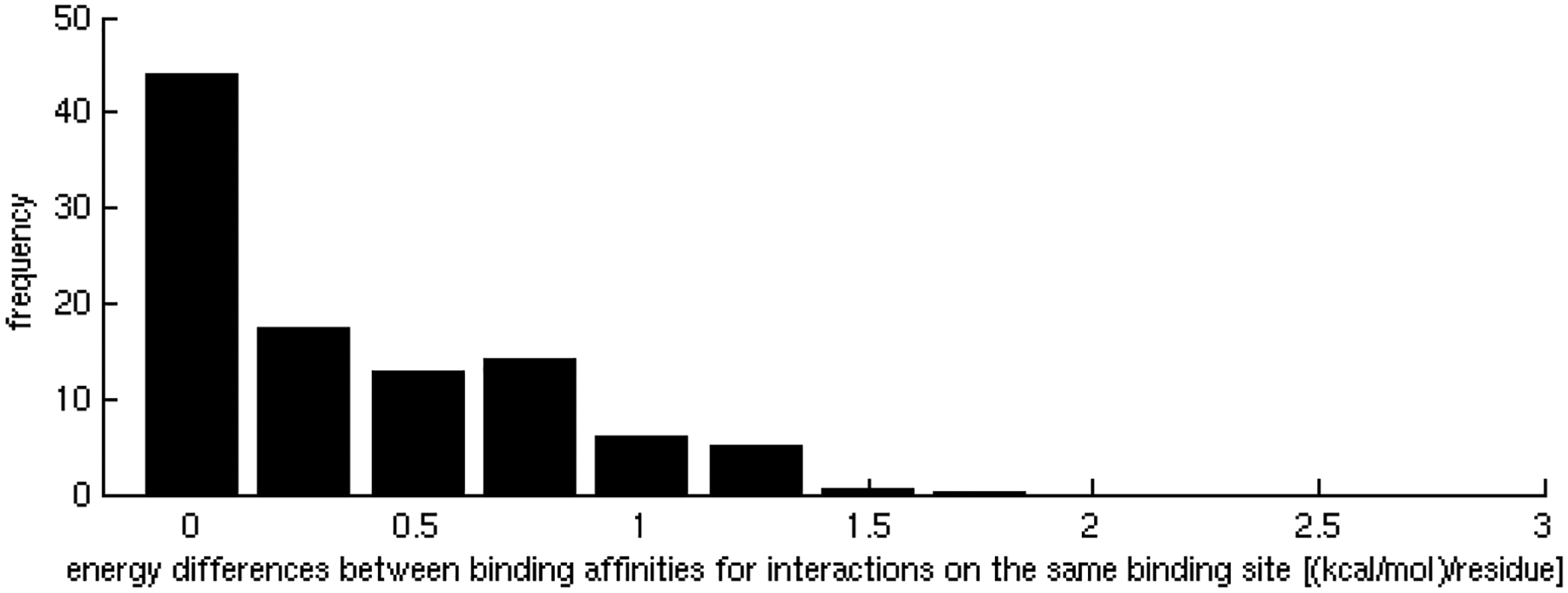
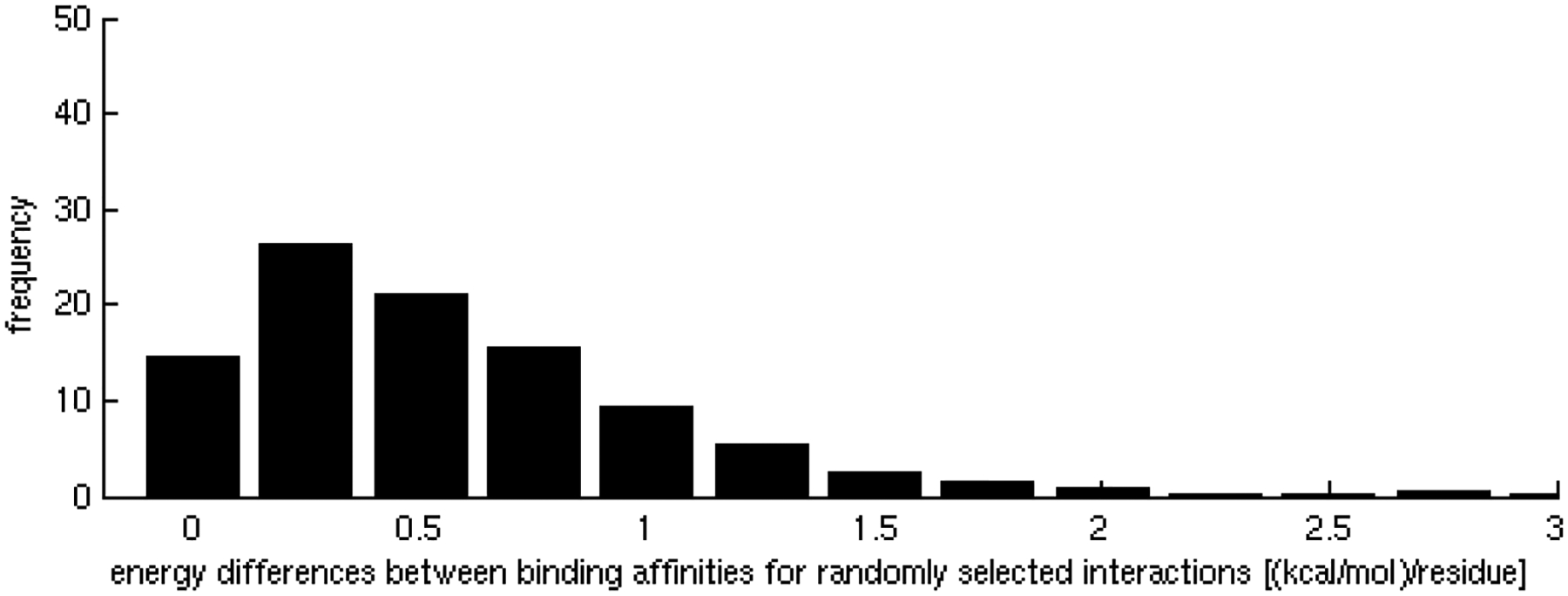
Homogeneity in affinities of interactions of promiscuous binding sites.
A) Distribution of the differences between binding affinities corresponding to interactions occurring through the same binding site. Energy differences between partners are mainly distributed between 0 and 0.2 kcal/mol per residue.
B) Random test for homogeneity of affinities of the interactions. The distribution of energy differences between interactions occurring through distinct binding sites is centered at 0.5 kcal/mol per residue.
These regularities in binding site interactions characterize protein interaction networks responding to different biological processes. For example, promiscuous binding sites mainly participate in weak transient interactions, which are involved in processes such as signaling and regulation. These weak interactions are advantageous for the dynamic association and dissociation of complexes [9]. In contrast, interactions between proteins that are initially not co-localized, such as hormone-receptor and antibody-antigen, are generally highly specific, and display high affinity [39].
3.2. Hot spot distribution on interfaces
We next investigated how binding hot spots were distributed on hub binding sites and the relationship between this distribution and binding site specificity and affinity. The hot spots were predicted by computational alanine scanning [31] for all interactions. Based on these calculations, we observed that only 18% of the residues were predicted as hot spots in two interactions, and approximately 6% in more than two interactions (Figure 4). Thus, the majority of the residues identified as hot spots fulfill this role in only a single interaction. Next, we focused our attention on partners that interact with the same hot spots, and identified sequence motifs in their binding sites [33]. Interestingly, we observed that there is a direct correlation between the number of interactions where the residue act as hot spot and the average number of common motifs with which the hot spot interacts (Table 2). Hence, our result suggests that binding sites have evolved for binding partners via common motifs. This result is supported by previous studies showing that binding site motifs of interacting partners can act as determinants of specificity [40,41,12,42].
Figure 4.
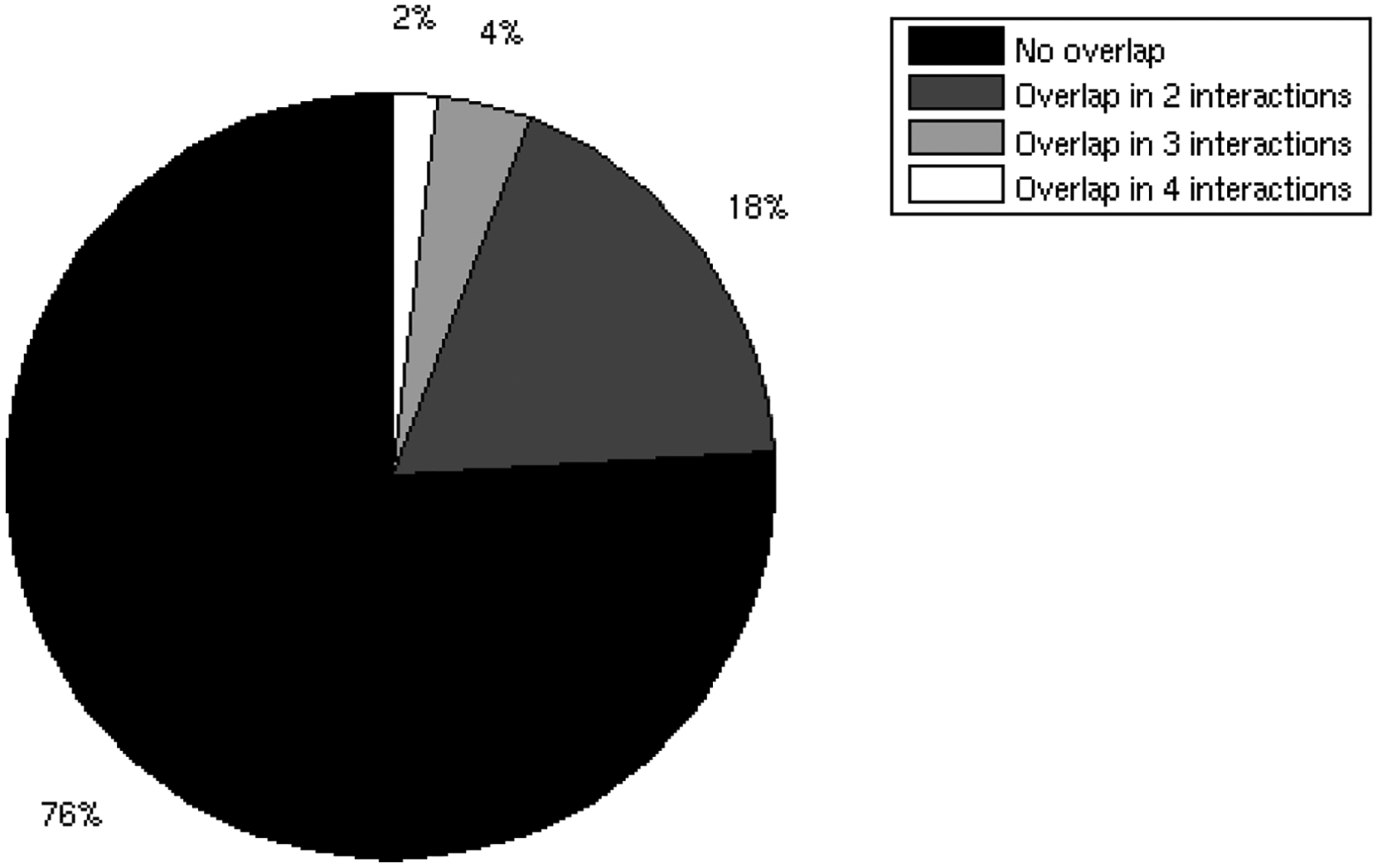
Overlap of binding sites hot spots.
Frequency distribution of predicted hot spot residues in promiscuous binding sites respect to the number of interactions where they act as hot spots. Only 18% of residues were predicted as hot spots in two interactions, and approximately 6% in more than two interactions.
Table 2.
Number of interactions in which a residue is a hot spot versus the average number of common motifs with which the hot spot interacts.
| Number of interactions of hot spots | Average number of common motifs interacting with hot spots |
|---|---|
| 1 | 1.4 |
| 2 | 2.5 |
| 3 | 3.0 |
| 4 | 4.0 |
Our next goal was to study the role of hot spots in the interplay between binding affinity and specificity. Although hot spot density on the binding site surface remains uniformly around 10% of the binding site residues (see Figure S1), it would be reasonable to assume that the hot spot distribution could be related to binding specificity. To examine the relationship between hot spot distribution and specificity, we represented the structures of hub proteins as residue interacting networks, and divided them into modules. Modules contain densely connected residues with few connections with other modules [43]. We have previously observed an energetic independence of hot spots located in different modules and cooperativity of those residing within the same modules [28]. Now, we focused on the modular distribution of hot spots in the binding site by comparing the number of modules containing hot spots with the total number of modules in the binding site. Interestingly, we found that the relative number of binding site modules containing hot spots increases with the number of partners, suggesting that binding site hot spots tend to be more distributed in different modules as the number of interacting partners increases. In binding sites involved in only one interaction, on average 35% of the modules contain hot spots, whereas in binding sites involved in 5 interactions on average more than 60% contain hot spots (Figure 5A). This result was tested by the generation of 500 random placements of hot spots on the binding sites (Figure 5B), and was found to be statistically significant (z-score = 5.24, p-value = 8.03 × 10−8). Therefore, hot spot residues corresponding to different interactions seem to be mainly distributed in different binding site modules, whereas a small part of them participate in more than one interaction, possibly acting as binding site anchors (Figure 6B) [20].
Figure 5.
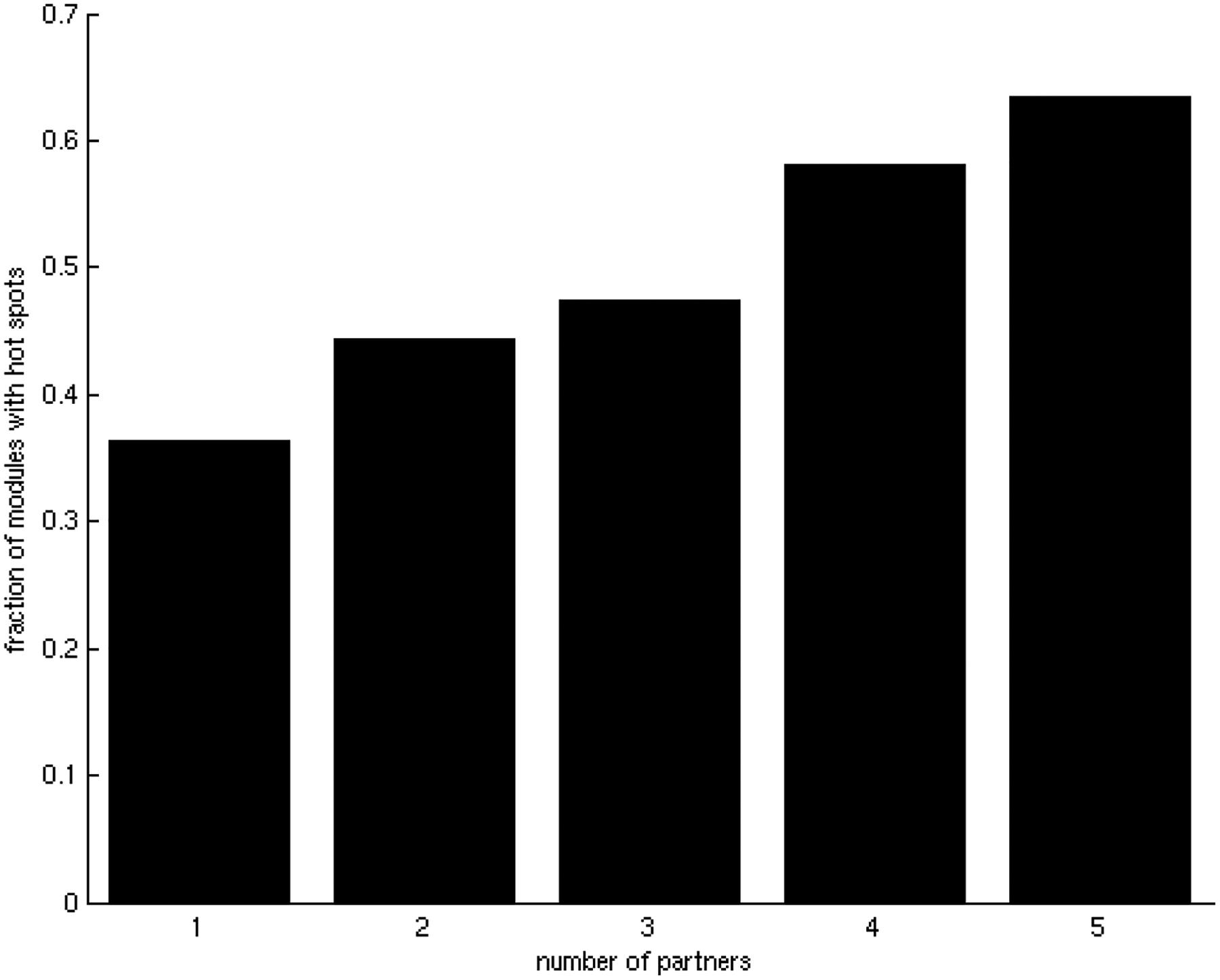
Relationship between the averaged fraction of modules with hot spots and specificity in the binding sites.
A) Relationship between the averaged fraction of modules containing hot spots in binding sites and specificity. There is a clear tendency for the modular distribution of hot spots to increase with binding site promiscuity.
B) Z-score frequency distribution of the correlation coefficients for 500 randomizations of the binding energies. In the dataset of this study, the tendency, marked with a black triangle, was found to have a statistically significant z-score = 5.24 (p-value = 8.03 × 10−8).
Figure 6.
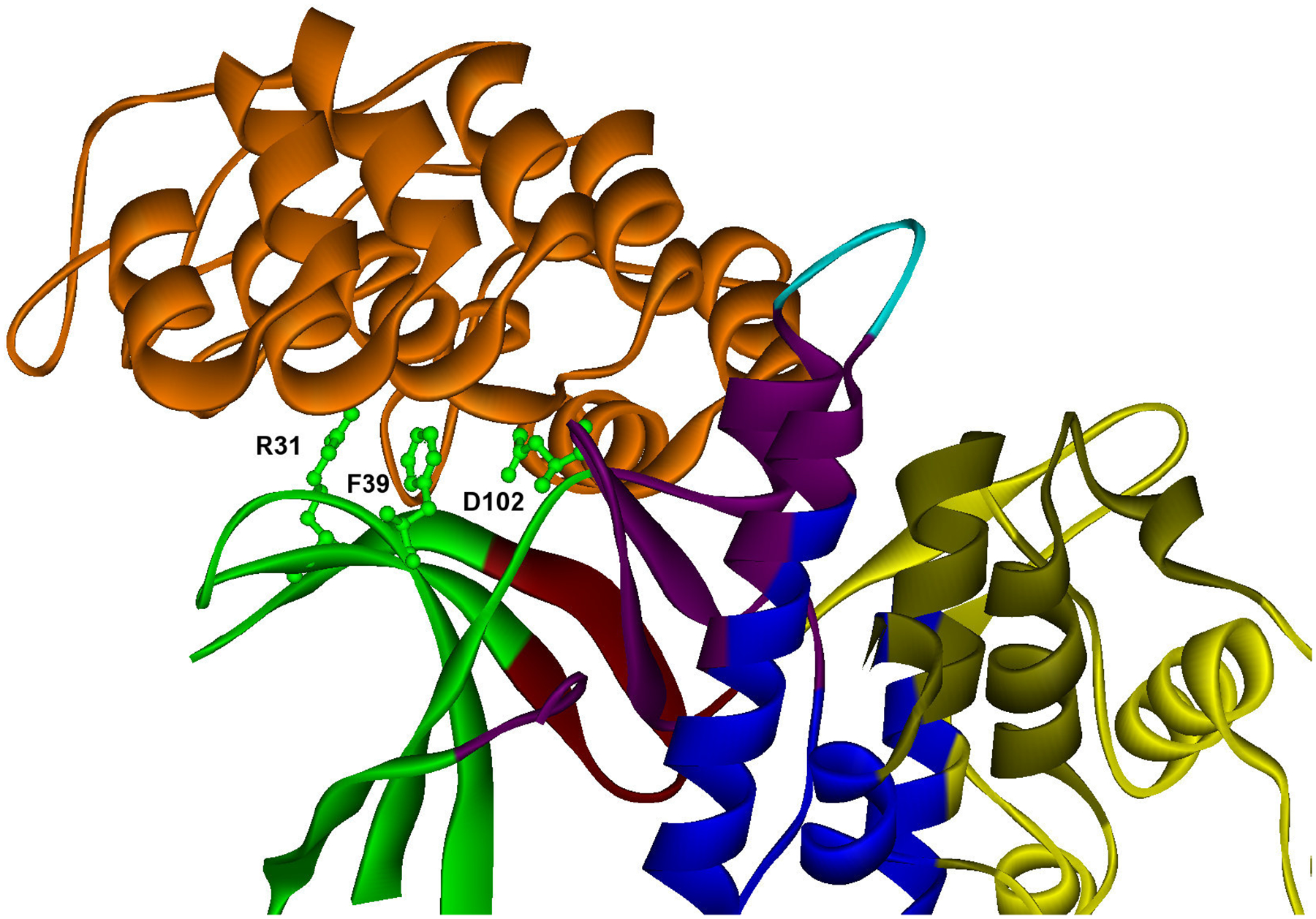
Different examples of modular distribution of hot spots in promiscuous and specific binding sites.
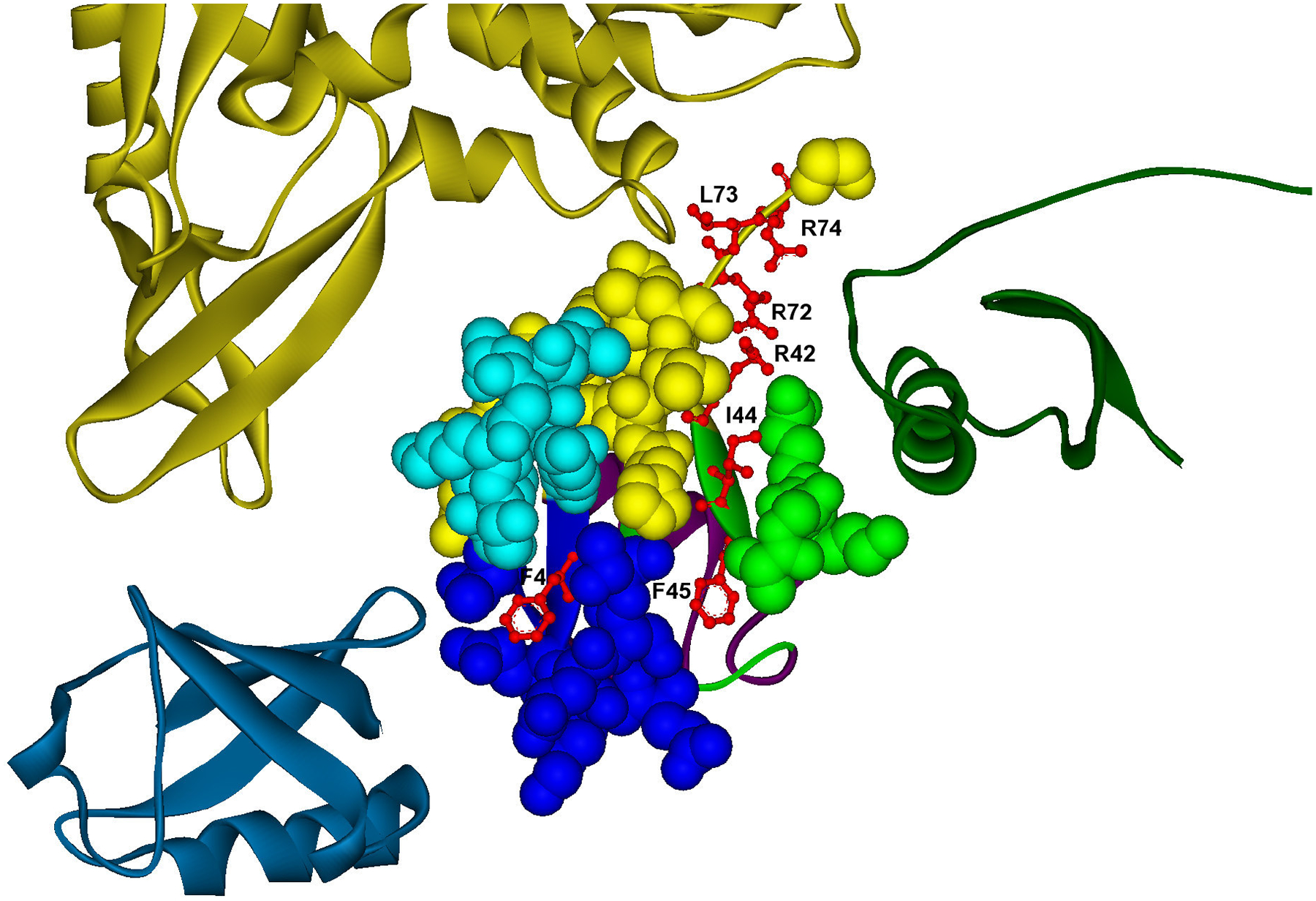
A) Example of promiscuous binding site with specific hot spots to different interactions, which are located in different modules. Predicted hot spots are shown in red ball and stick representation in a promiscuous binding site (cpk residues) of ubiquitin (YLR167W). The modular decomposition of the protein structure is represented by colors. The binding site is divided into four modules (yellow, blue, green, dark blue), three of them containing hot spots (F4; I44,F45; R42,R72,L73,L74), which are energetic determinants in specific interactions: a) YMR276W (F4), PDB: 1p3q V U (dark blue); b) YEL037C (I44,F45), PDB: 1gjz A B (green); c) YOR124C (R42,R72,L73,L74), PDB: 1nbf C B (yellow). PDB template for ubiquitin: 1nbf C.
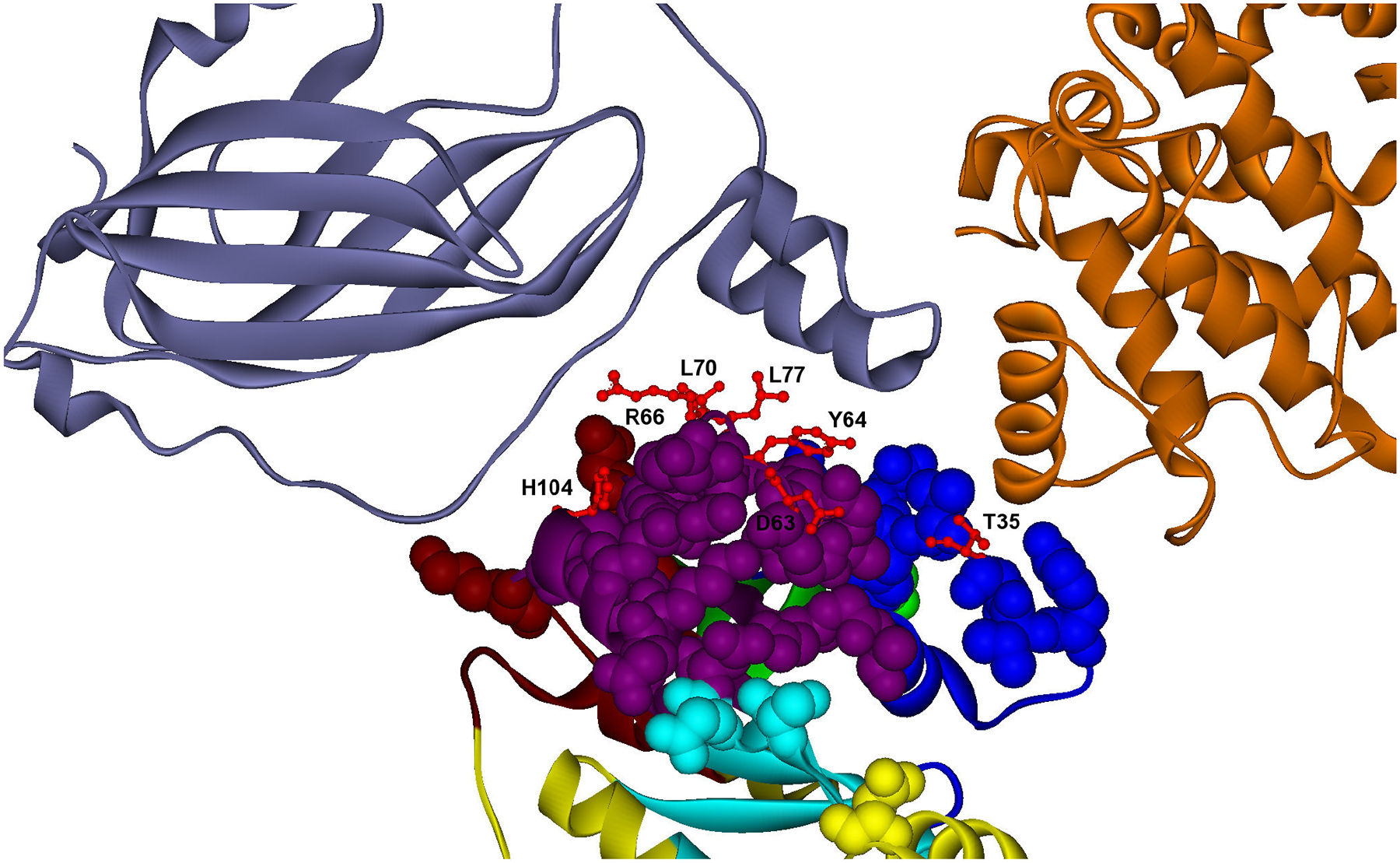
B) Example of promiscuous binding site with specific and common hot spots to different interactions. Specific and common hot spots are located in different modules. Predicted hot spots are shown in red ball and stick representation in a promiscuous binding site (cpk residues) of cdc42 (YLR229C). The modular decomposition of the protein structure is represented by colors. The binding site is divided into five modules (crimson, purple, blue, dark blue, yellow), two of them containing hot spots (D63,Y64,R66,L67,L70;T35), and participate as hot spots in different combination for the partners: a) YDL135C (D63,Y64,R66,L67,L70;T35), PDB: 1doa A B (dark blue); b) YDL240W (D63,Y64,R66,L67,L70), PDB: 2ngr A B (orange). PDB template for cdc42: 1kz7 B.
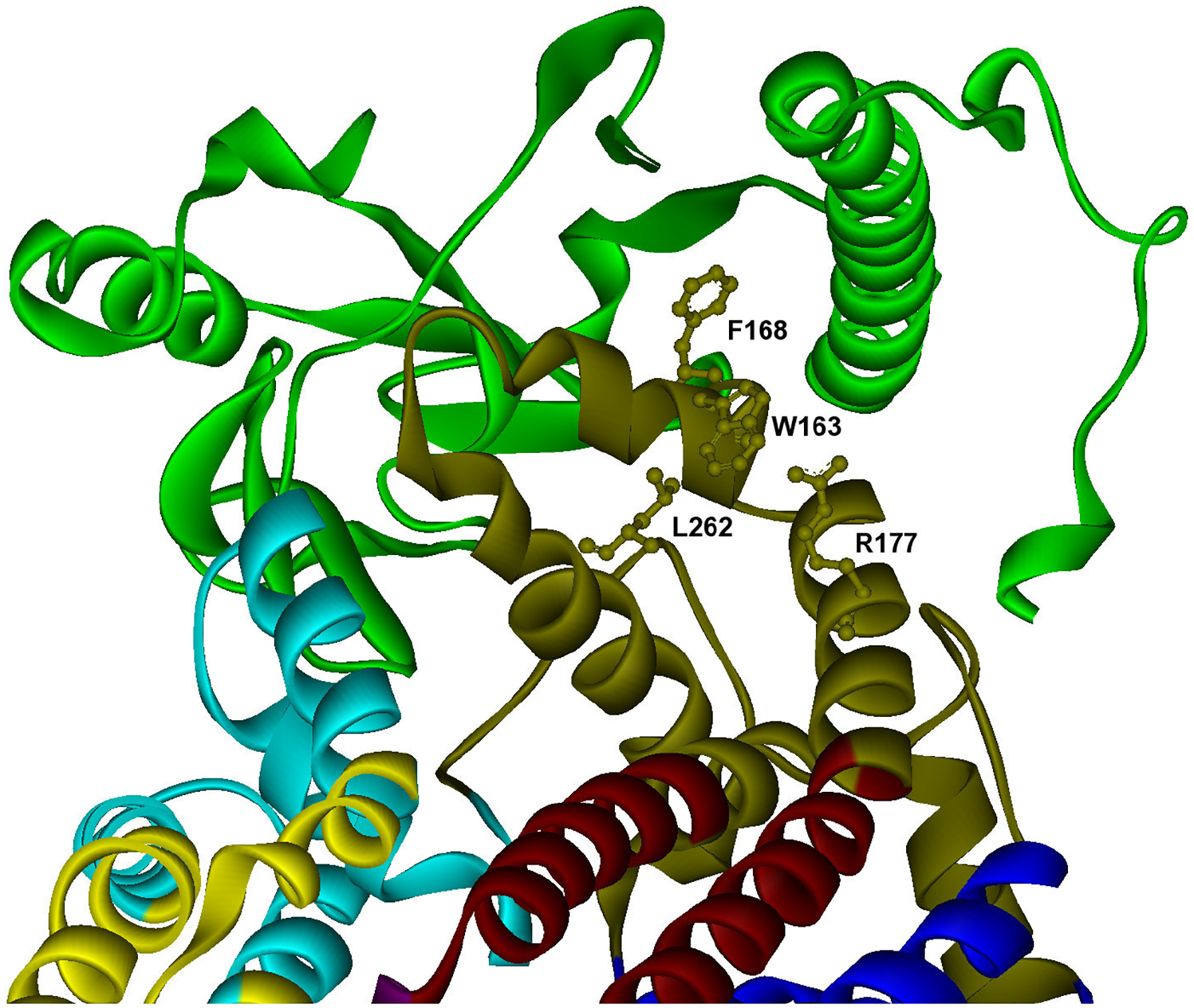
C) Example of specific binding site with hot spots contained in one module. Predicted hot spots are shown in ball and stick representation in a specific binding site of cytochrome b (Q0105), which interacts specifically with the Rieske iron-sulfur protein (YEL024W). The modular decomposition of the protein structure is represented by colors. The binding site is divided into two modules (dark green, light blue). Hot spots are located in a central dark green module (W163,F168,R177,L262), PDB: 2a06 P E (orange). PDB template for cytochrome b: 2bcc C.
D) Example of specific binding site with hot spots contained in one module. Predicted hot spots are shown in ball and stick representation in a specific binding site of cmk2 (YOL016C), which interacts specifically with akr1 (YDR264C). The modular decomposition of the protein structure is represented by colors. The binding site is divided into four modules (green, dark red, purple, yellow). The four predicted hot spots are located in the central green module (R31,F39,D102), PDB: 1bi8 A B (orange). PDB template for cmk2: 1bi8 A.
In addition to hot spot distribution, several other characteristics of binding sites have been observed to contribute to the mechanism of protein specificity. Using our dataset, here, we tested two of these, and obtained expected results. First, most structurally disordered binding sites were promiscuous (see Figure S2, Table S2), suggesting that flexibility might facilitate binding to different partners [44]. Similarly, binding sites containing a large fraction of hydrophobic patches were also observed to be predominantly promiscuous (see Figure S3, Table S3).
3.3. Examples of energetic determinants of specificity in protein-protein interactions
We selected four examples that illustrate our findings on how the hot spot distribution relates to specificity/promiscuity (ubiquitin, rho-like GTPase, cytochrome b, calmodulin-dependent protein kinase).
Ubiquitin.
Ubiquitin (Uniprot: P61864), a highly conserved 76 amino acid residue protein, is an example of a promiscuous protein whose interactions are typically weak (Kd of 50–100 μM) [45]. Ubiquitin-like proteins are covalently attached to a substrate protein via lysine side chains, altering protein location and activity. Ubiquination regulates many biological processes. Here, we studied a promiscuous binding site in ubiquitin (ubi3, ORF: YLR167W) that mediates three different interactions. Three out of four binding site modules contain predicted hot spot regions (F4; I44,F45; R42,R72,L73,L74) that are specific for each interaction (Figure 6A). Table 3 illustrates that two of the modules containing hot spots are involved in specific interactions, whereas one of them interacts with two partners; however, each of these modules contains a group of hot spots specific for each interaction. Predicted hot spots have been experimentally determined to be critical for cellular processes: For example, our predicted hot spots have been identified as essential for the vegetative growth of yeast [46]. In particular, the group of hot spots (R42,R72,L73,L74) contained in one module located at the ubiquitin tail, corresponds to a region where residues are important for ubiquitin conjugation and deubiquination [46]. This fact is in accord with our predictions of their role as hot spots in the interaction with ubiquitin protease ubp2 (YOR124C). The other two predicted modules are located in two hydrophobic patches, the first contains two hot spots (I44,F45) that has been related to endocytosis and proteasome degradation [47], and were predicted to interact with rad23 (YEL037C), a protein involved in DNA repair that interacts with the proteasome [48], while the second one contains a single hot spot (F4), which is mainly involved in internalization [49], and interacts with the deubiquitinating enzyme dsk2 (YMR276W).
Table 3.
List of modules containing hot spots in the four examples, predicted hot spots, interactions where the residues participate as hot spots, partners that interact with the module, and references to experimentally verified hot spots located in the module (a)[43]; b)[44]; c)[46]; d)[49]; e)[51]; f)[52]; g)[53]).
| Module | Protein | Hot spots | Interaction | Module partners | Experimentally verified |
|---|---|---|---|---|---|
| A1 | ubi3 | R42, R72, L73, L74 | ubp2 | ubp2 | R42a), R72a), L73 a), L74 a) |
| A2 | ubi3 | I44, F45 | rad23 | rad23 | I44b), F45b) |
| A3 | ubi3 | F4 | dsk2 | ubp2,dsk2 | F4c) |
| B1 | cdc42 | D63, Y64, R66, L67, L70 | rdi1, lrg1 | rdi1, lrg1 | R66d) |
| B2 | cdc42 | T35 | rdi1 | rdi1 | T35e) |
| C1 | cob | W163, F168, R177, L262 | isp | isp | G167f),g), G252f),g) |
| D1 | cmk2 | R31, F39, D102 | akr1 | akr1 | - |
Cdc42 GTPase.
Another example of a protein with a promiscuous binding site is the cell division control protein 42 precursor cdc42 (Uniprot: P19073), a small rho-like GTPase, involved in the maintenance of cell polarity (ORF: YLR229C). This protein contains a promiscuous binding site that mediates two different interactions, its regulator rho-GDI (rdi1) [50], and the GTPase-activating protein lrg1 [51]. This interface comprises four modules (Figure 6B), with one central module that contains five hot spots (D63,Y64,R66,L67,L70) common to both interactions, and therefore acting as a binding site anchor. Hot spot R66 has been experimentally determined to be involved in multiple interactions [52], and in particular it is essential for the proper positioning of the cdc42p-rdi1p complex at the membrane [53]. In addition, there is another module located around the rdi1 loop binding, which contains a single specific hot spot T35 that has been experimentally identified as a key residue for this interaction [54].
Cytochrome b.
Cytochrome b (Uniprot: POO613) provides an example of a protein with a specific binding site. Yeast cytochrome b (cob, ORF: Q0105) is a protein with several obligate interactions related to its role as a subunit of the ubiquinol-cytochrome c reductase (bc1) complex. One of its partners is rip1 (ORF: YEL024W), the Rieske iron-sulfur protein (isp) of the mitochondrial cytochrome bc1 complex, which transfers electrons from ubiquinol to cytochrome c1 during respiration. We observed that this interaction is mediated through a specific binding site, which contains all hot spot residues (W163,F168,R177,L262) located in a single module (Figure 6C). This binding site comprises the cytochrome b extramembranous cd2 helix, important for maintaining the structure of the hinge region of the sulfur protein, and the E-ef loop on the P side of the membrane. The specific module includes residues G167 and G252, both involved in disease-related mutations of the highly similar human cytochrome b [55], and having a severe effect on the stability of the binding of the iron-sulfur complex protein on the complex in yeast [56].
Calmodulin-dependent kinase.
Finally, another illustrative example of a protein with a specific binding site is the calmodulin-dependent protein kinase cmk2 (UniProt: Q05436, ORF: YOL016C), which plays a role in stress response. This specific binding site is divided into four modules (Figure 6D), and there are three predicted hot spots (R31,F39,D102), all contained in one of these modules.
Concluding remarks
Most studies of protein networks do not consider molecular details of the interacting proteins. Yet, analysis of structural and biophysical properties of protein-protein interactions is expected to provide insight into the systems biology description of protein interaction networks. In this work, we focused on the relationship between two characteristics which are related to the systems and molecular aspects of protein interaction networks, respectively- binding specificity and affinity. We carried out a large-scale calculation of binding free energies of all interactions contained in our structural protein-protein interaction network. Our analysis shows that interactions involving promiscuous binding sites are generally weaker than those including specific ones. This finding supports the general idea that promiscuity must come at a cost of affinity. To further investigate the thermodynamic determinants of the interplay between affinity and specificity, we performed an in-silico alanine scanning analysis of all interacting complexes. Our results revealed that although some residues can be hot spots of binding free energy in more than one interaction of a given binding site, in general distinct hot spots participate in different interactions. Interestingly, binding site residues acting as hot spots in several interactions tend to interact with binding partners sharing common motifs, whereas distinct hot spots generally interact with partners with different motifs. Our analysis further considered the modular division of protein structures into energetically independent modules containing highly cooperative residues, which exhibit few connections with other modules. We observed that hot spots in promiscuous binding sites tend to be more distributed over different modules, whereas specific binding sites generally contain hot spots within one module. These findings show that the modular distribution of hot spots in binding sites relates to binding specificity. Thus, despite the complexity of mechanisms by which binding sites modulate binding affinity and specificity for biological function, consistent with experimental data [25], it appears that the modular distribution of hot spots plays a major role in achieving this goal. This binding site architecture might have been designed by evolution to generate a wide range of binding affinities and specificities. Knowledge of the modular distribution of hot spots involved in different interactions is important in drug design since it would allow us to rationally modify binding specificity and affinity.
Supplementary Material
Table S1. Full list of protein interfaces and their partners. This table contains the list of binding sites used in this study for each ORF, its structure template, as well as the list of non-redundant partners that interact through each binding site.
Figure S1. Percentage of predicted hot spots in binding sites.
Averaged percentage of predicted hot spots in binding sites for different number of partners.
Figure S2. Percentage of promiscuous binding sites containing disordered regions.
Binding sites containing a high degree of disorder regions are predominantly promiscuous.
Figure S3. Percentage of promiscuous binding sites containing hydrophobic patches.
Binding sites containing a large fraction of hydrophobic patches are predominantly promiscuous.
Table S2. Enrichment of promiscuous binding sites in disordered binding sites.
Table S3. Enrichment of promiscuous binding sites in binding sites containing more than three hydrophobic patches.
Table S4. Comparison between the analysis of complex structures from yeast (experimental) and from their closest homologue in another organism (modeled). A) RMSD of the structural alignment of the complexes; B) Percentage of overlap in the modular decomposition of the aligned protein structures; C) Number of modules that contain predicted hot spots in both structures compared with the total number of modules that contain hot spots in the experimental structure; D) Differences in the estimation of the free binding energy for both structures of the complex in kcal/mol per residue.
Acknowledgements
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
The authors declare they have no conflict of interest.
4 References
- [1].He X, Zhang J, Why do hubs tend to be essential in protein networks? PLoS Genet. 2006, 2, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Beckett D, Multilevel regulation of protein-protein interactions in biological circuitry. Phys Biol. 2005, 2, S67–73. [DOI] [PubMed] [Google Scholar]
- [3].Kuriyan J, Eisenberg D, The origin of protein interactions and allostery in colocalization. Nature 2007, 450, 983–990. [DOI] [PubMed] [Google Scholar]
- [4].Han JD, Bertin N, Hao T, Goldberg DS et al. , Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 2004, 430, 88–93. [DOI] [PubMed] [Google Scholar]
- [5].Kim PM, Lu LJ, Xia Y, Gerstein MB, Relating three-dimensional structures to protein networks provides evolutionary insights. Science 2006, 314, 1938–1941. [DOI] [PubMed] [Google Scholar]
- [6].Beltrao P, Kiel C, Serrano L, Structures in systems biology. Curr. Opin. Struct. Biol 2007, 17, 378–384. [DOI] [PubMed] [Google Scholar]
- [7].Kiel C, Beltrao P, Serrano L, Analyzing Protein Interaction Networks Using Structural Information. Annu. Rev. Biochem 2008, 77, doi: 10.1146/annurev.biochem.77.062706.133317. [DOI] [PubMed] [Google Scholar]
- [8].Humphris EL, Kortemme T, Design of multi-specificity in protein interfaces. PLoS Comput. Biol 2007, 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li SS, Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem. J 2005, 390, 641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li W, Keeble AH, Giffard C, James R et al. , Highly discriminating protein-protein interaction specificities in the context of a conserved binding energy hotspot. J. Mol. Biol 2004, 337, 743–759. [DOI] [PubMed] [Google Scholar]
- [11].Reichmann D, Rahat O, Cohen M, Neuvirth H, Schreiber G, The molecular architecture of protein-protein binding sites. Curr. Opin. Struct. Biol 2007, 17, 67–76. [DOI] [PubMed] [Google Scholar]
- [12].Keskin O, Nussinov R, Favorable scaffolds: proteins with different sequence, structure and function may associate in similar ways. Protein Eng. Des. Sel 2005, 18, 11–24. [DOI] [PubMed] [Google Scholar]
- [13].Jones S, Thornton JM, Prediction of protein-protein interaction sites using patch analysis. J. Mol. Biol 1997, 272, 133–143. [DOI] [PubMed] [Google Scholar]
- [14].Shulman-Peleg A, Nussinov R, Wolfson HJ, Recognition of functional sites in protein structures. J. Mol Biol 2004, 339, 607–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].DeLano WL, Ultsch MH, de Vos AM, Wells JA, Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [DOI] [PubMed] [Google Scholar]
- [16].Keskin O, Ma B, Nussinov R, Hot regions in protein--protein interactions: the organization and contribution of structurally conserved hot spot residues. J. Mol. Biol 2005, 345, 1281–1294. [DOI] [PubMed] [Google Scholar]
- [17].Keskin O, Gursoy A, Ma B, Nussinov R, Principles of Protein-Protein Interactions: What are the Preferred Ways For Proteins To Interact? Chem. Rev 2008, 108, 1225–1244. [DOI] [PubMed] [Google Scholar]
- [18].Reichmann D, Cohen M, Abramovich R, Dym O et al. , Binding hot spots in the TEM1-BLIP interface in light of its modular architecture. J. Mol. Biol 2007, 365, 663–679 [DOI] [PubMed] [Google Scholar]
- [19].Ma B, Elkayam T, Wolfson H, Nussinov R, Protein-protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci 2003, 100, 5772–5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Keeble AH, Kirkpatrick N, Shimizu S, Kleanthous C, Calorimetric dissection of colicin DNase--immunity protein complex specificity. Biochemistry 2006, 45, 3243–3254. [DOI] [PubMed] [Google Scholar]
- [21].Pál G, Kouadio JL, Artis DR, Kossiakoff AA, Sidhu SS, Comprehensive and quantitative mapping of energy landscapes for protein-protein interactions by rapid combinatorial scanning. J. Biol. Chem 2006, 281: 22378–22385. [DOI] [PubMed] [Google Scholar]
- [22].Fernandez-Ballester G, Blanes-Mira C, Serrano L, The tryptophan switch: changing ligand-binding specificity from type I to type II in SH3 domains. J. Mol. Biol 2004, 335, 619–629. [DOI] [PubMed] [Google Scholar]
- [23].Kraich M, Klein M, Patiño E, Harrer H et al. , A modular interface of IL-4 allows for scalable affinity without affecting specificity for the IL-4 receptor. BMC Biol. 2006, 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Moza B, Buonpane RA, Zhu P, Herfst CA et al. , Long-range cooperative binding effects in a T cell receptor variable domain. Proc. Natl. Acad. Sci 2006, 103, 9867–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Reichmann D, Rahat O, Albeck S, Meged R et al. , The modular architecture of protein-protein binding interfaces. Proc. Natl. Acad. Sci 2005, 102, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bolon DN, Grant RA, Baker TA, Sauer RT, Specificity versus stability in computational protein design. Proc. Natl. Acad. Sci 2005, 102, 12724–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Güldener U, Münsterkötter M, Oesterheld M, Pagel P et al. , MPact: the MIPS protein interaction resource on yeast. Nucleic Acids Res. 2006, 34(Database issue), D436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].del Sol A, Carbonell P, The modular organization of domain structures: insights into protein-protein binding. PLoS Comput. Biol 2007, 3, e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xenarios I, Salwiński Ł, Duan XJ, Higney P et al. , DIP, the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Teyra J, Paszkowski-Rogacz M, Anders G, Pisabarro MT, SCOWLP classification: structural comparison and analysis of protein binding regions. BMC Bioinformatics 2008, 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schymkowitz J, Borg J, Stricher F, Nys R et al. , The FoldX web server: an online force field. Nucleic Acids Res. 2005, 33(Web Server issue), W382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tokuriki N, Stricher F, Schymkowitz J, Serrano L, et al. , The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol 2007, 369, 1318–32. [DOI] [PubMed] [Google Scholar]
- [33].Carter P, Liu J, Rost B, PEP: Predictions for Entire Proteomes. Nucleic Acids Res. 2003, 31, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Girvan M, Newman ME, Community structure in social and biological networks. Proc. Natl. Acad. Sci 2002, 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Olmea O, Straus CE, Ortiz AR, MAMMOTH (matching molecular models obtained from theory): an automated method for model comparison. Protein Sci. 2002, 11, 2606–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Janin J, Henrick K, Moult J, Eyck LT, et al. , CAPRI: a Critical Assessment of PRedicted Interactions. Proteins 2003, 52, 2–9. [DOI] [PubMed] [Google Scholar]
- [37].Cohen J, Cohen P, West SG, Aiken LS, Applied Multiple Regression/Correlation Analysis for the Behavioral Sciences. Lawrence Erlbaum Assoc., Hillsdale, New Jersey: 2002. [Google Scholar]
- [38].Linding R, Jensen LJ, Diella F, Bork P et al. , Protein disorder prediction: implications for structural proteomics. Structure 2003, 11, 1453–1459. [DOI] [PubMed] [Google Scholar]
- [39].Nooren IM, Thornton JM, Diversity of protein-protein interactions. EMBO J. 2003, 22, 3486–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zarrinpar A, Park SH, Lim WA, Optimization of specificity in a cellular protein interaction network by negative selection. Nature 2003, 426, 676–680. [DOI] [PubMed] [Google Scholar]
- [41].Aragues R, Sali A, Bonet J, Marti-Renom MA, Oliva B, Characterization of protein hubs by inferring interacting motifs from protein interactions. PLoS Comput. Biol 2007, 3, 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Keskin O, Nussinov R, Similar binding sites and different partners: implications to shared proteins in cellular pathways. Structure 2007, 15, 341–354. [DOI] [PubMed] [Google Scholar]
- [43].del Sol A, Araúzo-Bravo MJ, Amoros D, Nussinov R, Modular architecture of protein structures and allosteric communications: potential implications for signaling proteins and regulatory linkages. Genome Biol. 2007, 8, R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dyson HJ, Wright PE, Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol 2005, 6, 197–208. [DOI] [PubMed] [Google Scholar]
- [45].Hicke L, Schubert HL, Hill CP, Ubiquitin-binding domains. Nat. Rev. Mol. Cell. Biol 2005, 6, 610–621. [DOI] [PubMed] [Google Scholar]
- [46].Sloper-Mould KE, Jemc JC, Pickart CM, Hicke L, Distinct functional surface regions on ubiquitin. J. Biol. Chem 2001, 276, 30483–30489. [DOI] [PubMed] [Google Scholar]
- [47].Swanson KA, Kang RS, Stamenova SD, Hicke L, Radhakrishnan I, Solution structure of Vps27 UIM-ubiquitin complex important for endosomal sorting and receptor downregulation. EMBO J. 2003, 22, 4597–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schauber C, Chen L, Tongaonkar P, Vega I et al. , Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 1998, 391, 715–718. [DOI] [PubMed] [Google Scholar]
- [49].Shih SC, Sloper-Mould KE, Hicke L, Monoubiquitin carries a novel internalization signal that is appended to activated receptors. EMBO J. 2000, 19, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Su LF, Knoblauch R, Garabedian MJ, Rho GTPases as modulators of the estrogen receptor transcriptional response. J. Biol. Chem 2001, 276, 3231–3237. [DOI] [PubMed] [Google Scholar]
- [51].Roumanie O, Weinachter C, Larrieu I, Crouzet M, Doignon F Functional characterization of the Bag7, Lrg1 and Rgd2 RhoGAP proteins from Saccharomyces cerevisiae. FEBS Lett. 2001, 506, 149–156. [DOI] [PubMed] [Google Scholar]
- [52].Dransart E, Morin A, Cherfils J, Olofsson B, Uncoupling of inhibitory and shuttling functions of rho GDP dissociation inhibitors. J. Biol. Chem 2005, 280, 4674–4683. [DOI] [PubMed] [Google Scholar]
- [53].Cole KC, McLaughlin HW, Johnson DI, Use of bimolecular fluorescence complementation to study in vivo interactions between Cdc42p and Rdi1p of Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6: 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dovas A, Couchman JR, RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem. J 2005, 390, 1–9.16083425 [Google Scholar]
- [55].Fisher N, Castleden CK, Bourges I, Brasseur G et al. , Human disease-related mutations in cytochrome b studied in yeast. J. Biol. Chem 2004, 279: 12951–12958. [DOI] [PubMed] [Google Scholar]
- [56].Fisher N, Bourges I, Hill P, Brasseur G, Meunier B, Disruption of the interaction between the Rieske iron-sulfur protein and cytochrome b in the yeast bc1 complex owing to a human disease-associated mutation within cytochrome b. Eur. J. Biochem 2004, 271, 1292–1298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Full list of protein interfaces and their partners. This table contains the list of binding sites used in this study for each ORF, its structure template, as well as the list of non-redundant partners that interact through each binding site.
Figure S1. Percentage of predicted hot spots in binding sites.
Averaged percentage of predicted hot spots in binding sites for different number of partners.
Figure S2. Percentage of promiscuous binding sites containing disordered regions.
Binding sites containing a high degree of disorder regions are predominantly promiscuous.
Figure S3. Percentage of promiscuous binding sites containing hydrophobic patches.
Binding sites containing a large fraction of hydrophobic patches are predominantly promiscuous.
Table S2. Enrichment of promiscuous binding sites in disordered binding sites.
Table S3. Enrichment of promiscuous binding sites in binding sites containing more than three hydrophobic patches.
Table S4. Comparison between the analysis of complex structures from yeast (experimental) and from their closest homologue in another organism (modeled). A) RMSD of the structural alignment of the complexes; B) Percentage of overlap in the modular decomposition of the aligned protein structures; C) Number of modules that contain predicted hot spots in both structures compared with the total number of modules that contain hot spots in the experimental structure; D) Differences in the estimation of the free binding energy for both structures of the complex in kcal/mol per residue.