

Abstract
Hemoglobinopathies are among the most common monogenic diseases worldwide. Approximately 1–5% of the global population are carriers for a genetic thalassemia mutation. The thalassemias are characterized by autosomal recessive inherited defects in the production of hemoglobin. They are highly prevalent in the Mediterranean, Middle East, Indian subcontinent, and East and Southeast Asia. Due to recent migrations, however, the thalassemias are now becoming more common in Europe and North America, making this disease a global health concern. Currently available conventional therapies in thalassemia have many challenges and limitations. A better understanding of the pathophysiology of β-thalassemia in addition to key developments in optimizing transfusion programs and iron-chelation therapy has led to an increase in the life span of thalassemia patients and paved the way for new therapeutic strategies. These can be classified into three categories based on their efforts to address different features of the underlying pathophysiology of β-thalassemia: correction of the globin chain imbalance, addressing ineffective erythropoiesis, and improving iron overload. In this review, we provide an overview of the novel therapeutic approaches that are currently in development for β-thalassemia.
Key Points
| A better understanding of the pathophysiology of β-thalassemia has led to an increase in the life span of thalassemia patients and paved the way for new therapeutic strategies. |
| Gene therapy approaches using globin lentiviral vectors and genome-editing approaches to inhibit the BCL11A gene are currently under investigation. |
| Targeting ineffective erythropoiesis through the activin II receptor trap luspatercept has been shown to decrease the transfusion requirement in transfusion-dependent thalassemia. |
| Therapeutic strategies aimed at improving iron dysregulation such as minihepcidin and TMPRSS6 inhibitors are also showing promise, especially in non-transfusion-dependent thalassemia patients. |
Introduction
Hemoglobinopathies are the most common monogenic diseases worldwide, and 1–5% of the global population are carriers for a genetic thalassemia mutation [1]. β-Thalassemias are highly prevalent in the Mediterranean, Middle East, and the Indian subcontinent; however, due to recent migrations, they are becoming more common worldwide, making their management and care an increasing concern for health care systems [2].
The imbalance in the α/β-globin chain ratio leads to ineffective erythropoiesis, chronic hemolytic anemia, and compensatory hemopoietic expansion [3]. We classify thalassemia syndromes as non-transfusion-dependent thalassemia (NTDT) and transfusion-dependent thalassemia (TDT) according to their clinical features and transfusion requirement. NTDT patients spontaneously maintain hemoglobin (Hb) values between 7 and 10 g/dL, and may require transfusion occasionally, mainly during pregnancy, surgery, and infections [2]. Due to chronic anemia, the absorption of iron in the duodenum is increased, and patients develop iron overload, mainly in the liver. This process is mediated by the hepcidin-ferroportin axis [4]. Hepcidin is the master regulator of iron metabolism [5], and despite the presence of iron overload, its levels are low in β-thalassemia patients [6, 7] due to the continuous erythropoietic stimuli mediated by GDF 15 [8] and erythroferrone (ERFE) [9, 10]. Conversely, TDT patients require chronic red blood cell (RBC) transfusions to survive, and iron chelation therapy is necessary to counterbalance the iron intake and prevent iron overload and subsequent organ damage [11, 12].
Different conventional modalities for the management of TDT and NTDT patients exist today. These include, and still are being used, blood transfusion, splenectomy, hydroxyurea, iron chelation therapy, and, for a subgroup of patients, hematopoietic stem-cell transplantation (HSCT). These conventional modalities remain the mainstay of treatment and they form the basis of the currently available guidelines [13, 14]. There are, however, many challenges and limitations in the currently available conventional therapies. In the last few decades there have been considerable advances in understanding the pathophysiology of β-thalassemia in addition to key developments in optimizing transfusion programs and iron-chelation therapy [15, 16]. These in turn have not only led to an increase in the life expectancy of thalassemia patients but have also paved the way for new therapeutic strategies. Emerging therapies in thalassemia can be classified into three major categories based on their efforts to address different features of the underlying pathophysiology of β-thalassemia: correction of the globin chain imbalance, addressing ineffective erythropoiesis, and improving iron overload. At the end of 2019, a first-in-class investigational erythroid maturation agent that promotes late-stage erythropoiesis was approved by the US Food and Drug Administration (FDA) for the treatment of TDT patients [17]. Bone marrow transplantation was the only available curative option for TDT until June 2019, when the first gene therapy product was approved by the European Medicine Agency (EMA) for TDT patients who do not entirely lack β-globin and who are eligible for stem cell transplantation but do not have a matching related donor [18]. In this review, we provide an overview of the novel therapeutic approaches that are currently in development.
Correction of the Globin Chain Imbalance
Bone Marrow Transplantation
The rationale of bone marrow transplantation in a TDT patient is to restore the tissue's capability of producing functional hemoglobin. Data from the European Bone Marrow Transplant (EBMT) registry on 1493 consecutive patients with thalassemia major transplanted between 2000 and 2010 showed that the 2-year overall survival (OS) and event-free survival (EFS) were 88 ± 1% and 81 ± 1%, respectively, after a median observation period of 2 years. OS and EFS were 90%, 81%, and 93% (P < 0.001), and 82%, 76%, and 85% (P = 0.003) in patients who had received bone marrow, peripheral blood, or cord blood (alone or combined), respectively [19]. The pre-transplant evaluation includes the Pesaro risk assessment [20], which stratifies the outcome of transplantation into three classes based on hepatomegaly, portal fibrosis, and irregular chelation history [21]. Class 1 includes patients who have none of the risk factors, class 2 patients who have one or two risk factors, and class 3 patients who have all three. The thalassemia-free survival (TFS) was respectively 85–90% for class 1, 80% for class 2, and 65–70% for class 3, while the transplant-related mortality (TRM) increased from class 1 to 3 [22]. Thus, HSCT should be offered to a young TDT patient with a matched sibling donor (MSD) before the development of iron overload and subsequent tissue damage.
According to EBMT data, MSD transplants give the safest outcome with a 2-year OS of 91%, compared to 88% of matched family donors and 77% of matched unrelated donors [19]. A recent study by Li et al. on children and young adults with TDT compared the use of alternative donors and HLA-matched related donors in China, India, and the USA. The 5-year probabilities of overall survival (OS) after HLA-matched relative, HLA-mismatched relative, HLA-matched unrelated, and HLA-mismatched unrelated donor transplants were reported to be 89%, 73%, 87%, and 83%, respectively. The 5-year probabilities of EFS after HLA-matched relative, HLA-mismatched relative, HLA-matched unrelated, and HLA-mismatched unrelated donor transplants were 86%, 70%, 82%, and 78%, respectively [23]. This report shows comparable event-free and OS after HLA-matched related and HLA-matched unrelated donor transplantation. Of note, the availability of a suitable donor, patient fitness, and procedure-related toxicity are the main limitations to the use of hematopoietic stem cell transplantation (HSCT). For this reason, there is the need to investigate new conditioning regimens or implement an autologous gene therapy approach to the most severe cases too.
Gene Therapy and Gene Editing
Several clinical trials are investigating the safety and efficacy of gene addition and gene editing to restore Hb synthesis in β-thalassemia (Table 1). Most of the products are gene addition-based technologies, while few are gene-editing strategies that aim to reactivate fetal Hb (HbF). The first gene addition product approved for TDT in July 2019 in Europe is Lentiglobin BB305 (Zynteglo), by bluebird bio. In the initial trials, transfusion independence was obtained in adult patients with a β+/β+ or β+/β0 genotype (ClinicalTrials.gov number NCT01745120 and NCT02151526). The level of integration [vector copy number (VCN)] proved to be insufficient at achieving transfusion independence for most of the β0/β0 adults [24]. Therefore, subsequent trials have aimed at optimizing transduction protocols to obtain higher VCN in the drug product and maximizing transgenic chimerism and thanks to new transduction regimens patients with β0/β0 achieved transfusion independence (ClinicalTrials.gov number NCT02906202 and NTC02140554).
Table 1.
Gene therapy and gene editing clinical trials for β-thalassemia
| Trial identifier | Phase/status | Enrolled patients | Myeloablative regimen | Mobilization protocol | Drug product | Sponsor/center |
|---|---|---|---|---|---|---|
|
[25] |
Phase 1–2 Active, not recruiting |
TDT 9 (3 adults, 6 children) |
Treosulfan and Thiotepa | G-CSF and Plerixafor | Autologous HSCs genetically modified with GLOBE lentiviral vector encoding for the human β-globin gene |
IRCCS San Raffaele Fondazione Telethon Institute for Gene Therapy (SR-TIGET), Milan, Italy |
|
NCT01745120 (HGB-204) [24] |
Phase 1–2 Completed |
TM 18 (between 12 and 35 years of age) |
Busulfan | G-CSF and Plerixafor | Autologous HSCs transduced with LentiGlobin BB305 lentiviral vector encoding the human βA-T87Q-globin gene |
Bluebird bio 6 international sites |
|
NCT02151526 (HGB-205) [24] |
Phase 1–2 Completed |
TDT 4 (between 5 and 35 years of age) |
Busulfan (adjusted based on daily PK monitoring) | G-CSF and Plerixafor (after 3 months of enhanced transfusion) | Autologous HSCs transduced with LentiGlobin BB305 lentiviral vector encoding the human βA-T87Q-globin gene |
Bluebird bio Necker Children’s Hospital, Paris, France |
| NCT02906202 (HGB 207) |
Phase 3 Active, not recruiting |
TDT non β0/β0, ≤ 50 years of age Estimated enrollment: 23 pts |
Busulfan | G-CSF and Plerixafor | Autologous HSCs transduced with LentiGlobin BB305 lentiviral vector encoding the human βA-T87Q-globin gene |
Bluebird bio 8 international sites |
| NCT03207009 (HGB 212) |
Phase 3 Recruiting |
TDT β0/β0, β0/IVS-I-110, or IVS-I-110/IVS-I-110 ≤ 50 years of age Estimated enrollment: 18 pts |
Busulfan | G-CSF and Plerixafor | Utologous HSCs transduced with LentiGlobin BB305 lentiviral vector encoding the human βA-T87Q-globin gene |
Bluebird bio 9 international sites |
| NCT03745287 |
Phase 1–2 Recruiting |
Estimated enrollment: 45 participants including SCD and other hematological disorders | Busulfan | NA | CTX001: autologous CD34 + HSPCs modified with CRISPR-Cas9 at the erythroid lineage-specific enhancer of the BCL11A gene |
Vertex Pharmaceuticals Incorporated/CRISPR Therapeutics 12 international sites |
| NCT03432364 |
Phase 1–2 Recruiting |
TDT ≥ 18 and ≤ 40 years of age Estimated enrollment: 45 participants |
Busulfan | NA | ST-400: autologous CD34 + HSPCs genetically modified with ZFN technology at the erythroid-specific enhancer of the BCL11A gene |
Sangamo Therapeutics 6 sites in the United States |
TDT transfusion dependent thalassemia, TM thalassemia major, G-CSF granulocyte-colony stimulating factor, PK pharmacokinetics, SCD sickle cell disease, NA not available, hHSPCs Human Hematopoietic Stem and Progenitor Cells, ZFN zinc finger nuclease
In the Italian trial conducted in Milan at IRCCS San Raffaele-Telethon Institute of gene Therapy (SR-TIGET) (ClinicalTrials.gov number NCT02453477), using the vector GLOBE, the best outcomes were observed in the pediatric cohort, while it was progressively less successful for the middle and oldest ones [25]. The observation that naturally occurring mutations of the binding site of BCL11A in the gamma-globin promoter are associated with high persistence of fetal Hb (HPFH) paved the way for strategies to reactivate HbF. A clinical trial enrolling patients with β-thalassemia (ClinicalTrials.gov number NCT03745287) is being conducted by CRISPR Therapeutics. The CRISPR Therapeutics approach relies on the use of CRISPR/CAS9 to edit patients’ hematopoietic stem cells (HSC). An additional trial by Sangamo (ClinicalTrials.gov number NCT03432364) also uses a zinc-finger nuclease approach to target the enhancer of BCL11A. Preliminary results, presented by Sangamo at ASCGT 2019, showed successful gene editing of the peripheral white blood cells collected from the first treated patient, a β0/β0, an increase of fetal Hb levels, and stable total Hb values around 9 g/dL at 50 days from the infusion. More extended follow-up periods for all these most recent studies are needed to further elucidate the extent of the success of the gene-editing technology for hemoglobinopathies.
Although autologous HSCT using genetically corrected cells would avoid the risk of graft-versus-host disease (GVHD) and overcome the need for a suitable donor, several challenges remain to be optimized in the field of gene therapy and gene editing [26]. These include:
The conditioning regimen: the genotoxicity in non-malignant disorders lessens the willingness of patients and healthcare providers to consider this therapy. The procedure requires intensive myeloablative conditioning regimens in order to reduce the risk of graft failure [27]. Myeloablative conditioning is associated with short- and long-term toxicity in hematopoietic and non-hematopoietic tissues [28]. The ideal regimen should combine an efficient depletion of HSC and minimizing toxicity. Novel strategies are based on the use of antibodies that specifically target HSC and other hematopoietic cells in the bone marrow niche, sparing non-hematopoietic cells [29–31].
The mobilization: granulocyte-colony stimulating factor (G-CSF) is the standard agent to mobilize hematopoietic stem and progenitor cells (HSPC) for transplantation. However, peculiar features of hemoglobinopathies, namely splenomegaly and thrombophilia, may represent risk factors for adverse events [32–35]. Moreover, in adult thalassemia patients, iron overload and consequent oxidative stress, the suppressive effect of long-term transfusions and chelation on the stem cell compartment, and the "aged" stem cells, could compromise the safety and success of HSC procurement [36]. Among the other compounds available, Plerixafor represents a good alternative. Plerixafor has proved to be safe and effective in mobilizing HSC in thalassemia patients [24, 25, 37]. Hydroxyurea has been evaluated as an agent to be administered prior to mobilization; however, it reduces the amount of circulating CD34+ [38], is associated with myelosuppression, and did not show any beneficial effect in thalassemia patients [36, 39].
The large-scale availability: gene therapy has high costs. Lentivirus-based gene and cell therapy products were estimated in 2017 to cost in the range of $500,000–$1,000,000 per patient [40].
Targeting Ineffective Erythropoiesis
Activin II Receptor Traps
Members of the transforming growth factor β (TGF-β) superfamily ligands, including growth differentiation factors and activins, have been shown to act as inhibitors of late-stage erythropoiesis [41–44]. Activin receptor ligand traps, namely sotatercept and luspatercept, through their ability to reduce SMAD2/3 signaling, improve anemia in disorders characterized by ineffective erythropoiesis, such as β-thalassemia [45] and myelodysplastic syndromes (MDS) [46] (Fig. 1a). GDF11-mediated signaling has been initially hypothesized as the pathway inhibited by activin receptor ligand traps [45, 47]. However, Guerra et al. recently showed that the lack of GDF11 does not improve anemia in a mouse model of β-thalassemia [48]. The authors showed that β-thalassemia mice with the deletion in the hematopoietic compartments or the pancellular deletion of GDF11 respond to RAP-536 treatment, suggesting that GDF11 is not the main target of the activin receptor ligand trap and most likely is not the dominant player of ineffective erythropoiesis [48].
Fig. 1.
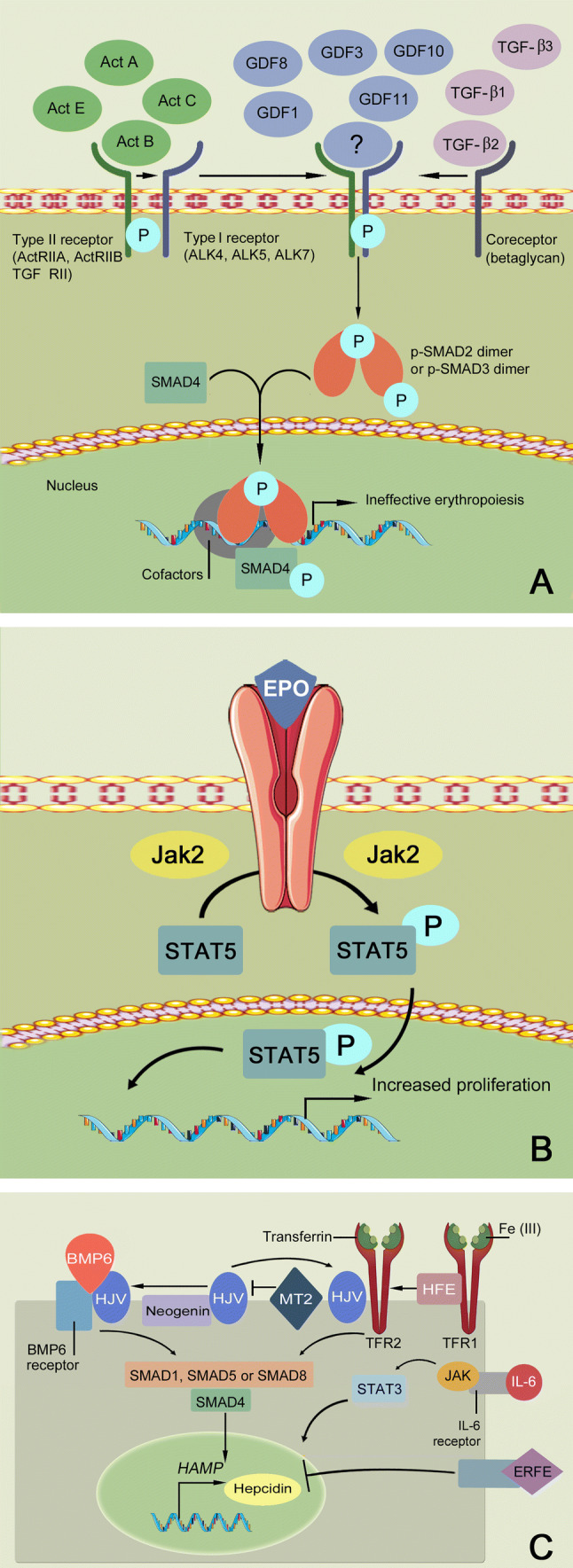
Targets of new therapeutic options in beta-thalassemia. a SMAD2/3 signaling pathway is the target of activin receptor ligand trap molecules. Members of the transforming growth factor β (TGF-β) superfamily ligands binding leads to the multimerization of type I and type II receptors. Upon the activation of the type I receptor, phosphorylation of SMAD2/SMAD3 takes place. This leads to dissociation from the type I receptor and oligomerization with SMAD4 to form a complex that translocates into the nucleus, thereby regulating the gene and promoting a cellular response with inhibition of late-stage erythropoiesis. Luspatercept and sotatercept prevent the binding of the ligand, thus inhibiting this pathway and promoting late-stage erythropoiesis. b JAK2/STAT5 signaling pathway is the target of JAK2 inhibitors, e.g., ruxolitinib. In β-thalassemia, erythropoietin (EPO) synthesis is increased, leading to activation of the JAK2/STAT5 pathway, thus altering the proliferation and differentiation of the erythroid progenitors. c Regulation of hepcidin expression in hepatocytes is the target of mini-hepcidin and TMPRSS6 inhibitors. The binding of bone morphogenetic protein 6 (BMP6) and hemojuvelin (HJV) to the BMP6 receptor leads to a downstream signaling mechanism via SMAD1, SMAD5, or SMAD8, which activate SMAD4. SMAD4 will then stimulate the transcription and expression of hepcidin (encoded by HAMP). The SMAD signaling pathway is also regulated by other molecules like transferrin receptor 2 (TFR2) upon its association with human hemochromatosis protein (HFE) and HJV. Transmembrane protease serine 6 (TMPRSS6) negatively modulates HAMP expression by cleaving HJV from the cell surface. The secretion of erythroferrone (ERFE) inhibits hepcidin expression, the signaling pathway of which is currently unknown. The administration of synthetic hepcidins and the inhibition of TMPRSS6, which ultimately increases hepcidin expression, result in ferroportin internalization and iron restriction. Ferroportin inhibitors (not shown) are another class of drugs that induce iron restriction (see Sect. 4.2)
Sotatercept (ACE-011)
Sotatercept or ACE-011 may correct ineffective erythropoiesis by acting as a ligand trap to inhibit negative regulators of late-stage erythropoiesis in the TGF-β superfamily. A phase 2 study using sotatercept was conducted on 16 TDT patients and 30 NTDT patients across seven centers around the world between November 2012 and November 2014 (ClinicalTrials.gov number NCT01571635) (Table 2) [49]. The median duration of treatment was 13.8 months for TDT patients and 19.6 months for NTDT patients. Patients were treated with sotatercept at doses of 0.1, 0.3, 0.5, 0.75, or 1.0 mg/kg. These were administered by subcutaneous injection every 3 weeks. In the TDT group, ten patients (63%) achieved a transfusion burden reduction of ≥ 20% sustained for ≥ 24 weeks; seven patients (44%) achieved a reduction of ≥ 33%, and two patients (13%) achieved a reduction of ≥ 50% [49]. In these patients, the mean change in Hb level from baseline to the end of treatment was 0.7 g/dL, and the active dose of sotatercept was ≥ 0.5 mg/kg [49]. In the NTDT group, on the order hand, 18 patients (60%) treated with sotatercept doses of 0.1–1.0 mg/kg achieved a mean Hb increase of ≥ 1.0 g/dL, and 11 (37%) had mean Hb increase of ≥ 1.5 g/dL sustained for ≥ 12 weeks [49]. Sotatercept exhibited an overall good safety profile, and was tolerated by most patients. Treatment discontinuation due to adverse events was rare, and the incidence of grade 3–4 adverse events was low.
Table 2.
Pharmacological treatments for β-thalassemia
| Trial identifier | Phase/status | Enrolled patients | Drug product | Route of administration | Target | Sponsor/center |
|---|---|---|---|---|---|---|
|
[49] |
Phase 2 Active, not recruiting |
16 TDT 30 NTDT |
Sotatercept (ACE-011) | Subcutaneous | Ineffective erythropoiesis |
Celgene Multicenter international sites |
|
[51] |
Phase 2 Completed |
31 TDT 33 NTDT |
Luspatercept (ACE-536) | Subcutaneous | Ineffective erythropoiesis |
Acceleron Pharma Multicenter international sites |
|
[52] |
Phase 3 Active, not recruiting |
336 TDT | Luspatercept (ACE-536) | Subcutaneous | Ineffective erythropoiesis |
Celgene and Acceleron Pharma Multicenter international sites |
| NCT03342404 |
Phase 2 Active, not recruiting |
145 NTDT | Luspatercept (ACE-536) | Subcutaneous | Ineffective erythropoiesis |
Celgene and Acceleron Pharma Multicenter international sites |
|
[56] |
Phase 2 Completed |
30 TDT | Ruxolitinib | Oral | Ineffective erythropoiesis through inhibition of JAK2 |
Novartis Pharmaceuticals Multicenter international sites |
| NCT03381833 |
Phase 2 Recruitingb,c |
100a TDT | LJPC-401 | Subcutaneous | Iron metabolism through synthetic human hepcidin |
La Jolla Pharmaceutical Company Multicenter international sites |
| NCT03802201 |
Phase 2 Recruitingb,c |
192a TDT and NTDT |
PTG-300 | Subcutaneous | Iron metabolism through synthetic human hepcidin | Protagonist Therapeutics |
| NCT04364269 |
Phase 2 Not yet recruiting |
36a NTDT | VIT-2763 | Oral | Iron metabolism through ferroportin inhibition |
Vifor Multicenter international sites |
| NCT04059406 |
Phase 2a Not yet recruiting |
36a NTDT | IONIS TMPRSS6-LRx | Subcutaneous | Iron metabolism through TMPRSS6 inhibition | Ionis Pharmaceuticals |
| NCT04176653 |
Phase 1 Withdrawn due to COVID-19b |
TDT and low-risk MDS | SLN124 | Subcutaneous | Iron metabolism through TMPRSS6 inhibition | Silence Therapeutics |
TDT transfusion-dependent thalassemia, NTDT non transfusion-dependent thalassemia, MDS myelodysplastic syndrome, COVID-19 Coronavirus disease 19
aEstimated enrollment
bAs of 31 May 2020, from ClinicalTrials.gov
cThis study was recently stopped and closed
Luspatercept
Luspatercept, or ACE-536, is also recognized as a recombinant fusion protein that acts as a trap for activin receptors. It has been recently approved by the FDA for the treatment of TDT patients at the recommended starting dose of 1 mg/kg once every 3 weeks by subcutaneous injection [17]. Luspatercept was overall very well tolerated and associated with a dose-dependent increase in Hb levels in healthy subjects [50]. A murine analog of luspatercept known as RAP-536 was also shown to reduce oxidative stress and anemia in a mouse model of β-thalassemia [45].
An initial phase 2 dose-finding study was designed in order to evaluate the effects of luspatercept in β-thalassemia patients (ClinicalTrials.gov number NCT01749540) (Table 2). Luspatercept was administered subcutaneously every 21 days at dose levels ranging from 0.2 to 1.25 mg/kg of body weight. Primary endpoints of the study included erythroid response, defined as a Hb increase from baseline of ≥ 1.5 g/dL for ≥ 2 weeks (in the absence of RBC transfusions) for NTDT and a reduction in RBC transfusion burden over a 12-week interval of ≥ 20% as compared with pretreatment for TDT patients. Eighteen out of 33 (58%) NTDT patients treated with luspatercept at a dose of 0.6–1.25 mg/kg achieved a mean Hb increase from baseline of ≥ 1.5 g/dL over 14 consecutive days [95% confidence interval (CI) 39.1–75.5]. Twenty-six out of 31 TDT patients (81%) achieved a transfusion burden reduction of ≥ 20% over any 12 weeks on study compared with the 12 weeks before baseline (95% CI 63.6–92.8), when treated with luspatercept at a dose of 0.6–1.25 mg/kg [51]. These findings, including the achievement of secondary endpoints, prompted a randomized phase 3 clinical trial (BELIEVE Trial) to assess efficacy and safety.
The BELIEVE study (ClinicalTrials.gov number NCT02604433) (Table 2) is a randomized, double-blind, placebo-controlled trial that was conducted in 65 sites across 15 countries, enrolling a total of 336 adult TDT patients (> 18 years) randomized in a 2:1 ratio to receive luspatercept or placebo subcutaneously every 21 days for at least 48 weeks. The primary endpoint was the percentage of patients who had an erythroid response, defined as a reduction in the transfusion burden of at least 33% from baseline (the 12 weeks before the first dose of luspatercept or placebo) during weeks 13 through 24 plus a reduction of at least 2 red-cell units over this 12-week interval. Key secondary endpoints included a reduction in the transfusion burden of at least 33% from baseline during weeks 37 through 48 plus a reduction of at least 2 red-cell units over this 12-week interval. Moreover, a reduction in the transfusion burden of at least 50% from baseline during weeks 13 through 24 plus a reduction of at least 2 red-cell units over this 12-week interval, a reduction in the transfusion burden of at least 50% from baseline during weeks 37 through 48, plus a reduction of at least 2 red-cell units over this 12-week interval, and a mean change from baseline in the transfusion burden during weeks 13 through 24 were also considered secondary endpoints.
Forty-eight out of 224 patients (21.4%) in the luspatercept group achieved the primary endpoints compared to the placebo group (5/112 patients: 4.5%); these results were statistically significant (P < 0.001). Similarly, a significantly higher percentage of patients in the luspatercept group than in the placebo group achieved the secondary endpoints. Indeed, three-quarters of patients (75%) had at least a 33% reduction in transfusion burden during any rolling 12-week interval with luspatercept, and they had a response within 86 days (four treatment cycles). Preliminary post hoc analyses indicate that the response was faster among the patients with a non-β0/β0 genotype than among those with a β0/β0 genotype. Transfusion independence was attained by 11% of the patients in the luspatercept group during any 8-week interval. Luspatercept was well tolerated, and safety was consistent with previous experience in phase 2 studies. In conclusion, all the results of all primary and key secondary efficacy analyses were in favor of luspatercept over placebo. A 5-year open-label extension phase is underway to provide long-term data on the safety of luspatercept and its effects on the transfusion burden and iron outcomes [52].
A phase 2 trial (BEYOND) in adults with NTDT is ongoing (ClinicalTrials.gov number NCT03342404) (Table 2). The primary endpoint is the proportion of patients with an increase in mean Hb concentration of ≥ 1 g/dL in the absence of RBC transfusion from weeks 13 to 24 versus baseline.
JAK2 Inhibitors
Several studies have provided evidence on the role of JAK2 as a potential target to treat disorders of ineffective erythropoiesis (Fig. 1b) [53]. The inhibition of JAK2 in TDT and NTDT mouse models not only improved ineffective erythropoiesis but also reversed splenomegaly [54, 55]. Based on these findings, it has been suggested that the use of ruxolitinib could benefit thalassemia patients. A phase 2a study (ClinicalTrials.gov number NCT02049450) (Table 2) assessed the efficacy and safety of ruxolitinib in TDT patients with spleen enlargement [56]. A total of 30 TDT patients received ruxolitinib at a starting dose of 10 mg twice daily. A decrease in spleen size from baseline was observed in ruxolitinib-treated patients. Mean change in spleen volume from baseline to week 12 (n = 26) and week 30 (n = 25) was − 19.7% and − 26.8%, respectively. At week 30, one patient who initially had a 15% decrease in spleen volume at week 12 demonstrated an increase in spleen volume [56]. No clinically significant improvements in pre-transfusion Hb were seen, thus there was no related reduction in transfusion needs. Moreover, although hepcidin levels increased in the ruxolitinib treatment group, no significant changes in serum iron or ferritin levels were observed over time [56]. For all the above-mentioned reasons, the study did not proceed into phase 3.
Modulating Iron Metabolism
Hepcidin is the master regulator of iron metabolism [5], and, despite the presence of iron overload, its levels are low in β-thalassemia patients (Fig. 1c) [6, 7]. A substantial number of agents that stimulate hepcidin expression or activity have been investigated in preclinical studies, demonstrating a beneficial activity [57–59]. A similar ferroportin-reducing effect can be achieved with agents that target ferroportin directly, blocking the binding of hepcidin and preventing internalization and degradation of the iron exporter protein. Owing to the homeostatic control of hepcidin expression, approaches that interfere with the underlying regulatory pathways may be a useful option [60]. These includes the blocking of matriptase 2 (MT2), interfering with the cleavage of haemojuvelin (Fig. 1c) [61, 62].
Minihepcidins
Minihepcidins are known to restrict iron absorption. They can, therefore, be used to target iron dysregulation. A 2016 study by Casu et al. showed that minihepcidins were able to ameliorate ineffective erythropoiesis, anemia, splenomegaly, and iron overload in young Hbbth3/+ mice, which resemble the human NTDT phenotype [57]. In old Hbbth3/+ mice, however, the adminsitration of minihepcidin in combination with the iron chelator deferiprone was able to improve ineffective erythropoiesis, anemia, and also reverse splenomegaly [57]. In order to assess the efficacy of minihepcidins in TDT, Casu et al. recently generated a new mouse model (Hbbth1/th2) that closely resembles the human TDT phenotype [58]. Hbbth1/th2BMC were treated with two doses of minihepcidin: 2.625 mg/kg (low dose) or 5.25 mg/kg (high dose) for a period of 2 months post-transplant. Administration of the high-dose minihepcidin ameliorated RBC lifespan, ineffective erythropoiesis, anemia and splenomegaly in untransfused Hbbth1/th2 BMC mice. Hbbth1/th2 BMC mice treated with minihepcidins also showed decreased serum erythroferrone levels, serum iron levels, and siginificant reduction of iron in the liver and spleen. The administration of minihepcidin therefore ameliorated iron overload in untransfused Hbbth1/th2 BMC mice [58].
Multiple ongoing clinical trials are assessing the efficacy and safety of minihepcidin. The first is a phase 2 study by La Jolla Pharmaceutical Company, using the molecule LJPC-401, for the treatment of myocardial iron overload in adult TDT patients (ClinicalTrials.gov number NCT03381833) (Table 2). As part of its primary objective, this study aimed at evaluating the effect of LJPC-401 on iron levels in adult TDT patients who suffer from myocardial iron overload. This trial, however, was prematurely terminated, as an interim analysis on the safety and efficacy data showed an absence of efficacy with the protocol treatment regimen, thus indicating an unfavorable risk–benefit profile for patients. Another phase 2 study by Protagonist Therapeutics aimed at assessing efficacy of PTG-300 on TDT and NTDT patients with chronic anemia (TRANSCEND trial) (ClinicalTrials.gov number NCT03802201) (Table 2). Primary outcome measures of this study include: (a) NTDT subjects who achieve an increase in Hb without transfusion and (b) TDT subjects who achieve a decrease in red blood cell units required over an 8-week period. Due to efficacy issues in the drug, however, this study was recently stopped.
Ferroportin Inhibitors
A more recent approach to target ineffective erythropoiesis through the modulation of iron metabolism involves the use of ferroportin inhibitors. A newly described compound in the field is VIT-2763, a small oral molecule that acts as a ferroportin inhibitor [63]. In cells, VIT-2763 blocked iron efflux, competed with hepcidin for ferroportin binding, and triggered ferroportin internalization and ubiquitination [63]. In Hbbth3/+ mice, VIT-2763 improved anemia and ameliorated erythropoiesis [63]. ROS levels were also subsequently lowered and overall oxidative damage was also decreased by the drug. Overall tissue oxygenation was also improved upon the administration of VIT-2763 in Hbbth3/+ mice, thus inhibiting the hypoxic cycle and lowering EPO production. Iron overload in the liver was also decreased with the use of this drug. Furthermore, a significant amelioration of myelopoiesis was noted in the spleen of Hbbth3/+ mice after 3 weeks of administration with VIT-2763 [63]. In rodents, the drug was well tolerated with no observed adverse effects for a dose above 600 mg/kg over 14 days, and no dose-limiting toxicity was noted in longer studies. All this evidence proves the therapeutic effect of the oral VIT-2763 ferroportin inhibitor for β-thalassemia patients.
In order to determine the safety, tolerability, pharmacokinetic properties, and pharmacodynamic effects of VIT-2763, a phase 1 randomized, double-blind, placebo-controlled, parallel-group, dose-escalation study, comprising a single ascending phase (SAD) and multiple ascending phases (MADs), was performed on healthy male and female volunteers aging between 18 and 65 years [64]. Patients were hospitalized on day-1 and were to receive VIT-2763 or placebo on the following day (day 1 in the SAD phase) or for the next 7 days (day 1 to day 7 on the MADs phase). During the SAD phase, five groups were recorded sequentially and were to receive either placebo or single oral doses (5, 15, 60, 120, or 240 mg) of VIT-2763. In the MAD phase, the drug was administered at multiple oral doses (60 or 120 mg, once daily or twice daily) or matching placebo, in sequential ascending groups, for 7 days [64]. Results showed that no adverse events or serious adverse events that led to drug discontinuation.
Moreover, most of the adverse events in the SAD (23/32) and MADs (68/96) were not related to the drug itself. The most reported adverse event was headache in both SAD and MAD phases, and it was more frequent in the MAD phase. There was a temporary decrease in mean serum iron levels with VIT-2763 single doses ≥ 60 mg and all multiple doses [64]. The mean calculated transferrin saturation (only assessed following multiple dosing) also temporarily decreased. A shift in mean serum hepcidin peaks followed the administration of all iron-lowering doses of VIT-2763 [64]. Based on the data from this study, a phase 2 study by Vifor Pharma will be soon initiated in NTDT patients, assessing the efficacy, safety, tolerability, pharmacokinetics, and pharmacodynamics of VIT-2763 in this patient population (ClinicalTrials.gov number NCT04364269) (Table 2).
Transmembrane Protease Serine 6 (TMPRSS6)
Other novel therapeutic approaches to target iron dysregulation include increasing the hepatic synthesis of hepcidin. This can be achieved by suppressing the gene TMPRSS6. Deletion of the TMPRSS6 gene in Hbbth3/+ mice has been shown to improve anemia, ineffective erythropoiesis, and splenomegaly [65]. The use of second-generation antisense oligonucleotides (ASOs) to target TMPRSS6 have also been described in Hbbth3/+ mice. A study by Guo et al. revealed that targeting TMPRSS6 by ASO can modulate hepcidin antimicrobial peptide (HAMP) expression [62]. Using hemochromatosis-affected mice (Hfe–/–), the ASO-treated group showed a significant decrease in iron parameters and liver iron accumulation, significant amelioration of ineffective erythropoiesis, lower erythropoietin levels, decrease in splenomegaly, and an increase in Hb levels [62]. Another study by Schmidt et al. showed that the use of small interfering RNAs (siRNA) can downregulate TMPRSS6. They showed that lipid nanoparticles-TMPRSS6 siRNA treatment of Hbbth3/+ mice induced hepcidin and decreased both tissue and serum iron levels [66].
A phase 2a study by Ionis Pharmaceuticals using TMPRSS6 inhibitors will soon be initiated in patients with NTDT ≥ 18 years of age. In this study patients will be subcutaneously administered IONIS TMPRSS6-LRx every 4 weeks (ClinicalTrials.gov number NCT04059406) (Table 2). Primary outcome measures of the study will include the percentage of participants with a ≥ 1.0 g/dL increase in plasma Hb from baseline at week 27. Another molecule currently under development by Silence Therapeutics is SLN124, a GalNAc conjugated double-stranded fully modified siRNA that targets TMPRSS6 messenger RNA (mRNA). A randomized, single-blind, placebo-controlled, phase 1b, single-ascending and multiple-dose study in adult patients with NTDT and very low- and low-risk myelodysplastic syndrome (MDS) is currently underway to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamic response of SLN124 (ClinicalTrials.gov number NCT04176653) (Table 2).
Conclusion
The improvement in the understanding of β-thalassemia pathophysiology has paved the way for new therapeutic approaches that aim to directly correct the imbalance between α- and β-globin chains through gene therapy or correct ineffective erythropoiesis acting on different pathways. On the other hand, the treatment of α-thalassemias is not as advanced as for β-thalassemias because of the complexity of α genes and the less ineffective erythropoiesis component. Nevertheless, the most severe form of α-thalassemia (absence of the four α genes), i.e., Hb Bart’s, which causes hydrops fetalis, requires in utero intervention, while the absence of three α genes, i.e., disease, is phenotypically an NTDT.
A new era of novel therapies beyond transfusion and iron chelation is emerging in the thalassemia realm, with the aim of improving outcomes and overall quality of life. Among the new options, activin receptor ligand trap molecules are, so far, the most promising, with significant improvement in Hb or transfusion requirements. Indeed, a reduction of 33% in transfusion burden is equivalent to a one-third reduction of transfused blood, corresponding to less blood per transfusion or less frequent transfusion, with fewer hospital visits. Ultimately, this produces less iron accumulation and subsequent reduction in iron chelation therapy. Nonetheless, these drugs have first been tested for osteoporosis, thus exhibiting a potential effect on an invalidating complication for β-thalassemia patients. Moreover, with some patients now on long-term treatment (up to 8 years), they present an excellent safety profile. The efficacy and safety of these novel therapies are still under study by several clinical trials, some in early phases. Ultimately, long-term, head-to-head, and comparison trials using these novel therapies are necessary for the future to determine the optimal use of these novel therapies. Moreover, whether these novel therapies will be used on their own or in combination with other conventional therapeutic agents, such as iron chelators, is yet to be determined.
Acknowledgements
We thank Daniela Canali for producing the figures.
Compliance with Ethical Standards
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
RB has nothing to disclose. IM reports receiving honoraria from Sanofi-Genzyme. ATT reports receiving consultancy, research funding, and honoraria from Novartis; research funding and consultancy from Celgene; consultancy from Vifor, research funding from La Jolla, research funding and consultancy from Protagonist Therapeutics; consultancy from IONIS Pharmaceuticals. MDC reports receiving consultancy, research funding and honoraria from Novartis; research funding and consultancy from Celgene; consultancy from Vifor, research funding from La Jolla, research funding from Protagonist Therapeutics; consultancy from IONIS Pharmaceuticals, and research funding from CRISPR Therapeutics.
References
- 1.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167. doi: 10.1016/S0140-6736(17)31822-6. [DOI] [PubMed] [Google Scholar]
- 3.Rivella S. beta-thalassemias: paradigmatic diseases for scientific discoveries and development of innovative therapies. Haematologica. 2015;100(4):418–430. doi: 10.3324/haematol.2014.114827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 5.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98(15):8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with beta-thalassemia major: a longitudinal study. Blood. 2013;122(1):124–133. doi: 10.1182/blood-2012-12-471441. [DOI] [PubMed] [Google Scholar]
- 7.Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Tsimirika K, MacFarlane J, et al. Hepcidin in iron overload disorders. Blood. 2005;105(10):4103–4105. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13(9):1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 9.Kautz L, Jung G, Du X, Gabayan V, Chapman J, Nasoff M, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood. 2015;126(17):2031–2037. doi: 10.1182/blood-2015-07-658419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camaschella C, Nai A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br J Haematol. 2016;172(4):512–523. doi: 10.1111/bjh.13820. [DOI] [PubMed] [Google Scholar]
- 11.Taher AT, Cappellini MD. How I manage medical complications of beta-thalassemia in adults. Blood. 2018;132(17):1781–1791. doi: 10.1182/blood-2018-06-818187. [DOI] [PubMed] [Google Scholar]
- 12.Taher AT. Thalassemia. Hematol Oncol Clin N Am. 2018;32(2):xv–xvi. doi: 10.1016/j.hoc.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V, editors. Guidelines for the management of transfusion dependent thalassaemia (TDT). Nicosia (CY); 2014. [PubMed]
- 14.Taher A, Musallam KM, Cappellini MD, editors. Guidelines for the management of non-transfusion dependent thalassaemia (NTDT). Nicosia (CY); 2017. [PubMed]
- 15.Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–1193. [PubMed] [Google Scholar]
- 16.Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet. 2000;355(9220):2051–2052. doi: 10.1016/S0140-6736(00)02357-6. [DOI] [PubMed] [Google Scholar]
- 17.Reblozyl. [cited April 15, 2020].https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761136lbl.pdf. Accessed 8 June 2020.
- 18.Zynteglo. [cited October 20, 2019]. https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo. Accessed 8 June 2020.
- 19.Baronciani D, Angelucci E, Potschger U, Gaziev J, Yesilipek A, Zecca M, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000–2010. Bone Marrow Transplant. 2016;51(4):536–541. doi: 10.1038/bmt.2015.293. [DOI] [PubMed] [Google Scholar]
- 20.Angelucci E, Pilo F, Coates TD. Transplantation in thalassemia: revisiting the Pesaro risk factors 25 years later. Am J Hematol. 2017;92(5):411–413. doi: 10.1002/ajh.24674. [DOI] [PubMed] [Google Scholar]
- 21.Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C, et al. Bone marrow transplantation in patients with thalassemia. N Engl J Med. 1990;322(7):417–421. doi: 10.1056/NEJM199002153220701. [DOI] [PubMed] [Google Scholar]
- 22.Lucarelli G, Isgro A, Sodani P, Gaziev J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med. 2012;2(5):a011825. doi: 10.1101/cshperspect.a011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C, Mathews V, Kim S, George B, Hebert K, Jiang H, et al. Related and unrelated donor transplantation for beta-thalassemia major: results of an international survey. Blood Adv. 2019;3(17):2562–2570. doi: 10.1182/bloodadvances.2019000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, et al. Gene therapy in patients with transfusion-dependent beta-thalassemia. N Engl J Med. 2018;378(16):1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 25.Marktel S, Scaramuzza S, Cicalese MP, Giglio F, Galimberti S, Lidonnici MR, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ss-thalassemia. Nat Med. 2019;25(2):234–241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 26.Motta I, Ghiaccio V, Cosentino A, Breda L. Curing hemoglobinopathies: challenges and advances of conventional and new gene therapy approaches. Mediterr J Hematol Infect Dis. 2019;11(1):e2019067. doi: 10.4084/MJHID.2019.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22(2):53–63. doi: 10.1016/j.blre.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Walters MC, Hardy K, Edwards S, Adamkiewicz T, Barkovich J, Bernaudin F, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16(2):263–272. doi: 10.1016/j.bbmt.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palchaudhuri R, Saez B, Hoggatt J, Schajnovitz A, Sykes DB, Tate TA, et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol. 2016;34(7):738–745. doi: 10.1038/nbt.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aiuti A, Naldini L. Safer conditioning for blood stem cell transplants. Nat Biotechnol. 2016;34(7):721–723. doi: 10.1038/nbt.3629. [DOI] [PubMed] [Google Scholar]
- 31.Hartigan AJ, Pearse BR, McDonough SM, Proctor JL, Adams HL, McShea MA, et al. A non-genotoxic antibody drug conjugate targeting C-kit for hematopoietic stem cell transplant conditioning. Biol Blood Marrow Transplant. 2018;24(3):S47–S48. [Google Scholar]
- 32.Falzetti F, Aversa F, Minelli O, Tabilio A. Spontaneous rupture of spleen during peripheral blood stem-cell mobilisation in a healthy donor. Lancet. 1999;353(9152):555. doi: 10.1016/S0140-6736(99)00268-8. [DOI] [PubMed] [Google Scholar]
- 33.Balaguer H, Galmes A, Ventayol G, Bargay J, Besalduch J. Splenic rupture after granulocyte-colony-stimulating factor mobilization in a peripheral blood progenitor cell donor. Transfusion. 2004;44(8):1260–1261. doi: 10.1111/j.1537-2995.2004.00413.x. [DOI] [PubMed] [Google Scholar]
- 34.Becker PS, Wagle M, Matous S, Swanson RS, Pihan G, Lowry PA, et al. Spontaneous splenic rupture following administration of granulocyte colony-stimulating factor (G-CSF): occurrence in an allogeneic donor of peripheral blood stem cells. Biol Blood Marrow Transplant. 1997;3(1):45–49. [PubMed] [Google Scholar]
- 35.Lindemann A, Rumberger B. Vascular complications in patients treated with granulocyte colony-stimulating factor (G-CSF) Eur J Cancer. 1993;29A(16):2338–2339. doi: 10.1016/0959-8049(93)90236-9. [DOI] [PubMed] [Google Scholar]
- 36.Pilo F, Angelucci E. Iron toxicity and hemopoietic cell transplantation: time to change the paradigm. Mediterr J Hematol Infect Dis. 2019;11(1):e2019030. doi: 10.4084/MJHID.2019.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yannaki E, Karponi G, Zervou F, Constantinou V, Bouinta A, Tachynopoulou V, et al. Hematopoietic stem cell mobilization for gene therapy: superior mobilization by the combination of granulocyte-colony stimulating factor plus plerixafor in patients with beta-thalassemia major. Hum Gene Ther. 2013;24(10):852–860. doi: 10.1089/hum.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richard RE, Siritanaratkul N, Jonlin E, Skarpidi E, Heimfeld S, Blau CA. Collection of blood stem cells from patients with sickle cell anemia. Blood Cells Mol Dis. 2005;35(3):384–388. doi: 10.1016/j.bcmd.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 39.Uchida N, Fujita A, Hsieh MM, Bonifacino AC, Krouse AE, Metzger ME, et al. Bone marrow as a hematopoietic stem cell source for gene therapy in sickle cell disease: evidence from rhesus and SCD patients. Hum Gene Ther Clin Dev. 2017;28(3):136–144. doi: 10.1089/humc.2017.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanna E, Remuzat C, Auquier P, Toumi M. Gene therapies development: slow progress and promising prospect. J Mark Access Health Policy. 2017;5(1):1265293. doi: 10.1080/20016689.2017.1265293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maguer-Satta V, Bartholin L, Jeanpierre S, Ffrench M, Martel S, Magaud JP, et al. Regulation of human erythropoiesis by activin A, BMP2, and BMP4, members of the TGFbeta family. Exp Cell Res. 2003;282(2):110–120. doi: 10.1016/s0014-4827(02)00013-7. [DOI] [PubMed] [Google Scholar]
- 42.Soderberg SS, Karlsson G, Karlsson S. Complex and context dependent regulation of hematopoiesis by TGF-beta superfamily signaling. Ann N Y Acad Sci. 2009;1176:55–69. doi: 10.1111/j.1749-6632.2009.04569.x. [DOI] [PubMed] [Google Scholar]
- 43.Shav-Tal Y, Zipori D. The role of activin a in regulation of hemopoiesis. Stem Cells. 2002;20(6):493–500. doi: 10.1634/stemcells.20-6-493. [DOI] [PubMed] [Google Scholar]
- 44.Blank U, Karlsson S. The role of Smad signaling in hematopoiesis and translational hematology. Leukemia. 2011;25(9):1379–1388. doi: 10.1038/leu.2011.95. [DOI] [PubMed] [Google Scholar]
- 45.Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408–414. doi: 10.1038/nm.3512. [DOI] [PubMed] [Google Scholar]
- 46.Verma A, Suragani RN, Aluri S, Shah N, Bhagat TD, Alexander MJ, et al. Biological basis for efficacy of activin receptor ligand traps in myelodysplastic syndromes. J Clin Investig. 2020;130(2):582–589. doi: 10.1172/JCI133678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suragani RN, Cawley SM, Li R, Wallner S, Alexander MJ, Mulivor AW, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine beta-thalassemia. Blood. 2014;123(25):3864–3872. doi: 10.1182/blood-2013-06-511238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerra A, Oikonomidou PR, Sinha S, Zhang J, Lo Presti V, Hamilton CR, et al. Lack of Gdf11 does not improve anemia or prevent the activity of RAP-536 in a mouse model of beta-thalassemia. Blood. 2019;134(6):568–572. doi: 10.1182/blood.2019001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cappellini MD, Porter J, Origa R, Forni GL, Voskaridou E, Galacteros F, et al. Sotatercept, a novel transforming growth factor beta ligand trap, improves anemia in beta-thalassemia: a phase II, open-label, dose-finding study. Haematologica. 2019;104(3):477–484. doi: 10.3324/haematol.2018.198887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Attie KM, Allison MJ, McClure T, Boyd IE, Wilson DM, Pearsall AE, et al. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am J Hematol. 2014;89(7):766–770. doi: 10.1002/ajh.23732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piga A, Perrotta S, Gamberini MR, Voskaridou E, Melpignano A, Filosa A, et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with beta-thalassemia. Blood. 2019;133(12):1279–1289. doi: 10.1182/blood-2018-10-879247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cappellini MD, Viprakasit V, Taher AT, Georgiev P, Kuo KHM, Coates T, et al. A phase 3 trial of luspatercept in patients with transfusion-dependent beta-thalassemia. N Engl J Med. 2020;382(13):1219–1231. doi: 10.1056/NEJMoa1910182. [DOI] [PubMed] [Google Scholar]
- 53.Melchiori L, Gardenghi S, Rivella S. beta-Thalassemia: HiJAKing ineffective erythropoiesis and iron overload. Adv Hematol. 2010;2010:938640. doi: 10.1155/2010/938640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Libani IV, Guy EC, Melchiori L, Schiro R, Ramos P, Breda L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875–885. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casu C, Presti VL, Oikonomidou PR, Melchiori L, Abdulmalik O, Ramos P, et al. Short-term administration of JAK2 inhibitors reduces splenomegaly in mouse models of beta-thalassemia intermedia and major. Haematologica. 2018;103(2):e46–e49. doi: 10.3324/haematol.2017.181511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taher AT, Karakas Z, Cassinerio E, Siritanaratkul N, Kattamis A, Maggio A, et al. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: results from a phase 2a study. Blood. 2018;131(2):263–265. doi: 10.1182/blood-2017-06-790121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Casu C, Oikonomidou PR, Chen H, Nandi V, Ginzburg Y, Prasad P, et al. Minihepcidin peptides as disease modifiers in mice affected by beta-thalassemia and polycythemia vera. Blood. 2016;128(2):265–276. doi: 10.1182/blood-2015-10-676742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casu C, Chessa R, Liu A, Gupta R, Drakesmith H, Fleming R, et al. Minihepcidins improve ineffective erythropoiesis and splenomegaly in a new mouse model of adult beta-thalassemia major. Haematologica. 2019. [DOI] [PMC free article] [PubMed]
- 59.Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. 2018;131(16):1790–1794. doi: 10.1182/blood-2017-11-737411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oikonomidou PR, Casu C, Rivella S. New strategies to target iron metabolism for the treatment of beta thalassemia. Ann N Y Acad Sci. 2016;1368(1):162–168. doi: 10.1111/nyas.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Casu C, Aghajan M, Oikonomidou PR, Guo S, Monia BP, Rivella S. Combination of Tmprss6- ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica. 2016;101(1):e8–e11. doi: 10.3324/haematol.2015.133348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Investig. 2013;123(4):1531–1541. doi: 10.1172/JCI66969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manolova V, Nyffenegger N, Flace A, Altermatt P, Varol A, Doucerain C, et al. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of beta-thalassemia. J Clin Investig. 2019;130(1):491–506. doi: 10.1172/JCI129382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richard F, van Lier JJ, Roubert B, Haboubi T, Gohring UM, Durrenberger F. Oral ferroportin inhibitor VIT-2763: first-in-human, phase 1 study in healthy volunteers. Am J Hematol. 2020;95(1):68–77. doi: 10.1002/ajh.25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nai A, Pagani A, Mandelli G, Lidonnici MR, Silvestri L, Ferrari G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119(21):5021–5029. doi: 10.1182/blood-2012-01-401885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidt PJ, Toudjarska I, Sendamarai AK, Racie T, Milstein S, Bettencourt BR, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121(7):1200–1208. doi: 10.1182/blood-2012-09-453977. [DOI] [PMC free article] [PubMed] [Google Scholar]