

Abstract
The malignant traits involved in tumor relapse, metastasis and the expansion of cancer stem-like cells are acquired via the epithelial–mesenchymal transition (EMT) process in the tumor microenvironment. In addition, the tumor microenvironment strongly supports the survival and growth of malignant tumor cells and further contributes to the reduced efficacy of anticancer therapy. Ionizing radiation can influence the tumor microenvironment, because it alters the biological functions of endothelial cells composing tumor vascular systems. However, to date, studies on the pivotal role of these endothelial cells in mediating the malignancy of cancer cells in the irradiated tumor microenvironment are rare. We previously evaluated the effects of irradiated endothelial cells on the malignant traits of human liver cancer cells and reported that endothelial cells irradiated with 2 Gy reinforce the malignant properties of these cancer cells. In this study, we investigated the signaling mechanisms underlying these events. We revealed that the increased expression level of IL-4 in endothelial cells irradiated with 2 Gy eventually led to enhanced migration and invasion of cancer cells and further expansion of cancer stem-like cells. In addition, this increased level of IL-4 activated the ERK and AKT signaling pathways to reinforce these events in cancer cells. Taken together, our data indicate that ionizing radiation may indirectly modulate malignancy by affecting endothelial cells in the tumor microenvironment. Importantly, these indirect effects on malignancy are thought to offer valuable clues or targets for overcoming the tumor recurrence after radiotherapy.
Keywords: ionizing radiation, endothelial cells, epithelial–mesenchymal transition, migration, invasion, cancer stem-like cells
INTRODUCTION
Although radiotherapy has been a fairly popular treatment for cancer patients until recently, patients treated with radiotherapy have had a significant risk of cancer recurrence following this treatment, impeding full recovery [1–3]. This nearly fatal event is accepted to be closely related to the tumor microenvironment generated by hypoxia and a diverse population of stromal cells, including fibroblasts, endothelial cells, perivascular cells and inflammatory cells [4]. In addition, the tumor microenvironment clearly affects the sensitivity of cancer cells to ionizing radiation and can lead to the development of aggressive tumor characteristics by triggering epithelial–mesenchymal transition (EMT) [4, 5]. Cancer cells undergoing EMT exhibit a decreased level of epithelial markers and an increased level of mesenchymal markers, thereby leading to the loss of cell–cell interactions [6, 7]. Consequently, these cells extravasate from the primary tumor sites and move to their new destination [5–7] and are thus easily detected in the regions of malignant tumors displaying aggressive characteristics [8, 9]. In addition, many lines of evidence show that populations of cancer cells possessing mesenchymal traits include a large population of cancer stem-like cells [10–12]. Furthermore, EMT, which leads to these traits, is recognized as a principal mechanism for generating cancer stem-like cells, which contribute to resistance to anticancer therapy [10–12].
The tumor vascular system in the tumor microenvironment contributes to the survival and growth of cancer cells [13, 14]. Furthermore, the endothelial cells composing this system have been reported to influence the induction of tumor malignancy related to cancer stem-like cells by activating signaling networks of cytokines or growth factors responsible for cell–cell communication [13–16]. The role and radioresponse of the diverse population of stromal cells composing the tumor microenvironment has been widely investigated, but, to date, the effects of these cells on tumor malignancy in the irradiated tumor microenvironment have been less well known [17, 18].
A variety of cytokines or growth factors are associated with the diverse events occurring in the tumor microenvironment [13–16]. Among these mediators, several interleukins (ILs) have been widely reported to play a fundamental role in regulating the migratory and invasive properties of cancer cells in the tumor microenvironment [19–21]. In addition, the activation of their associated signaling pathways is strongly related to an increased risk of tumor progression and poor survival [19, 21, 22].
Previously, we observed that irradiated endothelial cells can modulate the malignancy of liver cancer cells and further showed that 2 Gy–irradiated endothelial cells are associated with an increase in the migratory and invasive properties and the sphere size of liver cancer cells [23]. In this study, we performed additional research to identify the signaling pathways underlying the increased malignancy of liver cancer cells mediated by 2 Gy–irradiated endothelial cells. We found that activation of the IL-4/ERK/AKT signaling pathway led to increased malignancy of liver cancer cells treated with conditioned medium from 2 Gy–irradiated endothelial cells. Our findings suggest a mechanism or insight that may explain the means by which cancer recurs after radiotherapy.
MATERIALS AND METHODS
Reagents
AG490 was purchased from InvivoGen (San Diego, CA, USA). LY294002 was purchased from Cell Signaling Technology (Denver, MA, USA). PD98059, PD169316 and SP600125 were purchased from Millipore (Darmstadt, Germany). Mouse monoclonal antibodies against Vimentin (cat. no. sc-6260), E-cadherin (cat. no. sc-8426) and Zeb1 (cat. no. sc-81428) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). A mouse monoclonal antibody against N-cadherin (cat. no. 610920) was purchased from BD Bioscience (San Jose, CA, USA). Rabbit monoclonal antibodies against Slug (cat. no. 9585), phospho-Stat3 (Tyr705) (cat. no. 9145), phospho-p38 MAPK (Thr180/Tyr182) (cat. no. 9215) and Snail (cat. no. 3879) were purchased from Cell Signaling Technology (Denver, MA, USA). A rabbit polyclonal antibody against STAT-3 (cat. no. sc-7179) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit polyclonal antibodies against phospho-AKT (Ser473) (cat. no. 9271), AKT (cat. no. 9272), phospho-p44/42 MAPK (ERK1/2) (cat. no. 9101), ERK1/2 (cat. no. 9102), phospho-SAPK/JNK (Thr183/Tyr185) (cat. no. 9255) and p38 MAPK (cat. no. 9212) were purchased from Cell Signaling Technology (Denver, MA, USA). Mouse monoclonal anti-β-actin (cat. no. A5441), horseradish peroxidase–conjugated anti–mouse IgG (cat. no. A9044) and horseradish peroxidase–conjugated anti–rabbit IgG (cat. no. A0545) antibodies were purchased from Sigma–Aldrich (St Louis, MO, USA).
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from ScienCell (Carlsbad, CA, USA). HUVECs plated in gelatin-coated 60-mm dishes were cultured in complete endothelial cell culture medium (ScienCell Research Laboratories, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum, 1% antibiotics and 1% endothelial cell growth supplement in a humidified 5% CO2 incubator at 37°C. Cells from passages 2 to 5 were used for experiments. Human liver cancer HepG2, Hep3B and Huh7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). These cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Sigma–Aldrich, St Louis, MO, USA) supplemented with 10% (v/v) bovine calf serum, penicillin (50 units/ml), and streptomycin (50 μg/ml) (all from Gibco BRL, Grand Island, NY, USA).
Irradiation
HUVECs were exposed to γ-rays from a 137Cs irradiation source (Eckert & Ziegler, Berlin, Germany) at a dose rate of 2.6 Gy/min.
Preparation of conditioned medium
After the medium from endothelial cells was freshly replaced with new medium, HUVECs were irradiated with various doses of γ-rays and were then cultured for 24 h. Shortly thereafter, the endothelial cell–conditioned medium (ECM) was harvested and filtered through a 0.45-μm filter or centrifuged for 5 min at 700 × g to remove cells and debris and was then transferred to tubes.
Western blot analysis
Cells were treated with lysis buffer [40 mM Tris-HCl (pH 8.0), 120 mM NaCl, and 0.1% (v/v) NP40] supplemented with protease inhibitor cocktail tablets (1 tablet/50 ml; Boehringer, Mannheim, Germany) and centrifuged for 15 min at 12,000 × g and 4 C. SDS–PAGE was used to separate 30 μg of protein, which was transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% (w/v) nonfat dry milk in Tris-buffered saline and were then incubated for 1 h with all primary antibodies, each at a dilution of 1:1000 and at room temperature. All peroxidase-conjugated secondary antibodies were diluted 1:10,000 for the detection of specific reaction bands, which were visualized using an enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ, USA) at room temperature.
Small interfering RNA (siRNA) transfection
siRNA-mediated RNA interference was achieved using double-stranded RNA molecules. The siRNAs against IL-4 (sc-39623), IL-2 (sc-39619), IL-13 (sc-39642) and IL-16 (sc-39647) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Control siRNA-A (sc-37007) (Santa Cruz Biotechnology, CA, USA) was used as the control. Cells were grown to 30% confluency in 60-mm dishes and transfected with the siRNA duplexes (100 nM) using Lipofectamine 2000 (Gibco BRL, Grand Island, NY, USA) in accordance with the manufacturer’s instructions. Assays were performed 48 h after transfection.
Assessments of cytokine secretion
After HUVECs (~70% confluence) were washed with phosphate-buffered saline, serum-free medium was added to the cells. After 24 h, the levels of cytokines were measured using ELISA (enzyme-linked immunosorbent assay) kits [Human IL-4 Quantikine ELISA Kit (cat. no. D4050); Human IL-2 Quantikine ELISA Kit (cat. no. D2050); Human IL-13 Quantikine ELISA Kit (cat. no. D1300B); Human IL-16 Quantikine ELISA Kit (cat. no. D1600)] commercially purchased from R&D systems (Minneapolis, MN, USA).
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed using the SYBR Green reporter method. A sample of 1 μg of total RNA isolated using an RNeasy Mini kit (Qiagen, Valencia, CA, USA) was subsequently reverse transcribed to cDNA with the SuperScript First-Strand Synthesis System (Gibco Invitrogen Corp., Carlsbad, CA, USA). Then, 3 μl of cDNA was added to 17 μl of reaction mixture containing 5 μl of ddH2O, 10 μl of 2× SYBR Green Master Mix (Thermo Scientific, Waltham, MA, USA) and 1 μl of each primer. PCR amplification was carried out using a thermal cycling profile beginning at 94°C for 5 min, followed by 40 cycles at 94°C for 10 s, 58°C for 15 s and 72°C for 15 s, with a final extension step at 72°C for 5 min. The amplification specificity of each qRT-PCR analysis was confirmed by melting curve analysis. The transcripts of target genes were analyzed using the CFX96™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The primers used for qRT-PCR were as follows: IL-4 forward (5′-GTC TCA CCT CCC AAC TGC TT-3′) and reverse (5′-CTT GGA GGC AGC AAA GAT GT-3′); IL-2 forward (5′-CAA ACC TCT GGA GGA AGT GC-3′) and reverse (5′-AAT GGT TGC TGT CTC ATC AGC-3′); IL-13 forward (5′-CCA CTT CAC ACA CAG GCA AC-3′) and reverse (5′-ACT CCT GGT GTC CAC TGC TT-3′); IL-16 forward (5′-CTG TCA ACA CAG GCT GAG GA-3′) and reverse (5′-GAA GGC ACA GCA AGG ATT TC-3′); and GAPDH forward (5′-GAG CCC CAG CCT TCT CCA TG-3′) and reverse (5′-GAA ATC CCA TCA CCA TCT TCC AGG-3′). The GAPDH gene was used as the control to normalize the expression values of IL-4, IL-2, IL-13 and IL-16. The relative expression levels of the genes were calculated using the 2-△△Ct method [24].
Migration and invasion assays
Both migration and invasion assays were performed using Transwell chambers (8-μm pore size; BD Biosciences, San Jose, CA, USA). A total of 2 × 104 cells were resuspended in serum-free growth medium for these assays. For the invasion assay, the interior of the inserts was precoated with 10 mg/ml growth factor–reduced Matrigel (BD Biosciences, San Jose, CA, USA). For both assays, cells were seeded in the interior of the inserts. Growth medium supplemented with 10% (v/v) fetal bovine serum was added to the lower chambers. After incubation for 24 h, the cells attached to the upper surface of the filters were removed with a cotton swab. The cells on the lower surface of the filters were fixed and stained. The number of cells was determined by counting the cells in five microscopic fields per well. In addition, the cells were imaged by phase contrast microscopy (Nikon Eclipse 80i; Nikon, Tokyo, Japan).
Sphere formation assay
Cells (300 cells per well) were grown in serum-free DMEM/F12 (Gibco BRL, Grand Island, NY, USA) supplemented with B27 (Gibco BRL, Grand Island, NY, USA), N2 (Gibco BRL, Grand Island, NY, USA), 20 ng/ml basic fibroblast growth factor (bFGF; Peprotech, London, UK) and 20 ng/ml epidermal growth factor (EGF; Peprotech, London, UK) in 24-well ultralow attachment plates for 7 or 14 days, and the size and number of the spheres were determined using a phase contrast Nikon microscope (TS100; Tokyo, Japan). The size of 12 randomly selected spheres per group was measured.
Statistical analyses
The mean values and standard errors of the means were calculated using Microsoft Excel (2007 version, Microsoft Corporation, Redmond, WA, USA). All data presented are representative of at least three independent experiments and were statistically analyzed with GraphPad Prism version 8.0 (GraphPad Software Inc., San Diego, CA, USA). Differences between groups were analyzed using an unpaired Student’s t-test or with one-way or two-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test. P-values of <0.05 (indicated by the asterisks in the figures) were considered significant.
RESULTS
Irradiated endothelial cells reinforced the malignant traits of liver cancer cells in vitro
To further verify the results of our previous study indicating the effects of medium conditioned by 2 Gy–irradiated endothelial cells (2 Gy–irradiated ECM) on the malignancy of liver cancer cells [23], we performed a migration and invasion assay to observe whether treatment with 2 Gy–irradiated ECM led to an increase in the malignant potential of these cells, before identifying the signaling pathways. As shown in Supplementary Figure 1A–C, the ability of HepG2, Hep3B and Huh7 cells to migrate and invade was greatly increased by treatment with 2 Gy–irradiated ECM.
Tumor malignancy is closely associated with an increase in the population of cancer stem-like cells [10–12]. Therefore, we performed a sphere formation assay to investigate the effect of treatment with 2 Gy–irradiated ECM on the sphere-forming potential of cancer stem-like cells. As shown in Supplementary Figure 1D and E, 2 Gy–irradiated ECM led to the formation of larger spheres of HepG2, Hep3B or Huh7 cells than control ECM, indicating that 2 Gy–irradiated endothelial cells can readily lead to the increased sphere-forming potential of liver cancer cells.
These malignant properties of cancer cells appear after the acquisition of mesenchymal traits via EMT [10–12]. We thus examined whether 2 Gy–irradiated ECM affected the protein expression level of EMT markers in HepG2 cells. As shown in Supplementary Figure 1F, the expression level of the mesenchymal cell marker N-cadherin was greatly increased by treatment with 2 Gy–irradiated ECM, whereas that of the epithelial cell marker E-cadherin was unaffected. Furthermore, we measured the expression levels of the EMT-regulating transcription factors Slug, Snail and Zeb1 and found that 2 Gy–irradiated ECM increased the expression level of Slug but did not alter the levels of Snail and Zeb1.
Collectively, as demonstrated in our previous report [23], these data indicate that 2 Gy–irradiated endothelial cells contributed to the enhancement of tumor malignancy via triggering EMT and increasing the sphere-forming potential of liver cancer cells.
IL-4 secreted by irradiated endothelial cells contributed to the increase in liver cancer cell malignancy
Soluble proteins secreted from endothelial cells in the tumor microenvironment play a pivotal role in increasing tumor malignancy [17, 25–27]. In particular, a high level of Th2 cytokines has been reported to be closely connected with increased malignancy of cancer cells in the tumor microenvironment [19, 22, 28]. Thus, we examined whether ionizing radiation increases both the secretion and the mRNA expression of several cytokines in HUVECs. As shown in Fig. 1A and B, ionizing radiation led to increase in both the secretion and the mRNA expression of Th2 cytokines (IL-4 and IL-13) and Th1 cytokines (IL-2 and IL-16) in HUVECs. We further sought to determine which cytokines enhance the migration and invasive abilities of HepG2 cells. To this end, we transfected siRNAs targeting these cytokines into HUVECs and found that all treatments with these siRNAs efficiently attenuated the increased mRNA expression levels of these cytokines caused by 2 Gy irradiation (Fig. 1B).
Fig. 1.
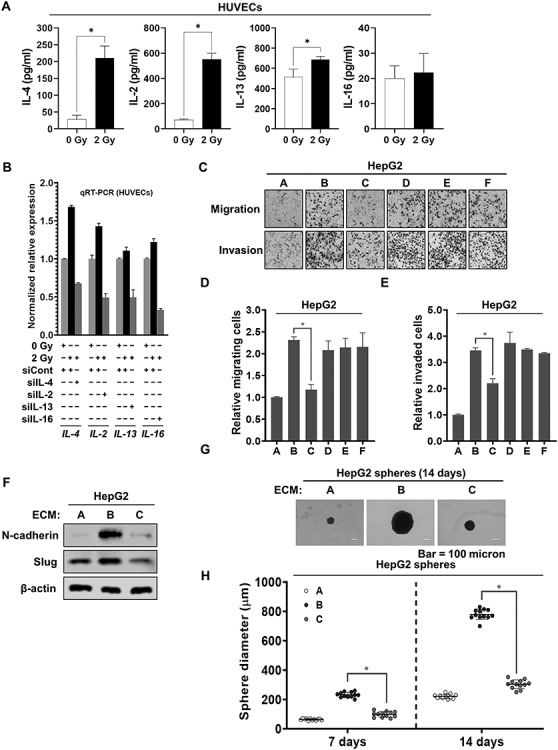
IL-4 released from 2 Gy–irradiated endothelial cells contributed to increasing the malignancy of liver cancer cells. (a) The secretion of IL-4, IL-2, IL-13 or IL-16 in 2 Gy–irradiated HUVECs. After HUVECs were irradiated with 2 Gy, and then cultured for 24 h, ELISA was performed to detect the secretion of interleukins, using conditioned medium obtained from HUVECs. (b) qRT-PCR analysis of the mRNA expression levels of IL-4, IL-2, IL-13 and IL-16 in the presence or absence of siRNAs targeting IL-4, IL-2, IL-13 and IL-16, respectively, in 2 Gy–irradiated HUVECs. (c–e) Migratory and invasive properties of HepG2 cells after treatment with conditioned medium from 2 Gy–irradiated HUVECs pretreated with siRNAs targeting IL-4, IL-2, IL-13 and IL-16. These properties of the cells were measured using Transwell chambers (magnification, ×200) (A = conditioned medium from unirradiated HUVECs pretreated with siCont; B = conditioned medium from 2 Gy–irradiated HUVECs pretreated with siCont; C = conditioned medium from 2 Gy–irradiated HUVECs pretreated with siIL-4; D = conditioned medium from 2 Gy–irradiated HUVECs pretreated with siIL-2; E = conditioned medium from 2 Gy–irradiated HUVECs pretreated with siIL-13; F = conditioned medium from 2 Gy–irradiated HUVECs pretreated with siIL-16). (f) Western blot analysis of the expression levels of N-cadherin and Slug after treatment of HepG2 cells with conditioned medium from 2 Gy–irradiated HUVECs pretreated with siRNA targeting IL-4 for 48 h. Experiments were performed in triplicate, and the data shown are representative of a typical experiment. (g and h) Quantification of the sphere-forming ability of HepG2 cells after treatment with conditioned medium from 2 Gy–irradiated HUVECs pretreated with siRNA targeting IL-4 (A = conditioned medium from unirradiated HUVECs pretreated with siCont; B = conditioned medium from 2 Gy–irradiated HUVECs pretreated with siCont; C = conditioned medium from 2 Gy–irradiated HUVECs pretreated with siIL-4). These cells (300 cells per well) were grown in DMEM/F12 supplemented with B27, N2, EGF and bFGF in 24-well ultralow attachment plates for 7 and 14 days, and the size of the spheres was determined. The size of 12 randomly selected spheres per group (n = 12/group) was measured. The average size of each sphere was quantified with the standard deviation and is shown in the representative graph. β-actin was used as the loading control. The results from three independent experiments are expressed as the means ±1 SEM. (*P < 0.05).
Thereafter, we harvested the conditioned medium from 2 Gy–irradiated HUVECs transfected with these siRNAs and treated HepG2 cells with this medium. As shown in Fig. 1C–E, only IL-4 siRNA effectively inhibited the migration and invasive abilities of HepG2 cells enhanced by the treatment with 2 Gy–irradiated ECM. Furthermore, IL-4 siRNA efficiently reduced the increased expression levels of N-cadherin and Slug in HepG2 cells treated with 2 Gy–irradiated ECM (Fig. 1F). These results clearly indicate that IL-4, a Th2 cytokine, is a key contributor to 2 Gy–irradiated ECM-induced malignant EMT in HepG2 cells. To further determine whether IL-4 siRNA affects the 2 Gy–irradiated ECM-enhanced sphere-forming ability of HepG2 cells, we performed a sphere formation assay. As shown in Fig. 1G and H, IL-4 siRNA effectively suppressed the sphere formation increased by treatment with 2 Gy–irradiated ECM.
Collectively, these data show that the expression of IL-4 in 2 Gy–irradiated endothelial cells leads to the enhancement of liver cancer cell malignancy.
JAK/STAT signaling pathway activation was required for the increase in liver cancer cell malignancy induced by irradiated endothelial cells
The Janus kinase/Signal transducer and activator of transcription protein (JAK/STAT) pathway is a principal mechanism of the signaling events induced by a variety of cytokines and soluble growth factors [29]. Thus, we examined whether this pathway contributes to the increased malignancy of liver cancer cells caused by treatment with 2 Gy–irradiated ECM. As shown in Fig. 2A, we found that the level of phosphorylated STAT3 increased early (by 10 min), with the increase typically being maintained for 12 h after treatment before decreasing. To further investigate the relevance of the JAK/STAT pathway to the enhanced EMT induced by 2 Gy-irradiated ECM in HepG2 cells, we treated HepG2 cells with a JAK inhibitor, AG490, are finding that treatment with AG490 efficiently suppressed the increased phosphorylation of STAT3 (Fig. 2B). As shown in Fig. 2C–F, pretreatment with AG490 effectively attenuated the 2 Gy–irradiated ECM-enhanced migratory and invasive abilities of HepG2 cells and greatly blocked the increase in the expression levels of N-cadherin and Slug. In addition, AG490 effectively suppressed the increase in the sphere-forming ability of HepG2 cells induced by treatment with 2 Gy–irradiated ECM (Fig. 2G–H).
Fig. 2.
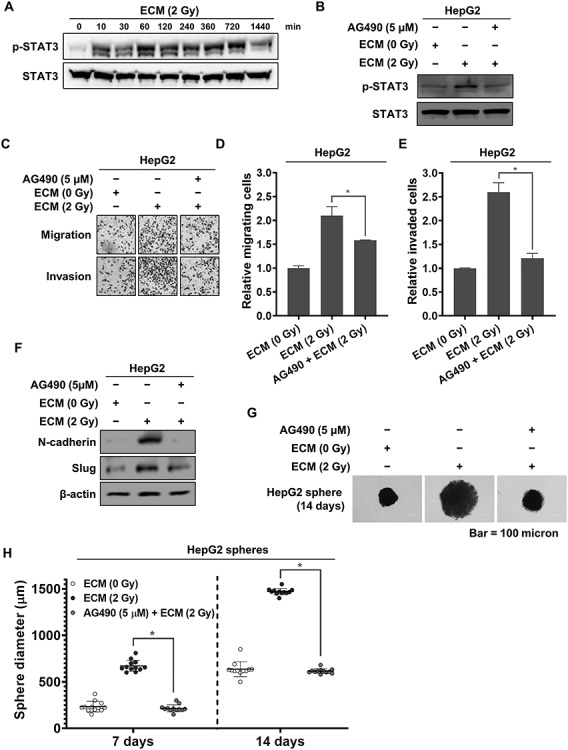
Activation of the JAK/STAT3 pathway in liver cancer cells induced by 2 Gy–irradiated ECM. (a) Western blot analysis of the phosphorylation of STAT3 in HepG2 cells treated with 2 Gy–irradiated ECM. (b) Effects of AG490 on the protein expression levels of phospho-STAT3. HepG2 cells treated with 2 Gy–irradiated ECM were grown in the presence or absence of AG490 (5 μM) for 24 h, and western blot analysis was performed. (c–e) Effects of AG490 on the migratory and invasive properties of HepG2 cells enhanced by treatment with 2 Gy–irradiated ECM. Cells were treated with 2 Gy–irradiated ECM in the presence or absence of AG490 (5 μM) and grown for 48 h. These properties of HepG2 cells were measured using Transwell chambers (magnification, ×200). (f) Effects of AG490 on the protein expression levels of N-cadherin and Slug in HepG2 cells treated with 2 Gy–irradiated ECM for 48 h. (f–g) Effects of AG490 on the sphere-forming abilities of HepG2 cells enhanced by treatment with 2 Gy–irradiated ECM. Cells (300 cells per well) were grown in DMEM/F12 supplemented with B27, N2, EGF and bFGF on 24-well ultralow attachment plates for 7 and 14 days, and the size of the spheres was determined. The size of 12 randomly selected spheres per group (n = 12/group) was measured. The average size of each sphere was quantified with the standard deviation and is shown in the representative graph. β-actin was used as the loading control. Western blotting was performed in triplicate, and the data shown are representative of a typical experiment. The results from three independent experiments are expressed as the means ±1 SEM. (*P < 0.05).
These results clearly indicate that 2 Gy–irradiated endothelial cells can lead to the increased malignancy of liver cancer cells via JAK/STAT pathway activation in liver cancer cells.
Irradiated endothelial cells enhanced malignancy by activating the ERK and JNK pathways in liver cancer cells
In addition to the JAK/STAT pathway, the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/AKT pathways can be activated by a variety of cytokines and their receptors, including IL-4 and its receptors [30–33]. Therefore, we further examined whether these pathways are involved in mediating the increased malignancy of HepG2 cells induced by 2 Gy–irradiated ECM. As shown in Fig. 3A, the level of phosphorylated AKT increased, peaking at 1 h then plateauing for 2 h after treatment, before steadily decreasing. In addition, the levels of both phosphorylated ERK1/2 and JNK1/2 were increased early, at 10 min, and then dramatically decreased, whereas the level of phosphorylated p38 MAPK was increased late.
Fig. 3.
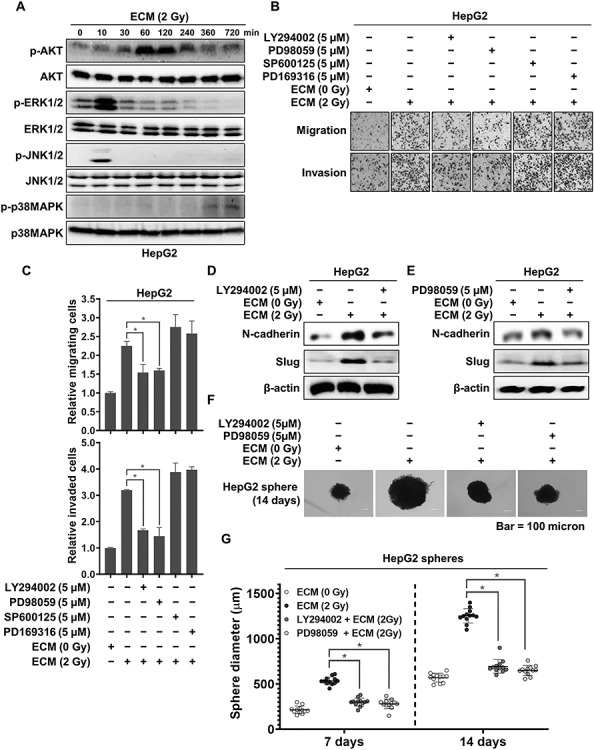
Activation of AKT and ERK induced by 2 Gy–irradiated ECM. (a) Western blot analysis of the phosphorylation of AKT and MAPKs in HepG2 cells treated with 2 Gy–irradiated ECM. (b and c) Effects of LY294002, PD98059, SP600125 and PD169316 on the migratory and invasive properties of HepG2 cells treated with 2 Gy–irradiated ECM. Cells were grown in the presence or absence of each inhibitor for 48 h, and migration and invasion assays were performed using Transwell chambers. (d and e) Effects of LY294002 and PD98059 on the protein expression levels of N-cadherin and Slug. HepG2 cells treated with 2 Gy–irradiated ECM were grown in the presence or absence of LY294002 (5 μM) or PD98059 (5 μM) for 48 h, and western blot analysis was performed. (f and g) Effects of LY294002 and PD98059 on the sphere-forming ability of HepG2 cells enhanced by treatment with 2 Gy–irradiated ECM. Cells (300 cells per well) were grown in DMEM/F12 supplemented with B27, N2, EGF and bFGF in 24-well ultralow attachment plates for 7 and 14 days, and the size of the spheres was determined. The size of 12 randomly selected spheres per group (n = 12/group) was measured. The average size of each sphere was quantified with the standard deviation and is shown in the representative graph. β-actin was used as the loading control. Western blotting was performed in triplicate, and the data shown are representative of a typical experiment. The results from three independent experiments are expressed as the means ±1 SEM. (*P < 0.05).
To further identify the factors involved in the increased migratory and invasive abilities of HepG2 cells induced by 2 Gy–irradiated ECM, we treated HepG2 cells with LY294002, PD98059, SP600125 and PD169316, inhibitors of the PI3K/AKT, ERK, JNK and p38 MAPK pathways, respectively. As shown in Fig. 3B and C, both LY294002 and PD98059 but not SP600125 or PD169316 effectively inhibited the increased migration and invasion. Furthermore, both LY294002 and PD98059 greatly suppressed the increased expression levels of N-cadherin and Slug in these cells and efficiently reduced their increased sphere-forming potential (Fig. 3D–G).
Collectively, these results demonstrated that the activation of the AKT or ERK pathway was required for the increase in tumor malignancy induced by 2 Gy–irradiated endothelial cells.
The IL-4/ERK/AKT signaling pathway was required for the increase in liver cancer cell malignancy induced by irradiated endothelial cells
To assess the effects of IL-4 on the activation of AKT or ERK in HepG2 cells treated with 2 Gy–irradiated ECM, we collected conditioned medium from 2 Gy–irradiated HUVECs transfected with siRNA targeting IL-4 and subsequently treated HepG2 cells with this medium. As shown in Fig. 4A, inhibition of IL-4 in HUVECs effectively attenuated the increased phosphorylation of AKT and ERK in HepG2 cells treated with 2 Gy–irradiated ECM, indicating that IL-4 secreted by 2 Gy–irradiated endothelial cells contributes to the activation of AKT and ERK in HepG2 cells. To further examine the interrelation between the AKT and ERK pathways in HepG2 cells treated with 2 Gy–irradiated ECM, we examined the changes in the phosphorylation of AKT and ERK after the treatment of HepG2 cells with LY294002 and PD98059. As shown in Fig. 4B, LY294002 did not affect the increased phosphorylation of ERK caused by 2 Gy–irradiated ECM. However, PD98059 greatly attenuated AKT phosphorylation, indicating that ERK acts upstream of AKT activation in HepG2 cells treated with 2 Gy–irradiated ECM (Fig. 4C).
Fig. 4.
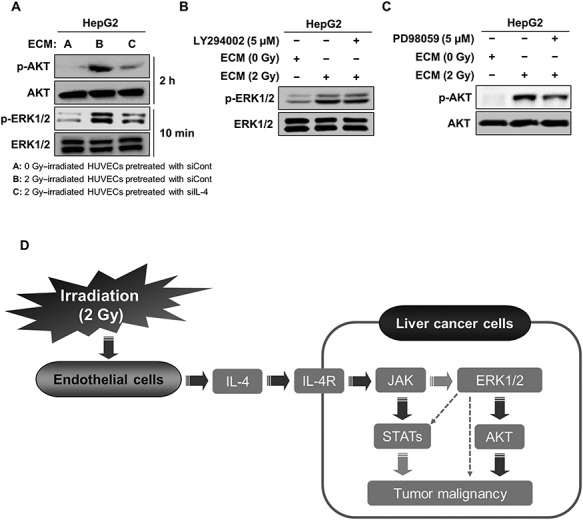
The activation of the IL-4/ERK/AKT pathway contributed to the increase in the malignancy of HepG2 cells treated with 2 Gy–irradiated ECM. (a) Western blot analysis of the levels of phosphorylated AKT and ERK after treatment of HepG2 cells with conditioned medium from 2 Gy–irradiated HUVECs pretreated with siRNA targeting IL-4 for 2 h and 10 min, respectively. (b) Effects of LY294002 on the protein level of phosphorylated ERK in HepG2 cells treated with 2 Gy–irradiated ECM. Cells were grown in the presence or absence of LY294002 (5 μM) for 10 min, and western blot analysis was performed. (c) Effects of PD98059 on the protein level of phosphorylated AKT in HepG2 cells treated with 2 Gy–irradiated ECM. Cells were grown in the presence or absence of PD98059 (5 μM) for 2 h, and western blot analysis was performed. (d) Proposed mechanism underlying the increase in the malignancy of liver cancer cells in response to 2 Gy–irradiated ECM. Western blotting was performed in triplicate, and the data shown are representative of a typical experiment.
Taken together, these results clearly suggest that the malignant behaviors of liver cancer cells induced by 2 Gy–irradiated ECM arise from the activation of the IL-4/ERK/AKT pathway (Fig. 4D).
DISCUSSION
The vasculature in the tumor microenvironment plays a fundamental role in contributing to the survival and growth of tumor cells by supplying them with nutrients and oxygen [13, 14]. In addition, the endothelial cells composing this tumor vasculature have been previously reported to increase the malignancy of diverse types of cancer cells by inducing EMT, which is also accepted as a mechanism leading to an increase in the number of cancer stem-like cells [13–16].
Ionizing radiation has been demonstrated to affect endothelial cells in both normal and tumor vasculature [34–36]. Furthermore, most of these studies have primarily considered the direct effects but not the indirect effects of irradiation on cells [37, 38]. Importantly, ionizing radiation can influence the unirradiated cells surrounding the irradiated cells, a phenomenon termed the bystander effect [4, 37]. Therefore, additional studies are required to characterize the bystander effect caused by ionizing radiation on the tumor microenvironment. As a contribution to this investigation, we previously found that irradiated endothelial cells can induce cancer cells to convert to a malignant phenotype [23]. In this study, we further investigated the signaling pathway underlying the role of irradiated endothelial cells in the increase in tumor malignancy. Consistent with our previous observation [23], we confirmed in this study that irradiated endothelial cells can increase the malignant potential of liver cancer cells.
A variety of cytokines are involved in the regulation of tumor malignancy [13–16, 19–22]. In particular, Th2 cytokines are closely associated with the induction of tumor growth and malignancy, whereas Th1 cytokines are involved in tumor suppression [19, 22, 28]. Among these cytokines, IL-4, a well-known Th2 cytokine, performs many immunoregulatory functions [39]. The high expression level of IL-4 in tumor tissues has been demonstrated to be strongly related to poor prognosis in tumor patients [39–41] and to play pivotal roles in the occurrence of malignancy in diverse tumor types [39, 41]. In addition, a variety of tumor cells exhibit a high expression level of the IL-4 receptor (IL-4R), and this high expression level is strongly correlated with an increase in tumor growth [39, 41]. In contrast, IL-4 has been reported to suppress angiogenesis and cancer cell growth by modulating immune cells in many types of tumors [41–45]. Thus, the role of IL-4 in tumor growth and malignancy remains controversial. However, in this study, treatment with siRNAs against various types of ILs revealed that the high expression of IL-4 in 2 Gy–irradiated endothelial cells greatly contributes to the increase in tumor malignancy, including migration, invasion and sphere formation behaviors, in liver cancer cells. In good agreement with our data, soluble factors released from endothelial cells have been indicated to influence tumor malignancy [17, 25–27]. Furthermore, several reports have already demonstrated that IL-4 is a key mediator of cancer progression and drug resistance in several cancer types, including colon, breast and prostate cancer [46–48]. Thus, targeting the signaling pathway induced by IL-4 may be a useful strategy for the suppression of tumor malignancy. Indeed, inflammatory cytokines can activate JAK/STAT signaling pathways to promote tumor malignancy [48]. Consistent with these reports, inhibition of these pathways significantly attenuated the malignancy increased by treatment with 2 Gy–irradiated ECM.
To activate IL-4–mediated signaling pathways, IL-4 first binds to IL-4R, its cognate receptor protein on the cell surface, which subsequently leads to the activation of JAK/STAT, AKT and MARKs [49]. More importantly, IL-13 and IL-4 share receptor proteins [20, 39, 40, 49]. Therefore, the downstream signaling molecules activated by IL-13 and the consequent cellular events are nearly identical to those induced by IL-4 [20, 39, 40, 49]. We found that 2 Gy irradiation also increased the expression level of IL-13 in endothelial cells, although inhibition of IL-13 expression in these cells did not attenuate the malignant traits of liver cancer cells. We presume that these differences may be due to IL-13Rα2, a subunit of the IL-13R complex. IL-13Rα2 has been reported to play a fundamental role in suppressing IL-13 as a decoy receptor possessing a higher binding affinity for IL-13 than does the other IL-13R [50]. Additionally, several papers have indicated that IL-4 can trigger the activation of signaling pathways to induce the expression of IL-13Rα2 [51]. Thus, the action of IL-13 may be blocked by the IL-13Rα2 expressed as a consequence of IL-4 secretion in this study. However, additional studies focusing on the role of IL-13Rα2 in this study are needed.
In addition, IL-4 has been reported to trigger the activation of PI3K/AKT or MAPKs in diverse types of cells [45, 47–49]. In this study, IL-4 induced the activation of AKT and ERK, thereby leading to tumor malignancy induced by 2 Gy–irradiated ECM. Crosstalk between the AKT and ERK pathways, including cross-activation or cross-inhibition, is frequently observed in the signaling pathways involved in various cellular events [52, 53]. In good agreement with several reports [54–56], we found that ERK positively regulates the activation of AKT. Although these reports did not clearly show the underlying mechanism by which ERK can affect the activation of this pathway, the following reports are thought to support our results.
Liver kinase B1 (LKB1), a well-known tumor suppressor, has recently been shown to possess protooncogenic abilities [57]. In this context, LKB1 has been reported to inactivate the phosphatase and tensin homology (PTEN) by phosphorylating it at Ser380, Thr382, Thr383 and Ser385 [58–60]. This inactivation of PTEN consequently results in the activation of the PI3K/AKT pathway [58–60]. In addition, LKB1 directly activates AKT independent of PI3K in liver cancer cells [61]. The increased kinase activity of LKB1 was reported to be influenced by its binding ability to phosphatidic acid (PA), one of the glycerophospholipids composing the plasma membrane and a second messenger modulating diverse signaling pathways in cells, suggesting a key role of PA in LKB1 activation [62]. PA is generated by phospholipase D (PLD)-mediated hydrolysis of phospholipids [62]. Interestingly, ERK has been demonstrated to contribute to the increase in the enzyme activity of PLD2 [63]. Therefore, the ERK-dependent activation of AKT in this study can be assumed to be due to the inhibition of PTEN caused by sequential signaling through the ERK/PLD2/PA/LKB pathway. Thus, further studies on this pathway are necessary.
After JAK phosphorylates tyrosine residues on the receptors for cytokines or growth factors, insulin receptor substrate-1 and -2 (IRS-1 and -2, respectively) bind to these phosphorylated tyrosine residues on the receptors, consequently leading to PI3K recruitment and activation [30–32]. Similarly, growth factor receptor–bound protein 2 (Grb2) has been reported to interact with the tyrosine residues phosphorylated by JAK on these receptors and to subsequently activate the Ras/Raf/MEK/ERK cascade [33]. In addition, ERK can directly phosphorylate serine residues on STATs [64]. Thus, because the activation of the JAK/STAT pathway in this study is reasonably thought to affect the PI3K/AKT or ERK pathways, further studies are needed to define the crosstalk between these signaling pathways activated by 2 Gy–irradiated ECM.
The signaling pathways leading to EMT have been well revealed by investigation of the functional role of TGF-β in a variety of biological events [65, 66]. In particular, the activation of ERK or JNK caused by TGF-β has been reported to increase the expression level of Snail and Slug, which are transcriptional repressors involved in downregulation of E-cadherin [67, 68]. In addition, TGF-β–mediated Ras activation triggers the activation of the Raf/MEK/ERK cascade, eventually leading to an increase in the expression level of a variety of genes responsible for cell motility [69]. AKT is also known to contribute to the induction of EMT via regulating the phosphorylation status of Twist1 or via increasing the expression level of Snail through the activation of NF-κB [70]. Furthermore, β-catenin has been shown to play its role in the induction of EMT, and its protein level can be downregulated by GSK3-β [71]. AKT directly phosphorylates GSK3β to suppress it. Therefore, active AKT can lead to an increase in the protein level of β-catenin via this mechanism [71].
Taken together, these findings indicate that 2 Gy–irradiated ECM effected an increase in liver cancer cell malignancy. In addition, we provide evidence that tumor malignancy was increased by the activation of the IL-4/ERK/AKT signaling pathway in liver cancer cells treated with 2 Gy–irradiated ECM (Fig. 4D).
Supplementary Material
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
Not applicable.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This work was supported by a National Research Foundation of Korea (DIRAMS) grant funded by the Korean government (MSIP) (50591–2019, 50595–2019).
References
- 1. Datta NR, Samiei M, Bodis S. Radiotherapy infrastructure and human resources in Europe – present status and its implications for 2020. Eur J Cancer 2014;50:2735–43. [DOI] [PubMed] [Google Scholar]
- 2. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nature Reviews Cancer 2011;11:239–53. [DOI] [PubMed] [Google Scholar]
- 3. Ogawa K, Yoshioka Y, Isohashi F et al. Radiotherapy targeting cancer stem cells: current views and future perspectives. Anticancer Res 2013;33:747–54. [PubMed] [Google Scholar]
- 4. Stapleton S, Jaffray D, Milosevic M. Radiation effects on the tumor microenvironment: implications for nanomedicine delivery. Adv Drug Deliv Rev 2017;109:119–30. [DOI] [PubMed] [Google Scholar]
- 5. Marie-Egyptienne DT, Lohse I, Hill RP. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: potential role of hypoxia. Cancer Lett 2013;341:63–72. [DOI] [PubMed] [Google Scholar]
- 6. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer 2013;13:97–110. [DOI] [PubMed] [Google Scholar]
- 7. Kim J-H, Shim J-W, Eum D-Y et al. Downregulation of UHRF1 increases tumor malignancy by activating the CXCR4/AKT-JNK/IL-6/snail signaling axis in hepatocellular carcinoma cells. Sci Rep 2017;7:2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008;27:6958–69. [DOI] [PubMed] [Google Scholar]
- 9. Vasko V, Espinosa AV, Scouten W et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc Natl Acad Sci U S A 2007;104:2803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010;29:4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asiedu MK, Beauchamp-Perez FD, Ingle JN et al. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014;33:1316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hwang-Verslues WW, Chang P-H, Wei P-C et al. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene 2011;30:2463–74. [DOI] [PubMed] [Google Scholar]
- 13. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 2011;121:3804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307:58–62. [DOI] [PubMed] [Google Scholar]
- 15. Zhu TS, Costello MA, Talsma CE et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res 2011;71:6061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu J, Ye X, Fan F et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013;23:171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leroi N, Lallemand F, Coucke P et al. Impacts of ionizing radiation on the different compartments of the tumor microenvironment. Front Pharmacol 2016;7:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menon H, Ramapriyan R, Cushman TR et al. Role of radiation therapy in modulation of the tumor stroma and microenvironment. Front Immunol 2019;10:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Setrerrahmane S, Xu H. Tumor-related interleukins: old validated targets for new anti-cancer drug development. Mol Cancer 2017;16:153–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Little AC, Pathanjeli P, Wu Z et al. IL-4/IL-13 stimulated macrophages enhance breast cancer invasion via rho-GTPase regulation of synergistic VEGF/CCL-18 signaling. Front Oncol 2019;9:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gocheva V, Wang H-W, Gadea BB et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev 2010;24:241–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldstein R, Hanley C, Morris J et al. Clinical investigation of the role of interleukin-4 and interleukin-13 in the evolution of prostate cancer. Cancers 2011;3:4281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim SD, Yi J-M, Park M-T. Irradiated endothelial cells modulate the malignancy of liver cancer cells. Oncol Lett 2019;17:2187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 25. Sharma A, Shiras A. Cancer stem cell–vascular endothelial cell interactions in glioblastoma. Biochem Biophys Res Commun 2016;473:688–92. [DOI] [PubMed] [Google Scholar]
- 26. Charles N, Ozawa T, Squatrito M et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010;6:141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Z, Dong Z, Lauxen IS et al. Endothelial cell–secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype. Cancer Res 2014;74:2869–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee HL, Jang JW, Lee SW et al. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci Rep 2019;9:3260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quintás-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res 2013;19:1933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heller NM, Qi X, Junttila IS et al. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci Signal 2008;1:ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rahaman SO, Vogelbaum MA, Haque SJ. Aberrant Stat3 signaling by interleukin-4 in malignant glioma cells: involvement of IL-13Rα2. Cancer Res 2005;65:2956–63. [DOI] [PubMed] [Google Scholar]
- 32. Heller NM, Gwinn WM, Donnelly RP et al. IL-4 engagement of the type I IL-4 receptor complex enhances mouse eosinophil migration to eotaxin-1 in vitro. PLoS ONE 2012;7:e39673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Angulo P, Kaushik G, Subramaniam D et al. Natural compounds targeting major cell signaling pathways: a novel paradigm for osteosarcoma therapy. J Hematol Oncol 2017;10:10–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li YQ, Ballinger JR, Nordal RA et al. Hypoxia in radiation-induced blood–spinal cord barrier breakdown. Cancer Res 2001;61:3348–54. [PubMed] [Google Scholar]
- 35. Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell 2005;8:89–91. [DOI] [PubMed] [Google Scholar]
- 36. García-Barros M, Paris F, Cordon-Cardo C et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003;300:1155–9. [DOI] [PubMed] [Google Scholar]
- 37. Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer 2009;9:351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Powathil GG, Munro AJ, Chaplain MAJ et al. Bystander effects and their implications for clinical radiation therapy: insights from multiscale in silico experiments. J Theor Biol 2016;401:1–14. [DOI] [PubMed] [Google Scholar]
- 39. Suzuki A, Leland P, Joshi BH et al. Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine 2015;75:79–88. [DOI] [PubMed] [Google Scholar]
- 40. Chang Y, Xu L, An H et al. Expression of IL-4 and IL-13 predicts recurrence and survival in localized clear-cell renal cell carcinoma. Int J Clin Exp Pathol 2015;8:1594–603. [PMC free article] [PubMed] [Google Scholar]
- 41. Li Z, Chen L, Qin Z. Paradoxical roles of IL-4 in tumor immunity. Cell Mol Immunol 2009;6:415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dehne N, Tausendschön M, Essler S et al. IL-4 reduces the proangiogenic capacity of macrophages by down-regulating HIF-1α translation. J Leukoc Biol 2014;95:129–37. [DOI] [PubMed] [Google Scholar]
- 43. Shankaranarayanan P, Nigam S. IL-4 induces apoptosis in A549 lung adenocarcinoma cells: evidence for the pivotal role of 15-hydroxyeicosatetraenoic acid binding to activated peroxisome proliferator–activated receptor gamma transcription factor. J Immunol 2003;170:887–94. [DOI] [PubMed] [Google Scholar]
- 44. Gooch JL, Lee AV, Yee D. Interleukin 4 inhibits growth and induces apoptosis in human breast cancer cells. Cancer Res 1998;58:4199–205. [PubMed] [Google Scholar]
- 45. Kim HD, Yu S-J, Kim HS et al. Interleukin-4 induces senescence in human renal carcinoma cell lines through STAT6 and p38 MAPK. J Biol Chem 2013;288:28743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Francipane MG, Alea MP, Lombardo Y et al. Crucial role of interleukin-4 in the survival of colon cancer stem cells. Cancer Res 2008;68:4022–5. [DOI] [PubMed] [Google Scholar]
- 47. Gaggianesi M, Turdo A, Chinnici A et al. IL4 primes the dynamics of breast cancer progression via DUSP4 inhibition. Cancer Res 2017;77:3268–79. [DOI] [PubMed] [Google Scholar]
- 48. Pencik J, Pham HTT, Schmoellerl J et al. JAK–STAT signaling in cancer: from cytokines to non-coding genome. Cytokine 2016;87:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chatila TA. Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol Med 2004;10:493–9. [DOI] [PubMed] [Google Scholar]
- 50. McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine 2015;75:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao Y, He D, Zhao J et al. Lysophosphatidic acid induces interleukin-13 (IL-13) receptor alpha2 expression and inhibits IL-13 signaling in primary human bronchial epithelial cells. J Biol Chem 2007;282:10172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rhim JH, Luo X, Gao D et al. Cell type–dependent Erk–Akt pathway crosstalk regulates the proliferation of fetal neural progenitor cells. Sci Rep 2016;6:26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mendoza MC, Er EE, Blenis J. The Ras–ERK and PI3K–mTOR pathways: cross-talk and compensation. Trends Biochem Sci 2011;36:320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Niba ETE, Nagaya H, Kanno T et al. Crosstalk between PI3 kinase/PDK1/Akt/Rac1 and Ras/Raf/MEK/ERK pathways downstream PDGF receptor. Cell Physiol Biochem 2013;31:905–13. [DOI] [PubMed] [Google Scholar]
- 55. Lee M, Sorn SR, Lee Y et al. Salt induces adipogenesis/lipogenesis and inflammatory adipocytokines secretion in adipocytes. Int J Mol Sci 2019;20:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matsuda Y, Wakai T, Kubota M et al. Valproic acid overcomes transforming growth factor-β–mediated sorafenib resistance in hepatocellular carcinoma. Int J Clin Exp Pathol 2014;7:1299–313. [PMC free article] [PubMed] [Google Scholar]
- 57. Lee S-W, Li C-F, Jin G et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol Cell 2015;57:1022–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Georgescu M-M. PTEN tumor suppressor network in PI3K–Akt pathway control. Genes Cancer 2010;1:1170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Milella M, Falcone I, Conciatori F et al. PTEN: multiple functions in human malignant tumors. Front Oncol 2015;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu W, Yang Z, Zhou S-F et al. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des Devel Ther 2014;8:1745–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martínez-López N, Varela-Rey M, Fernández-Ramos D et al. Activation of LKB1–Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology 2010;52:1621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dogliotti G, Kullmann L, Dhumale P et al. Membrane-binding and activation of LKB1 by phosphatidic acid is essential for development and tumour suppression. Nat Commun 2017;8:15747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Watanabe H, Yokozeki T, Yamazaki M et al. Essential role for phospholipase D2 activation downstream of ERK MAP kinase in nerve growth factor–stimulated neurite outgrowth from PC12 cells. J Biol Chem 2004;279:37870–7. [DOI] [PubMed] [Google Scholar]
- 64. Chung J, Uchida E, Grammer TC et al. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 1997;17:6508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xie L, Law BK, Chytil AM et al. Activation of the Erk pathway is required for TGF-beta1–induced EMT in vitro. Neoplasia 2004;6:603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Strippoli R, Benedicto I, Pérez Lozano ML et al. Epithelial-to-mesenchymal transition of peritoneal mesothelial cells is regulated by an ERK/NF-kappaB/Snail1 pathway. Dis Model Mech 2008;1:264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cano A, Pérez-Moreno MA, Rodrigo I et al. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000;2:76–83. [DOI] [PubMed] [Google Scholar]
- 68. Pérez-Mancera PA, González-Herrero I, Pérez-Caro M et al. SLUG in cancer development. Oncogene 2005;24:3073–82. [DOI] [PubMed] [Google Scholar]
- 69. Xie L, Law BK, Aakre ME et al. Transforming growth factor beta–regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res 2003;5:R187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xue G, Restuccia DF, Lan Q et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov 2012;2:248–59. [DOI] [PubMed] [Google Scholar]
- 71. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial–mesenchymal transition. Sci Signal 2014;7:re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.