

ABSTRACT
Radiation-induced rescue effect (RIRE) in cells refers to the phenomenon where irradiated cells (IRCs) receive help from feedback signals produced by partnered bystander unirradiated cells (UIRCs) or from the conditioned medium (CM) that has previously conditioned the UIRCs. In the present work, we explored the role of poly (ADP-ribose) polymerase 1 (PARP1) regulation in RIRE and the positive feedback loop between PARP1 and nuclear factor-kappa-light-chain-enhancer of activated B cell (NF-κB) in RIRE using various cell lines, including HeLa, MCF7, CNE-2 and HCT116 cells. We first found that when the IRCs (irradiated with 2 Gy X-ray) were treated with CM, the relative mRNA expression levels of both tumor suppressor p53-binding protein 1 (53BP1) and PARP1, the co-localization factor between 53BP1 and γH2AX as well as the fluorescent intensity of PARP1 were reduced. We also found that IRCs treated with the PARP1 inhibitor, Olaparib (AZD2281) had a higher 53BP1 expression. These results illustrated that PARP1 was involved in RIRE transcriptionally and translationally. We further revealed that treatment of IRCs with CM together with Olaparib led to significantly lower mRNA expression levels and fluorescent intensities of NF-κB, while treatment of IRCs with CM together the NF-κB inhibitor BAY-11-7082 led to significantly lower mRNA expression levels as well as fluorescent intensities of PARP1. These results illustrated that PARP1 and NF-κB were involved in the positive feedback loop transcriptionally and translationally. Thus, the results supported the occurrence of a PARP1–NF-κB positive feedback loop in RIRE. The present work provided insights into potential exploitation of inhibition of PARP1 and/or the PARP1–NF-κB positive feedback loop in designing adjuncts to cancer radiotherapeutics.
Keywords: radiation-induced rescue effect (RIRE), PARP1, NF-κB, TNF-α, BAY-11-7082; Olaparib
INTRODUCTION
Radiation-induced rescue effect (RIRE) in cells refers to the phenomenon where irradiated cells (IRCs) acquire aids from feedback signals produced by partnered bystander unirradiated cells (UIRCs) or from the conditioned medium (CM) that has previously conditioned the UIRCs [1–3]. RIRE was first reported by Chen et al. in 2011 [1]. RIRE is closely associated with the radiation-induced bystander effect (RIBE) which refers to a phenomenon where UIRCs behave as if they have been irradiated after being partnered with the IRCs or after being treated with the medium that has previously conditioned the IRCs. There were two widely adopted models for RIBE: (i) gap junction intercellular communication (GJIC) between cells that were in contact, and (ii) communication of soluble signal molecules secreted by IRCs into the medium [4]. Interestingly, bystander cells played a crucial role in regulating feedback signals for DNA repair, cell growth and cell proliferation to rescue IRCs through activation of the cell-signaling cascade [5].
Subsequent to the first report of RIRE [1], this radiobiological effect was observed by various research groups [6–10]. Different mechanisms were proposed for RIRE. For example, cyclic adenosine monophosphate (cAMP) [11] and nitric oxide (NO) [12–20] were proposed to be involved in RIRE. Lam et al. [21, 22] found that activation of the nuclear factor-kappa-light-chain-enhancer of activated B cell (NF-κB) pathway was responsible for triggering RIRE and speculated that tumor necrosis factor-α (TNF-α) could be a potential cytokine in activating the NF-κB pathway. Subsequently, Kong et al. [4] revealed that IRCs released bystander signals and induced autophagy in bystander cells to release interleukin 6 (IL-6), which could potentially activate the NF-κB pathway in IRCs. Interested readers are referred to the recent reviews on the different proposed mechanisms [2, 3].
In mammalian cells, DNA repair is known to be regulated by several signal transduction pathways [23]. TNF-α was identified as one such pro-inflammatory cytokine that induced cell-signaling pathways to promote cell survival [24]. It was established that TNF-α regulated biological responses through autocrine and paracrine signaling in the presence of oxidative stress [25, 26]. In fact, it was previously established that TNF-α was involved in bystander signaling when exposed to ionizing radiation [27]. Interestingly, TNF-α was also known to induce activation of a DNA repair enzyme, poly (ADP-ribose) polymerase 1 (PARP1) [28]. PARP1, a nuclear enzyme protein of 113 kDa, located in the human 1q41–42 chromosome, was known for catalysis of poly(ADP-ribosyl)ation of target proteins (PARylation) by using nicotinamide adenine dinucleotide (NAD+) as a substrate for generating adenosine diphosphate ribose (ADP-ribose) monomers [29–32]. In the presence of single-strand DNA (ssDNA) breaks, PARP1 was bound to the damaged DNA ends via its N-terminal DNA-binding domain and was found to catalyse PARylation through its C-terminal enzymatic domain [33–36]. It was also known to be involved in base excision repair (BER) and double-strand DNA (dsDNA) break repair pathways [34, 37]. During X-ray irradiation, generation of DNA strand breaks increased the activity of this DNA repair enzyme [38] by promoting PARylation [39]. In this regard, the first objective of the present work was to examine the involvement of PARP1 in RIRE. Four experiments were performed to achieve this objective. To begin with, we needed to explore whether manifestation of RIRE was correlated with the expression of PARP1 in the IRCs. Firstly, we used the relative mRNA expression levels of 53BP1 in IRCs as a surrogate for the manifestation of RIRE, and studied the correlation between the mRNA expression levels of 53BP1 and PARP1 in IRCs that were treated with or without the CM having previously conditioned the bystander cells (partnered with IRCs). Upon irradiation of cells, 53BP1 was known to hyperphosphorylate and co-localize with phosphorylated histone H2AX (γH2AX) in the megabase sites surrounding the regions of DNA lesions [40]. Since co-localization of γH2AX with 53BP1 was a widely accepted marker for detecting dsDNA breaks [41–46], it was also pertinent to study in the present work the co-localization of these proteins. In particular, we captured and compared images of IRCs treated with (i) fresh medium (FM) and (ii) CM to examine the co-localization, which formed our second experiment. In addition, we also captured and compared immunofluorescence images of IRCs stained with anti-PARP1 antibody that had been treated with (i) FM and (ii) CM to confirm the involvement of protein expression levels of PARP1 in RIRE, which formed our third experiment. Finally, we further examined whether IRCs treated with the PARP1 inhibitor, Olaparib (OLA), could still exhibit RIRE.
Upon confirmation of involvement of PARP1 in RIRE, it was then pertinent to study the relationship between the expression of PARP1 and the NF-κB pathway in the IRCs in RIRE, since activation of the NF-κB pathway was previously found to be responsible for triggering RIRE [4, 21, 22]. In fact, DNA-damage-induced PARP1 and TNF-α were known to be important cofactors in the activation cascade of the NF-κB complex [47], while interactions between PARP1 and both subunits of NF-κB (p50 and p65) were required for the co-activation of one another [48]. It was also established that the stress-inducible transcription factor NF-κB induced by TNF-α was required to enhance the activity of PARP1 in the presence of DNA damage [49, 50]. These results outlined a positive feedback loop of PARP1 and NF-κB in regulating DNA repair [51]. As such, the second objective of the present work was to explore the positive feedback loop between PARP1 and NF-κB in RIRE. Three experiments were performed to achieve this objective. Firstly, we compared the mRNA expression levels of NF-κB in IRCs treated with CM, having previously conditioned the bystander cells (partnered with IRCs), and in IRCs treated with CM together with the PARP1 inhibitor OLA (CM + OLA). Secondly, we compared the mRNA expression levels of PARP1 in IRCs treated with CM having previously conditioned the bystander cells (partnered with IRCs), and in IRCs treated with CM together with the NF-κB inhibitor BAY-11-7082 (CM+ BAY5). Finally, we also captured immunofluorescence images of (i) IRCs stained with anti-PARP1 antibody that had been treated with CM together with the NF-κB inhibitor (CM + BAY5), (ii) IRCs stained with anti-NF-κB p65 antibody that had been treated with CM together with the PARP1 inhibitor (CM + OLA), and compared these with the immunofluorescence images of IRCs that had only been treated with CM. If there was a positive feedback loop between PARP1 and NF-κB in RIRE, the first experiment would show that NF-κB was transcriptionally reduced in IRCs treated with the PARP1 inhibitor, the second experiment would show that PARP1 was transcriptionally reduced in IRCs treated with the NF-κB inhibitor, and the third experiment would show that (i) the fluorescent intensitiy of PARP1 in IRCs was significantly reduced when treated with CM together with the NF-κB inhibitor, and (ii) the fluorescent intensity of NF-κB in IRCs was significantly reduced when treated with CM together with the PARP1 inhibitor.
MATERIALS AND METHODS
Cell culture
In order to achieve generality of the results as far as possible, a total of four lines of cancer cells of different cancer types, including carcinoma and adenocarcinoma, were employed in the present work, namely, HeLa (cervical adenocarcinoma), MCF7 (mammary gland adenocarcinoma), CNE-2 (nasopharyngeal carcinoma) and HCT116 (colorectal carcinoma) cells. HeLa cells (ATCC* CCL-2™) and MCF7 cells (ATCC* HTB-22 ™) were purchased from American Type Culture Collection. CNE-2 cells were purchased from the nasopharyngeal carcinoma AoE Cell Line Repository. The HCT116 cells were supplied by Dr B. Vogelstein (Johns Hopkins University School of Medicine, Baltimore, MD, USA) [52]. Authentication of these cell lines was routinely performed by examining the morphology of cells under the microscope. The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco*), supplemented with 10% fetal bovine serum (FBS) (Gibco*) and 1% penicillin–streptomycin (Gibco*), incubated at 37°C in an environment containing humidified 5% CO2 and were routinely sub-cultured twice a week.
X-Ray irradiation
The X-RAD 320 irradiator (Precision X-ray Inc., North Branford, CT, USA) was employed to perform X-ray irradiation, with applied voltage and current of 200 kV and 10 mA, respectively, and a source-to-surface distance of 50 cm. The filter made of 2 mm thick aluminum was used to harden the X-ray beam, and the dose rate was set to ~ 0.9 Gy/min. For the experiments, 100 × 20 mm Petri dishes (Corning*) tailor-made with a hole (diameter = 7.5 mm) at the center and covered with 24 × 24 mm glass coverslips (Marienfeld Superior™) beneath the dishes were utilized.
A total of 5 × 104 cells were plated on an irradiation (IR) dish and recipient dishes, while 125 × 104 cells were plated on five 24 × 24 mm glass coverslips (Marienfeld Superior™), and these were incubated overnight before X-ray irradiation of cells in the IR dish. As illustrated in Fig. 1, the irradiation and partnering procedures were adopted and modified from a previous set-up [19, 21]. The irradiation experiments included three sets of dishes: (i) IR dishes, containing cells for irradiation with an X-ray dose of 2 Gy, which were partnered (i.e. co-cultured) with UIRCs; (ii) CM dishes, containing the CM that had conditioned the bystander cells previously co-cultured with IRCs; (iii) recipient dishes, containing cells first for irradiation with 2 Gy of X-ray, and then for different further treatments. For clarification, it should be noted that the IRCs in the recipient dishes were not the IRCs in the IR dish. Three different experimental sets were obtained for each irradiation experiment.
Fig. 1.
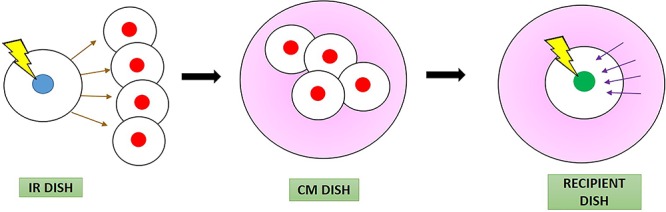
Schematic diagram showing procedures for studying the rescue effect by separating the rescue signals (represented by purple arrows) from the bystander signals (represented by brown arrows) through the preparation of the conditioned medium CM (represented in pink). The irradiated cells (blue) in IR dish sending out the bystander signals to bystander cells (red) are different from the irradiated cells (green) receiving the rescue signals. The experiments included three dishes, namely, the IR dish, CM dish and recipient dish.
Depending on the different objectives of the various experiments in the present work, different experimental set-ups were designed and are illustrated in Fig. 2. The first experimental set-up is illustrated in Fig. 2(a) and (b). Here, bystander cells were first partnered with IRCs in IR dishes for 2 h. The bystander cells (and not the CM) were then transferred into the CM dish with fresh medium, to make sure that bystander signals generated by IRCs were not transferred. After 3 h, CM from the CM dish was transferred to recipient dishes containing IRCs, and these IRCs were further incubated for 12 h. At the same time, control experiments were carried out through transferring fresh DMEM medium (FM) to recipient dishes containing IRCs instead of transferring CM from the CM dish to recipient dishes. The mRNA expression levels of various genes were quantified using real-time polymerase chain reaction (PCR), and after the cells were fixed, the protein expression levels were determined from immunofluorescent staining.
Fig. 2.
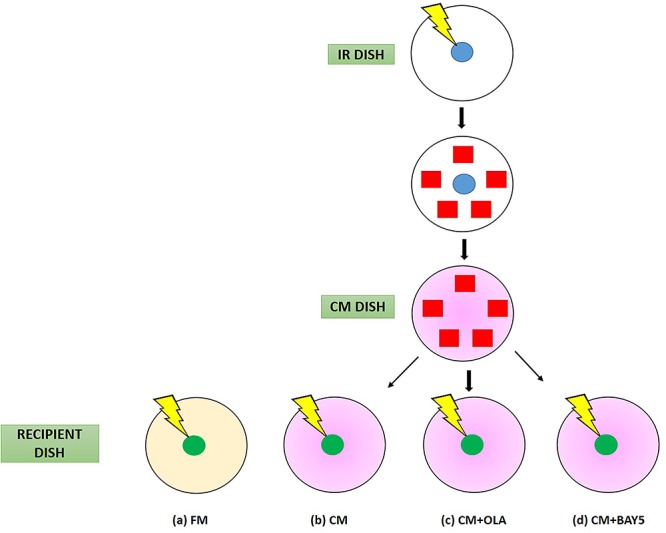
Schematic diagrams showing experimental set-up and procedures for (a) control experiments without treatment (FM; represented in light yellow), (b) experiments with transferred conditioned medium (CM; represented in pink), (c) experiments with CM plus PARP1 inhibitor (CM + OLA), and (d) experiments with CM plus NF-κB inhibitor (CM + BAY5). The cells involved are irradiated cells (blue) in the IR dish, bystander cells (red) in the IR dish and irradiated cells (green) in the recipient dish.
The second experimental set-up involved the PARP1 inhibitor Olaparib-AZD2281 (SelleckChem Inhibitors) at a concentration of 1 μM (hereafter referred to as OLA). The experiments were performed as illustrated in Fig. 2(b) and (c). IRCs were immediately treated post-irradiation with 15 mL of (1) CM and (2) CM + OLA. After the various treatments for 1 h, 15 mL of FM was used to replace the medium in the recipient dishes for 11.5 h (i.e. until 12 h post-irradiation). The mRNA expression levels were quantified using real-time PCR while the protein expression levels were determined from immunofluorescent staining after the cells were fixed.
The third experimental set-up involved the NF-κB inhibitor BAY-11-7082 (SelleckChem Inhibitors) at a concentration of 5 μM (hereafter referred to as BAY5), which followed the design in a previous study [21]. The experiments were performed as illustrated in Fig. 2(b) and (d). IRCs were immediately treated post-irradiation with 15 mL of (1) CM and (2) CM + BAY5. After the various treatments for 30 min, 15 mL of FM was used to replace the medium in the recipient dishes for 11.5 h (i.e. until 12 h post-irradiation). The mRNA expression levels were quantified using real-time PCR and protein expression levels were determined from immunofluorescent staining after the cells were fixed.
RNA isolation and cDNA synthesis
RNA was isolated by using TRIzol reagent (Invitrogen ™) and 1 μg of isolated RNA from each sample was transcribed to complementary DNA using a PrimeScript ™ RT reagent kit with gDNA Eraser (Takara Bio, USA).
Analysis and quantification of mRNA using real-time PCR
Quantitative PCR analysis in real-time was performed using SYBR* Premix Ex Taq™ (Tli RNaseH Plus) (Takara Bio, USA) according to the manufacturer’s instructions. Signals were detected with a StepOnePlus™Real-Time PCR System (Applied Biosystems, Biopolis, Singapore). One of the stable endogenous reference genes used was glyceraldehyde 3-phosphate dehydrogenase (GAPDH) which is a house-keeping gene known to express at similar levels under varying experimental conditions, or in different states of a similar tissue or cell-line [53–55]. The primer sequences for real-time PCR are shown in Table 1.
Table 1.
List of primer sequences for quantitative real-time polymerase chain reaction
Immunofluorescent staining
The cells were given a phosphate buffered saline (PBS)-wash and were fixed in 4% formaldehyde at room temperature (RT) at 12 h post-irradiation, then washed with PBS three times, permeabilized in 0.2% Triton X-100 at 37°C and finally incubated with 5% bovine serum albumin followed by immunostaining for several primary antibodies including anti-PARP1 (EPR18461) (abcam, ab191217) (1:500), anti-NF-κB p65 (phospho S536) (abcam, ab86299) (1:500), anti-53BP1 (abcam, ab21083) (1:200) and phospho-histone H2A.X (Ser139) (20E3) (Cell Signaling Technology, 9718) (1:200) at 4°C overnight. The cells were washed again with PBS three times and incubated with either secondary goat anti-rabbit IgG H&L (Alexa Fluor* 488) antibody (abcam, ab150077) (1:500) or secondary goat anti-mouse IgG H&L (Texas Red*) antibody (abcam, ab6787) (1:500) at 37°C for 1 h. A Nikon Ti-E motorized inverted fluorescence microscope (Nikon Instruments Inc., Melville, NY, USA) was used to capture fluorescent images with an objective magnification, analog gain and exposure of 40×, 6.2× and 500 ms, respectively. At least 100 cells in each sample were counted and analysed. The results for protein expression levels are quantitatively presented in terms of the fluorescent intensity, which was quantified using the NIS-Elements Advanced Research software (Nikon Instruments Inc., Melville, NY, USA). The fluorescent intensity was evaluated using automated measurement analysis by selecting a desired cell through a selection tool. On the other hand, the results from co-localization experiments for 53BP1 and γH2AX foci are quantitatively presented in terms of the co-localization factor, which was quantified using the NIS-Elements Advanced Research software (Nikon Instruments Inc., Melville, NY, USA). The co-localization factor is defined as [(fraction of co-localized cells) × (fraction of co-localized foci per cell)] × 100. For example, if 80% of the cells show co-localization, and on an average 60% of the foci in a cell show co-localization, the co-localization factor is given by (0.8 × 0.6) × 100 = 48.
Statistical analyses
Statistical analyses were performed on the means of data obtained from three individual experiments and performed in three sets (triplicates). All data are represented here as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was employed to analyse the differences among more than two groups of data. Student’s t-tests were employed to analyse the differences between two groups of data (assuming unequal variances). For all comparisons, those corresponding to P < 0.05 were considered statistically significant.
RESULTS
Involvement of PARP1 in RIRE
As per the first objective, through using the first experimental set-up, we found that IRCs treated with FM had higher amounts of 53BP1 and PARP1 proteins in comparison to CM treated IRCs, as shown in Figs 3 and 4. Figure 6 revealed co-localization factors between γH2AX and 53BP1 foci of ~ 85–90 in IRCs treated with FM, and ~ 70–74 in IRCs treated with CM. In other words, co-localized γH2AX and 53BP1 foci were found in larger amounts in IRCs treated with FM when compared to those treated with CM, as shown in Fig. 5. Furthermore, these results demonstrated that, for IRCs with similar treatment, the co-localization factor between γH2AX and 53BP1 foci, as well as the 53BP1 expression increased with PARP1 expression. The results shown in Figs 3–5 supported that PARP1 was involved in RIRE.
Fig. 3.
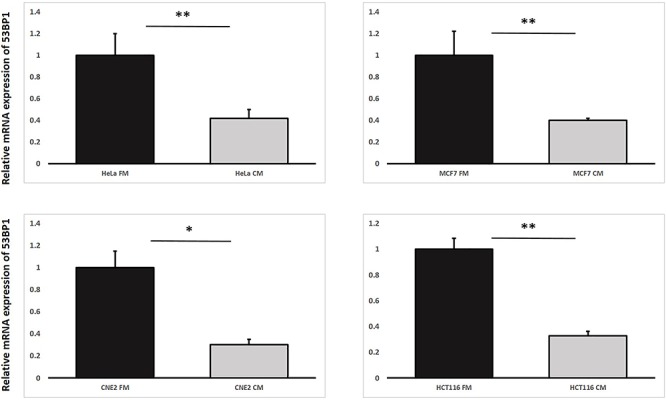
Relative mRNA expression levels of 53BP1 in irradiated HeLa, MCF7, CNE-2 and HCT116 cells treated with fresh medium (FM) or harvested conditioned medium (CM) having previously conditioned the bystander cells (partnered with IRCs) that were incubated until 12 h post-irradiation. Data were analysed using Student’s t-test: two-sample assuming unequal variances. *P < 0.05, **P < 0.01 and error bars represent mean ± SD.
Fig. 4.
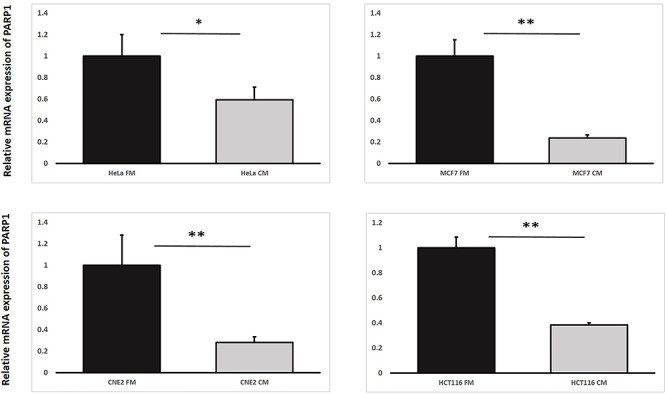
Relative mRNA expression levels of PARP1 in irradiated HeLa, MCF7, CNE-2 and HCT116 cells treated with fresh medium (FM) or harvested conditioned medium (CM) having previously conditioned the bystander cells (partnered with IRCs) that were incubated until 12 h post-irradiation. Data were analysed using Student’s t-test: two-sample assuming unequal variances. *P < 0.05, **P < 0.01 and error bars represent mean ± SD.
Fig. 6.
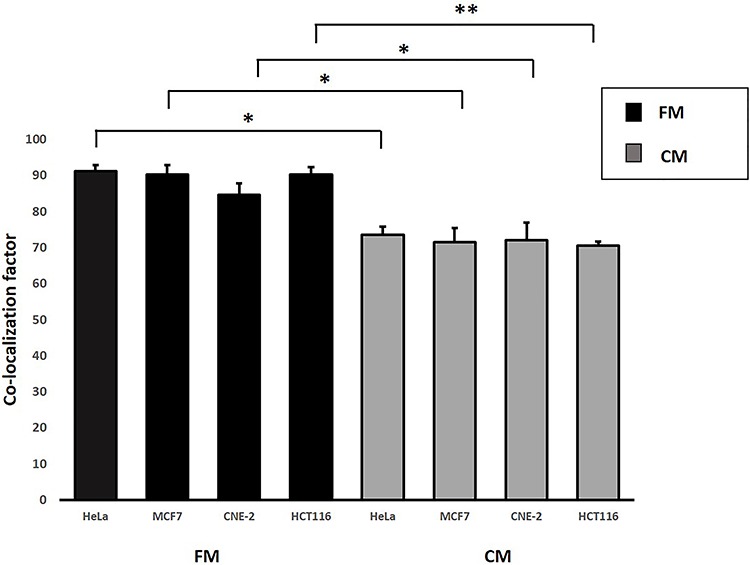
Co-localization factors of 53BP1 and γH2AX in HeLa, MCF7, CNE-2 and HCT116 cells treated with FM and CM that were incubated until 12 h post-irradiation and immunofluorescent staining was performed. Data were analysed using Student’s t-test: two-sample assuming unequal variances following one-way ANOVA. *P < 0.05, **P < 0.01 and error bars represent ± SD.
Fig. 5.
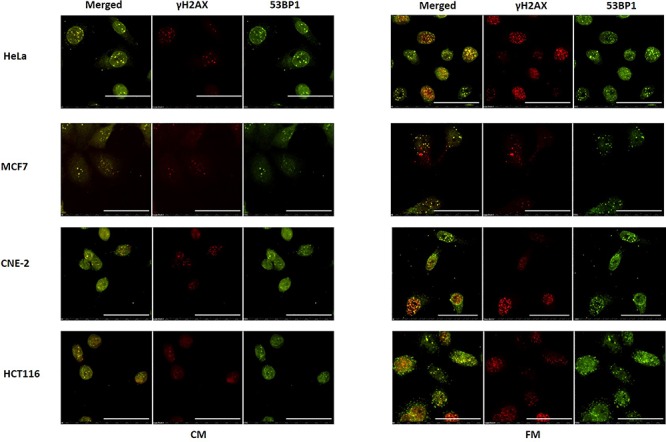
Representative images from immunofluorescence staining of co-localization between phospho-histone H2A.X and anti-53BP1 antibodies in HeLa, MCF7, CNE-2 and HCT116 cells treated with fresh medium (FM) or harvested conditioned medium (CM) having previously conditioned the bystander cells (partnered with IRCs) that were incubated until 12 h post-irradiation and immunofluorescent staining was performed. Scale bar = 50 μm.
We also captured and compared immunofluorescence images of IRCs stained with the anti-PARP1 antibody, which had been treated with (1) FM and (2) CM to further confirm the involvement of PARP1 in RIRE. Representative images are shown in Fig. 7(a). The PARP1 fluorescent intensities obtained from the captured immunofluorescence images are shown and compared in Fig. 7(b). The PARP1 fluorescent intensities were significantly different for IRCs treated with (1) FM and (2) CM, which further confirmed that PARP1 was involved in RIRE.
Fig. 7.
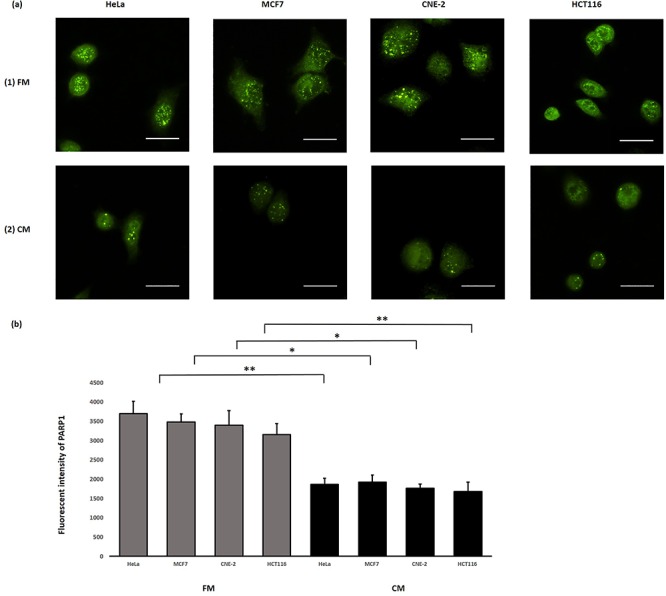
(a) Representative images from immunofluorescence staining with anti-PARP1 antibody in HeLa, MCF7, CNE-2 and HCT116 cells after X-ray irradiation. IRCs were treated with (1) FM and (2) CM, for 11.5 h (i.e. until 12 h post-irradiation) and immunofluorescent staining was performed. Scale bar = 25 μm. (b) Fluorescent intensities of PARP1 obtained after treatments with FM and CM in HeLa, MCF7, CNE-2 and HCT116 cells. Data were analysed using Student’s t-test: two-sample assuming unequal variances following one-way ANOVA. *P < 0.05, **P < 0.01 and error bars represent ± SD.
We further examined whether IRCs treated with the PARP1 inhibitor, OLA, could still exhibit RIRE. The results obtained using the second experimental set-up revealed that the IRCs treated with OLA (CM + OLA) led to a higher expression of 53BP1 as shown in Fig. 8 when compared to those IRCs treated with CM alone. These results demonstrated that in the absence of PARP1, the number of DNA breaks in the IRCs was increased and thus RIRE was attenuated, which again supported that PARP1 was involved in RIRE.
Fig. 8.
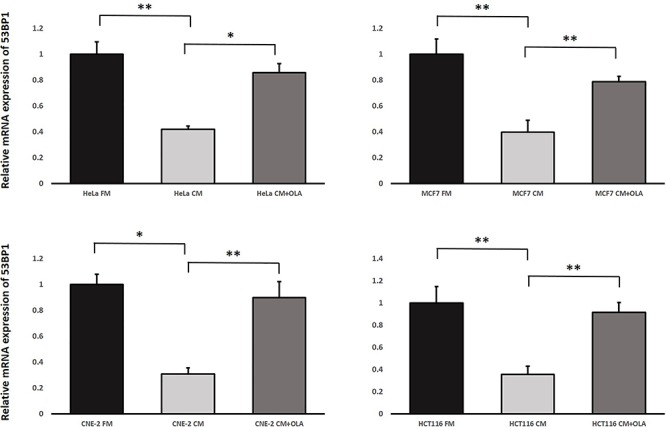
Relative mRNA expression levels of 53BP1 in irradiated HeLa, MCF7, CNE-2 and HCT116 cells treated with (1) FM, (2) CM, and (3) CM + OLA having previously conditioned the bystander cells (partnered with IRCs) for 1 h, followed by the replacement of media with FM and incubation until 12 h post-irradiation. Data were analysed using Student’s t-test: two-sample assuming unequal variances. *P < 0.05, **P < 0.01 and error bars represent mean ± SD.
Positive feedback loop between NF-κB and PARP1 in RIRE
As per the second objective, through using the second experimental set-up, our results shown in Fig. 9 revealed that the mRNA expression levels of NF-κB in IRCs treated with CM having previously conditioned the bystander cells (partnered with IRCs) together with the PARP1 inhibitor OLA (CM + OLA) were significantly lower than those in IRCs treated with CM only. In other words, NF-κB was transcriptionally reduced in IRCs treated with the PARP1 inhibitor, which supported the occurrence of the positive feedback loop between PARP1 and NF-κB in RIRE.
Fig. 9.
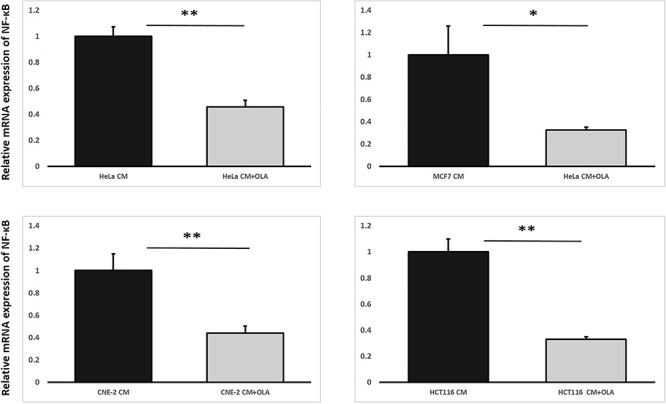
Relative mRNA expression levels of NF-κB in irradiated HeLa, MCF7, CNE-2 and HCT116 cells treated with harvested CM/CM + OLA having previously conditioned the bystander cells (partnered with IRCs) for 1 h, followed by the replacement of media with FM and incubation until 12 h post-irradiation. Data were analysed using Student’s t-test: two-sample assuming unequal variances. *P < 0.05, **P < 0.01 and error bars represent mean ± SD.
Furthermore, through using the third experimental set-up, our results shown in Fig. 10 revealed that the mRNA expression levels of PARP1 in IRCs treated with CM having previously conditioned the bystander cells (partnered with IRCs) together with the NF-κB inhibitor BAY5 (CM + BAY5) were significantly lower than those in IRCs treated with CM only. In other words, PARP1 was transcriptionally reduced in IRCs treated with the NF-κB inhibitor, which again supported the occurrence of the positive feedback loop between PARP1 and NF-κB in RIRE.
Fig. 10.
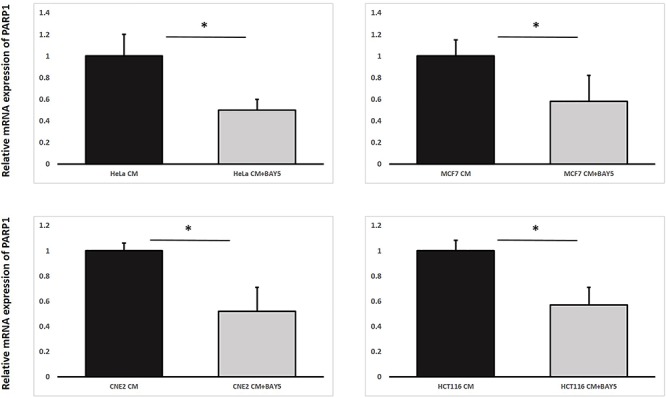
Relative mRNA expression levels of PARP1 in HeLa, MCF7, CNE-2 and HCT116 irradiated cells treated with harvested CM/CM + BAY5 having previously conditioned the bystander cells (partnered with IRCs) for 30 min, followed by the replacement of media with FM and incubation until 12 h post-irradiation. Data were analysed using Student’s t-test: two-sample assuming unequal variances. *P< 0.05 and error bars represent mean ± SD.
Finally, through using the second or third experimental set-up, we also captured immunofluorescence images of IRCs stained with (1) anti-PARP1 antibody that had been treated with CM together with the NF-κB inhibitor (CM + BAY5), and (2) anti-NF-κB p65 antibody that had been treated with CM together with the PARP1 inhibitor (CM + OLA). Representative images are shown in Figs 11 and 12. The PARP1 and NF-κB fluorescent intensities obtained from the captured immunofluorescence images are shown and compared in Fig. 13. The PARP1 fluorescent intensities were significantly lower for IRCs treated with CM + BAY5 and the NF-κB fluorescent intensities were significantly lower for IRCs treated with CM + OLA, when compared to those for IRCs treated with CM alone. In other words, PARP1 was translationally reduced in IRCs treated with the NF-κB inhibitor, and NF-κB was translationally reduced in IRCs treated with the PARP1 inhibitor, which further supported the occurrence of the positive feedback loop between PARP1 and NF-κB in RIRE.
Fig. 11.
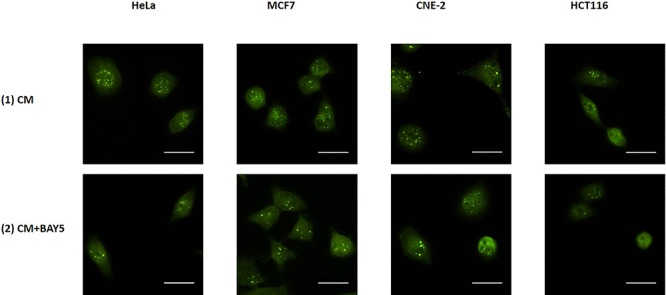
Representative images from immunofluorescence staining with anti-PARP1 antibody in HeLa, MCF7, CNE-2 and HCT116 cells after X-ray irradiation. IRCs were treated for 30 min with 15 mL of (1) CM and (2) CM + BAY5 post-irradiation. After the various treatments for 30 min, 15 mL of FM was used to replace the medium in the recipient dishes for 11.5 h (i.e. until 12 h post-irradiation) and immunofluorescent staining was performed. Scale bar = 25 μm.
Fig. 12.
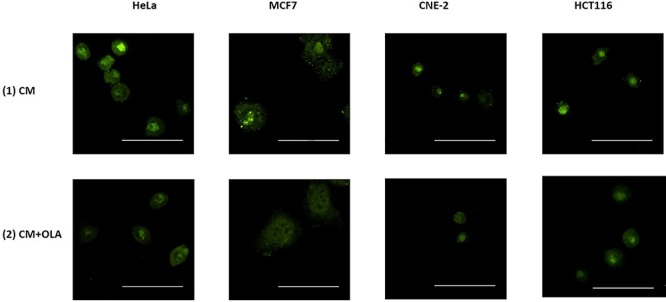
Representative images from immunofluorescence staining with anti-NF-κB p65 antibody in HeLa, MCF7, CNE-2 and HCT116 cells after X-ray irradiation. IRCs were treated for 1 h with 15 mL of (1) CM and (2) CM + OLA post-irradiation. After the various treatments for 1 h, 15 mL of FM was used to replace the medium in the recipient dishes for 11.5 h (i.e. until 12 h post-irradiation) and immunofluorescent staining was performed. Scale bar = 50 μm.
Fig. 13.
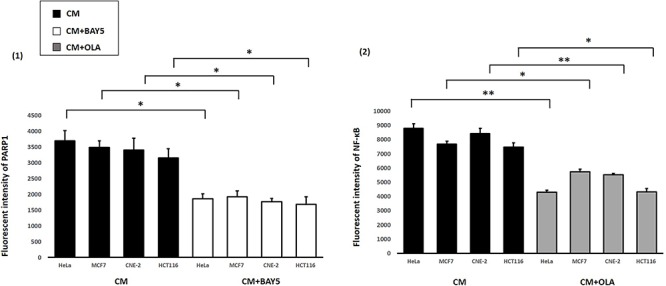
Fluorescent intensities of PARP1 and NF-κB obtained after treatments with (1) CM and CM + BAY5 and (2) CM and CM + OLA, in HeLa, MCF7, CNE-2 and HCT116 cells. Data were analysed using Student’s t-test: two-sample assuming unequal variances following one-way ANOVA. *P < 0.05, **P < 0.01 and error bars represent mean ± SD.
DISCUSSION
It was established that production of inflammatory cytokines in downstream signaling pathways was involved in RIBE [56]. For example, TNF-α, a pro-inflammatory cytokine, was found to be involved in bystander signaling mediated by high-dose ionizing radiation by regulating pro-survival signaling [27]. TNF-α, when released through autocrine or paracrine signaling, would be bound to its receptor, tumor necrosis factor receptor (TNFR), due to radiation-induced oxidative stress in cancer tumor cells [57]. The TNF-α–TNFR complex recruited several intracellular proteins by promoting the phosphorylation of inhibitor of nuclear factor kappa B (IκBα) and NF-κB activation [58]. Thus, the TNF-α–TNFR complex phosphorylated various downstream target proteins to enhance cell survival that led to co-activation of NF-κB-dependent gene expression [59]. In previous research, participation of the NF-κB response pathway in RIRE was revealed, presumably involving several anti-apoptotic proteins to promote cell survival [21]. However, TNF-α was also constitutively known to activate DNA repair enzymes like PARP1 [47]. During ssDNA breaks, PARP1 was bound to the damaged DNA and was found to catalyze PARylation [60, 61]. It was also known to be involved in ssDNA and dsDNA break repair pathways [62–64]. During X-ray irradiation, generation of DNA strand breaks increased the activity of this DNA repair enzyme [65] by promoting PARylation [39]. These results prompted us to examine the role of PARP1 regulation in RIRE.
The first objective of the present work was to examine the involvement of PARP1 in RIRE. We found, as shown in Fig. 3, that the relative mRNA expression levels of 53BP1 in IRCs treated with CM containing rescue signals were reduced by 70–75% when compared with those in IRCs not treated with CM, which revealed an increased accumulation of checkpoint proteins in response to DNA damage [66]. An increase in 53BP1, a mediator of the DNA damage checkpoint, increased the phosphorylation of several downstream checkpoint effector proteins that further recruited DNA repair proteins at the sites of DNA lesions [66]. The co-localization of 53BP1 and γH2AX foci upon irradiation of cells as illustrated in Fig. 5 was commensurate with similar findings at the sites of DNA lesions [67, 68]. In particular, phosphorylation of H2AX at Ser-140 due to DNA damage enhanced the interaction between γH2AX and 53BP1, which led to an increased accumulation of 53BP1 foci at the sites of dsDNA breaks in IRCs with or without treatment with CM [40]. In addition, the co-localization factors, which represented the degrees of co-localization [45], in IRCs treated or not treated with CM containing rescue signals were compared. Results shown in Fig. 6 indicate larger co-localization factors in IRCs not treated with CM, implying increased accumulation of DNA damage under this condition [40, 44]. This observation confirmed the presence of cellular-radiation-induced rescue effects in the cell lines.
We also found, see Fig. 4, that the relative mRNA expression levels of PARP1 in IRCs treated with CM were reduced when compared with those in IRCs not treated with CM. Since 53BP1 and γH2AX localized at DNA damage sites post-irradiation [40, 69], the localization of these proteins could act as a surrogate for the manifestation of RIRE, so our results illustrated that PARP1 was involved in RIRE. In addition, upregulation of PARP1 activity was also observed in cells [70, 71]. We further captured and compared immunofluorescence images of IRCs stained with the anti-PARP1 antibody, and the results, shown in Fig. 7, revealed significant reduction in the PARP1 fluorescent intensities of IRCs treated with CM, which further confirmed the involvement of protein expression levels of PARP1 in RIRE. In addition, our results shown in Fig. 8 demonstrated that IRCs treated with the PARP1 inhibitor, OLA, led to a higher expression of 53BP1 when compared to those IRCs treated with CM alone. OLA was known to selectively bind to and inhibit the PARP1-mediated repair of ssDNA breaks [72]. In fact, a related study found that the expression of 53BP1 was increased due to accumulation of DNA breaks in the absence of PARP1 [73]. PARP1 inhibition was also known to cause genomic instability and defective homologous repair that might lead to accumulation of unrepaired ssDNA and dsDNA breaks [74]. Our results here illustrated that in the absence of PARP1, the number of DNA breaks in the IRCs was increased and thus RIRE was attenuated, which again supported that PARP1 was involved in RIRE.
Upon confirmation of involvement of PARP1 in RIRE, it was pertinent to study the relationship between the expression of PARP1 and the NF-κB pathway in the IRCs in RIRE, since activation of the NF-κB pathway was previously found to be responsible for triggering RIRE [4, 21, 22]. The second objective of the present work was to explore the positive feedback loop between PARP1 and NF-κB in RIRE. Our results shown in Fig. 9 revealed that the mRNA expression levels of NF-κB in IRCs treated with CM having previously conditioned the bystander cells (partnered with IRCs) together with the PARP1 inhibitor OLA were significantly lower than those in IRCs treated with CM only. Similarly, the results shown in Fig. 10 revealed that the mRNA expression levels of PARP1 in IRCs treated with CM having previously conditioned the bystander cells (partnered with IRCs) together with the NF-κB inhibitor BAY5 were significantly lower than those in IRCs treated with CM only. In other words, NF-κB was transcriptionally reduced in IRCs treated with the PARP1 inhibitor, while PARP1 was transcriptionally reduced in IRCs treated with the NF-κB inhibitor. Furthermore, the PARP1 fluorescent intensities obtained from the captured immunofluorescence images shown in Figs 11 and 13 were significantly lower for IRCs treated with the NF-κB inhibitor BAY5. In addition, the NF-κB fluorescent intensities obtained from the captured immunofluorescence images shown in Figs 12 and 13 were significantly lower for IRCs treated with the PARP1 inhibitor OLA. In other words, PARP1 was translationally reduced in IRCs treated with the NF-κB inhibitor and NF-κB was translationally reduced in IRCs treated with the PARP1 inhibitor. All these results strongly supported the occurrence of a positive feedback loop between PARP1 and NF-κB in RIRE. PARP-1 proteins are known to form branched PAR polymers with various other proteins involved in DNA repair and cell survival [75, 76], while many nuclear proteins are known to express binding regions for these PAR polymers [77, 78]. Such protein–protein interactions that resulted from PAR formation and PAR binding modulated the activation of various transcription factors, like NF-κB [79–81]. In addition, it was observed that NF-κB regulated the activity of PARP1 in the presence of DNA damage [49, 50], while PARP1 mediated NF-κB activation [47]. It was also found that PARP1 could not help repair DNA damage in the absence of NF-κB [49, 50], while PARP1 was downregulated in the absence of RelA (p65 subunit) of NF-κB in the presence of DNA damage [51].
With the confirmation of the involvement of PARP1 as well as the presence of a positive feedback loop between PARP1 and NF-κB in RIRE, the TNF-α-induced PARP1–NF-κB positive feedback loop pathway for RIRE could be constructed as shown Fig. 14. The inhibition of NF-κB and PARP1 by the inhibitors BAY5 and OLA, respectively, are also depicted. RIRE has evoked extensive interest in understanding cancer radiotherapeutics in the past years due to its implication in oncogenesis. The capability of UIRCs in rescuing IRCs raised a concern on the efficacy of radiation therapy. The results in the present paper provide insights into potential exploitation of inhibition of PARP1 and/or the PARP1–NF-κB positive feedback loop in designing an adjunct to cancer radiotherapeutics. Interestingly, a combination of ionizing radiation and inhibition of PARP1 was proposed as a strategy for anti-cancer treatment [82]. Here, inhibition of PARP1 was aimed at causing genomic instability and defective homologous repair that resulted in cell death due to the accumulation of unrepaired ssDNA and dsDNA breaks [74]. The capability of PARP1 inhibition in preventing the rescue of irradiated cancer cells by the unirradiated healthy or cancer cells could provide another advantage of such a strategy.
Fig. 14.
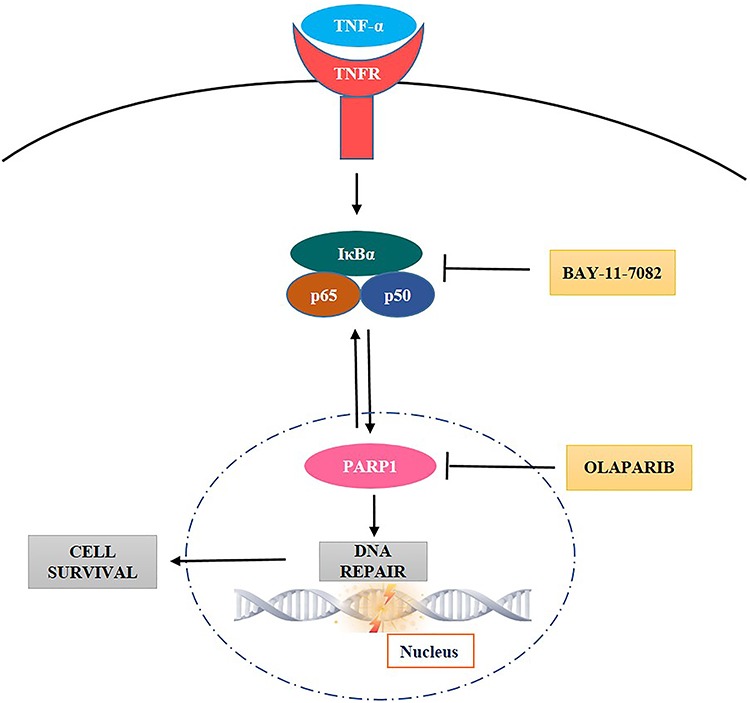
Schematic diagram representing the TNF-α-induced PARP1–NF-κB positive feedback loop pathway which promotes DNA repair and cell survival. Activation of NF-κB and PARP1 are inhibited by the inhibitors BAY-11-7082 and olaparib, respectively.
ACKNOWLEDGMENT
The present project was supported by the project IRF/0024 from the State Key Laboratory in Marine Pollution of the City University of Hong Kong. Funding for covering the cost to publish this article in open access was provided by the State Key Laboratory in Marine Pollution, City University of Hong Kong.
CONFLICT OF INTEREST
There are no conflicts of interest.
REFERENCES
- 1. Chen S, Zhao Y, Han W et al. Rescue effects in radiobiology: Unirradiated bystander cells assist irradiated cells through intercellular signal feedback. Mutat Res 2011;706:59–64. [DOI] [PubMed] [Google Scholar]
- 2. Lam RK, Fung YK, Han W. Rescue effects: Irradiated cells helped by unirradiated bystander cells. Int J Mol Sci 2015;16:2591–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu KN. Radiation-induced rescue effect. J Radiat Res 2019;60:163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kong EY, Cheng SH, Yu KN. Induction of autophagy and interleukin 6 secretion in bystander cells: Metabolic cooperation for radiation-induced rescue effect? J Radiat Res 2018;59:129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mukherjee S, Chakraborty A. Radiation-induced bystander phenomenon: Insight and implications in radiotherapy. Int J Radiat Biol 2019;95:243–63. [DOI] [PubMed] [Google Scholar]
- 6. Widel M, Przybyszewski WM, Pobuda AC et al. Bystander normal human fibroblasts reduce damage response in radiation targeted cancer cells through intercellular ROS level modulation. Mutat Res 2012;731:117–24. [DOI] [PubMed] [Google Scholar]
- 7. Pereira S, Malard V, Ravanat JL et al. Low doses of gamma-irradiation induce an early bystander effect in zebrafish cells which is sufficient to radioprotect cells. PLoS One 2014;9:e92974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desai S, Kobayashi A, Konishi T et al. Damaging and protective bystander cross-talk between human lung cancer and normal cells after proton microbeam irradiation. Mutat Res 2014;763:39–44. [DOI] [PubMed] [Google Scholar]
- 9. Liu Y, Kobayashi A, Fu Q et al. Rescue of targeted nonstem-like cells from bystander stem-like cells in human fibrosarcoma HT1080. Radiat Res 2015;184:334–40. [DOI] [PubMed] [Google Scholar]
- 10. Kobayashi A, Ahmad TAFT, Autsavapromporn N et al. Enhanced DNA double-strand break repair of microbeam targeted A549 lung carcinoma cells by adjacent WI38 normal lung fibroblast cells via bi-directional signaling. Mutat Res 2017;803:1–8. [DOI] [PubMed] [Google Scholar]
- 11. He M, Dong C, Xie Y et al. Reciprocal bystander effect between α-irradiated macrophage and hepatocyte is mediated by cAMP through a membrane signaling pathway. Mutat Res 2014;763:1–9. [DOI] [PubMed] [Google Scholar]
- 12. Matsumoto H, Hayashi S, Hatashita M et al. Induction of radioresistance to accelerated carbon-ion beams in recipient cells by nitric oxide excreted from irradiated donor cells of human glioblastoma. Int J Radiat Biol 2000;76:1649–57. [DOI] [PubMed] [Google Scholar]
- 13. Matsumoto H, Hayashi S, Hatashita M et al. Induction of radioresistance by a nitric oxide–mediated bystander effect. Radiat Res 2001;155:387–96. [DOI] [PubMed] [Google Scholar]
- 14. Shao C, Furusawa Y, Aoki M et al. Nitric oxide–mediated bystander effect induced by heavy-ions in human salivary gland tumour cells. Int J Radiat Biol 2002;78:837–44. [DOI] [PubMed] [Google Scholar]
- 15. Matsumoto H, Ohnishi T. Contribution of radiation-induced, nitric oxide-mediated bystander effect to radiation-induced adaptive response. Biol Sci Space 2004;18:108–9. [PubMed] [Google Scholar]
- 16. Matsumoto H, Hamada N, Takahashi A et al. Vanguards of paradigm shift in radiation biology: Radiation-induced adaptive and bystander responses. J Radiat Res 2007;48:97–106. [DOI] [PubMed] [Google Scholar]
- 17. Matsumoto H, Tomita M, Otsuka K et al. A new paradigm in radioadaptive response developing from microbeam research. J Radiat Res 2009;50:67–79. [DOI] [PubMed] [Google Scholar]
- 18. Matsumoto H, Tomita M, Otsuka K et al. Nitric oxide is a key molecule serving as a bridge between radiation-induced bystander and adaptive responses. Curr Mol Pharmacol 2011;4:126–34. [DOI] [PubMed] [Google Scholar]
- 19. Tomita M, Maeda M, Kobayashi K et al. Dose response of soft X-ray–induced bystander cell killing affected by p53 status. Radiat Res 2013;179:200–7. [DOI] [PubMed] [Google Scholar]
- 20. Maeda M, Kobayashi K, Matsumoto H et al. X-ray-induced bystander responses reduce spontaneous mutations in V79 cells. J Radiat Res 2013;54:1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lam RKK, Han W, Yu KN. Unirradiated cells rescue cells exposed to ionizing radiation: Activation of NF-κB pathway in irradiated cells. Mutat Res 2015;782:23–33. [DOI] [PubMed] [Google Scholar]
- 22. Lam RK, Fung YK, Han W et al. Modulation of NF-κB in rescued irradiated cells. Radiat Prot Dosimetry 2015;167:37–43. [DOI] [PubMed] [Google Scholar]
- 23. Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003;22:5792–812. [DOI] [PubMed] [Google Scholar]
- 24. Wang L, Reinach P, Lu L. TNF-alpha promotes cell survival through stimulation of K+ channel and NFkappaB activity in corneal epithelial cells. Exp Cell Res 2005;311:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr 2010;2:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pekalski J, Zuk PJ, Kochanczyk M et al. Spontaneous NF-κB activation by autocrine TNFα signaling: A computational analysis. PLoS One 2013;8:e78887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shareef MM, Cui N, Burikhanov R et al. Role of tumor necrosis factor-alpha and TRAIL in high-dose radiation-induced bystander signaling in lung adenocarcinoma. Cancer Res 2007;67:11811–20. [DOI] [PubMed] [Google Scholar]
- 28. Meloche J, Pflieger A, Vaillancourt M et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014;129:786–97. [DOI] [PubMed] [Google Scholar]
- 29. Burkle A, Diefenbach J, Brabeck C et al. Ageing and PARP. Pharmacol Res 2005;52:93–9. [DOI] [PubMed] [Google Scholar]
- 30. Woodhouse BC, Dianov GL. Poly ADP-ribose polymerase-1: An international molecule of mystery. DNA Repair 2008;7:1077–86. [DOI] [PubMed] [Google Scholar]
- 31. Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer 2011;105:1114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Andrabi SA, Umanah GK, Chang C et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A 2014;111:10209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Isabelle M, Moreel X, Gagne JP et al. Investigation of PARP-1, PARP-2, and PARG interactomes by affinity-purification mass spectrometry. Proteome Sci 2010;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Langelier MF, Planck JL, Roy S et al. Structural basis for DNA damage-dependent poly(ADP-ribosyl) ation by human PARP-1. Science 2012;336:728–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ali AAE, Timinszky G, Bosacoma AR et al. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat Struct Mol Biol 2012;19:685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirai T, Shirai H, Fujimori H et al. Radiosensitization effect of poly(ADP-ribose) polymerase inhibition in cells exposed to low and high liner energy transfer radiation. Cancer sci 2012;103:1045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ronson GE, Piberger AL, Higgs MR et al. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat Commun 2018;9:746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Villani P, Fresegna AM, Ranaldi R et al. X-ray induced DNA damage and repair in germ cells of PARP1(−/−) male mice. Int J Mol Sci 2013;14:18078–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wei H, Yu X. Functions of PARylation in DNA damage repair pathways. Genom Proteom Bioinf 2016;14:131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ward IM, Minn K, Jorda KG et al. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem 2003;278:19579–82. [DOI] [PubMed] [Google Scholar]
- 41. Noda A. Radiation-induced unrepairable DSBs: Their role in the late effects of radiation and possible applications to biodosimetry. J Radiat Res 2018;59(suppl_2):ii114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kurashige T, Shimamura M, Nagayama Y. Differences in quantification of DNA double-strand breaks assessed by 53BP1/γH2AX focus formation assays and the comet assay in mammalian cells treated with irradiation and N-acetyl-L-cysteine. J Radiat Res 2016;57:312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nikolova T, Dvorak M, Jung F et al. The γH2AX assay for genotoxic and nongenotoxic agents: Comparison of H2AX phosphorylation with cell death response. Toxicol Sci 2014;140:103–17. [DOI] [PubMed] [Google Scholar]
- 44. Sengupta S, Robles AI, Linke SP et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol 2004;166:801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakamura AJ, Rao VA, Pommier Y et al. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle 2010;9:389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Croco E, Marchionni S, Bocchini M et al. DNA damage detection by 53BP1: Relationship to species longevity. J Gerontol A Biol Sci Med Sci 2017;72:763–70. [DOI] [PubMed] [Google Scholar]
- 47. Vuong B, Cann ADH, Alano CC et al. NF-κB transcriptional activation by TNFα requires phospholipase C, extracellular signal-regulated kinase 2 and poly(ADP-ribose) polymerase-1. J Neuroinflammation 2015;12:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hassa PO, Covic M, Hasan S et al. The enzymatic and DNA binding activity of PARP-1 are not required for NF-κB coactivator function. J Biol Chem 2001;276:45588–97. [DOI] [PubMed] [Google Scholar]
- 49. Kraus W, Lis J. PARP goes transcription. Cell 2003;113:677–83. [DOI] [PubMed] [Google Scholar]
- 50. Oliva MD, Quesada AR, O’valle F et al. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res 2006;66:5744–56. [DOI] [PubMed] [Google Scholar]
- 51. Li D, Luo Y, Chen X et al. NF-κB and poly (ADP-ribose) polymerase 1 form a positive feedback loop that regulates DNA repair in acute myeloid Leukemia cells. Mol Cancer Res 2019;17:761–72. [DOI] [PubMed] [Google Scholar]
- 52. Bunz F, Dutriaux A, Lengauer C et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998;282:1497–501. [DOI] [PubMed] [Google Scholar]
- 53. Kozera B, Rapacz M. Reference genes in real-time PCR. J Appl Genet 2013;54:391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thellin O, Zorzi W, Lakaye B et al. Housekeeping genes as internal standards: Use and limits. J Biotechnol 1999;75:291–5. [DOI] [PubMed] [Google Scholar]
- 55. Vandesompele J, Preter DK, Pattyn F et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ivanov VN, Zhou H, Ghandhi SA et al. Radiation-induced bystander signaling pathways in human fibroblasts: A role for interleukin-33 in the signal transmission. Cell Signal 2010;22:1076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu H. Typical cell signaling response to ionizing radiation: DNA damage and extranuclear damage. Chin J Cancer Res 2012;24:83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bernitsas DGJ, Ichikawa H, Takada Y et al. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene 2007;26:1385–97. [DOI] [PubMed] [Google Scholar]
- 59. Hayden MS, Ghosh S. Regulation of NF-κB by TNF family cytokines. Semin Immunol 2014;26:253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chaudhuri AR, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 2017;18:610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leppard JB, Dong Z, Mackey ZB et al. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-ribose) polymerase 1 in DNA single-strand break repair. Mol Cell Biol 2003;23:5919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eustermann S, Videler H, Yang JC et al. The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J Mol Biol 2011;407:149–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Beck C, Robert I, Martin RSB et al. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp Cell Res 2014;329:18–25. [DOI] [PubMed] [Google Scholar]
- 64. Chen Y, Zhang H, Xu Z. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res 2019;47:8563–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Villanueva MM, Kramer A, Hammes T et al. Influence of acute exercise on DNA repair and PARP activity before and after irradiation in lymphocytes from trained and untrained individuals. Int J Mol Sci 2019;20:2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang B, Matsuoka S, Carpenter PB et al. 53BP1, a mediator of the DNA damage checkpoint. Science 2002;298:1435–8. [DOI] [PubMed] [Google Scholar]
- 67. Rappold I, Iwabuchi K, Date T et al. Tumor suppressor P53 binding protein 1 (53bp1) is involved in DNA damage–Signaling pathways. J Cell Biol 2003;153:613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schultz LB, Chehab NH, Malikzay A et al. P53 binding protein 1 (53bp1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 2000;151:1381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pryde F, Khalili S, Robertson K et al. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J Cell Sci 2005;118:2043–55. [DOI] [PubMed] [Google Scholar]
- 70. Ko HL, Ren EC. Functional aspects of PARP1 in DNA repair and transcription. Biomolecules 2012;2:524–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Caron MC, Sharma AK, Sullivan OJ et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat Commun 2019;10:2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. National Center for Biotechnology Information PubChem Database. Olaparib, CID=23725625, https://pubchem.ncbi.nlm.nih.gov/compound/Olaparib (accessed on Oct. 4, 2019).
- 73. Harvey A, Mielke N, Grimstead JW et al. PARP1 is required for preserving telomeric integrity but is dispensable for A-NHEJ. Oncotarget 2018;9:34821–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dziadkowiec KN, Gąsiorowska E, Markwitz NE et al. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny 2016;15:215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rouleau M, Aubin RA, Poirier GG. Poly(ADP-ribosyl)ated chromatin domains: Access granted. J Cell Sci 2004;117:815–25. [DOI] [PubMed] [Google Scholar]
- 76. Gagne JP, Isabelle M, Lo KS et al. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res 2008;36:6959–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Krishnakumar R, Kraus WL. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol Cell 2010;39:8–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pleschke JM, Kleczkowska HE, Strohm M et al. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem 2000;275:40974–80. [DOI] [PubMed] [Google Scholar]
- 79. Ullrich O, Diestel A, Eyupoglu IY et al. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol 2001;3:1035–42. [DOI] [PubMed] [Google Scholar]
- 80. Nakajima H, Nagaso H, Kakui N et al. Critical role of the automodification of poly(ADP-ribose) polymerase-1 in nuclear factor-kappaB-dependent gene expression in primary cultured mouse glial cells. J Biol Chem 2004;279:42774–86. [DOI] [PubMed] [Google Scholar]
- 81. Chang WJ, Gonzalez RA. The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J Biol Chem 2001;276:47664–70. [DOI] [PubMed] [Google Scholar]
- 82. Cesaire M, Thariat J, Candeias SM et al. Combining PARP inhibition, radiation, and immunotherapy: A possible strategy to improve the treatment of cancer? Int J Mol Sci 2018;19:3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Guillot C, Favaudon V, Herceg Z et al. PARP inhibition and the radiosensitizing effects of the PARP inhibitor ABT-888 in in vitro hepatocellular carcinoma models. BMC Cancer 2014;14:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Huang X, Pan Y, Cao D et al. UVA-induced upregulation of progerin suppresses 53BP1-mediated NHEJ DSB repair in human keratinocytes via progerin-Lamin a complex formation. Oncol Rep 2017;37:3617–24. [DOI] [PubMed] [Google Scholar]
- 85. Shiimura Y, Ohgusu H, Sato T et al. Regulation of the human ghrelin promoter activity by transcription factors, NF-κB and Nkx2.2. IntJ Endocrinol 2015;2015:580908. [DOI] [PMC free article] [PubMed] [Google Scholar]