

Abstract
Drugs produce their therapeutic effects by modulating specific targets, and there are 89 innovative targets of first-in-class drugs approved in 2004–17, each with information about drug clinical trial dated back to 1984. Analysis of the clinical trial timelines of these targets may reveal the trial-speed differentiating features for facilitating target assessment. Here we present a comprehensive analysis of all these 89 targets, following the earlier studies for prospective prediction of clinical success of the targets of clinical trial drugs. Our analysis confirmed the literature-reported common druggability characteristics for clinical success of these innovative targets, exposed trial-speed differentiating features associated to the on-target and off-target collateral effects in humans and further revealed a simple rule for identifying the speedy human targets through clinical trials (from the earliest phase I to the 1st drug approval within 8 years). This simple rule correctly identified 75.0% of the 28 speedy human targets and only unexpectedly misclassified 13.2% of 53 non-speedy human targets. Certain extraordinary circumstances were also discovered to likely contribute to the misclassification of some human targets by this simple rule. Investigation and knowledge of trial-speed differentiating features enable prioritized drug discovery and development.
Keywords: innovative targets, first-in-class drugs, clinical trial, druggability, clinical trial speed differentiating feature
Introduction
Intensive drug discovery effort [1–6] has led to the clinical trials of hundreds of new targets [7]. During 2004–17, 89 innovative targets (without approved drug before 2004) have become successful with a first-in-class drug approved during the period. These targets are the crown jewels of the recent decades’ scientific advances (e.g. disease mechanism and OMICS [8–12]), technological innovations (e.g. high-throughput screening [13], combinatorial chemistry [14], fragment-based design [15]), and new therapeutic approaches (e.g. monoclonal antibody [16], biomarker-guided therapeutics [17]). While these targets have successfully passed the rigorous clinical trial, some targets have advanced to drug approval more speedily than others. Following earlier study for prospective prediction of clinical success of the targets of clinical trial drugs [18], features of these targets with respect to their clinical timelines were analyzed for revealing their speed-differentiating characteristics.
The therapeutic potential of targets has been assessed by several such druggability characteristics as disease roles [1], drug-binding site features [3, 19–25], drug-binding domain relationships to pre-existing targets [1, 3, 26] and human system features for on-target and off-target collateral effects [3, 26–32]. Some drugs have benefited from the orphan drug act [33], biomarker-guided patient stratification [17] and selective targeting of disease-specific mutation [34]. Moreover, ~25% of the clinical trials have been significantly delayed, and ~10% of the trials were terminated [35, 36] due to recruitment shortfall [35], financial consideration [36] and trial design (e.g. the superiority or inferiority trials of anti-infectious drug are more difficult to evaluate than those randomized double-blind placebo-controlled trials [37]). These profiles were analyzed to reveal trial-speed differentiating features.
Materials and methods
First-in-class drugs approved in 2004–17 and their innovative targets
The drugs approved by the US Food and Drug Administration (FDA) during the period of 2004–17 were collected from the February issues of Nature Reviews Drug Discovery, and the approval year of each drug was further verified against the records in Drugs@FDA database [38], developers’ public releases and literatures. The first-in-class drugs were selected based on the descriptions in the FDA annual Novel Drug Summary (2011–17) and the literatures [39], wherein the drugs were explicitly described as the first-in-class or of the new therapeutic mechanism. For each drug with single reported target in the searched source, that target was tentatively recorded as its efficacy target. For each drug without target information or with ≥2 targets, its efficacy target(s) was (were) searched by literatures on the basis that it (they) is (are) linked to the clinically tested therapeutics using the established criterion of efficacy targets (Supplementary Table S1). For multi-target drugs, the primary efficacy targets were selected based on their essential roles in the targeted disease(s) or the availability of other drugs in the advanced development stages against the same target and the same disease(s).
Drugs of the innovative targets in clinical trials during 1981–2017
All clinical trial drugs during 1981–2017 were collected from such resources (Supplementary Table S2) as the PhRMA medicines in development reports, the drug pipeline reports, annual reports and announcements of 371 companies and 18 research organizations, therapeutic target database (TTD) [7], IUPHAR/BPS Guide to Pharmacology [40] and the reputable commercial databases (MDL® Drug Data Report 2004, CenterWatch Drugs in Clinical Trials 2007, Thomson Reuters Pharma™ 2010 and 2012, Springer AdisInsight 2015). Their annual trial status in the period of 1981–2017 was further searched or verified against the records in the NIH ClinicalTrials.gov [38], developer’s public releases and literatures. Synonyms reported in these sources were recorded and the duplicates were further removed. For each drug with ≥2 clinical trials within the same year, the highest trial phase was recorded for that year. These drugs were compared with the approved drugs in the official website of Drugs@FDA, popular databases (TTD, Drugbank and ChEMBL) and literatures to remove those approved before 2004. The targets of these drugs were searched and compared to the innovative targets of the first-in-class drugs approved during 2004–17 to select drug targeting one or more of these innovative targets. These drugs were subsequently used to determine the trial phase or approval status of each innovative target in every year in 1981–2017. For targets with ≥2 drugs, the highest phase or approval status was recorded for that year.
Druggability characteristics of the innovative targets
The disease role of each target was searched from the literatures by keyword combinations between target names and `disease’, `therapeutics’, `mechanism’ or `function’. The drug-binding site features were searched from the literatures by keyword combinations between target names and `binding site’ or `binding’. The relationship between the drug-binding domain of each target to that of pre-existing targets was determined as follows: (1) sequence similarity between the drug-binding domain of the target and the pre-existing targets was computed using BLAST [41] with E-value of ≤0.001 and 0.001–0.05 for high and fair similarity, respectively; (2) the affiliation of each target to drug-binding domain family of pre-existing drugs (drug approved before year 2004) was assigned by condition that the target is in a Pfam family [42] that contains ≥1 pre-existing target (without an approved drugbefore 2004).
The human systems features for the on-target collateral effects were determined as following: (1) the human protein-network of 8698 proteins and 45 932 protein-pairs was constructed based on the data of the STRING Database [43] with a confidence score of ≥0.95 [43–46], which was used for computing human biological network descriptors for each target based on Cytoscape [47]. For the protein-pairs with one protein paired to multiple subtypes of another protein, only one subtype was counted, and the computed 224 popular descriptors include the degree, betweenness centrality, clustering coefficient, average shortest path length, topological coefficient, neighborhood connectivity, closeness centrality, eccentricity, radiality, stress and so on [48]; (2) the number of the human pathways modulated by a target is the sum of target-affiliated and target immediate-downstream pathways in KEGG [49]; (3) the number of human tissues in which each target distributes is determined by tissue-distribution data from TissueDistributionDBs [50] (level-4), Uniprot [51] and literature search. A target was assumed to be distributed in a tissue if ≥8% of the total protein contents [3, 26, 50] are distributed in that tissue or the target concentration is higher than the average concentration of proteins in that tissue. The human system feature for probing off-target collateral effects is the number of human similarity proteins of a studied target outside the target families [3, 26], which was determined by the BLAST sequence similarity screening of the human proteins in Uniprot database [51] with a cutoff E-value of <0.005.
Target-relevant features and patient clinical profiles of the innovative targets
Besides the 227 human protein network and system features described in the above section, four target-relevant features (disease-specific target mutation [34], covalent drug binding [52], biomarkers [17] and orphan drug status [33]) were collected as follows: (1) the mode of actions of first-in-class drugs against their innovative target were literature-searched for discovering if that drug selectively-bind to its target with a disease-specific target mutation [34, 53] or covalently-bind to specific residue at the particular positions in target [54]; (2) the biomarker status of each target was collected from the latest version of FDA Pharmacogenomic Biomarkers in Drug Labeling, TTD and literature search based on the keyword combinations of `biomarker’, drug name and target name; (3) the orphan drug status of each target was discovered from the FDA Search Orphan Drug Designations and Approvals webpage and literature search based on keyword combinations of `orphan drug’, target name and drug name. The orphan status was assigned to a target if its corresponding drug is designated as an orphan drug for the approved treatment of the targeted disease. Moreover, the population-based disease characteristics (affected population size, death statistics, other life-threatening diseases and problems caused) of each target were found via a comprehensive literature search by keyword combinations of targeted disease name, `patient’, `statistics’, `death’, `risk’ and `cause’.
Figure 2.
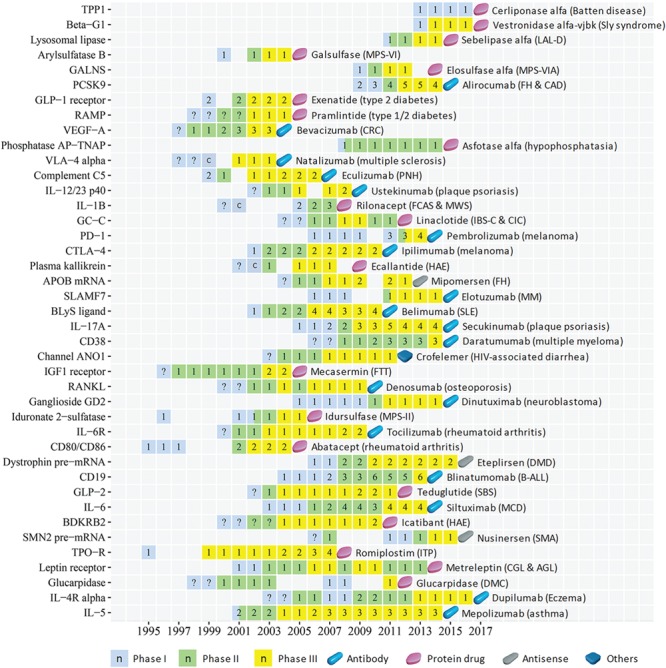
The CTP of the innovative targets of first-in-class biologics approved in 2004–17. The progression timeline of each target is from the year of 1st phase I to the year of 1st drug approval. The name of the 1st approved drug and the corresponding disease indications are provided. The clinical trial or approval status of each year is displayed as follows: phase I (light blue square), phase II (light green square), phase III (yellow square) and the 1st drug approval (blue capsule, violet red tablet, grey tablet and dark blue tablet for antibody, protein drug, antisense drug and proanthocyanidin oligomer, respectively). The number (n) in each square indicates the number of clinical trial drugs of all phases in each specific year, the question mark (?) inside the squares is a putatively estimated earlier trial phase and the letter `c’ inside the squares denotes that a completion of a trial phase reported in that year. Abbreviation for target: APOB mRNA, mRNA of apolipoprotein B; BDKRB2, Bradykinin B2 receptor; BLyS ligand: B-lymphocyte stimulator ligand; Beta-G1, beta-glucuronidase; CD19, B-lymphocyte antigen CD19; CD38, cADPr hydrolase 1; CTLA-4, cytotoxic T-lymphocyte protein-4; Channel ANO1, calcium-activated chloride channel; Dystrophin pre-mRNA, exon 51 of dystrophin pre-mRNA; GALNS, N-acetylgalactosamine 6 sulfatase; GC-C, guanylyl cyclase C; GLP-2, glucagon-like peptide 2; IGF1 receptor, insulin-like growth factor 1 receptor; IL-12/23 p40, interleukin-12/23 subunit p40; IL-17A, interleukin-17A ligand; IL-1B, interleukin-1 beta ligand; IL-4R alpha, IL-4 receptor alpha; IL-5, interleukin-5 ligand; IL-6, interleukin-6 ligand; IL-6R, interleukin-6 receptor; Lysosomal lipase, lysosomal acid lipase; PCSK9, proprotein convertase PC9; PD-1, programmed cell death protein 1; Phosphatase AP-TNAP, tissue non-specific alkaline phosphatase; Plasma kallikrein, plasma kallikrein; RAMP, receptor activity modifying protein; RANKL, receptor activator of nuclear factor kappa-B ligand; SLAMF7, SLAM family member 7; SMN2 pre-mRNA, pre-mRNA of survival of motor neuron 2; TPO-R, thrombopoietin receptor; TPP1, tripeptidyl-peptidase 1; VEGF-A, VEGF-A ligand; VLA-4 alpha, integrin alpha-4. Abbreviation for disease: AGL, acquired generalized lipodystrophy; B-ALL, B-cell precursor acute lymphoblastic leukemia; CAD, coronary artery disease; CGL, congenital generalized lipodystrophy; CIC, chronic idiopathic constipation; CRC, colorectal cancer; DMC, delayed methotrexate clearance; DMD, Duchenne muscular dystrophy; FCAS, familial cold autoinflammatory syndrome; FH, familial hypercholesterolemia; FTT, failure to thrive; HAE, hereditary angioedema; IBS-C, irritable bowel syndrome with constipation; ITP, immune thrombocytopenic purpura; LAL-D, lysosomal acid lipase deficiency; MCD, multicentric Castleman disease; MM, multiple myeloma; MPS-II, mucopolysaccharidosis II; MPS-VI, mucopolysaccharidosis VI; MPS-VIA, mucopolysaccharidosis IVA; MWS, Muckle–Wells syndrome; PNH, paroxysmal nocturnal hemoglobinuria; SBS, short bowel syndrome; SLE, systemic lupus erythematosus; SMA, spinal muscular atrophy.
Results and discussion
Clinical trial progression of the targets
The clinical trial progression (CTP) timelines of the 89 targets from the earliest reported trial (dated back up to 1984) to the 1st drug approval are shown in Figures 1–3 for three pharmaceutical groups and Figures 4–6 for three disease groups. Their features are summarized, which include (1) targets, the corresponding first-in-class drugs and CTP timeline (Supplementary Table S3); (2) the disease role and drug binding-site profiles (Supplementary Table S4); (3) target-associated biomarker status, orphan drug status, drug-binding domain sequence similarity and family affiliation to pre-existing targets (with an approved drug before 2004), human system features of on-target (human protein-network topologies, modulated pathways and distributed tissues) and off-target (similarity proteins) collateral effects (Supplementary Table S5); and (4) population-based disease characteristics including the affected population size, death population per year and threat level (Supplementary Tables S6 and S7). The CTP times are from the earliest phase I to the 1st drug approval. There are 8, 28, 1 and 2 targets with their earliest reported trial traced only up to phase I completion, phase II (II/III), phase II completion and phase III, respectively. Their CTP time was thus putatively estimated by adding the average time of the missing trial(s) (1.5 years for phase I and 2.5 years for phase II [55]) to the time from the earliest reported trial to the drug approval. The CTP time of 13.5%, 21.3%, 34.8% and 30.3% of the 89 targets are ≤5, 6–8, 9–10 and >10 years, respectively. In particular, there are eight infectious species targets (Supplementary Table S6) such as HIV integrase, HCV NS5B, Anthrax PA, TB ATP synthase, HCV NS3/4A, Fungal LeuRS, Clostridium difficile toxin B and CMV-terminase with their CTP times of 6, 7, 7, 9, 9, 10, 10 and 12 years, respectively.
Figure 1.
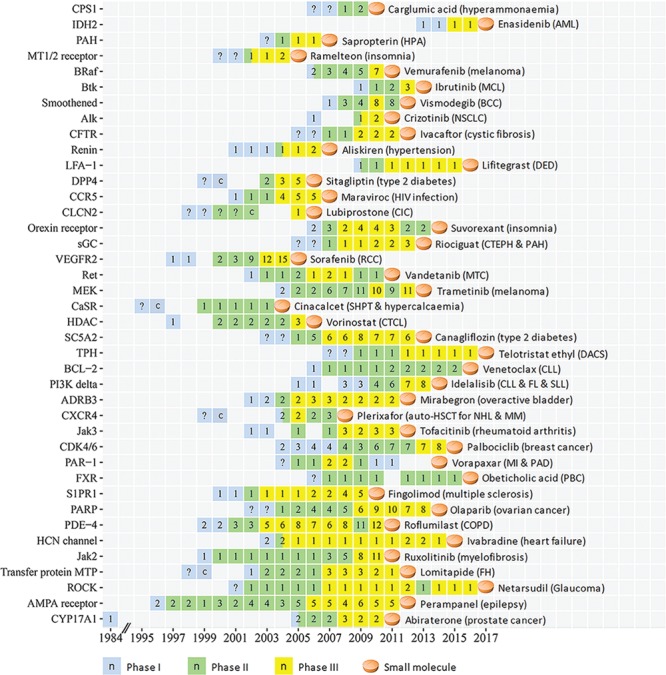
The CTP of innovative targets of first-in-class small molecule drugs approved in 2004–17. The progression timeline of each target is from the year of 1st phase I to the year of 1st drug approval. The name of the 1st approved drug and the corresponding disease indication are provided. Clinical trial or approval status of each year is represented as follows: phase I (light blue square), phase II (light green square), phase III (yellow square) and the 1st small molecule drug approval (light orange tablet). The number (n) in each square indicates the number of clinical trial drugs of all phases in each specific year, the question mark (?) inside the squares is a putatively estimated earlier trial phase and the letter `c’ inside the squares denotes that a completion of a trial phase reported in that year. Abbreviations for target: ADRB3, Beta 3 adrenoceptor; Alk, ALK tyrosine kinase receptor; BCL-2, Apoptosis regulator Bcl-2; BRaf, proto-oncogene B-Raf; Btk, Btk tyrosine kinase; CCR5, CC-chemokine receptor 5; CDK4/6, cyclin-dependent kinase-4/6; CFTR, cystic fibrosis transmembrane conductance regulator; CLCN2, chloride channel protein 2; CPS1, carbamoyl phosphate synthetase 1; CXCR4, C-X-C chemokine receptor type 4; CYP17A1, cytochrome P450 17A1; CaSR, extracellular calcium sensing receptor; DPP4, dipeptidyl peptidase IV; FXR, Farnesoid X receptor; HCN channel, hyperpolarization-activated cyclic nucleotide-gated channel; HDAC, histone deacetylase; IDH2, isocitrate dehydrogenase 2; Jak2, Jak2 tyrosine kinase; Jak3, Jak3 tyrosine kinase; LFA-1, lymphocyte function-associated antigen-1; MEK, MEK protein kinase; MT1/2 receptor, melatonin MT1/2 receptor; Orexin receptor, OX1/2 orexin receptor; PAH, phenylalanine hydroxylase; PAR-1, protease-activated receptor-1; PARP, poly ADP ribose polymerase; PDE-4, phosphodiesterase 4; PI3K delta, phosphoinositide-3 kinase delta; ROCK, rho kinase; Ret, Ret tyrosine kinase receptor; S1PR1, sphingosine 1-phosphate receptor 1; SC5A2, sodium glucose transporter-2; sGC, soluble guanylyl cyclase; TPH, tryptophan hydroxylase; Transfer protein MTP, microsomal triglyceride transfer protein; VEGFR2, VEGF-2 receptor. Abbreviation for disease: AML, acute myeloid leukemia; BCC, basal cell carcinoma; CIC, chronic idiopathic constipation; CLL, chronic lymphocytic leukemia; COPD, chronic obstructive pulmonary disease; CTCL, cutaneous T-cell lymphoma; CTEPH, chronic thromboembolic pulmonary hypertension; DACS, diarrhea associated with carcinoid syndrome; DED, dry eye disease; FH, familial hypercholesterolemia; FL, follicular lymphoma; HPA, hyperphenylalaninemia; “”MCL, mantle cell lymphoma; MI, myocardial infarction; MM, multiple myeloma; MTC, medullary thyroid cancer; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung carcinoma; PAD, peripheral arterial disease; PAH, pulmonary arterial hypertension; PB,: primary biliary cholangitis; RCC, renal cell carcinoma; SHP,: secondary hyperparathyroidism; SLL, small lymphocytic lymphoma.
Figure 3.
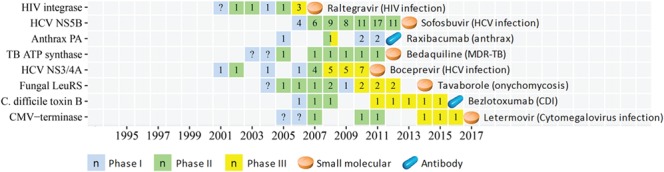
The CTP of the innovative infectious disease specie target of the first-in-class drugs approved in 2004–17. The progression timeline of each target is from the year of 1st phase I to the year of the 1st drug approval. The name of the 1st approved drug and the corresponding disease indications are provided. The clinical trial or approval status of each year is shown as follows: phase I (light blue square), phase II (light green square), phase III (yellow square) and 1st drug approval (light orange tablet and blue capsule for small molecule and antibody, respectively). The number (n) in each square indicates the number of clinical trial drugs of all phases in each specific year, and the question mark (?) inside the squares indicates a putatively estimated earlier trial phase. Abbreviation for target: Anthrax PA, anthrax protective antigen; C. difficile toxin B, Clostridium difficile toxin B; CMV-terminase, cytomegalovirus DNA terminase complex; Fungal LeuRS, fungal aureus leucyl-tRNA synthetase; HCV NS3/4A, hepatitis C virus NS3/4A protease; HCV NS5B, hepatitis C NS5B polymerase; TB ATP synthase, mycobacterial ATP synthase. Abbreviation for disease: CDI, Clostridium difficile infection; MDR-TB, multidrug-resistant tuberculosis.
Figure 4.
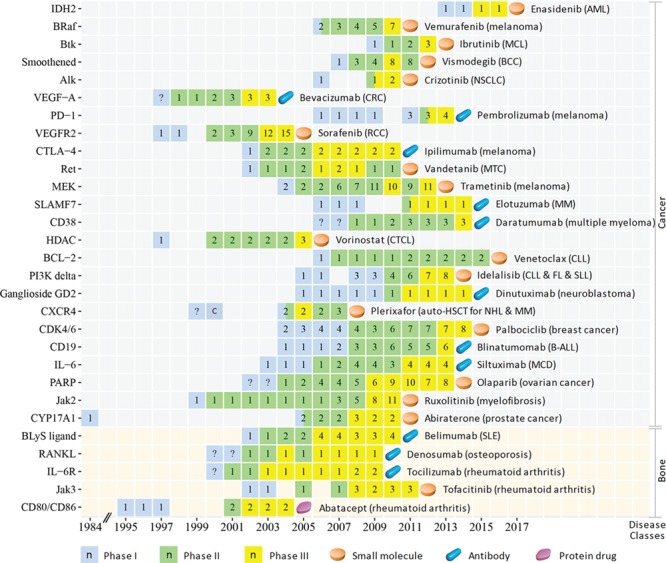
The CTP of the innovative targets of the first-in-class drugs approved in 2004–17 for treating cancers (Cancer) and diseases of musculoskeletal system and connective tissue (Bone). The progression timeline of each target is from the year of 1st phase I to the year of the 1st drug approval. The name of the 1st approved drug and the corresponding disease indications are provided. The clinical trial or approval status of each year is represented as follows: phase I (light blue square), phase II (light green square), phase III (yellow square) and the 1st drug approval (light orange tablet, blue capsule and violet red tablet for small molecule, antibody and protein drug, respectively). The number (n) in each square indicates the number of clinical trial drugs of all phases in each specific year, the question mark (?) inside the squares indicates the putatively estimated earlier trial phase and the letter `c’ inside the squares denotes that a completion of a trial phase reported in that year. Abbreviation for target: Alk, ALK tyrosine kinase receptor; BCL-2, apoptosis regulator Bcl-2; BLyS ligand, B-lymphocyte stimulator ligand; BRaf, proto-oncogene B-Raf; Btk, Btk tyrosine kinase; CD19, B-lymphocyte antigen CD19; CD38, cADPr hydrolase 1; CDK4/6, cyclin-dependent kinase-4/6; CTLA-4, cytotoxic T-lymphocyte protein-4; CXCR4, C-X-C chemokine receptor type 4; CYP17A1, cytochrome P450 17A1; HDAC, histone deacetylase; IDH2, isocitrate dehydrogenase 2; IL-6, interleukin-6 ligand; IL-6R, interleukin-6 receptor; Jak2, Jak2 tyrosine kinase; Jak3, Jak3 tyrosine kinase; MEK, MEK protein kinase; PARP, poly ADP ribose polymerase; PD-1, programmed cell death protein 1; PI3K delta, phosphoinositide-3 kinase delta; RANKL, receptor activator of nuclear factor kappa-B ligand; Ret, Ret tyrosine kinase receptor; SLAMF7, SLAM family member 7; VEGF-A, VEGF-A ligand; VEGFR2, VEGF-2 receptor. Abbreviation for disease (class): AML: acute myeloid leukemia; B-ALL, B-cell precursor acute lymphoblastic leukemia; BCC, basal cell carcinoma; CLL, chronic lymphocytic leukemia; CRC, colorectal cancer; CTCL, Cutaneous T cell lymphoma; FL, Follicular lymphoma; MCD, multicentric Castleman disease; MCL, mantle cell lymphoma; MM, multiple myeloma; MTC, medullary thyroid cancer; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung carcinoma; RCC, renal cell carcinoma; SLE, systemic lupus erythematosus; SLL, small lymphocytic lymphoma.
Figure 6.
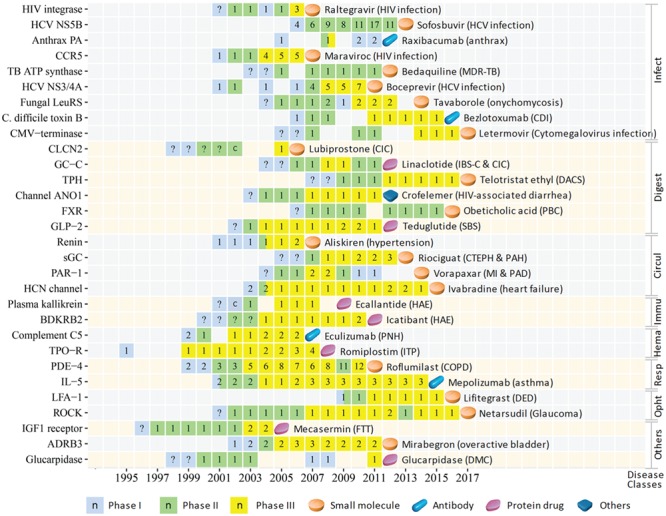
The CTP of the innovative targets of the first-in-class drugs approved in 2004–17 for treating infectious and parasitic diseases (Infect), diseases of the digestive system (Digest), diseases of the circulatory system (Circul), disorders involving the immune mechanism (Immu), diseases of the blood and blood-forming organs (Hema), diseases of the respiratory system (Resp), diseases of the eye and adnexa (Opht) and other diseases (Others). The progression timeline of each target is from the year of 1st phase I to the year of 1st drug approval. The name of the 1st approved drug and the corresponding indications are provided. The clinical trial or approval status of each year is represented as follows: phase I (light blue square), phase II (light green square), phase III (yellow square) and 1st drug approval (light orange tablet, blue capsule, violet red tablet and grey tablet for small molecule, antibody, protein and peptide and antisense drugs, respectively). The number (n) in each square indicates the number of clinical trial drugs of all phases in each specific year, the question mark (?) inside the squares indicates a putatively estimated earlier trial phase and the letter `c’ inside the squares denotes that a completion of a trial phase reported in that year. Abbreviation for target: ADRB3, beta 3 adrenoceptor; Anthrax PA, anthrax protective antigen; BDKRB2, bradykinin B2 receptor; C. difficile toxin B, Clostridium difficile toxin B; CCR5, CC-chemokine receptor 5; CLCN2, chloride channel protein 2; CMV-terminase, cytomegalovirus DNA terminase complex; Channel ANO1, calcium-activated chloride channel; FXR, Farnesoid X receptor; Fungal LeuRS, fungal aureus leucyl-tRNA synthetase; GC-C, guanylyl cyclase C; GLP-2, glucagon-like peptide 2; HCN channel, hyperpolarization-activated cyclic nucleotide-gated channel; HCV NS3/4A, hepatitis C virus NS3/4A protease; HCV NS5B, hepatitis C virus NS5B polymerase; IGF1 receptor, insulin-like growth factor 1 receptor; IL-5, interleukin-5 ligand; LFA-1, lymphocyte function-associated antigen-1; PAR-1, protease-activated receptor-1; PDE-4, phosphodiesterase 4; ROCK, rho kinase; sGC, soluble guanylyl cyclase; TB ATP synthase, mycobacterial ATP synthase; TPH, tryptophan hydroxylase; TPO-R, thrombopoietin receptor. Abbreviation for disease (class): CDI, Clostridium difficile infection; CIC, chronic idiopathic constipation; COPD, chronic obstructive pulmonary disease; CTEPH, chronic thromboembolic pulmonary hypertension; DACS, diarrhea associated with carcinoid syndrome; DED, dry eye disease; DMC, delayed methotrexate clearance; FTT, failure to thrive; HAE, hereditary angioedema; IBS-C, irritable bowel syndrome with constipation; ITP, immune thrombocytopenic purpura; MDR-TB, multidrug-resistant tuberculosis; MI, myocardial infarction; PAD, peripheral arterial disease; PAH, pulmonary arterial hypertension; PBC, primary biliary cholangitis; PNH, paroxysmal nocturnal hemoglobinuria; SBS, short bowel syndrome.
Figure 5.

The CTP of the innovative targets of the first-in-class drugs approved in 2004–17 for treating endocrine, nutritional and metabolic diseases (Metabo), diseases of nervous system (Nerve) and diseases of the skin and subcutaneous tissue (Skin). The progression timeline of each target is from the year of 1st phase I to year of 1st drug approval. The name of the 1st approved drug and the corresponding disease indications are provided. The clinical trial or approval status of each year is represented as follows: phase I (light blue square), phase II (light green square), phase III (yellow square) and the 1st drug approval (light orange tablet, blue capsule, violet red tablet, grey tablet and dark blue tablet for small molecule, antibody, protein drug, antisense drug and proanthocyanidin oligomer, respectively). The number (n) in each square is the number of clinical trial drugs of all phases in each specific year, the question mark (?) inside the squares indicates putatively estimated earlier trial phase and the letter `c’ inside the squares denotes that a completion of a trial phase reported in that year. Abbreviation for target: APOB mRNA, mRNA of apolipoprotein B; Beta-G1, beta-glucuronidase; CFTR, cystic fibrosis transmembrane conductance regulator; CPS1, carbamoyl phosphate synthetase 1; CaSR, extracellular calcium sensing receptor; DPP4, dipeptidyl peptidase IV; Dystrophin pre-mRNA, exon 51 of dystrophin pre-mRNA; GALNS, N-acetylgalactosamine 6 sulfatase; IL-12/23 p40, interleukin-12/23 subunit p40; IL-17A, interleukin-17A ligand; IL-1B, interleukin-1 beta ligand; IL-4R alpha, IL-4 receptor alpha; Lysosomal lipase, lysosomal acid lipase; MT1/2 receptor, melatonin MT1/2 receptor; Orexin receptor, OX1/2 orexin receptor; PAH, phenylalanine hydroxylase; PCSK9, proprotein convertase PC9; Phosphatase AP-TNAP, tissue non-specific alkaline phosphatase; RAMP, receptor activity modifying protein; S1PR1, sphingosine 1-phosphate receptor 1; SC5A2, sodium glucose transporter-2; SMN2 pre-mRNA, pre-mRNA of survival of motor neuron 2; TPP1, tripeptidyl-peptidase 1; Transfer protein MTP, microsomal triglyceride transfer protein; VLA-4 alpha, integrin alpha-4. Abbreviation for disease (class): AGL, acquired generalized lipodystrophy; CAD, coronary artery disease; CGL, congenital generalized lipodystrophy; DMD, Duchenne muscular dystrophy; FCAS, familial cold autoinflammatory syndrome; FH, familial hypercholesterolemia; HPA, hyperphenylalaninemia; LAL-D, lysosomal acid lipase deficiency; MPS-II, mucopolysaccharidosis II; MPS-VI, mucopolysaccharidosis VI; MPS-VIA, mucopolysaccharidosis IVA; MWS, Muckle–Wells syndrome; SHPT, secondary hyperparathyroidism; SMA, spinal muscular atrophy.
Common druggability features
The established druggability features [1, 3, 19, 20] remain important for the 89 targets. Although the common druggbility characteristics, such as critical disease role, have been described in literatures, it is worthwhile to summarize the actual descriptions about these innovative targets, so as to aid the future effort in the literature-based drug target discovery and investigation. Based on literature reports (Supplementary Table S4), all 89 targets play key roles as a driver (ALK), fundamental reliance (VEGFR2), required (HIV-1 integrase), pivotal (BRaf V600E), essential (CCR5), critical (IL-12/23 p40), crucial (anthrax PA), the major contributor (Jak2), unique mechanism (guanylyl cyclase C) and cause (GALNS) of the targeted disease. Their drug-binding sites are distinguished in their structure, conformation, residues, binding pocket or mRNA sequence. Their drug-binding domains are of high (65.5%) or fair (34.5%) similarity to those of pre-existing targets. Most (86.5%) targets are members of pre-existing drug-binding domain families. For the 81 human targets, the mean values of their human protein-network druggability descriptors degree, betweenness and clustering coefficient (9.17, 0.0012 and 0.13) are comparable to those of pre-2008 targets (3.12–11.51, 0.0006–0.0012 and 0.0066–0.1512) [27]. Most (81.5%, 85.2% and 74.1%) of the human targets are affiliated with <5 human pathways, distributed in <5 human tissues and similar to <15 human proteins outside the target family, which are in broader ranges than the pre-2006 targets (<3 human pathways, <5 human tissues and < 15 human proteins) partly due to the emergence of orphan drugs [33], mAbs [16] and biomarker-guided therapeutics [17], as well as significantly expanded KEGG database (120 pathways in 2008 to 496 pathways in 2017). Overall, the 89 targets generally occupy a narrow niche within the subspaces of the established druggability features.
Clinical trial speed differentiating features
By adding the average time for completing phases I, II, III and new drug application (1.5, 2.5, 2.5 and 1.5 years) [55], the cumulative average of phase I to drug approval is 8 years. Thus, the 89 targets can be divided into 31 speedy and 58 non-speedy ones with CTP time ≤8 and >8 years (Figures 7–9; Supplementary Tables S3–S5). The speedy targets are mostly from the preexisting target families (e.g. tyrosine kinases, GPCRs and proteases), while the non-speedy targets are from more diverse, underexplored families/subfamilies, pathway sectors and drug molecular types (e.g. serine/threonine kinases, pathway downstream and immunoglobulins targeted by mAb). The majority (71.4%) of 14 biomarker-guided targets and the minority (28.0%) of 75 non-biomarker ones are speedy (Figures 7–9). The median CTP time of speedy biomarker and non-biomarker targets is 5.1 and 6.8 years, respectively, and that of the non-speedy biomarker and non-biomarker ones equal to 8.9 and 10 years, respectively. Biomarker accelerates clinical trial with higher impact on speedy targets possibly by synergistic combination of improved therapeutic targeting and reduced chances of collateral effects.
Figure 7.
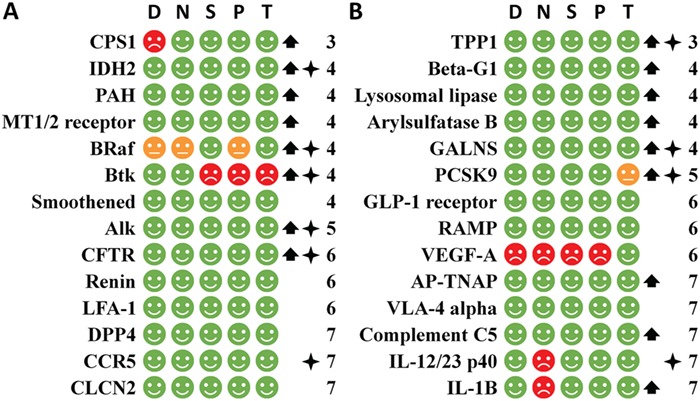
The speedy human targets of the first-in-class small molecule drugs (part A) and biologics (part B) arranged by the CTP time,and judged by the five criteria (D, N, S, P and T) of the simple rule for identifying the speedy human targets together with the orphan drug and biomarker status. The CTP time is the number of years for a target to proceed from the 1st phase I to the first-in-class drug approval. The speedy targets are with CTP time ≤8. D, N, S, P and T represents the criterion of the degree in the human protein-interaction network, neighborhood connectivity in the human protein-interaction network, human pathway affiliation and human tissue distribution respectively. A green, red or orange circle indicates the pass of a criterion, violation of a criterion or violation of a criterion that may be circumvented by selective targeting of disease specific target mutation. An upward arrow or star on the right indicates the orphan drug or biomarker status. The number on the further right indicates the CTP time.
Figure 9.
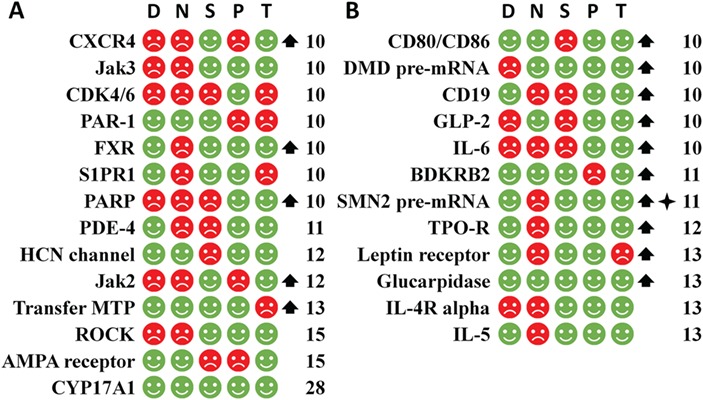
The non-speedy human targets of the first-in-class small molecule drugs (part A) and biologics (part B) arranged by the CTP time and judged by the five criteria (D, N, S, P and T) of the simple rule for identifying the speedy human targets together with the orphan drug and biomarker status. The non-speedy targets are with CTP time >8 years. The targets of this figure are with CTP time >10 years. The labels and legends are the same as Figure.7.
Figure 8.

The non-speedy human targets of the first-in-class small molecule drugs (part A) and biologics (part B) arranged by the CTP time and judged by the five criteria (D, N, S, P and T) of the simple rule for identifying the speedy human targets together with the orphan drug and biomarker status. The non-speedy targets are with CTP time >8 years. The CTP time of the targets in this figure is within the range of 8 to 10 years. The labels and legends are the same as Figure 7.
The minority (34.6%) of 52 orphan drug targets and that (35.1%) of 37 non-orphan ones are speedy (Figures 7–9). The median CTP time for the speedy orphan and non-orphan drug targets equals to 4.8 and 6.9 years, respectively, and that for the non-speedy orphan and non-orphan drug targets is 9.7 and 10.1 years, respectively. Orphan drug status accelerate clinical trials of speedy targets by shortened trials [33] (the 2.1 years reduction of the median CTP time is comparable to the 2.5 years average for completing phase III trial [55]), but it has minor impacts on non-speedy targets. Orphan status does not accelerate the non-speedy targets partly because the collateral effects like adverse drug reaction may result in patient dropout [56–58], this dropout can have a higher impact on the recruitment or replacement difficulties for orphan diseases [59], leading to prolonged trial times.
CTP speed differentiating features were systematically searched from a pool of 227 human systems and protein network descriptors, four target-relevant features and three population-based disease characteristics (Materials and methods). Based on the comprehensive analysis on all these features, Student’s t-test was applied to determine if the speedy and non-speedy targets can be significantly differentiated from each other using a given feature. As a result, 7 features exhibit high to moderate variations between the speedy and non-speedy targets, which are the neighborhood connectivity, degree, stress, number of human similarity proteins, radiality, number of affiliated human pathways and topological coefficient with unpaired t-test P-values of 0.0005, 0.0028, 0.0070, 0.01, 0.0412, 0.0422 and 0.0669, respectively (Figure 10). The neighborhood connectivity of a given target denotes the average number of the human interacting proteins of its own neighbors (interacting proteins). The degree refers to the total number of the interacting proteins of a given target. The stress indicates the number of the shortest paths passing through a target. The number of human similarity protein denotes the number of human similarity proteins of a target outside the corresponding target family. The radiality is a centrality index of a studied target in the human protein network. The number of affiliated pathways refers to the total number of target-affiliated and target immediate-downstream human pathways. The topological coefficient is a relative measure for the extent to which a studied target shares neighbors with other proteins. However, each feature individually cannot satisfactorily differentiate speedy and non-speedy targets. Hence, combinations of these features were evaluated.
Figure 10.
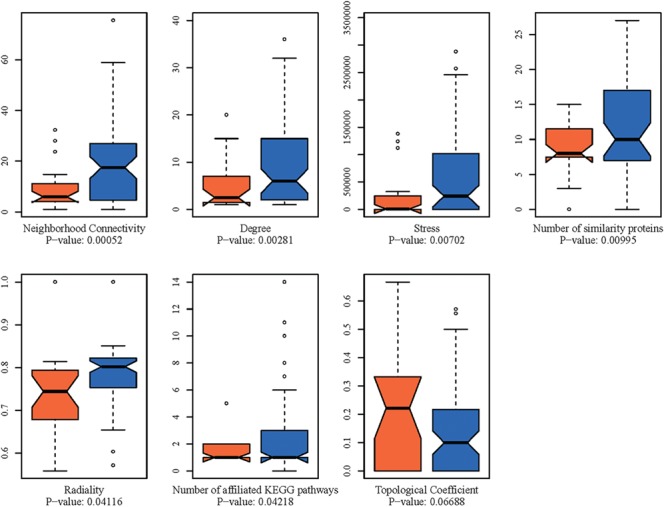
The box plot comparisons of seven features exhibiting high to moderate levels of variations between the speedy (orange color marked bars) and non-speedy targets (blue color marked bars). The P-values are provided at the bottom of each box plot.
Simple rule for identifying the speedy human targets
Our comprehensive analysis led to a combination of 5 features together with a simple rule for identifying the speedy human targets, which is obeyed by 21 (75.0%) of the 28 speedy and only unexpectedly misclassified 7 (13.2%) of 53 non-speedy human targets [95% confidence interval (CI) = 2.00–4.22, P = 6.06 × 10–7, Figures 7–9]. This simple rule states that a human target possessing the common druggability features may progress speedily through clinical trials if it has no violation of all the following criteria: (1) degree <15 in the human protein-interaction network, (2) neighborhood connectivity <15 in the human protein-interaction network, (3) affiliated with <5 human signal pathways, (4) distributed in <5 human tissues and (5) similar to <15 human similarity proteins outside the target family. Criteria (1)–(4) weigh the likelihood of on-target collateral effect based on the essentiality levels of the target and its nearest neighbors in the human protein-network and the numbers of the drug-affected pathways and tissues. Criterion (5) considers the chances of off-target collateral drug interactions with human similarity proteins outside the target families. These human physiology-related effects are usually inadequately evaluated in preclinical studies. The schematic workflow of the discovery of the clinical trial speed differentiating features in this study was illustrated in Figure 11. As shown, the 1st four criteria weigh the likelihood of on-target collateral effects, while the last criterion considers the off-target effects.
Figure 11.

Schematic workflow of the discovery of the clinical trial speed differentiating feature in this study. The 1st four criteria weigh the likelihood of on-target collateral effects based on the essentiality level of the target and its nearest neighbors in the human protein-network and the number of drug-affected pathways and tissues. The last criterion considers the chance of off-target collateral drug interactions with human similarity proteins outside the target family.
Violation of any of these five criteria likely increases the chances of on-target and off-target collateral effects of the targeted drugs, which may be circumvented in some extraordinary circumstances. In one circumstance, disease-specific target mutations may be selectively targeted with reduced interference of the wild-type target, thereby avoiding the collateral effects of the violation of the simple rule. For instance, BRaf V600E mutation, which drives melanoma, is selectively targeted by vemurafenib [60]. The PCSK9 gain-of-function mutations, causing autosomal dominant hypercholesterolemia, are specifically targeted by Evolocumab [61]. Another circumstance is target dispensability outside disease tissue. Btk, selectively inhibited by the covalent-binding ibrutinib against B cell cancers, is dispensable outside the B-cell compartment [62], among few kinases with a suitably positioned cysteine for selective covalent drug binding [54], and with certain on-target effects (e.g. toll-like receptor signaling and B-cell adhesion) helpful to drug therapeutic effect [62]. A third circumstance is drug selective evading of collateral effects. IL-12/IL-23, targeted by ustekinumab against psoriasis [63], has distinct binding epitope for enabling selective drug binding without affecting normal immune responses and has conformational flexibility to prevent drug binding to receptor-bound targets of Fc effector functions [63]. Among the seven non-speedy targets unexpectedly misclassified by simple rules, four targets have had clinical trial drugs undergone auction (APOB mRNA with Mipomersen), acquisition (BLyS ligand with Blisibimod and TPH with Telotristat) or licensing deal (Channel ANO1 with Crofelemer) and one target with the long trial absences (CYP17A1 with 20 years absence between the 1st drug Ketoconazole and next drug Abiraterone). These events occur typically for financial reasons, a key factor for trial delay [36].
Among those eight infectious species targets (Supplementary Table S6), there are three speedy ones including HIV integrase, HCV NS5B and Anthrax PA (CTP time of 6, 7 and 7 years, respectively) and 5 non-speedy ones such as TB ATP synthase, HCV NS3/4A, Fungal LeuRS, C. difficile toxin B and CMV-terminase (TCP time of 9, 9, 10, 10 and 12 years, respectively). The median CTP time of the speedy infectious species targets (7.1 years) is longer than that (5.7 years) of the speedy human targets, partly because anti-infection trials are more difficult to evaluate [37]. It is noted that the targets of CTP time <10 years are associated with a higher threat level (bioterror attack, higher death rate and cause of life-threatening problems) affecting larger populations than the target of CTP time ≥10 years (Supplementary Table S6). Moreover, those population-based disease characteristics (affected population size, death population per year and threat level) of non-infectious species targets were also collected (Supplementary Table S7) and analyzed. As shown, there is no clear difference in any of the population-based disease characteristics between the speedy and the non-speedy human targets.
Conclusion
Drug development processes, particularly the clinical trials, are costly and time consuming. Investigations of the druggability and clinical features of targets [1, 3, 20, 26–29, 33–37] and the drug-like and clinical profiles of drugs [39, 64–66] facilitate better understanding of the key factors that affect drug development and CTP speed, therefore providing useful clues for improving the efficiencies of drug discovery and development process. Our study showed that such a study is able to reveal the CTP speed differentiating features of the targets of the first-in-class drugs, from which a simple rule can be derived for identifying a speedy human target. As more innovative therapeutic targets [67] and agents [68, 69] are being explored, more extensive knowledge may be gained about the factors that affect the progression speed of the drug/target development and clinical trials.
Supplementary Material
Ying Hong Li, Xiao Xu Li, Jia Jun Hong and Yun Xia Wang are Ph.D. or master candidates of the College of Pharmaceutical Sciences, Zhejiang University, China. They are interested in bioinformatics and target discovery.
Yu Zong Chen is a professor of Bioinformatic and Drug Design Group, Department of Pharmacy, National University of Singapore. He is interested in bioinformatics, computation biology and computer-aided drug design.
Feng Zhu is a professor of the College of Pharmaceutical Sciences, Zhejiang University, China. His research group (https://idrblab.org/) has been working in the fields of bioinformatics, OMIC-based drug discovery, system biology and medicinal chemistry. Readers are welcome to visit his personal website at https://idrblab.org/Peoples.php.
Key Points
An outstanding issue in drug development is the high cost in clinical trials, and no exploration has been conducted to find the features affecting the drug/target CTP.
This study developed a method that can identify the targets of speedy progression through clinical trials and discovered and validated a simple rule for identifying targets of speedy clinical progression.
This work added significant knowledge to the key factors governing the CTP speed and enabled targets assessment and the prioritization of drug development efforts in clinical trials.
Funding
The National Natural Science Foundation of China (81872798), the National Key Research and Development Program of China (2018YFC0910500), the Innovation Project on Industrial Generic Key Technologies of Chongqing (cstc2015zdcy-ztzx120003) and the Fundamental Research Funds for the Central Universities (2018QNA7023, 10611CDJXZ238826, 2018CDQYSG0007, CDJZR14468801, CDJKXB14011).
References
- 1. Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov 2002;1:727–730. [DOI] [PubMed] [Google Scholar]
- 2. Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov 2006;5:821–834. [DOI] [PubMed] [Google Scholar]
- 3. Zheng CJ, Han LY, Yap CW, et al. Therapeutic targets: progress of their exploration and investigation of their characteristics. Pharmacol Rev 2006;58:259–279. [DOI] [PubMed] [Google Scholar]
- 4. Li XX, Yin J, Tang J, et al. Determining the balance between drug efficacy and safety by the network and biological system profile of its therapeutic target. Front Pharmacol 2018;9:1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang H, Qin C, Li YH, et al. Therapeutic target database update 2016: enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res 2016;44:D1069–D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li X, Li X, Li Y, et al. What makes species productive of anti-cancer drugs? Clues from drugs' species origin, druglikeness, target and pathway. Anticancer Agents Med Chem 2018. doi: 10.2174/1871520618666181029132017. [DOI] [PubMed] [Google Scholar]
- 7. Li YH, Yu CY, Li XX, et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res 2018;46:D1121–D1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dollery CT. Beyond genomics. Clin Pharmacol Ther 2007;82:366–370. [DOI] [PubMed] [Google Scholar]
- 9. Li B, Tang J, Yang Q, et al. NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res 2017;45:W162–W170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fu J, Tang J, Wang Y, et al. Discovery of the consistently well-performed analysis chain for SWATH-MS based pharmacoproteomic quantification. Front Pharmacol 2018;9:681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li B, Tang J, Yang Q, et al. Performance evaluation and online realization of data-driven normalization methods used in LC/MS based untargeted metabolomics analysis. Sci Rep 2016;6:38881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang J, Fu JB, Wang YX, et al. ANPELA: analysis and performance-assessment of the label-free quantification workflow for metaproteomic studies. Brief Bioinform 2018. doi: 10.1093/bib/bby127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shoemaker RH, Scudiero DA, Melillo G, et al. Application of high-throughput, molecular-targeted screening to anticancer drug discovery. Curr Top Med Chem 2002;2:229–246. [DOI] [PubMed] [Google Scholar]
- 14. Huwe CM. Synthetic library design. Drug Discov Today 2006;11:763–767. [DOI] [PubMed] [Google Scholar]
- 15. Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov 2007;6:211–219. [DOI] [PubMed] [Google Scholar]
- 16. Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010;9:767–774. [DOI] [PubMed] [Google Scholar]
- 17. Frank R, Hargreaves R. Clinical biomarkers in drug discovery and development. Nat Rev Drug Discov 2003;2:566–580. [DOI] [PubMed] [Google Scholar]
- 18. Zhu F, Li XX, Yang SY, et al. Clinical success of drug targets prospectively predicted by in silico study. Trends Pharmacol Sci 2018;39:229–231. [DOI] [PubMed] [Google Scholar]
- 19. Hajduk PJ, Huth JR, Fesik SW. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem 2005;48:2518–2525. [DOI] [PubMed] [Google Scholar]
- 20. Cheng AC, Coleman RG, Smyth KT, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol 2007;25:71–75. [DOI] [PubMed] [Google Scholar]
- 21. Xue W, Yang F, Wang P, et al. What contributes to serotonin-norepinephrine reuptake inhibitors' dual-targeting mechanism? The key role of transmembrane domain 6 in human serotonin and norepinephrine transporters revealed by molecular dynamics simulation. ACS Chem Neurosci 2018;9:1128–1140. [DOI] [PubMed] [Google Scholar]
- 22. Xue W, Wang P, Tu G, et al. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys Chem Chem Phys 2018;20:6606–6616. [DOI] [PubMed] [Google Scholar]
- 23. Zheng G, Yang F, Fu T, et al. Computational characterization of the selective inhibition of human norepinephrine and serotonin transporters by an escitalopram scaffold. Phys Chem Chem Phys 2018;20:29513–29527. [DOI] [PubMed] [Google Scholar]
- 24. Wang P, Fu T, Zhang X, et al. Differentiating physicochemical properties between NDRIs and sNRIs clinically important for the treatment of ADHD. Biochim Biophys Acta Gen Subj 1861;2017:2766–2777. [DOI] [PubMed] [Google Scholar]
- 25. Wang P, Zhang X, Fu T, et al. Differentiating physicochemical properties between addictive and nonaddictive ADHD drugs revealed by molecular dynamics simulation studies. ACS Chem Neurosci 2017;8:1416–1428. [DOI] [PubMed] [Google Scholar]
- 26. Zhu F, Han L, Zheng C, et al. What are next generation innovative therapeutic targets? Clues from genetic, structural, physicochemical, and systems profiles of successful targets. J Pharmacol Exp Ther 2009;330:304–315. [DOI] [PubMed] [Google Scholar]
- 27. Yao L, Rzhetsky A. Quantitative systems-level determinants of human genes targeted by successful drugs. Genome Res 2008;18:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yildirim MA, Goh KI, Cusick ME, et al. Drug-target network. Nat Biotechnol 2007;25:1119–1126. [DOI] [PubMed] [Google Scholar]
- 29. Zhang XD, Song J, Bork P, et al. The exploration of network motifs as potential drug targets from post-translational regulatory networks. Sci Rep 2016;6:20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan CG, Xie HT, Chen JJ, et al. A fast uyghur text detector for complex background images. IEEE Trans Multimedia 2018;20:3389–3398. [Google Scholar]
- 31. Yan CG, Li L, Zhang CJ, et al. Cross-modality bridging and knowledge transferring for image understanding. IEEE Trans Multimedia 2018. [Google Scholar]
- 32. Zhu F, Han B, Kumar P, et al. Update of TTD: Therapeutic Target Database. Nucleic Acids Res 2010;38:D787–D791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Braun MM, Farag-El-Massah S, Xu K, et al. Emergence of orphan drugs in the United States: a quantitative assessment of the first 25 years. Nat Rev Drug Discov 2010;9:519–522. [DOI] [PubMed] [Google Scholar]
- 34. Holderfield M, Deuker MM, McCormick F, et al. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 2014;14:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kasenda B, Elm E, You J, et al. Prevalence, characteristics, and publication of discontinued randomized trials. JAMA 2014;311:1045–1051. [DOI] [PubMed] [Google Scholar]
- 36. CenterWatch Contract and budget process top cause of study delay. The CenterWatch Monthly 2005;12:1–3. [Google Scholar]
- 37. Hill J, Bird HA. Failure of selenium-ace to improve osteoarthritis. Br J Rheumatol 1990;29:211–213. [DOI] [PubMed] [Google Scholar]
- 38. Zarin DA, Tse T, Williams RJ, et al. The ClinicalTrials.gov results database—update and key issues. N Engl J Med 2011;364:852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eder J, Sedrani R, Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov 2014;13:577–587. [DOI] [PubMed] [Google Scholar]
- 40. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 2018;46:D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boratyn GM, Camacho C, Cooper PS, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 2013;41:W29–W33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 2016;44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ghosh S, Kumar GV, Basu A, et al. Graph theoretic network analysis reveals protein pathways underlying cell death following neurotropic viral infection. Sci Rep 2015;5:14438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu J, Wang P, Yang H, et al. Comparison of FDA approved kinase targets to clinical trial ones: insights from their system profiles and drug-target interaction networks. Biomed Res Int 2016;2016:2509385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li YH, Wang PP, Li XX, et al. The human kinome targeted by FDA approved multi-target drugs and combination products: A comparative study from the drug-target interaction network perspective. PLoS One 2016;11:e0165737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang P, Tao L, Zeng X, et al. A protein network descriptor server and its use in studying protein, disease, metabolic and drug targeted networks. Brief Bioinform 2017;18:1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kogenaru S, Val C, Hotz-Wagenblatt A, et al. TissueDistributionDBs: a repository of organism-specific tissue-distribution profiles. Theor Chem Acc 2010;125:651–658. [Google Scholar]
- 51. The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018;46:2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Turetsky A, Kim E, Kohler RH, et al. Single cell imaging of Bruton's tyrosine kinase using an irreversible inhibitor. Sci Rep 2014;4:4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jia P, Jin H, Meador CB, et al. Next-generation sequencing of paired tyrosine kinase inhibitor-sensitive and -resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res 2013;23:1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garuti L, Roberti M, Bottegoni G. Irreversible protein kinase inhibitors. Curr Med Chem 2011;18:2981–2994. [DOI] [PubMed] [Google Scholar]
- 55. Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov 2010;9:203–214. [DOI] [PubMed] [Google Scholar]
- 56. Machado M, Iskedjian M, Ruiz I, et al. Remission, dropouts, and adverse drug reaction rates in major depressive disorder: a meta-analysis of head-to-head trials. Curr Med Res Opin 2006;22:1825–1837. [DOI] [PubMed] [Google Scholar]
- 57. Scoyni RM, Aiello L, Trani I, et al. Drug adverse events and drop-out risk: a clinical case. Arch Gerontol Geriatr 2007;44:359–364. [DOI] [PubMed] [Google Scholar]
- 58. Weinblatt ME, Reda D, Henderson W, et al. Sulfasalazine treatment for rheumatoid arthritis: a metaanalysis of 15 randomized trials. J Rheumatol 1999;26:2123–2130. [PubMed] [Google Scholar]
- 59. DeWard SJ, Wilson A, Bausell H, et al. Practical aspects of recruitment and retention in clinical trials of rare genetic diseases: the phenylketonuria (PKU) experience. J Genet Couns 2014;23:20–28. [DOI] [PubMed] [Google Scholar]
- 60. Jang S, Atkins MB. Which drug, and when, for patients with BRAF-mutant melanoma? Lancet Oncol 2013;14:e60–e69. [DOI] [PubMed] [Google Scholar]
- 61. Farnier M. PCSK9: from discovery to therapeutic applications. Arch Cardiovasc Dis 2014;107:58–66. [DOI] [PubMed] [Google Scholar]
- 62. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer 2014;14:219–232. [DOI] [PubMed] [Google Scholar]
- 63. Benson JM, Peritt D, Scallon BJ, et al. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs 2011;3:535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 2004;1:337–341. [DOI] [PubMed] [Google Scholar]
- 65. Pregelj L, Verreynne ML, Hine D. Changes in clinical trial length. Nat Rev Drug Discov 2015;14:307–308. [DOI] [PubMed] [Google Scholar]
- 66. Tao L, Zhu F, Xu F, et al. Co-targeting cancer drug escape pathways confers clinical advantage for multi-target anticancer drugs. Pharmacol Res 2015;102:123–131. [DOI] [PubMed] [Google Scholar]
- 67. Kodama M, Kodama T, Newberg JY, et al. In vivo loss-of-function screens identify KPNB1 as a new druggable oncogene in epithelial ovarian cancer. Proc Natl Acad Sci U S A 2017;114:E7301–E7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Martinko AJ, Truillet C, Julien O, et al. Targeting RAS-driven human cancer cells with antibodies to upregulated and essential cell-surface proteins. Elife 2018;7:e31098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bhardwaj G, Mulligan VK, Bahl CD, et al. Accurate de novo design of hyperstable constrained peptides. Nature 2016;538:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.