

Abstract
In metagenomic studies of microbial communities, the short reads come from mixtures of genomes. Read assembly is usually an essential first step for the follow-up studies in metagenomic research. Understanding the power and limitations of various read assembly programs in practice is important for researchers to choose which programs to use in their investigations. Many studies evaluating different assembly programs used either simulated metagenomes or real metagenomes with unknown genome compositions. However, the simulated datasets may not reflect the real complexities of metagenomic samples and the estimated assembly accuracy could be misleading due to the unknown genomes in real metagenomes. Therefore, hybrid strategies are required to evaluate the various read assemblers for metagenomic studies. In this paper, we benchmark the metagenomic read assemblers by mixing reads from real metagenomic datasets with reads from known genomes and evaluating the integrity, contiguity and accuracy of the assembly using the reads from the known genomes. We selected four advanced metagenome assemblers, MEGAHIT, MetaSPAdes, IDBA-UD and Faucet, for evaluation. We showed the strengths and weaknesses of these assemblers in terms of integrity, contiguity and accuracy for different variables, including the genetic difference of the real genomes with the genome sequences in the real metagenomic datasets and the sequencing depth of the simulated datasets. Overall, MetaSPAdes performs best in terms of integrity and continuity at the species-level, followed by MEGAHIT. Faucet performs best in terms of accuracy at the cost of worst integrity and continuity, especially at low sequencing depth. MEGAHIT has the highest genome fractions at the strain-level and MetaSPAdes has the overall best performance at the strain-level. MEGAHIT is the most efficient in our experiments. Availability: The source code is available at https://github.com/ziyewang/MetaAssemblyEval.
Keywords: metagenomics, assembly, next-generation sequencing, performance comparison
Introduction
Microorganisms occur as complex communities in all natural habitats, including ocean, soil and different parts of the human body. Investigations on what microbial organisms there are, their abundance levels and how they interact with each other to influence the function of the communities are essential problems in many different fields of scientific research. Previous studies have shown that microbiome composition is highly associated with human diseases [1–6], effectiveness of medical treatments [7–9], and functional changes in marine [10] and soil environments [11]. Therefore, research aimed at revealing the microbial composition, abundance and interactions have great significance.
Next-generation sequencing (NGS) technologies make it possible to rapidly sequence a large number of short reads efficiently and economically. Faced with massive and complex metagenomic sequencing reads, the first step in most of the metagenomic studies is to assemble the reads into contigs consisting of overlapping reads. Metagenomic assembly is essential for follow-up studies on microbial composition, abundance and interactions, as well as microbial associations with complex traits. Many metagenomic assembly algorithms have been developed based on various principles. While preprocessing the metagenomic data, researchers usually use sequence assemblers to link NGS sequence reads into longer sequences, generally called contigs, whose quality is essential for further analysis. The assembly quality and usefulness in practice are usually measured by contiguity, integrity, accuracy, running time and memory cost. Several research groups have evaluated the quality of various assemblers [12–17]. The current computational experimental designs for evaluating metagenomic assemblers fall into two main categories.
The first category of studies evaluates metagenomic assemblers using simulated metagenomes. Since real metagenomes are generally complex and their microbial compositions are unknown, it is challenging to compare different assemblers on real metagenomic datasets. Therefore, most studies evaluating metagenomic assemblers used simulation approaches [13, 15, 18, 19]. In these studies, the investigators first simulated the datasets using metagenome simulation programs by selecting a set of known genomes and their abundance profiles, then fragmented the genomes into simulated NGS reads, then used metagenomic assembly programs to assemble these simulated reads and finally evaluated the quality of the assembled contigs by comparing the assembly results with the original reference sequences. With the known reference genomes, the investigators were able to accurately assess the assembly accuracy and integrity. However, it is difficult to simulate the complexity of real metagenome datasets with pure simulated data and it is not clear whether the evaluation results based on the simulated data are consistent with that from complex real metagenomic datasets. Of particular concern is the fact that natural communities are often composed of many very close relatives, where extensive sequence variations interfere with the creation of long contigs.
The second category of studies evaluates metagenomic assemblers using real metagenomic datasets. Without the known genomes in the microbial community, it is difficult to evaluate assembly accuracy and integrity. Under such scenarios, the investigators first assembled the real metagenomic datasets and then used the resulting assembly to evaluate contiguity, running time and memory cost. Nurk et al. [20] used the genomes identified by metaQUAST [21] as the reference genomes to evaluate metagenomic assemblers. For example, they selected the reference genomes as those with a large proportion covered by the assembled contigs. The assembly integrity and accuracy of these genomes can then be evaluated. Due to the lack of known reference genomes, such an approach can only pick up the most likely genomes for alignment and the misassemblies and mismatches may be caused by the incorrect assembly or the incorrect reference genomes [20].
Evaluating the performance of different metagenomic assembly programs under realistic scenarios remains an important yet challenging problem. In this study, we used a hybrid strategy for evaluating the metagenomic assemblers by adding simulated reads of the known genomes into the sequencing reads of the real metagenomic datasets. We then evaluated the metagenomic assemblers using the sequence of the known genomes. The strategy has two obvious advantages. First, the reads we added into the metagenomic datasets were from the known genomes, that is, the references are known, which avoids the problem of unknown reference genomes using completely real metagenomes. Second, adding a small amount of simulated data into the real metagenomic datasets can construct an environment that is more similar to the real metagenomes. Such a strategy also avoids the problem that the simulated metagenomes are not as complex as real metagenomes.
Here we first review the major de novo sequence assembly algorithms and the metagenomic assemblers. Then, we introduce our experimental design including the real datasets and known genomes used for our study, the experimental variables and the evaluation criteria. Next, we present the results of the preliminary studies to show the necessity of using a hybrid approach to evaluate read assemblers and the results of our evaluation strategy. Finally, we discuss the comparison results, which provides guidelines for the choice of metagenomic assemblers in practical studies.
Overview of metagenomic assembly programs
In metagenomic studies, NGS is usually applied directly to sequence all the genetic materials from a certain environment resulting in a large number of short reads each with length ranging from 100–400 bp. Therefore, the data consists of a mixture of reads from different microbial organisms. For many analyses, the first step in metagenomic data analysis is sequence assembly that links overlapping reads into relatively long fragments called contigs. Many metagenomic assembly programs have been developed based on different principles [20, 22–25]. In the following, we briefly review some of the metagenomic assembly programs investigated in this study.
Genome and metagenomic assembly algorithms can be grouped into three categories based on their assembly principles [26]: greedy-extension, overlap–layout–consensus (OLC) and de Bruijn graph-based approaches. Figure 1 shows an overview of the different groups of de novo assembly algorithms. The greedy-extension algorithms start with some reads as seeds, followed by extending the seeds using the reads with the highest-scoring overlap or the reads whose prefix or suffix have overlap length longer than a given threshold. The algorithms then take the extension of the sequence as new seed sequences and make the next joint until no more reads can be merged [27]. The assembly algorithms based on the OLC principle mainly consist of three steps. First, the overlap step finds the overlaps among all the reads. Then, the layout step uses a graph to represent a layout of all the reads and the overlap information obtained in the first step. Finally, the consensus step infers the consensus sequence according to the layout. The greedy-extension algorithms just make the best choice in each step, potentially leading to local optimal solutions, which usually result in a relatively high number of incorrect assemblies. The computational costs of the OLC algorithms are usually very high, making it not practical to deal with large amounts of metagenomic shotgun sequence data. On the other hand, the metagenomic assembly algorithms based on the de Bruijn graph principle are usually much faster and memory efficient, resulting in their wide applications in metagenomic assembly.
Figure 1.
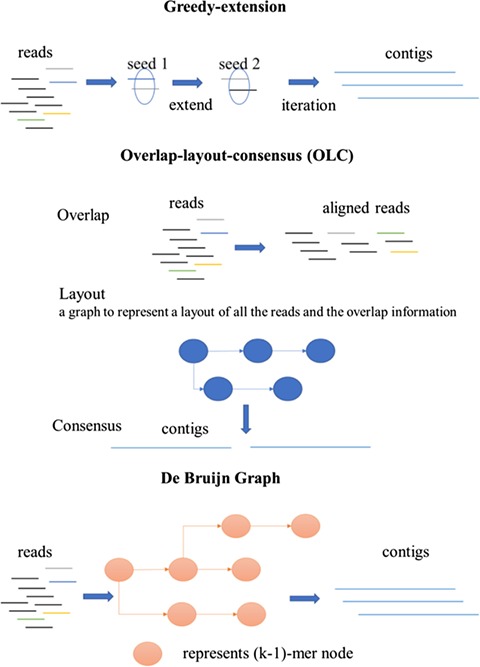
Overview of three different categories of de novo assembly programs: greedy extension, OLC and the de Bruijn graph.
Assembly algorithms based on the de Bruijn graph
The assembly algorithms based on the de Bruijn graph use the relationship between 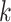 -length substrings (
-length substrings (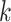 -mers) derived from the reads to construct the graph. First, the
-mers) derived from the reads to construct the graph. First, the
 prefix and suffix of each
prefix and suffix of each 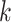 -mer are extracted and used as two nodes in the graph. If there are no corresponding identified nodes in the graph already, create a new node for the (
-mer are extracted and used as two nodes in the graph. If there are no corresponding identified nodes in the graph already, create a new node for the ( )-mer, and then establish a directional connection between the prefix and suffix. The resulting graph is called the de Bruijn graph. Next, sequence assembly might be achieved by finding a path that contains all edges from the de Bruijn graph, that is, identifying an Eulerian path in the de Bruijn graph. Since sequencing errors can be present in many substrings, the accuracy of the resulting assembly based on the de Bruijn graph is sensitive to sequencing errors, while such algorithms are generally highly efficient and are insensitive to sequencing depth. Many assembly algorithms based the de Bruijn graph have been developed for individual genomes [28–31] and metagenomes [20, 22, 23, 32–34].
)-mer, and then establish a directional connection between the prefix and suffix. The resulting graph is called the de Bruijn graph. Next, sequence assembly might be achieved by finding a path that contains all edges from the de Bruijn graph, that is, identifying an Eulerian path in the de Bruijn graph. Since sequencing errors can be present in many substrings, the accuracy of the resulting assembly based on the de Bruijn graph is sensitive to sequencing errors, while such algorithms are generally highly efficient and are insensitive to sequencing depth. Many assembly algorithms based the de Bruijn graph have been developed for individual genomes [28–31] and metagenomes [20, 22, 23, 32–34].
Challenges of using the de Bruijn graph for genome assembly
There are several challenges of using the de Bruijn graph approach for genome assembly. First, repeat regions with length greater than 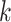 within a genome can cause branches in the graph that can be hard to resolve [35]. There are two kinds of branches: tips and bubbles. A tip is a chain of nodes that is disconnected on one end [28] as shown in Figure 2B. A bubble appears if two paths start and end at the same nodes [28] as shown in Figure 2B. Large
within a genome can cause branches in the graph that can be hard to resolve [35]. There are two kinds of branches: tips and bubbles. A tip is a chain of nodes that is disconnected on one end [28] as shown in Figure 2B. A bubble appears if two paths start and end at the same nodes [28] as shown in Figure 2B. Large 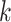 can potentially decrease the number of branches caused by repeats, because the lengths of some short repeat regions may be shorter than the read length.
can potentially decrease the number of branches caused by repeats, because the lengths of some short repeat regions may be shorter than the read length.
Figure 2.
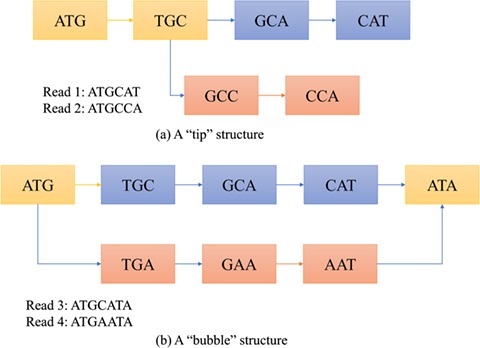
Examples of tip and bubble structures in the de Bruijn graph.
Sequencing errors can generate extra vertices in the de Bruijn graph and we call these extraneous ones ‘false positive vertices’. These false positive vertices not only take more storage space and influence the computational efficiency, but also bring more branches, thus, making it more prone to have short and sometimes erroneous contigs. When the sequencing coverage is low, some genomic positions may not be sequenced resulting in ‘gaps’ in assembly [35].
Challenges of using the de Bruijn graph for metagenomic assembly
Metagenomic assembly is even more challenging. First, assembly is a complicated problem with high computational complexity to process large volumes of data. The scale of metagenomic data is usually large and each sample contains GB-level or even TB-level of data. Increasing amount of data poses significant challenges to the existing assemblers. Second, a microbial community may contain hundreds to thousands of unknown microbial organisms and the abundance levels of the different genomes vary widely. For rare microbial organisms, it is challenging to distinguish the rare variants from sequencing errors in a complex microbial community. Moreover, due to uneven abundance, some rare species may not be well assembled because of low coverage [36]. Third, there may be repeat regions within the same genome or across multiple genomes, which makes metagenomic assembly especially challenging.
Selected metagenomic assemblers
Several metagenomic assemblers are extensions of the corresponding assemblers for individual genomes including Meta-IDBA [22], IDBA-UD [32], Ray Meta [23] and MetaSPAdes [20]. They are all based on the de Bruijn graph approach. Meta-IDBA [22] and IDBA-UD [32] are extensions of IDBA [30] that was originally developed for assembling individual genomes. Meta-IDBA partitions the de Bruijn graph into isolated subgraphs and merges the similar subgraphs using a consensus approach. IDBA-UD uses an iterative de Bruijn graph approach iterating from small to large 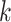 . When constructing the graph, instead of using a fixed
. When constructing the graph, instead of using a fixed 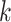 , IDBA-UD sets a minimum and a maximum value of
, IDBA-UD sets a minimum and a maximum value of 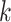 for iteration step by step. Small
for iteration step by step. Small 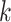 values can handle low coverage better while large
values can handle low coverage better while large 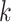 values can better handle the repeat issue. Ray Meta is based on a parallel short-read assembler developed to assemble reads obtained from different sequencing platforms [29]. It is a scalable metagenome assembler coupled with Ray Communities, which profiles microbiomes by adding colors to the de Bruijn graph utilizing bacterial genomes. However, Ray Meta does not modify the de Bruijn subgraph while producing the assembly, which may be the main reason for its low accuracy compared with other assemblers [20]. MetaSPAdes is an extension from a popular genome assembler SPAdes [31]. It develops a new process for processing branches in the graph caused by repeats compared with the SPAdes, using rare strain variants to improve assembly. In order to reduce the time required for assembly and save memory, a perfect hashing method is used to construct and simplify the graph structure in MetaSPAdes. MEGAHIT [25, 34] uses the succinct de Bruijn graph (SDBG) and is efficient for assembling large and complex metagenomic datasets. This assembler compresses the de Bruijn graph using the method similar to the Burrows–Wheeler transformation, constructing a sorted list of edges. Faucet [37] is a two-pass streaming algorithm for assembly graph construction that can optimize resource efficiency and can be used for metagenomic assembly.
values can better handle the repeat issue. Ray Meta is based on a parallel short-read assembler developed to assemble reads obtained from different sequencing platforms [29]. It is a scalable metagenome assembler coupled with Ray Communities, which profiles microbiomes by adding colors to the de Bruijn graph utilizing bacterial genomes. However, Ray Meta does not modify the de Bruijn subgraph while producing the assembly, which may be the main reason for its low accuracy compared with other assemblers [20]. MetaSPAdes is an extension from a popular genome assembler SPAdes [31]. It develops a new process for processing branches in the graph caused by repeats compared with the SPAdes, using rare strain variants to improve assembly. In order to reduce the time required for assembly and save memory, a perfect hashing method is used to construct and simplify the graph structure in MetaSPAdes. MEGAHIT [25, 34] uses the succinct de Bruijn graph (SDBG) and is efficient for assembling large and complex metagenomic datasets. This assembler compresses the de Bruijn graph using the method similar to the Burrows–Wheeler transformation, constructing a sorted list of edges. Faucet [37] is a two-pass streaming algorithm for assembly graph construction that can optimize resource efficiency and can be used for metagenomic assembly.
We selected three advanced metagenomic assemblers, IDBA-UD, MEGAHIT and MetaSPAdes, as in many recent comparative studies [14–16, 19, 36] and Faucet for evaluation. IDBA-UD was selected due to its good performance shown in other development of metagenomic assemblers [20, 24]. MEGAHIT was selected due to its good performance shown in the first CAMI challenge [15]. MetaSPAdes was selected due to its good performance in a comparative study of metagenomic assemblers [19]. Faucet was selected due to its good performance in accuracy compared with other advanced metagenomic assemblers in [37]. Table 1 summarizes the assembly strategies each assembler adopted to deal with the major difficulties in metagenomic assembly. More details can be found in their corresponding papers and Table S1 in the Supplementary Material.
Table 1.
The strategies that the assemblers MEGAHIT, MetaSPAdes and IDBA-UD, used to overcome major assembly difficulties including false positive vertices, branches, repeats and gaps
| Major difficulties | Assembly program | |||
|---|---|---|---|---|
| MEGAHIT | MetaSPAdes | IDBA-UD | Faucet | |
| False positive vertices | Filter 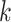 -mers -mers |
Disconnect weak edges | Use multiple depth-relative thresholds; Remove dead-end contigs | Use distances between junctions |
| Branches | Use multiple 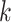 -mer sizes (large -mer sizes (large 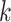 ); merge long-bubbles ); merge long-bubbles |
Use multiple 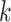 -mer sizes (large -mer sizes (large 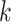 ); detect and mask strain variations ); detect and mask strain variations |
Use multiple 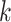 -mer sizes (large -mer sizes (large 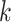 ); Remove dead-end contigs and bubbles ); Remove dead-end contigs and bubbles |
Introduce artificial dummy junctions |
| Repeats | Use multiple 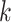 -mer sizes (large -mer sizes (large 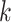 ); local assembly ); local assembly |
Use multiple 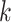 -mer sizes (large -mer sizes (large 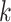 ); resolve repeats with exSPAnder ); resolve repeats with exSPAnder |
Use multiple 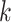 -mer sizes (large -mer sizes (large 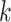 ); Local assembly ); Local assembly |
Clean and refine the graph structure iteratively |
| Gaps | Use Multiple 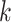 -mer sizes (small -mer sizes (small 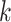 ); local assembly ); local assembly |
Use multiple 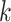 -mer sizes (small -mer sizes (small 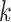 ) ) |
Use multiple 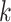 -mer sizes (small -mer sizes (small 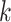 ); Local assembly ); Local assembly |
Clean and refine the graph structure iteratively |
| Efficiency | Use SDBG | Use perfect hashing technique; use parallelization | Filter 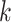 -mers -mers |
A two-pass streaming approach |
Experimental design and evaluation criteria
The objective of our study is to evaluate several metagenomic assembly programs using a combination of real metagenomic reads and simulated reads from known microbial organisms. This study differs from previous metagenomic assembly evaluation studies since previous studies used either pure simulated metagenomes or real metagenomes.
In order to investigate how the reads from known genomes are assembled within the real environment, we developed the following strategy that mixes real metagenomic data with the simulated reads from a few known completely sequenced genomes. First, using the complete genome sequences of the selected species, we applied Mason [38], a software tool for generating short reads using the complete genomic sequences, to obtain the simulated reads of the selected genomes. After mixing the simulated reads with the real metagenomic data, we assembled the hybrid reads with different metagenomic assemblers. Next, taking the selected genomes as references, we applied the metagenomic assembly evaluation tool, metaQUAST [21], to obtain the statistics of the contigs for subsequent analyses. Figure 3 shows the framework of our metagenomic reads assembly evaluation strategy.
Figure 3.
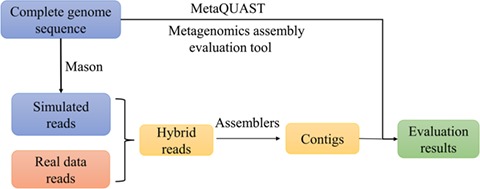
The framework of evaluating metagenomic sequence assembly programs using hybrid of real and simulated reads.
Many factors can affect the quality of assembling the reads from the newly added genomes. If the newly added genome is genetically highly similar to one of the genomes in the real microbial community, it will be challenging to assemble the reads from the newly added genome. On the other hand, if the newly added genome is highly different from the microbial organisms in the real metagenome, the reads from the newly added genome should be relatively easy to assemble. Therefore, the first parameter we investigated was the minimum distance between the newly added genome and the genomes in the real metagenome data.
Low sequencing coverage would result in many gaps in the assembly. Therefore, sequencing depth can influence the quality of the assembly. Thus, the second parameter we investigated was the sequencing depth of the added genomes. We used the following formulas to calculate the sequencing depth.
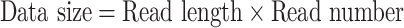 |
(1) |
 |
(2) |
Datasets
To evaluate the assembly quality as a function of the genetic difference of the newly added simulated genomes with the genome sequences in the real metagenome and the sequencing depth of metagenome, we chose two real metagenomic datasets with different data sizes and generated simulated reads from five and eight different genomes, respectively, as shown in Table 2.
Table 2.
The living environments and the genome size of the individual genomes chosen for generating simulated reads
| Species name | Abbreviation | Major living environment | Genome size |
|---|---|---|---|
| Individual genomes chosen for being added in the SRR769529 dataset | |||
| Porphyromonas gingivali | P. gingivali | human oral cavity | 2 339 898 bp |
| Streptococcus salivarius | S. salivarius | human oral cavity and upper respiratory tract | 2 259 227 bp |
| Aggregatibacter actinomycetemcomitans | A. actinomycetemcomitans | human localized aggressive periodontitis | 2 382 853 bp |
| Rahnella aquatilis | R. aquatilis | fresh water | 4 861 101 bp |
| Methanococcus maripaludis | M. maripaludis | wetland | 1 661 137 bp |
| Individual genomes chosen for being added in the ERR2762185 dataset | |||
| Synechococcus elongatus | S. elongatus | ocean | 2 696 255 bp |
| Loktanella vestfoldensis | L. vestfoldensis | ocean | 3 836 950 bp |
| Saccharomonospora marina | S. marina | ocean | 5 965 593 bp |
| Thermaerobacter marianensi | T. marianensi | ocean (isolated at a depth of >10000 meters) | 2 844 696 bp |
| Shewanella sediminis | S. sediminis | ocean | 5 517 674 bp |
| Alpha proteobacterium HIMB59 |
 -proteobacterium HIMB59
-proteobacterium HIMB59
|
ocean surface | 1 410 127 bp |
| Oceanithermus profundus | O. profundus | ocean (isolated from a hydrothermal vent in the Pacific Ocean) | 2 303 940 bp |
| Salinispora arenicola | S. arenicola | ocean | 5 786 361 bp |
Real metagenomic datasets
Dataset 1, human gut metagenome (SRR769529), contains Illumina HiSeq 101bp paired-end reads. The size of raw sequence reads of the sample is 7.9Gb. The reads were downloaded from the NCBI website (https://www.ncbi.nlm.nih.gov/).
Dataset 2, marine metagenome (ERR2762185), one of Tara Oceans Polar Circle DNA samples. It contains Illumina HiSeq 101bp paired-end reads. The size of raw sequence reads of the sample is 145.8Gb. The reads were downloaded from the NCBI website (https://www.ncbi.nlm.nih.gov/).
Individual genomes used for simulation
Table 2 shows the individual genomes chosen for generating simulated reads, their major living environments and genome sizes. Since the microbial organisms in the metagenomes are not completely known, it is difficult to know the genetic distance between the chosen individual genomes with the metagenome. As a proxy, we used the major living environment of the chosen individual genome for the closeness of the genome with the metagenome. We assume that the genetic difference between the metagenome and the species increase with their environmental difference.
For Dataset 1, S. salivarius is from the human saliva, and there is a high possibility that S. salivarius and its similar species appear in the human gut metagenome. P. gingivali and A. actinomycetemcomitans are from the human mouth that is relatively close to the human intestinal environment. R. aquatilis generally lives in fresh water, and is occasionally separated from human clinical specimens, which is comparatively far from the human intestinal environment. M. maripaludis lives in wetland that is quite different from the human gut. For Dataset 2, all the species are from oceans, but they are isolated from different parts of the oceans. We find that only around 16% of the reads from the human gut dataset (https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR769529) and the marine dataset (https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=ERR2762185) can be identified by the NCBI SRA Taxonomy Analysis Tool (STAT). The organisms that have strong signals in the datasets are summarized in Tables S2 and S3 in the Supplementary Material. According to the taxonomy analysis results based on the identified reads, there are no species in the same family with the references used in the human gut dataset. On the other hand, by using STAT to help identify close related genomes in the marine dataset to the references, we find that there are five species belonging to Shewanella genus with S. sediminis, four species belonging to Rhodobacteraceae family with L. vestfoldensis and one species belonging to Pelagibacteraceae family with  -proteobacterium HIMB59. All identified closely related organisms of the references in the metagenomes are summarized in Table S4 in the Supplementary Material.
-proteobacterium HIMB59. All identified closely related organisms of the references in the metagenomes are summarized in Table S4 in the Supplementary Material.
We used Mason [38] with default parameters to simulate Illumina reads from the chosen individual genomes.
Experimental variables
We set the sequencing depth of the simulated data as 5x, 20x and 50x. The size of the genomes selected for simulation are shown in Table 2. The calculation method of sequencing depth is shown in Equations (1) and (2).
The genetic difference between the simulated data and real metagenomes is described in Individual genomes used for simulation.
Evaluation criteria
The objective of genome and metagenomic assembly is to obtain genomes as complete and accurate as possible. Many different criteria have been used to evaluate genome assembly and they can be grouped into three broad categories: integrity, contiguity and accuracy. The integrity measures attempt to assess whether the aligned contigs can recover the complete genomes. The contiguity measures attempt to assess the length of the contigs, and one contig per chromosome is the ideal aim [16]. The accuracy measures attempt to assess how well the contigs correspond to the real genomes. The definitions of the corresponding evaluation criteria are given in Table 3. Most assembly programs try to balance integrity, contiguity and accuracy [39] and we follow this tradition in our evaluation. Given an assembly, metaQUAST [21] presents such measures and we used the outputs from metaQUAST to evaluate the different assembly programs.
Table 3.
Different measures for evaluating the assembly quality of individual genomes. The following introductions of the measures are adapted from the definitions in QUAST [40].
| Measures | Introductions | |
|---|---|---|
| Integrity | Genome fraction | The proportion of the aligned bases in the reference. |
| Duplication ratio | The total bases of the alignment in the assembly divided by the total bases of the alignment in the reference. | |
| Contiguity | N50 | The contig length for which 50% of the bases of the assembly are represented in longer or equal length contigs. |
| NGx | The contig length for which x% of the bases of the reference genome are represented in longer or equal length contigs. | |
| NGAx | NGAx is NGx where the contig lengths are replaced with the lengths of aligned blocks. | |
| L50 | The number of contigs of length at least N50. | |
| LGx | The number of contigs of length at least NGx. | |
| LGAx | LGAx is LGx where the contig lengths are replaced with the lengths of aligned blocks. | |
| Largest alignment | The total bases of the largest continuous alignment in the assembly. | |
| Total aligned length | The total bases of alignment in the assembly. | |
| Accuracy | # misassemblies | The number of misassemblies. |
| Misassembled contig length | The number of total bases contained in all contigs with one or more misassemblies. | |
| # mismatches | The number of mismatches in the alignment. | |
| # indels | The number of indels in the alignment. | |
| Indels length | The number of total bases that indels contain. | |
| # mismatches per 100 kbp | The average number of mismatches per 100 kbp (aligned bases). | |
| # indels per 100 kbp | The average number of indels per 100 kbp (aligned bases). |
Genome fraction and duplication ratio have both been used to measure the integrity of an assembly and their definitions are given in Table 3. The more complete the assembly is, the closer to 1 the genome fraction will be. The duplication ratio reflects whether the assembly contains many contigs that cover the same regions of the reference, which may be caused by over-estimating repeat multiplicities and small overlaps between contigs [40].
Eight different measures have been used to measurecontiguity of an assembly. N50 is a traditional assembly metric to assess the contiguity of genome assembly. Although some researchers pointed out that it is less informative and may frequently misrepresent the quality of the assembly for metagenomes [41], it is reasonable to use the N50 value to measure the contiguity of individual genomes. In addition, we also chose the NGx, NGAx, L50, LGx, LGAx, the size of the largest alignment and the total aligned length metrics for evaluation as shown in Table 3.
Seven other measures have been used to measure the accuracy of an assembly. ‘# misassemblies’ stands for the number of positions identified as wrong assembly in assembled contigs. ‘# mismatches’ and ‘# indels’ reflect whether the assembly can be aligned to the reference genomes perfectly. We also chose four corresponding measures for evaluation and the details are shown in Table 3. Because the references for evaluation are exactly the genomes in the dataset assembled, the ‘# misassemblies’ reported by metaQUAST are true ‘# misassemblies’ or sequencing errors, not due to structural variations.
Results
We evaluated MEGAHIT-1.1.3, SPAdes-3.12.0, IDBA-1.1.1 and Faucet in this study. All experiments were done on a machine with 4-way 6-core 1.87 GHz Intel Xeon CPUs and 1T memory. We ran all the evaluated assemblers with multiple threads except Faucet, which does not currently support multi-threaded execution. MEGAHIT was launched with default parameters. SPAdes was launched with 48 threads on the mode for metagenomic assembly (with ‘- -meta -t 48’ options). IDBA-UD was launched with read pre-correction part as recommended for metagenomic assembly (with ‘- -pre_correction’ options). Faucet was launched with a 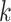 -mer size of 31 and ntCard [42] was used to extract the number of estimated
-mer size of 31 and ntCard [42] was used to extract the number of estimated 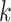 -mers (F0) and singletons (f1) in the datasets as recommended in [37] (with ‘-size_kmer 31 -max_read_length 101 -estimated_kmers F0 - singletons f1 –paired_ends’ options). We also varied the parameters of the assemblers to see the effect, and the results are shown in Table S12 in the Supplementary Material.
-mers (F0) and singletons (f1) in the datasets as recommended in [37] (with ‘-size_kmer 31 -max_read_length 101 -estimated_kmers F0 - singletons f1 –paired_ends’ options). We also varied the parameters of the assemblers to see the effect, and the results are shown in Table S12 in the Supplementary Material.
The necessity of using a hybrid approach to evaluate metagenomic assemblers
Limitations of evaluating metagenomic assemblers using real metagenomic data
Nurk et al. [20] compared the performance of MetaSPAdes with other metagenomic assemblers using real metagenomes. They first used metaQUAST [21] to automatically search for the reference genomes in the NCBI database and then chose the genomes with genome fractions greater than a certain threshold to evaluate the different metagenomic assembly programs. To see the validity of this approach, we assembled the reads from the five genomes chosen for being added in SRR769529 dataset and evaluated the assembly using metaQUAST [21] without providing known reference genomes. Then, we compared the assembly accuracy based on the metaQUAST recruited genomes with the corresponding values based on known reference genomes.
At 20-fold coverage, three genomes: Porphyromonas gingivalis TDC60 (P. gingivalis TDC60), Aggregatibacter actinomycetemcomitans D7S-1 (A. actinomycetemcomitans D7S-1) and Methanococcus maripaludis X1 (M. maripaludis X1), were recruited by metaQUAST and the average genome fractions using the four assembly programs were 96%, 93% and 85%, respectively. Only one genome, P. gingivalis TDC60, was the true genome used in the simulation.
Since the other two metaQUAST recruited genomes were different from the true genomes in the simulation, the assembly accuracy values based on the recruited genomes were highly different from that based on the true genomes. Table 4 shows some of the assembly accuracy values based on the metaQUAST recruited genomes and the most similar true genomes, respectively. Taking M. maripaludis as an example, the average ‘# misassemblies’ of the four assemblers based on the metaQUAST recruited reference genome was around 20, while that based on the true reference genome was zero. The comparison underlines that based on the metaQUAST-recruited genomes the numbers of misassemblies, mismatches, etc. can be much higher than that based on the true genomes.
Table 4.
The average accuracy statistics of the assembly of the simulated data based on the metaQUAST recruited and true reference genomes (20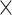 )
)
| Accuracy statistics | metaQUAST recruited versus corresponding true reference genomes | |
|---|---|---|
| A. actinomycetemcomitans D7S-1 | A. actinomycetemcomitans | |
| # Misassemblies | 6.50 | 1.00 |
| Misassembled contig length | 545106.50 | 24138.75 |
| # Mismatches per 100 kbp | 228.51 | 2.70 |
| # Indels per 100 kbp | 34.45 | 0.95 |
| M. maripaludis X1 | M. maripaludis | |
| # Misassemblies | 18.75 | 0.00 |
| Misassembled contig length | 1194508.00 | 0.00 |
| # Mismatches per 100 kbp | 2496.36 | 1.29 |
| # Indels per 100 kbp | 59.50 | 0.20 |
Limitations of evaluating metagenomic assemblers using purely simulated metagenomic data
Many simulation studies have been carried out to compare the performance of metagenomic assemblers. Since microbial communities are generally much more complex than simulated data, the results from simulation studies may be better than that for real metagenomic data. To test this, we took five known genomes chosen for being added in the human gut dataset as reference genomes. We assembled the hybrid reads of simulated genomes and real metagenomes, and then we compared the assembly with the assembly based on the simulated reads only. The corresponding results are shown in Table 5.
Table 5.
Summary statistics of the assembly of the datasets with different hybrid modes. The optimal numbers for each sequencing coverage and hybrid approach are in bold
| Assembly | Genome fraction (%) | Duplication ratio | Total aligned length | #misassemblies | #mismatches |
|---|---|---|---|---|---|
Simulated data (5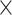 ) only ) only |
71.84 | 1.003 | 9546850.25 | 38.00 | 5623.00 |
SRR769529+Simulation(5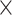 ) ) |
70.00 | 1.009 | 9343325.50 | 46.00 | 7330.75 |
Simulated data (20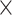 ) only ) only |
98.70 | 1.002 | 13168349.75 | 3.75 | 749.50 |
SRR769529+Simulation(20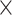 ) ) |
98.49 | 1.004 | 13157784.50 | 3.50 | 1636.00 |
Simulated data (50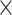 ) only ) only |
98.26 | 1.001 | 13109553.50 | 2.75 | 667.25 |
SRR769529+Simulation(50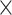 ) ) |
98.66 | 1.003 | 13172616.50 | 4.50 | 1419.75 |
The table shows that the assembly of the simulated data has better performance compared to the assembly of the mixture of the simulated data and real metagenomes at low sequencing depth. As for the high sequencing depths, the results are quite close except for the ‘# mismatches’ criterion. So if we simply use simulated data for evaluation, the assemblers will have better performance which does not reflect their performance on real metagenomic datasets, especially for the genomes at low sequencing depth. Therefore, it is useful to use the hybrid sequences of simulation and real metagenomes for the evaluation of metagenomic assemblers.
Comparison of metagenomic assemblers based on the hybrid approach
We also evaluated if the two metagenome datasets contain any of the added genomes used for simulation. To answer this question, we assembled the metagenomic reads from the two metagenomes using MEGAHIT, MetaSPAdes, IDBA-UD and Faucet and then used metaQUAST [21] to obtain the genome fractions of the genomes used for simulation. For the first dataset, the genome fractions for P. gingivali and S. salivarius were estimated to be 0.12% and 0.36%, respectively, and others were not present in the dataset. For the second dataset, the genome fractions for L. vestfoldensis, S. sediminis were estimated to be 0.37% and 0.26%, respectively, and the estimated genome fractions for the other genomes are all below 0.03%.
MetaSPAdes has the highest assembly integrity
We first evaluated assembly integrity measured by genome fraction and duplication ratio for the different genomes using the four assembly programs. The corresponding results are given in Tables S5 and S6 in the Supplementary Material. For Dataset 1, under all the simulated scenarios, the genome fractions for R. aquatilis and M. maripaludis are generally higher than that of P. gingivali, S. salivarius and A. actinomycetemcomitans, and the duplication ratios for P. gingivali, S. salivarius and A. actinomycetemcomitans are generally higher than that of the other organisms. For Dataset 2, the genome fractions for L. vestfoldensis and S. sediminis are generally lower than that of the other organisms and the duplication ratios for L. vestfoldensis and S. sediminis are generally higher than of the other organisms for all the assemblers. These results indicate that the genomes relatively far away from the real metagenomes are easier to assemble than genomes that are close to the metagenomes.
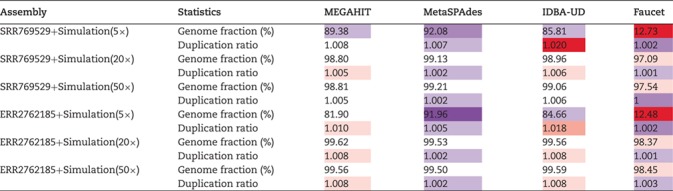
Table 6 shows the average genome fractions and duplication ratios of all the references in the assembly of the hybrid sequences. At all the sequencing depths, the assembly using MetaSPAdes has the highest genome fractions and comparable duplication ratios with others. At low sequencing coverage of 5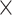 , Faucet has the lowest genome fractions. The genome fractions from MEGAHIT are close to that from MetaSPAdes in the human gut dataset, while the genome fractions from IDBA-UD were somewhat lower compared to that from the other two programs. For the marine dataset, the genome fractions from MetaSPAdes are much higher than that from other programs. At high sequencing coverage of 20 or higher, all the metagenomic assemblers have high genome fractions and low duplication ratios. For example, the genome fractions from three of the programs increased over 10% when the sequencing coverage changes from 5
, Faucet has the lowest genome fractions. The genome fractions from MEGAHIT are close to that from MetaSPAdes in the human gut dataset, while the genome fractions from IDBA-UD were somewhat lower compared to that from the other two programs. For the marine dataset, the genome fractions from MetaSPAdes are much higher than that from other programs. At high sequencing coverage of 20 or higher, all the metagenomic assemblers have high genome fractions and low duplication ratios. For example, the genome fractions from three of the programs increased over 10% when the sequencing coverage changes from 5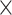 to 20
to 20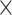 .
.
Table 6.
The assembly integrity measured by genome fraction and duplication ratio of the simulated genomes based on the hybrid approach using four assembler programs: MEGAHIT, MetaSPAdes, IDBA-UD and Faucet. Two real metagenomic datasets with different sequencing amounts (SRR769429 of data size 7.9Gb and ERR2762185 of data size 145.8Gb) were used (purple/red cells indicate that the results improve/deteriorate compared with the median value)
|
|
MetaSPAdes has the highest assembly contiguity and MEGAHIT follows
We next evaluated the assembly continuity using the continuity measures in Table 3. The complete results are given in the Supplementary Material as Tables S7 and S8 using real metagenome datasets SRR769429 and ERR2762185, respectively. For the NGA50 criterion, if a contig has a misassembly with respect to the reference, the contig will be broken into smaller pieces. Therefore, the NGA50 criterion is more reliable than N50 for evaluating assembly continuity. Figure 4 shows the NGA50 results for individual genomes of the six mixed datasets with different combinations of sequencing coverage and real metagenome datasets. MetaSPAdes performs best on most individual genomes in terms of the most contiguity statistics. MEGAHIT and IDBA-UD have comparable performance in terms of assembly contiguity, and Faucet has worst assembly contiguity in general. With the increase of sequencing depth, the assembly contiguity of each assembler is improved greatly, from about 2000 bp to over 19 000 bp for all the four assemblers in terms of N50.
Figure 4.
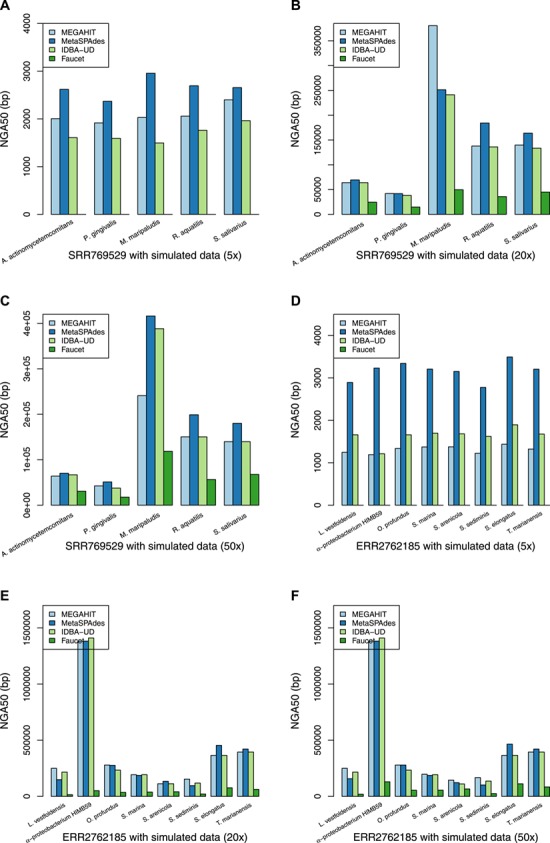
The NGA50 statistics of the assembly for the individual genomes based on different combinations of sequencing coverage and real metagenome datasets. (A) The NGA50 statistics of the mixture of the SRR769529 dataset and the simulation (5x). (B) The NGA50 statistics of the mixture of the SRR769529 dataset and the simulation (20x). (C) The NGA50 statistics for the individual genomes of the assembly of the mixture of the SRR769529 dataset and the simulation (50x). (D) The NGA50 statistics of the mixture of the ERR2762185 dataset and the simulation (5x). (E) The NGA50 statistics of the mixture of the ERR2762185 dataset and the simulation (20x). (F) The NGA50 statistics for the individual genomes of the assembly of the mixture of the ERR2762185 dataset and the simulation (50x).
The individual genomes that have greater genetic difference from the genomes in the real metagenome often obtain the assembly with better contiguity at high sequencing depth, however, the difference is not obvious at low sequencing depth. For instance, the NGA50 of the assembly of M. maripaludis is over 6 those of other genomes at high sequencing depth but close to others at low sequencing depth while using MetaSPAdes. For the individual genomes that have smaller genetic difference from the genomes in the ERR2762185 dataset, L. vestfoldensis and S. sediminis, MetaSPAdes performs best at low sequencing depth while MEGAHIT performs best at high sequencing depth.
those of other genomes at high sequencing depth but close to others at low sequencing depth while using MetaSPAdes. For the individual genomes that have smaller genetic difference from the genomes in the ERR2762185 dataset, L. vestfoldensis and S. sediminis, MetaSPAdes performs best at low sequencing depth while MEGAHIT performs best at high sequencing depth.
Faucet has best performance in assembly accuracy and MetaSPAdes follows
Finally, we evaluated the assembly accuracy of the different genomes using the different assembly programs and the complete results based on the accuracy measures in Table 3 are given in Tables S9 and S10 in the Supplementary Material. The aggregated results for the reference genomes based on the number of misassemblies, misassembled contig length, number of mismatches per 100 kbp and the number of indels per kbp are shown in Table 7. For each sequencing coverage, the assembly from Faucet has the best performance in accuracy and the assembly from MetaSPAdes and MEGAHIT have overall comparable performance in accuracy on the human gut dataset. At high sequencing coverage, the assembly accuracies from MetaSPAdes and Faucet are similar on the marine dataset. The misassembled contig length and number of mismatches from MetaSPAdes are close to that from MEGAHIT, but MetaSPAdes often has the highest number of indels at high sequencing depth. IDBA-UD has the highest number of misassemblies, but the number of mismatches and the number of indels decrease rapidly with the increase of sequencing coverage, almost the same as that of MEGAHIT. Since some rare species may not be well assembled because of low coverage, we present the whole results of mixture of real metagenome ERR2762185 and simulated data with low sequencing depth in Figure 5. Although the assembly from Faucet has high accuracy, the genome fractions were far lower than that of others. Therefore, the results of the assembly from Faucet are not shown in Figure 5. IDBA-UD has the worst performance with respect to the number of misassemblies, misassembled contig length and the number of mismatches per 100 kbp, meanwhile MetaSPAdes and MEGAHIT have comparable performance in these three criteria. However, MetaSPAdes has the worst performance in the number of indels per 100 kbp.
Table 7.
The assembly accuracy of the simulated genomes based on the hybrid approach using four assembler programs: MEGAHIT, MetaSPAdes, IDBA-UD and Faucet. Two real metagenomic datasets with different sequencing amounts (SRR769429 of data size 7.9Gb and ERR2762185 of data size 145.8Gb) were used (purple/red cells indicate that the results improve/deteriorate compared with the median value)
|
|
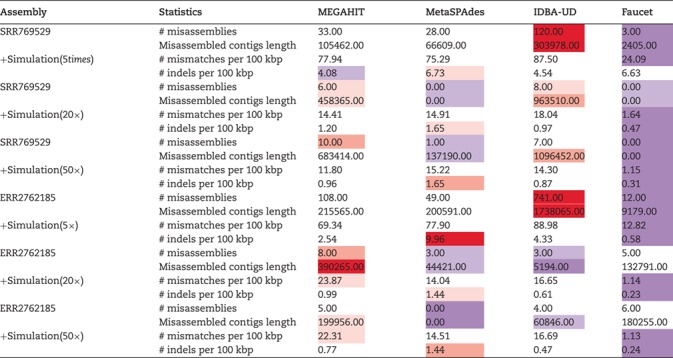
Figure 5.
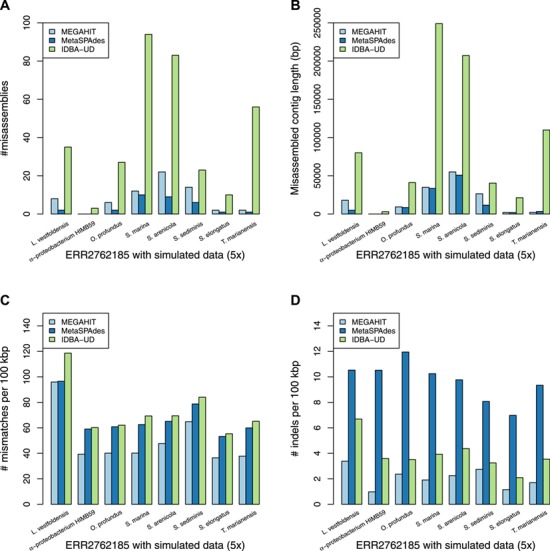
The assembly accuracy results of the simulated genomes with sequencing coverage of 5x and ERR2762185 metagenome dataset. (A) The numbers of the misassemblies of the assembly of the ERR2762185 dataset with the simulated data (5x). (B) The misassembled contig lengths of the assembly of the ERR2762185 dataset with the simulated data (5x). (C) The numbers of the mismatches per 100 kbp of the assembly of the ERR2762185 dataset with the simulated data (5x). (D) The numbers of the indels per 100 kbp of the assembly of the ERR2762185 dataset with the simulated data (5x).
In terms of individual genomes, the assembly of M. maripaludis has the best accuracy but the assembly of R. aquatilis does not have obvious advantages at low sequencing depth based on the human gut metagenomic data. The assembly of L. vestfoldensis from all the assemblers has the worst number of mismatches per 100 kbp based on the marine metagenomic data.
MetaSPAdes has the best performance on strain-level genomes
There might be similar strains in real metagenomes, and it is important to evaluate whether the metagenomic assemblers can handle strain micro-diversity. To test this, we included five Escherichia coli strains in the human dataset with simulated data. The complete results of the individual strains are given in Table S11 in the Supplementary Material. The aggregated results for the references are shown in Table 8. Although the assembly from Faucet has high accuracy, the genome fractions and assembly contiguity were far lower than that of others. At low sequencing depth, the assembly from MEGAHIT, MetaSPAdes and IDBA-UD have comparable genome fractions and assembly contiguity but MetaSPAdes performs best on the duplication ratio and the accuracy criteria. At high sequencing depth, the assembly from MEGAHIT has highest genome fractions and assembly contiguity, but the assembly accuracy were far lower than that of MetaSPAdes and Faucet.
Table 8.
The statistics of the assembly of the five simulated E. coli strains based on the hybrid approach using four assembler programs: MEGAH IT, MetaSPAdes, IDBA-UD and Faucet (purple/red cells indicate that the results improve/deteriorate compared with the median value)
|
|
MEGAHIT has the shortest computational time
Table 9 shows the running time of the compared programs. MEGAHIT has the shortest computational time. Even though Faucet does not currently support multi-threaded execution, its running time is comparable with that of the multi-threaded assemblers, MetaSPAdes and IDBA-UD.
Table 9.
Running time of the different assembly programs (purple/red cells indicate that the results improve/deteriorate compared with the median value)
|
|

aRuns of Faucet were performed with a single thread, as it does not currently support multi-threaded execution.
Discussion
Metagenomic sequence assembly is an essential while challenging problem in metagenomic studies. The characteristics of the metagenomic data, such as uneven depth, genomic variants of the different microbial organisms and the large volume of data, undoubtedly increase the difficulty of metagenomic reads assembly. Previous comparative studies of metagenomic assemblers either used purely simulated or completely real metagenomes. We showed in this paper that the purely simulation-based studies tend to yield better performance than in real metagenomic studies, while real metagenome based studies tend to give more conserved performance because the genomes recovered from metaQUAST [21] may not be the true genomes in the metagenome resulting in much lower assembly accuracy than the true accuracy.
In this study, we used a hybrid simulation approach to evaluate metagenomic reads assemblers by adding simulated sequence reads from multiple genomes to real metagenomes, assembling the combined reads and evaluating the assemblers for the simulated genomes. Such a hybrid approach can yield more realistic assembly quality including integrity, continuity and accuracy overcoming the problems using either the purely simulated or real metagenomes. We evaluated the performance of four popular metagenomic assemblers: MEGAHIT, MetaSPAdes, IDBA-UD and Faucet. When the sequencing coverage is relatively high (20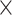 or higher), all four programs perform quite well on species-level. However, there are some marked differences when the sequencing coverage is low. At low sequencing coverage, MetaSPAdes performs best in terms of integrity and continuity. Faucet performs best in terms of accuracy at a cost of low integrity and continuity. MEGAHIT has the highest genome fractions on the strain-level genomes and MetaSPAdes has the best overall performance at the strain-level. We also showed that isolated genomes tend to be easily assembled than genomes that are similar to the others in the metagenome. MEGAHIT is the most efficient metagenomic assembler compared with other assemblers due to the use of SDBG. Our study provides useful guidelines for the choice of metagenomic assemblers in practical studies.
or higher), all four programs perform quite well on species-level. However, there are some marked differences when the sequencing coverage is low. At low sequencing coverage, MetaSPAdes performs best in terms of integrity and continuity. Faucet performs best in terms of accuracy at a cost of low integrity and continuity. MEGAHIT has the highest genome fractions on the strain-level genomes and MetaSPAdes has the best overall performance at the strain-level. We also showed that isolated genomes tend to be easily assembled than genomes that are similar to the others in the metagenome. MEGAHIT is the most efficient metagenomic assembler compared with other assemblers due to the use of SDBG. Our study provides useful guidelines for the choice of metagenomic assemblers in practical studies.
Key Points
Metagenomic assembly is challenging and essential for the follow-up studies. There are many metagenomic assemblers developed.
Benchmarking metagenomic assemblers based on hybrid reads of real and simulated metagenomic sequences would better reflect the performance of the metagenomic assemblers on real metagenomic datasets.
Four advanced metagenome assemblers, MEGAHIT, MetaSPAdes, IDBA-UD and Faucet, are selected for evaluation.
Overall, MetaSPAdes performs best in terms of integrity and continuity on the species-level and MEGAHIT follows. Faucet performs best in terms of accuracy at a cost of low integrity and continuity at low sequencing depth. MEGAHIT has the highest genome fractions on strain-level genomes and MetaSPAdes has the best overall performance on strain-level genomes. MEGAHIT is the most computationally efficient in our experiments.
Supplementary Material
Funding
ZW and SZ are partially supported by the National Natural Science Foundation of China (Nos. 61572139 and 61872094) JF and FS are partially supported by US National Science Foundation (NSF) [DMS-1518001] and National Institutes of Health (NIH) [R01GM120624]. WY is supported by the National Natural Science Foundation of China (61673324) and the Natural Science Foundation of Fujian (2016 J01316). SZ is also supported by the 111 Project (NO. B18015), the key project of Shanghai Science & Technology (No. 16JC1420402), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01) and ZJLab.
Ziye Wang is a graduate student in the School of Mathematical Sciences and the Institute of Science and Technology for Brain-inspired Intelligence, Fudan University, Shanghai, China. Her research interests include machine learning, data mining and their applications in metageomics.
Ying Wang is a professor in the Department of Automation, Xiamen University, Xiamen, China. Her research interests include machine learning, data mining and their applications in bioinformatics, especially in metageomics.
Jed A. Fuhrman is a professor in the Department of Biological Sciences and Wrigley Institute for Environmental Studies, University of Southern California, Los Angeles, California, United States of America. His research interests include marine metagenomics, microbial biodiversity, aquatic microbial ecology, biological oceanography and water quality.
Fengzhu Sun is a professor in the Quantitative and Computational Biology Program, Department of Biological Sciences, University of Southern California, Los Angeles, California, United States of America. He is also a guest professor of the Institute of Science and Technology for Brain-inspired Intelligence, Fudan University, Shanghai, China. His recent research interests include metagenomics, alignment-free genome and metagenome comparison, and molecular networks.
Shanfeng Zhu is an associate professor in Shanghai Key Lab of Intelligent Information Processing, the School of Computer Science and the Institute of Science and Technology for Brain-inspired Intelligence, Fudan University, Shanghai, China. His research interests include machine learning, data mining and their applications in bioinformatics.
References
- 1. Ley RE, Turnbaugh PJ, Klein S, et al. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022–1023. [DOI] [PubMed] [Google Scholar]
- 2. Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013; 500:541–546. [DOI] [PubMed] [Google Scholar]
- 3. Gagnière J, Raisch J, Veziant J,. et al Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 2016;22:501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qin J. Li Yand Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012; 490:55–60. [DOI] [PubMed] [Google Scholar]
- 5. Qin N, Yang F, Li A, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014;513:59–64. [DOI] [PubMed] [Google Scholar]
- 6. Dicksved J, Halfvarson J, Rosenquist M, et al. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J 2008;2:716–727. [DOI] [PubMed] [Google Scholar]
- 7. Zitvogel L, Ma Y, Raoult D,. et al The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–1370. [DOI] [PubMed] [Google Scholar]
- 8. Hartmann N, Kronenberg M. Cancer immunity thwarted by the microbiome. Science 2018;360:858–859. [DOI] [PubMed] [Google Scholar]
- 9. Ma C, Han M, Heinrich B,. et al Gut microbiome-mediated bile acid metabolism regulates liver cancer via nkt cells. Science 2018;360:eaan5931. [DOI] [PMC free article] [PubMed]
- 10. Cram JA, Xia LC, Needham DM,. et al Cross-depth analysis of marine bacterial networks suggests downward propagation of temporal changes. ISME J 2015;9:2573–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schloter M, Nannipieri P, Sørensen SJ,. et al. Microbial indicators for soil quality. Biol Fertil Soils 2018;54:1–10. [Google Scholar]
- 12. Charuvaka A, Rangwala H. Evaluation of short read metagenomic assembly. BMC Genomics 2011;12:S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pignatelli M, Moya A. Evaluating the fidelity of De Novo short read metagenomic assembly using simulated data. PLoS One 2011;6:e19984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quince C, Walker AW, Simpson JT,. et al Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 2017;35:833–844. [DOI] [PubMed] [Google Scholar]
- 15. Sczyrba A, Hofmann P, Belmann P,. et al Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat Methods 2017;14:1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olson ND, Treangen TJ, Hill CM,. et al Metagenomic assembly through the lens of validation: recent advances in assessing and improving the quality of genomes assembled from metagenomes. Brief Bioinform 2017, doi: 10.1093/bib/bbx098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. White DJ, Wang J, Hall RJ. Assessing the impact of assemblers on virus detection in a de novo metagenomic analysis pipeline. J Comput Biol 2017; 24:874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mende DR, Waller AS, Sunagawa S,. et al Assessment of metagenomic assembly using simulated next-generation sequencing data. PLoS One 2012;7:e31386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greenwald WW, Klitgord N, Seguritan V,. et al Utilization of defined microbial communities enables effective evaluation of meta-genomic assemblies. BMC Genomics 2017;18:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nurk S, Meleshko D, Korobeynikov A,. et al MetaSPAdes: a new versatile metagenomic assembler. Genome Res 2017; 27:824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mikheenko A, Saveliev V, Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 2016; 32:1088–1090. [DOI] [PubMed] [Google Scholar]
- 22. Peng Y, Leung HC, Yiu SM,. et al Meta-IDBA: a de Novo assembler for metagenomic data. Bioinformatics 2011;27:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boisvert S, Raymond F, Godzaridis E,. et al Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol 2012;13:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haider B, Ahn TH, Bushnell B,. et al Omega: an overlap-graph de novo assembler for metage-nomics. Bioinformatics 2014; 30:2717–2722. [DOI] [PubMed] [Google Scholar]
- 25. Li D, Liu CM, Luo R,. et al MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31:1674–1676. [DOI] [PubMed] [Google Scholar]
- 26. Miller JR, Koren S, Sutton G,. et al Assembly algorithm for next-generation sequencing data. Genomics 2010;95:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pop M. Genome assembly reborn: recent computational challenges. Brief Bioinform 2009;10:354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boisvert S, Laviolette F, Corbeil J. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol 2010; 17:1519–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peng Y, Leung HCM, Yiu SM, et al IDBA—a practical iterative de Bruijn graph de novo assembler. In: Proceedings of the 14th Annual International Conference on Research in Computational Molecular Biology (RECOMB 2010), 2010, p. 426–40. ACM Press, Lisbon. [Google Scholar]
- 31. Bankevich A, Nurk S, Antipov D,. et al SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peng Y, Leung HC, Yiu SM,. et al IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012;28:1420–1428. [DOI] [PubMed] [Google Scholar]
- 33. Namiki T, Hachiya T, Tanaka H,. et al MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res 2012;40:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li D, Luo R, Liu CM. et al MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016;102:3–11. [DOI] [PubMed] [Google Scholar]
- 35. Chin FY, Leung HC, Yiu SM. Sequence assembly using next generation sequencing datachallenges and solutions. Sci China Life Sci 2014;57:1140–1148. [DOI] [PubMed] [Google Scholar]
- 36. Ghurye JS, Cepeda-Espinoza V, Pop M. Metagenomic assembly: overview, challenges and applications. Yale J Biol Med 2016;89:353–362. [PMC free article] [PubMed] [Google Scholar]
- 37. Rozov R, Goldshlager G, Halperin E,. et al Faucet: streaming de novo assembly graph construction. Bioinformatics 2018;34:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holtgrewe M. Mason—a read simulator for second-generation sequencing data. Technical Report, FU Berlin, 2010. [Google Scholar]
- 39. Salzberg SL, Phillippy AM, Zimin A,. et al GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res 2012;22:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gurevich A, Saveliev V, Vyahhi N,. et al QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013;29(8):1072–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scholz M, Lo CC, Chain PS. Improved assemblies using a source-agnostic pipeline for metagenomic assembly by merging (MeGAMerge) of contigs. Sci Rep 2014;4:6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mohamadi H, Khan H, Birol I. ntCard: a streaming algorithm for cardinality estimation in genomics data. Bioinformatics 2017;33:1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.