

Abstract
Hypoxemia immediately following traumatic brain injury (TBI) has been observed to exacerbate injury. However, it remains unclear whether delayed hypoxemia beyond the immediate postinjury period influences white matter injury. In a retrospective clinical cohort of children aged 4–16 years admitted with severe TBI, 28/74 (35%) patients were found to experience delayed normocarbic hypoxemia within 7 days of admission. Based on these clinical findings, we developed a clinically relevant mouse model of TBI with delayed hypoxemia by exposing 5-week old (adolescent) mice to hypoxic conditions for 30 minutes starting 24 hours after moderate controlled cortical impact (CCI). Injured mice with hypoxemia had increased axonal injury using both β-amyloid precursor protein and NF200 immunostaining in peri-contusional white matter compared with CCI alone. Furthermore, we detected increased peri-contusional white matter tissue hypoxia with pimonidazole and augmented astrogliosis with anti-glial fibrillary acidic protein staining in CCI + delayed hypoxemia compared with CCI alone or sham surgery + delayed hypoxemia. Microglial activation as evidenced by Iba1 staining was not significantly altered by delayed hypoxemia. These clinical and experimental data indicate the prevention or amelioration of delayed hypoxemia effects following TBI may provide a unique opportunity for the development of therapeutic interventions to reduce axonal injury and improve clinical outcomes.
Keywords: Axonal injury, Brain hypoxia, Controlled cortical impact, Delayed hypoxemia, Secondary injury, Traumatic brain injury
INTRODUCTION
Despite advances in care, morbidity following traumatic brain injury (TBI) remains high ( 1 , 2 ). Following the primary direct mechanical injury to brain parenchyma, important secondary injuries develop over hours to days from multiple addressable mechanisms including hypoxemia, ischemia, and excitotoxicity ( 3 ). Clinical observational studies have found a strong association between early hypoxemia (pre-hospital or emergency room setting) following TBI and poor clinical outcomes ( 4–7 ). Preclinical investigations of hypoxemia immediately after experimental TBI have demonstrated exacerbations in brain edema and ischemia, hippocampal neuronal cell death, neuroinflammation, axonal injury, and behavioral deficits ( 8–12 ).
Patients with TBI in the intensive care setting are also at high risk for delayed secondary hypoxemia due to pulmonary contusions, aspiration, pneumonia, atelectasis, acute respiratory distress syndrome, and as a result of procedural interventions such as endotracheal tube displacement, emergent diagnostic imaging, and surgical interventions. However, little is known about the role that delayed hypoxemia (beyond the pre-hospital and emergency room setting) plays in secondary brain injury following TBI. A small prospective study found that any hypoxemia, defined as O 2 saturation < 90%, within the first 24 hours after TBI was associated with an increase in mortality ( 13 ). The majority of previous animal studies investigating post-TBI hypoxemia have utilized a 15- to 30-minute exposure of low inspired oxygen (8%–12%) occurring within 1 hour of experimental TBI ( 8–12 ). No studies to our knowledge have investigated delayed hypoxemia after TBI in an animal model.
Although traumatic axonal injury (TAI) is considered to be the major contributor to long-term cognitive deficits in TBI patients ( 14–18 ), only 1 preclinical study has investigated the contribution of immediate postinjury hypoxemia to axonal injury ( 11 ). Axonal injury following TBI is most commonly characterized histologically by immunohistochemical staining of amyloid precursor protein (APP) and neurofilament. APP under normal conditions traverses the length of the axon, but in response to injury will accumulate at axonal varicosities and retraction bulbs ( 19–22 ). Changes in structural integrity of the axonal cytoskeleton can result in increased neurofilament immunoreactivity ( 16 , 23 , 24 ). These 2 approaches, when combined, can provide a more complete histological characterization of axonal injury following TBI ( 19 , 25 , 26 ). To our knowledge, the impact of delayed hypoxemia on axonal injury has not been investigated in preclinical models of TBI.
Based on retrospective pediatric intensive care data, we developed a clinically relevant mouse model of delayed systemic hypoxemia following TBI. We used this model to test the hypothesis that delayed posttraumatic hypoxemia following controlled cortical impact (CCI) in young mice would exacerbate white matter injury.
MATERIALS AND METHODS
Patient Data
The Institutional Review Board at Washington University approved the retrospective clinical study and waived the requirement for obtaining written informed consent. Data were abstracted from registry of pediatric patients with severe TBI (postresuscitation Glasgow Coma Score (GCS) of 8 or less who were admitted to the pediatric intensive care unit (PICU) at Saint Louis Children’s Hospital between July 1, 1999 and December 31, 2011. Patients were excluded if they had a GCS of 3 with fixed and dilated pupils at time of admission, cardiac arrest prior to arrival in the PICU, abusive head trauma, and gunshot wounds to the head. Age at time of injury, sex, postresuscitation GCS, pediatric risk of mortality score (PRISM III), injury severity score, injury mechanism, PICU length of stay, hospital length of stay, PICU-free days at 28 days of admission, Glasgow outcome scale score at hospital discharge, and destination at hospital discharge were recorded. Initial head computed tomography scan was categorized by using the Marshall classification of initial head computed tomography ( 27 ). PICU-free days at 28 days of admission was defined as the total numbers of days alive outside the PICU at 28 days after admission. Complete arterial blood gas, hourly pulse oximetry, and end tidal CO 2 data were abstracted from the medical record. Hypoxemia was defined as a pulse oximetry reading < 90% or an arterial PO 2 <60 mm Hg. A discreet episode of hypoxemia was defined as complete with the first subsequent normal blood gas or pulse oximetry reading.
Animal Studies
Injury
All procedures were approved by the Washington University Animal Studies Committee, and are consistent with the National Institutes of Health guidelines for the care and use of animals. Five week-old C57BL/6J male mice (Jackson Laboratory, Bar Harbor, ME) weighing 16–20 g were used in these experiments. For studies of axonal injury, mice were killed at 2 different time points: 48 hours and 1 week after injury (n = 20 total for each time point). Two additional cohorts of 20 animals each were utilized for the same 2 time points (48 hours and 1 week after injury) to evaluate the response of microglia (1 week time point only) and astrocytes (48 hours and 1 week). For each time point, 8 mice underwent sham injury and 12 mice underwent CCI ( 28–30 ). The mice were anesthetized with 5% isoflurane at induction, followed by maintenance at 2% isoflurane for the duration of the procedure. The head was shaved and head holders were used to stabilize the head within the stereotaxic frame (MyNeurolab, St. Louis, MO). Then, a single 5-mm craniotomy was performed by an electric drill on the left lateral side of the skull centered 2.7 mm lateral from the midline and 3 mm anterior to lambda. Animals were randomized to sham or injury after craniotomy utilizing a computer generated numbers randomization. For injured animals, the 3-mm electromagnetic impactor tip was then aligned with the craniotomy site at 1.2 mm left of midline, 1.5 mm anterior to the lambda suture. The impact was then delivered at 2 mm depth (velocity 5 m/s, dwell time 100 ms). The head holders were released immediately after the injury. All animals then received a loose fitting plastic cap secured over the craniotomy with Vetbond (3M, St. Paul, Minnesota). The skin was closed with interrupted sutures and treated with antibiotic ointment before removing the mouse from anesthesia and allowing recovery on a warming pad.
Delayed Hypoxemia
One day after injury or sham surgery, animals were randomized to normoxemia (room air) or hypoxemia (8% O 2 , 4% CO 2 ) for 30 minutes. During normoxemia or hypoxemia animals were placed in fresh cages with littermates randomized to the same treatment arm and all mice were subjected to identical transport and handling. Animals randomized to hypoxemia were placed in a Coy Labs Hypoxia Chamber (6′ × 3′ × 4′) (Coy Laboratory, Grass Lake, MI), fitted with airlock, oxygen sensor, carbon dioxide sensor, and gas controllers.
Arterial Blood Gas Sampling
A separate cohort of 18 mice was utilized for the arterial blood gas experiments. Animals were randomized to injury or sham surgery (n = 9 for each group). One day after injury or sham surgery, animals were anesthetized with 5% isoflurane at induction, followed by maintenance at 2% isoflurane for placement of right carotid artery tunneled catheters. Once completely recovered and awake, animals underwent 30 minutes of low inspired oxygen (8% O 2 ,) and arterial blood samples were drawn at the end of the 30 minutes. Arterial blood gas analysis was performed on a Bayer Rapidlab 845 blood gas analyzer (Siemens Medical Diagnostics, Bayer, Tarrytown, NY). Initial screening was performed on 1 sham and 1 injured mouse at each of the following CO2 concentrations: 0.5%, 2%, 4%, and 6%. The conditions that produced normocarbia were then repeated with the 10 remaining mice (n = 5 for each group).
Immunohistochemistry
Mice were killed under isoflurane anesthesia by transcardial perfusion with 0.3% heparin in phosphate-buffered saline. Whole brains were removed and fixed in 4% paraformaldehyde for 48 hours, followed by equilibration in 30% sucrose for at least 48 hours prior to sectioning. Serial 50-μm-thick coronal slices were cut on a freezing microtome starting with the appearance of a complete corpus callosum and caudally to bregma −3.08 mm. Sets of 12 sections spaced every 300 μm were mounted on glass slides and used for immunohistochemical studies.
Staining was performed on free-floating sections washed in Tris-buffered saline (TBS) between applications of primary and secondary antibodies. Endogenous peroxidase was blocked by incubating the tissue in TBS + 3% hydrogen peroxide for 10 minutes. Normal goat serum (3%) in TBS with 0.25% Triton X (Tris-buffered saline (TBS)-X) was used to block nonspecific staining for all antibodies. Slices were then incubated at 4 °C overnight with one of the following primary antibodies: polyclonal rabbit anti-β-APP (Invitrogen, Carlsbad, CA) at a concentration of 1:1000; polyclonal rabbit anti-neurofilament-200 (Sigma, St. Louis, MO) at a concentration of 1:8000; polyclonal rabbit anti-Iba1 (Wako Chemicals USA, Richmond, VA) at a concentration of 1:1000; or polyclonal rabbit anti-glial fibrillary acidic protein (GFAP) (Dako North America, Carpinteria, CA) at a concentration of 1:1000. Biotinylated goat anti-rabbit secondary antibodies (Vector Laboratories, Burlingame, CA) in TBS-X were used at a 1:1000 concentration to detect bound primary antibodies. Colorization was achieved using the Vectastain ABC Elite Kit (Vector Laboratories) followed by the application of 3-3′ diaminobenzidine (DAB).
Detection of Brain Tissue Hypoxia
To determine if brain tissue hypoxia was occurring during delayed hypoxemia in regions where TAI was occurring, we utilized Hypoxyprobe TM -omni kit (Hypoxyprobe, Burlington, Massachusetts) (pimonidazole) to quantify white matter tissue hypoxia ( 31–33 ). Pimonidazole selectively adducts with thiol groups in proteins, peptides, and amino acids under hypoxic conditions (PO 2 <10 mm Hg) ( 34 ). Twenty mice (CCI, n = 12, sham, n = 8) were administered 60 mg/kg of pimonidazole intraperitoneally and then immediately randomized to either 30 minutes of hypoxemia or room air. Seventy-five minutes after injection of pimonidazole, mice were killed and brains were prepared for immunohistochemistry as described above. Affinity purified anti-pimonidazole rabbit antiserum (PAb2627AP, Hypoxyprobe) at a concentration of 1:1000 was utilized as the primary antibody.
Quantification of Immunohistochemistry
The extent of tissue loss of the ipsilateral hemisphere for each animal was quantified using images of β-APP stained slices acquired using Hamamatsu NanoZoomer 2.0-HT System (Hamamatsu Corporation, Middlesex, New Jersey). Tissue loss in the injured hemisphere was calculated as a percentage of the tissue volume in the contralateral hemisphere as described by others ( 35 ). Stereological analysis was performed using StereoInvestigator software version 8.2 (MBF Bioscience, Williston, Vermont). Assessments were made by an investigator blinded to group assignment. The optical fractionator function was used to quantify target markers per cubic millimeter of tissue. For quantification of axonal injury, a grid size of 250 × 250 µm, a counting frame of 40 × 40 µm, and a dissector height of 15 µm with a guard zone of 5 µm were used for all quantifications, resulting in 3% of the regions of interest (ROI) being randomly sampled. All ROIs were traced at 4× magnification and markers were counted at 60× magnification. The ipsilateral corpus callosum and external capsule spanning 12 sections starting with the appearance of a complete corpus callosum and caudally to bregma −3.08 mm were used as the ROI for the β-APP and neurofilament-200 (NF200). This region was defined as the white matter area between midline and the lateral edge of the cingulum in rostral sections; in caudal sections, a horizontal line drawn laterally from the end of the fimbria served as the end boundary of the ROI. Injured axons were identified by β-APP-positive varicosities greater than 5 µm. Similarly, NF200-positive axonal varicosities greater than 5 µm in diameter were counted as injured axons during stereological assessment. For stereological quantification of Iba1-positive cells, the optical fractionator function was again used, with a grid size of 180 × 180 µm and a counting frame of 80 × 80 µm. The ROI began with the most anterior slice containing hippocampal dentate gyrus and ended with the most posterior section containing corpus callosum fibers that cross midline which yielded 3–4 sections for analysis per animal. The midline served as the medial boundary for the ROI, whereas the lateral boundary was formed by drawing a horizontal line between the ventral hippocampus and dorsal thalamus in each coronal section. Gunderson’s coefficients of error were <0.1 for all stereological quantifications.
Automated quantification of axonal injury was performed with Visiopharm software version 4.3.1.0 (Visiopharm, Broomfield, Colorado) on images acquired using Hamamatsu NanoZoomer 2.0-HT System. Using the same ROI described above, 3 labels were generated for β-APP stained slices: 2 labels for positively stained axons (APP puncta, for cross sectional and APP axons for longitudinal), and 1 label for background white matter. The classification method used was threshold, and the ranges for the 2 stained axon labels were 0–7, and 7.01–190 for background white matter. The post process parameters were as follows, in order: APP puncta with an area <12 µm 2 and APP axons with an area <30 µm 2 were transformed to background tissue, APP puncta were separated into discreet objects, APP puncta with an area greater than 50 µm 2 were transformed into APP axons, APP axons were separated into discreet objects, APP axons label with an area greater than 120 µm 2 were transformed to background tissue, and APP puncta labeled areas with an irregularity greater than 3 were transformed to background tissue. For NF200 staining, the classification method used was threshold, and the ranges for NF-200 puncta, NF200 axons, and tissue were 0–30, 0–30, and 30.01–200, respectively. The post processes applied were as follows, in order: NF200 puncta were separated into discreet objects, NF200 puncta with an area <15 µm 2 were transformed into background tissue, NF200 puncta with an irregularity greater than 4 were transformed into NF200 axons, NF200 axons with an area <25 µm 2 were transformed into tissue, NF200 axons were then separated into discrete objects, and NF-200 axons with an area greater than 200 µm 2 were transformed into background tissue.
Quantification of brain tissue hypoxia staining was performed in the same ROI for axonal injury. Two labels, 1 for hypoxic cells and 1 for background white matter tissue were utilized. Threshold was again used as the classification method, and the ranges for hypoxic cells and background tissue were 0–90 and 90.01–210, respectively. The post processes were as follows: Separate objects for hypoxic cells and change hypoxic cells with an area of <6 µm 2 to background tissue. Test–retest reliability was evaluated by staining a second set of 12 slices from each brain analyzed and having the same blinded operator repeat the image analysis on these stained slices. Interrater reliability was assessed by having a second blinded operator perform the image analysis on the same set of images.
For GFAP measurements, 5× images of the ipsilateral corpus callosum and external capsule were obtained using the Nanozoomer. Using ImageJ, files were converted to 32-bit images and positive signal on each image was selected using the Max Entropy auto thresholding method. ROIs were selected and drawn as described for stereological assessment of microglia in ipsilateral white matter. Total area of ROIs and area of positive thresholding from each slice were calculated and summed to determine total percent area of positive GFAP staining.
All postprocessing and automated quantification algorithms are freely available upon request.
Statistical Analysis
All data were analyzed using Statistica version 12 (Dell Inc., Tulsa, OK). Clinical continuous data is presented as median and interquartile ranges. The association of variables and hypoxemia was performed using Kruskal–Wallis for continuous data and Fisher exact test for categorical data. Animal data results are presented as mean ± SD. For all animal data sets there was no evidence for significant deviations from normal distribution (p > 0.05 by Shapiro–Wilk tests). Quantitative histologic data were analyzed with 2-way ANOVA, followed by Tukey tests for multiple comparisons with a significance level of p < 0.05. Spearman correlations were utilized to assess the correlation between stereological and Visiopharm-based quantification of axonal injury.
RESULTS
Delayed Hypoxemia After Severe TBI Is Common
Seventy-four patients met study inclusion criteria and had complete arterial blood gas and hourly pulse oximetry data available. Twenty-eight patients (35%) were found to have experienced hypoxemia (PaO 2 <60 mm Hg or pulse oximetry <90%) within in the first 7 days of admission. The length of all hypoxemic episodes was observed to be no >1 hour, but their exact length could not be determined due to the resolution of the physiologic data in the medical record (hourly recordings). The majority of these episodes occurred in the first 48 hours ( Fig. 1 ). All patients were observed to be normocarbic during episodes of hypoxemia. Patients with hypoxemia were more commonly female; otherwise there were no differences in demographic data or injury severity between the groups ( Table 1 ). Patients who experienced hypoxemia were more likely to have a longer PICU stay. Patients with delayed hypoxemia trended toward poorer outcome (GOS of 3 or less) at hospital discharge (54% vs 29%) but this did not reach statistical significance. However, when GOS at hospital discharge was dichotomized, patients with delayed hypoxemia had poorer outcome (54% vs 29%, p = 0.047).
FIGURE 1.

Delayed hypoxemia (defined as O 2 saturation of <90% or PaO 2 <60 mm Hg) in pediatric patients with severe traumatic brain injury (TBI). Frequency and timing of hypoxemia in 74 pediatric patients admitted to the ICU with severe TBI. Forty-eight discreet episodes of hypoxemia were observed in 26 (35%) patients.
TABLE 1.
Demographic Characteristics, Severity of Injury, Injury Mechanisms, and Raw Outcomes
| Hypoxemia | Normoxemia | p Value | |
|---|---|---|---|
| n = 26 | n = 48 | ||
| Age (months) | 121 [55–189] | 144 [109–179] | 0.31 |
| Sex, male | 11 (42%) | 28 (58%) | 0.14 |
| Post resuscitation GCS | 4 [3–5] | 5 [3–6] | 0.25 |
| PRISM III | 6 [2–14] | 5 [2–8] | 0.26 |
| ISS | 28 [16 –41] | 28 [21 –35] | 0.61 |
| Injury mechanism | 0.088 | ||
| Motor vehicle accident | 18 (69%) | 34 (71%) | |
| Pedestrian accident | 4 (15%) | 9 (19%) | |
| Fall | 1 (4%) | 5 (10%) | |
| Other | 3 (12%) | 0 (0%) | |
| Marshall CT classification | 0.33 | ||
| Diffuse Injury I | 0 | 1 (2%) | |
| Diffuse Injury II | 24 (92%) | 38 (79%) | |
| Diffuse Injury III | 1 (4%) | 6 (13%) | |
| Diffuse Injury IV | 0 | 1 (2%) | |
| Evacuated mass legion | 0 | 2 (4%) | |
| Nonevacuated mass legion | 1 (4%) | 0 | |
| PICU LOS | 15.1 [12.2–23.0] | 12.3 [5.0–15.5] | 0.014 |
| PICU free days at 28 days | 11.6 [4.0–15.8] | 15.1 [11.8–22.7] | 0.017 |
| Hospital LOS | 45.0 [15.0–89.0] | 26.5 [16.5–37.5] | 0.19 |
| GOS at hospital discharge | 0.12 | ||
| 1 death | 1 (4%) | 2 (4%) | |
| 2 persistent vegetative state | 0 (0%) | 0 (0%) | |
| 3 severe disability | 13 (50%) | 12 (25%) | |
| 4 moderate disability | 11 (42%) | 26 (54%) | |
| 5 good recovery | 1 (4%) | 8 (17%) | |
| Dichotomized GOS at hospital discharge | 0.047 | ||
| Poor (GOS 1–3) | 14 (54%) | 14 (29%) | |
| Good (GOS 4–5) | 12 (46%) | 34 (71%) | |
| Destination | 0.87 | ||
| Home, no assistance | 18 (69%) | 30 (63%) | |
| Rehab facility | 7 (27%) | 15 (31%) | |
| Acute care hospital | 0 (0%) | 1 (2%) | |
| Death | 1 (4%) | 2 (4%) |
Data are presented as median and interquartile range.
GCS, Glasgow coma score; PRISM III, pediatric risk of mortality score; ISS, injury severity score; LOS, length of stay; GOS, Glasgow outcome scale score; PICU, pediatric intensive care unit, PRISM, pediatric risk of mortality score.
Mouse Model of Clinical Relevant Delayed Hypoxemia After TBI
To model delayed normocarbic hypoxemia following TBI explicitly, we exposed awake, spontaneously breathing mice 1 day after injury or sham surgery to 8% oxygen. As part of our experimental design, we avoided the use of inhaled anesthetics during delayed hypoxemia because they are not routinely utilized in the intensive care setting and may in fact be neuroprotective ( 36–39 ). However, allowing mice to breathe spontaneously in a low oxygen environment results in tachypnea and hypocarbia, which produce decreases in cerebral blood flow, and would cloud the clinical relevance of our model. Utilizing mice that had undergone tunneled arterial cannulation, ambient carbon dioxide concentration was increased stepwise from 0.5% to 6%. We found that 4% carbon dioxide maintained normocarbia (despite hypoxemia-related hyperventilation) in awake, spontaneously breathing mice during 30 minutes of 8% oxygen ( Table 2 ).
TABLE 2.
Arterial Blood Gas Samples From Awake Spontaneously Breathing Mice After 30 Minutes of 8% O 2 and 4% CO 2 24 Hours After Controlled Cortical Impact or Sham Surgery
| Group | pH | PaCO 2 (mm Hg) | PaO 2 (mm Hg) |
|---|---|---|---|
| Sham (n = 5) | 7.36 ± 0.02 | 40.8 ± 2.0 | 50.1 ± 1.4 |
| CCI (n = 5) | 7.35 ± 0.03 | 40.6 ± 1.9 | 49.8 ± 1.1 |
CCI, controlled cortical impact injury.
Pericontusional White Matter Hypoxia Is Worsened by Delayed Hypoxemia
As expected, hypoxemia 24 hours after CCI resulted in increased white matter immunohistochemical staining in the ipsilateral corpus callosum and external capsule for a marker of tissue hypoxia compared with CCI alone ( Fig. 2 ). Minimal white matter tissue hypoxia was observed in sham or sham + hypoxemia. Quantification of white matter tissue hypoxia with Visiopharm software was performed in the ipsilateral corpus callosum and external capsule with strong interrater and test–retest reliability ( Fig. 3 ). Two-way ANOVA revealed a significant injury (F 1,16 =561.8, p < 0.0001), hypoxemia (F 1,16 =26.8 p < 0.0001), and injury + hypoxemia interaction effect (F 1,16 =17.5, p < 0.0007). Posthoc Tukey tests demonstrated significantly more extensive tissue hypoxia in injured mice with hypoxemia compared with injury alone (p < 0.001) ( Fig. 2Q ). Interestingly, there was no difference in Hypoxyprobe staining in the white matter when comparing sham with sham + hypoxemia. Thus, this moderate degree of hypoxemia does not appear to cause white matter brain tissue hypoxia in isolation but substantially worsens tissue hypoxia in peri-contusional white matter.
FIGURE 2.
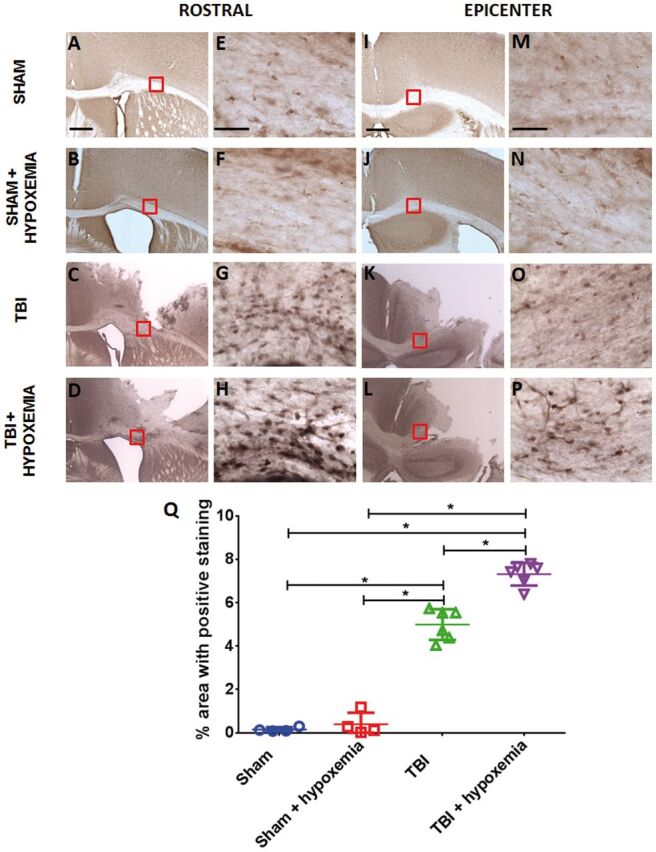
Immunohistochemical staining with the hypoxia marker pimonidazole. (A–D) Pimonidazole staining of white matter rostral to injury epicenter 24 hours after controlled cortical impact (CCI). Scale bar, 250 µm. (E–H) Higher magnification of the white matter. Scale bar, 50 µm. (I–L) Pimonidazole staining white matter at injury epicenter 24 hours after CCI. Scale bar, 250 µm. (M–P) Higher magnification of the white matter. Scale bar, 50 µm. (Q) Image quantification of white matter hypoxia of the ipsilateral corpus callosum and external capsule. *p < 0.0001, ANOVA followed by post hoc Tukey test.
FIGURE 3.

(A) Hypoxyprobe staining of white matter. (B) Overlay of Visiopharm labeling. Green represents hypoxyprobe staining; blue represents normoxic white matter in the region of interest. Scale bar, 100 µm. (C) Interrater reliability. Visomorph quantification of pimondazole staining of ipsilateral corpus callosum and external capsule by 2 blinded operators. R2 = 0.9953. (D) Test–retest reliability. Visiomorph quantification of pimonidazole staining of ipsilateral corpus callosum and external capsule. Two sets of 12 slices spaced 300 μm apart were stained for pimonidazole binding in the ipsilateral corpus callosum and external capsule and quantified by blinded operators. R2 = 0.9597. Each data point in (C) and (D) represents quantification of 12 slices spaced 300 μm apart from a single brain.
Delayed Hypoxemia Following TBI Is Associated With an Increase in Peri-Contusional White Matter TAI
Following CCI in rodent models, extensive TAI has been reported in the peri-contusional white matter ( 40 , 41 ). To determine the effects of delayed hypoxemia after TBI on TAI, we assessed TAI in the ipsilateral corpus callosum and external capsule using 2 different markers: β-APP and NF200. As previously reported, we observed increased β-APP-positive varicosities in the peri-contusional corpus callosum and external capsule of injured mice ( 25 , 41 ). We also noted a reduction in immunohistochemical staining for both markers over time. Injured mice that experienced hypoxemia 1 day after CCI appeared to have increased β-APP-positive staining compared with injured mice without hypoxemia ( Fig. 4 ). Hypoxemia did not affect nonspecific staining characteristics of the tissue ( Fig. 4 ). We did not observe β-APP stained axonal swellings in the contralateral white matter of any animals at either time point ( Fig. 5 ). Additionally, we observed a small but statistically significant increase in tissue loss in mice that experienced delayed hypoxemia following CCI compared with CCI alone ( Fig. 6 ).
FIGURE 4.
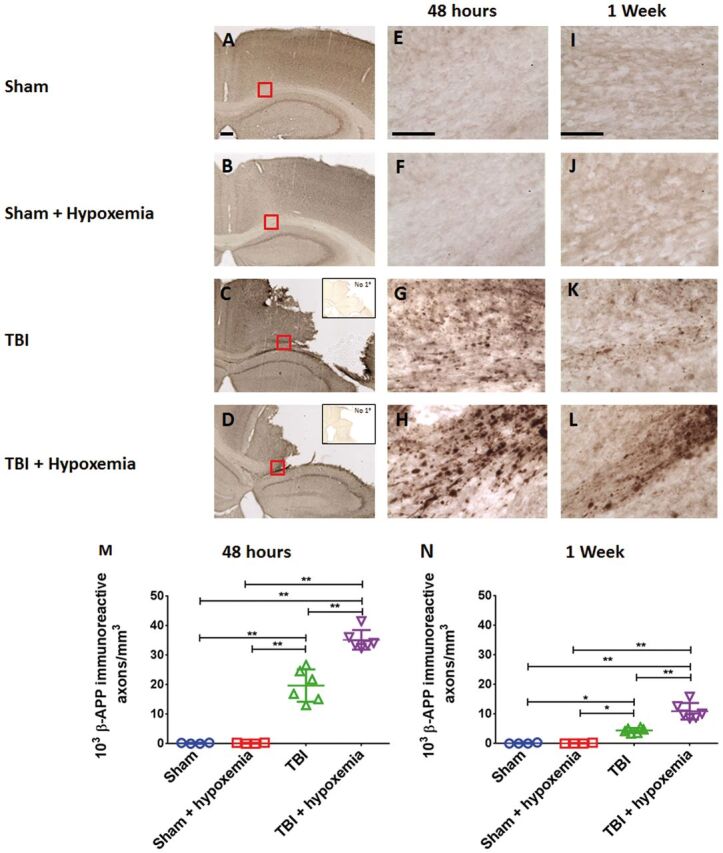
Controlled cortical impact (CCI) followed by delayed hypoxemia resulted in increased β-APP stained axonal swellings at both 48 hours and 1 week after CCI. (A–D) β-APP staining of the corpus callosum and external capsule at impact epicenter 48 hours postinjury. Scale bar, 250 µm. Insets in (C) and (D) show no primary antibody nonspecific staining at impact epicenter. Scale bar, 250 µm. (E–L) Higher magnification (of red squares in corresponding image) of the white matter at 48 hours (E–H) and 1 week postinjury (I–L) . Scale bar, 50 µm. (M, N) Stereological quantification of β-APP-positive swellings per cubic millimeter of the ipsilateral corpus callosum and external capsule. *p < 0.01, **p < 0.001 ANOVA followed by post hoc Tukey tests.
FIGURE 5.

β-APP staining of the contralateral corpus callosum or external capsule did not reveal any axonal swellings at either 48 hours or 1 week after controlled cortical impact (CCI). (A–D) β-APP staining of the contralateral white matter at 48 hours postinjury. Scale bar, 250 µm. (E–L) Higher magnification of the contralateral white matter at 48 hours (E–H) and 1 week (I–L) postinjury. Scale bar, 50 µm.
FIGURE 6.
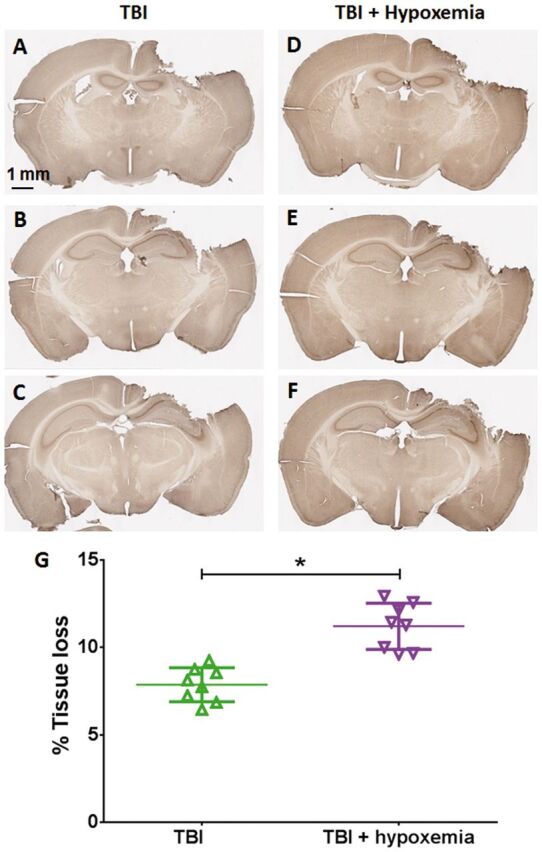
Tissue loss 1 week after injury. β-APP staining of whole brain slices. (A–F) TBI alone (A–C) and TBI + delayed hypoxemia (D–F) . (G) Quantification of tissue loss of the ipsilateral hemisphere. *p < 0.0001 unpaired t-test.
Our qualitative observations were confirmed by blinded stereological quantification of β-APP staining in the ipsilateral corpus callosum and external capsule ( Fig. 4M, N ). At 2 days postinjury, 2-way ANOVA revealed a significant injury (F 1,16 =279.7, p < 0.0001), hypoxemia (F 1,16 =22.5 p <0.0002), and injury + hypoxemia effect (F 1,16 =22.6, p <0.0002). Posthoc Tukey tests demonstrated a significantly higher number of β-APP positive varicosities in injured mice with delayed hypoxemia compared with injured mice without hypoxemia (p < 0.001). Two-way ANOVA at 7 days postinjury revealed similar results with significant injury (F 1,16 =104,5, p < 0.001), hypoxemia (F 1,16 =19.1, p < 0.005), and injury + hypoxemia effect (F 1,16 =19.3, p < 0.005). Posthoc Tukey tests also demonstrated a significantly higher number of β-APP-positive varicosities in injured mice with hypoxemia compared with injured mice without hypoxemia at 7 days postinjury (p < 0.001).
Immunohistochemical staining for NF200 was observed in injured mice in a more rostral aspect of the ipsilateral corpus callosum and external capsule. There was an increase in NF200 axonal swellings noted in injured mice with hypoxemia compared with injured mice without hypoxemia ( Fig. 7 ). Two-way ANOVA of stereological quantification of NF200-positive axonal swellings in the ipsilateral corpus callosum and external capsule at 2 days postinjury revealed a significant injury (F 1,16 =481.3, p < 0.0001), hypoxemia (F 1,16 =16.5, p < 0.0009), and injury + hypoxemia interaction effect (F 1,16 =19.6, p < 0.0004). Posthoc Tukey tests demonstrated a significantly higher number of NF200-positive swellings in injured mice with hypoxemia compared with injured mice alone (p < 0.001). Similarly, 2-way ANOVA at 7 days postinjury revealed a significant injury (F 1,16 =199.4, p < 0.0001), hypoxemia (F 1,16 =26.5, p < 0.0001), and injury + hypoxemia effect (F 1,16 =24.9, p < 0.0001). Posthoc Tukey tests also demonstrated a significantly higher number of NF200 varicosities in injured mice with hypoxemia compared with injured mice alone (p < 0.0001).
FIGURE 7.
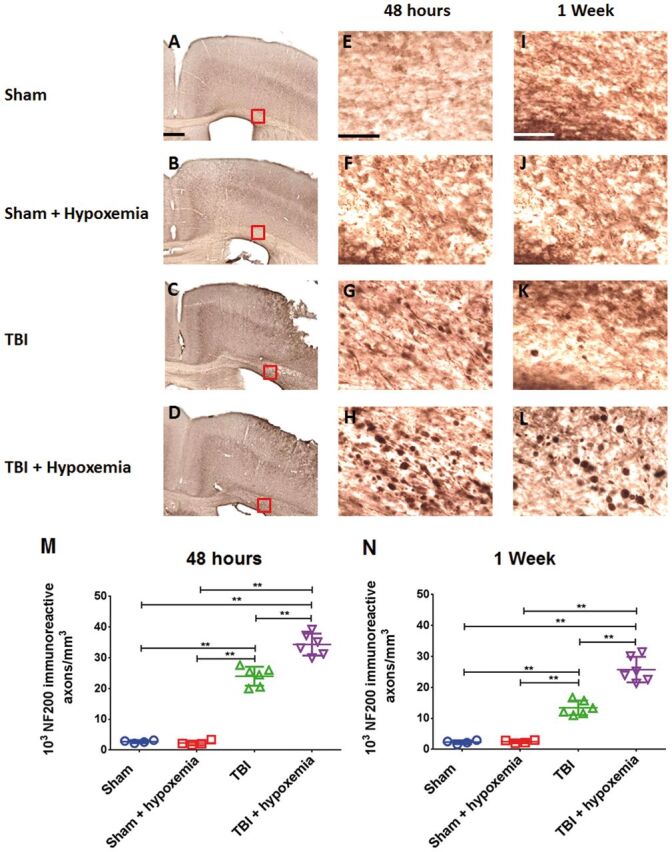
Controlled cortical impact (CCI) followed by delayed hypoxemia resulted in increased neurofilament-200 (NF200) stained axonal swellings at both 48 hours and 1 week after CCI. (A–D) NF200 staining of the white matter rostral to impact epicenter at 48 hours postinjury. Scale bar, 250 µm. (E–L) Higher magnification (of red squares in corresponding images) of the white matter at 48 hours (E–H) and 1 week (I–L) postinjury. Scale bar, 50 µm. (M, N) Stereological quantification of NF-200-positive axonal swellings per cubic millimeter of the ipsilateral corpus callosum and external capsule. **p < 0.001, ANOVA followed by post hoc Tukey tests.
Automated Quantification of Axonal Injury
Quantification of white matter injury immunohistochemistry with stereology is a powerful and accurate method to assess treatment effects on axonal injury. However, stereological quantification of axonal injury is laborious, expensive, and temporally limits experimental throughput (eg, for screening of candidate therapeutics for efficacy in axonal injury, assessments of effects of genotype, gender, age, etc.). To improve experimental efficiency, we developed a higher throughput image analysis protocol using the Visiopharm Integrator System software for quantification of axonal injury. Axonal swellings are identified by first thresholding the ROI, and then further distinguished by user developed parameters based on size, shape, and object separation ( Fig. 8 ). Using these parameters, 2 populations of positively stained axonal swellings can be identified: short axis (green) and long axis (red) ( Fig. 8B, E ). The sum of these 2 populations identified by this method correlated very well with traditional stereological quantification for both β-APP and NF200 stained axons ( Fig. 8C, F ). Throughput was approximately 7-fold faster than traditional stereology.
FIGURE 8.

Image quantification of axonal injury strongly correlates with stereological quantification. (A) β-APP staining of ipsilateral corpus callosum and external capsule. (B) Overlay of Visiopharm labeling of β-APP-positive axonal swellings. Green represents axonal swellings in the short axis; red represents axonal swellings in the long axis. Scale bar, 100 µm. (C) Correlation of stereological quantification and Visiopharm image analysis of ipsilateral corpus callosum and external capsule. R2 = 0.9502. Each data point represents quantification of 12 slices spaced 300 μm apart from 1 brain 48 hours or 1 week after injury or sham surgery. (D) NF200 staining of ipsilateral corpus callosum and external capsule. (E) Overlay of Visiopharm labeling of β-APP-positive axonal swellings. Green represents axonal swellings in the short axis; red represents axonal swellings in the long axis. Scale bar, 100 µm. (F) Correlation of stereological quantification and Visiopharm image analysis of ipsilateral corpus callosum and external capsule. R2 = 0.9227. Each data point represents quantification of 12 slices spaced 300 μm apart from 1 brain 48 hours or 1 week after injury or sham surgery.
Delayed Hypoxemia Following CCI Does Not Significantly Alter Microglial Response
To determine if increase microglial activation was associated with the increase in axonal injury following delayed hypoxemia, we examined the peri-contusional white matter with Iba1 immunohistochemical staining at 7 days postinjury. We selected the 7 day time point as it has been previously reported that microglial activation increases over the first 7 days after CCI in the mouse ( 42 ). Following CCI, we observed increased prominence of Iba1-positive cells in the ipsilateral corpus callosum and external capsule 7 days after initial injury ( Fig. 9 ). This injury effect was confirmed by stereological analysis ( Fig. 9I ) (F 1,16 =217, p < 0.0001). Stereological quantification of Iba1-positive cells did not demonstrate a statistically significant effect of delayed hypoxemia (F 1,16 =1.51, p = 0.24) or the interaction between hypoxemia and injury (F 1,16 =1.94, p = 0.18). No difference in microglial activation was observed in sham animals with or without hypoxemia.
FIGURE 9.
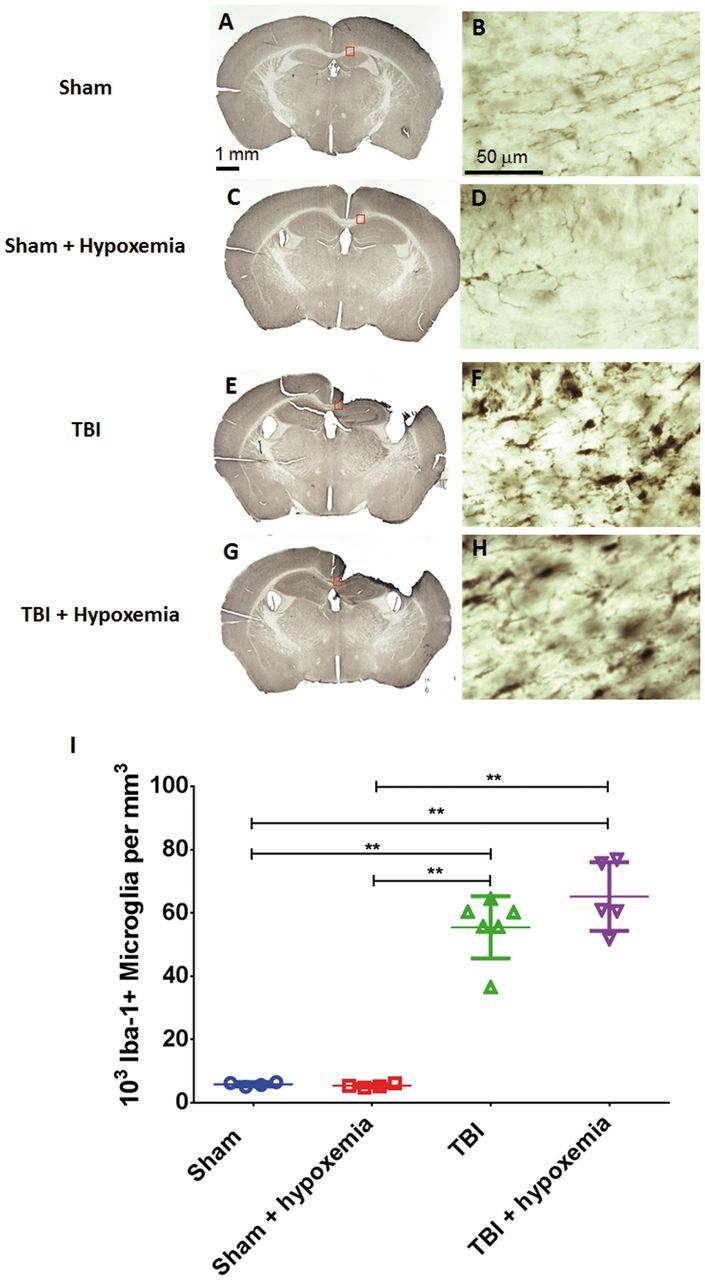
Microglial activation 1 week after traumatic brain injury (TBI). (A–H) Iba-1 stained whole sections (A, C, E, G) and higher magnification of the ipsilateral corpus callosum (B, D, F, H) at 1 week postinjury. (I) Stereological quantification of Iba-1-positive microglia in the ipsilateral corpus callosum and external capsule did not demonstrate a statistically significant difference between the 2 injury groups. **p < 0.001, ANOVA followed by post hoc Tukey tests.
Delayed Hypoxemia Augments Astrocyte Response Following CCI
To assess effects of delayed hypoxemia on astrocytic response to TBI, we performed GFAP immunohistochemical staining. As expected, we observed increased GFAP-positive cells in the ipsilateral corpus callosum and external capsule in injured animals compared with shams at 48 hours and 1 week after CCI ( Fig. 10 ). Hypoxemia alone did not increase GFAP staining in shams at 48 hours or 1 week postinjury. However, delayed hypoxemia after CCI did increase GFAP staining in the ipsilateral corpus callosum and external capsule compared with CCI alone at 48 hours and 1 week after injury ( Fig. 10 ). Two-way ANOVA of percent area of GFAP staining in the ipsilateral corpus callosum and external capsule demonstrated a significant injury (F 1,16 =359.6, p < 0.0001), hypoxemia (F 1,16 =8.7, p < 0.01), and injury + hypoxemia interaction effects (F 1,16 =4.8, p < 0.05) at 2 days postinjury. Posthoc Tukey test confirmed a higher percent area of GFAP staining in mice with delayed hypoxemia and CCI compared with CCI alone (23% vs 18%, p < 0.001). Similarly at 1 week postinjury, there were significant injury (F 1,16 =181.2, p < 0.0001), hypoxemia (F 1,16 =12.7, p < 0.003), and injury + hypoxemia interaction effects (F 1,16 =12.1, p < 0.003). Posthoc Tukey test demonstrated a higher percent area of GFAP staining with delayed hypoxemia compared with CCI alone (45% vs 24%, p < 0.01).
FIGURE 10.
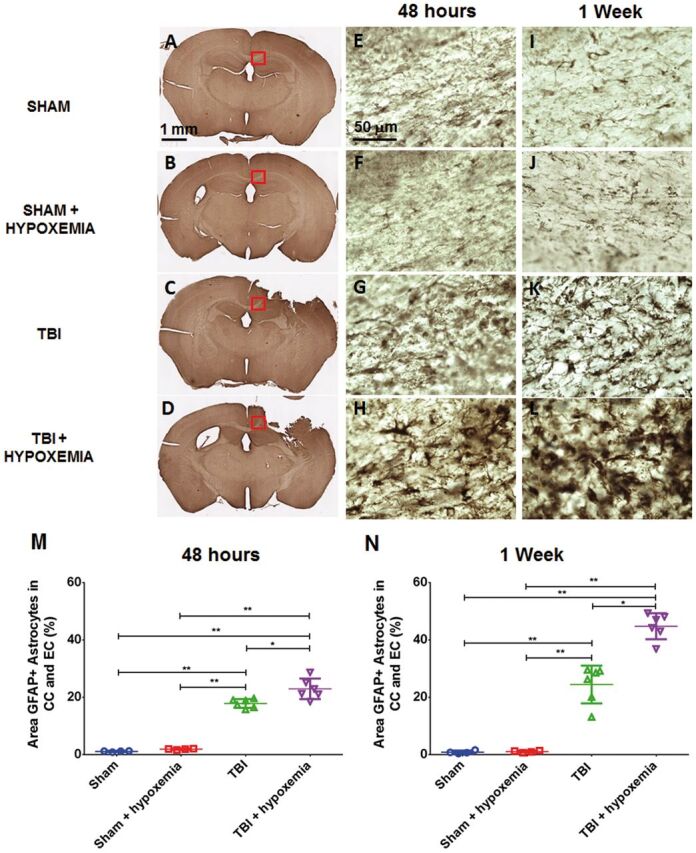
Increased astrocytosis following delayed hypoxemia after traumatic brain injury (TBI). (A–L) Anti-GFAP stained whole sections 48 hours after TBI (A–D) . Higher magnification of the corpus callosum (red squares in corresponding images) 48 hours after TBI (E–H) and 1 week after TBI (I–L) . (M, N) Percent area of GFAP staining in the ipsilateral corpus callosum (CC) and external capsule (EC). *p < 0.01, **p < 0.001 ANOVA followed by post hoc Tukey tests.
DISCUSSION
In summary, hypoxemia was common in children with severe head injury during the first 7 days of intensive care. To model this clinical phenomenon, we exposed mice to 30 minutes of systemic hypoxemia 24 hours after moderate CCI and observed increased peri-contusional axonal injury. We detected increased axonal injury in mice experiencing hypoxemia after TBI utilizing 2 different immunohistochemical markers of axonal injury, β-APP and NF200, at 48 hours and 1 week postinjury in the ipsilateral corpus callosum and external capsule. Our findings are further supported by increased white matter hypoxia and astrogliosis in mice undergoing CCI followed by delayed systemic hypoxemia compared with CCI alone. Together, these data support the hypothesis that delayed hypoxemia following CCI in mice exacerbates axonal injury in peri-contusional white matter.
Our model has several strengths. The electromagnetic CCI device utilized produces precise reproducible injury severity reducing injury variability and the sample size of mice required ( 28 ). It should be noted, however, that modeling human severe TBI in the rodent is technically challenging. Unlike in the clinical setting, the animals in this study were able to feed and care for themselves without medical intervention within 1 hour of injury. We utilized retrospective clinical data as a foundation for our model design of hypoxemia to ensure clinical relevance and maximize the translatability of our findings. As part of our experimental design, we avoided the use of inhaled anesthetics during delayed hypoxemia because they are not routinely utilized in the intensive care setting and may in fact be neuroprotective, thereby confounding results ( 36–39 ). By adding CO 2 in a controlled manner to the hypoxia chamber, we were able to achieve normocarbia, permitting us to examine the effects of hypoxemia without the confounding influence of hypocarbia. Because children with TBI and hypoxemia were normocarbic, this approach also supported fidelity between clinical data and our preclinical model.
We assessed white matter axonal injury utilizing 2 different immunohistochemical markers of axonal injury, β-APP a marker of impaired axoplasmic transport (IAT) and NF200, a marker of neurofilament compaction (NFC). IAT and NFC are thought to occur in different subset of axon populations following TBI ( 43–45 ). IAT detected by anti-β-APP can be found in both larger and smaller axons whereas NFC is typically detected by immunohistochemistry in larger axons ( 19 ). Both markers of axonal injury had greater densities in the ipsilateral corpus callosum and external capsule at 48 hours and 1 week after CCI in animals that had experienced delayed hypoxemia versus normoxemia, thereby demonstrating that a delayed systemic insult can exacerbate TAI. We also observed variations in IAT and NFC density based on location. The majority of NFC detection in our CCI model was observed in white matter rostral to the impact epicenter with little or no NF200 staining at the center of the impact. β-APP immunostaining was more diffuse with the greatest amount observed in white matter at the impact epicenter.
Our innovative approach to axonal injury quantification with a high throughput image analysis protocol using the Visiopharm software correlated well with traditional stereological quantification ( Fig. 8C, F ), and had excellent interrater and test–retest reliability ( Fig. 3C, D ). However, unlike stereology, our method does not estimate density of axonal pathology through the entire thickness of the ROI in each slice. Our image analysis is also highly dependent on high quality and consistent immunohistochemistry, DAB colorization, and imaging to achieve accurate quantification. Avenues for future development could include alterations in the histological approach, such as thinner sections spaced closer together. Nevertheless, this quantification technique with the stated caveats provides the opportunity for fast and accurate unbiased quantification of histopathology. This technique is very attractive as an initial screening tool for future investigations assessing the efficacy of therapeutic candidates targeting axonal injury.
We also assessed the microglial response in the ipsilateral corpus callosum and external capsule following delayed hypoxemia after CCI using Iba-1 immunohistochemistry and observed a strong microglial response 1 week after CCI ( Fig. 9 ), consistent with previous reports ( 42 , 46 ). By contrast, the addition of delayed hypoxemia did not result in a significant increase in the microglial response in the ipsilateral corpus callosum and external capsule. It should be noted that our evaluation of the microglia response was limited to Iba1 immunohistochemical staining and we did not investigate any influence of delayed hypoxemia on activations of M1 and M2 phenotypes. Previous investigations in a weight-drop rat model of TBI with immediate postinjury hypoxemia did not demonstrate an increase in microglial activation compared with TBI alone ( 10 ). Furthermore, when microglia are reduced prior to and after injury in a murine repetitive concussion model, no alterations in axonal injury were observed ( 47 ). These data are most consistent with the hypothesis that microglia do not play a prominent contributory role to the increase axonal injury observed after delayed hypoxemia following TBI. Conversely, GFAP immunohistochemistry demonstrated more prominent astrogliosis following delayed hypoxemia after CCI versus CCI alone at 48 hours postinjury with further increases observed at the 1 week postinjury interval. Astrocyte proliferation after CCI has been reported to peak at 3–4 days postinjury ( 46 , 48 , 49 ). We observed similar levels of reactive astrocytes at 2 days and 1 week postinjury in mice experiencing CCI alone but the addition of delayed hypoxemia resulted in a marked increase in reactive astrocytes at 1 week post-CCI. It is unclear if this astrocyte response is simply a marker of the increased axonal injury observed following delayed hypoxemia, plays a protective role, or contributes in a negative fashion to injury.
There are many other logical directions for future research on this topic. Our investigations into the effects of delayed hypoxemia after TBI were limited to a focal injury model. It would be of interest to see if similar exacerbations of axonal injury could be observed in a more diffuse mouse model of TBI. We have demonstrated axonal susceptibility to systemic hypoxemia 24 hours after primary TBI but in these experiments we did not determine the limits of this temporal window of vulnerability. Future experiments are planned to investigate how long after the initial TBI can axonal injury be exacerbated by systemic hypoxemia. Immediate systemic hypoxemia following experimental TBI has been observed to exacerbate hippocampal neuronal injury and we plan to investigate the effects of delayed hypoxemia on hippocampal injury following CCI. We did not observe any immunohistochemical evidence of TAI in the contralateral white matter with the markers described above. However, prominent silver staining contralateral to the impact site following CCI has been observed by others ( 40 , 50 ). Further investigations evaluating the effects of delayed hypoxemia on white matter regions remote to the impact site are planned. Our evaluation of TAI in white matter also did not include investigations into the effects of delayed hypoxemia on myelination of axons or changes in oligodendrocytes or their progenitor cells. Future investigations are planned including the use of electron microscopy to assess changes in axon myelination and Wallerian degeneration. Additional future directions include investigating the effect of age on the brain’s response to delayed hypoxemia. We utilized 5-week-old male mice, the developmental equivalent of a school age/adolescent patient to model the median age of our clinical retrospective data. It is not known whether younger children would be more or less vulnerable to delayed hypoxemia following severe TBI. Likewise, it is not known whether female mice would be affected similarly by delayed hypoxemia, though males and females are affected similarly by CCI in isolation ( 51 ). Future studies are planned to assess the effects of delayed hypoxemia on long-term behavioral deficits and neuropathology such as hippocampal neurons, white matter atrophy, and microglial activation.
In conclusion, clinically relevant hypoxemia 24 hours after CCI in mice resulted in white matter tissue hypoxia and increased axonal injury as determined by 2 different immunohistochemical markers. Targeting delayed hypoxemia after TBI in the ICU setting provides a unique opportunity for therapeutic interventions, permitting rapid administration of neuroprotective agents with short temporal windows of efficacy and the prospect to administer exceptionally safe neuroprotective agents prior to episodes of secondary brain hypoxia in high risk patients with severe TBI.
ACKNOWLEDGMENTS
The authors thank Ronaldo Perez for technical assistance, T.J. Esparza for methodological assistance, and Tina Day for assistance with the clinical data set.
REFERENCES
- 1. Heron M, Sutton PD, Xu J , et al. . Annual summary of vital statistics: 2007 . Pediatrics 2010. ; 125 : 4 – 15 [DOI] [PubMed] [Google Scholar]
- 2. Fisher MD. Pediatric traumatic brain injury . Crit Care Nurs 1997. ; 20 : 36 – 51 [DOI] [PubMed] [Google Scholar]
- 3. Kochanek PM, Clark RS, Ruppel RA , et al. . Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: Lessons learned from the bedside . Pediatr Crit Care Med 2000. ; 1 : 4 – 19 [DOI] [PubMed] [Google Scholar]
- 4. Chesnut RM, Marshall LF, Klauber MR , et al. . The role of secondary brain injury in determining outcome from severe head injury . J Trauma 1993. ; 34 : 216 – 22 [DOI] [PubMed] [Google Scholar]
- 5. Chi JH, Knudson MM, Vassar MJ , et al. . Prehospital hypoxia affects outcome in patients with traumatic brain injury: A prospective multicenter study . J Trauma 2006. ; 61 : 1134 – 41 [DOI] [PubMed] [Google Scholar]
- 6. Davis DP, Dunford JV, Poste JC , et al. . The impact of hypoxia and hyperventilation on outcome after paramedic rapid sequence intubation of severely head-injured patients . J Trauma 2004. ; 57 : 1 – 8 discussion -10 [DOI] [PubMed] [Google Scholar]
- 7. Davis DP, Meade W, Sise MJ , et al. . Both hypoxemia and extreme hyperoxemia may be detrimental in patients with severe traumatic brain injury . J Neurotrauma 2009. ; 26 : 2217 – 23 [DOI] [PubMed] [Google Scholar]
- 8. Huang RQ, Cheng HL, Zhao XD , et al. . Preliminary study on the effect of trauma-induced secondary cellular hypoxia in brain injury . Neurosci Lett 2010. ; 473 : 22 – 7 [DOI] [PubMed] [Google Scholar]
- 9. Feng JF, Zhao X, Gurkoff GG , et al. . Post-traumatic hypoxia exacerbates neuronal cell death in the hippocampus . J Neurotrauma 2012. ; 29 : 1167 – 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yan EB, Hellewell SC, Bellander BM , et al. . Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury . J Neuroinflam 2011. ; 8 : 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hellewell SC, Yan EB, Agyapomaa DA , et al. . Post-traumatic hypoxia exacerbates brain tissue damage: Analysis of axonal injury and glial responses . J Neurotrauma 2010. ; 27 : 1997 – 2010 [DOI] [PubMed] [Google Scholar]
- 12. Clark RS, Kochanek PM, Dixon CE , et al. . Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats . J Neurotrauma 1997. ; 14 : 179 – 89 [DOI] [PubMed] [Google Scholar]
- 13. Fang R, Markandaya M, DuBose JJ , et al. . Early in-theater management of combat-related traumatic brain injury: A prospective, observational study to identify opportunities for performance improvement . J Trauma Acute Care Surg 2015. ; 79 : S181 – 7 [DOI] [PubMed] [Google Scholar]
- 14. Adams JH. Diffuse axonal injury in non-missile head injury . Injury 1982. ; 13 : 444 – 5 [DOI] [PubMed] [Google Scholar]
- 15. Blumbergs PC, Scott G, Manavis J , et al. . Staining of amyloid precursor protein to study axonal damage in mild head injury . Lancet 1994. ; 344 : 1055 – 6 [DOI] [PubMed] [Google Scholar]
- 16. Grady MS, McLaughlin MR, Christman CW , et al. . The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans . J Neuropathol Exp Neurol 1993. ; 52 : 143 – 52 [DOI] [PubMed] [Google Scholar]
- 17. Povlishock JT, Erb DE, Astruc J. Axonal response to traumatic brain injury: reactive axonal change, deafferentation, and neuroplasticity . J Neurotrauma 1992. ;( Suppl 1) : S189 – 200 [PubMed] [Google Scholar]
- 18. Smith DH, Chen XH, Iwata A , et al. . Amyloid beta accumulation in axons after traumatic brain injury in humans . J Neurosurg 2003. ; 98 : 1072 – 7 [DOI] [PubMed] [Google Scholar]
- 19. Stone JR, Singleton RH, Povlishock JT. Intra-axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons . Exp Neurol 2001. ; 172 : 320 – 31 [DOI] [PubMed] [Google Scholar]
- 20. Gentleman SM, Nash MJ, Sweeting CJ , et al. . Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury . Neurosci Lett 1993. ; 160 : 139 – 44 [DOI] [PubMed] [Google Scholar]
- 21. Smith DH, Chen XH, Nonaka M , et al. . Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig . J Neuropathol Exp Neurol 1999. ; 58 : 982 – 92 [DOI] [PubMed] [Google Scholar]
- 22. Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein . Acta Neuropathol 1994. ; 87 : 55 – 62 [DOI] [PubMed] [Google Scholar]
- 23. Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury . J Head Trauma Rehabil 2005. ; 20 : 76 – 94 [DOI] [PubMed] [Google Scholar]
- 24. Yaghmai A, Povlishock J. Traumatically induced reactive change as visualized through the use of monoclonal antibodies targeted to neurofilament subunits . J Neuropathol Exp Neurol 1992. : 51 ; 158 – 76 [DOI] [PubMed] [Google Scholar]
- 25. Friess SH, Lapidus JB, Brody DL. Decompressive craniectomy reduces white matter injury after controlled cortical impact in mice . J Neurotrauma 2015. ; 32 : 791 – 800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marmarou CR, Povlishock JT. Administration of the immunophilin ligand FK506 differentially attenuates neurofilament compaction and impaired axonal transport in injured axons following diffuse traumatic brain injury . Exp Neurol 2006. ; 197 : 353 – 62 [DOI] [PubMed] [Google Scholar]
- 27. Marshall LF, Marshall SB, Klauber MR , et al. . A new classification of head injury based on computerized tomography . J Neurosurg 1991. ; 75 : S14 – 20 [Google Scholar]
- 28. Brody DL, Mac Donald C, Kessens CC , et al. . Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury . J Neurotrauma 2007. ; 24 : 657 – 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lighthall JW. Controlled cortical impact: A new experimental brain injury model . J Neurotrauma 1988. ; 5 : 1 – 15 [DOI] [PubMed] [Google Scholar]
- 30. Dixon CE, Clifton GL, Lighthall JW , et al. . A controlled cortical impact model of traumatic brain injury in the rat . J Neurosci Methods 1991. ; 39 : 253 – 62 [DOI] [PubMed] [Google Scholar]
- 31. Bernsen HJ, Rijken PF, Peters H , et al. . Hypoxia in a human intracerebral glioma model . J Neurosurg 2000. ; 93 : 449 – 54 [DOI] [PubMed] [Google Scholar]
- 32. Laurent F, Benard P, Canal P , et al. . Autoradiographic distribution of [14C]-labelled pimonidazole in rhabdomyosarcoma-bearing rats and pigmented mice . Cancer Chemother Pharmacol 1988. ; 22 : 308 – 15 [DOI] [PubMed] [Google Scholar]
- 33. Samoszuk MK, Walter J, Mechetner E. Improved immunohistochemical method for detecting hypoxia gradients in mouse tissues and tumors . J Histochem Cytochem 2004. : 52 ; 837 – 9 [DOI] [PubMed] [Google Scholar]
- 34. Raleigh JA, Chou SC, Arteel GE , et al. . Comparisons among pimonidazole binding, oxygen electrode measurements, and radiation response in C3H mouse tumors . Radiat Res 1999. ; 151 : 580 – 9 [PubMed] [Google Scholar]
- 35. Huh JW, Raghupathi R. Chronic cognitive deficits and long-term histopathological alterations following contusive brain injury in the immature rat . J Neurotrauma 2007. ; 24 : 1460 – 74 [DOI] [PubMed] [Google Scholar]
- 36. Todd MM, Weeks J. Comparative effects of propofol, pentobarbital, and isoflurane on cerebral blood flow and blood volume . J Neurosurg Anesthesiol 1996. ; 8 : 296 – 303 [DOI] [PubMed] [Google Scholar]
- 37. Statler KD, Alexander H, Vagni V , et al. . Isoflurane exerts neuroprotective actions at or near the time of severe traumatic brain injury . Brain Res 2006. ; 1076 : 216 – 24 [DOI] [PubMed] [Google Scholar]
- 38. Goren S, Kahveci N, Alkan T , et al. . The effects of sevoflurane and isoflurane on intracranial pressure and cerebral perfusion pressure after diffuse brain injury in rats . J Neurosurg Anesthesiol 2001. ; 13 : 113 – 9 [DOI] [PubMed] [Google Scholar]
- 39. Cucchiara RF, Theye RA, Michenfelder JD. The effects of isoflurane on canine cerebral metabolism and blood flow . Anesthesiology 1974. ; 40 : 571 – 4 [DOI] [PubMed] [Google Scholar]
- 40. Jiang Y, Brody DL. Administration of COG1410 reduces axonal amyloid precursor protein immunoreactivity and microglial activation after controlled cortical impact in mice . J Neurotrauma 2012. ; 29 : 2332 – 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mac Donald CL, Dikranian K, Song SK , et al. . Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury . Exp Neurol 2007. ; 205 : 116 – 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury . Exp Neurol 2008. ; 213 : 372 – 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stone JR, Okonkwo DO, Dialo AO , et al. . Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury . Exp Neurol 2004. ; 190 : 59 – 69 [DOI] [PubMed] [Google Scholar]
- 44. DiLeonardi AM, Huh JW, Raghupathi R. Impaired axonal transport and neurofilament compaction occur in separate populations of injured axons following diffuse brain injury in the immature rat . Brain Res 2009. ; 1263 : 174 – 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marmarou CR, Walker SA, Davis CL , et al. . Quantitative analysis of the relationship between intra- axonal neurofilament compaction and impaired axonal transport following diffuse traumatic brain injury . J Neurotrauma 2005. ; 22 : 1066 – 80 [DOI] [PubMed] [Google Scholar]
- 46. Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury . Exp Neurol 2003. ; 182 : 87 – 102 [DOI] [PubMed] [Google Scholar]
- 47. Bennett RE, Brody DL. Acute reduction of microglia does not alter axonal injury in a mouse model of repetitive concussive traumatic brain injury . J Neurotrauma 2014. ; 31 : 1647 – 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Villapol S, Byrnes KR, Symes AJ. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury . Front Neurol 2014. ; 5 : 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Susarla BT, Villapol S, Yi JH , et al. . Temporal patterns of cortical proliferation of glial cell populations after traumatic brain injury in mice . ASN Neuro 2014. ; 6 : 159 – 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hall ED, Bryant YD, Cho W , et al. . Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods . J Neurotrauma 2008. ; 25 : 235 – 47 [DOI] [PubMed] [Google Scholar]
- 51. Hall ED, Gibson TR, Pavel KM. Lack of a gender difference in post-traumatic neurodegeneration in the mouse controlled cortical impact injury model . J Neurotrauma 2005. ; 22 : 669 – 79 [DOI] [PubMed] [Google Scholar]