

Abstract
BACKGROUND:
The etiology of cervical dystonia (CD) is unknown. Cholinergic abnormalities have been identified in dystonia animal models and human imaging studies. Some animal models have cholinergic neuronal loss in the striatum and increased acetylcholinesterase activity in the pedunculopontine nucleus (PPN).
OBJECTIVES:
To determine the presence of cholinergic abnormalities in the putamen and PPN in CD human brain donors.
METHODS:
Formalin-fixed brain tissues were obtained from 8 CD and 7 age-matched control brains (controls). PPN was available in only 6 CD and 5 controls. Neurodegeneration was evaluated pathologically in putamen, PPN, and other regions. Cholinergic neurons were detected using choline acetyltransferase (ChAT) immunohistochemistry in putamen and PPN. Putaminal cholinergic neurons were quantified. 6 CD patients and 6 age-matched healthy controls (HC) underwent diffusion tensor imaging (DTI) to determine if there were white matter microstructural abnormalities around the PPN.
RESULTS:
Decreased or absent ChAT staining was identified in all 6 PPN samples in CD. In contrast, strong ChAT staining was present in 4 out of 5 PPN controls. There were no differences in PPN DTI between CD and HC. There was no difference in numbers of putaminal cholinergic neurons between CD and controls.
CONCLUSIONS:
Our findings suggest that PPN ChAT deficiency represents a functional cholinergic deficit in CD. Structural lesions and confounding neurodegenerative processes were excluded by absence of neuronal loss, gliosis, DTI abnormalities, and beta-amyloid, tau and alpha-synuclein pathologies.
Keywords: acetylcholine, cervical dystonia, MRI, diffusion tensor imaging, neuropathology
Introduction
Dystonia is a movement disorder in which patients are affected by involuntary muscle contractions resulting in unusual postures. Cervical dystonia (CD) is a form of primary focal dystonia of the neck region that is associated with significant disability and pain. Although secondary forms of CD have been described, most cases of CD are idiopathic or of unknown etiology.1 Some animal models of dystonia, though not of CD, show cholinergic abnormalities in the striatum and pedunculopontine nucleus, including loss of striatal cholinergic neurons.2,3 Further evidence for the role of acetylcholine in dystonia includes decreased putaminal cholinergic tracer uptake in single photon emission computed tomography (SPECT) in patients with CD, response to anticholinergic medications, and cases of CD provoked by cholinesterase inhibitors.4,5
The striatum, consisting of the caudate nucleus and putamen, has intrinsic and extrinsic cholinergic innervation. Intrinsic cholinergic innervation is predominant and consists of cholinergic interneurons; an extrinsic source of acetylcholine is from the pedunculopontine nucleus (PPN.)6 Although the anterior striatum has been described to be organized with striosomes and patches with cholinergic interneurons predominantly in patches,7 cholinergic interneurons are evenly distributed between striosomes and patches in the posterior (postcommissurral) putamen.8
In this study, we evaluated brain tissue from donors with a diagnosis of CD to determine if there were cholinergic abnormalities, such as neuronal loss, in the putamen and the PPN. In addition, we performed diffusion tensor imaging (DTI) in living CD patients to examine white matter microstructure around the PPN. DTI has been thought to be sensitive for detecting white matter abnormalities associated with neuronal loss in various diseases.9
Materials and Methods
Subjects
Brain tissues from 8 CD and 7 control brains were obtained from the University of Maryland Brain and Tissue Bank (BTB), a part of the NIH NeuroBiobank. Medical records were reviewed by a neurologist (KM) to ascertain history of CD and excluded history of neurological disorders, psychotic disorders, and substance abuse in both CD and control groups. The National Institutes of Health Office of Human Subjects Research Protection determined that the portion of this study involving human brain tissue from deceased individuals is exempt from review by the Institutional Review Board.
A separate group of 6 living patients with isolated CD and 6 healthy controls (HC) participated in MRI scanning as described below. The CD patients were recruited from the movement disorders clinic at the National Institutes of Health (NIH). HC’s were recruited from the local community. Both CD patients and HC’s had neurological examinations performed by neurologists. CD patients were also evaluated with the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS).10 CD patients had a history of botulinum toxin injections so MRI scanning in CD patients occurred at least 11 weeks after their last injection. The living subjects provided written informed consent to participate in this study, which was approved by the NIH Institutional Review Board.
Neuropathological Methods
In both CD and control groups, tissue sections from posterior putamen, midbrain at the level of the inferior colliculus for PPN (where available), frontal and parietal cortices, hippocampus, and cerebellum were procured from BTB in person by one investigator (KM). The PPN was identified according to Olszewski and Baxter.11 Briefly, the PPN is located at the midbrain-pons junction and is bordered by the superior cerebellar peduncle, medial lemniscus, and cuneiform nucleus. Midbrain sections at the level of the inferior colliculus and the inferior extent of the substantia nigra were available in 6 CD and 5 controls. Of note, only samples from the right hemisphere were available. One 3 mm thick block was cut from each region. Tissue blocks were processed and embedded in paraffin. Consecutive 5 μm-thick sections were cut from blocks and mounted on glass slides for subsequent histochemical and immunohistochemical staining. Routine hematoxylin and eosin (H&E) staining was performed on one section in each case of both groups. Bielschowsky silver stain and immunohistochemistry with β-amyloid (Cell Signaling, 2450), phospho-tau (AT8, Pierce, MN1020), alpha-synuclein (α-syn) (Cell Signaling, 2647), and ubiquitin (Millipore, MAB1510) antibodies were used to screen for neurodegenerative diseases, including Alzheimer’s disease (AD), other tauopathies, and synucleinopathies.12–15 Luxol fast blue (LFB) was used to stain myelin. Immunostaining for cholinergic neurons was performed with choline acetyltransferase antibodies (ChAT) (Millipore, AB1444P) at 1:100 on putamen and PPN sections with heat-induced and/or proteinase K antigen retrieval. In the putamen, there was additional ChAT immunostaining at 1:33 antibody concentration. The ChAT antibody is well-characterized and has been used in another study in our lab.16 Diaminobenzidine (DAB) was the chromogen. An automated immunostainer (Leica Bond-Max, Buffalo Grove, IL) was used for reproducible staining. Slides were visually evaluated independently by two neuropathologists (ARC, DI), who were blinded to the subjects’ diagnoses, and one neurologist (KM).
Quantitative Immunohistochemistry
Posterior putamen ChAT-stained slides were scanned with a whole slide imager (Aperio XT, Leica, Buffalo Grove, IL) at 0.25 μm/pixel. A single ChAT-stained slide from each putamen was scanned and used for neuronal counting. Positively stained neurons were manually enumerated on digitized slides in ImageScope version 12.3 (Leica, Buffalo Grove, IL) using the Counter tool with blinding to diagnoses. The cell counts in the annotations layer were verified on a separate day. The putaminal area on each slide was manually traced and automatically calculated in ImageScope.
MRI Acquisition and DTI Analysis
Diffusion MRI was performed on a 3-Tesla MRI scanner (Siemens Skyra, Munich, Germany) using a 32-channel receive head coil with following parameters: repetition time (TR) 12300 ms, echo time (TE) 92 ms, flip angle 90 degrees, voxel size 2.0 mm × 2.0 mm × 2.0 mm, and 30 directions at b-value=1100 s/mm3 with 5 reference b0 volumes. Acquisition included direct and reverse phase encoding (“blip up/blip down”) for distortion correction. Scanning time was approximately 15 minutes. For registration with diffusion MRI, a T2-weighted sequence with a fat saturation pulse was acquired: TR 11.72 ms, TE 90 ms, flip angle 120 degrees, and voxel size 1.7 mm × 1.7 mm × 1.7 mm. For anatomical segmentation, a T1-weighted multi-echo magnetization-prepared rapid gradient echo (MEMPRAGE) was acquired: TR 2530 ms; TE 1.69 ms, 3.55 ms, 5.41 ms, and 7.27 ms; inversion time 1100 ms, flip angle 7 degrees; and voxel size 1 mm × 1 mm × 1 mm.
Diffusion images were processed in Tolerably Obsessive Registration and Tensor Optimization Indolent Software Ensemble (TORTOISE).17 The DIFF_PREP module was used for registration of diffusion images to midsagittal and anterior commissure-posterior commissure (AC-PC) aligned T2 weighted images for motion correction. Diffeomorphic Registration for Blip-Up Blip-Down Diffusion Imaging (DR BUDDI) was used for eddy current and susceptibility artifact correction.18 Tensor calculations were made in DIFF_CALC followed by nonlinear tensor fitting in 3dTrackID, a tractography tool within FATCAT and a component of Analysis of Functional Neuroimaging (AFNI) software.19,20 Uncertainty intervals for DTI parameters were estimated with 300 jackknife iterations.
Skull-stripped T1 weighted images were aligned to T2 weighted images using an affine transformation in AFNI. The aligned MEMPRAGE images were then registered to the MNI152 template with nonlinear transformations.
PPN masks were prepared from histologically-confirmed postmortem MRI as described in Alho et al.21 The location of the PPN is based on Olszewski and Baxter.11 The deceased subject for postmortem MRI and PPN mask creation was a 66 year old woman whose cause of death was myocardial infarction. Absence of neurologic disease was confirmed pathologically. The PPN masks in MNI space were warped to the individual subject space. Probabilistic tractography was seeded from left and right PPN separately to the rest of the brain. Each tract is at least 2 mm long.
Statistics
SPSS 21 (IBM, Armonk, NY) and Real Statistics release 4.3 (Charles Zaiontz, www.real-statistics.com) were used for descriptive statistics, Fisher’s exact test, t-tests, and linear regression. P-value < 0.05 is considered statistically significant.
Results
Subject Characteristics
The baseline demographics of CD and control brains were similar (Table 1). Specifically, there were no statistically significant differences in the mean age at death (73.7 ± 11.1 years for CD and 72.9 ± 12.1 years for controls), and mean postmortem interval (12.0±8.4 hours for CD and 16.6 ± 7.3 hours for controls) between the two groups (p=0.88 and p=0.28, respectively). The demographics of the living patients with CD and HC’s were also similar (Table 2).
Table 1:
Subjects with Brain Tissue
| Subject # | Sex | Age at Death (years) | Cause of Death | Postmortem Interval (hours) | Disease Duration (years) | Dystonia Description |
|---|---|---|---|---|---|---|
| Cervical Dystonia | ||||||
| 1 | F | 52 | Pneumonia | 7 | 5 | Left torticollis |
| 2 | F | 65 | ASCD | 25 | 25 | Right laterocollis |
| 3 | M | 70 | COPD | 9 | Unknown | Unknown |
| 4 | F | 75 | Natural cause | 20 | 22 | Unknown |
| 5 | F | 78 | COPD | 6 | Unknown | Unknown |
| 6 | F | 81 | Natural cause | 7 | Unknown | Dystonic head tremor |
| 7 | F | 84 | ASCD | 20 | 43 | Left laterocollis |
| 8 | F | 85 | Natural cause | 2 | >5 | Dystonic head tremor |
| Controls | ||||||
| 1 | F | 54 | Pulmonary embolism | 27 | N/A | N/A |
| 2 | F | 59 | ASCD | 17 | N/A | N/A |
| 3 | F | 75 | ASCD | 24 | N/A | N/A |
| 4 | M | 75 | ASCD | 18 | N/A | N/A |
| 5 | F | 77 | ASCD | 8 | N/A | N/A |
| 6 | F | 82 | Neck injury | 14 | N/A | N/A |
| 7 | F | 88 | CHF | 8 | N/A | N/A |
Abbreviations: ASCD, atherosclerotic cardiovascular disease; CHF, congestive heart failure; COPD, chronic obstructive pulmonary disease
Table 2:
Subjects with MRI
| Cervical Dystonia | Healthy Controls | |
|---|---|---|
| Age at MRI scanning (years) | 47.6 ± 9.1 | 48.6 ± 9.2 |
| Sex | 3 males/3 females | 5 males/1 female |
| Age at Diagnosis (years) | 33.0 ± 13.9 | - |
| Disease Duration (years) | 14.6 ± 7.8 | - |
| TWSTRS score | 29.4 ± 10.7 | - |
Abbreviations: TWSTRS, Toronto Western Spasmodic Torticollis Rating Scale
Neuropathological Evaluation
There were no histopathological differences in CD and control groups in the posterior putamen, PPN, frontal and parietal cortices, hippocampus, and cerebellum on H&E staining. Of note, in the cerebellum, there was mild Bergmann gliosis and mild Purkinje cell loss in all cases from both groups. There was no gliosis in any other region. There was no myelin pallor on LFB staining of PPN sections. In evaluation for neurodegenerative disease, a negligible amount of neuritic amyloid plaques and neurofibrillary tangles (NFTs) were present if at all. Specifically, there were no Lewy bodies, glial or neuronal cytoplasmic inclusions, neuropil threads, tufted astrocytes, or oligodendroglial coiled bodies. There were no inclusions on ubiquitin immunohistochemistry. See Supplementary Materials for further neuropathology details.
Cholinergic Neuronal Density
ChAT antibodies were used to detect cholinergic neurons.22 The distribution and staining intensity of ChAT-positive neurons in the posterior putamen appeared similar in both CD and control groups by visual analysis (see Figures 1A–1D). When these neurons were enumerated, there was no difference in their densities between CD and control groups (p=0.17). The density of cholinergic neurons in the posterior putamen in CD was 2.35 ± 0.80 cells/mm2 and in controls was 1.81 ± 0.58 cells/mm2 (Figure 1E). By linear regression, age and postmortem interval did not affect cholinergic neuronal density (R2=0.34, p=0.083).
Figure 1.
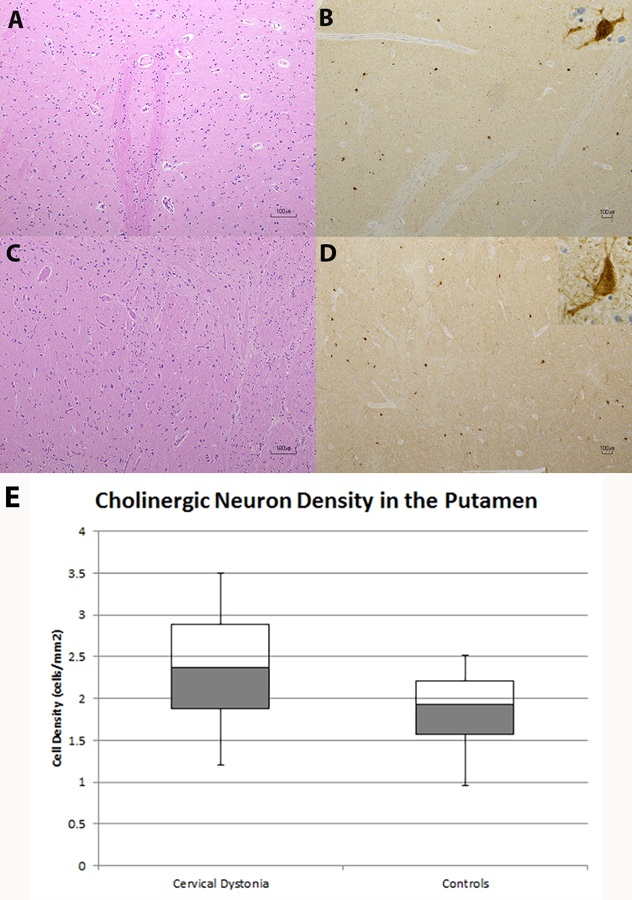
Cholinergic Neurons in the Putamen
A–D. Putamen with hematoxylin and eosin (H&E) staining (A) and choline acetyltransferase (ChAT) immunochemistry (B) from a control and putamen with H&E (C) and ChAT (D) staining from a CD brain appear similar. Inset in (B) and (D) show individual cholinergic neuron at 40x magnification. Scale bars in A-D are 100 µm. As shown in (E), there is no difference in the cholinergic neuronal density in the putamen between CD and control specimens.
Extrastriosome patches and striosomes could not be visualized with ChAT immunostaining at two different ChAT antibody concentrations. In other words, the background surrounding ChAT-positive neurons was homogeneous in both CD and control putamen samples.
Pedunculopontine Nucleus Immunohistochemistry
There were differences in ChAT immunostaining in the PPN between CD and control groups. These differences were present regardless of antigen retrieval method used. The differences in staining occurred without gliosis. In two out of six PPN samples in midbrain sections from the CD group, there was weakly positive cytoplasmic staining with ChAT; the remaining four samples were negative for ChAT staining. In contrast, four out of five PPN samples from the control group exhibited strong ChAT immunoreactivity. There was skewed sectioning in one of the five control samples so that the PPN could not be identified on H&E or ChAT immunostaining. There is a statistically significant difference in ChAT immunoreactivity between the CD and control groups by Fisher’s exact test (p=0.0095). Examples of PPN ChAT staining are shown in Figure 2. We did not count cholinergic neurons in the PPN because a variable proportion of cholinergic neurons among other types of PPN neurons was described in Mesulam et al.23 Changes in the numbers of cholinergic neurons along the length of the PPN have also been reported.24
Figure 2.
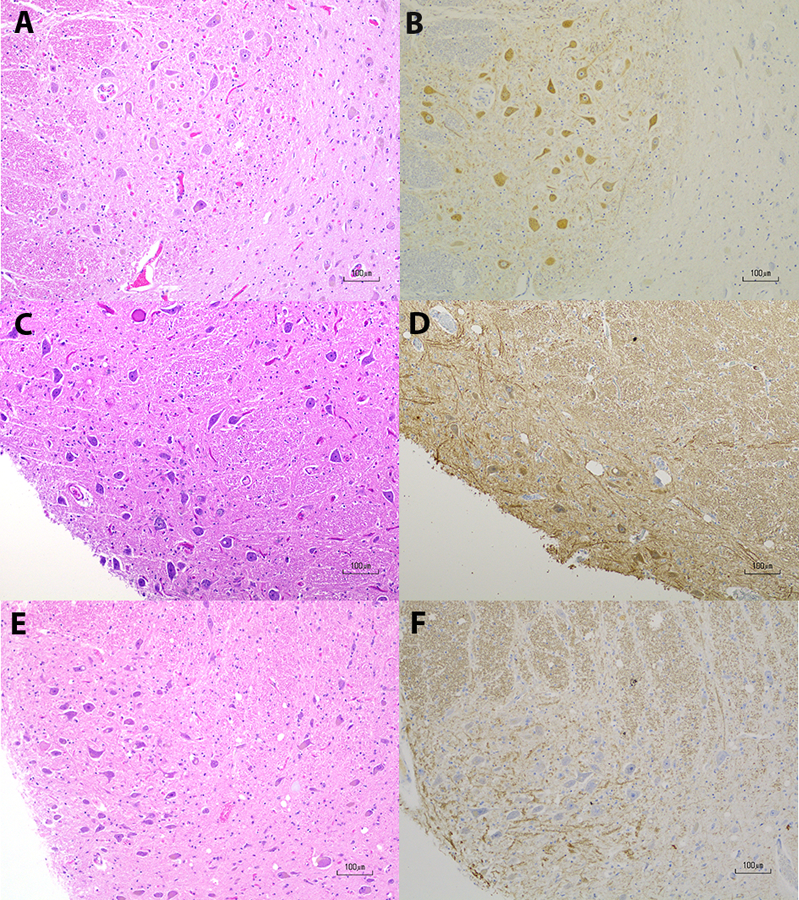
Choline Acetyltransferase Immunohistochemistry in the Pedunculopontine Nucleus
A & B. Control pedunculopontine nucleus (PPN) with hematoxylin and eosin (H&E) staining in A and choline acetyltransferase (ChAT) immunohistochemistry in B. There is positive ChAT immunoreactivity in B; C& D. CD PPN with H&E staining in C and ChAT staining in D. While background staining is slightly higher in and around the PPN in D, there is a population of neurons with weakly positive immunoreactivity in the cytoplasm, which is the expected pattern of ChAT staining; E & F. CD PPN with H&E staining in E and ChAT staining in F. F is an example of negative immunoreactivity in PPN neurons. Scale bars in A-F are 100 μm.
Pedunculopontine Nucleus DTI
DTI was used to evaluate the white matter within and surrounding the PPN in living subjects (Figure 3). The fractional anisotropy of the white matter within the PPN, which is part of the PPN pars dissipata, is the same in both CD patients and HC’s (p=0.65 for left PPN; p=0.68 for right PPN). When tractography was seeded from the PPN to evaluate the microstructure of white matter tracts connecting the PPN to the rest of the brain, there were also no differences in fractional anisotropy between the groups (p=0.27 for left PPN; p=0.63 for right PPN).
Figure 3.
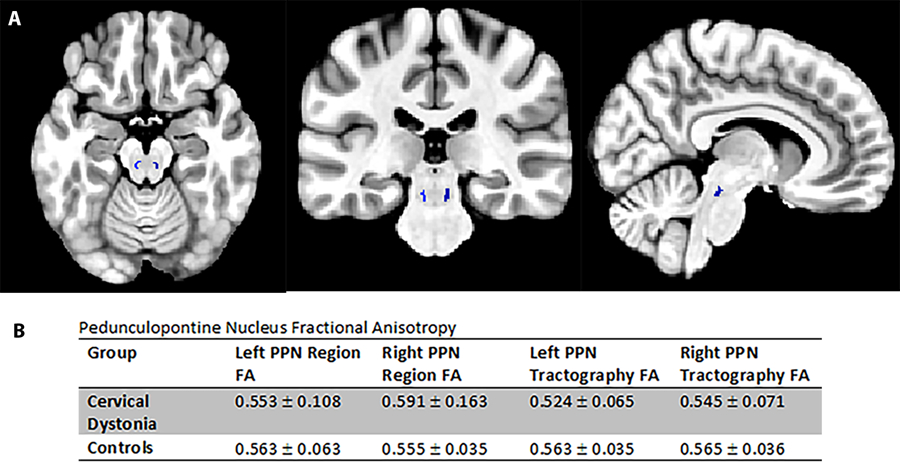
Pedunculopontine Nucleus Diffusion Tensor Imaging
A. Axial, coronal and sagittal brain MRI in MNI space showing the location of the pedunculopontine nucleus (PPN) in blue; B. This table shows fractional anistropy values in and around the PPN in CD and control subjects. There is no difference between the groups.
Discussion
Using immunohistochemical methods, we have identified loss of ChAT positivity in the pedunculopontine nucleus of CD brains. This ChAT deficiency occurs without reactive gliosis. Decreased ChAT immunostaining is compatible with decreased ChAT protein production in the PPN. Our finding is supported by the work of Clement et al. showing a cholinergic abnormality with increased acetylcholinesterase enzyme activity in the PPN of a dystonia mouse model with a dystonin gene loss of function mutation.3,25
The PPN in living CD patients and HC’s was evaluated with DTI, which is sensitive to white matter microstructure. There were no differences in fractional anistropy of white matter tracts surrounding the PPN or in white matter within the PPN. The lack of PPN DTI abnormalities in CD is supported by the similar findings in a larger DTI study in CD.26 These findings, in keeping with LFB staining, show intact myelin in histological sections containing the PPN from both CD and controls. In light of decreased ChAT staining in CD with absence of neuronal loss, gliosis, and myelin loss, and lack of differences in fractional anisotropy in DTI, it seems that there is a cholinergic deficiency at the cellular level in the PPN of CD subjects. This deficiency likely represents a functional disturbance in the protein level in neurons rather than neurodegeneration. In other words, there is an absence of reactive gliosis and associated Wallerian degeneration/secondary white matter loss in the PPN of CD cases. Furthermore, it is also unlikely that the cholinergic neurons were replaced with a different cell type because cellular morphology appeared similar.
While the PPN is a conserved structure across species, its function in humans is not completely clear.27 The relationship between dystonia and the PPN may be through one of the PPN’s proposed roles in motor control. Cholinergic neurons are the most studied neurons among various types of neurons in the PPN. These neurons are thought to be differentially connected to other brain regions, but this has not been established in humans.28 Following this line of thinking, the impact of decreased ChAT presence in the PPN in CD brains on the function of another brain region is unknown.
Contrary to previous studies in dystonia that showed striatal cholinergic deficiencies,2,5 our study did not detect cholinergic abnormalities in posterior putamen neurons of CD cases. CD and controls did not show obvious differences in cholinergic interneuron densities. This region of the putamen is known to play a role in motor control,29 and its lesions can be associated with development of CD.30
Although there are various causes of dystonia, the findings of this study strengthen the role of acetylcholine in dystonia, which has been demonstrated in numerous studies. Alterations in cholinergic pathway markers in animal models have been described.3,31 In human DYT1 dystonia, there were deficiencies of cholinergic markers in frozen putamen tissue lysates. One study showed cholinergic neuronal loss in a conditional torsinA deletion mouse model.2 In a human CD imaging study, Albin et al. found decreased striatal binding of vesicular acetylcholine transporter, a marker of cholinergic vesicles.5 Further evidence for acetylcholine’s significance in CD comes from cases of CD triggered by cholinesterase inhibitors that reverse with drug discontinuation.4
In spite of the acetylcholine’s importance in dystonia, the etiology of decreased ChAT protein in the PPN in CD is unclear. A decreased protein level may be the result of increased degradation or decreased production. Increased ChAT degradation occurs in congenital myasthenic syndrome because of mutations leading to increased ubiquitination.32 This is unlikely to be the case in CD because of normal staining in the putamen and the absence of ubiquitin inclusions. The alternative explanation is decreased protein production, either by decreased mRNA transcription or protein translation. While ChAT transcription is regulated by transcription factors, epigenetic factors, such as histone modifications, also affect ChAT transcription.33,34 Histone modifications are stable in frozen postmortem brain tissues, and can be assayed using chromatin immunoprecipation and DNA sequencing of isolated neuronal nuclei from the PPN.35 The other level of regulation, which is at time of translation, can be investigated when more technologies are developed for identifying RNA modifications, or epitranscriptomics.36
This study is not without limitations. First, the number of CD brain donors is limited, and these tissues could only be successfully procured from a single brain bank despite requests made to numerous other brain banks. At this brain bank, formalin-fixed samples were only available from the right hemisphere. The left hemisphere could not be evaluated because it was frozen and would possibly exhibit freeze artifacts in histological staining. In MRI, the inherent limitation is difficulty of localizing the PPN. In Alho et al., the challenge of distinguishing the PPN from the cuneiform nucleus was recognized.21 Finally, neuronal cell counting was not based on unbiased quantitative methods, such as unbiased stereology, which require larger amounts of tissue. Further studies need to be perfomed in the future to definitively establish cholinergic dysfunction in PPN as well as in other CD-associated brain regions.
Supplementary Material
Acknowledgements
We are appreciative of the individuals (and their families) who chose to donate their brains to BTB. We also thank Dr. Drew Pratt for providing staining and manuscript suggestions.
Funding: This research is in part supported by the Intramural Research Program of the National Institutes of Health and the National Institute of Neurological Disorders and Stroke.
Full Financial Disclosures of All Authors (Preceding 12 Months)
Dr. Mente received a research fellowship from the Dystonia Medical Research Foundation (unrelated to this study). Dr. Amaro is supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (unrelated to this study). Dr. Hallett serves as Chair of the Medical Advisory Board for and may receive honoraria and funding for travel from the Neurotoxin Institute (unrelated to this study). He may accrue revenue on US Patent #6,780,413 B2 (Issued: August 24, 2004): Immunotoxin (MAB-Ricin) for the treatment of focal movement disorders, and US Patent #7,407,478 (Issued: August 5, 2008): Coil for Magnetic Stimulation and methods for using the same (H-coil); in relation to the latter, he has received license fee payments from the NIH (from Brainsway) for licensing of this patent. He is on the Editorial Board of approximately 20 journals, and received royalties and/or honoraria from publishing from Cambridge University Press, Oxford University Press, and Elsevier (unrelated to this study). Supplemental research funds have been granted by UniQure, Merz, Allergan, and Medtronic (unrelated to this study).
Footnotes
Financial Disclosure/Conflict of interest: The authors declare no conflict of interest.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Nutt JG, Muenter MD, Aronson A, Kurland LT, Melton LJ. Epidemiology of focal and generalized dystonia in Rochester, Minnesota. Mov Disord. 1988;3:188–194. [DOI] [PubMed] [Google Scholar]
- 2.Pappas SS, Darr K, Holley SM, Cepeda C, Mabrouk OS, Wong J-MT, LeWitt TM, Paudel R, Houlden H, Kennedy RT, Levine MS, Dauer WT, Albin R, Cross D, Cornblath W, Wald J, Wernette K, Frey K, Minoshima S, Alexander G, DeLong M, Strick P, Berke J, Bressman S, Sabatti C, Raymond D, Leon D de, Klein C, Kramer P, Brin M, Fahn S, Breakefield X, Ozelius L, Risch N, Burke R, Fahn S, Marsden C, Burke R, Karanas A, Carbon M, Argyelan M, Ghilardi M, Mattis P, Dhawan V, Bressman S, Eidelberg D, Cepeda C, Galvan L, Holley S, Rao S, Andre V, Botelho E, Chen J, Watson J, Deisseroth K, Levine M, Chen P, Burdette A, Porter J, Ricketts J, Fox S, Nery F, Hewett J, Berkowitz L, Breakefield X, Caldwell K, Caldwell G, Chesselet M, Plotkin J, Wu N, Levine M, Dauer W, Jaunarajs KE, Bonsi P, Chesselet M, Standaert D, Pisani A, Gage G, Stoetzner C, Wiltschko A, Berke J, Geneser F, Gerace L, Gittis A, Leventhal D, Fensterheim B, Pettibone J, Berke J, Kreitzer A, Goodchild R, Kim C, Dauer W, Grundmann K, Glockle N, Martella G, Sciamanna G, Hauser T, Yu L, et al. Forebrain deletion of the dystonia protein torsinA causes dystonic-like movements and loss of striatal cholinergic neurons. Elife. 2015;4:e08352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clément C, Lalonde R, Strazielle C. Acetylcholinesterase activity in the brain of dystonia musculorum (Dst(dt-J)) mutant mice. Neurosci Res. 2012;72:79–86. [DOI] [PubMed] [Google Scholar]
- 4.Ikeda K, Yanagihashi M, Sawada M, Hanashiro S, Kawabe K, Iwasaki Y. Donepezil-induced cervical dystonia in Alzheimer’s disease: a case report and literature review of dystonia due to cholinesterase inhibitors. Intern Med. 2014;53:1007–1010. http://www.ncbi.nlm.nih.gov/pubmed/24785894. Accessed November 24, 2016. [DOI] [PubMed] [Google Scholar]
- 5.Albin RL, Cross D, Cornblath WT, Wald JA, Wernette K, Frey KA, Minoshima S. Diminished striatal [123I]iodobenzovesamicol binding in idiopathic cervical dystonia. Ann Neurol. 2003;53:528–532. [DOI] [PubMed] [Google Scholar]
- 6.Dautan D, Huerta-Ocampo I, Witten IB, Deisseroth K, Bolam JP, Gerdjikov T, Mena-Segovia J. A Major External Source of Cholinergic Innervation of the Striatum and Nucleus Accumbens Originates in the Brainstem. J Neurosci. 2014;34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holt DJ, Hersh LB, Saper CB. Cholinergic innervation in the human striatum: a three-compartment model. Neuroscience. 1996;74:67–87. http://www.ncbi.nlm.nih.gov/pubmed/8843078. Accessed November 27, 2016. [DOI] [PubMed] [Google Scholar]
- 8.Bernácer J, Prensa L, Giménez-Amaya JM, Parent A, Haber S, DiFiglia M, Pasik P, Pasik T, Koos T, Tepper J, Yelnik J, Francois C, Percheron G, Tande D, Roberts R, Gaither L, Peretti F, Lapidus B, Chute D, Graveland G, Williams R, DiFiglia M, Kubota Y, Inagaki S, Shimada S, Kito S, Eckenstein F, Chang H, Cragg S, Lapper S, Bolam J, Wilson C, Chang H, Kitai S, Galarraga E, Hernandez-Lopez S, Reyes A, Miranda I, Bermudez-Rattoni F, Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G, Perez-Rosello T, Figueroa A, Salgado H, Vilchis C, Tecuapetla F, Pakhoti P, Bracci E, Tepper J, Bolam J, Vizi S, Ronai A, Jr LH, Knoll J, Bertorelli R, Consolo S, Boer P De, Damsma G, Schram Q, Stoof J, Zaagsma J, Robertson G, Staines W, DeBoer P, Abercrombie E, Rakovska A, Javitt D, Raichev P, Ang R, Balla A, Rakovska A, Kiss J, Raichev P, Lazarova M, Kalfin R, Kawaguchi Y, Wilson C, Augood S, Emson P, Apicella P, Wang Z, Kai L, Day M, Ronesi J, Yin H, Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel A, Warren N, Piggott M, Perry E, Burn D, Selden N, et al. Cholinergic Interneurons Are Differentially Distributed in the Human Striatum. Waldvogel H, ed. PLoS One. 2007;2:e1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMillan CT, Irwin DJ, Avants BB, Powers J, Cook PA, Toledo JB, McCarty Wood E, Van Deerlin VM, Lee VM-Y, Trojanowski JQ, Grossman M. White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2013;84:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Consky ES, Basinsky A, Belle L, Ranawaya R, Lang AE. The Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS):assessment of validity and inter-rater reliability. Neurology. 1990;40(suppl 1:445. [Google Scholar]
- 11.Olszewski J, Baxter DW. Cytoarchitecture of the Human Brainstem. 3rd, revis ed. (Büttner-Ennever JA, Horn AKE, eds.). Karger Publishers; 2014. [Google Scholar]
- 12.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 14.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 15.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L, neuropathologists participating C. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 16.Pratt D, Mente K, Rahimpour S, Edwards NA, Tinaz S, Berman BD, Hallett M, Ray-Chaudhury A. Diminishing evidence for torsinA-positive neuronal inclusions in DYT1 dystonia. Acta Neuropathol Commun. 2016;4:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierpaoli C, Walker L, Irfanoglu MO, Barnett A, Basser P, Chang L, Koay C, Pajevic S, Rohde G, Sarlls J, Wu M. TORTOISE: an integrated software package for processing of diffusion MRI data. In: International Society of Magnetic Resonance in Medicine 18th Annual Meeting Stockholm; 2010. [Google Scholar]
- 18.Irfanoglu MO, Modi P, Nayak A, Hutchinson EB, Sarlls J, Pierpaoli C. DR-BUDDI (Diffeomorphic Registration for Blip-Up blip-Down Diffusion Imaging) method for correcting echo planar imaging distortions. Neuroimage. 2015;106:284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162–173. http://www.ncbi.nlm.nih.gov/pubmed/8812068. Accessed September 29, 2017. [DOI] [PubMed] [Google Scholar]
- 20.Taylor PA, Saad ZS. FATCAT: (an efficient) Functional and Tractographic Connectivity Analysis Toolbox. Brain Connect. 2013;3:523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alho ATDL, Hamani C, Alho EJL, da Silva RE, Santos GAB, Neves RC, Carreira LL, Araújo CMM, Magalhães G, Coelho DB, Alegro MC, Martin MGM, Grinberg LT, Pasqualucci CA, Heinsen H, Fonoff ET, Amaro E. Magnetic resonance diffusion tensor imaging for the pedunculopontine nucleus: proof of concept and histological correlation. Brain Struct Funct. March 2017:1–12. [DOI] [PMC free article] [PubMed]
- 22.Mesulam MM, Geula C. Overlap between acetylcholinesterase-rich and choline acetyltransferase-positive (cholinergic) axons in human cerebral cortex. Brain Res. 1992;577:112–120. [DOI] [PubMed] [Google Scholar]
- 23.Mesulam M ‐Marsel, Geula C, Bothwell MA, Hersh LB. Human reticular formation: Cholinergic neurons of the pedunculopontine and laterodorsal tegmental nuclei and some cytochemical comparisons to forebrain cholinergic neurons. J Comp Neurol. 1989;283:611–633. [DOI] [PubMed] [Google Scholar]
- 24.Manaye KF, Zweig R, Wu D, Hersh LB, De Lacalle S, Saper CB, German DC. Quantification of cholinergic and select non-cholinergic mesopontine neuronal populations in the human brain. Neuroscience. 1999;89:759–770. [DOI] [PubMed] [Google Scholar]
- 25.Kato K, Hayako H, Ishihara Y, Marui S, Iwane M, Miyamoto M. TAK-147, an acetylcholinesterase inhibitor, increases choline acetyltransferase activity in cultured rat septal cholinergic neurons. Neurosci Lett. 1999;260:5–8. http://www.ncbi.nlm.nih.gov/pubmed/10027686. Accessed April 2, 2017. [DOI] [PubMed] [Google Scholar]
- 26.Burciu RG, Hess CW, Coombes SA, Ofori E, Shukla P, Chung JW, McFarland NR, Wagle Shukla A, Okun MS, Vaillancourt DE. Functional activity of the sensorimotor cortex and cerebellum relates to cervical dystonia symptoms. Hum Brain Mapp. 2017;38:4563–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gut NK, Winn P. The pedunculopontine tegmental nucleus-A functional hypothesis from the comparative literature. Mov Disord. 2016;31:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mena-Segovia J, Bolam JP. Rethinking the Pedunculopontine Nucleus: From Cellular Organization to Function. Neuron. 2017;94:7–18. [DOI] [PubMed] [Google Scholar]
- 29.Draganski B, Kherif F, Klöppel S, Cook PA, Alexander DC, Parker GJM, Deichmann R, Ashburner J, Frackowiak RSJ. Evidence for Segregated and Integrative Connectivity Patterns in the Human Basal Ganglia. J Neurosci. 2008;28 http://www.jneurosci.org/content/28/28/7143.long. Accessed April 11, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molho ES, Factor SA. Basal ganglia infarction as a possible cause of cervical dystonia. Mov Disord. 1993;8:213–216. [DOI] [PubMed] [Google Scholar]
- 31.Sciamanna G, Hollis R, Ball C, Martella G, Tassone A, Marshall A, Parsons D, Li X, Yokoi F, Zhang L, Li Y, Pisani A, Standaert DG. Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA. Neurobiol Dis. 2012;47:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morey TM, Albers S, Shilton BH, Rylett RJ. Enhanced ubiquitination and proteasomal degradation of catalytically deficient human choline acetyltransferase mutants. J Neurochem. 2016;137:630–646. [DOI] [PubMed] [Google Scholar]
- 33.Quirin-Stricker C, Mauvais C, Schmitt M. Transcriptional activation of human choline acetyltransferase by AP2- and NGF-induced factors. Mol Brain Res. 1997;49:165–174. [DOI] [PubMed] [Google Scholar]
- 34.Aizawa S, Teramoto K, Yamamuro Y. Histone deacetylase 9 as a negative regulator for choline acetyltransferase gene in NG108–15 neuronal cells. Neuroscience. 2012;205:63–72. [DOI] [PubMed] [Google Scholar]
- 35.Kundakovic M, Jiang Y, Kavanagh DH, Dincer A, Brown L, Pothula V, Zharovsky E, Park R, Jacobov R, Magro I, Kassim B, Wiseman J, Dang K, Sieberts SK, Roussos P, Fromer M, Harris B, Lipska BK, Peters MA, Sklar P, Akbarian S. Practical Guidelines for High-Resolution Epigenomic Profiling of Nucleosomal Histones in Postmortem Human Brain Tissue. Biol Psychiatry. 2017;81:162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. 2017;18:275–291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.