

DAP3 represses adenosine-to-inosine (A-to-I) RNA editing in cancer cells and promotes cancer progression.
Abstract
RNA editing introduces nucleotide changes in RNA sequences. Recent studies have reported that aberrant A-to-I RNA editing profiles are implicated in cancers. Albeit changes in expression and activity of ADAR genes are thought to have been responsible for the dysregulated RNA editome in diseases, they are not always correlated, indicating the involvement of secondary regulators. Here, we uncover DAP3 as a potent repressor of editing and a strong oncogene in cancer. DAP3 mainly interacts with the deaminase domain of ADAR2 and represses editing via disrupting association of ADAR2 with its target transcripts. PDZD7, an exemplary DAP3-repressed editing target, undergoes a protein recoding editing at stop codon [Stop →Trp (W)]. Because of editing suppression by DAP3, the unedited PDZD7WT, which is more tumorigenic than edited PDZD7Stop518W, is accumulated in tumors. In sum, cancer cells may acquire malignant properties for their survival advantage through suppressing RNA editome by DAP3.
INTRODUCTION
RNA editing is a widespread co- or posttranscriptional modification process that introduces changes in RNA sequences encoded by the genome, contributing to “RNA mutations.” Editing of adenosine to inosine (A-to-I) in double-stranded RNA (dsRNA), catalyzed by adenosine deaminase acting on RNA (ADAR) family of enzymes, is the most common type of RNA editing in mammals (1). In vertebrates, a family of three ADAR proteins, ADAR1, ADAR2, and ADAR3, has been characterized (1, 2). ADAR1 and ADAR2 (ADARs) catalyze all currently known A-to-I editing sites. In contrast, ADAR3 has no documented deaminase activity. Inosine (I) essentially mimics guanosine (G); therefore, ADAR proteins introduce a virtual A-to-G substitution in transcripts. These changes can lead to specific amino acid substitutions, alternative splicing, microRNA-mediated gene silencing, or changes in transcript localization and stability (1).
Aberrant editing on specific transcripts and their association with cancer progression have been found in many cancer types (3). Protein-recoding type of RNA editing contributes to tumorigenesis mainly through enhancing the activity of oncogenes or reducing the activity of tumor suppressors (4–6). AZIN1 (antizyme inhibitor 1) is one of the most well-studied ADAR1 target in cancer. Editing of AZIN1 results in a serine (S) to glycine (G) substitution at residue 367, and the edited form AZIN1S367G may undergo protein conformational change and thus has much stronger tumorigenic capabilities than the wild-type (WT) form. RNA editing of AZIN1 was significantly higher in different cancer types such as hepatocellular carcinoma (HCC) (5), esophageal squamous cell carcinoma (ESCC) (7), and colorectal cancer (CRC) (8), which predicted worse prognosis of cancer patients. Editing of noncoding regions or noncoding RNAs can also promote cancer progression. One example is FAK (focal adhesion kinase), which harbors a specific editing site in intronic region, leading to the stabilization of FAK transcripts and lung adenocarcinoma cell migration and invasion (9). In addition, editing of microRNAs is also linked to cancer progression. For instance, RNA editing of a tumor-suppressive microRNA pri-let-7d, which disrupts its biogenesis, is implicated in chronic myelogenous leukemia (10).
Although the causal relationship between the dysregulated A-to-I RNA editome and cancer has been well studied in the past decade, the causes of RNA editing dysregulation in cancer remain to be investigated. Most studies attempted to simply correlate altered RNA editing profiles with changes in expression level and/or activity of ADARs during cancer progression. However, they are not always correlated with each other in numerous diseases (11), indicative of the involvement of non-ADAR editing regulators. It is therefore critical to uncover key secondary regulators that manipulate another layer of editing regulation and understand their role in cancer. To date, a couple of regulatory mechanisms of A-to-I RNA editing have been reported. CREB1 (cyclic adenosine 5′-monophosphate–responsive element–binding protein 1) (12) and JNK1 (c-Jun N-terminal kinase 1) (13) can induce ADAR2 expression in neurons and pancreatic β cells, respectively. Self-editing of ADAR2 pre-mRNA modulates its own alternative splicing, forming an autoregulatory loop to tightly control editing level (14). At posttranslational level, degradation of ADAR proteins can be modulated by PIN1 (peptidylprolyl cis/trans isomerase NIMA-interacting 1), WWP2 (WW domain containing E3 ubiquitin protein ligase 2), and AIMP2 (aminoacyl TRNA synthetase complex interacting multifunctional protein 2) (15, 16), while sumoylation of ADAR1 by SUMO1 (small ubiquitin-like modifier 1) alters the editing activity of ADAR1 (17). Because the A-to-I RNA editing of pre-mRNAs takes place in the nucleus, changes in subcellular localization of ADARs affect the editing process. Transportin-1 (TNPO1) and Exportin-5 (XPO5) mediate the import and export of ADAR1 to the nucleus, respectively (18). In addition, homodimerization of ADAR1 or ADAR2 (ADAR1/2) monomers through their dsRNA binding domain (dsRBD) is also essential for editing to occur (19–21). A recent study reported that SRSF9 (serine/arginine-rich splicing factor 9) could selectively suppress brain-specific A-to-I RNA editing by disrupting ADAR2 homodimerization (22). Recognition of editing substrates by ADARs may depend on the former’s dsRNA secondary structures. RNA binding proteins, which are capable of modulating RNA structure, may therefore affect A-to-I RNA editing. More recently, our group found DHX9 (DEAH box helicase 9) as the first bidirectional regulator of editing in human, and editing substrate specificity determines the opposing effects of DHX9 on A-to-I editing (23). All these observations suggested that the regulation of A-to-I RNA editing is much more complex than previously appreciated, and identifying non-ADAR regulators would provide new insights into understanding the interwoven regulatory network of A-to-I RNA editing and its contribution to cancer.
In this study, we uncovered a previously unidentified ADAR-interacting protein death associated protein 3 (DAP3) as a potent repressor of A-to-I RNA editome, and its negative regulation of editing is possibly one mechanism by which DAP3 promotes cancer development. DAP3 is also named as MRPS29 (mitochondrial 28S ribosomal protein S29). As a subunit of mitoribosome, it is involved in the mitochondrial homeostasis and protein synthesis (24, 25). It also functions as a positive mediator of apoptosis induced by IFN-γ (interferon-γ), tumor necrosis factor–α, and Fas ligand as well as anoikis induced by detachment from extracellular matrix (26). Our detailed mechanistic studies indicated that DAP3 localizes in the nucleus and preferentially interacts with the deaminase domain of ADAR2 rather than ADAR1, thereby disrupting the binding of ADAR2 to its target dsRNA substrates. Despite the controversial role of DAP3 in human cancers as reported previously (26), we found that DAP3 was overexpressed in 17 types of cancers and functioned as a strong oncogene. Suppression of A-to-I RNA editome by DAP3 is possibly one mechanism that contributes to the oncogenic effect of DAP3. An exemplary DAP3-repressed editing of PDZD7 (PDZ domain containing 7) gene was found to contribute to tumorigenesis because of the accumulation of a more aggressive form PDZD7WT (unedited) than PDZD7Stop518W (edited). Together, our study demonstrates that DAP3 can serve as a potent editing repressor, featuring another layer of RNA editing modulation in cancer. Cancer cells may acquire malignant properties for their survival advantage through suppressing RNA editome by DAP3, which is likely to be one of the critical mechanisms in driving cancer development.
RESULTS
DAP3 interacts with ADARs in the nucleus
Our previous ADARs coimmunoprecipitation coupled with mass spectrometry (ADARs co-IP–MS) identified DAP3 as a high-confidence ADAR2-interacting protein in human embryonic kidney (HEK) 293T cells (23). We further validated their interaction in an ESCC cell line, EC109, which has been used in our previous RNA editing studies (7, 23). In agreement with the co-IP–MS data, V5 or Flag-tagged ADAR2 could be pulled down by either endogenous or green fluorescent protein (GFP)–tagged DAP3, respectively (Fig. 1, A and B), and their interaction was also observed endogenously in EC109 cells (Fig. 1C). Although DAP3 was not identified as an ADAR1-interacting protein by co-IP–MS because of the low peptide coverage, our co-IP assays indicated a rather weak binding of ADAR1 (the p110 isoform) to DAP3, particularly endogenously in EC109 cells (Fig. 1, A to C). Despite the fact that DAP3 is known to be a mitochondrial ribosomal protein, cell fractionation assays confirmed that DAP3 could be expressed in both nucleus and cytoplasm of EC109 cells (Fig. 1D). Immunofluorescence staining further confirmed that DAP3 colocalizes with the mitochondrial outer membrane protein TOMM20 and ADAR2 in the mitochondrion and nucleus of EC109 cells, respectively (fig. S1, A and B). Moreover, co-IP assays after cell fractionation indicated that the interaction of DAP3 to ADARs occurs only in the nucleus where the pre-mRNA editing takes place (Fig. 1E).
Fig. 1. DAP3 interacts with ADARs in the nucleus.
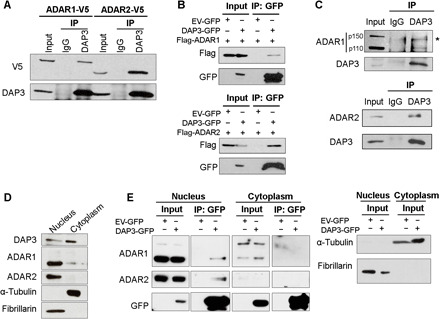
(A) Co-IP analysis of protein extracts from EC109 cells transfected with V5-tagged ADAR1 or ADAR2. Western blot (WB) analysis of DAP3-pulldown products was conducted using V5 and DAP3 antibodies. IgG, immunoglobulin G. (B) Co-IP analysis of protein extracts from EC109 cells cotransfected with the indicated tagged ADAR1/2 and DAP3 constructs, using a GFP-trap system. WB analysis of GFP-pulldown products was conducted using Flag and GFP antibodies. EV-GFP, GFP empty vector. (C) Co-IP analysis of protein extracts from EC109 cells. IP was performed with a DAP3 antibody, followed by WB analysis of DAP3-pulldown products using ADAR1, ADAR2, and DAP3 antibodies. *, nonspecific band. (D) WB analysis of the nuclear and cytoplasmic fractions of EC109 cells. (E) Co-IP analysis of the nuclear and cytoplasmic fractions of EC109 cells transfected with DAP3-GFP or EV-GFP. WB analysis of GFP-pulldown products was conducted using the indicated antibodies (left). α-Tubulin (cytoplasmic control) and fibrillarin (nucleic control) were analyzed in the input after fractionation (right). (A to C and E) One percent of the total cell lysate was loaded as an input control.
The dsRBD or deaminase domain of ADAR1 or ADAR2 is essential for its binding to DAP3, respectively
To study the exact domain of ADAR1/2 protein responsible for its interaction with DAP3, we performed co-IP analysis by cotransfecting GFP-tagged DAP3 and V5-tagged ADAR1/2 truncated mutants (Fig. 2, A and B). The C3 mutant of ADAR1, which lacks the second dsRBD, and the C4 mutant, which is depleted of the first and second dsRBDs, could not be pulled down, indicating that the second dsRBD of ADAR1 is required for its binding to DAP3 (Fig. 2A). In contrast, C1 and C2 mutants of ADAR2 lacking the deaminase domain failed to be pulled down (Fig. 2B), suggesting that DAP3 interacts with ADAR2 though the latter’s deaminase domain. Since the removal of nuclear localization signal in the N1 and N2 mutants of ADAR2 may affect the subcellular localization of ADAR2 protein, we further confirmed that the interaction between the N1 or N2 mutant and DAP3 still occurred in the nucleus (fig. S1C). We then examined whether the interaction between ADAR1/2 and DAP3 is dependent on RNA molecules. Ribonuclease A (RNase A) treatment markedly reduced the interaction between DAP3 and ADAR2, but not ADAR1 (Fig. 2, C and D). To confirm whether the interaction between DAP3 and ADAR2 is dependent on RNA, we generated an ADAR2 EAA mutant that carries point mutations introduced in the lysine-lysine-x-x-lysine (KKxxK) motifs within the dsRBDs, rendering the EAA mutants incapable of dsRNA binding (27), and found that endogenous DAP3 protein only pulled down WT ADAR2, but not the EAA mutant, suggesting that their interaction is truly dependent on RNA molecules (Fig. 2E).
Fig. 2. ADAR1 and ADAR2 interact with DAP3 through different domains.
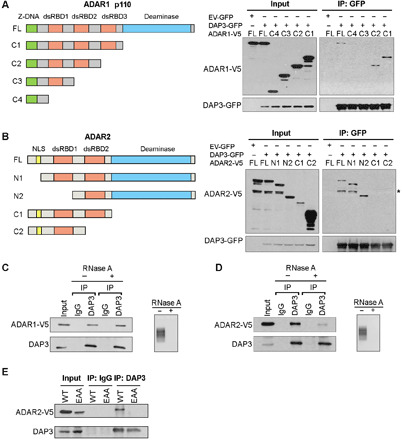
(A) Schematic diagrams of full-length (FL), C1, C2, C3, and C4 deletion mutants of ADAR1 p110 isoform (left). Co-IP analysis of protein lysates from EC109 cells transfected with V5-tagged deletion mutants and DAP3-GFP or EV-GFP, using GFP-trap system, followed by WB analysis of GFP-pulldown products using GFP and V5 antibodies (right). (B) Schematic diagrams of FL, N1, N2, C1, and C2 mutants of ADAR2 (left). Co-IP and WB analyses were conducted as described in (A). *, nonspecific band; NLS, nuclear localization signal. (C and D) Co-IP analysis was conducted in EC109 cells transfected with (C) ADAR1-V5 and (D) ADAR2-V5. Before IP assays, protein lysates were treated with (+) or without (−) RNase A, followed by WB analysis of DAP3-pulldown products using V5 and DAP3 antibodies. Agarose gel demonstrating the successful digestion of total RNA. (E) Co-IP analysis was conducted in EC109 cells transfected with the indicated construct, followed by WB analysis using V5 and DAP3 antibodies. (A to E) One percent of the total cell lysate was loaded as an input control.
Although ADAR1 and ADAR2 are homologous, they are not identical, particularly their dsRBDs. We conducted a sequence alignment using the BLAST (Basic Local Alignment Search Tool) Sequence Analysis Tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and found that only the first dsRBDs of ADAR1 and ADAR2 proteins could be aligned, sharing 38% amino acid sequence identity, while the deaminase domains of two ADAR proteins share 37% amino acid sequence identity. Therefore, involvement of distinct domains of ADARs in DAP3-ADARs interaction may be attributed to differences in sequences and structures of ADAR1 and ADAR2 proteins. These differences may eventually contribute to the differential mechanisms by which DAP3 represses ADAR1- and ADAR2-regulated editing.
DAP3 mainly functions as a potent repressor of A-to-I RNA editing in cancer cells
It has been reported by us that the dysregulated A-to-I RNA editome is implicated in ESCC, HCC, and gastric cancer, which might be partially attributed to the differentially expressed ADARs in tumors (6, 7, 28). It remains unexplored whether non-ADAR regulators control another layer of editing regulation during cancer progression. To this end, we stably knocked down DAP3 in two ESCC cell lines, EC109 and KYSE180, using a lentiviral system and observed that the depletion of DAP3 had no obvious effect on two isoforms (p110 and p150) of ADAR1 and ADAR2 (Fig. 3A and fig. S2). We went on to perform the strand-specific RNA sequencing (RNA-seq) analysis to elucidate the regulatory effect of DAP3 on the global editome in an unbiased and transcriptome-wide manner. Using our RNA editing analysis pipeline with two filter criteria, (i) a read coverage of ≥ 20 and (ii) editing frequency > 10%, we first identified high-confidence A-to-I RNA editing sites in three RNA-seq datasets from our scramble control (shScr) EC109 and KYSE180 cells (n = 2), HEK293T cells (n = 6) (16) and randomly selected normal esophagus mucosa samples from the Genotype-Tissue Expression (GTEx) database (n = 10) (29). We found that the extent of A-to-I RNA editing in our shScr cells was largely comparable with that in normal esophagus mucosa samples and HEK293T cells (fig. S3). We next identified DAP3-affected editing sites using our RNA-seq dataset with two additional criteria: (i) |% editing change| ≥10% between both of DAP3-knockdown (sh1 and sh2) and the shScr samples and (ii) a read coverage of ≥20 in all samples. Most of the affected sites resides in 3′ untranslated regions (3′UTRs), followed by the intronic regions (Fig. 3B). In EC109 and KYSE180 cells, 1109 and 2322 affected sites distributed across 337 and 526 genes, respectively (Fig. 3C and tables S1 and S2). Ingenuity pathway analysis of these genes harboring affected editing sites showed that they were functionally enriched in many key processes relevant to cancer development and progression, such as cell cycle, DNA replication, recombination and repair, and cell death and survival (fig. S4A). Intriguingly, depletion of DAP3 drastically enhanced the A-to-I editing, as reflected by the fact that approximately 95% (1053 of 1109) and 97% (2261 of 2322) of affected sites were overedited in DAP3-depleted EC109 and KYSE180 cells, respectively (Fig. 3D and tables S1 and S2). We randomly selected 27 overedited sites for validation, and 20 of 27 (74%) were validated in both DAP3-depleted EC109 cells (Fig. 3E and table S3), while among 9 inferred underedited sites, 7 sites were confirmed to be either unaffected or overedited sites (fig. S4B). CRISPR-Cas9–mediated DAP3 knockout (KO) in EC109 cells further confirmed a strong repressive effect of DAP3 on A-to-I RNA editome (Fig. 3F and fig. S4C). Of note, the DAP3-mediated repression on editing is not limited to ESCC, as DAP3 was also found to repress editing in two HCC cell lines, SNU398 and Huh7, and a glioblastoma cell line, U251 (fig. S5, A and B). All these findings support that DAP3 mainly functions as a potent repressor of A-to-I RNA editing in cancer cells.
Fig. 3. DAP3 functions as a potent editing repressor in ESCC cells.
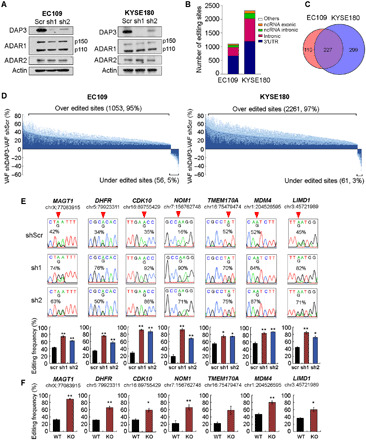
(A) WB analysis of the indicated proteins in stable DAP3-knockdown EC109 and KYSE180 cells. β-Actin (Actin) was used as a loading control. (B) Distribution of DAP3-affected editing sites over annotated genomic regions in EC109 and KYSE180 cells. (C) Venn diagram showing genes containing DAP3-affected sites in EC109 and KYSE180 cells. (D) Transcriptome-wide RNA editing analysis uncovers DAP3-knockdown–mediated changes in A-to-I editome of EC109 and KYSE180 cells. Changes in editing level are calculated by subtracting VAF (variant allele frequency) of shDAP3 with VAF of shscr (VAFshDAP3 − VAFshscr). Data are shown as the average editing change (mean; deep blue) induced by DAP3 sh1 and sh2 with the SE (SEM; light blue). (E) Sanger sequencing chromatograms illustrate editing of randomly selected sites (top). Quantification of editing frequency of each site is shown in the bottom. Data are presented as mean ± SEM. of technical triplicates. (F) Quantification of editing frequency of DAP3-affected sites in WT and DAP3-KO EC109 cells. Data are presented as mean ± SEM. of three individual clones generated by the CRISPR-Cas9 method. (E and F) Percentage represents the editing frequency calculated by taking the peak area of “G” peak over the sum of “A” and “G” peaks. Arrow indicates position of editing. Statistical analysis is conducted using unpaired, two-tailed Student’s t test (*P < 0.05; **P < 0.01).
DAP3 disturbs ADAR1 homodimerization and inhibits the binding of ADAR2 protein to its dsRNA substrates
It is critical to decipher the underlying mechanism by which DAP3 represses A-to-I RNA editing without affecting ADAR expression. HTR2C is a well-characterized editing target, and its dsRNA structure has been well delineated in many studies (30). In addition, MAGT1, one of validated editing targets, has multiple editing sites within its 3′UTR, and silencing of DAP3 increased the editing level of these sites (Fig. 3, E and F). Therefore, both HTR2C and MAGT1 were chosen as editing substrates for the following mechanistic study. Since binding of ADARs to editing substrates and ADAR homodimerization are two critical factors required for A-to-I RNA editing, we first examined whether DAP3 could affect ADAR1/2-dsRNA binding. RNA immunoprecipitation (RIP) assays indicated that overexpression of DAP3 significantly repressed the association of ADAR2 protein with HTR2C and MAGT1 RNA transcripts in vivo (Fig. 4A). In contrast, DAP3 had no obvious effect on the association of ADAR1 with their substrates (Fig. 4B). Furthermore, we performed RNA electrophoretic mobility shift assay to examine the effect of DAP3 on the binding of ADAR2 to MAGT1 and HTR2C RNA duplex probes (Fig. 4, C and D, and fig. S6A). Purified ADAR2 protein could bind to HTR2C and MAGT1 RNA duplexes, and the amount of ADAR2-bound RNA duplexes decreased gradually by the addition of increasing amounts of DAP3–glutathione S-transferase (GST) but not the control GST protein (Fig. 4, C and D). Of note, we found that DAP3 could not directly bind to these RNA duplexes (fig. S6B), indicative of the fact that DAP3 represses editing via affecting the binding of ADAR2 to dsRNAs, rather than competing with ADAR2 for substrate binding. Moreover, we conducted enhanced cross-linking and immunoprecipitation followed by high-throughput sequencing (eCLIP-Seq) analysis and mapped the binding sites of DAP3 on its target RNAs. We observed that DAP3-binding peaks on edited RNAs were not in close proximity to the edited sites (the median distance from DAP3-binding sites to editing sites is 2.5 kb) (fig. S7), suggesting that DAP3-mediated editing suppression is unlikely because of the blockage of ADAR2-binding sites or a competitive binding to the editing regions between DAP3 and ADAR2. In addition, by conducting co-IP assays, we found that DAP3 inhibited ADAR1 homodimerization (Fig. 4E, lane 6 versus lane 8) but had no obvious effect on ADAR2 homodimerization (Fig. 4F, lane 6 versus lane 8). Because of the fact that DAP3 preferentially interacts with ADAR2 endogenously (Fig. 1C) and DAP3 tends to regulate ADAR1- and ADAR2-regulated editing via differential mechanisms, we focused on the functional consequences of DAP3-repressed ADAR2 editing in the remainder of our study.
Fig. 4. DAP3 affects ADAR1 homodimerization and inhibits ADAR2-dsRNA association.
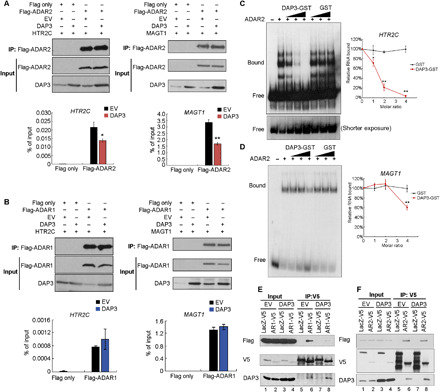
(A and B) RIP–quantitative polymerase chain reaction (qPCR) analysis of the binding of HTR2C or MAGT1 transcripts to ADAR2 (A) or ADAR1 (B) protein in EC109 cells. HTR2C and MAGT1 transcripts were introduced into EC109 cells to reach a detectable level for RIP assays. The WB and qPCR analyses of Flag-RIP immunoprecipitates are shown in the top and bottom, respectively. Data are presented as mean ± SD of technical triplicates. (C and D) RNA electrophoretic mobility shift assay analysis of the binding of HTR2C or MAGT1 RNA duplexes to ADAR2 protein in the presence of DAP3-GST (or GST) protein with an increasing molar ratio of DAP3-GST (or GST) to ADAR2 protein (1:1, 2:1, and 4:1). Relative RNA bound to ADAR2 was calculated as [intensity of bound probe in the presence of ADAR2 and DAP3-GST or GST]/[intensity of bound probe in the presence of ADAR2 alone]. Data are presented as mean ± SD from three independent RNA electrophoretic mobility shift assay experiments. (E and F) Co-IP analysis of protein extracts from EC109 cells cotransfected with V5-tagged ADAR1 (E) or ADAR2 (F) or LacZ control (LacZ-V5), Flag-tagged ADAR1 or ADAR2, and DAP3 or empty vector (EV) control. (A, B, E, and F) Input indicates 1% of the total cell lysate. (A to D) Statistical analysis is conducted using unpaired, two-tailed Student’s t test (*P < 0.05; **P < 0.01).
DAP3 is overexpressed in many cancer types and characterized as an oncogene
To understand the clinical relevance of DAP3 in cancer, we first analyzed DAP3 expression using the RNA-seq data from the Cancer Genome Atlas (TCGA) database (31) and observed that DAP3 was significantly up-regulated in tumor samples as compared to their corresponding nontumor (NT) tissues across 17 of 22 cancer types (Fig. 5). Of note, the expression level of DAP3 in esophageal carcinoma is 1.4-fold of that in NT sample tissues (P = 0.00151) (Fig. 5). Furthermore, stable knockdown of DAP3 by two short hairpin RNAs (shRNAs; sh1 and sh2) in EC109 and KYSE180 cells significantly reduced the frequency of foci formation and decreased the number of colonies formed in soft agar compared with the scrambled shRNA (shScr) control (Figs. 6, A to D, and 3A). Subcutaneous injection of DAP3-depleted EC109 cells into mice led to much smaller tumors when compared to that of shScr cells during a 17-day observation period (Fig. 6, E and F). At end point, we confirmed that DAP3 was expressed at very low level or absent in the xenograft tumors derived from DAP3-depleted cells (Fig. 6G). Knockdown DAP3 in two HCC cell lines, Huh7 and SNU398, was also found to inhibit tumorigenesis (fig. S8, A to D).
Fig. 5. DAP3 is overexpressed in many cancer types.
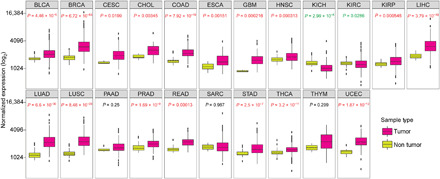
Expression of DAP3 gene in 22 cancer types from TCGA. Wilcoxon rank sum test was performed to compute the statistical significance. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; STAD, stomach adenocarcinoma; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma.
Fig. 6. DAP3 functions as an oncogene.
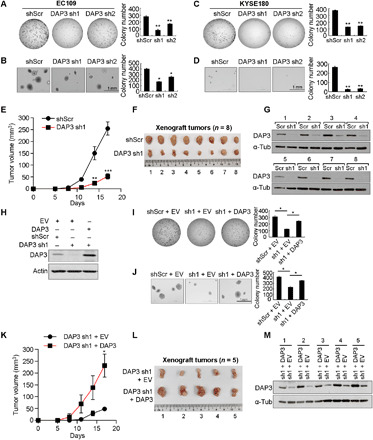
(A and B) Quantification of foci formation (A) or soft agar colony formation (B) in the indicated stable EC109 cells. (C and D) Quantification of foci formation (C) or soft agar colony formation (D) in the indicated stable KYSE180 cells. (E) Growth curve of tumors derived from the indicated stable cells in mice. (F) Tumors derived from the indicated stable cell lines at end point (n = 8 mice per group). (G) WB analysis of DAP3 protein in xenograft tumors at end point. (H) WB analysis of DAP3 expression in stable DAP3-depleted EC109 cells without (DAP3 sh1 + EV) or with the restored DAP3 expression (DAP3 sh1 + DAP3). (I and J) Quantification of foci formation (I) or soft agar colony formation (J) in the indicated stable cells. (K) Growth curve of tumors derived from the indicated stable cells in mice. (L) Tumors derived from the indicated stable cell lines at end point (n = 5 mice per group). (M) WB analysis of DAP3 protein in xenograft tumors at end point (A to D, I, and J) Data are presented as the mean ± SD of duplicate or triplicate wells from a representative experiment. (E and K) Data are presented as the mean ± SEM. Statistical significance is determined by unpaired, two-tailed Student’s t test (*P < 0.05; **P < 0.01; ***P < 0.001).
To eliminate the off-target effects of shRNAs against DAP3, we conducted rescue assays by reintroducing DAP3 into DAP3-depleted EC109 cells (Fig. 6H). Both in vitro and in vivo tumorigenicity assays indicated that the tumor-suppressive phenotypes caused by DAP3 knockdown were effectively rescued by overexpressing a DAP3 mutant that preserves the native amino acid sequence but contains seven point mutations within the DAP3 sh1 targeting site (Fig. 6, I to M). Together, these results suggest that DAP3 is implicated as an oncogene in cancers, which may not be specific to ESCC and HCC, but many types of cancers.
The oncogenic role of DAP3 is associated with its regulation of A-to-I RNA editome
To ascertain whether the negative regulation of RNA editome by DAP3 contributes to the oncogenic role of DAP3, we first knocked down ADAR2 before the overexpression of DAP3 in EC109 cells and found that silencing ADAR2 alone significantly increased tumorigenesis (Fig. 7, A to C), which is consistent with previous studies (6, 32). Among all groups of cells, shScr cells overexpressing DAP3 showed the strongest oncogenic phenotypes, while in ADAR2-knockdown cells with DAP3 overexpression, editing of ADAR2-regulated sites was repressed upon ADAR2 knockdown, such that the oncogenic effect of DAP3 overexpression through its editing suppression was significantly attenuated, indicating that the oncogenic ability of DAP3 is at least partially attributable to its ability to suppress RNA editing (Fig. 7, A to C). Moreover, we also observed that cells with both ADAR2 knockdown and DAP3 overexpression demonstrated slightly but significantly stronger oncogenic phenotypes than cells with ADAR2 knockdown alone (Fig. 7, A to C), suggesting that DAP3 might also have editing-independent oncogenic function. Together, these observations suggested that repressing RNA editing is possibly one mechanism by which DAP3 promotes tumorigenesis.
Fig. 7. Editing of PDZD7 reduces its oncogenic ability.
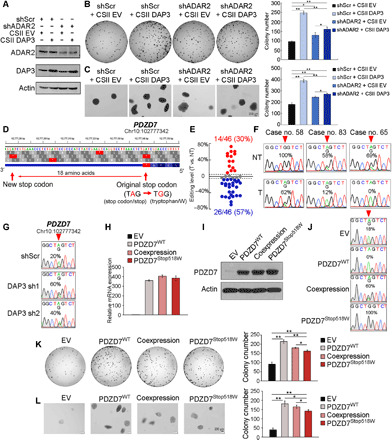
(A) WB analysis of ADAR2, DAP3, and actin expression in the indicated EC109 cells. (B and C) Quantification of foci formation (B) or soft agar colony formation (C) induced in the indicated cells. (D) Diagram of PDZD7 gene locus indicates an A-to-I RNA editing at Chr10:102777342 leads to a stop codon to tryptophan substitution, generating an 18–amino acid extension at its C terminus. (E) Dot plots showing editing level changes of PDZD7 transcripts between tumors (T) and their corresponding nontumor (NT) samples (n = 46). Red dot: ≥5%; blue dot: ≤−5%; gray dot: between −5 and 5%. (F) Sequence chromatograms show the editing of PDZD7 in three representative pairs of ESCC tumors and NT samples. (G) Sequence chromatograms illustrate the editing of PDZD7 in the indicated cells. (H to J) PDZD7 RNA expression, protein expression, and editing level are analyzed by quantitative reverse transcription PCR (H), WB (I), and Sanger sequencing (J) respectively, in EC109 cells stably expressing PDZD7WT and PDZD7Stop518W or coexpressing PDZD7WT and PDZD7Stop518W. (K and L) Quantification of foci formation (K) or soft agar colony formation (L) induced in the indicated stable cells. (B, C, K, and L) Data are presented as the mean ± SD of triplicate wells from a representative experiment. Statistical significance is determined by unpaired, two-tailed Student’s t test (*P < 0.05; **P < 0.01).
It has been reported that protein-recoding type of RNA editing, which generates protein variants, makes a remarkable contribution to cancer progression (33). On the basis of our RNA-seq analysis of DAP3-depleted EC109 cells, we identified a DAP3-affected editing site within a stop codon (Chr10:102777342) of PDZD7 gene, which leads to a substitution of Stop codon (Stop) to tryptophan (Trp/W). Although PDZD7 editing, the first and perhaps the only A-to-I editing event found in vertebrate that impairs stop codon, has been recently reported (34), the function of PDZD7 editing remains unknown. Arising from RNA editing, the original stop codon was bypassed and the edited form PDZD7Stop518W had an 18–amino acid extension at its C terminus (Fig. 7D). Although PDZD7 was found to be regulated by both ADAR1 and ADAR2 (fig. S9), we found that 57% (26 of 46) of primary ESCC tumors demonstrated lower editing level than their corresponding NT samples (Fig. 7, E and F, and fig. S10), presumably due to down-regulation of ADAR2 and further editing repression by DAP3 in tumors. We then confirmed that knockdown of DAP3 was able to restore the editing of PDZD7 (Fig. 7G). Next, to study the functional difference between PDZD7WT and PDZD7Stop518W, we generated EC109 cells stably expressing PDZD7WT (100%) and PDZD7Stop518W (100%) or coexpressing PDZD7WT (40%) and PDZD7Stop518W (60%) (Fig. 7, H to J). Albeit both forms of PDZD7 were found to enhance tumorigenicity of ESCC cells, EC109 cells expressing more PDZD7 Stop518W became less oncogenic than cells expressing PDZD7WT only (Fig. 7, K and L). We conducted the same experiment in KYSE180 cells and observed similar phenotypic changes to those in EC109 cells (fig. S11). These observations suggested that cancer cells might acquire malignant properties for their survival advantage through repressing RNA editome by DAP3, and PDZD7 serves as an exemplary target for which DAP3 represses the PDZD7 editing and prevents cancer cells from expressing the less oncogenic edited form.
DISCUSSION
A-to-I RNA editing plays an important role in development and physiology in various organisms from Drosophila to human. Dysregulated A-to-I RNA editing is implicated in multiple diseases in human including cancer. As reported by us and others in the past decade, dysregulated A-to-I editing is a key driver in the pathogenesis of various cancers, such as glioma (3, 35), HCC (5, 6), CRC (8), gastric cancer (28), and ESCC (7, 23). What are the causes of RNA editing dysregulation remains to be one of long-standing questions in the area. A poor correlation between the expression level of principal catalytic enzymes (ADAR1 and ADAR2) and A-to-I editing frequency suggests an intricate spatiotemporal mode of regulation governed by secondary regulators. As previously reported (19–21), the formation of dsRNA structure, binding of ADARs to dsRNA substrates, homodimerization of ADAR monomers, and a recently described mechanism whereby ADAR2 flips the reactive adenosine out of the target RNA duplex into the active site (36), are essential for A-to-I RNA editing to take place. A number of previously identified editing regulators (e.g., PIN1, SRSF9, and DHX9) modulate RNA editing through affecting expression of ADARs (15–17), disrupting ADAR homodimerization (22), altering subcellular localization of ADARs (18), or remodeling structure of target dsRNA (23). Distinct from these regulators, here, we showed a novel editing repressor, DAP3, that negatively regulates RNA editome mainly through interacting with the deaminase domain of ADAR2 protein and inhibiting the association of ADAR2 to its dsRNA substrates. It has been reported that the deaminase domain of ADAR2 requires the binding and recognition of duplex RNA for efficient RNA editing reaction (37), suggesting that DAP3 may outcompete dsRNA substrates for binding to the deaminase domain of ADAR2 and thus function as an editing “eraser.” DAP3 was also found to inhibit ADAR1 homodimerization in a forced overexpression system; however, as evidenced by the fact that DAP3 barely binds to ADAR1 endogenously in ESCC cells, DAP3-repressed ADAR1 editing might not play a major role in ESCC development. Recent studies have shown that, upon the ablation of ADAR1, unedited endogenous dsRNAs can be detected by MDA5 (melanoma differentiation associated gene 5) and PKR (protein kinase R), which triggers IFN induction and transcription of a wide array of IFN-stimulated genes (38–41). These findings could explain why mutations of ADAR1 lead to the autoimmune disorder Aicardi-Goutières syndrome, which is a fatal childhood encephalopathy with aberrant IFN expression (42). To further investigate the possible involvement of DAP3 in regulating the innate immune response in cancer cells through its repression of ADAR1-regulated editing, we conducted Gene Ontology analysis of differentially expressed genes after DAP3 depletion using our DAP3 KD RNA-seq data. As a result, the vast majority (159 of 161; 98.8%) of IFN-responsive genes were not affected. Therefore, DAP3 is unlikely to have a major effect on IFN signaling under the experimental conditions of this study. Nevertheless, involvement of DAP3 and other regulators in the additional layer of RNA editing regulation helps to ascertain that the A-to-I RNA editing frequency is not solely dependent on the expression and activity of ADARs. Besides ADARs, many trans-acting enhancers, repressors, and bidirectional regulators work in a synergistic or antagonistic fashion to ensure proper execution of A-to-I RNA editing machinery and fine-tune the RNA editing level in different physiological settings.
DAP3 is known to be expressed in mitochondrion and important for mitochondrial physiology and cell apoptosis (43, 44). However, the role of DAP3 in human cancers is still controversial (26). In our study, using the RNA-seq data from TCGA (31), DAP3 was found to be overexpressed in tumors when compared to their matched NT samples, across 17 cancer types. Furthermore, knockdown of DAP3 significantly reduced the malignant properties of ESCC and HCC cells, and this tumor-suppressing phenotype could be markedly rescued by recovering DAP3 expression in cancer cells. These findings support that DAP3 is a strong oncogene in cancer, which is unlikely to be limited to ESCC and HCC. To relate the tumor-promoting role of DAP3 with its negative regulation of RNA editome, our gene ontology analysis suggested that DAP3 tends to affect editing targets that are involved in cancer-related signaling pathways and processes, such as cell cycle, DNA replication, recombination and repair, and cell death and survival. Various forms of genomic, epigenetic, and transcriptomic alternations have been described in cancer, among which RNA editing is a unique epigenetic mechanism, but its role in human cancer development has only recently started to be understood. Several mouse studies suggested that no obvious spontaneous tumor development was observed in Adar1/Adar2 single- or double-KO mice and that their phenotypic abnormalities could be largely rescued by introducing a mutation in both Gria2 alleles (Gria2R/R) or knocking out the innate immunity genes Ifih1 or Mavs gene, albeit to different extents (38, 39, 45–48). However, these studies have focused heavily on defects in nervous system and innate immune responses, rather than cancer development, and one cannot rule out the possibility that spontaneous tumors may develop during a long-term observation. Moreover, editing dysregulation mediated by aberrantly expressed ADARs and/or other editing regulators such as DAP3 in healthy or premalignant cells may trigger cancer initiation and development by altering the responsiveness to environmental factors and cancer-causing signals and thus acquiring alterations leading to cancer. Although the in vivo evidence for the role of RNA editing in cancer development is still lacking, numerous studies have demonstrated that dysregulated A-to-I RNA editing is associated with cancer progression. ADAR1 is overexpressed in many types of cancers and characterized as an oncogene; as for ADAR2, it is down-regulated in many types of cancers and less abundantly expressed in many tissues than ADAR1. Because of down-regulation of ADAR2 in tumors, decreased editing pattern was also observed in in ESCC, HCC, and gastric cancer and associated with cancer progression (6, 28, 32). As abovementioned, we found that DAP3 preferentially interacts with ADAR2 in ESCC cells and contributes to cancer progression at least partially through repressing ADAR2-regulated RNA editome, which was further confirmed by our observation that DAP3 demonstrated a much less oncogenic effect when ADAR2 was silenced to repress RNA editome. On the basis of our observations, we could not deny other mechanisms involved in DAP3-driven tumorigenesis considering its role in mitochondrial physiology and cell apoptosis (24, 43, 44). Given that we observed thousands of editing events repressed by DAP3, it is possible that some of the DAP3-repressed editing events may have opposing effects; however, the overall effect mediated by DAP3-repressed editing contributes to tumorigenesis.
It is well accepted that protein-recoding RNA editing events, which can alter codon usage, are highly conserved and biologically important. Although only few recoding-type editing events that markedly alter protein functions were found so far, they have been demonstrated to be essential for normal development or associated with human diseases (1). In cancer, aberrant recoding editing on specific transcripts [e.g., AZIN1 (5, 7), AR (4), COPA (6), GluA2 (49), and PODXL (28)] is proved to be exploited by tumor cells to promote cancer progression. On the basis of our RNA-seq analysis, a previously functionally uncharacterized recoding editing target, PDZD7, was found to be regulated by DAP3. RNA editing of PDZD7 gene leads to a substitution of stop codon (TAG) to tryptophan (TGG) at residue 518, and the edited form PDZD7Stop518W has an 18–amino acid extension at the C terminus of PDZD7 protein. As the less tumorigenic form PDZD7Stop518W was suppressed by DAP3, the more malignant WT form PDZD7WT was accumulated in the majority of ESCC tumors, contributing to tumor progression. This suggests that cancer cells might acquire malignant properties for their survival advantage through repressing RNA editome by DAP3. However, the mechanism responsible for the functional difference between PDZD7WT and PDZD7Stop518W is still under our investigation. Nevertheless, PDZD7 may only represent as one of the DAP3-regulated editing targets, and we believe that DAP3 plays a pivotal role in cancer at least partially via reshaping the cancer RNA editome. Functional contributions of other DAP3-repressed editing targets to ESCC or other types of cancer remain for our further investigation.
In this study, we revealed the role of a novel ADAR-interactor DAP3 in reshaping the A-to-I RNA editome and its contribution to cancer. Mechanistically, we demonstrated that DAP3 disrupted ADAR2-dsRNA substrates association to function as a potent RNA editing repressor. Our findings highlight the importance of the additional layer of RNA editing regulation in cancer and identify key editing events, such as the PDZD7 stop codon editing, which drive cancer progression. The next step is to investigate whether the intervention of DAP3 expression, the disruption of DAP3-ADARs interaction, or the restoration of tumor-suppressive form of editing targets in tumors could block cancer progression.
MATERIALS AND METHODS
Cell culture
EC109, KYSE180, and SNU398 cells were cultured in HyClone RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified incubator containing 5% CO2. Huh7, HEK293T, and U251cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (Biowest) supplemented with 10% FBS at 37°C in a humidified incubator containing 5% CO2.
Generation of stable knockdown and overexpression cells
The DAP3-knockdown stable EC109, KYSE180, SNU398, and Huh7 cell lines were established using lentiviral transduction, followed by puromycin selection. The pLKO-DAP3-sh1 (5′GCTTATCCAGCTATACGATAT3′) and pLKO-DAP3-sh2 (5′ATCCTGGTTTCCAACTATAAC3′) constructs were used for the lentivirus packaging. For the DAP3 rescue experiment, the DAP3 mutant (DAP3 sh1-resistant) construct was generated by introducing seven synonymous mutations (5′ATACCCTGCAATCAGGTAT3′) at the DAP3 sh1-targeting sites using site-directed polymerase chain reaction (PCR) mutagenesis. EC109 cell lines stably expressing DAP3 after ADAR2 knockdown were established by the transduction of packaged lentiviral pLKO shScr or ADAR2 shRNA constructs followed by transduction of CSII-CMV-DAP3 constructs. EC109 and KYSE180 cells stably expressing WT and edited PDZD7 were established by the transduction of packaged lentiviral CSII-CMV-PDZD7WT and CSII-CMV-PDZD7Stop518W constructs, followed by the puromycin selection.
Coimmunoprecipitation
For the pulldown of DAP3 and V5-tagged protein, EC109 cells were lysed with prechilled lysis buffer [50 mM tris-HCl, (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 1× EDTA-free cOmplete protease inhibitor (Roche)]. The lysates were precleared with Dynabeads Protein G (Invitrogen) at 4°C overnight. The precleared lysates with incubated with anti-DAP3 (Abcam, ab2637) or anti-V5 (Bio-Rad, MCA1360) antibodies for 4 hours at 4°C and subsequently with Dynabeads Protein G at 4°C overnight. The Dynabeads Protein G (Invitrogen) with bound proteins were washed with 150 mM NaCl with 1× EDTA-free cOmplete protease inhibitor six times and boiled with 2× protein loading buffer for 10 min at 95°C to elute bound proteins. Western blot (WB) analysis was performed to detect co-IP products. For the RNase A treatment before the immunoprecipitation, the total lysates were incubated with RNase A (0.1 μg/ml) (Thermo Fisher Scientific) at 37°C for 10 min.
For the pulldown of GFP-tagged protein, a GFP-trap system (ChromoTek) was used according to the manufacturer’s protocol. EC109 cells were lysed with prechilled lysis buffer [10 mM tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.5% Nonidet P-40, and 1× cOmplete protease inhibitor] and incubated with GFP-trap beads at 4°C for 1 hour. The GFP-trap (ChromoTek) beads with bound proteins were washed with washing buffer [10 mM tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, and 1× cOmplete protease inhibitor] six times and boiled with 2× protein loading buffer for 10 min at 95°C to elute bound proteins. WB was performed to detect co-IP products.
WB analysis
Protein lysates were denatured and separated on SDS–polyacrylamide gel electrophoresis gels, transferred onto polyvinylidene difluoride membranes, and immunoblotted with a primary antibody at 4°C overnight, followed by incubation with a secondary antibody at room temperature for 1 hour. The following antibodies are used in this study: anti-DAP3 (1:1000; Abcam, ab2637), anti-ADAR1 (1:1000; Abcam, ab88574), anti-ADAR2 (1:500; Sigma-Aldrich, SAB1405426), anti–β-actin (1:5000; Santa Cruz Biotechnology, sc-47778), anti-GFP (1:1000; Santa Cruz Biotechnology, sc-9996), anti-V5 (1:1000; Bio-Rad, MCA1360), anti–Flag–horseradish peroxidase (1:10,000; Sigma-Aldrich, A8592), anti-fibrillarin (1:1000; Abcam, ab4566), anti–α-tubulin (1:1000; Santa Cruz Biotechnology, sc-5286), and anti-PDZD7 (1:1000; Abcam, ab169060).
Cell fractionation assays
Nuclear and cytoplasmic fractions of EC109 cells were isolated using the Nuclear Extract Kit (Active Motif, Carlsbad, CA) following the manufacturer’s protocol. Equal amount of nuclear and cytoplasmic fractions was used for WB and co-IP analysis. The anti-fibrillarin (Abcam, ab4566) and anti–α-tubulin (Santa Cruz Biotechnology, sc-5286) antibodies were used to check fractionation purity.
Generation of DAP3-KO cells by CRISPR-Cas9 system
DAP3 single-guide RNAs (sgRNAs) were designed using the CRISPR design tool from The Massachusetts Institute of Technology (http://crispr.mit.edu). The DAP3 sgRNA (ATAGCTCTCGGACTCTCAAC) targeting exon 3 of DAP3 was cloned into the pX330A vector. EC109 cells were transfected with either empty vector or vector expressing DAP3 sgRNA and split into single cell. Clones grew from single cell were lysed by DirectPCR Lysis Reagent (Viagen Biotech) followed by the detection of indels in each single clone by T7EI assays. PCR products were TA-cloned and Sanger-sequenced to confirm the biallelic KO of DAP3 in each clone. WB was performed to confirm DAP3 KO at protein level.
DAP3 expression profiling in TCGA samples
RNA-seq data (fastq files) of 11,374 samples across 33 cancer types from TCGA were downloaded from the dbGaP repository, under accession phs000178.v11.p8 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000178.v11.p8). Each sample was processed as follows: Raw reads were aligned to the reference human genome (hg19) by using STAR (v2.5.2a) (50). The gene expression quantification was performed by using featureCounts (v1.5.0-p3) (51) to obtain raw counts, which were then normalized by dividing with the total number of uniquely mapped reads in the corresponding sample and multiplying by a factor of 100 million. The distribution of normalized and log2-scaled expression levels for DAP3 was compared by using Wilcoxon rank sum test between tumors and NT samples, selecting the cancer types represented by at least five NT samples (22 of 33 cancer types). A P value less than 0.05 was regarded as significant up/down-regulation of DAP3 between tumors and NT samples.
RNA electrophoretic mobility shift assay
The HTR2C and MAGT1 RNA probes were generated by in vitro transcription using RiboMAX Large Scale RNA Production Systems (Promega). ADAR2 proteins were purified by M2 beads pulldown and eluted by 3xFlag peptides. Recombinant human GST protein (ab81793, Abcam) and DAP3-GST fusion protein (H00007818, Novus) were purchased from Abcam and Novus, respectively. RNA probe was dephosphorylated using Shrimp Alkaline Phosphatase (New England Biolabs) and then 5′-end–labeled with 32P using γ-32P-ATP (PerkinElmer) and T4 PNK (New England Biolabs) followed by G25 column purification (GE Healthcare). Labeled RNA probes were heated for 5 min at 80°C and placed on ice immediately to release secondary structure. Labeled RNA probes (0.5 pmol) were incubated with proteins [40 ng of ADAR2 and GST or DAP3-GST proteins with an increasing molar ratio (1:1, 1:2, and 1:4)] in binding buffer containing 10 mM Hepes (pH 7.3), 20 mM KCl, 1 mM MgCl2, 1 mM dithiothreitol, transfer RNA (100 ng/μl), and SUPERase·In RNase Inhibitor (0.2 U/μl) (Invitrogen) at room temperature for 30 min. Samples were subjected to electrophoresis on 4 or 6% native acrylamide gels, followed by gel drying and gel exposure to BioMax MS film (Carestream Health). Probe sequences can be found in table S4.
RIP–quantitative PCR analysis
The HTR2C and MAGT1 minigenes were cloned into pcDNA3.1 plasmid and cotransfected with Flag only, Flag-ADAR2, CSII-CMV-EV, or CSII-CMV-DAP3 plasmids into EC109 cells. Cells were lysed by prechilled lysis buffer [50 mM tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 × cOmplete protease inhibitor (Roche), and SUPERase·In RNase Inhibitor (0.1 U/μl) (Invitrogen)] and incubated overnight with M2 magnetic beads at 4°C. Then, the M2 magnetic beads were washed six times with tris-buffered saline buffer [50 mM tris (pH 7.5), 150 mM NaCl, and SUPERase·In RNase Inhibitor (0.02 U/μl) (Invitrogen)]. Bound proteins were eluted with 2× protein loading buffer after boiling at 95°C for 10 min. WB was performed to examine pull-down efficiency. RNAs bound to the M2 magnetic beads were eluted with buffer RLT and purified with the RNeasy Mini Kit (Qiagen). Complementary DNA (cDNA) was synthesized using the Advantage RT-for-PCR Kit (Takara), and quantitative PCR (qPCR) was performed using GoTaq qPCR Master Mix (Promega). Enrichment of pulled-down RNAs was normalized to the input RNA expression levels. Primer sequences can be found in table S4.
RNA editing detection by Sanger sequencing
The cDNA was PCR-amplified using FastStart Taq DNA Polymerase (Roche) and purified using the PCR Purification Kit (Qiagen). The purified PCR products were Sanger-sequenced and visualized by SnapGene Viewer. The RNA editing frequency was calculated on the basis of the peak area of adenosine and guanosine determined by ImageJ. Validated DAP3-affected editing sites are listed in table S3. Primer sequences can be found in table S4.
Foci and soft agar colony formation assay
For foci formation assay, EC109, KYSE180, and SNU398 cells were seeded at a density of 1 × 103 per well in six-well plates. Huh7 cells were seeded at a density of 3 × 103 per well in six-well plates. Medium was replaced every 3 days. Visible colonies in each well were stained with crystal violet solution (0.1% crystal violet; 25% methanol) and quantified. A representative image of a stained well for each treatment was shown.
For soft agar assay, EC109 (1 × 103 per well), KYSE180 (1 × 103 per well), SNU398 (5 × 103 per well), and Huh7 (9 × 103 per well) cells resuspended in 0.4% low-melting agarose were seeded on top of 0.6% low-melting agarose in six-well plates and incubated for 2 weeks. Visible colonies were stained with crystal violet solution (0.005% crystal violet; 25% methanol) and quantified. A representative image viewed under a microscope for each treatment was shown.
In vivo tumorigenicity assay
For in vivo tumorigenicity assay, 0.5 × 106 EC109 cells were subcutaneously injected into the left or right flank of 4- to 6-week-old nonobese diabetic scid gamma mice (n ≥ 5). Tumor growth was monitored and tumor length (L) and width (W) were measured at indicated time points. Tumor volume was calculated by the formula V = 0.5 × L × W × W. All animal experiments were approved by and performed in accordance with the Institutional Animal Care and Use Committees of National University of Singapore.
RNA-seq and identification of editing events
A bioinformatics pipeline adapted from a previously published method (52) was used to identify RNA editing events as previously described (23). For each sample, raw reads were mapped to the reference human genome (hg19) and a splicing junction database generated from transcript annotations derived from University of California Santa Cruz, RefSeq, Ensembl, and GENCODE (v19) by using Burrows-Wheeler aligner with default parameters (BWA-MEM algorithm, v0.7.15-r1140) (53). To retain high-quality data, PCR duplicates were removed (samtools rmdup function, v1.4.1) (53) and the reads with mapping quality score <20 were discarded. Junction-mapped reads were then converted back to the genomic-based coordinates. An in-house Perl script was used to call the variants from samtools pileup data, and the sites with at least two supporting reads were retained. The candidate events were filtered by removing the single-nucleotide polymorphisms (SNPs) reported in different cohorts [1000 Genomes Project (54), NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and dbSNP v138 (55)] and excluding the sites within the first six bases of the reads caused by imperfect priming of random hexamer during cDNA synthesis. For the sites not located in Alu elements, the candidates within the four bases of a splice junction on the intronic side and those residing in the homopolymeric regions and in the simple repeats were all removed. Candidate variants located in the reads that map to the nonunique regions of the genome by using the BLAST-like alignment tool (56) were also excluded. At last, only A-to-G editing sites based on the strand information from the strand-specific RNA-seq data were considered for all the downstream analyses. The genomic regions of the editing variants and the associated genes were annotated by using ANNOVAR (v2016) (57) with the refGene table.
Supplementary Material
Acknowledgments
We thank and acknowledge X.-Y. Guan (The University of Hong Kong, Hong Kong, China) and Y. Qin (the First Affiliated Hospital, Zhengzhou University, China) for providing the cDNA samples of ESCC cases. We thank and acknowledge A. D. Jeyasekharan (Cancer Science Institute of Singapore, National University of Singapore, Singapore) for providing the antibody against TOMM20. We thank and acknowledge G. W. Yeo (Department of Physiology, National University of Singapore, Singapore; Department of Cellular and Molecular Medicine, University of California San Diego, USA) for providing help with the eCLIP experiment. Funding: This project was supported by National Research Foundation Singapore, Singapore Ministry of Education under its Research Centres of Excellence initiative, Singapore Ministry of Education’s Tier 2 grants [MOE2018-T2-1-005 and MOE2019-T2-2-008], NMRC Clinician Scientist-Individual Research Grant (CS-IRG, grant award number: MOH-CIRG18nov-0007), and Singapore Ministry of Education’s Tier 3 grants [MOE2014-T3-1-006]. Author contributions: L.C. conceived and supervised the study. L.C. and J.H. designed and performed the experiments. H.Y. and O.A. conducted all the bioinformatics analyses. T.H.M.C. and Y.S. assisted in conducting mouse-related experiments. H.S. assisted in validation of RNA editing sites. H.H., J.S.L., V.H.E.N., D.J.T.T., S.J.T., Y.S., F.B.M., H.S., and P.P. provided insightful suggestions and experimental materials. H.Q.T. contributed to the eCLIP-Seq experiments. J.H. wrote the manuscript. O.A. and L.C. edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The datasets supporting the conclusions of this article are available in the GEO repository; accession ID: GSE123020 and GSE144318. Data bank URL: www.ncbi.nlm.nih.gov/geo/. Bioinformatics codes are available upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaba5136/DC1
REFERENCES AND NOTES
- 1.Nishikura K., A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 17, 83–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walkley C. R., Li J. B., Rewriting the transcriptome: Adenosine-to-inosine RNA editing by ADARs. Genome Biol. 18, 205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paz N., Levanon E. Y., Amariglio N., Heimberger A. B., Ram Z., Constantini S., Barbash Z. S., Adamsky K., Safran M., Hirschberg A., Krupsky M., Ben-Dov I., Cazacu S., Mikkelsen T., Brodie C., Eisenberg E., Rechavi G., Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 17, 1586–1595 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez H. D., Jasavala R. J., Hinkson I., Fitzgerald L. D., Trimmer J. S., Kung H. J., Wright M. E., RNA editing of androgen receptor gene transcripts in prostate cancer cells. J. Biol. Chem. 283, 29938–29949 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L., Li Y., Lin C. H., Chan T. H. M., Chow R. K. K., Song Y., Liu M., Yuan Y. F., Fu L., Kong K. L., Qi L., Li Y., Zhang N., Tong A. H. Y., Kwong D. L. W., Man K., Lo C. M., Lok S., Tenen D. G., Guan X. Y., Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 19, 209–216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan T. H. M., Lin C. H., Qi L., Fei J., Li Y., Yong K. J., Liu M., Song Y., Chow R. K. K., Ng V. H. E., Yuan Y. F., Tenen D. G., Guan X. Y., Chen L., A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut 63, 832–843 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin Y.-R., Qiao J. J., Chan T. H. M., Zhu Y. H., Li F. F., Liu H., Fei J., Li Y., Guan X. Y., Chen L., Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 74, 840–851 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Shigeyasu K., Okugawa Y., Toden S., Miyoshi J., Toiyama Y., Nagasaka T., Takahashi N., Kusunoki M., Takayama T., Yamada Y., Fujiwara T., Chen L., Goel A., AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight 3, –99976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amin E. M., Liu Y., Deng S., Tan K. S., Chudgar N., Mayo M. W., Sanchez-Vega F., Adusumilli P. S., Schultz N., Jones D. R., The RNA-editing enzyme ADAR promotes lung adenocarcinoma migration and invasion by stabilizing FAK. Sci. Signal. 10, eaah3941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zipeto M. A., Court A. C., Sadarangani A., Delos Santos N. P., Balaian L., Chun H. J., Pineda G., Morris S. R., Mason C. N., Geron I., Barrett C., Goff D. J., Wall R., Pellecchia M., Minden M., Frazer K. A., Marra M. A., Crews L. A., Jiang Q., Jamieson C. H. M., ADAR1 activation drives leukemia stem cell self-renewal by impairing let-7 biogenesis. Cell Stem Cell 19, 177–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deffit S. N., Hundley H. A., To edit or not to edit: Regulation of ADAR editing specificity and efficiency. Wiley Interdiscip. Rev. RNA 7, 113–127 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Peng P. L., Zhong X., Tu W., Soundarapandian M. M., Molner P., Zhu D., Lau L., Liu S., Liu F., Lu Y. M., ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 49, 719–733 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Yang L., Huang P., Li F., Zhao L., Zhang Y., Li S., Gan Z., Lin A., Li W., Liu Y., c-Jun amino-terminal kinase-1 mediates glucose-responsive upregulation of the RNA editing enzyme ADAR2 in pancreatic beta-cells. PLOS ONE 7, e48611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rueter S. M., Dawson T. R., Emeson R. B., Regulation of alternative splicing by RNA editing. Nature 399, 75–80 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Marcucci R., Brindle J., Paro S., Casadio A., Hempel S., Morrice N., Bisso A., Keegan L. P., del Sal G., O’Connell M. A., Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 30, 4211–4222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan M. H., Li Q., Shanmugam R., Piskol R., Kohler J., Young A. N., Liu K. I., Zhang R., Ramaswami G., Ariyoshi K., Gupte A., Keegan L. P., George C. X., Ramu A., Huang N., Pollina E. A., Leeman D. S., Rustighi A., Goh Y. P. S.; GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration &Visualization—EBI; Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz, Chawla A., del Sal G., Peltz G., Brunet A., Conrad D. F., Samuel C. E., O’Connell M. A., Walkley C. R., Nishikura K., Li J. B., Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254, 254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desterro J. M. P., Keegan L. P., Jaffray E., Hay R. T., O'Connell M. A., Carmo-Fonseca M., SUMO-1 modification alters ADAR1 editing activity. Mol. Biol. Cell 16, 5115–5126 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritz J., Strehblow A., Taschner A., Schopoff S., Pasierbek P., Jantsch M. F., RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol. Cell. Biol. 29, 1487–1497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho D.-S. C., Yang W., Lee J. T., Shiekhattar R., Murray J. M., Nishikura K., Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J. Biol. Chem. 278, 17093–17102 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Poulsen H., Jorgensen R., Heding A., Nielsen F. C., Bonven B., Egebjerg J., Dimerization of ADAR2 is mediated by the double-stranded RNA binding domain. RNA 12, 1350–1360 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valente L., Nishikura K., RNA-Binding independent dimerization of ADAR and dominant negative effects of nonfunctional subunits on dimer functions. J. Biol. Chem. 282, 16054–16061 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shanmugam R., Zhang F., Srinivasan H., Charles Richard J. L., Liu K. I., Zhang X., Woo C. W. A., Chua Z. H. M., Buschdorf J. P., Meaney M. J., Tan M. H., SRSF9 selectively represses ADAR2-mediated editing of brain-specific sites in primates. Nucleic Acids Res. 46, 7379–7395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong H., An O., Chan T. H. M., Ng V. H. E., Kwok H. S., Lin J. S., Qi L., Han J., Tay D. J. T., Tang S. J., Yang H., Song Y., Bellido Molias F., Tenen D. G., Chen L., Bidirectional regulation of adenosine-to-inosine (A-to-I) RNA editing by DEAH box helicase 9 (DHX9) in cancer. Nucleic Acids Res. 46, 7953–7969 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H.-R., Chae H. J., Thomas M., Miyazaki T., Monosov A., Monosov E., Krajewska M., Krajewski S., Reed J. C., Mammalian dap3 is an essential gene required for mitochondrial homeostasis in vivo and contributing to the extrinsic pathway for apoptosis. FASEB J. 21, 188–196 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Xiao L., Xian H., Lee K. Y., Xiao B., Wang H., Yu F., Shen H. M., Liou Y. C., Death-associated protein 3 regulates mitochondrial-encoded protein synthesis and mitochondrial dynamics. J. Biol. Chem. 290, 24961–24974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wazir U., Orakzai M. M. A. W., Khanzada Z. S., Jiang W. G., Sharma A. K., Kasem A., Mokbel K., The role of death-associated protein 3 in apoptosis, anoikis and human cancer. Cancer Cell Int. 15, 39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valente L., Nishikura K., RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J. Biol. Chem. 282, 16054–16061 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan T. H., Qamra A., Tan K. T., Guo J., Yang H., Qi L., Lin J. S., Ng V. H., Song Y., Hong H., Tay S. T., Liu Y., Lee J., Rha S. Y., Zhu F., So J. B., Teh B. T., Yeoh K. G., Rozen S., Tenen D. G., Tan P., Chen L., ADAR-mediated RNA editing predicts progression and prognosis of gastric cancer. Gastroenterology 151, 637–650.e10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration &Visualization—EBI; Genome Browser Data Integration &Visualization—UCSC Genomics Institute; University of California Santa Cruz; Lead analysts; Laboratory, Data Analysis &Coordinating Center (LDACC); NIH program management; Biospecimen collection; Pathology; eQTL manuscript working group, Battle A., Brown C. D., Engelhardt B. E., Montgomery S. B., Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).29022597 [Google Scholar]
- 30.Burns C. M., Chu H., Rueter S. M., Hutchinson L. K., Canton H., Sanders-Bush E., Emeson R. B., Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387, 303–308 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., Stuart J. M., The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y.-B., Liao X. Y., Zhang J. B., Wang F., Qin H. D., Zhang L., Shugart Y. Y., Zeng Y. X., Jia W. H., ADAR2 functions as a tumor suppressor via editing IGFBP7 in esophageal squamous cell carcinoma. Int. J. Oncol. 50, 622–630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.K. Fritzell, L.-D. Xu, J. Lagergren, M. Öhman, in Seminars in Cell & Developmental Biology (Elsevier, 2018), vol. 79, pp. 123–130. [DOI] [PubMed] [Google Scholar]
- 34.Hwang T., Park C. K., Leung A. K. L., Gao Y., Hyde T. M., Kleinman J. E., Rajpurohit A., Tao R., Shin J. H., Weinberger D. R., Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 19, 1093–1099 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Maas S., Patt S., Schrey M., Rich A., Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. U.S.A. 98, 14687–14692 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matthews M. M., Thomas J. M., Zheng Y., Tran K., Phelps K. J., Scott A. I., Havel J., Fisher A. J., Beal P. A., Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 23, 426–433 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phelps K. J., Tran K., Eifler T., Erickson A. I., Fisher A. J., Beal P. A., Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res. 43, 1123–1132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mannion N. M., Greenwood S. M., Young R., Cox S., Brindle J., Read D., Nellåker C., Vesely C., Ponting C. P., McLaughlin P. J., Jantsch M. F., Dorin J., Adams I. R., Scadden A. D. J., Öhman M., Keegan L. P., O’Connell M. A., The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pestal K., Funk C. C., Snyder J. M., Price N. D., Treuting P. M., Stetson D. B., Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liddicoat B. J., Piskol R., Chalk A. M., Ramaswami G., Higuchi M., Hartner J. C., Li J. B., Seeburg P. H., Walkley C. R., RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chung H., Calis J. J. A., Wu X., Sun T., Yu Y., Sarbanes S. L., Thi V. L. D., Shilvock A. R., Hoffmann H. H., Rosenberg B. R., Rice C. M., Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824. e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice G. I., Kasher P. R., Forte G. M. A., Mannion N. M., Greenwood S. M., Szynkiewicz M., Dickerson J. E., Bhaskar S. S., Zampini M., Briggs T. A., Jenkinson E. M., Bacino C. A., Battini R., Bertini E., Brogan P. A., Brueton L. A., Carpanelli M., de Laet C., de Lonlay P., del Toro M., Desguerre I., Fazzi E., Garcia-Cazorla À., Heiberg A., Kawaguchi M., Kumar R., Lin J. P. S. M., Lourenco C. M., Male A. M., Marques W. Jr., Mignot C., Olivieri I., Orcesi S., Prabhakar P., Rasmussen M., Robinson R. A., Rozenberg F., Schmidt J. L., Steindl K., Tan T. Y., van der Merwe W. G., Vanderver A., Vassallo G., Wakeling E. L., Wassmer E., Whittaker E., Livingston J. H., Lebon P., Suzuki T., McLaughlin P. J., Keegan L. P., O’Connell M. A., Lovell S. C., Crow Y. J., Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 44, 1243–1248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kissil J. L., Cohen O., Raveh T., Kimchi A., Structure–function analysis of an evolutionary conserved protein, DAP3, which mediates TNF-α-and Fas-induced cell death. EMBO J. 18, 353–362 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kissil J. L., Deiss L. P., Bayewitch M., Raveh T., Khaspekov G., Kimchi A., Isolation of DAP3, a novel mediator of interferon-γ-induced cell death. J. Biol. Chem. 270, 27932–27936 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Chalk A. M., Taylor S., Heraud-Farlow J. E., Walkley C. R., The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol. 20, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higuchi M., Maas S., Single F. N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P. H., Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406, 78–81 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Horsch M., Seeburg P. H., Adler T., Aguilar-Pimentel J. A., Becker L., Calzada-Wack J., Garrett L., Götz A., Hans W., Higuchi M., Hölter S. M., Naton B., Prehn C., Puk O., Rácz I., Rathkolb B., Rozman J., Schrewe A., Adamski J., Busch D. H., Esposito I., Graw J., Ivandic B., Klingenspor M., Klopstock T., Mempel M., Ollert M., Schulz H., Wolf E., Wurst W., Zimmer A., Gailus-Durner V., Fuchs H., de Angelis M. H., Beckers J., Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J. Biol. Chem. 286, 18614–18622 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bajad P., Ebner F., Amman F., Szabó B., Kapoor U., Manjali G., Hildebrandt A., Janisiw M. P., Jantsch M. F., An internal deletion of ADAR rescued by MAVS deficiency leads to a minute phenotype. Nucleic Acids Res. 48, 3286–3303 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cenci C., Barzotti R., Galeano F., Corbelli S., Rota R., Massimi L., di Rocco C., O’Connell M. A., Gallo A., Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J. Biol. Chem. 283, 7251–7260 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Ramaswami G., Zhang R., Piskol R., Keegan L. P., Deng P., O’Connell M. A., Li J. B., Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 10, 128–132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.1000 Genomes Project Consortium, Auton A., Brooks L. D., Durbin R. M., Garrison E. P., Kang H. M., Korbel J. O., Marchini J. L., McCarthy S., McVean G. A., Abecasis G. R., A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., Sirotkin K., dbSNP: The NCBI database of genetic variation. Nucleic Acids Res 29, 308–311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kent W. J., BLAT—The BLAST-like alignment tool. Genome Res. 12, 656–664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang K., Li M., Hakonarson H., ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.E. L. Van Nostrand, T. B. Nguyen, C. Gelboin-Burkhart, R. Wang, S. M. Blue, G. A. Pratt, A. L. Louie, G. W. Yeo, Robust, Cost-Effective Profiling of RNA Binding Protein Targets with Single-end Enhanced Crosslinking and Immunoprecipitation (seCLIP), in mRNA Processing, Y. Shi, Ed. (Springer, 2017), pp. 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Nostrand E. L., Pratt G. A., Shishkin A. A., Gelboin-Burkhart C., Fang M. Y., Sundararaman B., Blue S. M., Nguyen T. B., Surka C., Elkins K., Stanton R., Rigo F., Guttman M., Yeo G. W., Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.An Ö., Tan K.-T., Li Y., Li J., Wu C.-S., Zhang B., Chen L., Yang H., CSI NGS Portal: An online platform for automated NGS data analysis and sharing. Int. J. Mol. Sci. 21, 3828 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaba5136/DC1