

Abstract
Extracellular vesicles (EVs) released by neural cells play an essential role in brain homeostasis and the crosstalk between neural cells and the periphery. EVs are diverse, nano-sized vesicles, which transport proteins, nucleic acids, and lipids between cells over short and long expanses and hence are proficient for modulating the target cells. EVs released from neural cells are implicated in synaptic plasticity, neuron-glia interface, neuroprotection, neuroregeneration, and the dissemination of neuropathological molecules. This review confers the various properties of EVs secreted by astrocytes and their potential role in health and disease with a focus on evolving concepts. Naïve astrocytes shed EVs containing a host of neuroprotective compounds, which include fibroblast growth factor-2, vascular endothelial growth factor, and apolipoprotein-D. Stimulated astrocytes secrete EVs with neuroprotective molecules including heat shock proteins, synapsin 1, unique microRNAs, and glutamate transporters. Well-characterized astrocyte-derived EVs (ADEVs) generated in specific culture conditions and ADEVs that are engineered to carry the desired miRNAs or proteins are likely useful for treating brain injury and neurogenerative diseases. On the other hand, in conditions such as Alzheimer’s disease (AD), stroke, Parkinson’s disease, Amyotrophic lateral sclerosis (ALS), and other neuroinflammatory conditions, EVs released by activated astrocytes appear to mediate or exacerbate the pathological processes. The examples include ADEVs spreading the dysregulated complement system in AD, mediating motoneuron toxicity in ALS, and stimulating peripheral leukocyte migration into the brain in inflammatory conditions. Strategies restraining the release of EVs by activated astrocytes or modulating the composition of ADEVs are likely beneficial for treating neurodegenerative diseases. Also, periodic analyses of ADEVs in the blood is useful for detecting astrocyte-specific biomarkers in different neurological conditions and for monitoring disease progression and remission with distinct therapeutic approaches.
Keywords: Astrocytes, Astrocyte-derived extracellular vesicles, Alzheimer’s disease, Amyotrophic lateral sclerosis, Exosomes, Microvesicles, Neurological disorders, Neuroinflammation, Parkinson’s disease, Stroke
1. Introduction
Astrocytes, one of the glial cell types, play a critical role in maintaining the architecture and the function of the central nervous system (CNS). Accumulative research in the past several decades has improved our understanding of mechanisms underlying the function of astrocytes in physiological and pathological conditions [1, 2]. In sharp contrast to the initial description of these cells as passive players, it is now clear that astrocytes occupy a strategic position in the CNS because they are involved in multiple processes in health and disease. Such involvement can be gleaned from the fact that a single astrocyte can tightly wrap around soma, dendrites, axons, and synapses of several neurons [3, 4]. Moreover, tight associations between astrocytic processes and pre- and post-synaptic neuronal elements form a tripartite synapse [5]. Intricate neuron-astrocyte connections facilitate upholding of multiple vital functions by astrocytes, which include the formation and maintenance of blood-brain barrier (BBB), neuron-glia communication, recycling of neurotransmitters, structural support and nutrients to neurons and other glial cells [6–12].
Moreover, astrocytes potently influence stem cells and stem cell niches [13–18]. The ability to perform such diverse functions explains the occurrence of different astrocyte subtypes observed in the CNS, which includes astrocytes with long, short, straight, crooked, or highly-branched processes. Astrocytes also play a role in immunodefense by their ability to switch from a pro-inflammatory phenotype to an anti-inflammatory phenotype and vice versa [19–21]. Such dynamic properties facilitate their contribution to the neuroimmune state of the CNS, repair, and disease prevention. Furthermore, mounting evidence has demonstrated the active participation of astrocytes as information processors in the CNS [22]. Astrocytes accomplish these tasks by regulating the release of various molecules which comprise neurotrophic factors, peptides, hormones, growth factors and cytokines, and through selective uptake of neurotransmitters in the synaptic cleft [23–25]. Astrocytes perform these functions through various means, which include exocytosis, diffusion, and active and passive transport.
Recent studies have highlighted that one of the significant forms of communication between astrocytes and neurons occurs through extracellular vesicles (EVs) [26–30]. The EVs shed by astrocytes in normal conditions are believed to have neurotrophic and neuroprotective properties whereas EVs released from astrocytes in ischemic, oxidative stress, nutrient-deprived, or thermal stress conditions have been shown to carry various factors that are involved in neuronal survival, guarding neurons against neurotransmitter toxicity, and promoting neurite outgrowth [30–34]. However, in neurodegenerative disease conditions such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Amyotrophic Lateral Sclerosis (ALS), astrocyte-derived EVs (ADEVs) have been suggested to have a role in spreading neuropathology or exacerbating the extent of neurodegeneration [35, 36]. This review article is focused on discussing the properties of ADEVs, their role in maintaining or adversely affecting the function of CNS in physiological and pathological conditions, and their utility as a source of brain biomarkers in the circulating blood. Furthermore, EVs derived from stem cells have shown efficacy for improving brain function in several neurodegenerative diseases [37–48]. Likewise, ADEV therapy may also be useful for treating neurodegenerative diseases. Therefore, we also discussed the potential use of ADEVs as a biologic for improving brain function in disease conditions.
2. Extracellular vesicles
EVs are double membrane-enclosed, nano-sized vesicles exuded into the extracellular space by a variety of cells, including astrocytes [49]. There are two major types of EVs, which include smaller exosomes (EXs) with a size of 30–150 nm, and relatively larger microvesicles (MVs) ranging in size from 100–1000 nm [50]. The EVs either originate from the endosomal pathway (EXs) or shed through outward blebbing of the plasma membrane (MVs). Inward invaginations of the endosomal membrane lead to the formation of intraluminal vesicles (ILVs) within endosomes. Such endosomes then evolve into multi-vesicular bodies (MVBs) displaying a collection of EXs. Although most MVBs containing EXs undergo degradation through fusion with lysosomes, some MVBs secrete EXs into the extracellular space through fusion with the plasma membrane. The manner of EV biogenesis is believed to be related to their physiologic function and/or to the physio-pathologic status of the generating cell [50]. The markers of EVs include tetraspanins CD9, CD63, CD81, the endosomal protein tumor susceptibility gene 101 (TSG101), and ALG-2-interacting protein X (ALIX). The lipids that are commonly found in EVs include cholesterol, phosphatidylserine and phosphatidylinositol, sphingomyelin, and phosphatidylcholine. The classification of EVs as either EXs or MVs requires a painstaking characterization of EV properties because of the overlap in their size range. It was thought earlier that, to classify EVs as EXs, the occurrence of three specific markers out of five well-characterized EV markers (CD9, CD63, CD81, TSG101 or ALIX) may be sufficient [51]. Nonetheless, recent guidelines from the International Society for Extracellular Vesicles (ISEV) recommend employing operational terms unless the release of EXs by cells is captured in the act by live imaging approaches because similar EV markers may also be seen in some MVs [52, 53]. The operational terms are based on features such as cells of origin (e.g., astrocyte-derived EVs or neuron-derived EVs), the size of EVs (small EVs, medium/large EVs) or the composition (e.g., CD63+/CD81+-/CD9+- EVs) [53]. Hence, the phrase “EVs” is used for all EV or EX studies that are conferred in this review.
The EXs are formed through both endosomal sorting complex required for transport (ESCRT)-dependent and ESCRT-independent pathways. The creation of EXs via ESCRT-dependent pathway comprises several steps, which include enrichment of the endosomal membrane with tetraspanins such as CD9 and CD63 [54], recognition and sorting of ubiquitinated proteins into specific domains of the endosomal membrane by ESCRT-0 [55], and the formation of ILVs by ESCRT I-III complex [56, 57]. The involvement of ESCRT in these processes is supported by the presence of ESCRT proteins TSG101, and CHMP4 among the cargo proteins of EXs. In ESCRT-independent pathway, ceramide resulting from the hydrolysis of sphingomyelin by neutral sphingomyelinase 2 (nSMase2) causes inward blebbing in endosomes leading to the formation of ILVs. It has been suggested that the cone-shaped structure of ceramide causes the endosomal membrane to develop a negative curvature, which eventually leads to the formation of ILVs [58, 59]. Tetraspanins (CD81, CD63 & CD9) also participate in EX biogenesis and protein loading [60–64]. The MVs are formed through outward budding of the plasma membrane and fission. Re-distribution of phospholipids and contraction of the actin-myosin machinery are involved in MV biogenesis [65]. The presence of TSG101 in MVs also suggested a role for ESCRT proteins in MV biogenesis [66, 67].
Although EVs were initially portrayed as waste carriers through which cells dispose-off cellular wastes, their role in exchanging materials between cells, signal transduction, maintaining cellular homeostasis, and the spread of pathological proteins and miRNAs in neurodegenerative disease conditions have received enormous interest [50, 68, 69]. EVs enable intercellular communication, which facilitates cells to interchange protein, lipids, and nucleic acids (RNAs and DNAs). Following the attachment of EVs to the plasma membrane of target cells, EV’s are internalized through endocytosis, macropinocytosis, or phagocytosis depending on the type of target cell involved. EVs can also dispense their cargo into the cytosol by directly fusing with the target cell’s plasma membrane. In the brain, EVs are actively involved in neuroimmune crosstalk, brain cancer progression and propagation of tau in AD [70–72]. Besides, neural cell-derived EVs crossing the BBB serve as valuable biomarkers of neurodegenerative diseases [73–75].
3. Properties of Astrocyte-derived EVs
The composition and biological properties of EVs derived from astrocytes in physiological and pathological conditions are listed in Table 1 and described below.
Table 1.
Biological properties of ADEVs
| Source of EVs | Culture Conditions | Purification Method | Composition of EVs and Beneficial or Adverse Effects | References |
|---|---|---|---|---|
| Astrocytes from PND2 rat cerebral cortex | Normal | Ultracentrifugation | EVs contain FGF-2 and VEGF Potential function: Tissue regeneration, Cell Survival and Differentiation, Neurogenesis, Angiogenesis and Neuroprotection |
[33, 76–93] |
| Astrocytes from PND0/1 mouse cerebral cortex | Normal | Ultracentrifugation | EVs contain apolipoprotein-D. Beneficial effects: Neuroprotection against oxidative stress Potential other functions: Anti-aging and longevity |
[94–100] |
| Astrocytes from E12 chick lumbar spinal cords | Normal | Ultracentrifugation | EVs contain HSP70. Potential function: Neuroprotection and immunoregulatory effects |
[30, 101–113] |
| Astrocytes from PND1/2 mouse cerebral cortex or whole brain | Stimulated with KCl (75–80mM) | Ultracentrifugation | EVs contain synapsin I Potential function: Neuroprotection against stress, neurite outgrowth, neural survival, maturation of synapses and synaptic plasticity |
[34, 114–122] |
| Astrocytes from PND2 rat cerebral cortex | Stimulated with PKC | Ultracentrifugation | EVs contain glutamate transporters EAAT-1/2 Potential function: Neural homeostasis and neuroprotection against glutamate toxicity |
[31, 123–125] |
| Astrocytes from rat cerebral cortex | Stimulated through oxidative stress | Ultracentrifugation | EVs contain PrP and STI-1. Beneficial effects: Neuroprotection against oxidative stress, ischemia, hypoxia and hypoglycemia Potential other functions: Neuronal differentiation, neurite growth, neuronal homeostasis |
[126–133] |
| Astrocytes from PND2 cerebral cortex | Stimulated with IL-10 or ATP | Ultracentrifugation | Beneficial effects: Increased neurite length and the overall dendritic complexity, synaptic transmission and neuronal survival | [27, 134] |
| Astrocytes from PND2 cerebral cortex | Stimulated with IL-1β or TNF-α | Ultracentrifugation | EVs enriched with miR-125a-5p and miR-16–5p Adverse effects: Reduced neurite formation, diminished dendritic complexity, and exacerbation of neurodegeneration |
[27, 134] |
| Astrocytes from PND2 rat cerebral cortex | Stimulated with LPS | Ultracentrifugation | EVs enriched with miR-34a Adverse effects: Enhanced vulnerability of dopaminergic neurons to neurotoxins |
[136] |
Abbreviations: E, embryonic day; EAAT, excitatory amino acid transporter; FGF-2, fibroblast growth factor-2; IL-1β, interleukin-1 beta; IL-10, interleukin-10; KCl, potassium chloride; LPS, lipopolysaccharide; miR, micro-RNA; PKC, protein kinase C; PND, postnatal day; PrP, prion protein; STI-1, Stress inducible protein-1; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor.
3.1. Naïve astrocytes shed EVs containing FGF-2 and VEGF
Proia and associates demonstrated that EVs shed by astrocytes, varying in size from 150–500 nm, carry angiogenic factors, fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor (VEGF) [33]. These EVs were thought to be MVs because of the expression of beta-1 integrin by them. The authors confirmed the occurrence of FGF-2 and VEGF through immunofluorescence, western blot, and electron microscopic analyses. The VEGF was found in two isoforms (22 and 45 kDa) whereas FGF-2 was observed as a series of bands ranging in size from 21 to 43 kDa. The presence of these trophic factors in ADEVs has implications for brain repair in conditions such as injury or disease.
The growth factor FGF-2 signaling through FGF receptors (FGFRs) can mediate a broad spectrum of biological functions, which comprise proliferation, survival, migration, and differentiation of a variety of cells including endothelial cells and neural stem cells, and axon regeneration (Fig. 1 [A]). Activation of FGFRs by FGF-2 results in recruitment of specific molecules that bind to phosphorylated tyrosine at the cytosolic part of the receptor, which triggers many signaling pathways and leads to specific cellular responses. As a result of this, FGF-2 plays a significant role in tissue regeneration [76–79]. The main pathway mediated by FGF-2 is the mitogen-activated protein (MAP) kinase pathway, which regulates various cellular activities such as gene expression, mitosis, differentiation and cell survival [80]. The effectors of MAP kinase include c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 mitogen-activated kinase [81]. Indeed, a multitude of studies have shown that FGF-2 increases hippocampal neurogenesis [82–84] and is neuroprotective in adverse conditions [85–87]. On the other hand, VEGF is well known for promoting angiogenesis [88], neurogenesis [89–90], synaptogenesis and synaptic plasticity [91] and neuroprotective activity [92–93] in the brain. Thus, EVs containing FGF-2 and VEGF shed by astrocytes can support a wide variety of functions in the naïve brain and also promote repair following injury or disease. Besides, exogenous administration of ADEVs in neurological conditions exhibiting a deficiency of FGF-2 and/or VEGF, such as aging, stroke, epilepsy, AD, and PD, is likely to be therapeutic.
Figure 1:
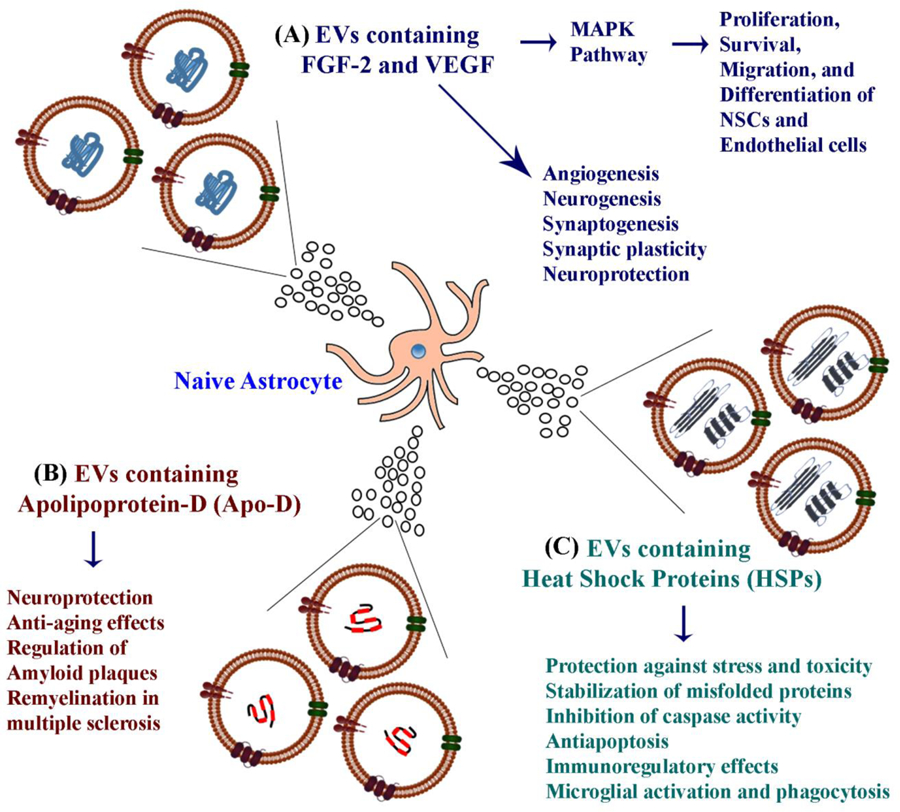
Schematic representation of the possible functions mediated by EVs shed from naïve astrocytes. Astrocyte-derived EVs (ADEVs) contain fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor (VEGF) (A), apolipoprotein-D (Apo-D) (B), and heat shock proteins (HSPs) (C). These proteins can mediate a variety of functions, as depicted in the figure.
3.2. ADEVs carry Apolipoprotein-D
ADEVs contain apolipoprotein-D (Apo-D), which promotes the functional integrity and survival of neurons in adverse conditions [94] (Fig. 1 [B]. Apo-D containing ADEVs get incorporated into neighboring neurons and protect in conditions such as increased oxidative stress. The protective role of Apo-D in ADEVs was apparent from only partial autocrine protection against oxidative stress when EVs from Apo-D knock out astrocytes were employed [94]. ApoD has beneficial effects on aging and neurodegenerative diseases too [95–97]. For example, deficiency of ApoD triggers premature aging of the brain without modifying the lifespan [98]. Such changes were apparent from markers of glial reactivity, proteostasis, oxidative and inflammatory damage, reduction in neuronal calcium-dependent functionality markers and signs of early loss of neurons in the cortex of the Apo-D knockout brain. Also, in AD, Apo-D seems to be involved in the regulation of amyloid plaque pathology [99]. Moreover, Apo-D is also involved in myelination and remyelination processes in multiple sclerosis [100]. Thus, administration of Apo-D containing ADEVs is likely therapeutic in conditions such as aging and neurodegenerative diseases.
3.3. Astrocytes shed EVs containing heat shock proteins
Taylor and colleagues suggested that metabolically active astrocytes release the cytoprotective heat shock protein 70 (HSP70) into the extracellular space primarily through EVs [30]. The study also suggested that the release of HSP70 containing EVs by astrocytes is enhanced in stressful conditions, which is mediated in part by the increased activity of ERK1/1 and P13K/Akt. The presence of HSP70 in ADEVs has considerable significance because HSP70 can mediate protective effects on neurons against physical trauma or toxicity [101–105] (Fig. 1 [C]). First, in stressful conditions, HSPs can protect cells through stabilization of unfolded or misfolded peptides, which, in turn, can facilitate the repair or re-synthesis of damaged proteins by cells. Second, HSPs can exert neuroprotective effects through inhibition of caspase activation. HSPs are known to interact with pro-apoptotic proteins such as Bcl-2, disrupt apoptosome complex, and thereby prevent the activation of apoptosis [106–107]. Third, HSPs can mediate immunoregulatory effects through interaction and binding with Toll-like receptors TLR2 and TLR4 on antigen-presenting cells, and CD94 on natural killer cells [108–112]. Fourth, HSPs can induce microglial activation, production of cytokines, and stimulate phagocytosis of debris [101, 102, 113]. Thus, HSPs in ADEVs is likely useful for modulating neuroinflammation in injury or disease conditions.
3.4. Astrocytes stimulated with KCl release EVs containing synapsin 1
An investigation by Wang and colleagues suggested that in conditions of neuronal hyperactivity or increased oxidative stress, synapsin I is released by astrocytes through EVs [34] (Fig. 2 [A]). Synapsin I is a glycoprotein and an oligomannose-binding lectin capable of modulating neurite outgrowth in an oligomannose- and neural cell adhesion molecule (NCAM)-dependent manner. The authors uncovered the release of synapsin-containing EVs when cortical astrocytes were cultured in the presence of 75–80 mM potassium chloride (KCl). Such increased concentration of KCl in the CNS are typically seen during the propagation of spreading depression (SD) waves, in conditions such as focal ischemia, stroke or migraine [114–115]. It has been suggested that SD waves promote the activation of microglia and astrocytes, which can mediate neuroprotective functions through secretion of neurotrophic factors and make the brain more resistant to subsequent insults [114–116]. Indeed, hypoxia or ischemic conditions are associated with increased expression of synapsin in different brain regions [117], implying that synapsin is involved in the brain’s response to stressful conditions. Synapsin promotes neurite outgrowth, neuronal survival, functional maturation of synapses, regulation of neurotransmitter release, neurotransmission and synaptic plasticity [118–122]. Thus, the release of ADEVs containing synapsin by astrocytes is beneficial for brain repair in injury or neurodegenerative conditions. Also, the administration of ADEVs in conditions exhibiting synapse loss and diminished synaptic plasticities, such as aging and early stage of the AD, would likely to have a therapeutic value.
Figure 2:
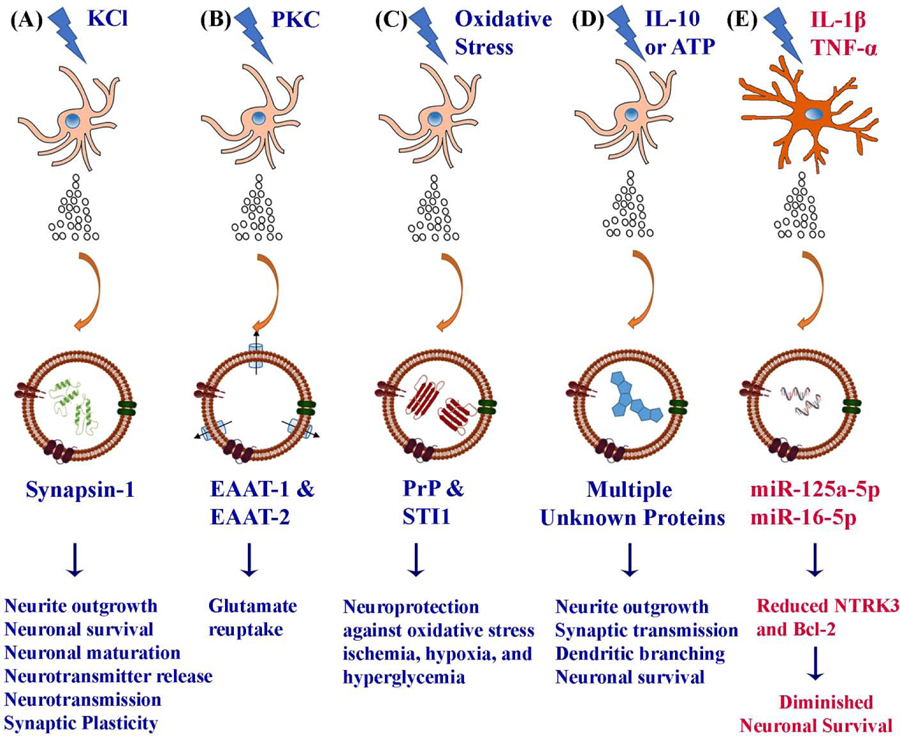
Astrocytes shed EVs containing different proteins when subjected to mild to moderate stress. These include EVs containing synapsin-1 from astrocytes cultured with potassium chloride (KCl) (A), glutamate transporters EAAT-1 and EAAT-2 from astrocytes activated with protein kinase C (PKC) (B), functional prion protein (PrP) from astrocytes subjected to oxidative stress (C), multiple beneficial proteins from astrocytes treated with the antiinflammatory cytokine, interleukin-10 (IL-10) or adenosine triphosphate (ATP) (D). The various beneficial effects mediated by these proteins are mentioned at the bottom of the figure. In contrast, astrocytes challenged with interleukin-1 beta (IL-1β) or tumor necrosis factor-alpha (TNF-α) (E) secrete EVs containing specific miRNAs that suppress factors involved in neuronal survival, differentiation, and neurite outgrowth.
3.5. PKC activation in astrocytes leads to release of EVs with glutamate transporters
A study by Gosselin and colleagues showed the presence of functional glutamate transporters, EAAT-1, and EAAT-2 in the ADEVs [31] (Fig. 2 [B]). The authors demonstrated that activation of protein kinase C in cultured rat primary astrocytes through phorbol myristate acetate leads to the packaging of EAATs into ADEVs. The EAATs are membrane transporters found in both neurons and astrocytes. They are involved in glutamate reuptake, which keeps the extracellular glutamate at low concentrations and reduces excitotoxicity in the brain. Because of the involvement of EAATs in scavenging the extracellular glutamate released at synapses, the release of EAAT-containing EVs by astrocytes has considerable significance for maintaining neural homeostasis. Dysfunctional glutamate transport can cause neurotoxicity, which is apparent in numerous neuropathological conditions, including stroke/ischemia, temporal lobe epilepsy, AD, ALS, and Huntington’s disease (HD) [123–125]. Thus, exogenous administration of EAAT-containing ADEVs into the brain is likely advantageous in neurological conditions that exhibit increased glutamate levels in the brain.
3.6. Astrocytes exposed to stress shed EVs carrying prion protein:
Guitart and associates showed that EVs released from cultured rat primary cortical astrocytes contain functional prion protein (PrP), which promoted neuroprotection to cerebellar neurons in conditions of oxidative stress, ischemia, hypoxia and hypoglycemia [126]. Interestingly, wild-type neurons survived better when co-cultured with stressed wild-type astrocytes in comparison to PrP-deficient astrocytes exposed to hypoxia (Fig. 2 [C]). This observation implied that neuronal defense by astrocytes against oxidative stress involved PrP [127–130]. Indeed, additional analysis revealed increased expression of PrP in EVs shed from wild-type astrocytes, and the selective uptake of EVs by the cerebellar neurons, implying the role of ADEVs in neuroprotection against oxidative stress. Another study showed that ADEVs also carry a protein called stress-inducible protein 1 (STI-1) [131], which binds to PrPc (a normal cell surface glycoprotein) with high affinity and causes the activation of ERK1/2 signaling to promote neuronal differentiation [132–133]. An enriched occurrence of PrP in EVs shed by astrocytes in response to stress has significance because PrP contributes to several biological processes, which comprise neuritogenesis, neuronal homeostasis, cell signaling, cell adhesion, and a protective role against stress. Thus, administration of PrP-containing ADEVs into the brain may be useful in conditions presented with increased oxidative stress in the brain.
3.7. Astrocytes exposed to IL-1β, TNF-α or IL-10 shed EVs that modulate synapses:
ADEVs carry multiple miRNAs and proteins [27, 134–135] by which astrocytes regulate essential functions particularly in synaptic clefts where they are involved in the monitoring or altering synaptic function to control the synaptic transmission. Chaudhuri and co-workers examined changes in the miRNA and protein composition of ADEVs in inflammatory conditions [27, 134]. When astrocytes were treated with two pro-inflammatory cytokines IL-1β and TNF-α, enriched expression of several sets of miRNAs were found in ADEVs, in comparison to EVs shed by astrocytes treated with ATP (Fig. 2 [D–E]). Two enriched miRNAs (miR-125a-5p and miR-16–5p) found in EVs shed by IL-1β or TNF-α stimulated astrocytes (ADEV- TNF-α or ADEV-IL-1β) are known to decrease neurotrophic receptor tyrosine kinase 3 (NTRK3) and B cell lymphoma-2 (Bcl-2) in neurons, which are involved in neuronal survival, differentiation, and neurite outgrowth. At the translational level, ADEV-IL-1β contained proteins involved in regulating the movement of immune cells to CNS. Exposure of primary hippocampal neurons to EVs released by ATP or IL-10 stimulated astrocytes (ADEV-ATP or ADEV- IL-10) increased neurite length, the overall dendritic complexity, synaptic transmission and neuronal survival [27, 134]. In contrast, exposure of hippocampal neurons to ADEV-IL-β or ADEV-TNF-α dose-dependently diminished neurite formation and the extent of dendritic complexity. Interestingly, hippocampal neurons treated with ADEV-IL-1β following knock-down of 125a-5p and miR-16–5p through antisense oligonucleotide inhibitors prevented not only decreases in NTRK3 and Bcl-2 proteins but also the adverse effects on neurite growth and dendritic complexity.
Thus, in inflammatory conditions, astrocytes modify the miRNA and protein cargo of ADEVs which can considerably weaken target neuron activity through modification of the translational expression of proteins or directly by proteins that control synaptic stability and neuronal excitability [27, 134]. Another study has shown that modulation of miRNA composition of ADEVs results in loss of the ability of EVs to support neuronal survival or even exacerbation of neurodegeneration [136], implying that individual miRNAs within ADEVs promote neuroprotection through activation of specific signaling pathways. Therefore, investigation of the miRNA cargo of EVs shed by astrocytes in different microenvironment conditions is necessary to discern the specific effects of astrocytes on neurons or brain repair in diverse contexts. Also, culturing of astrocytes in different conditions may yield ADEVs with highly variable miRNA signature. Thus, for generating ADEVs for therapeutic applications, standardization of culture conditions in which astrocytes shed a large number of EVs enriched with beneficial miRNAs would be necessary.
4. Role of ADEVs in the pathogenesis of neurological/neurodegenerative conditions:
The potential composition and adverse effects of EVs shed from astrocytes in conditions such as AD, stroke, PD, ALS, inflammatory brain injury, drug abuse, HIV-, ZIKA-, or alcohol-induced brain dysfunction are listed in Table 2 and detailed below.
Table 2.
Role of ADEVS in Neurological and Neurodegenerative Diseases
| Neurological Disease/Model | Known or Potential Composition of ADEVs | Potential Adverse Effects mediated by ADEVs | References |
|---|---|---|---|
| Alzheimer’s disease | EVs enriched with Aβ42 protofibrils and ApoE ɛ4 | Neurotoxicity of neighboring neurons, less effective clearing of Aβ42 peptides | [137–143] |
| Alzheimer’s disease | EVs enriched with BACE-1, sAPPβ, and various complement proteins | Neuroinflammatory cascade and neurodegeneration | [145, 147–148] |
| Stroke or ischemia | EVs enriched with SEMA3A | Activation of astrocytes, diminished axon growth, neurodegeneration | [154–157] |
| Parkinson’s Disease | EVs enriched with miR-34a following exposure to LPS | Increased dopaminergic neurodegeneration following exposure to a neurotoxin | [136] |
| Amyotrophic lateral sclerosis | EVs enriched with mutant SOD1 | Increased oxidative stress, neuroinflammation, and death of motoneurons | [158] |
| Amyotrophic lateral sclerosis | EVs enriched with mutant TDP-43 | Neurodegeneration | [1, 161–163] |
| Amyotrophic lateral sclerosis | EVs enriched with FUS | Juvenile-onset of ALS | [1, 161] |
| Amyotrophic lateral sclerosis | EVs enriched with DRPs | Neurodegeneration | [165] |
| Amyotrophic lateral sclerosis | EVs with depleted miR-49–3p | Upregulation of SEMA3A and neurodegeneration | [157] |
| Inflammatory Brain Injury using IL-1β injection | EVs containing unknown cargo | Inhibition of PPARα, secretion of cytokines in the liver and mobilization of peripheral immune cells to the brain | [166] |
| Opiate abuse (morphine induced prototype) | EVs enriched with lincRNA-Cox2 | Impaired phagocytosis by microglia, activation of TLR-7 signaling pathway | [167–169] |
| HIV-associated brain dysfunction | EVs enriched with nef | Increased oxidative stress, reduced neurite growth, and cognitive impairment | [170–173] |
| HIV-associated brain dysfunction | EVs enriched with HIV-1 Tat | Reduced neurite growth, and neurodegeneration | [174] |
| miR-9 | EVs enriched with miR-9 | Enhanced microglial migration | [175] |
| Zika virus mediated brain dysfunction | EVs enriched with Zika viral particles | Neurodegeneration | [177] |
| Alcohol-induced brain dysfunction | EVs enriched with pro-inflammatory cytokines and miRs | Neuroinflammation and brain dysfunction | [178] |
Abbreviations: Aβ42, amyloid-beta 42; ApoE ɛ4, apolipoprotein E allele 4; BACE-1, β-site amyloid precursor protein-cleaving enzyme 1; DRPs, dipeptide repeat proteins; FUS, fused in sarcoma; HIV, human immunodeficiency virus; IL-1β, interleukin-1 beta; lincRNA-Cox2, long intergenic non-coding RNA-cyclooxygenase-2; LPS, lipopolysaccharide; miR, micro-RNA; PPARα, nef, negative regulatory factor; peroxisome proliferator-activated receptor alpha; SEMA3A, semaphorin 3A; SOD1, superoxide dismutase 1; Tat, trans-activator of transcription; TDP-43, transactive response (TAR) DNA binding protein; TLR-7, toll-like receptor-7.
4.1. ADEVs in Alzheimer’s Disease
There is considerable evidence supporting the involvement of ADEVs as carriers of aggregated proteins like amyloid beta oligomers and protofibrils (Aβ) in AD pathogenesis [137] (Fig. 3). In the early stages of AD, astrocytes are known to engulf massive amounts of Aβ aggregates [2, 138–139]. A study by Sollvander and co-workers showed that astrocytes, when co-cultured with neurons and oligodendrocytes, engulfed a more significant proportion of fluorescently labeled Aβ42 (Aβ42–555) protofibrils [137]. Interestingly, no labeled Aβ42 was found in neurons, but oligodendrocytes engulfed a smaller proportion of Aβ42, which confirmed the role of astrocytes in engulfing large amounts of Aβ42 protofibrils. As a consequence of this, a large amount of Aβ gets accumulated within the astrocytes for prolonged periods without undergoing degradation, which induces a severe endosomal/lysosomal defects in astrocytes. This faulty degradation pathway leads to astrocytes releasing the engulfed Aβ42 protofibrils through EVs, which in turn causes severe neurotoxicity in neighboring neurons [137]. Additional studies demonstrated that the release of ApoE (particularly apoE ɛ4) by Aβ42 protofibrils exposed astrocytes through EVs increases by three folds [140]. These findings have implications because, there are three apoE isoforms in humans and those carrying apoE ɛ4 isoform display a strong susceptibility for developing late-onset AD where apoE ɛ4 has been shown to interact with Aβ42 peptides leading to a less effective clearing of Aβ42 peptides [141–143].
Figure 3:
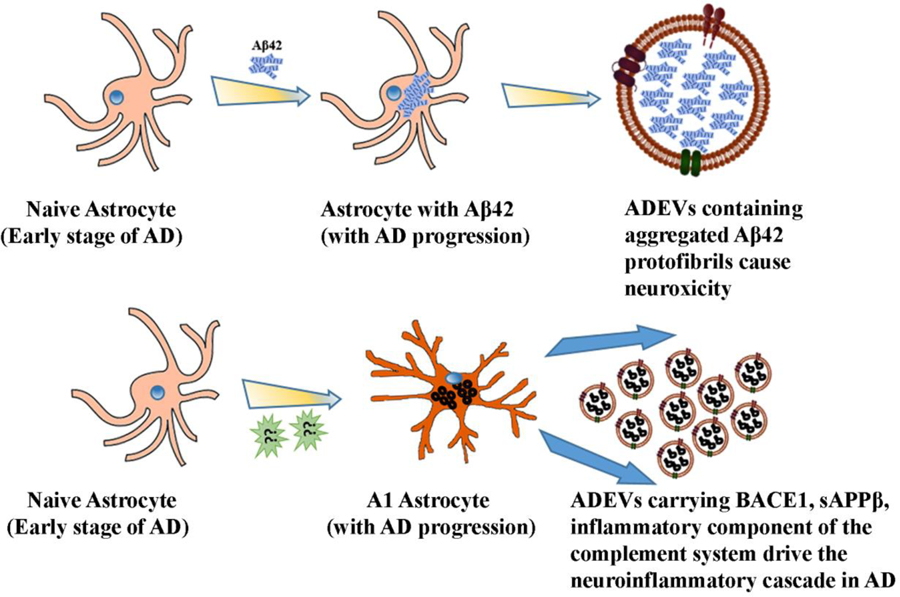
Schematic representation of mechanisms by which astrocyte-derived EVs (ADEVs) contribute to neuroinflammation and neurotoxicity in Alzheimer’s disease (AD). In the early phase of AD, astrocytes engulf massive amounts of Aβ42 aggregates, which leads to endosomal/lysosomal defects in astrocytes (upper panel). Such astrocytes then release EVs containing Aβ42, causing neurotoxicity. Moreover, inflammatory and neurodegenerative conditions in the early phase of AD transform astrocytes into the toxic A1 type, which shed EVs containing neurotoxic mediators such as BACE-1, soluble APPβ (sAPPβ) and the inflammatory component of the complement system.
Another study demonstrated that shedding of EVs by cultured primary astrocytes increases in response to Aβ exposure due to the involvement of the enzyme nSMase2. This finding implied an increased release of EVs containing toxic protein aggregates by astrocytes in AD and the proposition that the progression of AD could be slowed down through inhibition of nSMase2 using a small molecule inhibitor [144]. A recent study by Goetzl and associates investigated the role of ADEVs in carrying the components of the complement system during inflammation and neurodegeneration in AD [145]. The authors suggested that certain inflammatory and neurodegenerative reactions elicit a coordinated response that leads to hyperplasia of astrocytes and their conversion into reactive phenotypes, which are now recognized as inflammatory type A1 astrocytes. Such astrocytes display increased expression of pro-inflammatory elements that damage both synapses and neurons [146]. The precise neurotoxic mediators secreted by A1 astrocytes are yet to be discovered. However, several studies imply the presence of higher levels of β-site amyloid precursor protein-cleaving enzyme 1 (BACE-1), soluble amyloid precursor protein β (sAPPβ) of the Aβ42 peptide-generating system and inflammatory complement proteins of classical and alternative systems such as C1q, C4b, C3d, factor B, factor D, Bb, C3b, and C5b-C9 terminal complement complex in ADEVs isolated from the plasma of AD patients [145, 147–148] (Fig. 3). The finding that diminished levels of complement regulatory proteins are typically seen in preclinical AD supports the notion that the loss of normal inhibition function of the classical and alternative complement pathway drives neuroinflammatory cascade in AD and ADEVs seem to have a role in this process [145].
Overall, it appears that, neuroinflammation and neurodegeneration in AD are associated with activation and transformation of astrocytes into A1 phenotype through their interactions with various forms of Aβ, which leads to shedding of the dysregulated complement system through EVs released by astrocytes making ADEVs as a pathogenic factor [149–152]. The presence of abnormally high levels of mRNA transcripts of the complement pathway observed in several regions of the postmortem AD brains supports this hypothesis [146, 153].
4.2. ADEVs in Stroke
An investigation by Hira and colleagues suggested the harmful effects of ADEVs in a rat model of ischemia, induced through middle cerebral artery occlusion [154]. The adverse effects of ADEVs in this prototype were linked to the overexpression of semaphorin 3A (SEMA3A), which is an ECM protein implicated in several degenerative conditions [155–157]. Additional analysis suggested that SEMA3A promoted the activation of astrocytes after ischemia, which resulted in the shedding of toxic EVs by activated astrocytes and reduced axonal growth (Fig. 4 [A]). Interestingly, enhanced axonal growth occurred when ischemic astrocytes were treated with SEMA3A inhibitor SEMA3A-I. Further studies suggested the role of prostaglandin D2 synthase (PTGDS) in enhanced axonal outgrowth, as enhanced expression of PTGDS was seen in EVs derived from oxygen-glucose deprived astrocytes treated with the SEMA3A-I. Thus, ADEVs generated in conditions such as stroke can mediate adverse effects and impede spontaneous brain repair, but the modification of reactive A1 astrocytes into antiinflammatory phenotypes through drug/biologics therapy would likely induce beneficial effects.
Figure 4:
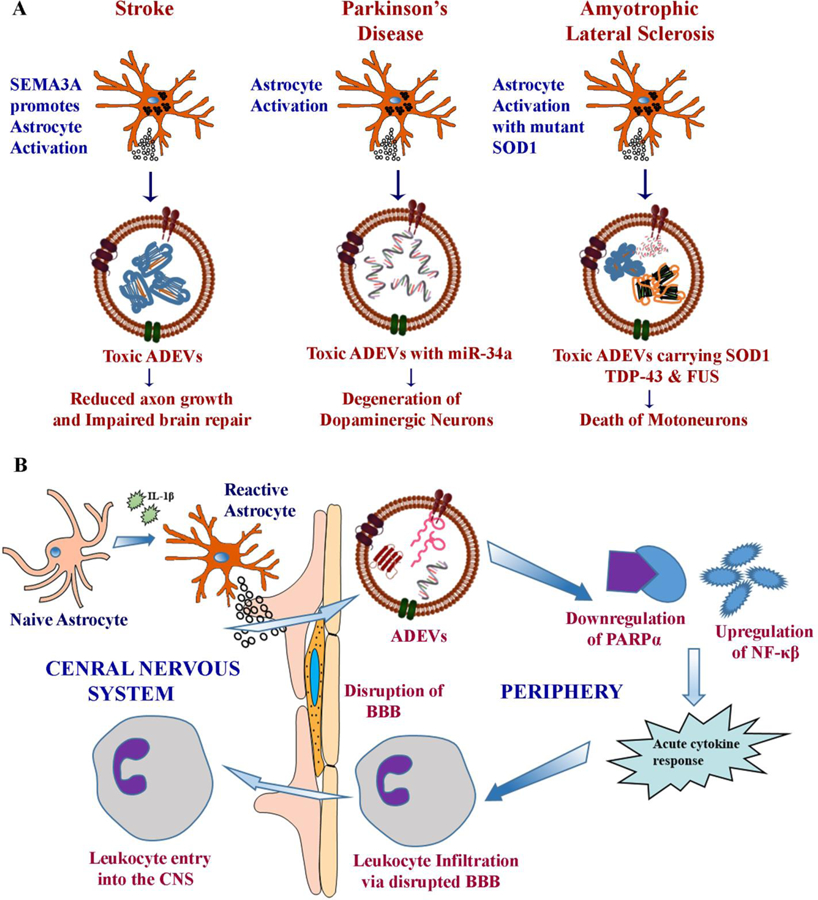
Diagrammatic representation of potential mechanisms by which astrocyte-derived EVs (ADEVs) contribute to neuroinflammation and neurotoxicity in stroke, Parkinson’s disease (PD), and Amyotrophic lateral sclerosis (ALS) (A). Toxic ADEVs reduce axonal growth and impair brain repair in stroke, likely contribute to the degeneration of dopaminergic neurons through miR-34a and induce motoneuron death through mutant superoxide dismutase 1 (SOD1), transactive response DNA binding protein (TDP-43) and fused in sarcoma (FUS). Figure B illustrates the involvement of ADEVs in propagating inflammation in other neuroinflammatory conditions. When challenged with pro-inflammatory cytokine IL-1β, astrocytes in the brain shed EVs carrying specific proteins and miRNAs, which migrate rapidly into the liver through circulation and cause acute cytokine response by downregulating PARPα and upregulating NF-κB. Such a response leads to the transmigration of peripheral leukocytes into the brain through the disrupted blood-brain barrier.
4.3. ADEVs in Parkinson’s Disease and Amyotrophic Lateral Sclerosis
Pertaining to Parkinson’s disease (PD), a cell culture study suggested that ADEVs in PD likely carry a specific miRNA that plays a crucial role in PD pathogenesis [136]. When an astroglial cell line was challenged with LPS, selective enrichment of miR34a occurred in EVs shed by astrocytes, which enhanced the sensitivity of dopaminergic neurons to a neurotoxin. It appeared that miR-34a carried by ADEVs entered the dopaminergic neurons and targeted the anti-apoptotic Bcl-2 protein (Fig. 4 [A]). Indeed, inhibition of the miR-34a through siRNA technique postponed dopaminergic neuron death against the stress [136]. However, it remains to be seen whether ADEVs in PD is indeed enriched with miR-34a and whether silencing of miR-34a in PD models would prevent/reduce dopaminergic neuron loss and promote functional recovery.
Regarding Amyotrophic lateral sclerosis (ALS), an in vitro study suggested a role for ADEVs in the progression of this disease [158] (Fig. 4 [A]). In this study, overexpression of the mutant SOD1 in primary astrocytes activated unconventional secretory pathways and the release of a higher number of EVs containing mutant SOD1. Considerable death of motoneurons was observed when the conditioned medium containing EVs from cultures of mutant SOD1 overexpressing astrocytes was added to motoneuron cultures. The process involved the transfer and aggregation of the mutant SOD1 in motoneurons and the release of proinflammatory cytokines and free radicals through microglia-mediated injury [158]. Interestingly, motoneuron death did not occur when the conditioned media devoid of EVs was added to cultures, implying the role of ADEVs in the progression of ALS. However, a few other studies, while confirming the role of astrocytes in ALS progression, did not find a role for ADEVs. Instead, the studies suggested the involvement of an alternative secretory mechanism behind ALS propagation [159–160].
A recent study by Sproviero and associates suggested the possibility of EVs carrying another ALS causative element called transactive response (TAR) DNA binding protein (TDP-43) [1] (Fig. 4 [A]). TDP43 is a nuclear protein having many roles, including the regulation of transcription and translation [161]. The mutated form of TDP-43 is frequently seen in the cytosol as an aggregated form [162] and behaves as a prion-like structure due to its tendency to oligomerize and aggregate. Feiler and co-workers demonstrated that the mutant TDP-43 oligomers get loaded onto EVs and delivered to neuronal soma and synaptic clefts, which likely triggers neurodegeneration [163]. Authors speculated the involvement of ADEVs in delivering the mutant form of TDP-43 since astrocytic processes wrap around the pre- and post-synaptic elements and the synaptic cleft. Another study found an increased accumulation of toxic TDP-43 in cell lysates when U251 cells were treated with cerebrospinal fluid (CSF) enriched with TDP-43 from ALS-FD patients for 21 days. Authors suggested a possible role for EVs in mediating this process [164]. However, definitive evidence of the role of EVs is not available, and the cell origin of EVs mediating disease progression in ALS is yet to be discovered. Another ALS protein that is suspected to be transported via EVs is Fused in Sarcoma (FUS). FUS is an RNA binding protein, and the mutant form of FUS is associated with one of the most aggressive, juvenile-onset of ALS [161]. A study showed the presence of FUS in EVs from the plasma of ALS patients, but the cell origin of EVs was not investigated [1]. A recent addition to the causative element underlying the ALS pathology is the aberrant hexanucleotide repeat expansions found in the C9orf72 gene. RNA transcripts containing these hexanucleotide repeat form a dipeptide repeat proteins (DRPs) through repeat-associated non-ATG translation (RAN-T) and the aggregated form of DRPs known to cause neurodegeneration in vitro and in vivo [165]. This study showed cell to cell propagation of DRPs via EV-dependent and EV-independent pathways but did not provide the cell source of EVs. In this context, performing selective enrichment of EVs using antibodies specific to astrocytes and neurons [73] would be useful for definitive conclusions on the type of EVs mediating disease progression in ALS.
Apart from proteins, miRNAs carried by ADEVs are likely also involved in ALS disease progression. A study by Varcianna and associates suggested that astrocytes generated from fibroblasts of ALS patients are toxic to motoneurons and that toxicity is chiefly transmitted through ADEVs [157]. Furthermore, the miRNA profiling of ADEVs revealed a significant downregulation of miR-494–3p, which is a miRNA involved in the regulation of SEMA3A. SEMA3A is typically upregulated in several neurological disorders including ALS and higher levels of SEMA3A in motoneurons caused reduced neurite growth and motoneuron death. However, exogenous supply of miR-494–3p reduced SEMA3A concentration and rescued motoneurons from death. Thus, reduced miR-494–3p in EVs shed by astrocytes likely contributes to ALS pathogenesis [157].
4.4. ADEVs in other neuroinflammatory conditions
ADEVs are likely also involved in the propagation of neuroinflammation in several other inflammatory conditions as they can induce transmigration of peripheral leukocytes into the brain under pathological conditions (Fig. 4 [B]). By employing a mouse model of inflammatory brain injury, Dickens and associates demonstrated that when challenged with acute inflammation such as after intracerebral injection of IL-1β, astrocytes release EVs with a highly modified cargo [166]. Such modified ADEVs rapidly crossed the blood-brain barrier and moved to the liver through peripheral circulation. In the liver, the modified ADEVs stimulated the secretion of cytokines that mobilized peripheral immune cells to infiltrate the brain. Characterization of the protein and microRNA cargo in modified ADEVs recognized peroxisome proliferator-activated receptor α (PPARα) as the principal molecular target of ADEVs (Fig. 4 [B]). Indeed, inhibition of PPARα augmented nuclear factor κB (NF-κB) activity that initiated increased production of cytokines in the liver. Overall, this study confirmed that ADEVs are involved in regulating the bidirectional exchange between the brain and immune system and that modifications in the cargo of EVs may regulate disease-specific biological responses. Also, the study implied that inhibition of the distant interaction between the brain and the liver might considerably enhance functional recovery after brain injuries [166].
4.5. ADEVs in opiate abuse
A study examining the role of morphine-induced changes in astrocytes and composition of EVs secreted by astrocytes showed upregulation of specific long intergenic noncoding RNAs (lincRNAs), namely long intergenic non-coding RNA-cyclooxygenase-2 (lincRNA-Cox2) in ADEVs (Fig. 5 [A]) [167]. LincRNAs are regulatory RNA molecules and play a pivotal role in regulating many cellular processes [168–169]. When microglia internalized ADEVs from morphine-exposed animals, their phagocytic ability was impaired with a significant upregulation of lincRNA-Cox2 through the activation of the TLR-7 signaling pathway [167]. Interestingly, the intranasal administration of lincRNA-Cox2 siRNA restored microglial phagocytic activity in morphine-administered mice [167]. Thus, ADEVs seem to be involved in drug abuse induced brain abnormalities and hence are potential targets for drug abuse treatment.
Figure 5:
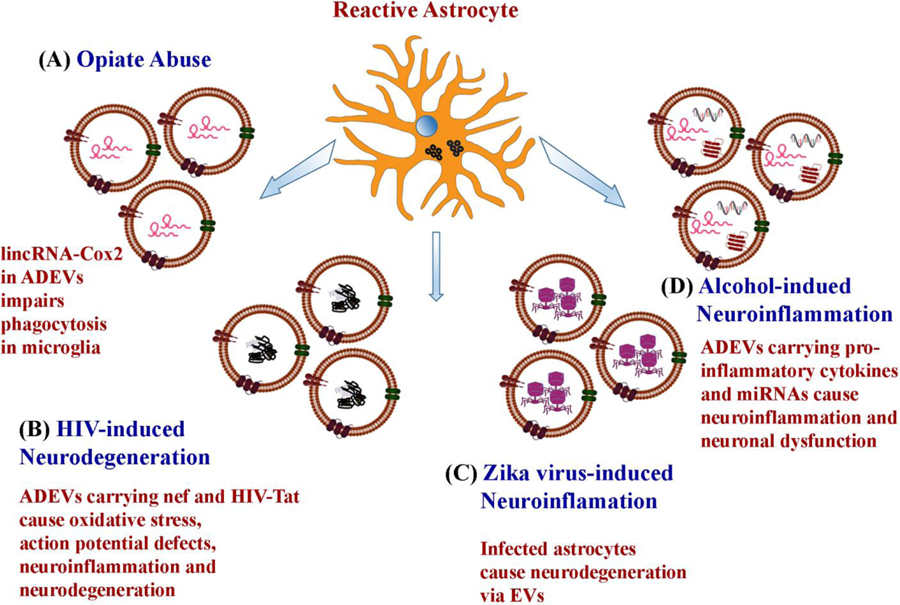
Potential mechanisms by which astrocyte-derived EVs (ADEVs) mediate neurotoxicity in opiate abuse, human immunodeficiency virus (HIV)- and Zika virus-induced neurodegeneration, and alcohol intake-induced neuroinflammation. Opiates such as morphine upregulate long intergenic noncoding RNA lincRNA-Cox2 in ADEVs, which impairs the phagocytic behavior of microglia when internalized. In HIV associated neurological disorder, ADEVs carry an HIV associated protein negative regulator factor (nef) and HIV-1 transactivator of transcription (HIV-Tat), which cause increased oxidative stress, reduced neurite and axonal growth, impaired action potential and neurodegeneration by inducing the production of pro-inflammatory cytokines and other toxic proteins. In Zika virus infection, astrocytes get infected first and promote neurodegeneration through EVs secreted by them. In alcohol-induced neuroinflammation, EVs secreted by alcohol-exposed astrocytes carry pro-inflammatory cytokines and miRNAs, which cause neuroinflammation and neuronal dysfunction.
4.6. ADEVs in HIV-associated neurological disorders
Sami and colleagues investigated the role ADEVs in carrying (negative regulatory factor (nef), an HIV associated protein that causes oxidative stress to neurons and subsequent HIV-associated neurological disorders and cognitive impairment [170–172] (Fig. 5 [B]). It has been reported that HIV-1 mediated toxicity to neurons involves several other viral proteins and induction of a spike in proinflammatory cytokines. One such protein that is believed to be responsible for HIV-1 induced neuronal death is the Nef [173]. HIV-infected astrocytes secrete nef via ADEVs. Studies have shown that internalization of such ADEVs by neurons causes oxidative stress, reduction in neurite and axonal growth, and impaired action potential [172]. Similarly, cell to cell movement of HIV-1 trans-activator of transcription (Tat), a transactivator for HIV gene transcription and replication likely occurs through EVs (Fig. 5 [B]). A study demonstrated that Tat expressing primary astrocytes release HIV-1 Tat through EVs and cause neurite shortening and neurodegeneration [174]. In another study, astrocytes stimulated by HIV-1 Tat shed EVs enriched with miR-9, which caused increased microglial migration [175]. Another study demonstrated reduced expression of platelet-derived growth factor-B (PDGF-B) and release of miR-29b via EVs when astrocytes were exposed to morphine and HIV-1 Tat in vitro [176]. Thus, ADEVs are significantly involved in HIV-mediated neurodegeneration and brain dysfunction.
4.7. ADEVs in Zika virus propagation, and alcohol intake-induced neuroinflammation
ADEVs are also known to carry the Zika virus, which causes fatal brain abnormalities in human fetal development. Huang and co-workers observed that Zika virus specifically infected the primary human astrocytes than neural progenitor cells of fetal origin [177]. The infected primary astrocytes released viral particles through EVs, which induced neuronal cell death (Fig. 5 [C]). It was also observed that the Zika infection of astrocytes resulted in increased production and release EVs. These results imply that ADEVs are also a target for treating Zika virus-mediated neurodegeneration. Another study examined the role of ADEVs in spreading ethanol-induced neuroinflammation in a cell culture model. Ethanol is known to activate glial cells and cause neuroinflammation through Toll-like receptor 4 (TLR-4) by triggering the release of neuroinflammatory cytokines via EVs. This study showed that ethanol treatment leads to increased shedding of EVs from astrocytes containing pro-inflammatory proteins and miRNAs, which induce neuroinflammation and neuronal dysfunction [178] (Fig. 5 [C]).
5. ADEVs in the blood as a source of brain biomarkers in neurological disorders
A biomarker is a molecular attribute that can be quantitatively measured and evaluated to gauge normal physiology, pathologic progression, or reaction to a therapeutic intervention [179]. Identification of appropriate biomarkers in neurodegenerative disorders is a challenge because, in most instances, neurodegenerative disorders are confined to a subset of neural cells located in distinct regions of the CNS. From this perspective, molecular characterization of EVs shed from neurons and glia found in the CSF, and the blood has considerable promise for biomarker identification in neurodegenerative disorders. As the configuration of EVs imitates the physiological or pathological state of cells from which they are derived at the time of secretion [180], characterization of EVs derived from neurons or glia in the blood would help in identification of consistent biomarkers in neurodegenerative disorders. Also, since the cargo of EVs is often protected from protease and RNase degradation, molecular characterization of EVs derived from specific brain cells (e.g., NDEVs, ADEVs) facilitate the identification of robust biomarkers of neurological disorders [73, 181]. The enrichment of specific markers in EVs would also facilitate the detection of biomarkers that are typically expressed at relatively low levels in body fluids. Thus, biomarkers of neurological or neurodegenerative disorders could be tracked consistently via analyses of EVs shed from neurons and glia in the CSF or the circulating blood in different stages of the disease or following therapeutic interventions [182]. The following section describes the usefulness of analyses of ADEVs isolated from the blood of patients with different neurodegenerative conditions.
Goetzl and colleagues have identified several specific proteins in ADEVs derived from plasma of patients with AD and frontotemporal dementia (FTD) [147]. They demonstrated that ADEVs contained 20-fold higher levels of multiple disease-related proteins than NDEVs. These include β-site amyloid precursor protein-cleaving enzyme 1 (BACE-1), γ-secretase, soluble Aβ-42, soluble APP-α and APP-β, glial cell-line derived neurotrophic factor (GDNF) and phosphorylated tau. In comparison to ADEVs from controls, ADEVs from both AD and FTD patients displayed higher levels of BACE-1 and soluble APP-β. ADEVs from AD patients, besides, exhibited a reduced concentration of GDNF [147]. Another study by the same research group showed high complement levels in ADEVs derived from the plasma of AD patients. Notably, ADEVs contained higher levels of C1q, C4b of the classical complement pathway, C3d, factor B, factor D, fragment Bb of the alternative complement pathway, and C3b, and C5b-C9 terminal complement complex of both pathways [145]. A recent study compared the protein composition of ADEVs from patients with mild cognitive impairment (MCI) converting to dementia within three years (MCIC) and patients with MCI remaining stable over three years (MCIS). The study demonstrated that ADEVs in the circulating blood contained higher levels of C1q, C4b, factor D, fragment Bb, C5b, C3b, and C5b-C9 in patients with MCIC than those with MCIS. The study also revealed that ADEVs in patients with MCIC displayed lower concentrations of multiple inhibitory complement proteins than those with MCIS [148]. Thus, studies on ADEV cargo proteins are useful for understanding mechanisms contributing to dementia in MCI and AD. It is likely that the complement proteins in ADEVs damage neurons and facilitate the progression of dementia in these conditions.
Another study, using an animal model of Gulf War Illness (GWI), demonstrated that biomarkers of neuroinflammation and complement activation in the brain could be tracked reliably via analyses of NDEVs and ADEVs in the circulating blood [73]. The authors showed that elevated levels of multiple proinflammatory and complement proteins related to neuroinflammation found in the cerebral cortex of GWI rats could also be seen in NDEVs (e.g., HMGB1, TNF-α, and IL-1β) and ADEVs (e.g., C3 and TccC5b-9) isolated from the blood of GWI rats [73]. Overall, the above studies particularly underscore that characterization of ADEVs in the blood is a highly useful strategy for discerning the extent of complement activation in the brain in different neurological and neurodegenerative conditions as well as for monitoring changes with different treatment approaches. In addition to the analysis of proteins, ADEVs from the blood may also be investigated for the type of miRNAs they carry, as the miRNA composition of EVs is known to undergo modifications in neurodegenerative disease conditions. Such analysis may uncover distinct miRNAs as biomarkers of specific neurodegenerative conditions. For example, significant downregulation of miR-494–3p found in EVs secreted by astrocytes derived from fibroblasts of ALS patients [157] might also be seen in ADEVs in the blood of ALS patients.
6. Future perspectives
6.1. Promise of ADEVs as biologics for treating brain injury or disease
ADEVs released in physiological conditions likely modulate the immediate microenvironment as they carry a cargo of neurotrophic compounds. The compounds identified hitherto include FGF-2, VEGF, Apo-D, and HSP [30, 33, 82, 94]. These proteins carried by ADEVs are capable of activating MAPK signaling pathway, stimulating the proliferation, survival, migration, and differentiation of neural stem cells and endothelial cells, regulating the extent of neurogenesis and angiogenesis, as well as contributing to synaptic plasticity and cognitive function [80–82, 84, 88, 91]. Besides, astrocytes, in conditions of increased oxidative stress, neuronal hyperactivity or activation of PKC shed EVs loaded with several beneficial proteins. These include synapsin 1 [34], and glutamate transporters [31]. These molecules are involved in neuroprotection, neurite outgrowth, synapse formation, synaptic plasticity, regulation of neurotransmitter release, and glutamate reuptake. On the other hand, EVs shed by astrocytes in chronic inflammatory conditions can contain adverse miRNAs such as miR-125a-5p and miR-16–5p123, which can promote synaptic loss, inhibit neuroregeneration as well as promote neurodegeneration. Thus, it appears that ADEVs shed from cultured naïve astrocytes or astrocytes exposed to mild to moderate oxidative stress or subjected to PKC activation are likely suitable as biologics to treat brain injury or neurodegenerative diseases.
Administration of EVs shed by naïve or mildly stimulated astrocytes into the brain through intravenous or intranasal routes in conditions such as aging, the early stages of the AD, ALS, and PD, traumatic brain injury, stroke, and epilepsy would likely to have significant therapeutic benefits. Direct grafting of ADEVs to brain regions afflicted with age-related changes, injury or disease may not be required as intranasally administered EVs quickly target multiple regions of the brain including those afflicted with acute inflammation [183–184]. In such conditions, ADEVs may restrain neurodegeneration and synapse loss, promote neurogenesis, neosynaptogenesis and angiogenesis, improve synaptic plasticity, and reduce neuronal hyperexcitability. Nonetheless, the biological activity and therapeutic properties of ADEVs generated in various conditions will need to be analyzed thoroughly using proteomic and small RNA sequencing approaches to evaluate their cargo. Furthermore, they need to be investigated rigorously for antioxidant, antiinflammatory, and neuroprotective properties through specific in vitro and short-term in vivo assays before testing their long term efficacy in animal prototypes of neurodegenerative diseases. Such careful and sequential approaches would pave the way for clinical application of ADEVs containing a consistent set of cargo for treating neurodegenerative disorders in the future. Comprehending the composition of ADEVs and the development of advanced approaches to modulate the cargo of ADEVs to include the desired miRNAs and proteins would further enhance the therapeutic efficacy of ADEVs for different neurological and neurodegenerative conditions.
6.2. ADEVs as targets for improving brain function in disease conditions
In disease conditions, EVs shed by reactive, proinflammatory or toxic A1 astrocytes mediate or exacerbate the pathological processes. EVs shed by the reactive astrocytes in AD are involved in the dissemination of Aβ oligomers, protofibrils and the components of the complement system [137, 145]. Likewise, astrocytes activated through SEMA3A, an ECM protein that exhibits upregulation after stroke, shed EVs that hinder axonal regeneration [154]. There is also speculation that miR-34a carried by EVs secreted by proinflammatory astrocytes promote degeneration of dopaminergic neurons in PD [136]. Several studies have also suggested that ADEVs promote motoneuron death in ALS through the transfer of mutant SOD1 [158], TDP-43 [163], or downregulation of miR-494–3p [157]. Furthermore, ADEVs trigger the migration of leukocytes into the brain through modulation of peripheral cytokine response in neuroinflammatory conditions [166], and dampen microglial phagocytic activity through the activation of the TLR-7 signaling in conditions such as drug abuse [167]. Moreover, in HIV-associated neurological disorders, ADEVs are contributing to brain dysfunction through the propagation of a viral protein nef that causes oxidative stress in neurons and cognitive impairment [173]. From the above perspectives, strategies that restrain the release of ADEVs or modulate the composition of EVs secreted by reactive astrocytes are likely beneficial for treating neurodegenerative diseases. In this context, the study by Dinkins and colleagues showing that inhibition of nSMase2 with a small molecule GW4869 can moderate the release of EVs by neural cells in a model of AD is highly relevant [144]. Such approaches have promise to reverse or at least slow down disease progression.
7. Conclusions
ADEVs, akin to EVs secreted by other cells, are biological nanoparticles carrying a cargo of proteins, nucleic acids, and lipids. They likely play an essential role in a two-way communication of astrocytes with neurons, other glia, or brain endothelial cells. Such interactions likely contribute to the maintenance of brain homeostasis. In conditions such as after an injury, ADEVs are likely neuroprotective and contribute to neural regeneration and plasticity because multiple neuroprotective proteins and miRNAs are found in EVs released from mild-moderately stimulated astrocytes. In contrast, EVs secreted by reactive, proinflammatory, or toxic A1 astrocytes appear to partake in the spread pathological molecules as well as the recruitment of leukocytes from the periphery into the brain. The use of well-characterized EVs secreted by naïve or mildly stimulated astrocytes in controlled culture conditions or ADEVs that are engineered specifically to carry the desired miRNAs or proteins has great promise for treating neurogenerative diseases. Mainly, explicitly targeting therapeutic ADEVs to neurons or microglia using advanced techniques may ease the loss of neurons and synapses, suppress neuroinflammation, promote neurogenesis, angiogenesis, and neosynaptogenesis, and improve brain function in neurodegenerative disease conditions. Furthermore, approaches that curb the release of ADEVs or regulate the composition of EVs secreted by reactive astrocytes are likely valuable for slowing down the progression of neurodegenerative diseases. Additionally, the characterization of ADEVs in the blood is a highly useful strategy for discerning the progression of neuroinflammation in the CNS in different conditions and for monitoring anti-inflammatory effects with different therapeutic interventions.
Acknowledgments
Funding
Authors are supported by grants from the National Institute of Neurological Disorders and Stroke (1R01NS106907–01 to A.K.S.), the Department of Defense (W81XWH-14-1-0558 to A.K.S.), and the State of Texas (Emerging Technology Fund to A.K.S.)
Abbreviations
- Aβ
Amyloid beta
- AD
Alzheimer’s disease
- ADEVs
Astrocyte derived extracellular vesicles
- ALIX
ALG-2-interacting protein X
- ALS
Amyotrophic lateral sclerosis
- ApoD
Apolipoprotein- D
- ApoE
Apolipoprotein-E
- ApoE ɛ4
Apolipoprotein-E epsilon 4
- ATP
Adenosine triphosphate
- BACE-1
β-site amyloid precursor protein-cleaving enzyme 1
- BBB
Blood-brain barrier
- Bcl2
B cell lymphoma - 2
- C3
Complement 3
- CD9
Cluster of differentiations 9
- CD63
Cluster of differentiations 63
- CD81
Cluster of differentiations 81
- CHMP4
Charged multivesicular body protein 4
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- DRPs
Dipeptide repeat proteins
- EAAT-1/2
Excitatory amino acid transporters1/2
- ECM
Extracellular matrix
- ERK
Extracellular signal-regulated kinase
- ESCRT
Endosomal sorting complex required for transport
- EVs
Extracellular vesicles
- EXs
Exosomes
- FGF-2
Fibroblast growth factor-2
- FGFRs
Fibroblast growth factor receptors
- FUS
Fused in sarcoma
- GWI
Gulf war illness
- HD
Huntington’s disease
- HIV-1
Human immunodeficiency virus-1
- HIV-1 Tat
Human immunodeficiency virus- transactivator of transcription
- HMGB1
High mobility group box 1
- IL-1β
Interleukin-1 beta
- IL-6
Interleukin-6
- ILVs
Intraluminal vesicles
- ISEV
International Society for Extracellular Vesicles
- JNK
Jun N-terminal kinase
- KCl
Potassium chloride
- lincRNAs
Long intergenic noncoding RNAs
- LPS
Lipopolysaccharide
- MAP
Microtubule-associated protein
- miRNAs
MicroRNAs
- MV
Microvesicles
- MVBs
Multivesicular bodies
- NCAM
Neural cell adhesion molecule
- Nef
Negative regulator factor
- NF-κB
Nuclear factor kappa B
- nSMase2
Neutral sphingomyelinase 2
- NTRK3
Neurotrophic receptor tyrosine kinase 3
- P13k
Phosphatidylinositol 3-kinase
- PARP-α
Procyclic acidic repetitive protein-alpha
- PD
Parkinson disease
- PDGF-B
Platelet-derived growth factor subunit B
- PKC
Protein kinase C
- PrP
Prion protein
- PTGDS
Prostaglandin D2 Synthase
- RAN-T
Repeat-associated non-ATG translation
- sAPPβ
Soluble amyloid precursor protein-beta
- SEMA3A
Semaphorin 3A
- SD
Spreading depression
- SOD1
Superoxide dismutase 1
- STI1
Stress inducible protein
- TAR
Transactive response
- TCC-C5b-9
Terminal complement C-C5b-9
- TDP43
Transactive response DNA binding protein 43
- TLR2/4
Toll like receptor 2/4
- TLRs
Toll like receptors
- TNF-α
Tumor necrosis factor-alpha
- TSG-101
Tumor Susceptibility gene 101 protein
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declared that there is no conflict of interest.
References
- [1].Sproviero D, La Salvia S, Giannini M, Crippa V, Gagliardi S, Bernuzzi S, Diamanti L, Ceroni M, Pansarasa O, Poletti A, Cereda C, Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients, Front Neurosci, 12 (2018) 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J, Adult mouse astrocytes degrade amyloid-beta in vitro and in situ, Nat Med, 9 (2003) 453–457. [DOI] [PubMed] [Google Scholar]
- [3].Bushong EA, Martone ME, Jones YZ, Ellisman MH, Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains, J Neurosci, 22 (2002) 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG, Synaptic islands defined by the territory of a single astrocyte, J Neurosci, 27 (2007) 6473–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Araque A, Parpura V, Sanzgiri RP, Haydon PG, Tripartite synapses: glia, the unacknowledged partner, Trends Neurosci, 22 (1999) 208–215. [DOI] [PubMed] [Google Scholar]
- [6].Abbott NJ, Ronnback L, Hansson E, Astrocyte-endothelial interactions at the blood-brain barrier, Nat Rev Neurosci, 7 (2006) 41–53. [DOI] [PubMed] [Google Scholar]
- [7].Ransom BR, Ransom CB, Astrocytes: multitalented stars of the central nervous system, Methods Mol Biol, 814 (2012) 3–7. [DOI] [PubMed] [Google Scholar]
- [8].Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF, Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate, Neuron, 16 (1996) 675–686. [DOI] [PubMed] [Google Scholar]
- [9].Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW, Localization of neuronal and glial glutamate transporters, Neuron, 13 (1994) 713–725. [DOI] [PubMed] [Google Scholar]
- [10].Sofroniew MV, Vinters HV, Astrocytes: biology and pathology, Acta Neuropathol, 119 (2010) 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sontheimer H, Voltage-dependent ion channels in glial cells, Glia, 11 (1994) 156–172. [DOI] [PubMed] [Google Scholar]
- [12].Vasile F, Dossi E, Rouach N, Human astrocytes: structure and functions in the healthy brain, Brain Struct Funct, 222 (2017) 2017–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Casse F, Richetin K, Toni N, Astrocytes’ Contribution to Adult Neurogenesis in Physiology and Alzheimer’s Disease, Front Cell Neurosci, 12 (2018) 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hattiangady B, Shetty AK, Aging does not alter the number or phenotype of putative stem/progenitor cells in the neurogenic region of the hippocampus, Neurobiol Aging, 29 (2008) 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hattiangady B, Shuai B, Cai J, Coksaygan T, Rao MS, Shetty AK, Increased dentate neurogenesis after grafting of glial restricted progenitors or neural stem cells in the aging hippocampus, Stem Cells, 25 (2007) 2104–2117. [DOI] [PubMed] [Google Scholar]
- [16].Luarte A, Cisternas P, Caviedes A, Batiz LF, Lafourcade C, Wyneken U, Henzi R, Astrocytes at the Hub of the Stress Response: Potential Modulation of Neurogenesis by miRNAs in Astrocyte-Derived Exosomes, Stem Cells Int, 2017 (2017) 1719050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schneider J, Karpf J, Beckervordersandforth R, Role of Astrocytes in the Neurogenic Niches, Methods Mol Biol, 1938 (2019) 19–33. [DOI] [PubMed] [Google Scholar]
- [18].Williams CA, Lavik EB, Engineering the CNS stem cell microenvironment, Regen Med, 4 (2009) 865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Balu DT, Pantazopoulos H, Huang CCY, Muszynski K, Harvey TL, Uno Y, Rorabaugh JM, Galloway CR, Botz-Zapp C, Berretta S, Weinshenker D, Coyle JT, Neurotoxic astrocytes express the d-serine synthesizing enzyme, serine racemase, in Alzheimer’s disease, Neurobiol Dis, 130 (2019) 104511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sofroniew MV, Astrocyte barriers to neurotoxic inflammation, Nat Rev Neurosci, 16 (2015) 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang SU, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee S, Dawson TM, Ko HS, Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease, Nat Med, 24 (2018) 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kastanenka KV, Moreno-Bote R, De Pitta M, Perea G, Eraso-Pichot A, Masgrau R, Poskanzer KE, Galea E, A roadmap to integrate astrocytes into Systems Neuroscience, Glia, 68 (2020) 5–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shetty AK, Hattiangady B, Shetty GA, Stem/progenitor cell proliferation factors FGF-2, IGF-1, and VEGF exhibit early decline during the course of aging in the hippocampus: role of astrocytes, Glia, 51 (2005) 173–186. [DOI] [PubMed] [Google Scholar]
- [24].Verkhratsky A, Matteoli M, Parpura V, Mothet JP, Zorec R, Astrocytes as secretory cells of the central nervous system: idiosyncrasies of vesicular secretion, EMBO J, 35 (2016) 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Volterra A, Meldolesi J, Astrocytes, from brain glue to communication elements: the revolution continues, Nat Rev Neurosci, 6 (2005) 626–640. [DOI] [PubMed] [Google Scholar]
- [26].Bianco F, Perrotta C, Novellino L, Francolini M, Riganti L, Menna E, Saglietti L, Schuchman EH, Furlan R, Clementi E, Matteoli M, Verderio C, Acid sphingomyelinase activity triggers microparticle release from glial cells, EMBO J, 28 (2009) 1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Datta Chaudhuri A, Dasgheyb RM, DeVine LR, Bi H, Cole RN, Haughey NJ, Stimulus-dependent modifications in astrocyte-derived extracellular vesicle cargo regulate neuronal excitability, Glia, 68 (2020) 128–144. [DOI] [PubMed] [Google Scholar]
- [28].Fruhbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, Kirchhoff F, Mobius W, Goebbels S, Nave KA, Schneider A, Simons M, Klugmann M, Trotter J, Kramer-Albers EM, Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication, PLoS Biol, 11 (2013) e1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Guescini M, Genedani S, Stocchi V, Agnati LF, Astrocytes and Glioblastoma cells release exosomes carrying mtDNA, J Neural Transm (Vienna), 117 (2010) 1–4. [DOI] [PubMed] [Google Scholar]
- [30].Taylor AR, Robinson MB, Gifondorwa DJ, Tytell M, Milligan CE, Regulation of heat shock protein 70 release in astrocytes: role of signaling kinases, Dev Neurobiol, 67 (2007) 1815–1829. [DOI] [PubMed] [Google Scholar]
- [31].Gosselin RD, Meylan P, Decosterd I, Extracellular microvesicles from astrocytes contain functional glutamate transporters: regulation by protein kinase C and cell activation, Front Cell Neurosci, 7 (2013) 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moidunny S, Vinet J, Wesseling E, Bijzet J, Shieh CH, van Ijzendoorn SC, Bezzi P, Boddeke HW, Biber K, Adenosine A2B receptor-mediated leukemia inhibitory factor release from astrocytes protects cortical neurons against excitotoxicity, J Neuroinflammation, 9 (2012) 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Proia P, Schiera G, Mineo M, Ingrassia AM, Santoro G, Savettieri G, Di Liegro I, Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor, Int J Mol Med, 21 (2008) 63–67. [PubMed] [Google Scholar]
- [34].Wang S, Cesca F, Loers G, Schweizer M, Buck F, Benfenati F, Schachner M, Kleene R, Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes, J Neurosci, 31 (2011) 7275–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schiera G, Di Liegro CM, Di Liegro I, Extracellular Membrane Vesicles as Vehicles for Brain Cell-to-Cell Interactions in Physiological as well as Pathological Conditions, Biomed Res Int, 2015 (2015) 152926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Silverman JM, Christy D, Shyu CC, Moon KM, Fernando S, Gidden Z, Cowan CM, Ban Y, Stacey RG, Grad LI, McAlary L, Mackenzie IR, Foster LJ, Cashman NR, CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)(G93A) ALS mice originate from astrocytes and neurons and carry misfolded SOD1, J Biol Chem, 294 (2019) 3744–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Doeppner TR, Herz J, Gorgens A, Schlechter J, Ludwig AK, Radtke S, de Miroschedji K, Horn PA, Giebel B, Hermann DM, Extracellular Vesicles Improve Post-Stroke Neuroregeneration and Prevent Postischemic Immunosuppression, Stem Cells Transl Med, 4 (2015) 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang Y, Chopp M, Meng Y, Katakowski M, Xin H, Mahmood A, Xiong Y, Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury, J Neurosurg, 122 (2015) 856–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim DK, Nishida H, An SY, Shetty AK, Bartosh TJ, Prockop DJ, Chromatographically isolated CD63+CD81+ extracellular vesicles from mesenchymal stromal cells rescue cognitive impairments after TBI, Proc Natl Acad Sci U S A, 113 (2016) 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Drommelschmidt K, Serdar M, Bendix I, Herz J, Bertling F, Prager S, Keller M, Ludwig AK, Duhan V, Radtke S, de Miroschedji K, Horn PA, van de Looij Y, Giebel B, Felderhoff-Muser U, Mesenchymal stem cell-derived extracellular vesicles ameliorate inflammation-induced preterm brain injury, Brain Behav Immun, 60 (2017) 220–232. [DOI] [PubMed] [Google Scholar]
- [41].Long Q, Upadhya D, Hattiangady B, Kim DK, An SY, Shuai B, Prockop DJ, Shetty AK, Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus, Proc Natl Acad Sci U S A, 114 (2017) E3536–E3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cui GH, Wu J, Mou FF, Xie WH, Wang FB, Wang QL, Fang J, Xu YW, Dong YR, Liu JR, Guo HD, Exosomes derived from hypoxia-preconditioned mesenchymal stromal cells ameliorate cognitive decline by rescuing synaptic dysfunction and regulating inflammatory responses in APP/PS1 mice, FASEB J, 32 (2018) 654–668. [DOI] [PubMed] [Google Scholar]
- [43].Otero-Ortega L, Gomez de Frutos MC, Laso-Garcia F, Rodriguez-Frutos B, Medina-Gutierrez E, Lopez JA, Vazquez J, Diez-Tejedor E, Gutierrez-Fernandez M, Exosomes promote restoration after an experimental animal model of intracerebral hemorrhage, J Cereb Blood Flow Metab, 38 (2018) 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [44].Vogel A, Upadhya R, Shetty AK, Neural stem cell derived extracellular vesicles: Attributes and prospects for treating neurodegenerative disorders, EBioMedicine, 38 (2018) 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Webb RL, Kaiser EE, Jurgielewicz BJ, Spellicy S, Scoville SL, Thompson TA, Swetenburg RL, Hess DC, West FD, Stice SL, Human Neural Stem Cell Extracellular Vesicles Improve Recovery in a Porcine Model of Ischemic Stroke, Stroke, 49 (2018) 1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Narbute K, Pilipenko V, Pupure J, Dzirkale Z, Jonavice U, Tunaitis V, Kriauciunaite K, Jarmalaviciute A, Jansone B, Klusa V, Pivoriunas A, Intranasal Administration of Extracellular Vesicles Derived from Human Teeth Stem Cells Improves Motor Symptoms and Normalizes Tyrosine Hydroxylase Expression in the Substantia Nigra and Striatum of the 6-Hydroxydopamine-Treated Rats, Stem Cells Transl Med, 8 (2019) 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Upadhya D, Shetty AK, Promise of extracellular vesicles for diagnosis and treatment of epilepsy, Epilepsy Behav, (2019) 106499. [DOI] [PMC free article] [PubMed]
- [48].Upadhya D, Shetty AK, Extracellular Vesicles as Therapeutics for Brain Injury and Disease, Curr Pharm Des, 25 (2019) 3500–3505. [DOI] [PubMed] [Google Scholar]
- [49].Holm MM, Kaiser J, Schwab ME, Extracellular Vesicles: Multimodal Envoys in Neural Maintenance and Repair, Trends Neurosci, 41 (2018) 360–372. [DOI] [PubMed] [Google Scholar]
- [50].Vidal M, Exosomes: revisiting their role as ‘garbage bags’, Traffic, 20 (2019) 815–828. [DOI] [PubMed] [Google Scholar]
- [51].Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, Witwer KW, Thery C, Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles, J Extracell Vesicles, 3 (2014) 26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Thery C, Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes, Proc Natl Acad Sci U S A, 113 (2016) E968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, Ayre DC, Bach JM, Bachurski D, Baharvand H, Balaj L, Baldacchino S, Bauer NN, Baxter AA, Bebawy M, Beckham C, Bedina Zavec A, Benmoussa A, Berardi AC, Bergese P, Bielska E, Blenkiron C, Bobis - Wozowicz S, Boilard E, Boireau W, Bongiovanni A, Borras FE, Bosch S, Boulanger CM, Breakefield X, Breglio AM, Brennan MA, Brigstock DR, Brisson A, Broekman ML, Bromberg JF, Bryl-Gorecka P, Buch S, Buck AH, Burger D, Busatto S, Buschmann D, Bussolati B, Buzas EI, Byrd JB, Camussi G, Carter DR, Caruso S, Chamley LW, Chang YT, Chen C, Chen S, Cheng L, Chin AR, Clayton A, Clerici SP, Cocks A, Cocucci E, Coffey RJ, Cordeiro-da-Silva A, Couch Y, Coumans FA, Coyle B, Crescitelli R, Criado MF, D’Souza-Schorey C, Das S, Datta Chaudhuri A, de Candia P, De Santana EF, De Wever O, Del Portillo HA, Demaret T, Deville S, Devitt A, Dhondt B, Di Vizio D, Dieterich LC, Dolo V, Dominguez Rubio AP, Dominici M, Dourado MR, Driedonks TA, Duarte FV, Duncan HM, Eichenberger RM, Ekstrom K, El Andaloussi S, Elie-Caille C, Erdbrugger U, Falcon-Perez JM, Fatima F, Fish JE, Flores-Bellver M, Forsonits A, Frelet-Barrand A, Fricke F, Fuhrmann G, Gabrielsson S, Gamez-Valero A, Gardiner C, Gartner K, Gaudin R, Gho YS, Giebel B, Gilbert C, Gimona M, Giusti I, Goberdhan DC, Gorgens A, Gorski SM, Greening DW, Gross JC, Gualerzi A, Gupta GN, Gustafson D, Handberg A, Haraszti RA, Harrison P, Hegyesi H, Hendrix A, Hill AF, Hochberg FH, Hoffmann KF, Holder B, Holthofer H, Hosseinkhani B, Hu G, Huang Y, Huber V, Hunt S, Ibrahim AG, Ikezu T, Inal JM, Isin M, Ivanova A, Jackson HK, Jacobsen S, Jay SM, Jayachandran M, Jenster G, Jiang L, Johnson SM, Jones JC, Jong A, Jovanovic-Talisman T, Jung S, Kalluri R, Kano SI, Kaur S, Kawamura Y, Keller ET, Khamari D, Khomyakova E, Khvorova A, Kierulf P, Kim KP, Kislinger T, Klingeborn M, Klinke DJ 2nd, Kornek M, Kosanovic MM, Kovacs AF, Kramer-Albers EM, Krasemann S, Krause M, Kurochkin IV, Kusuma GD, Kuypers S, Laitinen S, Langevin SM, Languino LR, Lannigan J, Lasser C, Laurent LC, Lavieu G, Lazaro-Ibanez E, Le Lay S, Lee MS, Lee YXF, Lemos DS, Lenassi M, Leszczynska A, Li IT, Liao K, Libregts SF, Ligeti E, Lim R, Lim SK, Line A, Linnemannstons K, Llorente A, Lombard CA, Lorenowicz MJ, Lorincz AM, Lotvall J, Lovett J, Lowry MC, Loyer X, Lu Q, Lukomska B, Lunavat TR, Maas SL, Malhi H, Marcilla A, Mariani J, Mariscal J, Martens-Uzunova ES, Martin-Jaular L, Martinez MC, Martins VR, Mathieu M, Mathivanan S, Maugeri M, McGinnis LK, McVey MJ, Meckes DG Jr., Meehan KL, Mertens I, Minciacchi VR, Moller A, Moller Jorgensen M, Morales - Kastresana A, Morhayim J, Mullier F, Muraca M, Musante L, Mussack V, Muth DC, Myburgh KH, Najrana T, Nawaz M, Nazarenko I, Nejsum P, Neri C, Neri T, Nieuwland R, Nimrichter L, Nolan JP, Nolte-’t Hoen EN, Noren Hooten N, O’Driscoll L, O’Grady T, O’Loghlen A, Ochiya T, Olivier M, Ortiz A, Ortiz LA, Osteikoetxea X, Ostergaard O, Ostrowski M, Park J, Pegtel DM, Peinado H, Perut F, Pfaffl MW, Phinney DG, Pieters BC, Pink RC, Pisetsky DS, Pogge von Strandmann E, Polakovicova I, Poon IK, Powell BH, Prada I, Pulliam L, Quesenberry P, Radeghieri A, Raffai RL, Raimondo S, Rak J, Ramirez MI, Raposo G, Rayyan MS, Regev-Rudzki N, Ricklefs FL, Robbins PD, Roberts DD, Rodrigues SC, Rohde E, Rome S, Rouschop KM, Rughetti A, Russell AE, Saa P, Sahoo S, Salas-Huenuleo E, Sanchez C, Saugstad JA, Saul MJ, Schiffelers RM, Schneider R, Schoyen TH, Scott A, Shahaj E, Sharma S, Shatnyeva O, Shekari F, Shelke GV, Shetty AK, Shiba K, Siljander PR, Silva AM, Skowronek A, Snyder OL 2nd, Soares RP, Sodar BW, Soekmadji C, Sotillo J, Stahl PD, Stoorvogel W, Stott SL, Strasser EF, Swift S, Tahara H, Tewari M, Timms K, Tiwari S, Tixeira R, Tkach M, Toh WS, Tomasini R, Torrecilhas AC, Tosar JP, Toxavidis V, Urbanelli L, Vader P, van Balkom BW, van der Grein SG, Van Deun J, van Herwijnen MJ, Van Keuren-Jensen K, van Niel G, van Royen ME, van Wijnen AJ, Vasconcelos MH, Vechetti IJ Jr., Veit TD, Vella LJ, Velot E, Verweij FJ, Vestad B, Vinas JL, Visnovitz T, Vukman KV, Wahlgren J, Watson DC, Wauben MH, Weaver A, Webber JP, Weber V, Wehman AM, Weiss DJ, Welsh JA, Wendt S, Wheelock AM, Wiener Z, Witte L, Wolfram J, Xagorari A, Xander P, Xu J, Yan X, Yanez-Mo M, Yin H, Yuana Y, Zappulli V, Zarubova J, Zekas V, Zhang JY, Zhao Z, Zheng L, Zheutlin AR, Zickler AM, Zimmermann P, Zivkovic AM, Zocco D, Zuba-Surma EK, Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines, J Extracell Vesicles, 7 (2018) 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pols MS, Klumperman J, Trafficking and function of the tetraspanin CD63, Exp Cell Res, 315 (2009) 1584–1592. [DOI] [PubMed] [Google Scholar]
- [55].Bebelman MP, Smit MJ, Pegtel DM, Baglio SR, Biogenesis and function of extracellular vesicles in cancer, Pharmacol Ther, 188 (2018) 1–11. [DOI] [PubMed] [Google Scholar]
- [56].Hurley JH, ESCRTs are everywhere, EMBO J, 34 (2015) 2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Villarroya-Beltri C, Baixauli F, Gutierrez-Vazquez C, Sanchez-Madrid F, Mittelbrunn M, Sorting it out: regulation of exosome loading, Semin Cancer Biol, 28 (2014) 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Airola MV, Hannun YA, Sphingolipid metabolism and neutral sphingomyelinases, Handb Exp Pharmacol, (2013) 57–76. [DOI] [PMC free article] [PubMed]
- [59].Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M, Ceramide triggers budding of exosome vesicles into multivesicular endosomes, Science, 319 (2008) 1244–1247. [DOI] [PubMed] [Google Scholar]
- [60].Mazurov D, Barbashova L, Filatov A, Tetraspanin protein CD9 interacts with metalloprotease CD10 and enhances its release via exosomes, FEBS J, 280 (2013) 1200–1213. [DOI] [PubMed] [Google Scholar]
- [61].Perez-Hernandez D, Gutierrez-Vazquez C, Jorge I, Lopez-Martin S, Ursa A, Sanchez-Madrid F, Vazquez J, Yanez-Mo M, The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes, J Biol Chem, 288 (2013) 11649–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].van den Boorn JG, Dassler J, Coch C, Schlee M, Hartmann G, Exosomes as nucleic acid nanocarriers, Adv Drug Deliv Rev, 65 (2013) 331–335. [DOI] [PubMed] [Google Scholar]
- [63].van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, Marks MS, Rubinstein E, Raposo G, The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis, Dev Cell, 21 (2011) 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Verweij FJ, van Eijndhoven MA, Hopmans ES, Vendrig T, Wurdinger T, Cahir-McFarland E, Kieff E, Geerts D, van der Kant R, Neefjes J, Middeldorp JM, Pegtel DM, LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-kappaB activation, EMBO J, 30 (2011) 2115–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Akers JC, Gonda D, Kim R, Carter BS, Chen CC , Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies, J Neurooncol, 113 (2013) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nabhan JF, Hu R, Oh RS, Cohen SN, Lu Q, Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein, Proc Natl Acad Sci U S A, 109 (2012) 4146–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson RJ, Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes, Methods, 56 (2012) 293–304. [DOI] [PubMed] [Google Scholar]
- [68].Vogel A, Upadhya R, Shetty AK, Neural stem cell derived extracellular vesicles: Attributes and prospects for treating neurodegenerative disorders, EBioMedicine, 38 (2018) 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].You Y, Ikezu T, Emerging roles of extracellular vesicles in neurodegenerative disorders, Neurobiol Dis, 130 (2019) 104512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Delpech JC, Herron S, Botros MB, Ikezu T, Neuroimmune Crosstalk through Extracellular Vesicles in Health and Disease, Trends Neurosci, 42 (2019) 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Morad G, Moses MA, Brainwashed by extracellular vesicles: the role of extracellular vesicles in primary and metastatic brain tumour microenvironment, J Extracell Vesicles, 8 (2019) 1627164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Perez M, Avila J, Hernandez F, Propagation of Tau via Extracellular Vesicles, Front Neurosci, 13 (2019) 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Madhu LN, Attaluri S, Kodali M, Shuai B, Upadhya R, Gitai D, Shetty AK, Neuroinflammation in Gulf War Illness is linked with HMGB1 and complement activation, which can be discerned from brain-derived extracellular vesicles in the blood, Brain Behav Immun, 81 (2019) 430–443. [DOI] [PubMed] [Google Scholar]
- [74].Shi M, Sheng L, Stewart T, Zabetian CP, Zhang J, New windows into the brain: Central nervous system-derived extracellular vesicles in blood, Prog Neurobiol, 175 (2019) 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Upadhya D, Shetty AK, Promise of extracellular vesicles for diagnosis and treatment of epilepsy, Epilepsy Behav, (2019) 106499. [DOI] [PMC free article] [PubMed]
- [76].Anderson MA, O’Shea TM, Burda JE, Ao Y, Barlatey SL, Bernstein AM, Kim JH, James ND, Rogers A, Kato B, Wollenberg AL, Kawaguchi R, Coppola G, Wang C, Deming TJ, He Z, Courtine G, Sofroniew MV, Required growth facilitators propel axon regeneration across complete spinal cord injury, Nature, 561 (2018) 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bikfalvi A, Klein S, Pintucci G, Rifkin DB, Biological roles of fibroblast growth factor-2, Endocr Rev, 18 (1997) 26–45. [DOI] [PubMed] [Google Scholar]
- [78].Dehghan S, Javan M, Pourabdolhossein F, Mirnajafi-Zadeh J, Baharvand H, Basic fibroblast growth factor potentiates myelin repair following induction of experimental demyelination in adult mouse optic chiasm and nerves, J Mol Neurosci, 48 (2012) 77–85. [DOI] [PubMed] [Google Scholar]
- [79].Reeve RL, Yammine SZ, Morshead CM, van der Kooy D, Quiescent Oct4(+) Neural Stem Cells (NSCs) Repopulate Ablated Glial Fibrillary Acidic Protein(+) NSCs in the Adult Mouse Brain, Stem Cells, 35 (2017) 2071–2082. [DOI] [PubMed] [Google Scholar]
- [80].Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH, Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions, Endocr Rev, 22 (2001) 153–183. [DOI] [PubMed] [Google Scholar]
- [81].Shao H, Wilkinson B, Lee B, Han PC, Kaye J, Slow accumulation of active mitogen-activated protein kinase during thymocyte differentiation regulates the temporal pattern of transcription factor gene expression, J Immunol, 163 (1999) 603–610. [PubMed] [Google Scholar]
- [82].Rai KS, Hattiangady B, Shetty AK, Enhanced production and dendritic growth of new dentate granule cells in the middle-aged hippocampus following intracerebroventricular FGF-2 infusions, Eur J Neurosci, 26 (2007) 1765–1779. [DOI] [PubMed] [Google Scholar]
- [83].Waldau B, Shetty AK, Behavior of neural stem cells in the Alzheimer brain, Cell Mol Life Sci, 65 (2008) 2372–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Woodbury ME, Ikezu T, Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration, J Neuroimmune Pharmacol, 9 (2014) 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dietrich WD, Alonso O, Busto R, Finklestein SP, Posttreatment with intravenous basic fibroblast growth factor reduces histopathological damage following fluid-percussion brain injury in rats, J Neurotrauma, 13 (1996) 309–316. [DOI] [PubMed] [Google Scholar]
- [86].Zhao YZ, Lin M, Lin Q, Yang W, Yu XC, Tian FR, Mao KL, Yang JJ, Lu CT, Wong HL, Intranasal delivery of bFGF with nanoliposomes enhances in vivo neuroprotection and neural injury recovery in a rodent stroke model, J Control Release, 224 (2016) 165–175. [DOI] [PubMed] [Google Scholar]
- [87].Zucchini S, Buzzi A, Barbieri M, Rodi D, Paradiso B, Binaschi A, Coffin JD, Marzola A, Cifelli P, Belluzzi O, Simonato M, Fgf-2 overexpression increases excitability and seizure susceptibility but decreases seizure-induced cell loss, J Neurosci, 28 (2008) 13112–13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Shim JW, Madsen JR, VEGF Signaling in Neurological Disorders, Int J Mol Sci, 19 (2018) E275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Fournier NM, Duman RS, Role of vascular endothelial growth factor in adult hippocampal neurogenesis: implications for the pathophysiology and treatment of depression, Behav Brain Res, 227 (2012) 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Newton SS, Duman RS, Regulation of neurogenesis and angiogenesis in depression, Curr Neurovasc Res, 1 (2004) 261–267. [DOI] [PubMed] [Google Scholar]
- [91].Tillo M, Ruhrberg C, Mackenzie F, Emerging roles for semaphorins and VEGFs in synaptogenesis and synaptic plasticity, Cell Adh Migr, 6 (2012) 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Greenberg DA, Jin K, Vascular endothelial growth factors (VEGFs) and stroke, Cell Mol Life Sci, 70 (2013) 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lange C, Storkebaum E, de Almodovar CR, Dewerchin M, Carmeliet P, Vascular endothelial growth factor: a neurovascular target in neurological diseases, Nat Rev Neurol, 12 (2016) 439–454. [DOI] [PubMed] [Google Scholar]
- [94].Pascua-Maestro R, Gonzalez E, Lillo C, Ganfornina MD, Falcon-Perez JM, Sanchez D, Extracellular Vesicles Secreted by Astroglial Cells Transport Apolipoprotein D to Neurons and Mediate Neuronal Survival Upon Oxidative Stress, Front Cell Neurosci, 12 (2018) 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dassati S, Waldner A, Schweigreiter R, Apolipoprotein D takes center stage in the stress response of the aging and degenerative brain, Neurobiol Aging, 35 (2014) 1632–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ganfornina MD, Do Carmo S, Lora JM, Torres-Schumann S, Vogel M, Allhorn M, Gonzalez C, Bastiani MJ, Rassart E, Sanchez D, Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress, Aging Cell, 7 (2008) 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Muffat J, Walker DW, Apolipoprotein D: an overview of its role in aging and age-related diseases, Cell Cycle, 9 (2010) 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sanchez D, Bajo-Graneras R, Del Cano-Espinel M, Garcia-Centeno R, Garcia-Mateo N, Pascua-Maestro R, Ganfornina MD, Aging without Apolipoprotein D: Molecular and cellular modifications in the hippocampus and cortex, Exp Gerontol, 67 (2015) 19–47. [DOI] [PubMed] [Google Scholar]
- [99].Li H, Ruberu K, Munoz SS, Jenner AM, Spiro A, Zhao H, Rassart E, Sanchez D, Ganfornina MD, Karl T, Garner B, Apolipoprotein D modulates amyloid pathology in APP/PS1 Alzheimer’s disease mice, Neurobiol Aging, 36 (2015) 1820–1833. [DOI] [PubMed] [Google Scholar]
- [100].Navarro A, Rioseras B, Del Valle E, Martinez-Pinilla E, Astudillo A, Tolivia J, Expression Pattern of Myelin-Related Apolipoprotein D in Human Multiple Sclerosis Lesions, Front Aging Neurosci, 10 (2018) 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Houenou LJ, Li L, Lei M, Kent CR, Tytell M, Exogenous heat shock cognate protein Hsc 70 prevents axotomy-induced death of spinal sensory neurons, Cell Stress Chaperones, 1 (1996) 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S, Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins, FASEB J, 16 (2002) 601–603. [DOI] [PubMed] [Google Scholar]
- [103].Novoselova TV, Margulis BA, Novoselov SS, Sapozhnikov AM, van der Spuy J, Cheetham ME, Guzhova IV, Treatment with extracellular HSP70/HSC70 protein can reduce polyglutamine toxicity and aggregation, J Neurochem, 94 (2005) 597–606. [DOI] [PubMed] [Google Scholar]
- [104].Robinson MB, Tidwell JL, Gould T, Taylor AR, Newbern JM, Graves J, Tytell M, Milligan CE, Extracellular heat shock protein 70: a critical component for motoneuron survival, J Neurosci, 25 (2005) 9735–9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Tidwell JL, Houenou LJ, Tytell M, Administration of Hsp70 in vivo inhibits motor and sensory neuron degeneration, Cell Stress Chaperones, 9 (2004) 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Beere HM, “The stress of dying”: the role of heat shock proteins in the regulation of apoptosis, J Cell Sci, 117 (2004) 2641–2651. [DOI] [PubMed] [Google Scholar]
- [107].Beere HM, Green DR, Stress management - heat shock protein-70 and the regulation of apoptosis, Trends Cell Biol, 11 (2001) 6–10. [DOI] [PubMed] [Google Scholar]
- [108].Becker T, Hartl FU, Wieland F, CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes, J Cell Biol, 158 (2002) 1277–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Beg AA, Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses, Trends Immunol, 23 (2002) 509–512. [DOI] [PubMed] [Google Scholar]
- [110].Gross C, Hansch D, Gastpar R, Multhoff G, Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94, Biol Chem, 384 (2003) 267–279. [DOI] [PubMed] [Google Scholar]
- [111].Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H, HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway, J Biol Chem, 277 (2002) 15107–15112. [DOI] [PubMed] [Google Scholar]
- [112].Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK, Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4, J Biol Chem, 277 (2002) 15028–15034. [DOI] [PubMed] [Google Scholar]
- [113].Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK, HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine, Nat Med, 6 (2000) 435–442. [DOI] [PubMed] [Google Scholar]
- [114].Enger R, Dukefoss DB, Tang W, Pettersen KH, Bjornstad DM, Helm PJ, Jensen V, Sprengel R, Vervaeke K, Ottersen OP, Nagelhus EA, Deletion of Aquaporin-4 Curtails Extracellular Glutamate Elevation in Cortical Spreading Depression in Awake Mice, Cereb Cortex, 27 (2017) 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Walz W, Role of astrocytes in the spreading depression signal between ischemic core and penumbra, Neurosci Biobehav Rev, 21 (1997) 135–142. [DOI] [PubMed] [Google Scholar]
- [116].Shibata M, Suzuki N, Exploring the role of microglia in cortical spreading depression in neurological disease, J Cereb Blood Flow Metab, 37 (2017) 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ploughman M, Granter-Button S, Chernenko G, Tucker BA, Mearow KM, Corbett D, Endurance exercise regimens induce differential effects on brain-derived neurotrophic factor, synapsin-I and insulin-like growth factor I after focal ischemia, Neuroscience, 136 (2005) 991–1001. [DOI] [PubMed] [Google Scholar]
- [118].Lu B, Greengard P, Poo MM, Exogenous synapsin I promotes functional maturation of developing neuromuscular synapses, Neuron, 8 (1992) 521–529. [DOI] [PubMed] [Google Scholar]
- [119].Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Sudhof TC, Short-term synaptic plasticity is altered in mice lacking synapsin I, Cell, 75 (1993) 661–670. [DOI] [PubMed] [Google Scholar]
- [120].Corradi A, Zanardi A, Giacomini C, Onofri F, Valtorta F, Zoli M, Benfenati F, Synapsin-I- and synapsin-II-null mice display an increased age-dependent cognitive impairment, J Cell Sci, 121 (2008) 3042–3051. [DOI] [PubMed] [Google Scholar]
- [121].Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P, Synapsins as regulators of neurotransmitter release, Philos Trans R Soc Lond B Biol Sci, 354 (1999) 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Melloni RH Jr., DeGennaro LJ, Temporal onset of synapsin I gene expression coincides with neuronal differentiation during the development of the nervous system, J Comp Neurol, 342 (1994) 449–462. [DOI] [PubMed] [Google Scholar]
- [123].Anderson CM, Swanson RA, Astrocyte glutamate transport: review of properties, regulation, and physiological functions, Glia, 32 (2000) 1–14. [PubMed] [Google Scholar]
- [124].Fontana AC, Current approaches to enhance glutamate transporter function and expression, J Neurochem, 134 (2015) 982–1007. [DOI] [PubMed] [Google Scholar]
- [125].Vandenberg RJ, Ryan RM, Mechanisms of glutamate transport, Physiol Rev, 93 (2013) 1621–1657. [DOI] [PubMed] [Google Scholar]
- [126].Guitart K, Loers G, Buck F, Bork U, Schachner M, Kleene R, Improvement of neuronal cell survival by astrocyte-derived exosomes under hypoxic and ischemic conditions depends on prion protein, Glia, 64 (2016) 896–910. [DOI] [PubMed] [Google Scholar]
- [127].Bertuchi FR, Bourgeon DM, Landemberger MC, Martins VR, Cerchiaro G, PrPC displays an essential protective role from oxidative stress in an astrocyte cell line derived from PrPC knockout mice, Biochem Biophys Res Commun, 418 (2012) 27–32. [DOI] [PubMed] [Google Scholar]
- [128].Guitart K, Loers G, Schachner M, Kleene R, Prion protein regulates glutathione metabolism and neural glutamate and cysteine uptake via excitatory amino acid transporter 3, J Neurochem, 133 (2015) 558–571. [DOI] [PubMed] [Google Scholar]
- [129].Kleene R, Loers G, Langer J, Frobert Y, Buck F, Schachner M, Prion protein regulates glutamate-dependent lactate transport of astrocytes, J Neurosci, 27 (2007) 12331–12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lima FR, Arantes CP, Muras AG, Nomizo R, Brentani RR, Martins VR, Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation, J Neurochem, 103 (2007) 2164–2176. [DOI] [PubMed] [Google Scholar]
- [131].Hajj GN, Arantes CP, Dias MV, Roffe M, Costa-Silva B, Lopes MH, Porto-Carreiro I, Rabachini T, Lima FR, Beraldo FH, Prado MA, Linden R, Martins VR, The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles, Cell Mol Life Sci, 70 (2013) 3211–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R, Martins VR, Interaction of cellular prion and stress -inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways, J Neurosci, 25 (2005) 11330–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MA, Linden R, Prion protein: orchestrating neurotrophic activities, Curr Issues Mol Biol, 12 (2010) 63–86. [PubMed] [Google Scholar]
- [134].Chaudhuri AD, Dastgheyb RM, Yoo SW, Trout A, Talbot CC Jr., Hao H, Witwer KW, Haughey NJ, TNFalpha and IL-1beta modify the miRNA cargo of astrocyte shed extracellular vesicles to regulate neurotrophic signaling in neurons, Cell Death Dis, 9 (2018) 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lafourcade C, Ramirez JP, Luarte A, Fernandez A, Wyneken U, MiRNAs in Astrocyte-Derived Exosomes as Possible Mediators of Neuronal Plasticity, J Exp Neurosci, 10 (2016) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mao S, Sun Q, Xiao H, Zhang C, Li L, Secreted miR-34a in astrocytic shedding vesicles enhanced the vulnerability of dopaminergic neurons to neurotoxins by targeting Bcl-2, Protein Cell, 6 (2015) 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Sollvander S, Nikitidou E, Brolin R, Soderberg L, Sehlin D, Lannfelt L, Erlandsson A, Accumulation of amyloid-beta by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons, Mol Neurodegener, 11 (2016) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jones RS, Minogue AM, Connor TJ, Lynch MA, Amyloid-beta-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE, J Neuroimmune Pharmacol, 8 (2013) 301–311. [DOI] [PubMed] [Google Scholar]
- [139].Nielsen HM, Mulder SD, Belien JA, Musters RJ, Eikelenboom P, Veerhuis R, Astrocytic A beta 1–42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins, Glia, 58 (2010) 1235–1246. [DOI] [PubMed] [Google Scholar]
- [140].Nikitidou E, Khoonsari PE, Shevchenko G, Ingelsson M, Kultima K, Erlandsson A, Increased Release of Apolipoprotein E in Extracellular Vesicles Following Amyloid-beta Protofibril Exposure of Neuroglial Co-Cultures, J Alzheimers Dis, 60 (2017) 305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Huang YA, Zhou B, Wernig M, Sudhof TC, ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion, Cell, 168 (2017) 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Winkler K, Scharnagl H, Tisljar U, Hoschutzky H, Friedrich I, Hoffmann MM, Huttinger M, Wieland H, Marz W, Competition of Abeta amyloid peptide and apolipoprotein E for receptor-mediated endocytosis, J Lipid Res, 40 (1999) 447–455. [PubMed] [Google Scholar]
- [143].Tai LM, Bilousova T, Jungbauer L, Roeske SK, Youmans KL, Yu C, Poon WW, Cornwell LB, Miller CA, Vinters HV, Van Eldik LJ, Fardo DW, Estus S, Bu G, Gylys KH, Ladu MJ, Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples, J Biol Chem, 288 (2013) 5914–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E, Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease, Neurobiol Aging, 35 (2014) 1792–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Goetzl EJ, Schwartz JB, Abner EL, Jicha GA, Kapogiannis D, High complement levels in astrocyte-derived exosomes of Alzheimer disease, Ann Neurol, 83 (2018) 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Liddelow SA, Barres BA, Reactive Astrocytes: Production, Function, and Therapeutic Potential, Immunity, 46 (2017) 957–967. [DOI] [PubMed] [Google Scholar]
- [147].Goetzl EJ, Mustapic M, Kapogiannis D, Eitan E, Lobach IV, Goetzl L, Schwartz JB, Miller BL, Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease, FASEB J, 30 (2016) 3853–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Winston CN, Goetzl EJ, Schwartz JB, Elahi FM, Rissman RA, Complement protein levels in plasma astrocyte-derived exosomes are abnormal in conversion from mild cognitive impairment to Alzheimer’s disease dementia, Alzheimers Dement, 11 (2019) 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Eikelenboom P, Stam FC, Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study, Acta Neuropathol, 57 (1982) 239–242. [DOI] [PubMed] [Google Scholar]
- [150].Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, et al. , Complement activation by beta-amyloid in Alzheimer disease, Proc Natl Acad Sci U S A, 89 (1992) 10016–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kolev MV, Ruseva MM, Harris CL, Morgan BP, Donev RM, Implication of complement system and its regulators in Alzheimer’s disease, Curr Neuropharmacol, 7 (2009) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Webster S, Rogers J, Relative efficacies of amyloid beta peptide (A beta) binding proteins in A beta aggregation, J Neurosci Res, 46 (1996) 58–66. [DOI] [PubMed] [Google Scholar]
- [153].Yasojima K, Schwab C, McGeer EG, McGeer PL, Up-regulated production and activation of the complement system in Alzheimer’s disease brain, Am J Pathol, 154 (1999) 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Hira K, Ueno Y, Tanaka R, Miyamoto N, Yamashiro K, Inaba T, Urabe T, Okano H, Hattori N, Astrocyte-Derived Exosomes Treated With a Semaphorin 3A Inhibitor Enhance Stroke Recovery via Prostaglandin D2 Synthase, Stroke, 49 (2018) 2483–2494. [DOI] [PubMed] [Google Scholar]
- [155].Hou ST, Keklikian A, Slinn J, O’Hare M, Jiang SX, Aylsworth A, Sustained up-regulation of semaphorin 3A, Neuropilin1, and doublecortin expression in ischemic mouse brain during long-term recovery, Biochem Biophys Res Commun, 367 (2008) 109–115. [DOI] [PubMed] [Google Scholar]
- [156].Kaneko S, Iwanami A, Nakamura M, Kishino A, Kikuchi K, Shibata S, Okano HJ, Ikegami T, Moriya A, Konishi O, Nakayama C, Kumagai K, Kimura T, Sato Y, Goshima Y, Taniguchi M, Ito M, He Z, Toyama Y, Okano H, A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord, Nat Med, 12 (2006) 1380–1389. [DOI] [PubMed] [Google Scholar]
- [157].Varcianna A, Myszczynska MA, Castelli LM, O’Neill B, Kim Y, Talbot J, Nyberg S, Nyamali I, Heath PR, Stopford MJ, Hautbergue GM, Ferraiuolo L, Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS, EBioMedicine, 40 (2019) 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, Spaltro G, Lidonnici D, Gensano F, Battaglia E, Bendotti C, Bonetto V, Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis, J Biol Chem, 288 (2013) 15699–15711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Ferraiuolo L, Higginbottom A, Heath PR, Barber S, Greenald D, Kirby J, Shaw PJ, Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis, Brain, 134 (2011) 2627–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S, Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons, Nat Neurosci, 10 (2007) 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Mackenzie IR, Rademakers R, Neumann M, TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia, Lancet Neurol, 9 (2010) 995–1007. [DOI] [PubMed] [Google Scholar]
- [162].Scotter EL, Chen HJ, Shaw CE, TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets, Neurotherapeutics, 12 (2015) 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC, Danzer KM, Weishaupt JH, TDP-43 is intercellularly transmitted across axon terminals, J Cell Biol, 211 (2015) 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Ding X, Ma M, Teng J, Teng RK, Zhou S, Yin J, Fonkem E, Huang JH, Wu E, Wang X, Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure, Oncotarget, 6 (2015) 24178–24191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D, Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD, Cell Rep, 17 (2016) 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, Megra B, Williams DW, Witwer KW, Gacias M, Tabatadze N, Cole RN, Casaccia P, Berman JW, Anthony DC, Haughey NJ, Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions, Sci Signal, 10 (2017) 473:eaai7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Hu G, Liao K, Niu F, Yang L, Dallon BW, Callen S, Tian C, Shu J, Cui J, Sun Z, Lyubchenko YL, Ka M, Chen XM, Buch S, Astrocyte EV-Induced lincRNA-Cox2 Regulates Microglial Phagocytosis: Implications for Morphine-Mediated Neurodegeneration, Mol Ther Nucleic Acids, 13 (2018) 450–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Batista PJ, Chang HY, Long noncoding RNAs: cellular address codes in development and disease, Cell, 152 (2013) 1298–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Briggs JA, Wolvetang EJ, Mattick JS, Rinn JL, Barry G, Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution, Neuron, 88 (2015) 861–877. [DOI] [PubMed] [Google Scholar]
- [170].Clifford DB, Ances BM, HIV-associated neurocognitive disorder, Lancet Infect Dis, 13 (2013) 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Masanetz S, Lehmann MH, HIV-1 Nef increases astrocyte sensitivity towards exogenous hydrogen peroxide, Virol J, 8 (2011) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Sami Saribas A, Cicalese S, Ahooyi TM, Khalili K, Amini S, Sariyer IK, HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity, Cell Death Dis, 8 (2017) e2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Lamers SL, Poon AF, McGrath MS, HIV-1 nef protein structures associated with brain infection and dementia pathogenesis, PLoS One, 6 (2011) e16659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Rahimian P, He JJ, Exosome-associated release, uptake, and neurotoxicity of HIV-1 Tat protein, J Neurovirol, 22 (2016) 774–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Yang L, Niu F, Yao H, Liao K, Chen X, Kook Y, Ma R, Hu G, Buch S, Exosomal miR-9 Released from HIV Tat Stimulated Astrocytes Mediates Microglial Migration, J Neuroimmune Pharmacol, 13 (2018) 330–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Hu G, Yao H, Chaudhuri AD, Duan M, Yelamanchili SV, Wen H, Cheney PD, Fox HS, Buch S, Exosome-mediated shuttling of microRNA-29 regulates HIV Tat and morphine-mediated neuronal dysfunction, Cell Death Dis, 3 (2012) e381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Huang Y, Li Y, Zhang H, Zhao R, Jing R, Xu Y, He M, Peer J, Kim YC, Luo J, Tong Z, Zheng J, Zika virus propagation and release in human fetal astrocytes can be suppressed by neutral sphingomyelinase-2 inhibitor GW4869, Cell Discov, 4 (2018) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Ibanez F, Montesinos J, Urena-Peralta JR, Guerri C, Pascual M, TLR4 participates in the transmission of ethanol-induced neuroinflammation via astrocyte-derived extracellular vesicles, J Neuroinflammation, 16 (2019) 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Biomarkers Definitions Working Group, Biomarkers and surrogate endpoints: preferred definitions and conceptual framework, Clin Pharmacol Ther, 69 (2001) 89–95. [DOI] [PubMed] [Google Scholar]
- [180].Boukouris S, Mathivanan S, Exosomes in bodily fluids are a highly stable resource of disease biomarkers, Proteomics Clin Appl, 9 (2015) 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Thompson AG, Gray E, Heman-Ackah SM, Mager I, Talbot K, Andaloussi SE, Wood MJ, Turner MR, Extracellular vesicles in neurodegenerative disease - pathogenesis to biomarkers, Nat Rev Neurol, 12 (2016) 346–357. [DOI] [PubMed] [Google Scholar]
- [182].Kapogiannis D, Boxer A, Schwartz JB, Abner EL, Biragyn A, Masharani U, Frassetto L, Petersen RC, Miller BL, Goetzl EJ, Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease, FASEB J, 29 (2015) 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Long Q, Upadhya D, Hattiangady B, Kim DK, An SY, Shuai B, Prockop DJ, Shetty AK, Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus, Proc Natl Acad Sci U S A, 114 (2017) E3536–E3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Kodali M, Castro OW, Kim DK, Thomas A, Shuai B, Attaluri S, Upadhya R, Gitai D, Madhu LN, Prockop DJ, Shetty AK, Intranasally Administered Human MSC-Derived Extracellular Vesicles Pervasively Incorporate into Neurons and Microglia in both Intact and Status Epilepticus Injured Forebrain, Int J Mol Sci, 21 (2020) E181. [DOI] [PMC free article] [PubMed] [Google Scholar]