

Abstract
Purpose
Glioblastoma multiforme is a highly aggressive form of brain cancer whose location, tendency to infiltrate healthy surrounding tissue, and heterogeneity significantly limit survival, with scant progress having been made in recent decades.
Experimental design
123I-MAPi (Iodine-123 Meitner-Auger PARP1 inhibitor) is a precise therapeutic tool composed of a PARP1 inhibitor radiolabeled with an Auger- and gamma-emitting iodine isotope. Here, the PARP inhibitor, which binds to the DNA repair enzyme PARP1, specifically targets cancer cells, sparing healthy tissue, and carries a radioactive payload within reach of the cancer cells’ DNA.
Results
The high relative biological efficacy of Auger electrons within their short range of action is leveraged to inflict DNA damage and cell death with high precision. The gamma ray emission of 123I-MAPi allows for the imaging of tumor progression and therapy response, and for patient dosimetry calculation. Here we demonstrated the efficacy and specificity of this small molecule radiotheranostic in a complex preclinical model. In vitro and in vivo studies demonstrate high tumor uptake and a prolonged survival in mice treated with 123I-MAPi when compared to vehicle controls. Different methods of drug delivery were investigated to develop this technology for clinical applications, including convection enhanced delivery (CED) and intrathecal injection.
Conclusions
Taken together, these results represent the first full characterization of an Auger-emitting PARP inhibitor which demonstrate a survival benefit in mouse models of GBM and confirm the high potential of 123I-MAPi for clinical translation.
Introduction
Glioblastoma multiforme (GBM) is one of the deadliest forms of solid tumors, with a 5-year survival rate as low as 5% (1). Clinical intervention typically consists of maximal safe surgical resection followed by adjuvant chemo-radiotherapy. This therapeutic regimen, unfortunately, imparts only limited improvements to survival (2). Additionally, the GBM molecular heterogeneity represents a robust challenge in need of better imaging tools that would allow for the monitoring of therapy response and lead to better and more personalized therapeutic plans. Furthermore, the presence of the blood-brain barrier (BBB) biochemically limits the pharmacokinetics of many GBM drug candidates.
A novel approach for treating GBM is therefore urgently needed, one that would address both pharmacodynamic as well as pharmacokinetic hurdles (3, 4). Promising delivery strategies to overcome these obstacles in the clinics include aligning novel targeting schemes with improved drug delivery approaches including convection enhanced delivery (CED) (5, 6) and intrathecal injection (7, 8).
A known molecular biomarker for most tumors, including GBM, is poly(ADP-ribose) Polymerase 1 (PARP1) (9–15). PARP1 is recruited to the nucleus of cancer cells and binds DNA as a single-strand break (SSB) repair enzyme (16). This central role has been successfully leveraged for the development of various PARP inhibitors, both as a monotherapy and in combination with other therapeutics (17, 18). Modified PARP inhibitors have also been widely used for tumor detection and imaging due to their cancer specificity and their high target-to-background contrast (14, 15, 19–25); more recently, they have found theranostic applications (26).
Here, we focus on PARP1-based radiotherapy with a more sporadically utilized type of radioactive emission: Auger radiation. Auger emitters are an extremely potent radioactive source for targeted radiotherapy, characterized by their greater linear energy transfer, incredibly short range, and ability to cause a complex, lethal DNA damage as compared to traditional X-rays or β-particles (27–32). Previous attempts to use Auger emitters as cancer therapies have not been successful, due to the limited range of the radiation emitted and the difficulty of reliably delivering the lethal electrons close enough to the DNA target (< 100 Å) (31, 33–35).
In this study, we developed and characterized 123I-MAPi, the first Auger-based theranostic PARP inhibitor able to directly deliver its lethal payload within a 50-angstrom distance of the DNA of GBM cancer cells (Fig. 1A). This distance is within the Auger radius of action, resulting in an effective preclinical cancer treatment drug and leading to improved survival in a preclinical GBM model. We used the 159 keV gamma ray to image tumor progression using SPECT imaging and calculate dosimetry and treatment efficacy using SPECT imaging.
Fig. 1.
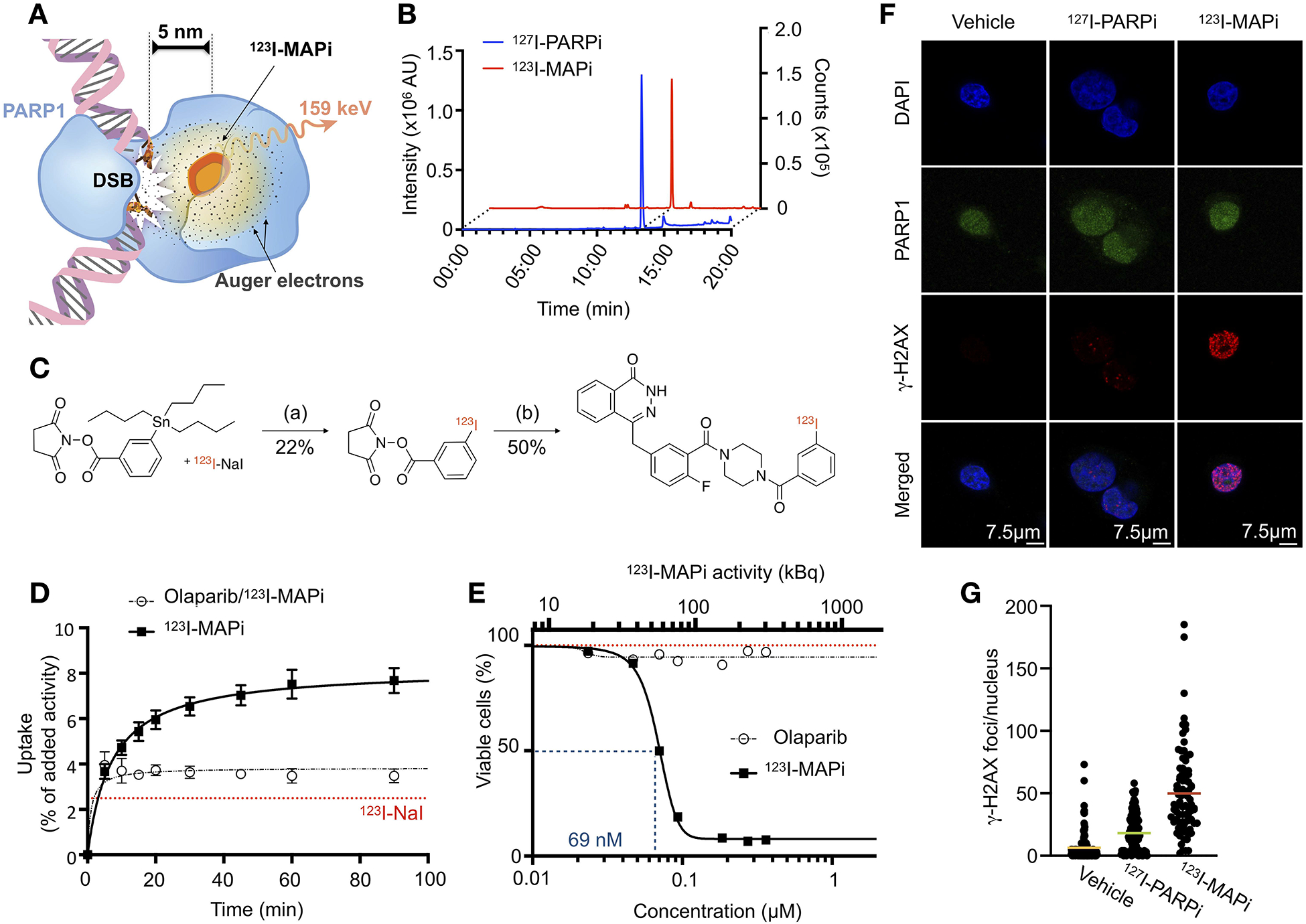
123I-MAPi Synthesis, binding, and efficacy in vitro. (A) 123I-MAPi binding to PARP1 can deliver lethal Auger radiation within ~100 Å, enough to affect DNA in cancer cells when bound. (B) RP-HPLC coelution of 123I-MAPi with nonradioactive I-127 analog. (C) Synthetic scheme of 123I-MAPi, (a) 0.1 M NaOH, Chloramine T, AcOH, MeOH 20 m, RT; (b) 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one, HBTU, DMAP, 2,6-Lutidine, DMSO, ACN, 2 h, 65 °C. (D) Cellular uptake of 123I-MAPi in U251 GBM cell line. Blocking performed with 100-fold incubation of Olaparib prior to treatment. Sodium Iodine-123 represented by red line. Michaelis-Menten curve fitting. (E) Alamar Blue assay comparing 123I-MAPi efficacy with Olaparib at equal molar concentrations. 123I-MAPi EC50 = 69 nM. Nonlinear fit four parameters variable slope. (F) DNA damage characterization in vitro showing immunofluorescence of γ-H2AX (red), DAPI nuclear staining (blue), PARP1 expression (green), and merged images. (G) Quantification of the number of γ-H2AX foci within the nucleus of cells after treatment with 123I-MAPi (n = 85), 127I-PARPi (n = 125), and vehicle control (n = 140). ***p-value < 0.001, Kruskal-Wallis test.
Taken together, these results illustrate the tremendous potential of 123I-MAPi as an Auger-emitting PARP inhibitor and a theranostic agent for GBM treatment.
Materials and Methods
General.
Sodium [123I]iodide in 0.1 N NaOH with a specific activity of 7.14 ×107 GBq/g was purchased from Nordion (Ottawa, Canada). 4-(4-fluoro-3-(4-(3-iodobenzoyl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one was synthesized as described previously (36). Olaparib (AZD2281) was purchased from LC Laboratories (Woburn, MA). PARPi-FL was synthesized as previously described (14, 37). Water (>18.2 MΩcm−1 at 25 °C) was obtained from an Alpha-Q Ultrapure water system (Millipore, Bedford, MA) and acetonitrile (AcN) as well as ethanol (EtOH) were of HPLC grade purity. Sterile 0.9% saline solution (Hospira, Lake Forest, IL) was used for all in vivo injections. High performance liquid chromatography (HPLC) purification and analysis was performed on a Shimadzu UFLC HPLC system equipped with a DGU-20A degasser, an SPD-M20A UV detector, a LC-20AB pump system, and a CBM-20A Communication BUS module. A LabLogic Scan-RAM radio-TLC/HPLC-detector was used to detect activity. HPLC solvents (Buffer A: Water, Buffer B: AcN) were filtered before use. Purification of 2,5-dioxopyrrolidin-1-yl 3-(iodo-123I)benzoate was performed with Method 1 (flow rate: 1 mL/min; gradient: 15 min 5–95% B; 17 min 100% B; 20 min 100%−5% B); QC analysis was performed with Method 1. Method 1 was performed on a reversed phase C18 Waters Atlantis T3 column (C18-RP, 5 μm, 6 mm, 250 mm). Purification of the final product was performed on a C6 Waters Spherisorb Column (C6, 5 μm, 4.6 mm × 250 mm) with Method 2 (flow rate: 1.5 mL/min; isocratic: 0–30 min 35% B.
Cell Culture.
The human glioblastoma cell line U251 was kindly provided by the laboratory of Dr. Blasberg (MSK, New York, NY). Cells were grown in Eagle’s Minimal Essential Medium (MEM), 10% (vol/vol) heat inactivated fetal bovine serum, 100 IU2 penicillin, and 100 μg/ml streptomycin, purchased from the culture media preparation facility at MSK (New York, NY). TS543 cells are a patient-derived glioblastoma stem line kindly provided by the laboratory of Dr. Mellinghoff (MSK, New York, NY). These cells were grown in suspension in NeuroCultTM NS-A Proliferation Kit with proliferation supplement (StemCell Technologies, Cat 05751), 20 ng/mL Recombinant Human Epidermal Growth Factor (EGF) (StemCell Technologies, Cat 02633), 10 ng/mL Recombinant Human Basic Fibroblast Growth Factor (Bfgf) (StemCell Technologies, Cat 02634), 2 μg/mL Heparin (StemCell Technologies, Cat 07980), 1× antibiotic-antimicotic (Life Technologies Gibco, Cat 15240–062), 2.5 mg/mL Plasmocin (InvivoGen, Cat ant-mpp). All cells were tested for mycoplasma contamination.
Immunofluorescence.
Cells were plated on a coverslip slide on the bottom of a 6-well plate and incubated overnight. 123I-MAPi, 127I-PARPi, or vehicle control were added to the media and cells were returned to the incubator overnight. Cells were then fixed in 4% paraformaldehyde/PBS, permeabilized and stained with anti γ-H2AX antibody (Millipore Sigma, 05–636) and anti PARP1 antibody (Invitrogen, PA5–16452). DAPI was used to localize nuclei. Coverslips were mounted on slides for microscopy imaging.
Immunoblotting.
Cells were lysed in RIPA (Thermofisher Scientific, cat #89900) buffer containing protease inhibitor at 4 °C. Lysates were run on an SDS-Page gel (BioRad). Bound antibodies were detected by developing film from nitrocellulose membranes exposed to chemiluminescence reagent (#34077, Thermo Scientific). PARP1 primary antibody (Santa Cruz #sc-7150, 0.2 μg/mL) and goat anti-rabbit IgG-HRP secondary antibody (1:10000 dilution, sc-2004, SantaCruz). An anti-β-actin antibody (Sigma #A3854, 1:1000) was used as loading control.
Synthesis of 123I-MAPi.
123I-MAPi was obtained similar to synthetic procedures reported before (26). Firstly, 2,5-dioxopyrrolidin-1-yl 3-(iodo-123I)benzoate was obtained by adding N-succinimidyl-3-(tributylstannyl) benzoate (250 μg, 0.5 μmol) in 10 μL of AcN to a solution containing methanol (40 μL), chloramine T (9 μg, 32 nmol), acetic acid (3 μL) and 123I-NaI in NaOH 0.1 M (2.5 mCi). After the reaction solution was driven for 20 min at room temperature, the reaction was purified by HLPC (Method 1), and 2,5-dioxopyrrolidin-1-yl 3-(iodo-123I)benzoate at 15.1 min. The collected purified fraction was concentrated to dryness in vacuum, reconstituted in a solution of 80 μL AcN and added to a solution of 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (0.3 mg, 0.9 μmol) in 20 μL DMSO, N,N,N’,N’-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) (0.3 mg, 0.8 μmol) in 20 μL DMSO, 4-Dimethylaminopyridine (DMAP) (0.1 mg, 0.8 μmol) in 20 μL DMSO and 10 μL triethylamine. The reaction mixture stirred in an Eppendorf ThermoMixer® for 2 h at 65 °C (500 rpm). Afterwards, the reaction mixture was injected and purified by HPLC (Method 2) and 123I-MAPi was collected at RT = 25.5 min (RY > 70%; RP > 99%) and concentrated to dryness under vacuum. 123I-MAPi was formulated with 30% PEG300 / 70% saline (0.9% NaCl) for both in vitro and in vivo assays. Co-elution with nonradioactive 127I-PARPi reference compound confirmed the identity of the radiotherapeutic. 123I-MAPi was synthesized with molar activity of 3.93 ± 0.10 GBq/μmol.
Internalization assay.
Uptake of 123I-MAPi was tested in vitro (3 replicates). 5×105 U251 cells were plated 24h prior to the experiment (n = 3). Media was changed and 1 h later 3.7 kBq of 123I-MAPi were added to the cells. For blocking, cells were incubated with a 100-fold molar excess of Olaparib 1 h before adding 123I-MAPi. Media was removed, and cells were washed with PBS and lysed (1 M NaOH) at different time points. The lysate was collected, and uptake was determined by radioactivity on a gamma counter.
Viability assay.
U251 GBM cells were incubated with 0 – 296 kBq of 123I-MAPi or equivalent dose of Olaparib overnight and then washed and incubated for 4 d in normal media (3 replicates, n = 3 each). Viability was determined by AlamarBlue assay as indicated by the manufacturer.
Apoptosis assay.
U251 GBM cells were incubated with 370 kBq of 123I-MAPi and apoptosis was detected at 1- and 24-hours post treatment using an in situ Direct DNA Fragmentation (TUNEL) Assay Kit (Abcam, ab66110) and following manufacturer’s instructions.
Colony formation assay.
Colony formation assay (CFA) was performed using U251 cells. Cells were plated as single cell suspensions and left to attach for 8 h prior to irradiation or treatment (n = 3). Colony formation was measured at two weeks post 123I-MAPi treatment and compared to EBIR. Cells were treated adding 0 – 23 kBq of compound in each well. Colony count was normalized on plating efficiency at 0 kBq for each treatment. External beam irradiation (EBIR) was performed using a Shepperd Irradiator Cs-137 at Memorial Sloan Kettering Cancer Center, New York, NY, USA. Radiobiological parameters of the linear-quadratic model were determined via non-linear regression within GraphPad software, and relative biological effectiveness was determined by interpolation, using 37% survival as the endpoint.
Animal Work.
All in vivo experiments were performed with female athymic nude CrTac:NCr-Fo mice were purchased from Taconic Laboratories (Hudson, NY) at age 6–8 weeks. During subcutaneous injections, mice were anesthetized using 2% isoflurane gas in 2 L/min medical air. 1×106 TS543 cells were injected in the right shoulder subcutaneously in 150 μL volume of 50% media/matrigel (BD Biosciences, San Jose, CA) and allowed to grow for approximately two weeks until the tumors reached about 8 mm in diameter (100 ± 8 mm3).
For intravenous injections, the lateral tail vein was used. Mice were warmed with a heat lamp, placed in a restrainer, and the tail was sterilized with alcohol pads before injection.
For orthotopic injections, TS543 cells (5×105 cells in 2 μL growth media) were injected 2 mm lateral and 1 mm posterior to the bregma using a Stoelting Digital New Standard Stereotactic Device and a 5 μL Hamilton syringe and allowed to grow for three weeks before treatment. All mouse experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of MSK and followed National Institutes of Health (NIH) guidelines for animal welfare.
SPECT/CT imaging.
SPECT/CT scans were performed using a small-animal NanoSPECT/CT from Mediso Medical Imaging Systems (Budapest, HU). For subcutaneous TS543 xenografts, 123I-MAPi (2.3 ± 0.5 MBq in 20 μL 30% PEG300 in 0.9% sterile saline) was administered intratumorally as single injection. At chosen time points post injection, the mice were anesthetized with 1.5–2.0% isoflurane (Baxter Healthcare) at 2 mL/min in oxygen and SPECT/CT data was acquired for 60 min.
For orthotopic TS543 tumor-bearing mice 123I-MAPi (614.2 kBq in 5 μL 30% PEG300 in 0.9% sterile saline) was injected intracranially using the same coordinates as for tumor cell injection (2 mm lateral and 1 mm posterior to the bregma using a Stoelting Digital New Standard Stereotactic Device and a 5 μL Hamilton syringe). At chosen time points post injection, the mice were anesthetized with 1.5–2.0% isoflurane (Baxter Healthcare) at 2 mL/min in oxygen. SPECT/CT data was collected for 60 min.
Ex-vivo biodistribution for intratumoral administration route.
Biodistribution studies were performed in subcutaneous TS543 xenograft-bearing mice. Mice were randomized and divided in two groups (blocked and unblocked, ntotal = 6) and 123I-MAPi was administered intratumorally (average injected activity 1.702 ± 0.629 MBq in 20 μL, 30% PEG300 in 0.9% sterile saline). The blocked group was pre-injected (1 mg/mouse in 100 μL 30% PEG300 / 70% NaCl 0.9%) 60 min prior to treatment with Olaparib (100mM stock in DMSO). Mice were sacrificed by CO2 asphyxiation at 18-h post injection and counted in a WIZARD2 automatic γ-counter (PerkinElmer, Boston, MA). Uptake was expressed as a percentage of injected dose per gram (% ID/g) using the following formula: [(activity in the target organ/grams of tissue)/injected dose].
Survival studies.
Mice were inoculated orthotopically at Week 0 with TS543 cells (5×105 cells in 2 μL growth media). Cells were injected 2 mm lateral and 1 mm posterior to the bregma using a Stoelting Digital New Standard Stereotactic Device and a 5 μL Hamilton syringe and allowed to grow for three weeks before treatment. At Week 3 mice were randomly grouped into cohorts, and intratumorally injected with 123I-MAPi or 30% PEG300 in 0.9% sterile saline vehicle using the same stereotactic coordinates as for tumor implantation. Mice were monitored daily thereafter. Study endpoint was determined based on animals’ sign of discomfort, pain, or significant weight loss.
Orthotopic CED model.
ALZET® Osmotic Pumps were implanted subcutaneously into the mice to slowly deliver an infusion into the brain using the same coordinates as for the tumor cell injection as previously described (26). Control mice received an ALZET® Osmotic Pump Model 1003D with Brain Infusion Kit 3 containing 30% PEG/PBS vehicle. Treatment mice received 123I-MAPi (average pump activity 481 ± 111 kBq in 100 μL, 30% PEG300 in 0.9% sterile saline. Delivery flow 1 μL/h, over 5 d). Pumps were surgically removed 5 days post implantation and mice monitored daily thereafter.
Intrathecal injection.
Brain-tumor-bearing mice were anesthetized and injected in the intrathecal space (injection site from L3/5 or L4/5 to prevent spinal cord injury) 2.0 MBq of 123I-MAPi in 50 μL 30% PEG300 in 0.9% sterile saline 123I-MAPi were injected and the mice imaged 1-h post injection.
Hematoxylin–Eosin (H&E) staining.
Brains were collected from mice at the time of death. The collected brains were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA), flash-frozen in liquid nitrogen and cut into 10 μm sections using a Vibratome UltraPro 5000 Cryostat (Vibratome, St. Louis, MO). Sections were subsequently subjected to H&E staining for morphological evaluation of tissue pathology.
In vivo γ-H2AX analysis.
Mice were injected intracranially at four weeks post tumor implantation with either 123I-MAPi or vehicle for control (n = 3 per cohort). Mice were perfused 1-hour post injection and brains were fixed in 4% paraformaldehyde. Brains were then sliced and stained for γ-H2AX as a marker for double-strand breaks.
Liver enzymes analysis.
Animals (n = 5/cohort) were injected systemically with either 123I-MAPi or Vehicle control. After 24 hours blood was collected via retro-orbital bleeding. Enzymes levels were determined by the Antitumor assessment core at Memorial Sloan Kettering Cancer Center.
Organ-level and cell level dosimetry.
Dosimetry calculations are described in detail in the supplemental material.
Results
Synthesis of 123I-MAPi and in vitro validation.
We previously showed that it is possible to successfully conjugate a PARP inhibitor with radioiodine without altering the high affinity to the target – maintaining an IC50 in the nanomolar range (11 ± 3 nM) and a logPCHI of 2.3 (38). 123I-MAPi is a novel, previously unreported isotopologue, and was synthesized with a final molar activity of 3.93 ± 0.10 GBq/μmol. Radiochemical purity was 99.1 ± 0.9% for all prepared compounds (Fig. 1B).
Pharmacological properties determined with 127I-PARPi suggest that 123I-MAPi (Fig. 1C, S1A), retains the same properties as 131I-PARPi, which have been shown to be similar to the FDA-approved PARP inhibitor Olaparib (26, 38).
In vitro internalization was tested on U251 cells expressing PARP1 (Fig. S1B). 123I-MAPi (37 kBq/well) was added to adherent cells in monolayer and uptake was calculated by measuring gamma radiation in cell lysates at different time points. We confirmed rapid cellular internalization, with 50% of the final total uptake being reached after 5 – 10 minutes post incubation and with an uptake plateau reached at 1-h post treatment (Fig. 1D). The final total uptake was ~8% of added activity, Vmax = 8.2 ± 0.2 % compared to blocked uptake, Vmax = 3.8 ± 0.1 % (Michaelis-Menten fit, R2 = 0.974 and R2 = 0.879, respectively). Uptake was blocked with a 100-fold excess dose of Olaparib to show target specificity. Na-123I was used as a control and showed significantly lower uptake, Vmax = 2.5 ± 0.1. The two dominant targets for 127I-PARPi, the stable-isotope labelled form of the molecule, are PARP1 and PARP2 (26), similar to what has been previously reported for Olaparib and other modified PARP inhibitors (15).
We tested the cancer-specific efficacy of 123I-MAPi in vitro by treating U251 GBM cells with 123I-MAPi. Comparing 123I-MAPi with the previously-published beta-emitting 131I-PARPi, we observed a 16-fold increase in EC50 potency (EC50 of 131I-PARPi being 1148 ± 1 nM, Fig. S1C). 123I-MAPi treatment proved to reduce viability at safe concentrations of 123I-NaI (Fig. S1D and S1E). Critically, in the molar concentration range used in these studies, Olaparib showed negligible effect in terms of cell viability, suggesting that clonogenic inactivation arises exclusively from I-123 radiotoxicity as opposed to PARP1 inhibition (Fig. 1E). 123I-MAPi proved to be capable of killing cancer cells with a sub-micromolar EC50 (EC50 = 68.9 ± 1.1 nM, R2 = 0.999). At the same concentrations, Olaparib did not show any effect in terms of cell viability, suggesting that the observed effect is due to the PARP1 inhibitor-mediated close proximity to the target DNA.
In order to characterize the induction of DNA damage in cancer cells after treatment with 123I-MAPi, we performed immunofluorescence analysis of the levels of γ-H2AX foci, a known marker for double-strand breaks (39). We treated U251 GBM cells with 740 kBq 123I-MAPi in 20 μL of 30%PEG/PBS added to the media in each well (2 mL) and compared it to the equivalent dose of non-radioactive 127I-PARPi and vehicle control (Fig. 1F, and S2A). 123I-MAPi treated cancer cells showed a significantly higher number of γ-H2AX foci in the cell nucleus (***p-value < 0.001, Kruskal-Wallis test), Fig 1G. We performed a TUNEL assay to detect apoptosis after treatment with 123I-MAPi in vitro and detected increased levels of apoptotic cells at 24-h post treatment as compared to control (Fig. S2B, *p-value < 0.05).
Efficacy of 123I-MAPi in a colony formation assay.
We tested 123I-MAPi in comparison with externally applied photon radiation, assessing clonogenic survival. Colony formation assays (CFAs) were performed in 6-well plates to compare efficacy of 123I-MAPi in cell killing when compared to standard external beam irradiation (EBIR). U251 GBM cells were plated and treated with Cs-137 γ-rays (662 keV) or by adding 123I-MAPi to the wells (Fig. S3A).
As expected with high LET radiation treatment, cells treated with the Auger-emitting small molecule showed a steep decline in survival, confirming the high efficacy in tumor cell killing when compared to EBIR (Fig. S3B). We derived first-order estimates of relative biological effectiveness of on-target 123I-MAPi through cell dosimetry calculations (Supplemental Information), which suggest an increase in therapeutic potency when 123I-MAPi is bound to PARP1 (i.e. when DNA is within reach of its Auger emissions). Radiobiological parameters were calculated after linear-quadratic model fit of the data: 123I-MAPi α = 19.8 Gy−1, β = N/A. EBIR α = 0.269 Gy−1, β = 0.0588 Gy−1. 123I-MAPi D37 = 0.05 Gy, EBIR D37 = 2.41 Gy. Relative biological effectiveness (RBE) was calculated for 123I-MAPi: RBE at D37 = 48.4, RBE (αI-123/ αEBIR) = 73.4 (Table S1).
Biodistribution and toxicity of 123I-MAPi in vivo.
TS543 patient-derived glioblastoma stem cells were used to grow tumors in athymic nude mice in order to investigate the biodistribution of 123I-MAPi. Tumors were grown subcutaneously on the animals’ right shoulder. Mice were then randomized and divided into two cohorts (n = 3/group) one of which was used for blocking. Blocking was performed systemically with an intravenous injection of Olaparib 1 h prior to 123I-MAPi treatment. 123I-MAPi was injected intratumorally for this model. SPECT/CT images showed that 123I-MAPi tumor uptake was retained at 1 h, 6 h and 18 h post injection in non-blocked animals, but was strongly reduced at 6 h and 18 h in blocked animals (Fig. 2A, Fig. S4A). Biodistribution at 18 h was also examined and showed higher tumor uptake (33.4 ± 28.0 % ID/g) compared to the tumor of blocked animals (0.4 ± 0.1 % ID/g) (Fig. 2B). Tumor-to-muscle ratios in 123I-MAPi treated mice versus blocked mice were > 500 and 5, respectively, with less than 1% ID/g in all clearing organs.
Fig. 2.
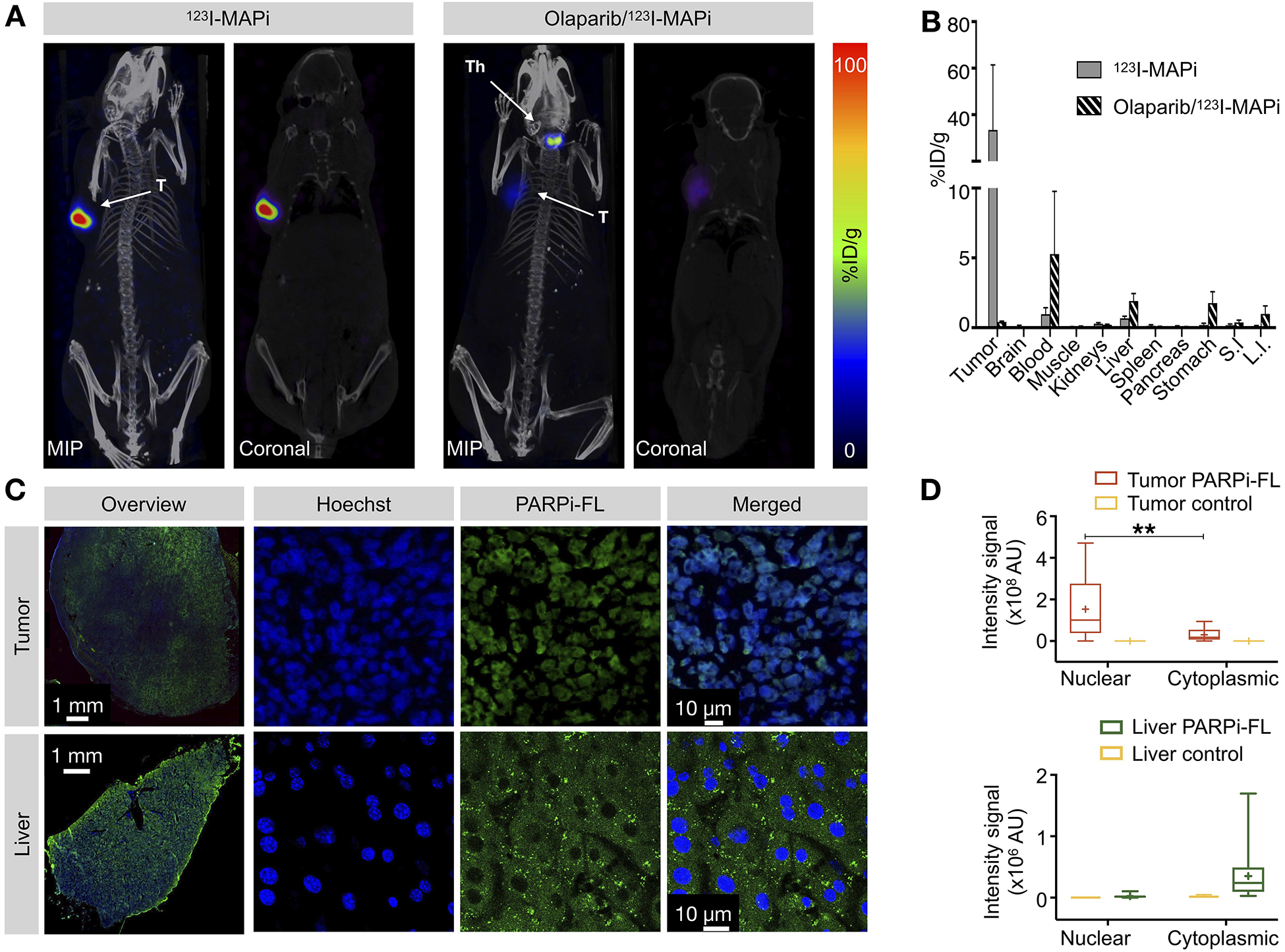
Imaging and biodistribution of 123I-MAPi in subcutaneous TS543 mouse model. (A) Specific tumor uptake of 123I-MAPi at 18 h after local injection. Blocking was performed intravenously 1 h before injection. T = tumor, Th = thyroid. (B) Ex vivo biodistribution of 123I-MAPi at 18 h after local injection. (C) Uptake of PARPi-FL in tumor and liver of TS543 tumor-bearing mice show nuclear uptake for tumor and cytoplasmic uptake for liver tissue. (D) Quantification of PARPi-FL uptake in the nucleus and cytoplasm of tumor and liver tissue. Tumor control was performed by IV injection of blocking Olaparib dose. Liver control was performed by injection of vehicle. Nuclear vs cytoplasmic uptake was automatically detected using an ImageJ script to detect colocalization of Hoechst (IV injected 5 minutes before extracting the organs) and PARPi-FL. Student t-test, **p-value < 0.01, ***p-value < 0.001.
Auger particles are highly cytotoxic when they can directly interact with the DNA and cause complex damage. However, they are significantly less so in the cellular cytosol, where they are beyond the reach of their DNA target (27). As liver clearance is the main route of excretion of 123I-MAPi that could cause dose-limiting problems in the clinic, we investigated potential clinical liver failure by observing specific liver toxicity in our preclinical study. We compared tumor and liver accumulation of PARPi-FL, a thoroughly characterized fluorescent analogue of the same PARP1 inhibitor with comparable biodistribution and tumor uptake after intravenous injection (14, 15, 20, 38, 40). PARPi-FL was injected intravenously (50 μg per mouse) 2 h before collecting the animals’ tumor and liver (Fig. 2C and Fig. S5A). Organs were then sectioned, and tissue sections digitalized. Hoechst (150 μL/mouse of 10 mg/mL) was used for nuclear counterstaining. PARPi-FL accumulated in the nucleus of GBM cells as confirmed by co-localization with Hoechst signal. Livers were collected and imaged with an inverted confocal microscope. PARPi-FL accumulation was observed in the cytoplasm of liver cells (Fig. 2D), suggesting that liver cells are protected from Auger-toxicity. In order to confirm this, we performed an enzyme analysis assay in the blood of mice injected with and without prior injection of 123I-MAPi. 1.09 ± 0.04 MBq were injected in n = 5 mice for the treated cohort, whereas the vehicle control cohort (n = 5) received 150 μL of 30% PEG/PBS. No significant variations were observed in treated mice compared with control (Fig. S5B), suggesting a limited systemic toxicity of the injected molecule, likely due to the large distance of the Auger emitter from the DNA (Fig. S5C).
Imaging of an orthotopic GBM model using 123I-MAPi.
To test 123I-MAPi in a more realistic model of GBM, we orthotopically implanted TS543 cells into the right brain hemisphere. This GBM model proved to be very consistent in growth and take rate, as we monitored by MRI imaging of the head. 50,000 cells were injected at Week 0, which resulted in rapid disease progression leading to animal death at Week 7 (Fig. 3A). Based on this data we decided to deliver 123I-MAPi treatment at Week 3. 123I-MAPi was injected intratumorally using the same stereotactic coordinates as for tumor implantation. Animals were then imaged at 1-h and 18-h post treatment. Full body SPECT/CT images were acquired, showing retention of 123I-MAPi in the tumor at 18-hours post-injection (Fig. 3B and Fig. S6A).
Fig. 3.
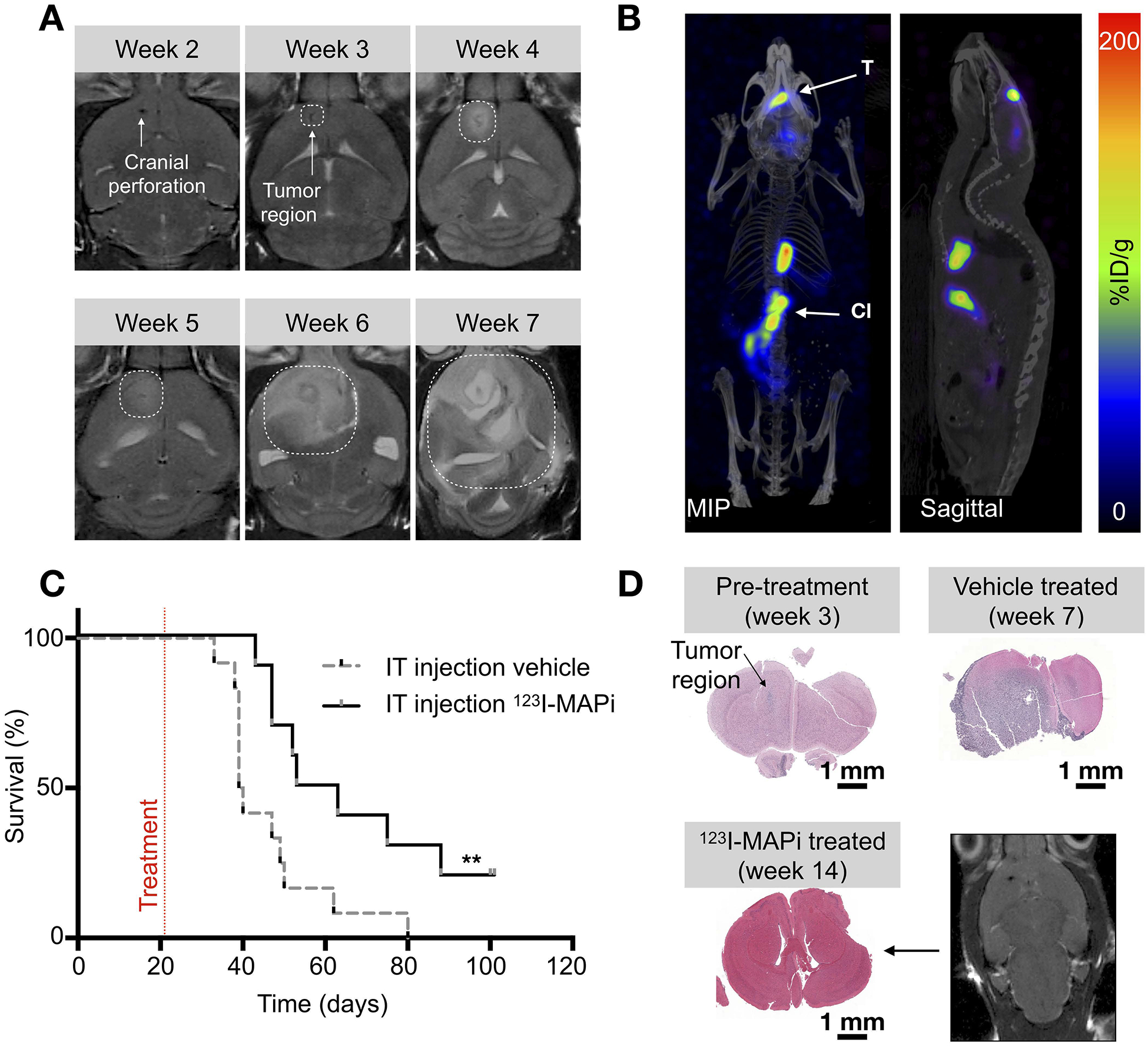
TS543 glioblastoma stem-cell mouse model and 123I-MAPi efficacy. (A) MRI monitoring of TS543 xenograft disease progression. (B) SPECT/CT imaging of GBM with 123I-MAPi single injection. Images were taken at 18 h after local injection. T = tumor, Cl = clearing organs. (C) Kaplan-Meier curve of mice injected with 123I-MAPi (local single injection 370 kBq - 1.11 MBq, n = 10) compared to vehicle injection (n = 12). Log-Rank (Mantel-Cox) test, **p-value < 0.01. (D) Cytology staining of untreated mice at Weeks 3 and 7 after tumor implantation and cytology staining and MRI imaging of treated mouse brain at 14 weeks post implantation.
Therapeutic efficacy of 123I-MAPi in a GBM mouse model.
We monitored the clinical potential utility of 123I-MAPi in treating GBM in the above described orthotopic mouse model. 123I-MAPi-treated mice were then monitored daily, using Day 98 (the end of Week 14), measured from the day of xenografting, as the study endpoint. We injected an intratumoral single dose (0.37 – 1.11 MBq) of 123I-MAPi for the treated cohort (n = 10) and the same volume of vehicle for the control cohort (n =12). Survival data confirmed an improved survival for the 123I-MAPi treatment cohort, with a median survival of 58 days as opposed to 40 days observed for the control cohort. Log-rank curve comparison showed a significant difference, with p-value = 0.009 (Fig. 3C, Table 1). Animals brains were imaged ex vivo with H&E staining and in vivo with MRI showing a diffuse presence of cancer cells in vehicle treated mice as opposed to the treatment (Fig. 3D). Mice brains (n = 3 per cohort) were also stained for the presence of γ-H2AX foci. We found a significantly increased number of DSB at 1-hour post intratumoral injection of 123I-MAPi when compared to vehicle (Fig. S6B and Fig. S6C, Kruskal-Wallis test ****p-value < 0.0001).
Table 1.
Survival of single dose treatment with 123I-MAPi.
| n | Median surv. (days) | p-value† | |
|---|---|---|---|
| IT injection vehicle | 12 | 50 | |
| IT injection 123I-MAPi | 10 | 58 | ** 0.0094 |
Log-Rank (Mantel-Cox) test.
Improved delivery of 123I-MAPi for clinical translation.
In clinics, to improve biodistribution toward a more effective targeted therapy, the radiopharmaceutical can be administered directly into the tumor compartment. For brain tumors this is achieved through intra-tumoral injection, intra-thecal injection, or convection enhanced delivery (CED), an approach that elevates the injection pressure so as to impel the agent across the BBB. To allow for clinical translation of 123I-MAPi, we first built a preclinical model of CED. We implanted an ALZET® osmotic delivery pump with a subcutaneous catheter in tumor-bearing mice. This delivered the content of the subcutaneous reservoir at a flow rate of 1 μL/h over the course of approximately 100 h through a cannula connected to the mouse brain. Subcutaneous reservoirs were filled with 100 μL of 123I-MAPi for the treatment cohort (n = 8) and with 100 μL of vehicle for the control cohort (n = 8). Five days post-implantation, we surgically removed the pumps, as per manufacturer instruction, and monitored mice survival. Kaplan-Meier survival plots confirmed the therapeutic efficacy of 123I-MAPi: treated mice presented a median survival of 72 d as opposed 48 d in the control cohort, a statistically significant difference of 50% (Log-rank p-value = 0.0361, Fig. 4A and Table 2). Dosimetry for the CED experiments (Fig. 4B, and S6D) confirmed accumulation in the tumor with minimal absorbed dose to healthy organs not in direct proximity to the implanted pump (Fig. 4C). Importantly, while not feasible in a mouse model, normal organ doses would be significantly reduced in a corresponding clinical scenario where the radionuclide reservoir would be external, shielded, and the administration performed over a shorter timescale.
Fig. 4.
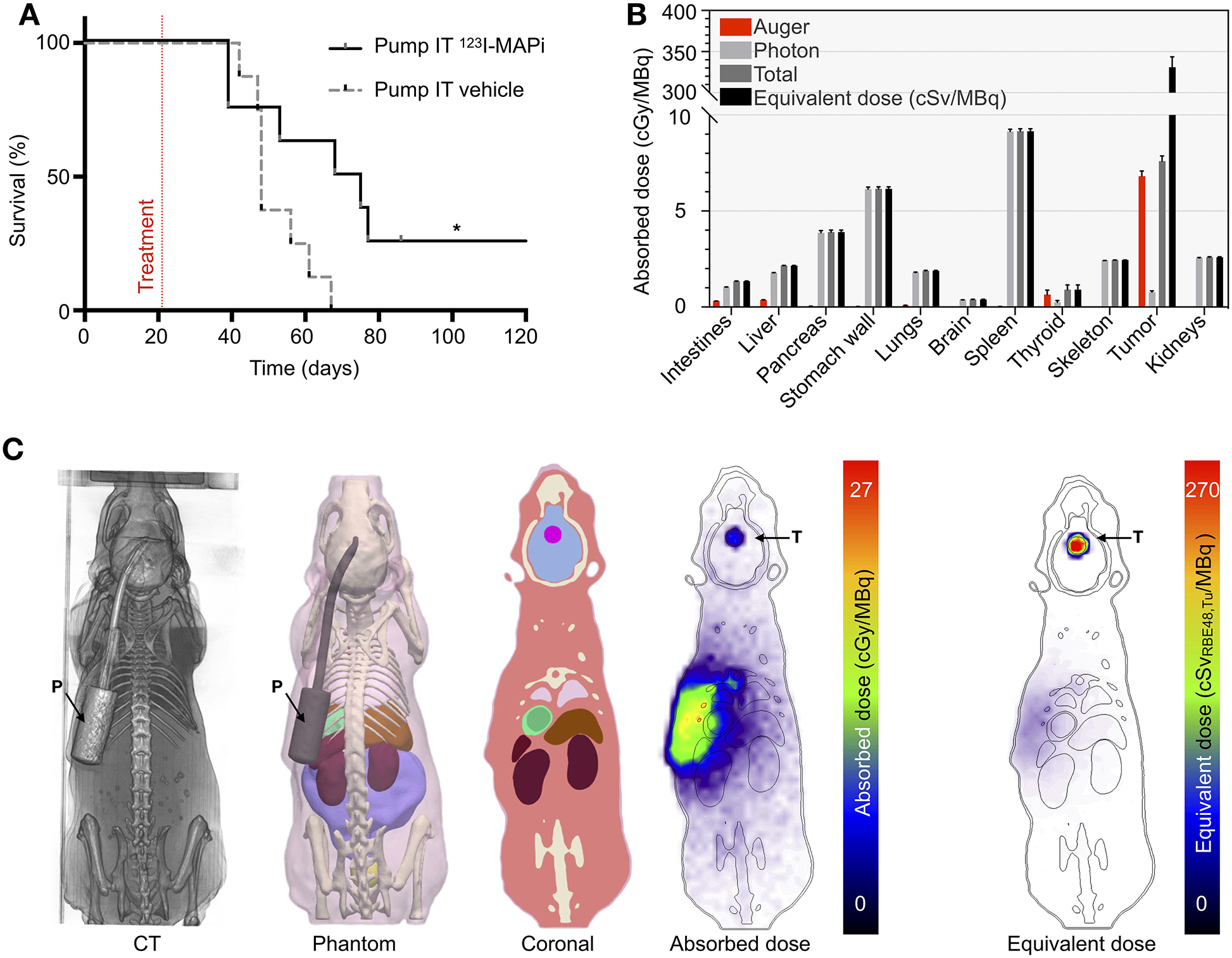
Improved delivery of 123I-MAPi with an in vivo CED model. (A) Kaplan-Meier survival study of pump implanted mice shows an improvement of survival of 123I-MAPi treated mice (n = 8) when compared to control (n = 8). Treatment mice osmotic pumps were loaded with 481 ± 111 kBq. Log-Rank (Mantel-Cox) test, *p-value < 0.05. (B) Organ-level and (C) 3D dosimetry estimates calculated by Monte Carlo simulation for subcutaneous pump administration. Equivalent doses assume the measured deterministic RBE of 48.4 for 123I-MAPi tumor tissue and assume RBE of 1.0 elsewhere. T = tumor, P = pump.
Table 2.
Survival of CED dose treatment with 123I-MAPi.
| n | Median surv. (days) | p-value† | |
|---|---|---|---|
| IT injection vehicle | 8 | 48 | |
| IT injection 123I-MAPi | 8 | 72 | * 0.0361 |
Log-Rank (Mantel-Cox) test.
Intrathecal injection of 123I-MAPi for a further simplified clinical translation.
We then investigated a more broadly used and technically feasible brain drug delivery method for delivering 123I-MAPi across the BBB. Tumor-bearing mice were injected with 50 μL of 123I-MAPi intrathecally at 3 weeks after orthotopic tumor implantation. SPECT/CT imaging was performed at 1-h post injection in order to visualize 123I-MAPi accumulation in the brain (Fig. S7A). Imaging confirmed favorable pharmacokinetics and specific tumor uptake, suggesting intrathecal delivery as a feasible technique for our small molecule. Unfortunately, this technique leads to surgery-related high levels of stress in the mice model preventing us from being able to monitor survival.
Discussion
In the present work, we show the results for the first preclinical characterization of an Auger-emitting theranostic PARP inhibitor. We present a functioning workflow for the synthesis of a stable 123I-MAPi compound which proved to be effective in vitro at reducing the viability of GBM cell lines. The lethality of 123I-MAPi suggests that the DNA is in range of the Auger electrons when it is complexed with PARP1. It was further established that GBM cell death is induced through radiogenic damage rather than PARP inhibition at these tracer levels.
In vivo studies with human tumor xenografts models also show 123I-MAPi drug treatment efficacy. The 159 keV gamma emission of I-123 allowed for quantification of drug uptake in tumor-bearing mice by SPECT/CT imaging. The isotope is ideal isotope for a proposed theranostic PARPi clinical agent because of its widespread use in nuclear medicine for thyroid disorders (Na123I) and pediatric tumors (123I-MIBG). Animal studies employing the non-radioactive drug Olaparib show prolongation of blood pool clearance and blocked tumor uptake, proving 123I-MAPi binding specificity.
To illustrate the potential advantages of clinical translation of 123I-MAPi, and to better illustrate the impact of Auger therapeutics, we looked at the agent’s potential subcellular biodistribution. In vitro γ-H2AX analysis showed a significantly increased number of double-strand breaks in cells treated with 123I-MAPi when compared to 127I-PARPi or vehicle control (***p-value < 0.001, Kruskal-Wallis test, Fig. S2D). Furthermore, by looking at the foci morphology, it is possible to observe larger and more clustered foci in the 123I-MAPi treated samples as compared to spontaneously-induced and background foci (Fig. S2A, S2B, S2C). These observations were also confirmed in vivo, showing elevated levels of γ-H2AX foci post 123I-MAPi treatment in GBM cells when compared to control (Fig. S6C). In this study, we did not notice any variation in the levels of PARP1 (green staining) as expected due to a positive feedback loop of self-amplification of PARP1 expression (21). For this reason, we decided to not include a potential PARP1 amplification effect in the kinetic modelling for dosimetry studies. We performed a colony formation assay to test efficacy of 123I-MAPi in U251 cells. The obtained data shows a steep drop in survival, which was expected due to the high-LET nature of Auger electrons. Comparison with EBIR allowed us to calculate the RBE, which corroborated the potential of Auger-emitting molecules for targeted radiotherapy, a fast-growing and promising field (41, 42). This proved to be a significant improvement compared to a previously-published version of the molecule, 131I-PARPi, where the RBE of the emitted electron is significantly lower. There has been a prior Phase I/II clinical trial in which the therapeutic effects of two antibodies targeting colon cancer were studied (131I-A33 (43) and 125I-A33 (29)). I-125 is an Auger electron emitting radionuclide with comparable properties to I-123 (44). This study showed that the maximum tolerated activity for 131I-A33 was 2.78 GBq/m2 (75 mCi/m2) in heavily pretreated patients, whereas for 125I-A33, bone marrow toxicity was not seen after administered activities as high as 12.95 GBq/m2 (350 mCi/m2). In this study as in the A33 trial, it is expected that the short-range emissions from I-123 will result in far lower normal tissue toxicity (including dose limiting organs). In vivo, Olaparib-based PARP radiotherapeutics are likely going to be cleared via the hepatobiliary pathway (15) and the liver is therefore a potential organ for Auger-specific radiotoxicity. In preparation for clinical translation of 123I-MAPi, we performed studies to investigate liver toxicity. We found near-exclusive extracellular or perinuclear localization of 123I-MAPi in liver cells (i.e. the DNA of the liver parenchymal cells is out of range of most of the emitted low-range Auger electrons) as shown with the fluorescent analog PARPi-FL. This is in stark contrast to the observed nuclear accumulation of 123I-MAPi in GBM cells (Fig. 2C). The cytoplasmic liver accumulation is also corroborated by a liver enzyme analysis we performed on injected mice. We did not find significant changes of enzyme levels when compared to control mice (Fig. S5B). The radiobiological effectiveness of Auger-emitting compounds is critically dependent on the proximity of the electron emitter to the cellular DNA (45, 46). This distance-dependent relationship for double strand break production has been shown for I-125 (47). Cell-level dosimetry analysis showed a noteworthy increase of cell sterilization as a function of absorbed dose with 123I-MAPi in comparison to external photon irradiation, resulting in high estimates of relative biological effectiveness and suggesting that intranuclearly delivered radiation doses from PARP1-bound 123I-MAPi are nearly 50 times as potent as that from externally directed photons. This estimation, despite dependent on the parameters of the Monte Carlo simulation geometry, is in agreement with the expected equivalent dose for Auger electron emitters, which is comparable to that of intracellularly incorporated 5.3 MeV alpha particles (35, 48). The dose was calculated to the tumor cells using PARaDIM v1.0, a Monte Carlo method, using as input the time-integrated activity coefficients from measured data (49). These cellular dose estimates were confirmed against standard cellular methods from MIRDcell (50). We do, however, caution that these cell dose and RBE estimates strongly depend on the measured cell size, end point, reference radiation, and model assumptions. A limitation of this study is that the RBE is calculated starting from in vitro observations. This is because the orthotopic mouse model made it challenging to dose-paint the tumor with external beam irradiation without simultaneously irradiating the whole brain. Based on the limited literature on the potential use of I-123 labeled pharmaceuticals for therapy (51, 52) (although this could be different in using I-123 labeled PARP agents), projected in-human dose could start from an imaging dose of 8 mCi and collect biodistribution data for dosimetry calculations, guiding a Phase I study in patients.
To force a more favorable biodistribution toward tumor-targeting in clinical targeted radionuclide studies directed at brain tumors, investigators have proposed intra-tumor injection (53), intrathecal injection (54) and more recently convection enhanced delivery (CED) to administer the antibody across the BBB and into tumor tissue – e.g. to treat diffuse intrinsic pontine glioma (55). The above approaches have shown the ability to significantly improve the ratio of the radiation dose delivered to tumor relative to normal dose-limiting tissues. Many clinics do have the capability to perform intratumoral and intrathecal injection with or without a CED system (56–58). This delivery method, albeit not simple, is becoming more widely used as more patients present with tumors growing in the central nervous system; a consequence of the improved efficacy of chemotherapy for the treatment of vascular-accessible disease. To emulate these studies in a pre-clinical setting, we adopted an orthotopic GBM model in mice and performed a local injection of 123I-MAPi using an intratumoral osmotic pump delivery system. This system allowed for constant and prolonged delivery of the drug directly to the tumor and its microenvironment. Dosimetry estimates based on imaging, tissue specimen counting, and treatment response shown by Kaplan-Meier survival data are suggestive of the high potential clinical impact of this approach.
CEDs and Ommaya reservoirs are emerging as effective chemotherapy and targeted radionuclide delivery methods for patients with previously inaccessible metastatic disease. We have developed a method to perform CED injection of novel radiopharmaceuticals in pre-clinical models of cancer. We also performed intrathecal injection which would allow tumor imaging and therapy in clinical settings. This technique is routinely performed in most hospitals for chemotherapy drug delivery and allows clinicians to access the brain without opening the patient’s skull. SPECT/CT imaging can visualize and quantify radiolabeled drug uptake in tumor and its dispersion throughout the tissues of the body. We have demonstrated this capability with a newly developed theranostic agent, 123I-MAPi. We have shown that it can access the brain when injected intrathecally and using the intratumoral osmotic pump delivery system. While the favorable pharmacokinetics was accompanied by stressful side-effects in some mice (on account of the comparatively small volumes of the murine skull), this has not been, nor is it expected to be, a limiting factor for human studies.
To conclude, we characterized the first Auger-emitting PARP inhibitor in vivo which presented promising therapeutic results in a pre-clinical glioma model. The physical properties of Auger emission, paired with the biological distribution of a PARP inhibitor, make it possible to speculate that a dose escalation in patients could achieve high tumoricidal doses with limited normal tissue toxicity. The 159 keV gamma ray emitted is at the sweet spot for gamma camera imaging in patients and will allow for monitoring of treatment delivery across the BBB, redistribution, and precise dose evaluation, leading to truly personalized treatment plans (Fig. S7B, and S7C). 123I-MAPi has the potential to be a new and potent clinical agent for the treatment of brain tumors when used in conjunction with new intrathecal/CED administration methods.
Supplementary Material
Translational relevance.
Glioblastoma is one of the deadliest forms of solid tumors. Its heterogeneity, invasiveness and location make it difficult to treat without significant side effects. We developed an Auger-emitting theranostic PARP inhibitor, 123I-MAPi, which is able to deliver a lethal dose of radiation specifically to tumor cells. The combined biophysical properties of iodine-123 and the small molecule PARP inhibitor convey a lethal payload in close proximity to cancer cells’ DNA with limited damage to healthy surrounding tissue. The emitted gamma rays of iodine-123 make 123I-MAPi suitable for SPECT imaging. We present different delivery methods in order to overcome the blood-brain barrier and reach tumor cells within the brain. This study could represent an essential milestone in the clinical development of PARP-targeted Auger therapies.
Acknowledgements
We thank Dr. Cameron Brennan and the Brain Tumor Center for the GBM cell line model. We thank Dr. Ingo Mellinghoff and Dr. Beatriz Salinas for their helpful discussions and for providing their expertise. We thank Dr. Pat Zanzonico and Valerie Longo for technical support and help with animal imaging. We thank Dr. Elisa de Stanchina and Dr. Vanessa R. Thompson from the Antitumor Facility Core. We thank the Molecular Cytology Core Facility, the Animal Imaging Core Facility and the Radiochemistry & Molecular Imaging Probes Core Facility at Memorial Sloan Kettering Cancer Center. We thank Dr Ronald A. Ghossein, MD for consultation on pathology slides. We also thank Garon Scott for editing the manuscript.
FUNDINGS. This work was supported by National Institutes of Health grants R01 CA204441, P30 CA008748, R43 CA228815, R35 CA232130, and K99 CA218875. The authors thank the Tow Foundation and MSK’s Center for Molecular Imaging & Nanotechnology and Imaging and Radiation Sciences Program. LMC acknowledges support from the Ruth L. Kirschstein Postdoctoral Fellowship (NIH F32-EB025050).
Footnotes
Data and materials availability: All data necessary for interpreting the manuscript have been included in the main manuscript or in the Supplementary Materials. Additional information may be requested from the authors.
Conflict of interest statement S.K. and T.R. are shareholders of Summit Biomedical Imaging, LLC. S.K. and T.R. are co-inventors on filed U.S. patent (WO2016164771) held by MSK that covers methods of use for PARPi-FL. T.R. is a co-inventor on U.S. patent (WO2012074840) held by the General Hospital Corporation that covers the composition of PARPi-FL. J.S.L. and T.R. are co-inventors on U.S. patent (WO2016033293) held by MSK that covers methods for the synthesis and use of 18F-PARPi, 131I-PARPi and 123I-MAPi. T.R. is a paid consultant for Theragnostics, Inc.
References
- 1.Alexander BM, Cloughesy TF. Adult Glioblastoma. J Clin Oncol 2017;35:2402–9. [DOI] [PubMed] [Google Scholar]
- 2.Beating the odds: extreme long-term survival with glioblastoma. [editorial]. Neuro Oncol 2014;16(9):1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong X Current Strategies for Brain Drug Delivery. Theranostics. 2018;8:1481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker NR, Khong P, Parkinson JF, Howell VM, Wheeler HR. Molecular heterogeneity in glioblastoma: potential clinical implications. Front Oncol. 2015;5:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anjum K, Shagufta BI, Abbas SQ et al. Corrigendum to “Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review” [Biomed. Pharmacother. 92 (2017) 681–689]. Biomed Pharmacother. 2018;101:820. [DOI] [PubMed] [Google Scholar]
- 6.Vogelbaum MA, Aghi MK. Convection-enhanced delivery for the treatment of glioblastoma. Neuro Oncol. 2015;17 Suppl 2:ii3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kommidi H, Guo H, Chen N et al. An [. Theranostics. 2017;7:2377–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oberoi RK, Parrish KE, Sio TT, Mittapalli RK, Elmquist WF, Sarkaria JN. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro Oncol. 2016;18:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galia A, Calogero AE, Condorelli R et al. PARP-1 protein expression in glioblastoma multiforme. Eur J Histochem. 2012;56:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer. 2010;1:812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Murcia JM, Niedergang C, Trucco C et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 1997;94:7303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouchard VJ, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol. 2003;31:446–54. [DOI] [PubMed] [Google Scholar]
- 13.Tang J, Salloum D, Carney B et al. Targeted PET imaging strategy to differentiate malignant from inflamed lymph nodes in diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2017;114:E7441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irwin CP, Portorreal Y, Brand C et al. PARPi-FL - a fluorescent PARP1 inhibitor for glioblastoma imaging. Neoplasia. 2014;16:432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carney B, Kossatz S, Reiner T. Molecular Imaging of PARP. J Nucl Med. 2017;58:1025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko HL, Ren EC. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules. 2012;2:524–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryant HE, Schultz N, Thomas HD et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. [DOI] [PubMed] [Google Scholar]
- 18.Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 19.Carney B, Kossatz S, Lok BH et al. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat Commun. 2018;9:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kossatz S, Weber WA, Reiner T. Optical Imaging of PARP1 in Response to Radiation in Oral Squamous Cell Carcinoma. PLoS One. 2016;11:e0147752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kossatz S, Carney B, Schweitzer M et al. Biomarker-Based PET Imaging of Diffuse Intrinsic Pontine Glioma in Mouse Models. Cancer Res. 2017;77:2112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzales J, Kossatz S, Roberts S et al. Nanoemulsion-Based Delivery of Fluorescent PARP Inhibitors in Mouse Models of Small Cell Lung Cancer. Bioconjug Chem. 2018;29:3776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donabedian PL, Kossatz S, Engelbach JA et al. Discriminating radiation injury from recurrent tumor with [18F]PARPi and amino acid PET in mouse models. EJNMMI Res. 2018;8:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson RC, Makvandi M, Xu K et al. Iodinated benzimidazole PARP radiotracer for evaluating PARP1/2 expression in vitro and in vivo. Nucl Med Biol. 2016;43:752–8. [DOI] [PubMed] [Google Scholar]
- 25.Makvandi M, Xu K, Lieberman BP et al. A Radiotracer Strategy to Quantify PARP-1 Expression In Vivo Provides a Biomarker That Can Enable Patient Selection for PARP Inhibitor Therapy. Cancer Res. 2016;76:4516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jannetti SA, Carlucci G, Carney B et al. PARP-1-Targeted Radiotherapy in Mouse Models of Glioblastoma. J Nucl Med. 2018;59:1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchegger F, Perillo-Adamer F, Dupertuis YM, Delaloye AB. Auger radiation targeted into DNA: a therapy perspective. Eur J Nucl Med Mol Imaging. 2006;33:1352–63. [DOI] [PubMed] [Google Scholar]
- 28.Kiess AP, Minn I, Chen Y et al. Auger Radiopharmaceutical Therapy Targeting Prostate-Specific Membrane Antigen. J Nucl Med. 2015;56:1401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welt S, Scott AM, Divgi CR et al. Phase I/II study of iodine 125-labeled monoclonal antibody A33 in patients with advanced colon cancer. J Clin Oncol. 1996;14:1787–97. [DOI] [PubMed] [Google Scholar]
- 30.Daghighian F, Barendswaard E, Welt S et al. Enhancement of radiation dose to the nucleus by vesicular internalization of iodine-125-labeled A33 monoclonal antibody. J Nucl Med. 1996;37:1052–7. [PubMed] [Google Scholar]
- 31.Bavelaar BM, Lee BQ, Gill MR, Falzone N, Vallis KA. Subcellular Targeting of Theranostic Radionuclides. Front Pharmacol. 2018;9:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill MR, Falzone N, Du Y, Vallis KA. Targeted radionuclide therapy in combined-modality regimens. Lancet Oncol. 2017;18:e414–23. [DOI] [PubMed] [Google Scholar]
- 33.Hofer KG, Hughes WL. Radiotoxicity of intranuclear tritium, 125 iodine and 131 iodine. Radiat Res. 1971;47:94–101. [PubMed] [Google Scholar]
- 34.Kassis AI. Cancer therapy with Auger electrons: are we almost there. J Nucl Med. 2003;44:1479–81. [PubMed] [Google Scholar]
- 35.Lee H, Riad A, Martorano P et al. PARP-1-targeted Auger emitters display high-LET cytotoxic properties in vitro but show limited therapeutic utility in solid tumor models of human neuroblastoma. J Nucl Med. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menear KA, Adcock C, Boulter R et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51:6581–91. [DOI] [PubMed] [Google Scholar]
- 37.Reiner T, Lacy J, Keliher EJ et al. Imaging therapeutic PARP inhibition in vivo through bioorthogonally developed companion imaging agents. Neoplasia. 2012;14:169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salinas B, Irwin CP, Kossatz S et al. Radioiodinated PARP1 tracers for glioblastoma imaging. EJNMMI Res. 2015;5:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. [DOI] [PubMed] [Google Scholar]
- 40.Carlucci G, Carney B, Brand C et al. Dual-Modality Optical/PET Imaging of PARP1 in Glioblastoma. Mol Imaging Biol. 2015;17:848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malcolm J, Falzone N, Lee BQ, Vallis KA. Targeted Radionuclide Therapy: New Advances for Improvement of Patient Management and Response. Cancers (Basel). 2019;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hennrich U, Kopka K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals. 2019;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welt S, Divgi CR, Kemeny N et al. Phase I/II study of iodine 131-labeled monoclonal antibody A33 in patients with advanced colon cancer. J Clin Oncol. 1994;12:1561–71. [DOI] [PubMed] [Google Scholar]
- 44.Howell RW. Radiation spectra for Auger-electron emitting radionuclides: report No. 2 of AAPM Nuclear Medicine Task Group No. 6. Med Phys. 1992;19:1371–83. [DOI] [PubMed] [Google Scholar]
- 45.Royle G, Falzone N, Chakalova R, Vallis K, Myhra S. Internalization of Auger electron-emitting isotopes into cancer cells: a method for spatial distribution determination of equivalent source terms. Int J Radiat Biol. 2016;92:633–40. [DOI] [PubMed] [Google Scholar]
- 46.Falzone N, Ackerman NL, Rosales LF et al. Dosimetric evaluation of radionuclides for VCAM-1-targeted radionuclide therapy of early brain metastases. Theranostics. 2018;8:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Humm JL, Charlton DE. A new calculational method to assess the therapeutic potential of Auger electron emission. Int J Radiat Oncol Biol Phys. 1989;17:351–60. [DOI] [PubMed] [Google Scholar]
- 48.Howell RW, Narra VR, Sastry KS, Rao DV. On the equivalent dose for Auger electron emitters. Radiat Res. 1993;134:71–8. [PMC free article] [PubMed] [Google Scholar]
- 49.Carter LM, Crawford TM, Sato T et al. PARaDIM - A PHITS-based Monte Carlo tool for internal dosimetry with tetrahedral mesh computational phantoms. J Nucl Med. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vaziri B, Wu H, Dhawan AP, Du P, Howell RW, SNMMI MIRDC. MIRD pamphlet No. 25: MIRDcell V2.0 software tool for dosimetric analysis of biologic response of multicellular populations. J Nucl Med. 2014;55:1557–64. [DOI] [PubMed] [Google Scholar]
- 51.Mariani G, Di Sacco S, Volterrani D et al. Tumor targeting by intra-arterial infusion of 5-[123I]iodo-2’-deoxyuridine in patients with liver metastases from colorectal cancer. J Nucl Med. 1996;37:22S–5S. [PubMed] [Google Scholar]
- 52.Li S, Peck-Radosavljevic M, Kienast O et al. Iodine-123-vascular endothelial growth factor-165 (123I-VEGF165). Biodistribution, safety and radiation dosimetry in patients with pancreatic carcinoma. Q J Nucl Med Mol Imaging. 2004;48:198–206. [PubMed] [Google Scholar]
- 53.Zalutsky MR, Reardon DA, Akabani G et al. Clinical experience with alpha-particle emitting 211At: treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med. 2008;49:30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kramer K, Pandit-Taskar N, Humm JL et al. A phase II study of radioimmunotherapy with intraventricular 131 I-3F8 for medulloblastoma. Pediatr Blood Cancer. 2018;65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Souweidane MM, Kramer K, Pandit-Taskar N et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: a single-centre, dose-escalation, phase 1 trial. Lancet Oncol. 2018;19:1040–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kramer K, Humm JL, Souweidane MM et al. Phase I study of targeted radioimmunotherapy for leptomeningeal cancers using intra-Ommaya 131-I-3F8. J Clin Oncol. 2007;25:5465–70. [DOI] [PubMed] [Google Scholar]
- 57.Kramer K, Kushner BH, Modak S et al. Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol. 2010;97:409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kramer K, Pandit-Taskar N, Zanzonico P et al. Low incidence of radionecrosis in children treated with conventional radiation therapy and intrathecal radioimmunotherapy. J Neurooncol. 2015;123:245–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.