

Abstract
TAp63 is a p53 family member and potent tumor and metastasis suppressor. Here, we show that TAp63−/− mice exhibit an increased susceptibility to UVR-induced cutaneous squamous cell carcinoma (cuSCC). A human-to-mouse comparison of cuSCC tumors identified miR-30c-2* and miR-497 as underexpressed in TAp63-deficient cuSCC. Reintroduction of these microRNAs significantly inhibited the growth of cuSCC cell lines and tumors. Proteomic profiling of cells expressing either microRNA showed downregulation of cell cycle progression and mitosis associated proteins. A mouse to human and cross-platform comparison of RNA-Seq and proteomics data identified a 7-gene signature, including AURKA, KIF18B, PKMYT1, and ORC1, which were overexpressed in cuSCC. Knockdown of these factors in cuSCC cell lines suppressed tumor cell proliferation and induced apoptosis. Additionally, selective inhibition of AURKA suppressed cuSCC cell proliferation, induced apoptosis, and showed anti-tumor effects in vivo. Finally, treatment with miR-30c-2* or miR-497 microRNA mimics was highly effective in suppressing cuSCC growth in vivo. Our data establishes TAp63 as an essential regulator of novel microRNAs that can be therapeutically targeted for potent suppression of cuSCC.
Keywords: Cancer, Squamous cell carcinoma, microRNAs, Cell Cycle, TAp63
Introduction
Cutaneous squamous cell carcinoma (cuSCC) is the second most common cancer type diagnosed in the United States, with an estimated incidence of 700,000 new cases each year (1). Cumulative exposure to ultraviolet radiation (UVR) is the most common environmental risk factor for the development of cuSCC, in part through its ability to induce mutagenesis in the epidermis. The most effective course of treatment for cuSCC is surgical excision, and adjuvant radiotherapy (2). While the management of cuSCC generally has a favorable outcome, there is a small but significant percentage of patients that do not respond to therapy, resulting in severe morbidity and reduced survival. Efforts to develop targeted therapies for treatment-resistant cuSCC have been limited in success (3). Therefore, a better understanding of the underlying genetic and molecular determinants of cuSCC is required for the development of more effective treatments.
Large percentages of cuSCC (3–5), as well as squamous cell cancers of the lung (LUSC), head and neck (HNSCC), esophagus (ESCC), and bladder (BSCC) share mutations in TP53, overexpression of TP63, and de-regulation of microRNA expression (6–8). Our lab and others have shown that a diverse group of TP63 isoforms, including TAp63 and ΔNp63, exhibit non-overlapping and antagonistic functions (9). ΔNp63 isoforms promote tumor cell survival (10–12), whereas TAp63 isoforms have potent tumor and metastatic suppressive activities (13). We have shown previously that TAp63 directly transactivates Dicer, an endoribonuclease that cleaves pre-microRNAs into short double-stranded RNA fragments called microRNAs, and the expression of primary microRNA transcripts encoding miR-130b and miR-34a, to suppress tumorigenesis and metastasis (13).
microRNAs are a class of small, highly conserved non-coding RNAs that inhibit the translation of target mRNAs (14). Since their discovery, microRNAs have been shown to have important roles in different biological processes and cellular contexts. These microRNAs are frequently disrupted in disease states, including cancer (15). Given their mechanism of action, elucidating the functions of a given microRNA requires the identification of its numerous mRNA targets, which may also be cell- and context-specific (16).
Given our previous observations regarding the activities of TAp63 as a suppressor of tumorigenesis and metastasis, we investigated the contribution of TAp63 in UVR-induced cuSCC cuSCC. Our results show that TAp63-deficient mice develop UVR-driven cuSCC at a significantly higher frequency compared to wild-type (WT) mice. Through the use of next generation RNA-sequencing, we identified deregulated expression of several microRNAs in TAp63−/− cuSCC, most notably miR-30c-2* and miR-497. We also found that several predicted and verified mRNA targets of these microRNAs are conversely overexpressed in human cuSCC, including AURKA, KIF18B, PKMYT1, and ORC1. Reintroduction of miR-30c-2* and miR-497, or inhibition of their validated targets was sufficient to halt tumor cell proliferation and promote survival. Among these targets, miR-497 targets AURKA, was frequently overexpressed in cuSCC, and was associated with poor survival in patients with SCC. Moreover, inhibition of AURKA was effective at suppressing cuSCC proliferation and inducing apoptosis. We therefore tested the effect of Alisertib, an investigational AURKA inhibitor, on TAp63−/− cuSCC and found that it had a modest anti-tumor effect, suggesting that inhibition of a larger network of miR-30c-2* and/or miR-497 targets may be a more effective therapeutic strategy. Accordingly, re-introduction of miR-30c-2* or miR-497 using microRNA mimics almost completely abolished cuSCC growth in vivo. In summary, our data indicate that the therapeutic delivery of miR-30c-2*/miR-497 may be an effective treatment strategy for advanced and aggressive cuSCC.
Materials and Methods
Animal studies
Animal studies were conducted in compliance with and with the approval of the Institutional Animal Care and Use Committee (IACUC) at The University of Texas MD Anderson Cancer Center or the IACUC at Moffitt Cancer Center. TAp63−/− mice (17) were backcrossed over 10 generations to generate TAp63−/− and WT mice on a pure C57BL/6 background. Athymic nu/nu mice were purchased from Envigo. Age- and sex-matched mice were used for all experiments.
UVR treatments
To model UVR-driven cuSCC, 4-week-old mice were shaved on the entire dorsum prior to irradiation, and as needed throughout the experiment. Mice were exposed to low-dose UVR (2.5kJ m−2, 3x a week) using a solar UV irradiator, consisting of a bank of 4 FS40 T12 fluorescent sunlamps (Westinghouse, NJ). Mice were irradiated for up to 60 weeks or until tumors exceeding 2mm in diameter developed, at which point the mice were euthanized and the tissues were collected for RNA extraction and/or histological processing.
Tumor xenograft studies
To model human cuSCC, we modified the use of a previously described xenograft model of cuSCC (18). For alisertib treatments, 2 × 105 COLO16 cells were subcutaneously injected into both flanks of athymic (nu/nu) mice. For microRNA mimic transfections, COLO16 cells stably expressing RFP and luciferase were reverse transfected with microRNA mimics or scrambled mimic control. Six hours later, the transfected cells were harvested and diluted in a 1:1 mixture of PBS and matrigel (Corning). 2 × 105 COLO16 cells transfected with either microRNA mimic was subcutaneously injected into both flanks of athymic (nu/nu) mice. When tumors became palpable, mice were randomized into treatment and control groups. Mice were monitored daily for signs of distress and weighed weekly. Alisertib was delivered to the treatment group once daily at 30mg/kg via oral gavage. Tumor growth was measured with calipers and tumor volume was calculated using the formula: tumor volume (mm3) = D × d2/2, where D and d are the longest and the shortest diameters of the tumor, respectively. Mice were euthanized when the largest tumors reached 1,000mm3 in volume, or when the mice exhibited signs of distress.
cuSCC cell lines and cell culture
Human cutaneous squamous cell carcinoma (cuSCC) cell lines (COLO16, SRB12, SRB1, RDEB2, and IC1) were kindly provided by Dr. K.Y. Tsai and maintained in culture conditions as previously reported (19). All cell lines were authenticated using STR profiling and routinely tested for mycoplasma contamination. Normal human epidermal keratinocytes (NHEKs) were purchased from Lonza and cultured in KGM-Gold™ Keratinocyte Growth media (Lonza).
RNA isolation
Tissue specimens were flash frozen in liquid nitrogen and homogenized with a mortar and pestle and total RNA was isolated and purified using the miRvana microRNA isolation kit (Ambion). For cell lines, total RNA was isolated and purified using the miRNeasy RNA isolation kit (Qiagen).
RNA sequencing and analysis
5 μg of polyA+ RNA per sample was used to construct RNA-seq libraries, which were submitted for 100 nt paired-end sequencing on an Illumina Hi-Seq 2000, to a depth of 20–50 million reads per sample. The RNA-Seq reads were aligned to the mouse reference genome build mm10 (GRCm38) using TopHat (v2.0.12) (20). Cufflinks (v2.2.1) (21) was used to assemble transcripts and calculate the fragments per kilobase of transcript per million mapped reads (FPKM).
Small RNA sequencing and analysis
The same RNA samples used for RNA sequencing were submitted to the laboratory of Preethi Gunaratne, PhD (University of Houston, Biology & Biochemistry) for small RNA sequencing. Illumina small RNA adapter sequences were removed from sequence reads. Reads shorter than 10 nt or ending in homopolymers greater than or equal to 9 nt were discarded. From this, the total number of usable reads for each sample was calculated. Sequence reads were then mapped to the miRBase reference (22) (http://www.mirbase.org/) using BLAST. The relative abundance of each individual microRNA sequenced in the experiment was counted as a fraction of the total usable reads (parts per million) .
Human Cancer Patient Survival Analysis
After gene activity score was computed for each patient, patients were stratified. Survival was evaluated using the log-rank test as implemented in the survival (23) library from the R statistical system.
Statistics
Results were reported as mean ± SD or ± SEM. Statistical significance was calculated using Student’s t test (two-tailed) for normally distributed data sets. Differentially expressed mRNAs and microRNAs were identified following a cut-off of absolute fold change>1.5, and a p-value<0.05 the R statistical software. Hierarchical clustering analysis of the RNA-seq and small RNA-seq was performed using the Pearson correlation coefficient as the distance metric. Gene expression heatmaps were constructed using the heatmap.2 function from the gplots package. Principal Component Analysis was performed using the Stats package from the R statistical software environment (http://www.r-project.org).
Data Availability
Raw RNA and small RNA sequencing data from all human and mouse samples that were used to support the findings of this study have been deposited in NCBI/GEO with Series accession code GSE146258. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (24) partner repository with the dataset identifier PXD014261 and 10.6019/PXD014261.
Results
Loss of TAp63 promotes UVR-induced tumorigenesis
To investigate the role of TAp63 in skin tumorigenesis, we treated mice that TAp63 (WT) versus homozygous deletion of TAp63 (TAp63−/−) (17) with 2.5kJ m−2 UVR, 3 times a week (Fig. 1A). As anticipated, this treatment regimen induced cutaneous tumors in both cohorts, including pre-malignant papillomas and fully malignant cuSCC (Figs. 1B and C). While the number of papillomas did not differ between the two genotypes, we observed a higher frequency of cuSCC in the TAp63−/− cohort (46.67% vs. 20%) (Fig. 1D). There was also a statistically significant increase in the average number of cuSCC tumors per mouse in the TAp63−/− cohort (0.80 vs. 0.20, p=0.0429) (Fig. 1E). Together, these observations indicate that the loss of TAp63 promote malignant progression of cuSCC.
Figure 1:
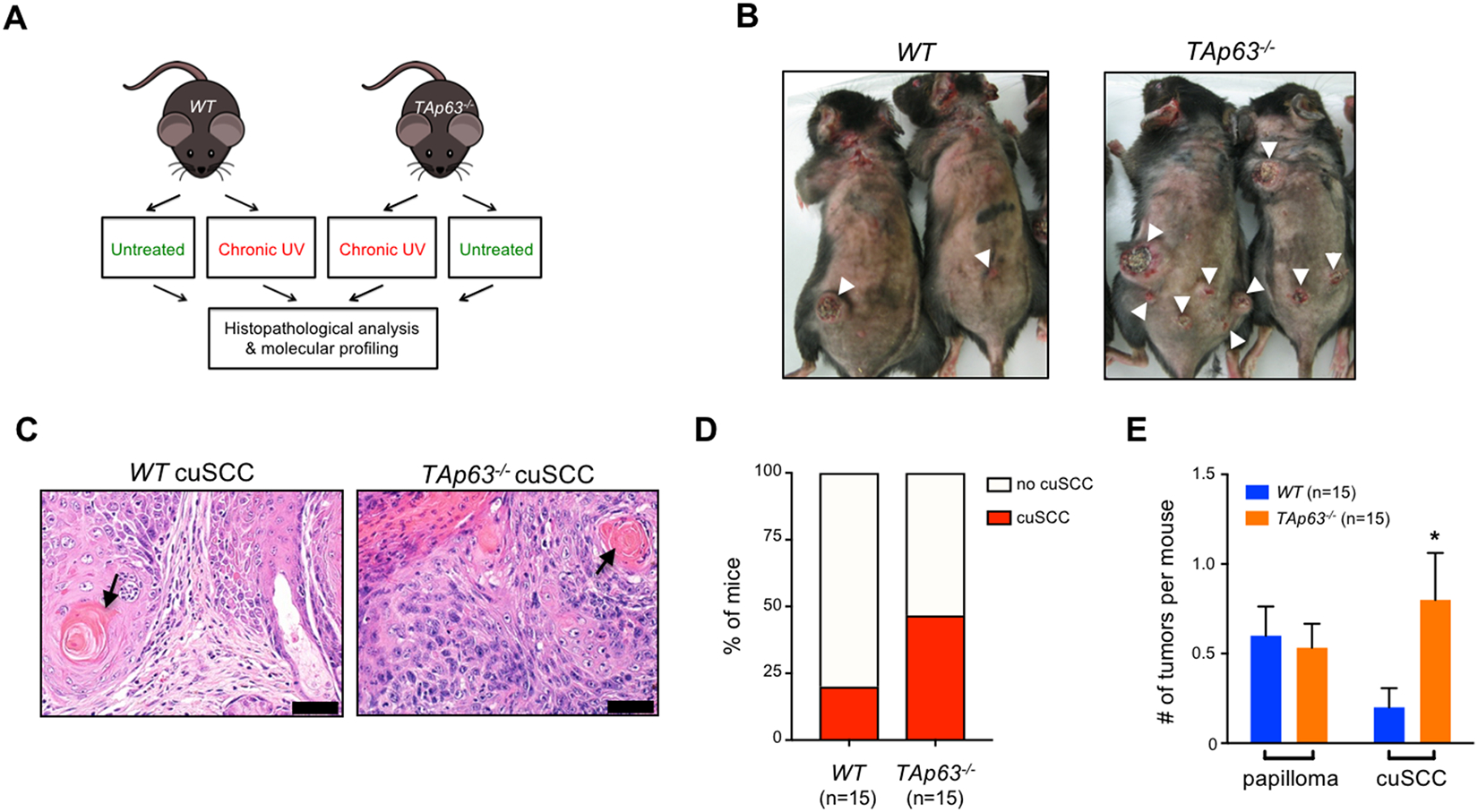
Loss of TAp63 promotes UVR-induced tumorigenesis. A, WT and TAp63−/− mice treated with UVR (5kJ/m2, 3x a week, for up to 60 weeks) (n=15 mice per group). B, Tumors from irradiated mice. White arrowheads indicate the presence of cuSCC. C, H&E stained cross-sections of well-differentiated cuSCC tumors from the indicated genotypes. Keratin pearls (black arrow) indicate squamous differentiation. Scale bars: 50 μm. D, Quantification of mice harboring cuSCC for both genotypes. E, The average number of pre-malignant papillomas and cuSCCs per mouse was quantified for each genotype. * p<0.05, Student’s t test (two-tailed).
An important prognostic feature of cuSCC is its differentiation status. Tumors with a poorly differentiated histology harbor an enhanced risk for recurrence and metastasis (25). Histopathological examination found that all of the identified tumors, regardless of genotype, were well-differentiated cuSCC (Fig. 1E). Since we have previously shown that TAp63-deficient mice are prone to metastatic carcinomas (13), we examined the organs of irradiated mice for evidence of metastasis. While no metastases were found in WT mice, multiple lung metastases, which stained positive for the squamous cell marker keratin 5, were found in TAp63−/− mice bearing cuSCC lesions (Supplementary Figs. 1A and B). Taken together, these results indicate that TAp63 is an important suppressor of tumor progression and metastasis of cuSCC in response to UVR treatment.
Human / mouse cross-species analysis revealed TAp63-regulated microRNA in cuSCC
Because TAp63 has been implicated in microRNA biogenesis (13), we asked whether the increased cuSCC development in TAp63−/− mice was due to deregulated microRNA expression. To answer this question, we performed RNA-seq and small RNA-seq on skin and cuSCC lesions from both WT and TAp63−/− irradiated mice. In total, RNA from 3 WT normal skin, 3 TAp63−/− normal skin, 2 WT cuSCC tumors, and 3 TAp63−/− cuSCC tumor samples were subjected to Illumina RNA=sequencing. Principal component analysis (PCA) of the RNA-seq (Supplementary Fig. 2A) and small RNA-seq (Supplementary Fig. 2B) showed that the global expression patterns of these transcripts stratified skin from cuSCC samples. Moreover, samples within each tissue type clustered according to genotype, indicating that the loss of TAp63 has global effects on the transcriptional landscape of normal skin and cuSCC. Unsupervised hierarchical clustering of genes (Supplementary Fig. 2C) and microRNAs (Supplementary Fig. 2D) also showed clustering of normal skin and cuSCC. These results suggest that the loss of TAp63 affects a significant proportion of the cuSCC transcriptome.
To identify TAp63-regulated mRNAs and microRNA networks, we generated a TAp63−/− cuSCC signature from the mRNA (Fig. 2A) and microRNA (Fig. 2B) expression datasets (fold change>1.5, p-value<0.05). To identify those targets that are dependent on TAp63, we focused on genes and microRNAs that were differentially expressed in the TAp63−/− cuSCC signatures but were not similarly affected in WT cuSCC (Fig. 2C, blue). From this comparison we identified 1993 mRNAs (Supplementary Table 1) and 90 microRNAs (Supplementary Table 2) that were differentially expressed in the TAp63−/− cuSCC signature.
Figure 2.
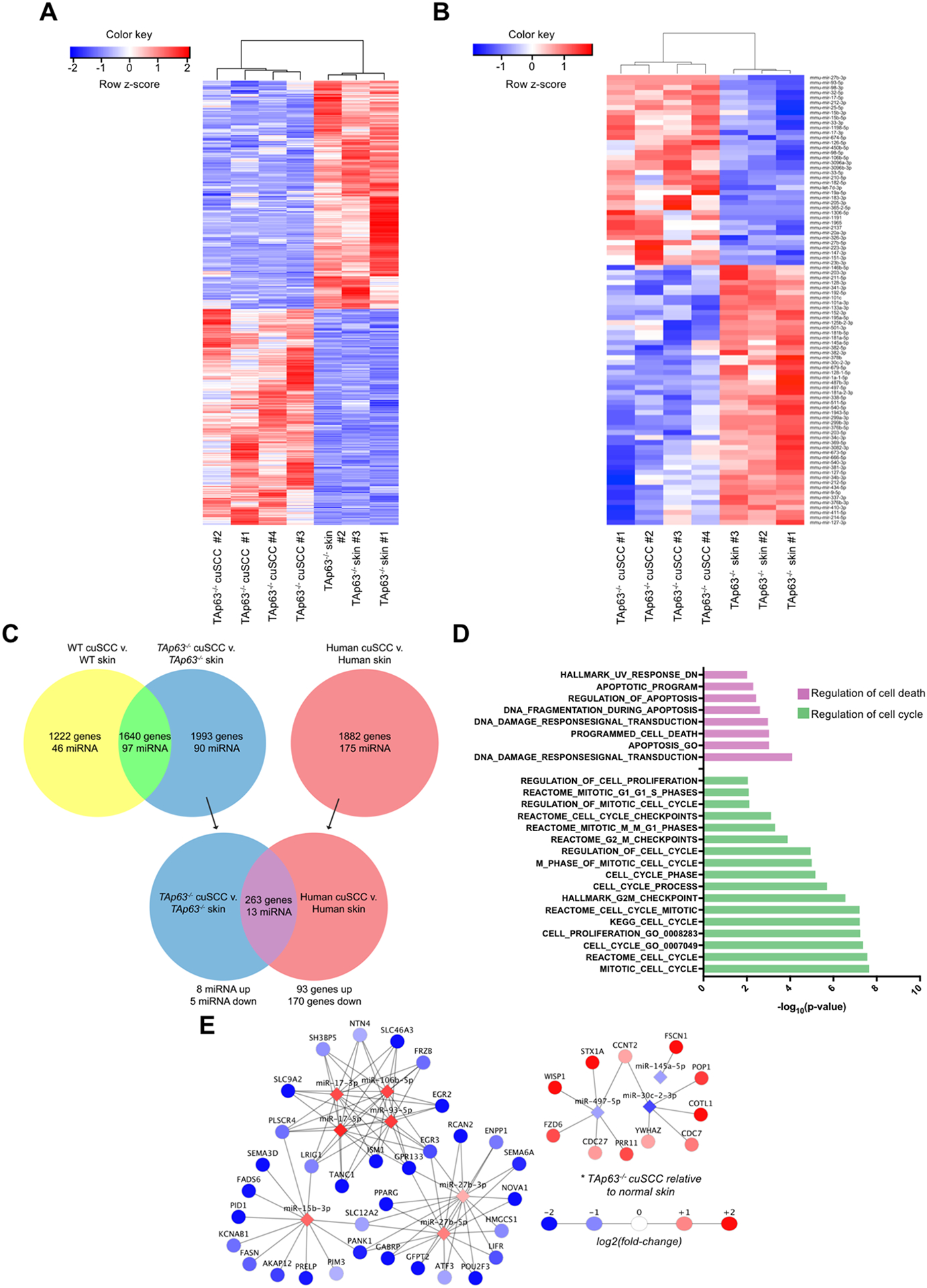
TAp63-deficient tumors exhibit deregulated mRNA and microRNA expression. A-B, Hierarchical clustering analysis based on differentially expressed mRNAs (A) and microRNAs (B) in TAp63−/− cuSCC vs. TAp63−/− skin samples. Each row represents a single mRNA or microRNA, while each column represents a sample. The Pearson correlation matrix is shown on top. The color scale illustrates the relative expression levels of mRNAs and microRNAs across each sample. Blue shades correspond to reduced expression and red shades represent increased expression levels. C, Comparison of the TAp63−/− cuSCC signature and human cuSCC identified similar differential expression of the indicated number of microRNAs and mRNAs. D, Pathway analysis of the overlapping targets (purple) identified in (C). E, Functional pair analysis identified a conserved microRNA/mRNA regulatory network in both TAp63−/− murine cuSCC and human cuSCC. Shown are microRNAs with fold change>1.5 in both comparisons.
To examine the TAp63-regulated microRNAs in the pathogenesis of cuSCC, we performed microRNA-mRNA pair analysis by identifying differentially expressed microRNAs that complement microRNA response elements (MREs) within the 3’ UTR of significantly anti-correlated mRNAs (26,27). Within the TAp63−/− cuSCC signature, we identified key functional microRNA-mRNA pairs that were associated with loss of TAp63 in cuSCC. We found that the TAp63−/− cuSCC signature contained 28 underexpressed microRNAs with 311 overexpressed mRNAs, and 25 overexpressed microRNAs with 333 underexpressed mRNAs (Supplementary Tables 3 and 4).
To determine if any of the TAp63-associated microRNAs and mRNAs are relevant to human cuSCC, we compared the TAp63−/− cuSCC signatures to previously published human cuSCC mRNA and microRNA signatures generated from 9 cuSCC and 7 normal skin samples obtained from human patients (3) (Fig. 2C). This comparison identified 263 mRNAs (93 upregulated, 170 downregulated) (Supplementary Table 5) and 13 microRNAs (8 upregulated, 5 downregulated) (Supplementary Table 6) that were differentially expressed in both mouse TAp63−/− cuSCC and human cuSCC. Pathway analysis revealed significant enrichment of genes related to cell proliferation and apoptosis (Fig. 2D and Supplementary Table 7). These observations are consistent with previous studies, which have demonstrated that TAp63 is a regulator of protein coding genes involved in these two processes (17,28), and that this regulation is a major component of the tumor suppressive functions of TAp63. Prior to this study, however the role of TAp63-regulated microRNAs in these two processes remained unexplored.
To prioritize microRNAs for further validation, we performed functional pair analyses in both the TAp63−/− cuSCC and human cuSCC microRNA and mRNA signatures. From this analysis, we identified 10 conserved differentially expressed microRNAs which had at least one predicted mRNA target expressed in the opposite direction (Fig. 2E). Several of the microRNAs that were found to be overexpressed in this analysis have been previously implicated in the pathogenesis of cuSCC, including miRs-15b, 17, and 27b (3,29,30). Conversely, several microRNAs that were underexpressed, including miRs-30c-2* and 497, were previously shown to be reduced in cuSCC and found to exhibit tumor suppressive functions (3,29).
TAp63-regulated miR-30c-2* and miR-497 suppress cuSCC through induction of apoptosis and cell cycle arrest
To determine the important microRNAs in mediating the tumor suppressive functions of TAp63 we tested the correlation of these microRNAs with TAp63 expression. Among the 10 differentially expressed microRNAs, we were able to consistently validate by qRT-PCR the downregulation of miR-30c-2* and miR-497 in TAp63−/− cuSCC compared to normal skin (Supplementary Fig. 3, A and B). We then profiled the expression of these 2 microRNAs in 5 human cuSCC cell lines (COLO16, IC1, RDEB2, SRB1, and SRB12) and compared it to their expression in normal human epidermal keratinocytes (NHEKs), the cell of origin for cuSCC (Supplementary Fig. 3C). Similar to TAp63−/− cuSCC, both miRs were significantly underexpressed in almost all of the cuSCC cell lines when compared to NHEKs. Interestingly, we also found reduced TAp63 levels in each of these cell lines, except for RDEB2, which showed very little difference with NHEKs (Supplementary Fig. 3D). These results suggest that reduced expression of iis associated with reduced expression of miR-30c-2* and miR-497.
To further probe the interaction between TAp63, miR-30c-2* and miR-497 in cuSCC, we used siRNAs to knockdown TAp63 in NHEKs (Fig. 3A–D). Knockdown of TAp63 resulted in diminished expression of miR-30c-2* (Fig. 3C) and miR-497 (Fig. 3D), supporting a model in which TAp63 activates the expression of these microRNAs. In line with previous observations, downregulation of TAp63 resulted in a significant reduction in Dicer mRNA expression (Fig. 3B). To determine if the loss of Dicer is responsible for the diminished miR-30c-2* and miR-497 expression following the downregulation of TAp63, we transfected NHEKs with siRNAs targeting Dicer (Supplementary Fig. 3E). Downregulation of Dicer resulted in significant reductions of miR-30c-2* (Supplementary Fig. 3F) and miR-497 (Supplementary Fig. 3G) expression, similar to what is observed upon TAp63 deletion.
Figure 3.
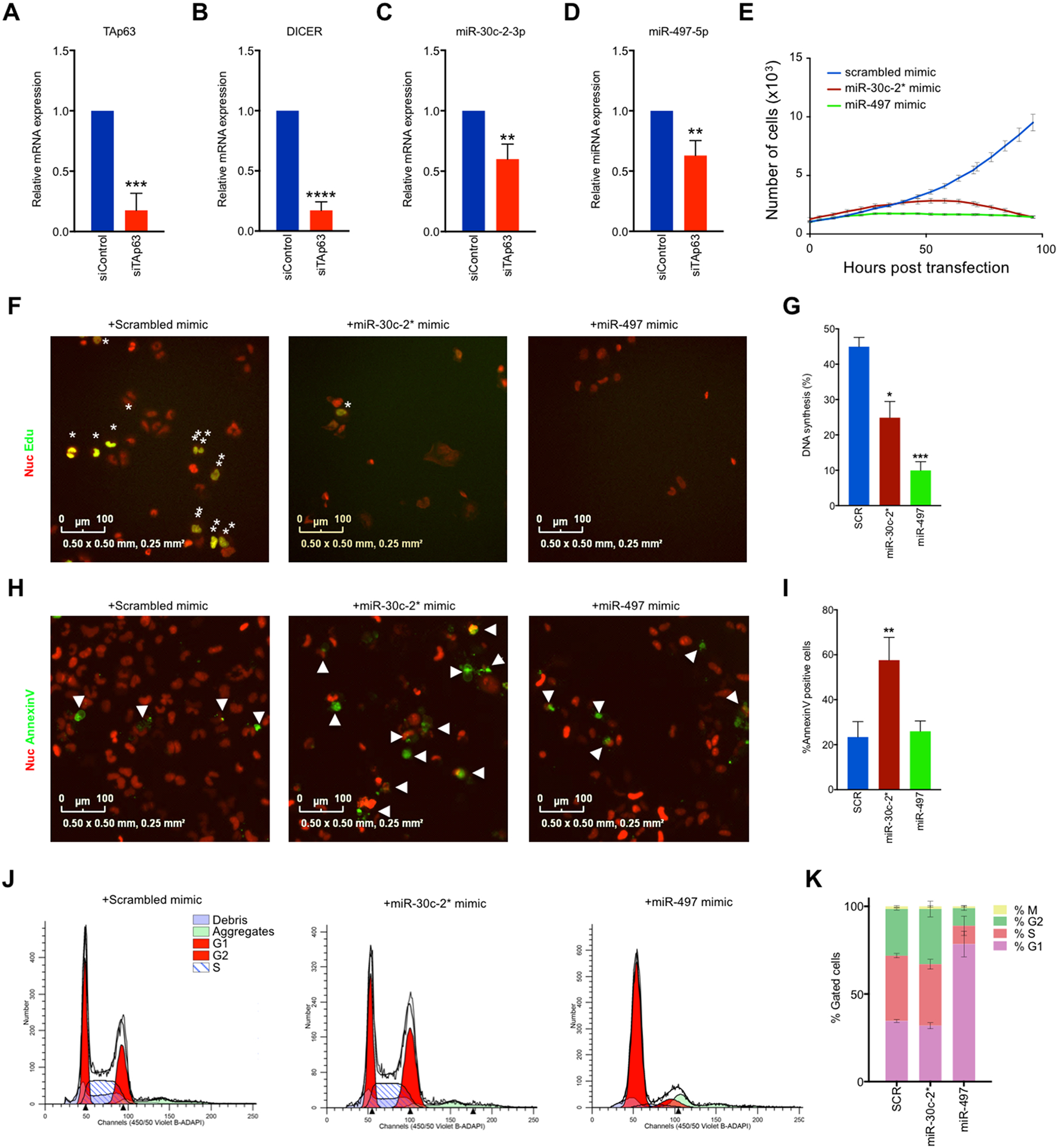
TAp63-regulated miR-30c-2* and miR-497–5p suppresses cuSCC through induction of cell death and cell cycle arrest. A-B, SYBR green qRTPCR of TAp63 (A) and Dicer (B) in NHEKs following transfection with the indicated siRNAs. C-D, Taqman qRTPCR of miR-30c-2* C, and miR-497–5p D in NHEKs following transfection with the indicated siRNAs. E, Representative growth curve of nucRed-mCherry-labeled COLO16 cells transfected with the indicated microRNA mimics. F-G, Immunofluorescence (F) and quantification (G) for Annexin V-488 (green)-positive nucRed-mCherry-labeled COLO16 cells transfected with the indicated microRNA mimics. H-I, Immunofluorescence (H) images and quantification (I) for Edu (green) incorporation in COLO16 cells transfected with the indicated microRNA mimics following a 3 hour Edu pulse. NucRed® dead 647 (red) was used as a counterstain. J-K, Cell cycle profiles (J) and quantification (K) of COLO16 cells 48 hours after transfection with the indicated microRNA mimic as measured by FACS analysis. M phase was measured as the percentage of cells staining positive for Histone H3-pS28-AF647. Data shown are mean ± SD, n=3, unless noted otherwise. * p<0.05, ** p<0.01, ***p<0.001, Student’s t test (two-tailed).
To test the putative anti-tumor effects of miR-30c-2* and miR-497 we transfected chemically-modified microRNA mimics into the human cuSCC cell lines, COLO16 and SRB12, and analyzed cell growth. Both miR-30c-2* and miR-497 significantly inhibited proliferation of COLO16 (Fig. 3E) and SRB12 (Supplementary Fig. 3H) cells when compared to a scrambled control mimic. To further dissect the anti-proliferative effects of these microRNAs, we assayed transfected cuSCC cells for DNA synthesis, apoptosis, and cell cycle profiling. miR-30c-2* led to a significant reduction of cell proliferation as assessed by Edu incorporation, indicating decreased DNA synthesis (Figs. 3F and G). We also saw a more striking induction of apoptosis in miR-30c-2*-transfected cells, as evidenced by increased annexin V staining (Figs. 3H and I). Likewise, miR-497-transfected cells showed a more significant reduction in DNA synthesis (Figs. 3F and G). miR-497-transfected cell did not however show increased apoptosis (Figs. 3H and I). Instead, cell cycle profiling demonstrated that miR-497-transfected cells exhibited a striking increase in the percentage of cells in G1, which is indicative of a G1/S arrest (Figs. 3J and K and Supplementary Fig. 3I). miR-30c-2*-transfected cells, on the other hand, were found to have a slight, non-significant increase in the percentage of cells in G2. These results suggest that miR-30c-2* and miR-497 suppress cuSCC cell growth through the induction of apoptosis and cell cycle arrest, respectively.
Proteogenomic analysis identifies multiple direct mRNA targets for miR-30c-2* and miR-497
The ultimate consequence of microRNA function is the translational inhibition of the mRNAs that it targets. To study the regulatory functions of miR-30c-2* and miR-497, we employed a shotgun proteomics approach to measure global protein expression following overexpression of either microRNA (Fig. 4A). COLO16 cells were transfected with miR-30c-2*, miR-497 or scrambled control mimics and collected 48 hours later. After lysis, reduction, and alkylation, proteins in whole cell lysates were subjected to trypsin digestion, followed by Tandem mass tag (TMT) labeling. Peptides were then identified via LC-MS/MS and proteins that were quantified by at least 2 unique peptides were selected for further analysis. Differential protein expression was determined in each mimic transfected sample compared to the scrambled mimic control.
Figure 4.
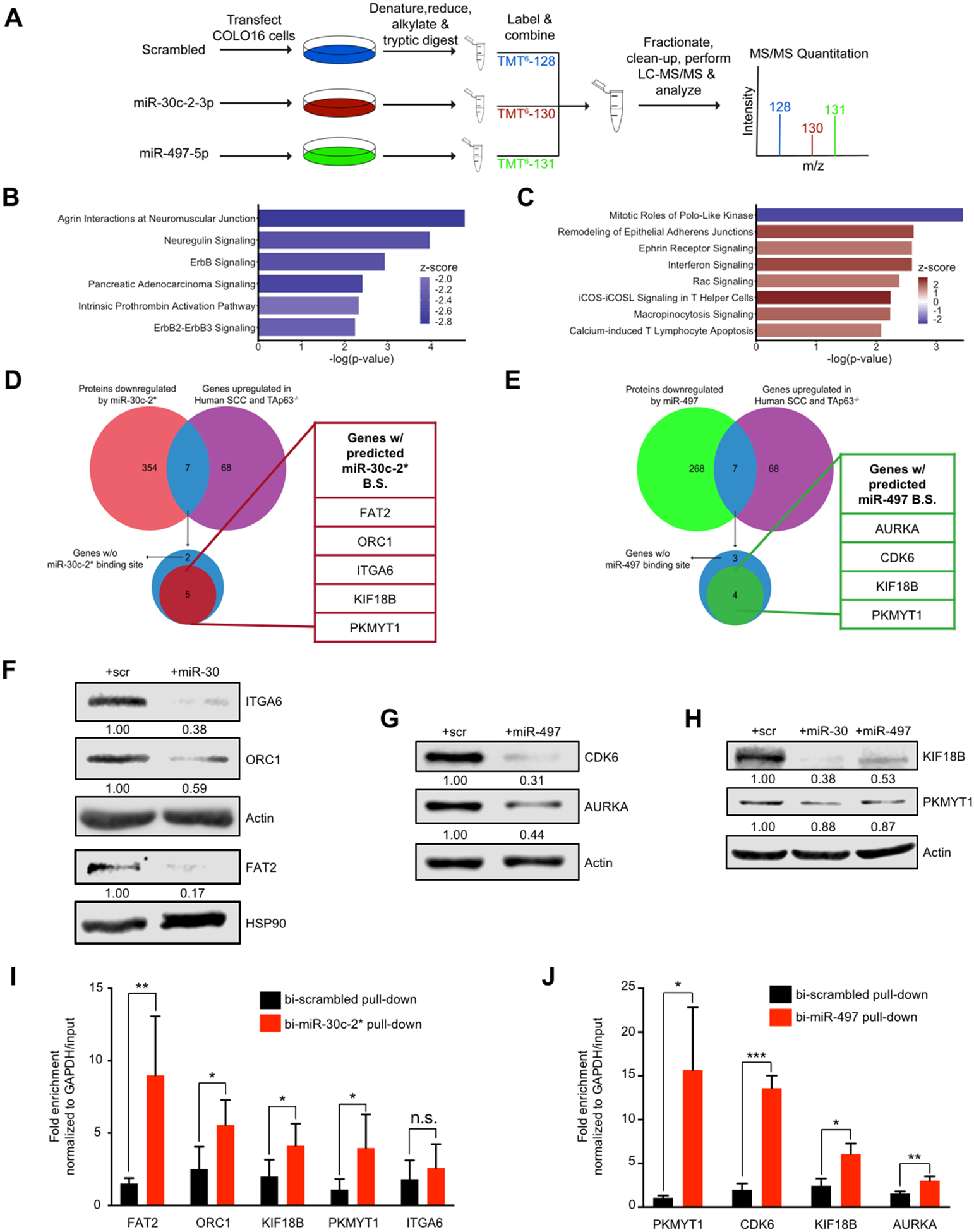
Proteogenomic analysis identifies multiple direct mRNA targets for miR-30c-2* and miR-497. A, Schematic representation of experimental design. COLO16 cells transfected with the indicated microRNA mimic were collected, denatured, and subjected to tryptic digest prior to labeling of each sample with the indicated tandem mass tag (TMT). Labeled peptides were then combined, fractionated and subjected to LC-MS/MS analysis to identify differentially expressed proteins. B-C, Ingenuity Pathway Analysis (IPA) of underexpressed proteins (fold change<0.67) in cells transfected with miR-30c-2–3p (B) and miR-497–5p (C) compared to scrambled control. The canonical pathways shown exhibited a -log(p-value) > 2, and an absolute z-score > 1.5. A positive (orange bars) or negative (blue bars) z-score indicates that pathway activity is predicted to be increased or reduced following overexpression of the corresponding microRNA mimic. D-E, Comparison of significantly underexpressed proteins following transfection with miR-30c-2* (D) and miR-497 (E) with the overexpressed mRNAs in murine TAp63−/− cuSCC and human cuSCC by RNA-Seq. Overlapping targets were examined for the presence of MREs for miR-30c-2* and miR-497. Note that KIF18B and PKMYT1 were found to be predicted targets of both miR-30c-2* and miR-497–5p and were underexpressed in both proteomics experiments. F-H, Western blot analysis for the indicated proteins following transfection with mimics of miR-30c-2* (F), miR-497 (G), and both miRs (H). I-J, COLO16 cells were transfected with the indicated biotinylated microRNA mimics (bi-miR) and collected 24 hrs after transfection. Fold enrichment for the indicated targets in the streptavidin pull-down of bi-miR-30c-2* (n=5) (I) and bi-miR-497–5p (n=3) (J) was calculated using qRT-PCR. Data shown represent the mean ± SD of at least 3 independent experiments, unless noted otherwise. * p<0.05, ** p<0.01, *** p<0.001, n.s. = not significant, Student’s t test (two-tailed).
A total of 1751 proteins were differentially expressed based on an absolute fold change greater than 1.5 in the miR-30c-2*-transfected sample relative to the scrambled mimic-transfected sample (Supplementary Fig. 4A). Among these proteins, 361 were underexpressed, while 1390 proteins were overexpressed relative to control-transfected cells (Supplementary Table 8). Alternatively, 587 proteins were differentially expressed in the miR-497-transfected sample relative to control with 275 proteins being underexpressed, and 312 proteins were overexpressed (Supplementary Fig. 4B and Table S9).
Ingenuity pathway analysis (IPA) identified significantly enriched pathways among the underexpressed proteins in both comparisons. Interestingly, the proteins that were underexpressed in the miR-30c-2*-transfected cells were significantly enriched for pathways related to ErbB signaling, including Agrin Interactions at Neuromuscular Junction, Neuregulin Signaling, and ErbB2-ErbB3 Signaling (Fig. 4B). Alternatively, the top pathway that was enriched in the underexpressed proteins in the miR-497 comparison was Mitotic Roles of Polo-like kinase (Fig. 4C). Importantly, the global changes in protein expression reflect the phenotypic changes observed following the overexpression of the corresponding microRNA.
To determine if the measured protein expression changes reflect direct regulatory effects of miR-30c-2* and miR-497, we profiled the corresponding mRNA for each of the differentially expressed proteins in each comparison for the presence of microRNA response elements (MREs) for the respective microRNA utilizing the miRwalk 2.0 software (31). From this analysis, we found that 241 of the 361 (66.7%) underexpressed proteins harbored a predicted MRE for miR-30c-2* (Supplementary Table 10). Likewise, 160 of the 275 underexpressed proteins (58.18%) also harbored predicted MRE for miR-30c-2* (Supplementary Table 11).
To prioritize targets for validation, we compared the list of underexpressed proteins (fold change<0.67) in the miR-30c-2* (Fig. 4D and Table S8) and miR-497 (Fig. 4E and Supplementary Table 9) proteomics signatures against the mRNAs that were conversely overexpressed (fold change>1.5, p<0.05) in both the mouse TAp63−/− cuSCC and human cuSCC RNA-Seq signatures (Supplementary Table 12). We then examined both lists of mRNAs for the presence of at least one MRE to focus our studies on likely targets of either microRNA. From this analysis we found 5 putative targets of miR-30c-2* (FAT2, ITGA6, KIF18B, ORC1, and PKMYT1) and 4 predicted targets of miR-497 (AURKA, CDK6, KIF18B, and PKMYT1) that are frequently overexpressed in both mouse TAp63−/− and human cuSCC. Interestingly, both microRNAs are predicted to target KIF18B and PKMYT1. We validated the LC-MS/MS observations for all 7 of the putative targets by Western blotting (Fig. 4F–4H). Interestingly, only AURKA and CDK6 appeared to be significantly reduced at the RNA level following overexpression of miR-497, whereas none of the miR-30c-2* targets were affected (Supplementary Fig. 4, B and C). These results confirm that all 7 targets are significantly downregulated following overexpression of the corresponding miR and suggest that they may be inhibited via microRNA-mRNA mediated targeting.
To determine if the putative targets of either microRNA are repressed via RISC-mediated microRNA-mRNA interactions we transfected COLO16 cells with microRNA mimics biotinylated at the 3’-end of the mature strand (bi-miR-30c-2*, bi-miR-497, or bi-scrambled). We then performed streptavidin pull-downs and qRT-PCR for each of the putative targets of miR-30c-2* and miR-497, normalized to GAPDH expression (32). Relative mRNA abundance in the streptavidin pull-downs and inputs in the bi-miR-30c-2*- and bi-miR497-transfected cells were separately normalized to their levels in cells transfected with a biotinylated scrambled mimic (bi-scrambled). The putative miR-30c-2* targets FAT2, ORC1, KIF18B, and PKMYT1 were significantly enriched in the bi-miR-30c-2* pull-down (Fig. 4I). Similarly, all 4 putative targets of miR-497 (AURKA, CDK6, KIF18B, and PKMYT1) were enriched in the bi-miR-497 pull-down (Fig. 4J). Taken together, these results suggest that FAT2, ORC1, KIF18B and PKMYT1 appear to be novel direct targets of miR-30c-2*, while AURKA, CDK6, KIF18B, and PKMYT1 appear to be bona fide targets of miR-497.
miR-30c-2* and miR-497 targets promote tumor cell proliferation and survival
To determine if the validated targets of miR-30c-2* and miR-497 are relevant to human tumors, we profiled their mRNA (Fig. 5A) and protein (Fig. 5B) levels in 5 human cuSCC cell lines (COLO16, SRB12, SRB1, IC1, and RDEB2) and NHEKs. From these experiments, each of these targets are overexpressed in the majority of the cuSCC cell lines profiled, with the exception of FAT2 and PKMYT1, which showed variable expression across the different cuSCC cell lines.
Figure 5:
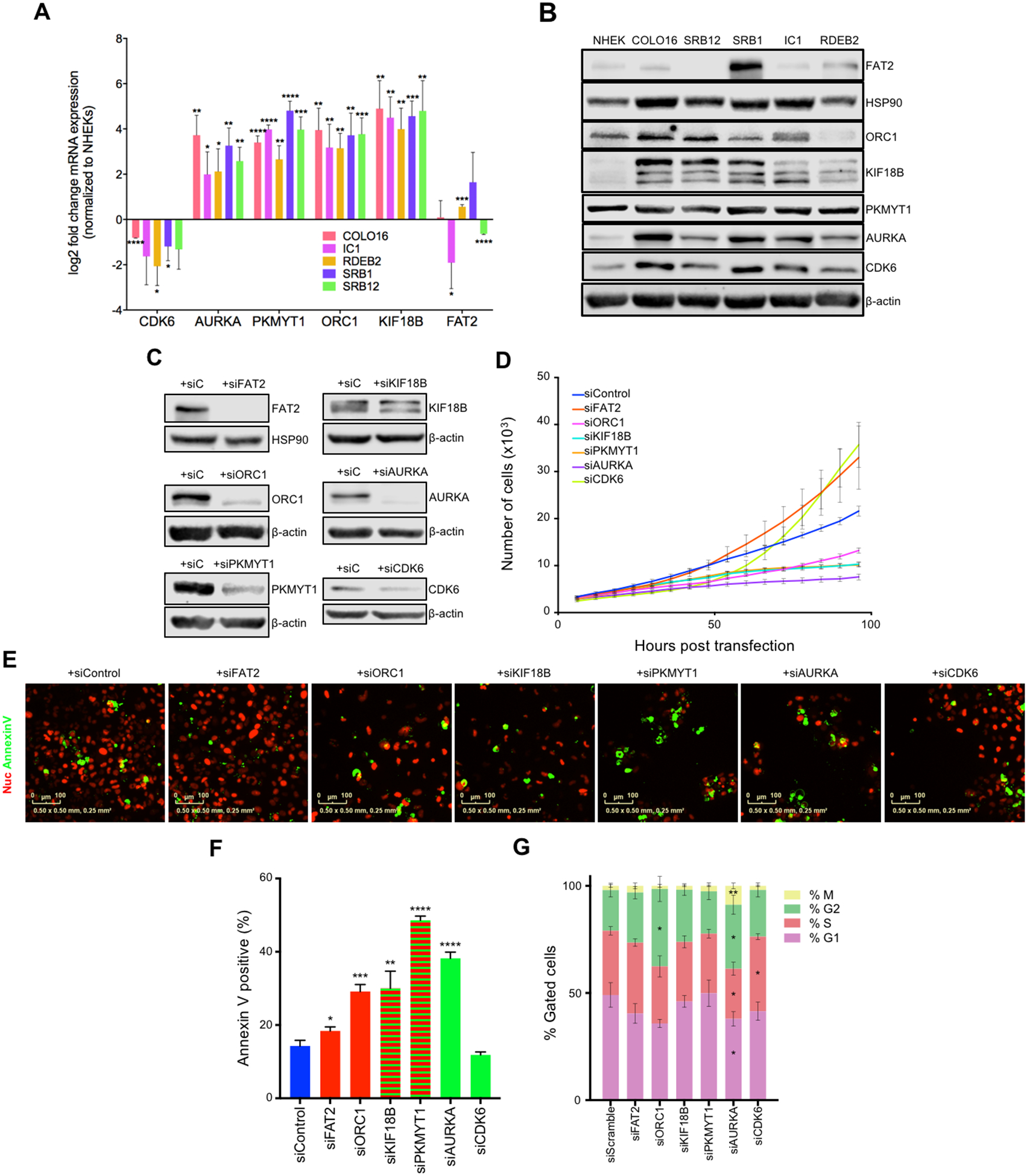
Inhibition of mIR-30c-2* and miR-497–5p targets affects cuSCC cell proliferation and survival. (A) SYBR green qRT-PCR of the validated miR-30c-2* and miR-497 targets in NHEKs and the cuSCC cell lines COLO16, SRB12, SRB1, IC1, and RDEB2. (B) Representative results of Western blotting of the validated miR-30c-2* and miR-497 targets in the same cell lines as in (A). (C) Western blotting of the indicated targets following siRNA-mediated knockdown in COLO16 cells. (D-F) COLO16 cells stably expressing nuclear mCherry transfected with the indicated siRNAs, incubated with Annexin V-488, and scanned every 4 hours using the Incucyte® high-content live-cell imaging platform. (D) Growth curve of COLO16 cells transfected with the indicated siRNAs. (E and F) Immunofluorescence (E) and quantification (F) for annexin V-488 (green)-positive cells transfected with the indicated siRNAs. (G) Cell cycle profiles of COLO16 cells were assessed by FACS 48 hours after siRNA transfection. Data shown are mean ± SD, of at least 3 independent experiments. * p<0.05, ** p<0.01, ***p<0.001, Student’s t test (two-tailed).
Given that overexpression of miR-30c-2* and miR-497 can impair cell proliferation and induce apoptosis, we asked whether depletion of these putative targets could produce the same biological effects. To do so, we utilized 2 independent siRNAs to knockdown each individual target in COLO16 cells (Supplementary Fig. 5A). After demonstrating that either siRNA could induce similar effects (Supplementary Fig. 5, A–C), we selected the siRNA that induced the greatest knockdown for further testing (Fig. 5C). Knockdown of all 7 targets in SRB12 cells caused decreased cell growth (Supplementary Fig. 5D). Conversely, knockdown of FAT2 and CDK6 caused an increase in cell proliferation in COLO16 cells, whereas knockdown of KIF18B, PKMYT1, ORC1, and AURKA resulted in significant reductions in COLO16 growth (Fig. 5D). Knockdown of FAT2, ORC1, KIF18B, PKMYT1, and AURKA led to significant increases in cell death in COLO16 cells, (Fig. 5, E and F). In SRB12 cells, however, only knockdown of AURKA induced apoptosis (Supplementary Fig. 5E). We next asked whether inhibition of these targets can phenocopy the effects that miR-30c-2* or miR-497 overexpression had on the cell cycle of COLO16 cells. Cell cycle profiling showed that ORC1 knockdown resulted in an apparent G2/M arrest, resembling the effects of miR-30c-2* overexpression (Fig. 5G and Supplementary Fig. 5F). Similarly, AURKA inhibition in both cell lines resulted in significant reductions in the percentage of cells in G1 and S phase, and increases in the percentage of cells in G2 and M phase, indicative of a G2/M arrest. Interestingly, the percentage of cells that stained positive for H3 S28 phosphorylation was significantly elevated in AURKA knockdown cells. This phenotype resembles mitotic catastrophe, a process in which aberrant mitotic activity results in cell death or irreversible cellular senescence (33).
Given the potent anti-tumor effect of AURKA inhibition, we hypothesized that miR-497 suppresses cuSCC cell growth primarily through AURKA inhibition. To test this hypothesis, we attempted to rescue the effects of miR-497 overexpression by simultaneously overexpressing AURKA. We generated stable AURKA overexpressing COLO16 cells, using a retroviral pBABE vector harboring a puromycin selection cassette (34) and the full-length AURKA CDS (35). After puromycin selection, we were able to obtain robust overexpression of AURKA in COLO16 cells (Supplementary Fig. 5G). We subsequently transfected COLO16 cells transduced with pBABE-empty or pBABE-AURKA with a miR-497 mimic or scrambled mimic control. While miR-497 suppressed AURKA expression in COLO16+pBABE-empty cells, AURKA protein levels remained high in COLO16+pBABE-AURKA cells transfected with miR-497. We subsequently performed cell cycle profiling and found that AURKA overexpression was unable to rescue the G1/S arrest induced by miR-497 (Supplementary Fig. 5H).
These results, along with the observation that miR-497 overexpression does not recapitulate the effects of AURKA inhibition, suggest that miR-497 suppresses tumorigenesis by targeting a network of genes and cellular processes. The apparent discordance in these phenotypes may be explained by previous studies, which have shown that miR-497 targets multiple genes involved in G1/S checkpoint regulation, including CDK4/6, CCNE1, CDC25A, and CCND3 (36–38). Accordingly, miR-497-mediated inhibition of G1/S progression precedes G2/M, thus preempting the mitotic catastrophe that occurs as a result of inhibiting AURKA expression. Nevertheless, these results suggest that KIF18B, ORC1, PKMYT1, and AURKA are oncogenic drivers and potentially viable therapeutic targets for the treatment of cuSCC.
AURKA is a viable therapeutic target in cuSCC
To further investigate this miRNA-mRNA axis human cuSCC, we performed correlation analyses of miR-30c-2* and miR-497 with the respective putative mRNA targets in the human cuSCC dataset from Tsai et al (3). There was a significant negative correlation between miR-497 and AURKA (Fig. 6A) (Pearson’s r = −0.79, p-value=2.44 × 10−4) and PKMYT1 (Supplementary Fig. 6A) (Pearson’s r = −0.58, p-value=0.019) mRNA expression. The expression of miR-497 and KIF18B was negatively correlated, however this trend was not statistically significant (Pearson’s r = −0.46, p-value=0.07) (Supplementary Fig. 6B). These results suggest that miR-497 may regulate the expression of AURKA and PKMYT1 in human cuSCC.
Figure 6:
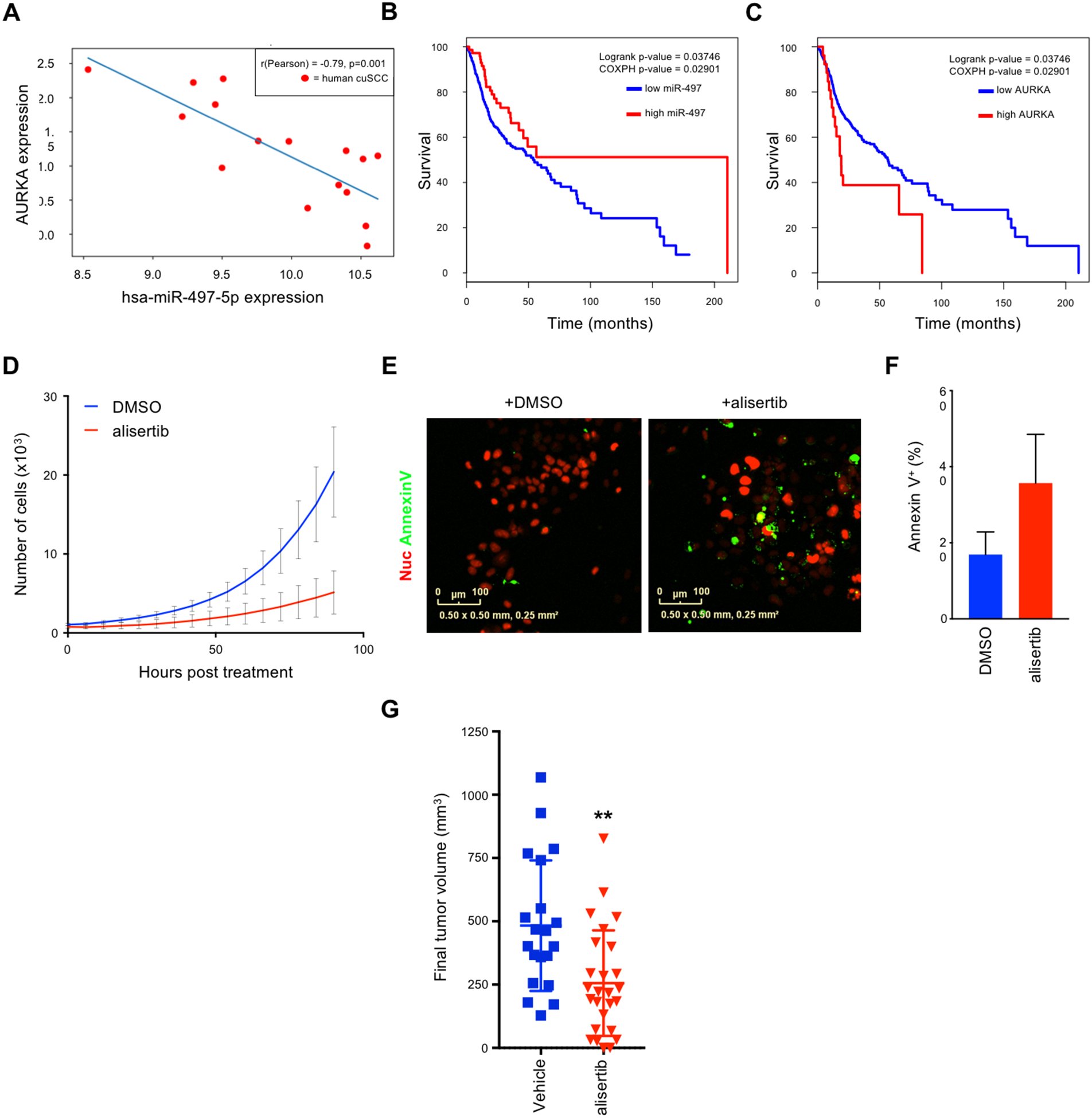
AURKA is a viable therapeutic target in cuSCC. A, Correlation analysis of miR-497 and AURKA in human cuSCC tumors. Pearson’s correlation coefficient (r) values and p values are listed. B-C, Kaplan-Meier survival curves from HNSC patients with high vs. low expression of miR-497 (B) and AURKA (C). D-F, COLO16 cells stably expressing nuclear mCherry treated with DMSO vs. alisertib (12nM), incubated with Annexin V-488, and scanned every 6 hours using the Incucyte® high-content live-cell imaging platform. D, Growth curve of COLO16 cells treated with alisertib vs. DMSO. E-F, Immunofluorescence (E) and quantification (F) for annexin V-488 (green)-positive cells following treatment with alisertib or DMSO. G, Final tumor volumes of xenograft mouse models composed of COLO16 cells subcutaneously injected into both flanks of athymic nu/nu mice. Tumor bearing mice were randomized into 2 groups and subsequently treated daily with either vehicle or alisertib (30mg/kg) via oral gavage. Data shown are mean ± SD, n=20 and 26 for vehicle and alisertib treated mice, ** p<0.01, Student’s t test (two-tailed).
We also performed correlation analysis for TAp63 and miR-30c-2* (Supplementary Fig. 6C), and miR-497 (Supplementary Fig. 6D) in the HNSCC samples profiled in The Cancer Genome Atlas (TCGA) (3). We did not find a significant correlation between TAp63 and miR-30c-2*, but we did find a modest, but significant negative correlation between TAp63 and miR-497 (Pearson’s r = −0.38, p-value=2.09 × 10−3).
We next asked whether the expression levels of miR-30c-2*, miR-497, or the targets validated in this study could predict patient survival. To do so, we leveraged the publicly available data from The Cancer Genome Atlas (TCGA). Since data for cuSCC is not available, we focused our analyses on HNSCC (8), which shares many of the same genetic features of human cuSCC (3). From this analysis, we found that high expression of miR-497 correlated with improved survival outcomes in patients with HNSCC (Fig. 6B). Conversely, HNSCC patients with high expression of AURKA showed significantly reduced survival (Fig. 6C). These observations suggest that miR-497-mediated regulation of AURKA is functionally and clinically important in cuSCC and HNSCC.
To pursue the potential therapeutic significance of these findings, we treated COLO16 cells with the AURKA-selective kinase inhibitor alisertib. Alisertib was particularly potent in its ability to reduce the viability of COLO16 cells (IC50 = 12nM) (Supplementary Fig. 6E). We then asked whether alisertib could reduce cell proliferation and induce cell death in COLO16 cells to the same extent as siRNA-mediated knockdown of AURKA. High content live cell imaging demonstrated that cell proliferation was almost completely inhibited by alisertib treatment (Fig. 6D). Similar to the siRNA-mediated knockdown of AURKA, alisertib treatment resulted in an increase in the percentage of apoptotic cells (Fig. 6, E and F).
To extend these observations to an in vivo setting, we assessed the anti-tumor effects of alisertib in a cuSCC xenograft mouse model, consisting of athymic (nu/nu) mice injected with COLO16 cells. Mice with palpable tumors were treated orally with alisertib (30mg/kg) or vehicle, daily for 14 days. Notably, alisertib treatment had an inhibitory effect on tumor growth, resulting in a 47% reduction in final tumor volume (p=0.0019) (Fig. 6G). These results indicate that AURKA inhibition may be effective in treating cuSCC, however the absence of complete tumor regression suggests that inhibition of other miR-30c-2* and/or miR-497 targets may augment this anti-tumor effect.
miR-30c-2* and miR-497 suppress cuSCC growth in vivo
Given the profound cytoxic effects of miR-30c-2* and miR-497 observed in vitro, we next set out to investigate the therapeutic potential of these microRNA mimics in an in vivo model of cuSCC. COLO16 cells stably expressing RFP-luciferase were transfected with mimics of miR-30c-2*, miR-497, or negative control, and subsequently xenografted into athymic nu/nu mice (Fig. 7A). In accordance with our in vitro observations, transfection of miR-30c-2* or miR-497 almost completely abolished the tumor growth of COLO16 cells compared to controls (Fig. 7, B-E). Notably, these microRNA mimics were far more effective in inhibiting tumor growth when compared to alisertib treatment alone. These results suggest that inhibition of AURKA alone may be insufficient to totally suppress tumor growth. Instead, treatment with miR-30c-2* and/or miR-497 themselves could be a more effective therapeutic strategy in the treatment of advanced cuSCC.
Figure 7:
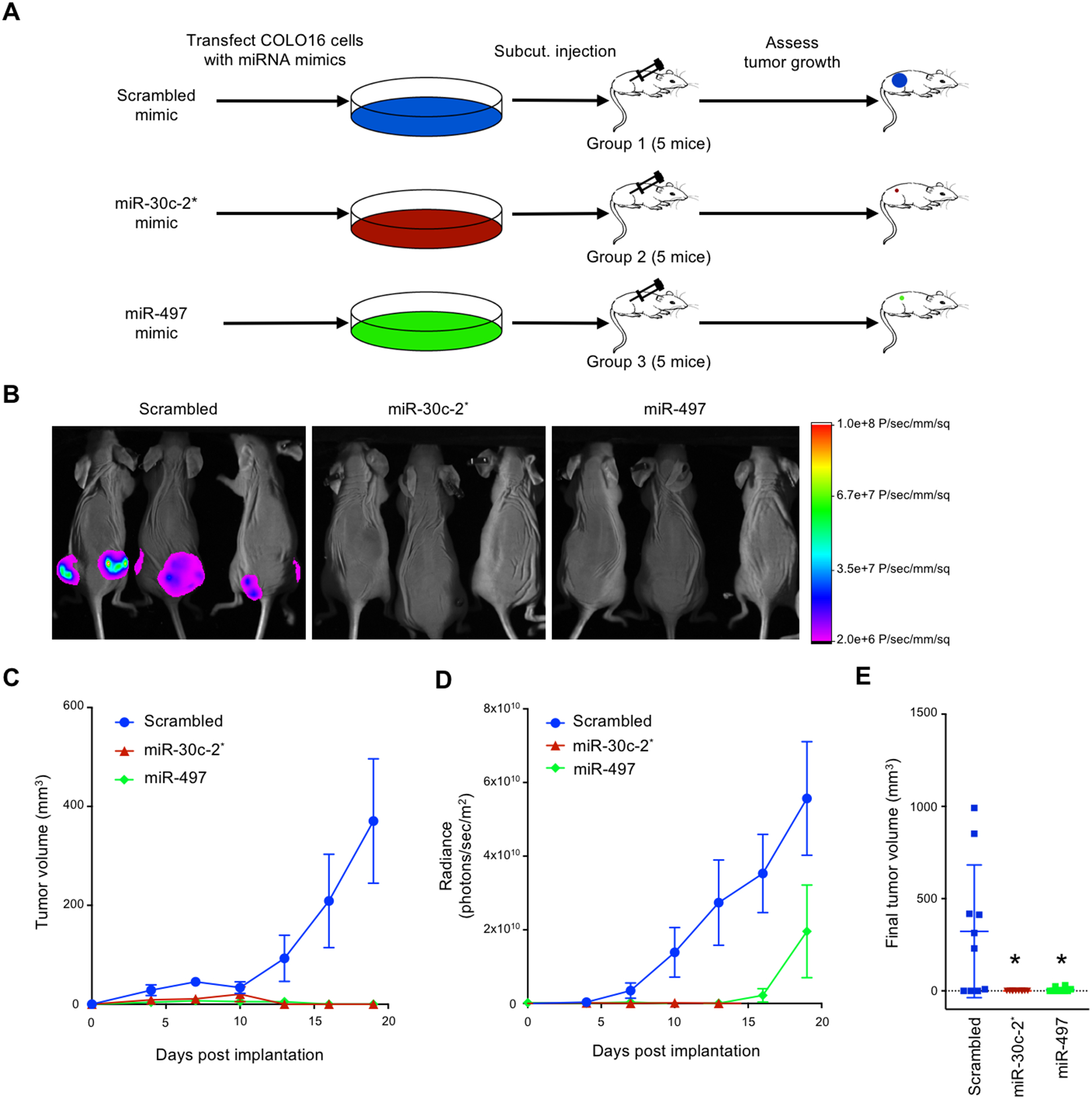
miR-30c-2* and miR-497 suppress cuSCC growth in vivo. A, COLO16 cells stably expressing RFP and luciferase were transfected with the indicated microRNA mimics and subcutaneously injected into both flanks of athymic nu/nu mice. B, Representative images of mice injected with COLO16 cells transfected with scrambled, miR-30c-2*, and miR-497–5p microRNA mimics on Day 19 prior to tumor harvest. C, Tumor volume assessed using caliper s. D, Time course of in vivo bioluminescence imaging of tumor xenografts. E, Quantification of extracted tumor volumes. Data shown are mean ± SEM, n=10. * p<0.05, Student’s t test (two-tailed).
Discussion
TAp63 exhibits tumor suppressive functions through transcriptional activation of genes involved in miRNA processing (13), nucleotide excision repair (39), cell death (40), and metabolism (41). In a previous study, we demonstrated that TAp63 functions as an important tumor suppressor through transcriptional activation of DICER (13). In accordance with this observation, the current study shows that deletion of TAp63 leads to reduced Dicer expression, and reduced expression of a number of tumor suppressive miRNAs (miR-10b, miR-34a, miR-130b, and miR-200b). While deletion of TAp63 disrupted miRNA expression, it did not lead to a global reduction of miRNA expression, as would be expected if Dicer were absolutely necessary for the biogenesis of all miRNAs. Instead, we observed varied effects on miRNA expression, with both upregulated and downregulated miRNA expression in UVR- treated - TAp63−/− skin and cuSCC. These results are consistent with a recent study, which demonstrated that Dicer is not absolutely required for the maturation of all miRNAs (42). Instead, while loss of Dicer leads to reduced biogenesis of most miRNAs, there are a number of miRNAs that are still properly processed and detected in Dicer -deficient cells. Intriguingly, loss of Dicer seemed to have a larger effect on miRNAs from the 3’ strand (i.e. −3p miRNAs) than miRNAs from the 5’ strand (i.e. −5p miRNAs), a phenotype also observed when DICER is deleted in both mouse (43) and zebrafish (44) cells. These results suggest that while DICER is required for most miRNAs, alternative mechanisms for the maturation of some miRNAs exist, which may explain why some, but not all miRNAs were found to be significantly reduced in TAp63−/− tissues. (13). Importantly, the TAp63−/− mouse expresses normal levels of ΔNp63 mRNA and protein (17), indicating that this deregulation of miRNA expression is not likely to be due to the disruption of ΔNp63-mediated regulation of DGCR8 (12).
In line with previous observations, our present study demonstrates that TAp63 is critical for the suppression of UVR-induced cuSCC. Specifically, we show that mice lacking TAp63 are more susceptible to UVR-induced SCC, and that these mice have a significantly altered microRNA and mRNA expression profile compared to WT mice. Through the integration of RNA-seq and microRNA-seq (ie small RNA-seq), we identified a network microRNAs and mRNAs that are differentially expressed and exhibit negative correlations in their expression, in both mouse and human cuSCC. Subsequent validation showed the downregulation of miR-30c-2* and miR-497 in both mouse TAp63−/− cuSCC and human cuSCC. The expression of these two miRNAs appear to be dependent on TAp63-mediated Dicer expression, as shown previously (13). Through the use of microRNA mimics we were able to establish tumor suppressive functions of miR-30c-2* and miR-497 in human cuSCC. miR-30c-2* overexpression led to reduced cell proliferation and significant apoptosis in multiple cell lines. On the other hand, miR-497 overexpression did not induce apoptosis, but rather, it induced a striking G1/S cell cycle arrest and significantly reduced cell proliferation. We further showed that re-introduction of miR-30c-2* and miR-497 into xenografts resulted in reduced tumor growth, demonstrating the therapeutic potential of these microRNAs.
Using shotgun proteomics, we identified putative targets of both miR-30c-2* and miR-497. Notably, many of the genes repressed by these miRNAs were also significantly overexpressed in both mouse TAp63−/− cuSCC and human cuSCC RNA-Seq signatures. Further validation identified five putative targets of miR-30c-2* (FAT2, ITGA6, KIF18B, and PKMYT1) and four targets of miR-497 (AURKA, CDK6, PKMYT1, and KIF18B). In line with their putative oncogenic functions, inhibition of AURKA, KIF18B, PKMYT1, and ORC1 resembled the phenotypes of the overexpression of the corresponding microRNA. The inhibition of AURKA was most effective in suppressing tumor cell proliferation and inducing apoptosis. AURKA inhibition resulted in a phenotype that was reminiscent of mitotic catastrophe, similar to previously reported phenotypes observed in other cancer cell types (45). These include reduced proliferation and defects in spindle pole assembly and chromosome condensation (46). This in turn results in defective chromosome segregation, ultimately causing mitotic cell death. Interestingly, AURKA is frequently overexpressed in human cuSCC and exhibits a negative correlation with miR-497 expression. Previous studies have shown that AURKA is frequently amplified and/or overexpressed in multiple human tumor types (47,48), and that high expression levels of AURKA correlate with late clinical stage and metastasis in HNSCC (49). Moreover, multiple AURKA inhibitors have been entered into clinical trials (50), including alisertib. Alisertib was highly effective in suppressing cuSCC cell growth and inducing apoptosis. These results are notable, as targeting AURKA using alisertib, or any other small molecule, has not been tested in the treatment of cuSCC. Alisertib treatment of cuSCC xenografts had modest anti-tumor effects, suggesting that inhibiting AURKA alone may not be sufficient to induce tumor regression. Alternatively, re-introduction of miR-30c-2* or miR-497 was highly effective in suppressing tumor growth in vivo. Together, these observations suggest that the therapeutic delivery of miR-30c-2* or miR-497 may be an even more effective therapeutic strategy for the treatment of advanced cuSCC than AURKA inhibition alone.
Our study establishes TAp63 as an essential regulator of microRNA expression during skin carcinogenesis and reveals a previously undescribed functional network of microRNAs and targeted mRNAs. These microRNA-mRNA pairs include viable, yet previously unexplored targets for the treatment of human cuSCC. These results are also significant in that they identify crucial targets for mutant p53 cuSCC. Given the lack of FDA approved targeted therapies for advanced cuSCC, our study provides preclinical evidence for the use of miR-30c-2*/miR-497 delivery or AURKA inhibition for the effective treatment of cuSCC.
Supplementary Material
Statement of significance.
This study provides preclinical evidence for the use of miR-30c-2*/miR-497 delivery and AURKA inhibition in the treatment of cuSCC, which currently has no FDA approved targeted therapies
Acknowledgements
We thank N. Bui, M. Napoli, B. Murphy, N. Montey, R. Berdeaux, N. Jenkins, L. Li, and J. Kroeger for scientific and technical advice. This work has been supported in part by the Proteomics Core, the Flow Cytometry Core Facility, the Small Animal Imaging Laboratory, and the Tissue Histology Core at Moffitt Cancer Center, an NCI-designated Comprehensive Cancer Center (P30-CA076292).
Financial Support: This work was supported by R35CA197452 to ERF and Moffitt Cancer Center Support Grant P30CA076292. ERF is a National Cancer Institute Outstanding Investigator, Moffitt Distinguished Scholar, and Scholar of the Leukemia and Lymphoma Society, the Rita Allen Foundation, and the V Foundation for Cancer Research. A.D. is an American Legion Auxiliary Fellow and a Center for Clinical and Translation Sciences TL1 Fellow (TL1TR000369). K.R. and CC. were partially supported by the Cancer Prevention Institute of Texas (CPRIT) RP170005, NIH P30 shared resource grant CA125123, NIEHS P30 Center grant 1P30ES030285
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Rogers HW, Weinstock MA, Harris AR, Hinckley MR, Feldman SR, Fleischer AB, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Archives of dermatology 2010;146:283–7 [DOI] [PubMed] [Google Scholar]
- 2.Parikh SA, Patel VA, Ratner D. Advances in the management of cutaneous squamous cell carcinoma. F1000Prime Rep 2014;6:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chitsazzadeh V, Coarfa C, Drummond JA, Nguyen T, Joseph A, Chilukuri S, et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nature communications 2016;7:12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20:6582–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li YY, Hanna GJ, Laga AC, Haddad RI, Lorch JH, Hammerman PS. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21:1447–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014;158:929–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nature reviews Cancer 2013;13:136–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer cell 2006;9:45–56 [DOI] [PubMed] [Google Scholar]
- 11.Venkatanarayan A, Raulji P, Norton W, Chakravarti D, Coarfa C, Su X, et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Napoli M, Venkatanarayan A, Raulji P, Meyers BA, Norton W, Mangala LS, et al. DeltaNp63/DGCR8-Dependent MicroRNAs Mediate Therapeutic Efficacy of HDAC Inhibitors in Cancer. Cancer cell 2016;29:874–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010;467:986–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell 2012;148:1172–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nature genetics 2011;43:854–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell stem cell 2009;5:64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park YW, Younes MN, Jasser SA, Yigitbasi OG, Zhou G, Bucana CD, et al. AEE788, a dual tyrosine kinase receptor inhibitor, induces endothelial cell apoptosis in human cutaneous squamous cell carcinoma xenografts in nude mice. Clinical cancer research : an official journal of the American Association for Cancer Research 2005;11:1963–73 [DOI] [PubMed] [Google Scholar]
- 19.Martins VL, Vyas JJ, Chen M, Purdie K, Mein CA, South AP, et al. Increased invasive behaviour in cutaneous squamous cell carcinoma with loss of basement-membrane type VII collagen. Journal of cell science 2009;122:1788–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 2012;7:562–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research 2014;42:D68–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Therneau TM, Grambsch PM. Modeling survival data : extending the Cox model. New York: Springer; 2000. xiii, 350 p. p. [Google Scholar]
- 24.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic acids research 2019;47:D442–D50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowe DE, Carroll RJ, Day CL Jr., Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol 1992;26:976–90 [DOI] [PubMed] [Google Scholar]
- 26.Creighton CJ, Nagaraja AK, Hanash SM, Matzuk MM, Gunaratne PH. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. Rna 2008;14:2290–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunaratne PH, Coarfa C, Soibam B, Tandon A. miRNA data analysis: next-gen sequencing. Methods in molecular biology 2012;822:273–88 [DOI] [PubMed] [Google Scholar]
- 28.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002;416:560–4 [DOI] [PubMed] [Google Scholar]
- 29.Mizrahi A, Barzilai A, Gur-Wahnon D, Ben-Dov IZ, Glassberg S, Meningher T, et al. Alterations of microRNAs throughout the malignant evolution of cutaneous squamous cell carcinoma: the role of miR-497 in epithelial to mesenchymal transition of keratinocytes. Oncogene 2018;37:218–30 [DOI] [PubMed] [Google Scholar]
- 30.Wang A, Landen NX, Meisgen F, Lohcharoenkal W, Stahle M, Sonkoly E, et al. MicroRNA-31 Is Overexpressed in Cutaneous Squamous Cell Carcinoma and Regulates Cell Motility and Colony Formation Ability of Tumor Cells. PloS one 2014;9:e103206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods 2015;12:697. [DOI] [PubMed] [Google Scholar]
- 32.Orom UA, Lund AH. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 2007;43:162–5 [DOI] [PubMed] [Google Scholar]
- 33.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell death and differentiation 2018;25:486–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic acids research 1990;18:3587–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crane R, Kloepfer A, Ruderman JV. Requirements for the destruction of human Aurora-A. Journal of cell science 2004;117:5975–83 [DOI] [PubMed] [Google Scholar]
- 36.Furuta M, Kozaki K, Tanimoto K, Tanaka S, Arii S, Shimamura T, et al. The tumor-suppressive miR-497–195 cluster targets multiple cell-cycle regulators in hepatocellular carcinoma. PloS one 2013;8:e60155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He XX, Kuang SZ, Liao JZ, Xu CR, Chang Y, Wu YL, et al. The regulation of microRNA expression by DNA methylation in hepatocellular carcinoma. Mol Biosyst 2015;11:532–9 [DOI] [PubMed] [Google Scholar]
- 38.Xie Y, Wei RR, Huang GL, Zhang MY, Yuan YF, Wang HY. Checkpoint kinase 1 is negatively regulated by miR-497 in hepatocellular carcinoma. Med Oncol 2014;31:844. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Lin M, Zhang C, Wang D, Feng Z, Hu W. TAp63gamma enhances nucleotide excision repair through transcriptional regulation of DNA repair genes. DNA repair 2012;11:167–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sen T, Sen N, Huang Y, Sinha D, Luo ZG, Ratovitski EA, et al. Tumor protein p63/nuclear factor kappaB feedback loop in regulation of cell death. The Journal of biological chemistry 2011;286:43204–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su X, Gi YJ, Chakravarti D, Chan IL, Zhang A, Xia X, et al. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell metabolism 2012;16:511–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YK, Kim B, Kim VN. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proceedings of the National Academy of Sciences of the United States of America 2016;113:E1881–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ravi A, Gurtan AM, Kumar MS, Bhutkar A, Chin C, Lu V, et al. Proliferation and tumorigenesis of a murine sarcoma cell line in the absence of DICER1. Cancer cell 2012;21:848–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 2010;328:1694–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Gandara DR. Aurora kinases as anticancer drug targets. Clinical Cancer Research 2008;14:1639–48 [DOI] [PubMed] [Google Scholar]
- 46.Tanaka E, Hashimoto Y, Ito T, Okumura T, Kan T, Watanabe G, et al. The clinical significance of Aurora-A/STK15/BTAK expression in human esophageal squamous cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2005;11:1827–34 [DOI] [PubMed] [Google Scholar]
- 47.Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer cell 2006;9:485–95 [DOI] [PubMed] [Google Scholar]
- 48.Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2003;9:991–7 [PubMed] [Google Scholar]
- 49.Reiter R, Gais P, Jutting U, Steuer-Vogt MK, Pickhard A, Bink K, et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2006;12:5136–41 [DOI] [PubMed] [Google Scholar]
- 50.Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J. Aurora kinases: novel therapy targets in cancers. Oncotarget 2017;8:23937–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw RNA and small RNA sequencing data from all human and mouse samples that were used to support the findings of this study have been deposited in NCBI/GEO with Series accession code GSE146258. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (24) partner repository with the dataset identifier PXD014261 and 10.6019/PXD014261.