

Abstract
The anti-inflammatory drug candidate, 6-shogaol, has demonstrated excellent efficacies in various in vitro studies. However, its rapid metabolism after oral administration results in poor bioavailability and undetectable in vivo pharmacokinetics. Here, we constructed a natural-lipid (NL) nanoparticle drug delivery system (NP-DDS) to encapsulate 6-shogaol and undertake its controlled release to the proposed drug target (colon). Our in vitro drug-release assay revealed that NL-encapsulated 6-shogaol (6-S-NL) exhibits a delayed drug-release profile compared to free 6-shogaol (free-6-S). Consistent with our expectations, orally administrated 6-S-NL exhibits a superior anti-inflammatory efficacy likely due to the controlled release compared to free 6-S in a dextran sulfate sodium (DSS)-induced mouse model of colitis. Although 6-S-NL treatment yields an enhanced concentration of 6-shogaol at the target site (colon), this concentration is still far below the effective level. We hypothesize that the released 6-shogaol undergoes rapid metabolism and that the metabolites of 6-shogaol may contribute to the anti-inflammatory efficacy of 6-S-NL. We thus examined the in vitro anti-inflammatory efficacies of two highly abundant colonic metabolites, M2 (a cysteine-conjugated metabolite) and M13 (a glutathione-conjugated metabolite), against macrophage cells. Reverse transcription-polymerase chain reaction (RT-PCR) data showed that both M2 and M13 (at 1.0 μg/mL) could down-regulate pro-inflammatory factors (TNF-α, IL-1β, and IL-6) and up-regulate an anti-inflammatory factor (IL-10) in inflamed Raw 264.7 cells. Subsequent in vitro wound-healing assays also confirmed that M2 and M13 accelerate the wound recovery process of Caco-2 cells at the concentrations seen in the colon (1.0 μg/mL). Further, in the DSS-induced mouse model of colitis, oral administration of M2- or M13-loaded NL nanoparticles (M2-NL, M13-NL) demonstrated excellent in vivo wound-healing effects, and these activities were better than those observed for 6-S-NL. Combined with the 6-S-NL’s bio-distribution assay, our data show that: the 6-shogaol metabolites, M2 and M13, are more potent anti-inflammatory compounds than 6-shogaol itself; NL nanoparticles can effectively deliver 6-shogaol to the colon, with little accumulation seen in the kidney or liver; and the actions of M2 and M13 mostly confer the anti-inflammatory effect of 6-S-NL. Our results explained the discrepancy between the low tissue concentrations of NL delivered 6-shogaol and its effectiveness against ulcerative colitis (UC) in a mouse model. This study paved the way for further developing the NL-loaded active metabolites, M2 or M13, as novel targeted therapeutic approaches for curing UC.
Keywords: natural-lipid nanoparticles, 6-shogaol, phase II metabolites, colon targeted drug delivery
Graphical abstract
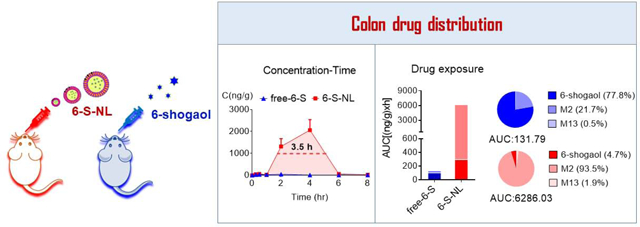
Natural-lipid nanoparticle encapsulation significantly increases the bio-distribution of 6-shogaol and its metabolites M2 and M13 to the colon.
1. Introduction
The United States has experienced a significant increase in the incidence of inflammatory bowel disease (IBD) in adults since 1999 (0.9% or 2 million in 1999 versus an estimated 1.3% or 3 million in 2015).[1, 2] Commonly used oral formations (such as tablets or capsules) have proven to be of limited use in treating IBD.[3] These approaches, which are generally designed to enhance the systemic absorption of the drug, are not specifically tuned to target the colon.[4] Thus, they have many drawbacks, including unspecific bio-distribution and ineffective control of drug release, and exhibit significant therapeutic variations due to the altered colonic environment related to IBD symptoms (e.g., diarrhea) and the limited targeting of inflamed areas.[5] The currently used medications, which include anti-inflammatory drugs (including amino-salicylates and corticosteroids), immunosuppressants, and antibiotics, can only temporarily induce and maintain remission. At present, 80% of Crohn’s disease (CD) and 45% of ulcerative colitis (UC) patients will require more than one surgical intervention in their lifetime.[6, 7] Furthermore, the enhanced permeability of the upper intestinal tract in IBD patients may mean that the utilized medicines will cause serious side effects.[4] Therefore, researchers are seeking to develop novel carrier systems capable of delivering drugs specifically to the inflamed regions for IBD treatment.
Among the novel drug-delivery systems (DDSs) introduced in recent years, nanoparticle (NP)-based DDSs are promising carriers for improving IBD therapy.[4, 8] Various NP-DDSs (such as liposomes, mesoporous silica, and polymer NPs) have been rationally designed and synthesized. After oral administration, these NPs can release the encapsulated drug at a specific pH value, resist digestive enzymes, and undergo microbiota-mediated cleavage for activation.[3, 9–11] In general, these engineered DDSs provide several substantial advantages over the conventional formulations; for example, they can protect the encapsulated drug against degradation, improve the solubility of the drug, enhance topical absorption, and enable sustained drug release.[5, 12] However, exposure of the gastrointestinal (GI) tract to these synthesized NPs also increases the possibility of acute and chronic toxicity [13–15] and may trigger detrimental immune responses in the digestive system. [16] Employing edible, nontoxic, natural NPs to deliver a drug to its specific target(s) may offer a better option than using the synthetic NPs.[17, 18]
Ginger (Zingiber officinale Roscoe) has been widely used as a spice, dietary supplement, and traditional medicine for centuries.[19–22] Our group recently demonstrated that a specific population of ginger-derived NPs could effectively reduce colitis.[18] These ginger-derived natural NPs contain pharmacologically active compounds of ginger (such as gingerols and shogaols) and can ameliorate colitis and colitis-associated colon cancer in established animal models. Importantly, these ginger-derived NPs are non-toxic at their therapeutic concentration.[18] Further, we extracted the lipids from these active ginger-derived NPs and reassembled them to liposomal NPs with a uniform diameter (around 180 nm). These naturally occurring lipids proved to be a superior resource for producing re-engineered novel lipid NPs that capable of encapsulating different drug candidates (such as 6-shogaol, siRNA-CD98) or commercialized drugs (doxorubicin). Orally delivering these lipid NPs can target the large intestine to improve the loaded drug’s treatment of colon diseases.[12, 23, 24]
6-shogaol has received a great deal of research attention because it is one of the most potent active components in ginger and has been shown to offer a variety of beneficial effects, such as anti-inflammatory, anti-oxidative, and anti-carcinogenic activities.[25–27] Since 6-shogaol is a hydrophobic phenolic compound and its rapid metabolism results in poor bioavailability, the concentrations of 6-shogaol in the target tissues and the circulation system are far below its therapeutic concentration observed in vitro.[28, 29] Although scientists have obtained many promising results in preclinical efficacy studies, 6-shogaol failed to enter clinical study due to its lack of a sufficient blood-drug concentration in human tests and consequent unavailability to yield measurable pharmacokinetics. [22, 28]
Recently, an important animal study confirmed that the anti-inflammatory activity of 6-shogaol was maintained when it was encapsulated in Poly(DL-lactide-coglycolide)/poly(DL-lactic acid)-Poly(ethylene glycol)-Folic acid (PLGA/PLA-PEG-FA) NPs.[12] In this study, we orally delivered ginger natural lipid encapsulated 6-shogaol NPs to the large intestine and observed that their therapeutic effect against UC was superior to that of free 6-shogaol. However, subsequent preliminary testing revealed that 6-shogaol was present in the blood and targeted tissue (colon) at deficient concentrations. Therefore, we investigated how this limited target tissue concentration of 6-shogaol can generate superior therapeutic effects when orally delivered using a natural-lipid NP-DDS.
2. Materials and methods
2.1. Chemicals
6-shogaol was purchased from Sigma-Aldrich (St. Louis, MO, USA), and two metabolites of 6-shogaol (M2 and M13) were purchased from Chembind LLC (Alpharetta, GA, USA). Carboxymethylcellulose sodium salt (CMC-Na, medium viscosity), KCl, methanol, dichloromethane, and acetonitrile (LC-MS grade) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Formic acid (98%, LC-MS grade) and phosphate buffer saline (Corning ™ PBS, 1x) were obtained from Fisher Scientific (Hampton, NH, USA). Ultrapure deionized water was supplied by a Milli-Q water system (Millipore, Bedford, MA).
2.2. Preparation of ginger-derived nanoparticles-2 (GDNPs-2) and total lipid extraction
Fresh ginger (Zingiber officinale) roots (~3.0 kg) were purchased from Your DeKalb farmers’ market (DeKalb, GA, USA). GDNPs-2 (~200 mg) was obtained using the ultracentrifugation method reported in our previous paper.[18] Total lipids extraction was extracted using a modified liquid-liquid extraction method reported by Bligh and Dyer.[30] In general, 60 mL of methanol/dichloromethane with a volume ratio of 2:1 (v/v) was added to 15 mL of GDNPs-2 suspension (1 mg/mL in PBS) in a glass separatory funnel. After mixing, dichloromethane (20 mL) and ddH2O (20 mL) were added sequentially into the funnel. The mixture was shaken thoroughly at room temperature for 5 to 10 times, and after standing for 15 mins, the bottom phase (organic phase) was separated and transferred into a fresh glass separatory funnel. Then the sample was washed once with 5 mL KCl solution (1 mol/L) and once with 5 mL of water. Finally, the collected organic phase was dried in a vacuum rotavapor at 45 °C.
2.3. Fabrication and characterization of 6-S-NL, M2-NL, and M13-NL
0.5 mL 6-shogaol (2.0 mg/mL) in methanol was mixed with total lipids (5.0 mg) in dichloromethane and dried under reduced pressure to obtain a thin lipids-complex film. The dried 6-shogaol GDNPs-2 lipids film was then suspended in 2.5 mL PBS buffer (1x, Fisher scientific). After a bath sonication for 5 minutes along with proper pipetting, an equal volume of PBS buffer was added and sonicated for another 5 minutes. Finally, the suspension was passed through AVESTIN liposomes extruder (Ottawa, ON) for 10 to 20 times (at 55 °C) with a 200 nm polycarbonate membrane. M2-NL and M13-NL were made by the identical protocol, as described above.
Particle sizes of prepared obtained NPs were measured using a Malvern Zetasizer Nano ZS90 Apparatus (Malvern Instruments, Worcestershire, UK) at room temperature. Atomic force microscopy (AFM) images of GDNPs-2 and 6-S-NL were captured by a nanosurf CoreAFM instrument (Nanosurf, Liestal, Switzerland).
2.4. Drug (6-shogaol, M2, and M13) loading efficiency
The loading efficiencies of 6-shogaol, M2, or M13 to lipid NPs were carried out using Slide-A-Lyzer mini dialysis devices (10 K molecular weight cutoff [MWCO], 15 mL). Briefly, dialysis devices were stabilized by adding 0.5 mL release medium (3% MeOH and 0.1% Polysorbate 80 in phosphate buffer saline [PBS], pH 7.4) to the apical side of the dialysis tube. The basal side of the tube was filled with a 12.5 mL release medium. All the mini dialysis devices were stabilized at room temperature for approximately 30 min before use. Then 1mL of drug-loaded NPs (1mg/mL) was added to the apical side of the tube. The samples were shaken at 200 rpm at room temperature, and aliquots (0.5 mL) were taken out respectively from the apical and basal sites of the dialysis inserts after 2 hr. All samples were mixed with the same volume of acetonitrile and filtered through a 0.22-μm syringe filter before injection. High-performance liquid chromatography (HPLC) analysis was performed to evaluate 6-shogaol loading efficiency using an Agilent® 1100 LC System (Agilent Technologies Inc.; CA, USA). A Zobax C18 column from Agilent (2.1 × 50 mm, 5 μm, 80 A) was employed for the separation, and the UV detection wavelength was set at 282 nm. The drug loading efficiency was calculated using the formula: (encapsulated drug/total drug) ×100%.
2.5. In vitro drug (6-shogaol) release
The 24 hr release kinetics of 6-shogaol from NPs were evaluated using a previously described dialysis method. Briefly, Slide-A-Lyzer mini dialysis devices (10 K MWCO, 15 mL) were stabilized by adding 0.5 mL release medium a (3% MeOH and 0.1% Polysorbate 80 in PBS, pH 7.4), b (3% MeOH and 0.1% Polysorbate 80 in simulated gastric fluid [SGF], pH 1.4), or c (3% MeOH and 0.1% Polysorbate 80 in simulated intestinal fluid [SIF], pH 6.9), respectively, to the apical side of the dialysis tube. The basic side of the tube was filled with 12.5 mL release medium a, b, or c accordingly. All the mini dialysis devices were incubated at 37°C for approximately 30 min before use. Subsequently, 1 mL of each of the reconstituted 6-S-NL (1 mg/mL in PBS) was added to the apical side of the dialysis tube. The release test was performed at 200 rpm and 37°C; 1 mL of the released sample was collected from the basal tube at each time point (1, 2, 4, 8, 12, and 24h). The release medium was continuously replenished to the basal tube to maintain its volume at 12.5 mL. To compare the release patterns of free-6-shogaol, we placed 1 mg/mL 6-shogaol CMC-Na suspension in the dialysis tube, and the release of 6-shogaol was measured using the same method as described for the 6-S-NL release measurements. The content of 6-shogaol in each sample was measured using the high-performance liquid chromatography (HPLC) method. All samples were filtered through a 0.22-μm syringe filter before injection. HPLC analysis was performed using an Agilent® 1100 LC System [Agilent Technologies Inc.; CA, USA]. A Zobax C18 column from Agilent (2.1 × 50 mm, 5 μm, 80 A) was employed for the separation, and the UV detection wavelength was set at 282 nm. The obtained concentration-time data was input to Windows excel (2013) to calculate the different modeling parameters [31], and the results were transferred to Graphpad Prism (Version 8.3.1) to visualize the drug release kinetics.
2.6. Visualization of intracellular 6-S-NL delivery
Confocal microscopy was used to visualize the intracellular delivery of 6-S-NL. First, 6-S-NL was labeled with a lipid dye (DIO, D275, Fisher Scientific, IL, USA) by incubating 50 μL DIO (0.1 mg/mL in DMSO) with 5 mL 6-S-NL suspension (0.2mg/mL in PBS) for 30 mins at room temperature. Then, CT-26.WT (fibroblast cells) or Raw 264.7 (macrophage cells) with a concentration of 0.5×105 per well were seeded in eight-chamber tissue culture glass slides (Falcon® Culture Slides, Corning, NY, USA) for overnight. Next, 50 μL DIO-labeled 6-S-NL (6-S-NL/DIO) was added to 0.5 mL of culture medium and incubated with cells for 0.5, 1, 1.5, 2, 3, 4, and 5 h. Cells with blank culture medium were used as a negative control. The cells were fixed with 4% paraformaldehyde (PFA) for 15 min and washed with PBS (three times). After being permeabilized with 0.1% Triton X-100 for 5 min, the plate was rewashed with PBS, and 100 μL of phalloidin-Tetramethylrhodamine B isothiocyanate (TRTIC, Sigma-Aldrich, MO, US) [1:50 dilution in PBS] was added. Then the plate was incubated for an additional 30 min. Finally, cells were washed with PBS twice, dried in the dark, and coverslip-mounted with mounting medium, which contains 4-,6-diamidino-2-phenylindole (DAPI). Images were taken using Zeiss (LSM 800) confocal fluorescence microscopy. To obtain comparable data, we adjusted all photomultipliers to the same sensitivity (gain/offset). The fluoresce intensities of 6-S-NL/DIO (green channel) were quantitated by ImageJ software (https://imagej.nih.gov/ij).
2.7. Cell culture for in vitro anti-inflammation assay
Raw 264.7 macrophage cells were purchased from the American Type Culture Collection (ATCC, VA, USA) and were cultured in Dulbecco’s Modified Eagle Medium (DMEM) medium. The culture medium was supplemented with penicillin (100 U/mL), streptomycin (100 U/mL), and 10% heat-inactivated fetal bovine serum (FBS, Atlanta Biologicals; GA, USA). Cells were cultured to 75% confluence in 6-well plates at 37°C in a humidified atmosphere containing 5% CO2. Different drug candidates (6-shogaol, M2, or M13) or control (blank medium) were incubated with cells for 12 to 16 hours (overnight). The activation of macrophages was optimized by replacing the drug-containing medium with lipopolysaccharide (LPS)-containing medium (at 250 ng/mL) for 4 hours of incubation times.
2.8. RNA extraction and quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from macrophage cells or homogenized colon tissues by an RNeasy Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Concentrations of extracted RNA were determined by a Synergy-2 microplate reader (BioTek, Winooski, VT, USA). cDNA was generated from the extracted total RNA using a Maxima cDNA Synthesis kit (Thermo Scientific, Lafayette, CO, USA). Expression level of target mRNAs was determined by qRT-PCR, using Maxima SYBR Green/ROX (6-carboxyl-X-rhodamine) qPCR Master Mix (Thermo Scientific) and the following primer pairs: TNF-α, 5’-AGG CTG CCC CGA CTA CGT-3’ (forward) and 5’-GAC TTT CTC CTG GTA TGA GAT AGC AAA-3’ (reverse); IL-1β, 5’-TCG CTCA GGG TCA CAA GAA A-3’ (forward) and 5’-CAT CAG AGG CAA GGA GGA AAA C-3’ (reverse); IL-6, 5’-ACA AGT CGG AGG CTT AAT TAC ACA T-3’ (forward) and 5’-TTG CCA TCC GCA CAA CTC TTT TC-3’ (reverse); IL-10, 5’-GGT TGC CAA GCC TTA TCG GA-3’ (forward) and 5’-CTT CTC ACC CAG GGA ATT CA-3’ (reverse); and 36B4, 5’-TCC AGG CTT TGG GCA TCA-3’ (forward) and 5’-CTT TAT CAG CTG CAC ATC ACT CAG A-3’ (reverse).
2.9. In vitro wound-healing assay
Healing of wounded intestinal epithelial monolayers (Caco-2 BBE) by 6-shogaol, 6-SNL, M2-NL, or M13-NL were performed using electric cell-substrate impedance sensing (ECIS) technology (ECIS model 1600R; Applied BioPhysics, NY, USA). An optimized concentration (at 1.0 μg/mL) was chosen for wound healing study based on previous in vitro experiments of ginger derived bioactive compounds and in vivo pharmacokinetic data. Caco-2 BBE cells (ATCC, VA, USA) were cultured in 8-well culture dishes (ECIS 8W1E plate) with 75% confluence in the measurement chamber consists of a humidified atmosphere containing 5% CO2. Once cells reached confluency, medium (with or without testing samples [6-shogaol, M2, or M13]) was added. For wounding, monolayers grown on ECIS 8W1E plates were subjected to a 30 s pulse with a frequency of 40 kHz and amplitude of 4.5 V. Basal resistance was measured at a frequency of 500 Hz and a voltage of 1 V. [18]
2.10. Pharmacokinetics (PK) of free 6-S versus 6-S-NL
2.10.1. Animal
Female C57BL/6J mice (7–8 weeks old) were purchased from the Jackson Laboratory. Animal experiments were performed in accordance with Georgia State University guidelines covering the humane care and use of animals in research. All animal procedures used in this study were approved by the University Committee on Use and Care Animals at Georgia State University (IACUC, Protocol # A17044).
Mice were divided into two groups, namely free 6-shogaol group and 6-S-NL group. PK studies were performed following a single oral (PO) administration of 6-shogaol CMC-Na suspension (5 mg/kg, 0.5% CMC-Na in PBS) and 6-S-NL (in PBS solution) at 5 mg/kg. All animals were acclimatized for more than three days in the experimental area. Mice were fasted for 4 hrs. before dose and water were provided ad libitum through the study period. Animals were marked and housed (three per cage) in polypropylene cages and maintained in controlled environmental conditions with 12 h light and dark cycles. The temperature and humidity of the room were maintained between 22–23°C and 30–70%, respectively. The sparse sampling design was used to collect blood samples from animals at 0 (pre-dose), 5, 10, 15, 30 and 45 min, and at 1, 1.5, 2, 4, 6, and 8 h into K2EDTA (200 mM, 20 μL/mL of blood) coated tubes. At the given time points, the mice (3 mice/time point) were under anesthesia by 3% isoflurane, and 0.3–0.4 mL blood was immediately collected via the retro-orbital sinus by blood-collecting tubes. Plasma was harvested from the blood by centrifugation of samples at 8 000–10 000g for 10 min. After the blood collection, all mice were euthanized, organs (stomach, small intestine, colon, liver, and kidney) from mice at 0 (pre-dose), 15, 30 min, and 1, 2, 4, 6, and 8 h were immediately collected, all the organs were rinsed extensively in phosphate-buffered saline (pH 7.4) to remove residual blood and internal contents and stored at −80°C until further analysis.
2.10.2. The stock solution, internal standard, calibration curve, and quality control
6-shogaol, M-2, and M-13 were accurately weighed and dissolved in MeOH to the concentration of 1.0 mg/mL as stock solutions and stored at −20°C. Dihydro-capsaicin (IS, internal standard) solution was prepared at a concentration of 1.0 mg/mL in MeOH and further diluted in acetonitrile to a final concentration of 100 ng/mL and stored at 4°C for sample preparation.
Calibrator standard solutions were prepared by mixing 50 μL blank plasma, or blank tissue suspension with 15 μL working solution and 85 μL IS. Standards so lutions contain blank, blank with internal standard and six non-zero calibration standards covering a lower limit of quantification (LLOQ) to 5.0 ng/mL for plasma and most tissues. A concentration range from 5.0 to 500 ng/mL was employed for the quantification of analytes. The calibration curves were generated by plotting the peak area ratios (6-shogaol, M2, and M13 peak area/IS peak area) against the concentrations of specific standards, using linear regression and with no weighting factor. Quality control (QC) working solutions at low, medium, and high were prepared using a separately weighed and prepared stock solution. QC samples were evenly distributed among samples.
2.10.3. Sample preparation
All samples were processed by the protein precipitation method. To prepare plasma samples, we dispensed an aliquot (50 μL) of plasma (from different time points) into a 96-well plate, then we added 100 μL of ice-cold acetonitrile (contain 100 μg/mL IS) to each well and vortex mixed the plate for 2 min. The plate was centrifuged at 4500 rpm for 10 min at 4 °C to precipitate protein. An aliquot of supernatant (100 μL) was transferred to 1.5 ml auto-sampler vials, and 5 μL was injected into LC-MS/MS for analysis. Tissue samples were weighed and suspended into 20% acetonitrile (80% water) with mass/volume of 1 to 10, then homogenized using a tissue homogenizer (Tissue miser Homogenizer, Fisher Scientific) for 10 min. The tissue suspension was then processed in the same manner as plasma samples for LC-MS/MS analysis. For the samples having a concentration over the upper limit of quantification, proper dilution with blank matrix was applied as needed.
2.10.4. LC-MS/MS analysis
An Agilent 6410 (Agilent Technology, Santa Clara, CA, USA) mass spectrometer with electrospray ionization source was interfaced with Agilent 1200 high-performance liquid chromatography (HPLC) system. The Agilent MassHunter (B.07.00) was used to control the liquid chromatography-tandem mass spectrometry system (LC-MS/MS) and for data acquisition and processing. The separation was performed on a Zobax C18 column, 50 mm length and 2.1 mm ID, 3.5 um (Agilent, Agilent Technology, Santa Clara, CA, USA) at a flow rate of 0.20 mL/min. The mobile phase consisted 100% H2O with 0.1% formic acid as A and 100% acetonitrile with 0.1% formic acid as B. The gradient started with 35%B for 4 min, linearly increased to 95%B at 8 min, kept at 95% B for 5 min, decreased to 35%B at 13.1 min and kept 35% for 5 min. The mass spectrometer was operated in a positive mode with multiple reaction monitoring (MRM) for analysis. The gas temperature was 300 °C with the ion spray voltage of 3500 V, drying gas flow of 10L/min, nebulizer of 30 psi, ion dwell time of 50ms, and cell accelerator voltage 4 V. The MRM transitions and retention time (RT) were monitored at m/z 277.2→137.1 for 6-shogaol, m/z 398.2→137.1 for M2, m/z 584.3→179.1 for M13, and m/z 308.1→137.1 for internal standard.
2.10.5. Pharmacokinetic (PK) data analysis
PK parameters of 6-shogaol, M2, and M13 from free 6-shogaol and 6-S-NL were calculated from the concentration-time data using the non-compartmental analysis tool of PKSolver.[32] We used the linear trapezoidal rule to calculate the area under the concentration-time curve (AUClast and AUCinf). Following oral administration, peak concentration (Cmax), and time for the peak concentration (Tmax) were the observed values. The elimination rate constant value (k) was obtained by linear regression of the log-linear terminal phase of the concentration-time profile using at least three declining concentrations in the terminal phase with a correlation coefficient of more than 0.8. The terminal half-life value (T1/2) was calculated using the equation ln2/k. [32] The tissues from the gastrointestinal tract (stomach, small intestine, and colon), plasma, liver, and kidney tissue concentration-time data were compiled and plotted using GraphPad Prism (Version 6.01, La Jolla, CA, USA).
2.11. In vivo efficacy study
Acute colitis was induced in C57BL/6 mice by adding 2.0% (w/v) dextran sodium sulfate (DSS; 36 to 50 kDa; MP Biomedicals (Santa Ana, CA, USA) in drinking water for 7 d. Then, normal drinking water with or without drug treatment (6-S-NL, free 6-shogaol, M2-NL, or M13-NL) was offered to mice for the subsequent seven days. The oral dose is deduced from a human clinical study of ginger extract and the quantification study of 6-shogaol in the ginger extract. [33, 34]. Bodyweight, feces, and physical activity were monitored daily or once every two days. After day 14, mice were euthanized by CO2 followed by cervical dislocation and dissection, colons were taken out, and colon lengths and weights were measured, spleens were weighed, and small pieces of distal colon were collected for mRNA (see section 2.7.) and MPO analysis.
2.12. Quantification of fecal Lcn-2
Pre-weighted frozen fecal samples (~100 mg) were reconstituted in PBS buffer (~1mL, containing 0.1% Tween 20) and vortexed for 20 min to yield a homogeneous fecal suspension. The suspensions were then centrifuged for 10 min at 4 °C at full speed, and clear supernatants were collected for analysis. Levels of Lcn-2 were evaluated using a Duoset mouse Lcn-2 ELISA kit (R&D Systems).
2.13. Myeloperoxidase (MPO) assay
Colon tissues were homogenized in pre-chilled potassium phosphate buffer (50 mM K2HPO4 and 50 mM KH2PO4, pH 6.0) containing 0.5% hexadecyl-trimethyl-ammonium bromide (HTAB; Sigma). The homogenates were then sonicated, subjected to three freeze-thaw cycles (10 min/10 min), and centrifuged at 14,000 rpm for 15 min. The clarified supernatants were collected. O-dianisidine dihydrochloride (1 mg/mL) and 0.0005% H2O2 were added to supernatants (50 μL), or myeloperoxidase (MPO) standards and the change in absorbance at 460 nm was measured before saturation. One unit of MPO activity was defined as the amount that degraded one μmol of peroxidase per minute.
2.14. Statistical analysis
One-way and two-way analyses of variance (ANOVA) and t-tests were used to determine statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001).
3. Results
3.1. Ginger NL NPs can be effectively loaded with 6-shogaol
6-shogaol, one of the primary active components of ginger, offers many health-benefiting activities, such as anti-inflammation, anti-cancer, amelioration of age-related neurological disorders (ANDs), and attenuation of cognitive deficits.[25, 35, 36] However, when orally administrated, 6-shogaol is very unstable and easily forms conjugated metabolites through phase II metabolism.[28] Moreover, the high hydrophobicity of 6-shogaol makes it very difficult to deliver this agent to the target organ without the aid of a proper pharmaceutical formulation.
Previous research suggested that a specific population of ginger-derived natural NPs (GDNPs-2) offers good anti-inflammatory efficacy in the DSS-induced colitis model and is safer (both in vitro and in vivo) than cationic liposomes.[18] Furthermore, natural lipids isolated from GDNPs-2 can be reassembled into stable nano-sized lipid particles and produced on a large scale.[17, 18, 23]
To develop more homogeneous and reproducible NPs and make full use of the most potent phytochemical from ginger (6-shogaol), we isolated the lipids from GDNPs-2, reorganized them into NPs, and loaded them with pure 6-shogaol. Briefly, sonication was used to generate self-assembled lipid NPs in PBS, and 6-shogaol was encapsulated into the lipid NPs by hydrophobic effect. The 6-shogaol-encapsulating natural lipid NPs (6-S-NL) were size-homogenized using an AVESTIN liposome extruder with a polycarbonate membrane (pore size: 200 nm). Representative AFM images showed that the 6-S-NL particles were nearly spherical (Figure 1A), which are similar to natural GDNPs-2 (Figure 1B). 6-S-NL particles also presented a more size-homogenized appearance (Figure 1A) compared to GDNPs-2 (Figure 1B). Size distribution and zeta potential of 6-S-NL were about 180.6 nm (PDI: 0.054) and −14.5 ± 1.9 mV, respectively (Figure 1C and S1A). These values are slightly different from the original GDNPs-2, which were 246.4 nm (PDI: 0.137) and −18.0 ± 2.1 mV (Figure 1D and S1B), respectively. The values of size and zeta potential of 6-S-NL were within the ranges that have been shown to support the in vivo structural stability of NPs critically. [17]
Figure 1. Preparation and characterization of 6-shogaol-loaded ginger lipid NPs.

(A) Atomic force microscopy (AFM) characterization (the morphology) of 6-shogaol-loaded ginger lipid NPs (6-S-NL). (B) AFM characterization (the morphology) of ginger-derived nanoparticle band 2 (GDNP-2). (C) The particle size of 6-S-NL, measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS90 Apparatus. (D) The particle size of GDNP-2. (E) Representative high-performance liquid chromatography (HPLC) profile for quantifying 6-shogaol in the apical side and basal side of the dialysis device. (F) Loading efficacy of 6-shogaol in lipid NPs (three batches).
We next measured the loading efficiency of 6-shogaol in NPs by testing the 6-shogaol concentrations inside (found on the apical side minus the basal side of the dialysis device) versus outside (on the basal side) the NPs. A typical HPLC chromatograph of the apical solution versus basal solution is shown in Figure 1E. As presented in Figure 1F, data from three batches demonstrated that the NL NPs could achieve a 6-shogaol loading efficiency of up to 89.1 ± 2.6% at a lipid concentration of 0.1 mg/mL.
3.2. In vitro drug release profile indicates a delayed drug release of 6-S-NL
To investigate whether our newly constructed 6-S-NL exhibits a propensity for controlled release, we examined the drug release profiles of 6-S-NL in PBS, simulated gastric fluid (SGF), and simulated intestinal fluid (SIF). Free 6-shogaol in CMC-Na suspension was used as the control. Since 6-shogaol is a hydrophobic compound, each solution (PBS, SGF, or SIF) was supplemented with 3% methanol and 0.5% polysorbate 80 as a solvent [37]. As shown in Figure 2A, 6-S-NL exhibited a delayed drug release (occurring over 24 h) compared to the control. 6-S-NL showed a slight preference for releasing 6-shogaol in the intestine versus the stomach or PBS solution after 8 h, as the cumulative release of 6-shogaol was higher in SIF than in SGF or PBS (Figure 2A).
Figure 2. In vitro 6-shogaol release profiles and kinetic modeling of 6-S-NL in simulated intestinal fluid (SIF).

(A) The drug release profiles of 6-S-NL in PBS (red color), simulated gastric fluid (SGF, green color), or SIF (blue color). Free 6-shogaol in CMC-Na suspension was used as the control (gray color) (n=3). HPLC was used to measure the concentration of released 6-shogaol. Kinetic modeling of the release of 6-shogaol from 6-S-NL in SIF solution was fitted to (B) Higuchi model, (C) Zero-order kinetic model, (D) First-order kinetic model, (E) Korsmeyer-Peppas model, and (F) Hixson-Crowell model.
Next, we used the dialysis method to study the kinetics of drug release in different solutions. The observed concentration-time data were fitted with various mathematical models, including the zero-order, first-order, Higuchi, Korsmeyer-Peppas, and Hixson-Crowell models [31]. As shown in Figure 2B–F, the drug release of 6-shogaol in SIF appeared to be the best fit with the Higuchi’s square root model (R2=0.968) and the Korsmeyer-Peppas model (R2=0.966), followed by the first-order kinetic model (R2=0.913). The zero-order kinetic model, which indicates a concentration-independent Fickian-diffusion, was fitted with a lower correlation coefficient (R2=0.865). These results suggest that the mechanism through which 6-shogaol is released from 6-S-NL is similar to the mechanism that a drug is released from control-released liposomes [38]. This may reflect that 6-S-NL NPs and these liposomal NPs share a similar structure [18, 23]. The kinetics constant (K) and the release exponent n value, as calculated from the Korsmeyer-Peppas model, were 12.94 and 0.424, respectively, confirming that 6-shogaol was released from spherical particles (n value close to 0.43, Table S1) [39, 40].
Importantly, the release of 6-shogaol is unlikely to be an immediate gradient-triggered release, as the zero-order kinetic model presented a much lower correlation coefficient than the Higuchi and Korsmeyer-Peppas models [39]. We observed similar fitting patterns of drug release in other solutions with different pH values (PBS and SGF) (Figure S3 and S4). Thus, the in vitro release mechanism of 6-shogaol in these solutions is likely to be identical to the mechanism by which the poorly water-soluble drug is released from liposomes [37, 38].
3.3. In vitro cell uptake and intracellular delivery of 6-S-NL
To explore whether colon cells and macrophages could internalize 6-S-NL, we visualized the intracellular uptake and delivery of 6-S-NL by confocal fluorescence microscope. As shown in Figure 3A, the control group (CT-26 cells without 6-S-NL treatment) displayed no green fluorescence, whereas strong green fluorescence was detected from cells treated with 6-S-NL for 60 min. The strongest intracellular green fluorescence was seen after 3 h of incubation. These observations indicate that a significant number of 6-S-NL NPs were taken up by CT-26 cells after 60 min, and the uptake reached a peak at 3 h (Figure 3A and 3C). Similarly, control Raw 264.7 cells (macrophages without 6-S-NL treatment) displayed no green fluorescence (Figure 3B), whereas intense green fluorescence was observed in macrophages treated with 6-S-NL for as little as 30 min. The strongest intracellular green fluorescence was observed at 1.5 h of incubation (Figure 3B and 3D). These findings suggest that macrophages internalize the 6-S-NL NPs much faster than CT-26 cells. Moreover, macrophages incubated with 6-S-NL NPs (from 30 min to 2 h) showed much stronger intracellular fluorescence than CT-26 cells incubated with 6-S-NL NPs for the same durations, indicating that macrophages internalize more 6-S-NL NPs than CT-26 cells did. As the hydrodynamic diameter of 6-S-NL NPs (~180 nm) exceeded the aperture of the nuclear pore complex (20 to 50 nm), it was not surprising that the green fluorescence was distributed only in the cytoplasm of macrophages and CT-26 cells, not within the nuclei (Figure 3A and 3B). Together, our results suggest that 6-S-NL NPs can be efficiently internalized by both CT-26 cells and macrophages, and that intracellularly delivered 6-S-NL NPs may be restricted to the cytoplasm and not enter the nucleus.
Figure 3. Cellular uptake profiles and intracellular delivery of 6-S-NL.

(A) Fluorescence images of the internalization of 6-S-NL by CT-26.WT cells at different time points. (B) Fluorescence images of the internalization of 6-S-NL by Raw 264.7 cells at different time points. (C) Fluorescence intensities of 6-S-NL/DIO (green channel) in CT-26 cells (quantitated with the ImageJ program). (D) Fluorescence intensities of 6-SNL/DIO (green channel) in Raw 264.7 cells (quantitated with the ImageJ program).
3.4. Oral administration of 6-S-NL demonstrates enhanced bioavailability of 6-shogaol in the colon tissue than oral administration of free 6-shogaol
After confirming that NP encapsulation delays the release of 6-shogaol in an in vitro environment, and the NPs are uptaken efficiently by colon cells and macrophages, we next checked whether NP encapsulation contributes to the targeted delivery of 6-shogaol to the colon in a drug tissue bio-distribution study. An LC-MS/MS method with multiple reaction monitoring (MRM) was used to monitor 6-shogaol in colon tissues following oral administration of free-6-S and 6-S-NL (5 mg/kg BW) to C57/BL6J mice (female, n = 3). We compared the colon tissue drug exposure of 6-shogaol between the 6-S-NL and free-6-S treated mouse groups. As shown in Figure 4A, concentration-time (C-t) profiles of 6-shogaol exhibited a delayed distribution of 6-shogaol in the colon of the 6-S-NL treated group compared to the free-6-S group. We also found a significantly higher maximum concentration of 6-shogaol (Cmax, Figure 4B) and significantly higher drug exposure of 6-shogaol (area under the curve, or AUC; Figure 4C) in the colon tissues of the 6-S-NL treated group compared to the free-6-S group. All these data reflect that NP encapsulation enhances the colon bioavailability of loaded 6-shogaol.
Figure 4. Comparison of pharmacokinetics of 6-shogaol in the colon tissues after mice orally administrated with free-6-shogaol (free-6-S) and 6-S-NL and comparison of the in vivo efficacy of free-6-S and 6-S-NL in a mouse model of colitis.
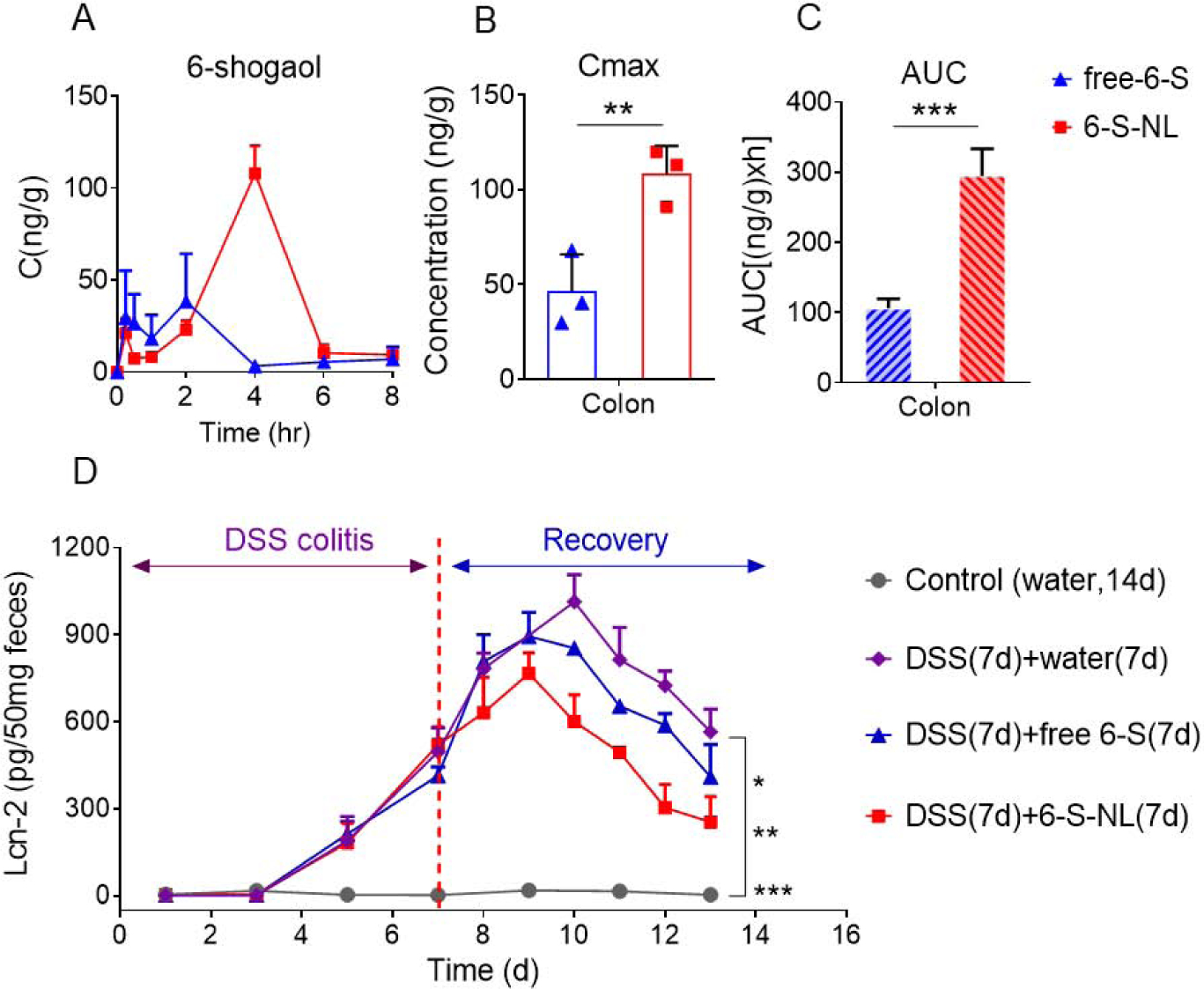
(A) C-t profiles of 6-shogaol in the colon after orally administrated with free-6-S or 6-S-NL. (n=3 per time point) (B) Cmax of 6-shogaol in the colon. (n=3 per time point) (C) AUCs of 6-shogaol in the colon. (n=3 per time point) (D) Fecal Lcn-2 levels from mice treated with water, seven days 2% DSS followed by seven days of water, seven days 2% DSS followed by seven days free-6-S treatment, and seven days 2% DSS followed by seven days 6-S-NL treatment. (n=3) (*P < 0.05, **P < 0.01, ***P < 0.001)
We also assessed whether NP-encapsulated 6-shogaol could present a better therapeutic effect than free-6-S on mucosal healing in the DSS-induced UC model, which is a well-established mouse model for studying human UC.[41, 42] We first provided mice with drinking water containing 2% DSS for 7 days (wounding phase), and then gave water alone (control group), water plus a once-daily dose of 6-S-NL (5 mg/kg), or water plus a once-daily dose of free-6-S (5 mg/kg) by gavage for an additional 7 days (healing phase). We detected the fecal biomarker, Lcn-2, for the noninvasive assessment of colitis development.[43] The control group (water only for the entire experimental period) showed no increase in the level of Lcn-2 (Figure 4D). DSS-wounded mice that did not receive any experimental formulation exhibited an increased Lcn-2 level across the 2% DSS treatment period; this was followed by a continuing increase in the Lcn-2 level from day 7 to 10 and a gradual decrease in the Lcn-2 level from day 7 to 13. As expected, the Lcn-2 levels of the 6-S-NL group demonstrated a much faster decrease from day 9 to 13 compared to the free 6-S group. These results indicate that NP-delivered 6-shogaol exhibits better mucosal healing efficacy than free 6-shogaol.
3.5. Orally administrated 6-S-NL enhanced the distribution of 6-shogaol metabolites, M2 and M13, into colon tissues
Although orally administrated 6-S-NL generated a satisfactory mucosal healing efficacy, the drug concentration of 6-shogaol in the colon was surprisingly low (Cmax: 100 ng/g, Figure 4B). This level of 6-shogaol is likely too low to generate therapeutic effects, given that an in vitro study indicated that the effective 6-shogaol concentration should be ten times higher (about 1 μg/mL).[12] A previous effort to identify the metabolites of 6-shogaol in an anticancer study revealed a high abundance of the conjugated metabolites, M2 (a cysteine-conjugated metabolite) and M13 (a glutathione-conjugated metabolite).[44, 45] Notably, these phase II metabolism-derived conjugates were subsequently found to maintain theirs in vitro anti-cancer activities.[46] We, therefore, hypothesized that M2 and M13 might be involved in the ability of 6-S-NL to facilitate mucosal healing. To test this possibility, we examined whether M2 and M13 were present in the colon tissues of the 6-S-NL treated group. An LC-MS/MS method with multiple reaction monitoring (MRM) was used to screen for M2 and M13 in colon tissues following oral administration of 6-S-NL or free-6-S (5 mg/kg BW) to C57/BL6J mice (female, n = 3 per group). Our results demonstrated that M2 and M13 were present in colon tissues at 2 hours post-administration (Figure 5A). M2 exhibited a very impressive Cmax at around 2000 ng/g of tissue (Figure 5B).
Figure 5. Orally administrated 6-S-NL enhanced the distribution of 6-shogaol metabolites, M2 and M13, into colon tissues.
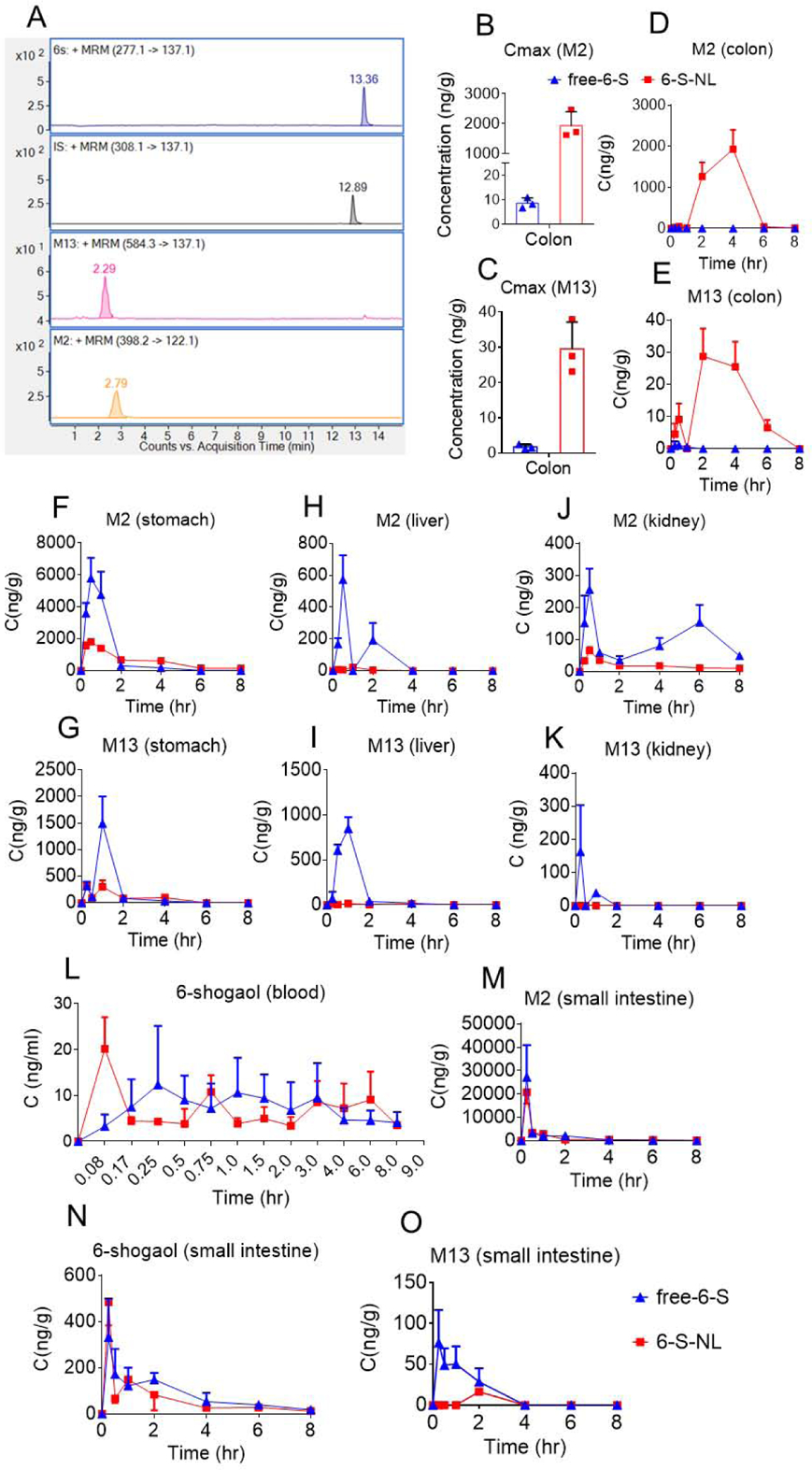
(A) Representative LC-MS/MS (ESI+, MRM) chromatograms of 6-shogaol, M2, M13, and internal standard (dihydrocapsaicin). (B-C) Cmax of M2 and M13 in colon tissues after oral delivery of free-6-S or 6-S-NL to mice. (n=3 per time point) (*P < 0.05, **P < 0.01, ***P < 0.001) (D-E) Concentration-time profiles of M2 and M13 in colon tissues after oral delivery of free-6-S, 6-S-NL to mice. (F-G) Concentration-time profiles of M2 and M13 in stomach tissues after oral delivery of free-6-S, 6-S-NL to mice. (H-I) Concentration-time profiles of M2 and M13 in liver tissues after oral delivery of free-6-S, 6-S-NL to mice. (J-K) Concentration-time profiles of M2 and M13 in kidney tissues after oral delivery of free-6-S, 6-S-NL to mice. (L) Concentration-time profiles of 6-shogaol blood after oral delivery of free-6-S, 6-S-NL to mice. (M-O) Concentration-time profiles of M2, 6-shogaol, and M13 in small intestine tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3 per time point)
To better understand how NP encapsulation improves the in vivo colon distribution of the 6-shogaol’s active metabolites, we also performed a tissue bio-distribution study to determine the levels of the M2 and M13 in the gastrointestinal tract, major metabolizing organs (liver and kidney), and blood (plasma). Using a sparse sampling design, we orally gavaged C57/BL6J mice (female, n = 3 per time point) with free-6-S or 6-S-NL (5 mg/kg BW) and used LC-MS/MS to evaluate tissues and plasma samples collected from the two groups for the presence of M2 and M13.
A comparison of the mean GI tissue concentration kinetics of free-6-S and 6-S-NL, as shown in Figure 5, confirmed that higher concentrations of M2 and M13 were achieved in the colon tissue (Figure 5B and 5C) after oral administration of 6-S-NL compared to free-6-S. This may reflect the enrichment effect of NPs in the colon.[23, 47] In the stomach, liver, and kidney, the levels of M2 and M13 were lower in the 6-S-NL group (Figure 5F–5K), indicating that lipid NPs could prevent 6-shogaol from being metabolized in non-target tissues. Concentration-time (C-t) profiles of 6-shogaol in the blood were similar between the free-6-S and 6-S-NL groups (Figure 5L), so as the M2 and 6-shogaol profiles in the small intestine (Figure 5M and 5N). However, higher M13 concentrations were observed in the small intestine of the free-6-S group (Figure 5O), confirming that, in the absence of NL loading, 6-shogaol was easier to be metabolized in the small intestine. In most of the blood samples, we were unable to detect M2 or M13 (below the LLOQ).
3.6. M2 and M13 show better in vitro anti-inflammatory activities and wound-healing effects than 6-shogaol
The above-described bio-distribution studies (e.g., the C-t profiles) revealed that M2 and M13 could accumulate in the colon when 6-shogaol was delivered by ginger lipid NPs. As M2 and M13 had previously been reported to show anti-cancer activities in vitro [46], we speculated that they might play essential roles in the efficacy of 6-S-NL. To address this hypothesis, we first used RT-PCR to determine the effects of M2 or M13 on the gene expression levels of pro-inflammatory (TNF-α, IL-1β, and IL-6) and anti-inflammatory (IL-10) factors in inflamed macrophage (Raw 264.7) cells. Our RT-PCR data showed that both M2 and M13 (at 1.0 μg/mL) could down-regulate TNF-α, IL-1β, and IL-6 and up-regulate IL-10 in inflamed Raw 264.7 cells. Moreover, both metabolites showed higher anti-inflammatory activities than 6-shogaol (Figure 6A–6D).
Figure 6. In vitro anti-inflammatory and wound-healing effects of M2 and M13.

(A) Gene expression fold changes of TNF-α, IL-1β, IL-6, and IL-10 in inflamed Raw 264.7 cells treated with 6-shogaol, M2, and M13 (1 μg/mL). (n=3) (B) Effects of 6-shogaol, M2, and M13 (1 μg/mL) treatment on the healing of wounded Caco-2 BBE cells. (n=3) (NS: not significant, *P < 0.05, **P < 0.01, ***P < 0.001)
Having confirmed that M2 and M13 can regulate pro- and anti-inflammatory cytokines of inflamed macrophage cells, we sought to uncover whether they can also accelerate the healing of wounded epithelial cells. We investigated the effects of M2 and M13 on wounded Caco2-BBE cells using ECIS technology, which calculates the impedance (Z) of a cell-coated electrode, as previously described.[48, 49] Confluent Caco2-BBE monolayers grown on ECIS 8W1E plates were wounded by a 30-s electrical pulse (40 kHz, 4.5V), and the wounded cells were treated with the control formulation, 6-shogaol, M2, or M13. ECIS measurements revealed that wounded Caco-2 epithelial monolayers treated with M2 or M13 (0.6 mL of 1.0 μg/mL solution) healed significantly faster than those treated with 6-shogaol at the same concentration (Figure 6E). These data suggest that M2 and M13 show more efficacy in promoting epithelial wound healing, compared to 6-shogaol.
3.7. In vivo anti-inflammatory efficacy of orally administrated 6-S-NL may reflect the actions of M2 or a combination of M2, M13, and 6-shogaol
Ideally, the effective in vivo drug-target concentration is consistent with the active concentration observed using in vitro assays. Our cellular anti-inflammatory and wound-healing studies showed that 6-shogaol, M2, and M13 exhibited satisfactory efficacy at a concentration of 1.0 μg/mL. Consistent with this, mice orally administrated with 6-S-NL maintained a drug-target (colon) concentration of M2 above 1.0 μg/g for about 3.3 hours (Figure 7A), reaching a maximum concentration (Cmax) of around 2.46 μg/g (Table S3). Although the concentrations of 6-shogaol and M13 were much lower than that of M2 in the 6-S-NL group, the total concentration of all three together (6-shogaol+M2+M13) remained above 1.0 μg/g for more than 3.5 hours (Figure 7B). These findings indicate that the in vivo anti-inflammatory efficacy of 6-S-NL may reflect the actions of M2 or a combination of M2, M13, and 6-shogaol. In contrast, very limited concentrations of 6-shogaol, M2, and M13 were observed in the colon of the free-6-S group, potentially explaining its relative lack of efficacy. Notably, the Cmax of 6-shogaol and its metabolites were significantly higher in colon tissues of the 6-S-NL group compared to the free-6-S group (Figure 7C, 7D), indicating that the active ingredients showed better accumulation at the colon site when 6-shogaol was loaded into lipid NPs.
Figure 7. Gastrointestinal C-t profile and maximum drug concentration (Cmax) comparison between orally administrated free-6-S and 6-S-NL.
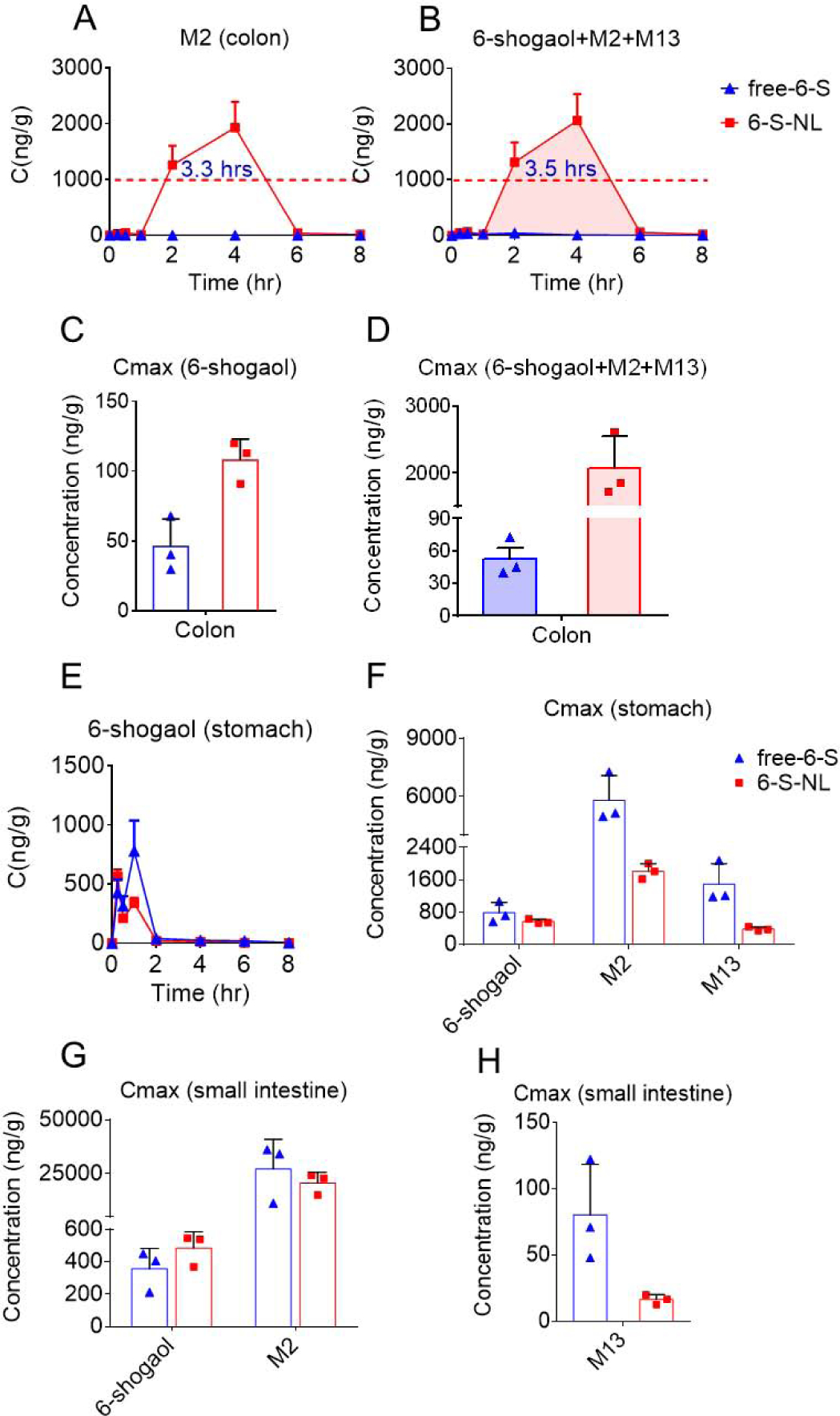
(A) Concentration-time profiles of M2 in colon tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3 per time point) (B) Concentration-time profiles of total active form (6 shogaol plus M2 plus M13) in colon tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3 per time point) (C) Cmax of 6-shogaol in the colon tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3) (D) Cmax of total active form (6 shogaol plus M2 plus M13) in colon tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3) (E) Concentration-time profiles of 6-shogaol in stomach tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3 per time point) (F) Cmax of 6-shogaol, M2, and M13 in stomach tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3) (G) Cmax of 6-shogaol and M2 in small intestine tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3) (H) Cmax of M13 in small intestine tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3) (*P < 0.05, **P < 0.01, ***P < 0.001)
Not surprisingly, stomach tissues exhibited the highest initial concentration of 6-shogaol in the GI tract in mice subjected to gavage feeding (Figure 7E, 5N, and 4A) of free-6-S and 6-S-NL. In this tissue, the Cmax of 6-shogaol was slightly lower in the 6-S-NL group than in the free 6-S group (Figure 7F), whereas the Cmax of M2 and M13 was significantly lower in the 6-S-NL group compared to the free-6-S group (Figure 7F). This suggests that the lipid NP formulation may protect 6-shogaol from being released and being metabolized in the stomach.
In the small intestine, the Cmax of 6-shogaol and M2 were similar between the free-6-S and 6-S-NL groups (Figure 7G), but the Cmax of M13 was much lower in the 6-S-NL group (Figure 7H). This seems to suggest that the NP formulation prevented the glutathione conjugation (phase II metabolism) of 6-shogaol in the small intestine.
3.8. Drug exposures of M2 and M13 are higher following oral administration of 6-S-NL versus free-6-S
The area under the curve (AUC) is an important pharmacokinetic parameter that reflects the actual body exposure to the drug.[50] It is typically calculated from the blood (plasma) drug concentration-time curve, but it can also be used to evaluate the tissue drug exposure. We observed that in the stomach and small intestine tissues, the exposure of 6-shogaol was significantly higher in the free-6-S group than in the 6-S-NL group (Figure 8A). In colon tissue, however, 6-shogaol was significantly higher in the 6-S-NL group. Moreover, in the colon, the drug exposures of M2 and M13 were also significantly higher in the 6-S-NL group than in the free-6-S group (Figure 8B, C). As a result, the overall drug exposure in colon tissue, as calculated by the AUC of total active components (6-shogaol+M2+M13) was significantly higher in the 6-S-NL group compared to the free-6-S group (Figure 8D). Also, as shown in Figure 8E, the ratios of metabolites to 6-shogaol in the colon tissue were significantly higher in the 6-S-NL group than in the free-6-S group. This indicates that the colon might be an excellent target tissue for taking up 6-S-NL and converting 6-shogaol to active M2 and M13.
Figure 8. Comparison of drug exposure in the gastrointestinal tract of mice orally administrated with free-6-S and 6-S-NL.
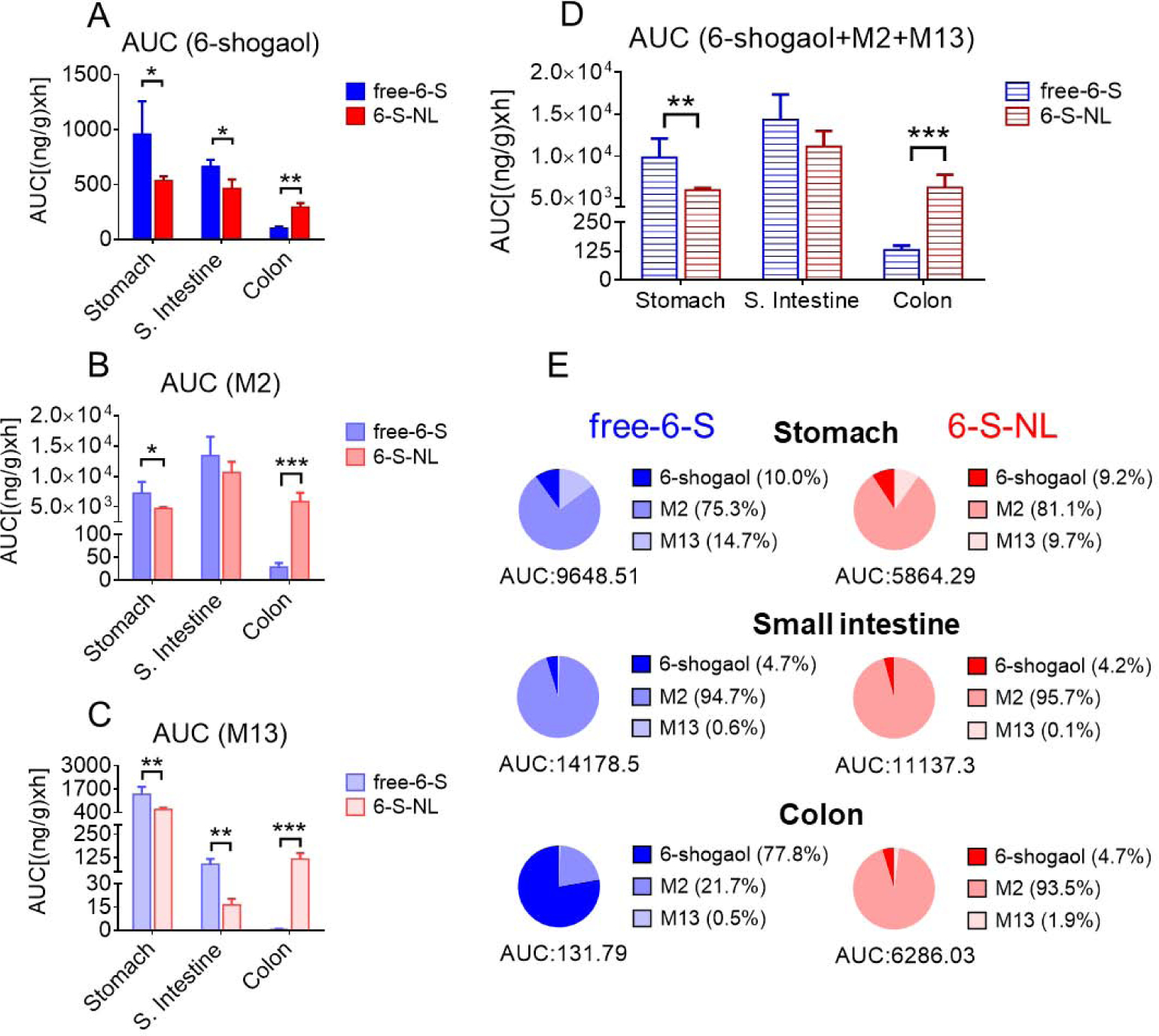
(A) AUC (area under the curve) of 6-shogaol in the gastrointestinal tract (stomach, small intestine, and colon). (n=3) (B) AUC of M2 in the gastrointestinal tract. (n=3) (C) AUC of M13 in the gastrointestinal tract. (n=3) (D) AUC of total active components (6-shogaol, M2, and M13) in the gastrointestinal tract. (n=3) (E) Percentages of 6-shogaol, M2, and M13 in the gastrointestinal tract of free-6-S and 6-S-NL treated mice. (Average, n=3) (*P < 0.05, **P < 0.01, ***P < 0.001)
3.9. Orally administrated 6-S-NL is less likely to be distributed to liver and kidney than free-6-S
Generally, orally administrated drugs are absorbed in the GI tract, metabolized by the liver, and eliminated by the kidney. In liver and kidney tissues, the AUC values of M2 and M13 were higher in the free-6-S group than in the 6-S-NL group (Figure 9A, B). Meanwhile, the AUC ratios of 6-shogaol to its metabolites were significantly higher in a 6-S-NL group than in the free-6-S group (Figure 9C). This indicates that 6-S-NL underwent less metabolism than free-6-S in liver and kidney tissues. We speculate that free 6-shogaol is more prone to be conjugated (converted to hydrophilic metabolites M2 and M13) in the liver and eliminated in the kidney, compared to encapsulated 6-shogaol. Thus, ginger lipid NPs can conceivably be used as a drug-delivery platform with the potential to decrease the toxicity and adverse effects of free 6-shogaol in the liver and kidney.
Figure 9. Comparison of drug exposure in the metabolizing organs (liver and kidney) of mice orally administrated with free-6-S and 6-S-NL.
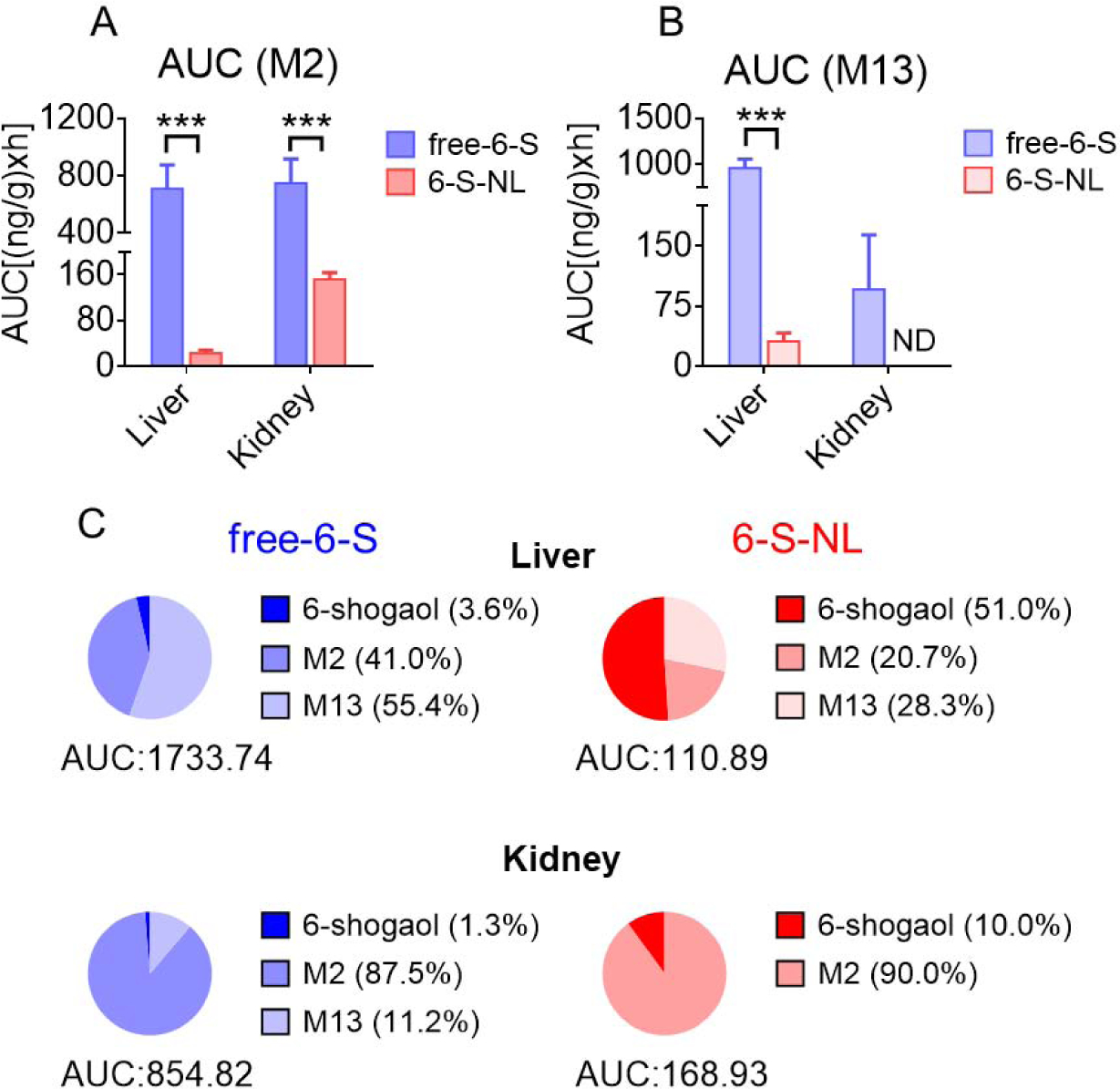
(A) AUC of M2 in the liver and kidney tissues after oral delivery of free-6-S, 6-S-NL to mice. (n=3) (B) AUC of M13 in the liver and kidney tissues. (n=3) (C) Percentages of 6-shogaol, M2, and M13 in the liver and kidney tissues of free-6-S and 6-S-NL treated mice. (Average, n=3) (*P < 0.05, **P < 0.01, ***P < 0.001, ND: not detected)
3.10. Orally administrated 6-S-NL may be retained in the colon for longer than orally administrated free-6-S
We compared the time to reach the Cmax (Tmax) between 6-shogaol and its metabolites in different tissues of the GI tract. Our data indicate that it took the free-6-S group 1.5 hours to reach the Cmax of 6-shogaol in the colon, whereas the 6-S-NL group required 4 hours to reach the Cmax of 6-shogaol (Figure 10A). Further, the mean residence times (MRTs) of M2 and M13 in colon tissues were around 4 hours in the 6-S-NL group, compared to less than 0.5 hours (or not detected) in the free-6-S group (Figure 10B, C). This indicates that the active metabolites stayed in the colon for a significantly longer time in the 6-S-NL group, suggesting that 6-S-NL may be retained in the colon for longer than free-6-S and that it may slowly release the loaded 6-shogaol to the colon tissue.
Figure 10. Time to reach the peak (Tmax), and Mean residence time (MRT) of 6-shogaol, M2, and M13 in the gastrointestinal tract of mice given a single administration of free-6-S or 6-S-NL.

(A) Tmax of 6-shogaol in the gastrointestinal tract (stomach, small intestine, and colon). (n=3) (B) MRT of M2 in the gastrointestinal tract. (C) MRT of M13 in the gastrointestinal tract. (n=3, ND: not detected)
3.11. Orally administrated M2-NL and M13-NL have more potent wound-healing effects than orally administrated 6-S-NL in an in vivo model of UC
As our data indicated that 6-S-NL-generated M2 or M13 have better pharmacokinetic features (AUC, Cmax, and MRT) than free 6-shogaol, and confirmed that M2 and M13 are potent anti-inflammatory agents in vitro, we next assessed whether NL-encapsulated M2 or M13 could benefit mucosal healing in the DSS-induced UC model. Using an identical method of constructing 6-S-NL, we obtained drug loading efficiencies of NPs to M2 and M13 can reach 91.3 ± 3.1% and 83.3 ± 1.9%, respectively (Figure S2). Mice were provided with drinking water containing 2% DSS for seven days (wounding phase), and then given water alone or water plus a once-daily dose of 6-SNL, M2-NL, or M13-NL (5 mg/kg) administered by gavage for an additional seven days (healing phase). The control group (water only for the entire experimental period) showed a slight increase in body weight (Figure 11A), but no increase in the level of Lcn-2 (Figure 11B), which is a marker that can be used for the noninvasive assessment of colitis development.[43] DSS-wounded mice that did not receive any experimental formulation maintained their body weight during the wounding phase, but their Lcn-2 levels increased across this period. During the healing phase (Figure 11A, B, purple dots), these mice lost up to 15% of their body weight and showed an increase in Lcn-2 until day 10. By Day 14, these mice had not recovered their initial body weight and displayed high levels of Lcn-2, indicative of unrecovered intestinal inflammation. In contrast, mice fed with 6-S-NL, M2-NL, or M13-NL lost significantly less body weight and exhibited reduced levels of Lcn-2 from Day 9 to Day 14, compared with control mice. In addition, mice treated with M2-NL and M13-NL almost completely regained their pretreatment body weight and Lcn-2 levels by Day 14, and their recovery progressed faster than the 6-S-NL treated group. Similarly, histological analysis (Figure 11E) and measurement of colonic MPO activities (Figure 11G) confirmed that intestinal mucosal ulceration was decreased in mice treated with M2-NL and M13-NL. We also found that the mRNAs for the studied pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) were more highly expressed in the recovery group (DSS followed by water) compared with the 6-S-NL, M2-NL, and M13-NL groups (Figure 11F). The apparent ability of 6-S-NL, M2-NL, and M13-NL to decrease the mRNA expression levels of TNF-α, IL-6, and IL-1β may explain their efficacy in accelerating the healing of intestinal mucosal injuries, as well as the ability of M2-NL and M13-NL to recover cytokine levels in DSS-treated mice (Figure 11F).
Figure 11. Effects of M2-NL (M2-loaded lipid NPs) and M13-NL (M13-loaded lipid NPs) on an in vivo wound-healing model.

(A) Bodyweight changes from mice treated with water (gray), seven days 2% DSS followed by seven days of water (purple), seven days 2% DSS followed by seven days 6-S-NL treatment (blue), seven days 2% DSS followed by seven days M2-NL treatment (green), and seven days 2% DSS followed by seven days M13 treatment (Red). (n=5) (B) Lcn-2 levels from different groups of mice. (n=5) (C) Colon lengths and colon-to-body weight ratios from different groups of mice. (D) Spleen-to-body weight ratio. (n=5) (E) H&E-staining. Arrowheads indicate inflammatory cells in the lamina propria. Scale bar = 100 μm. (F) Measurement of mRNA levels of cytokines in the DSS-induced mouse model of wound healing. (n=5) (G) MPO activity from different groups of mice. (n=5) (NS: not significant, *P < 0.05, **P < 0.01, ***P < 0.001)
3.12. Orally administrated 6-S-NL, M2-NL, and M13-NL are biocompatible
To investigate the in vivo toxicity of our formulations, we orally administrated 6-S-NL, M2-NL, M13-NL, or PBS (control) to healthy mice for seven days. Body weights were recorded daily, and changes were compared. On day 7, blood was collected for hematological and biochemical analyses. There was no significant difference in the body weight change, hematological parameters, or biochemical findings of treated mice compared with controls (Figure 12A–D). The safety of 6-S-NL, M2-NL, and M13-NL was further demonstrated by the similar spleen-to-body weight ratios seen in the treatment and control groups in the above-described in vivo wound healing experiment (Figure 11D, DSS-induced UC model). These findings were consistent with previous reports [18, 23] and indicated that 6-S-NL, M2-NL, M13-NL do not have any obvious in vivo toxicity.
Figure 12. In vivo toxicity evaluation of M2-NL and M13-NL.
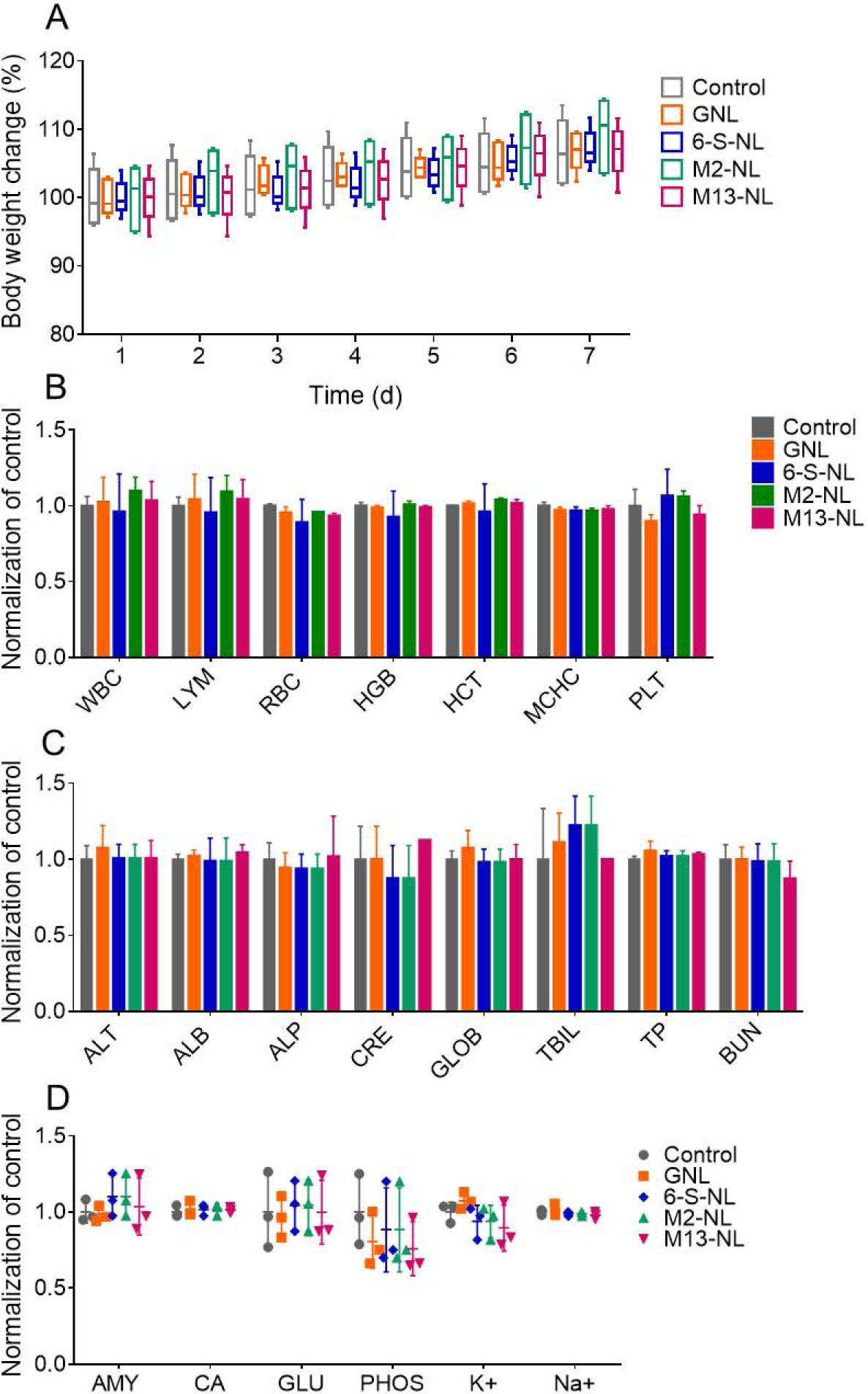
(A) Bodyweight change. Body weights were measured in each group and normalized by the corresponding initial body weight. (n=3) (B) Blood was collected by retro-orbital bleeding, and hematological analyses were performed using an automatic hematology analyzer (VetScan HM5; Abaxis, CA, USA). The following hematologic parameters are shown: WBC, total white blood cells; LYM, lymphocytes; RBC, red blood cells; HGB, hemoglobin; HCT, hematocrit; MCHC, mean corpuscular hemoglobin concentration; and PLT, platelets. (n=3) (C) Blood was collected, and biochemical analyses were performed using a biochemical analyzer (VetScan VS2; Abaxis, CA, USA) with a comprehensive diagnostic profile reagent rotor. The following biochemical parameters were assessed: ALT, alanine aminotransferase; ALB, albumin; ALP, alkaline phosphatase; CRE, creatinine; GLOB, globulin; TBIL, total bilirubin; TP, total protein; and BUN, urea nitrogen. (n=3) (D) The following additional biochemical parameters were assessed: AMY, amylase; CA, calcium; GLU, glucose; PHOS, phosphorus; K+, potassium; and Na+, sodium. (n=3)
4. Discussion
Natural products are important weapons in the fight to conquer the disease. More than 60% of the useful drugs sold today are derived from natural sources, such as plants, animals, microbes, and prebiotic sources.[51, 52] Since many biologically active natural products are isolable in only small quantities, the costs of these natural active ingredients are usually formidable.[52] Thus, natural product synthesis and the development of advanced drug formulations are two major goals in the pharmaceutical industry. Although synthesis or semi-synthesis can solve some problems, such as shortages and the high price of raw active ingredients, issues with drug dosage and side effects cannot be reduced without applying advanced DDSs. Moreover, contrary to popular belief, many natural products and natural product derived drugs can have severe toxicities, such as neutropenia induced by paclitaxel and the cardiotoxicity of doxorubicin. If used as conventional drug formulations or at an incorrect dose, some natural medicines can generate unpredictable systemic toxicity. Clearly, advanced drug formulations and/or targeted DDSs are needed to solve these issues.
Among the targeted DDSs developed to date, nanotechnology-based DDS represents the most promising path for delivering drugs to specific targets.[8, 53] Although enormous applications of synthetic NPs have emerged in this field, we have turned our attention to edible plant-derived NPs (PDNPs), such as natural liposomal vehicles or plant exosomal vesicles. Because they originate from edible plants and the human immune system had adapted to the stimuli from edible plants, PDNPs do not induce human toxicity or immunogenicity.[54] In this way, PDNPs out-perform their synthetic counterparts, such as liposomes and metal NPs. Another advantage of PDNPs over synthetic NPs is that the former are biodegradable and thus do not carry any risk for potential adverse environmental impacts. Here, we applied ginger-derived NPs (GDNPs).
Lipidomic analysis has revealed that GDNPs have high contents of phospholipids (~47% phosphatidic acid or PA) and glycolipids (~15% digalactosyl-diacylglycerol or DGDG and ~27% monogalactosyldiacylglycerol or MGDG).[18, 23] Interestingly, GDNPs do not contain cholesterol, which usually acts as a stabilizing component in liposomal structures and mammalian cell membranes.[55] It has been speculated that the non-bilayer glycolipids, DGDG and MGDG, are the crucial stabilizers in GDNPs.[56] Lipids isolated from GDNPs can self-assemble to spherical lipid NPs in phosphate-buffered saline (PBS); these NPs retain their stability for more than 25 days without any other additive,[23] and can easily encapsulate hydrophobic drugs. As the structure of self-assembled lipid NPs is similar to those of liposomes, it is reasonable to postulate that the drug release mechanism and kinetics of lipid NPs may also be similar to those of liposomes. Such drug release could be fitted with and predicted by the Higuchi and Korsmeyer-Peppas models. Similar to a drug releases from liposome, the release of 6-shogaol from ginger lipid NPs may include two steps: collisions between NPs, and diffusion of the drug molecules through the aqueous phase. Concentration and activity gradients may play important roles in affecting the release kinetics of 6-S-NL [38].
Broadly speaking, plant NP derived lipids are the natural products (fatty acids and their derivatives) that make up the bio-membranes of plant cells. Since lipids are inactive ingredients, they are often trashed (as impurities) by medicinal chemists during the isolation of active natural products. It is therefore notable that natural lipids are useful drug excipients and are generally recognized as safe (GRAS) compounds. Our studies have demonstrated that reorganized ginger lipid NPs are an ideal DDS for targeting diseased colon tissue.[18] In the present work, our tissue distribution study showed that mice fed 6-S-NL had significantly higher concentrations of active ingredients in their colon tissues than free 6-shogaol-fed mice. Thus, the bioavailability of the active ingredients was largely improved by NP encapsulation. It seems safe to say that encapsulation in lipid NPs would reduce the required dosage of 6-shogaol, given that this strategy was found to prevent the drug from penetrating and being metabolized by non-target tissues.
It is well known that phase II drug metabolism (or conjugation) is often a detoxification and deactivation step, during which the free or redox (phase I metabolized) forms of drugs are often converted to water-soluble metabolites (non-active polar compounds).[50] In rare cases, the conjugation reaction can produce active metabolites (e.g., morphine-6-glucuronide, which is a phase II conjugate of morphine).[57] Our present work demonstrates an unusual case, wherein a pair of phase II metabolites (M2 and M13) are more potent active anti-inflammatory ingredients than the parent drug, 6-shogaol. M2 is a cysteine-conjugated metabolite, and M13 is the glutathione-conjugated metabolite of 6-shogaol; M13 can degrade to M2 or 6-shogaol, and M2 can further degrade to 6-shogaol. The glutathione (GSH) metabolism pathway is involved, suggesting that modification of the oxidized/reduced glutathione ratio may contribute to the bio-activity of 6-shogaol, M2, and M13.
5. Conclusions
Collectively, our findings indicate that ginger lipid NPs are very efficient in delivering 6-shogaol to the colon, where the drug is conjugated to its more potent metabolites, M2 and M13, to execute anti-inflammatory effects and increase intestinal mucosal wound healing. More importantly, we show that ginger lipid NPs-encapsulated M2 or M13 could be very useful therapeutic nanomedicines for treating ulcerative colitis.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (RO1-DK-116306 and RO1-DK-107739 to D.M.), the Department of Veterans Affairs (Merit Award BX002526 to D.M.). D.M. is a recipient of a Senior Research Career Scientist Award (BX004476) from the Department of Veterans Affairs. The authors are grateful to Dr. Zhao-hua Cai (Center for Molecular and Translational Medicine, Georgia state university) for her assistance in the confocal microscopy study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors disclose no conflicts of interest.
References:
- [1].Nguyen GC, Chong CA, Chong RY, National estimates of the burden of inflammatory bowel disease among racial and ethnic groups in the United States, J Crohns Colitis, 8 (2014) 288–295. [DOI] [PubMed] [Google Scholar]
- [2].Croft JMDEPZBWWAGWJB, Prevalence of Inflammatory Bowel Disease Among Adults Aged ≥18 Years — United States, 2015, Morbidity and Mortality Weekly Report, 65 (2016) 4. [DOI] [PubMed] [Google Scholar]
- [3].Hua S, Marks E, Schneider JJ, Keely S, Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: selective targeting to diseased versus healthy tissue, Nanomedicine, 11 (2015) 1117–1132. [DOI] [PubMed] [Google Scholar]
- [4].Philip AK, Philip B, Colon targeted drug delivery systems: a review on primary and novel approaches, Oman Med J, 25 (2010) 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Date AA, Hanes J, Ensign LM, Nanoparticles for oral delivery: Design, evaluation and state-of-the-art, J Control Release, 240 (2016) 504–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bewtra M, Johnson FR, Assessing patient preferences for treatment options and process of care in inflammatory bowel disease: a critical review of quantitative data, Patient, 6 (2013) 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fakhoury M, Negrulj R, Mooranian A, Al-Salami H, Inflammatory bowel disease: clinical aspects and treatments, J Inflamm Res, 7 (2014) 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ulbrich W, Lamprecht A, Targeted drug-delivery approaches by nanoparticulate carriers in the therapy of inflammatory diseases, J R Soc Interface, 7 Suppl 1 (2010) S55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pridgen EM, Alexis F, Farokhzad OC, Polymeric nanoparticle drug delivery technologies for oral delivery applications, Expert Opin Drug Deliv, 12 (2015) 1459–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang Z, Gan J, Jia L, Guo G, Wang C, Zang Y, Ding Z, Chen J, Zhang J, Dong L, An orally administrated nucleotide-delivery vehicle targeting colonic macrophages for the treatment of inflammatory bowel disease, Biomaterials, 48 (2015) 26–36. [DOI] [PubMed] [Google Scholar]
- [11].Zhang S, Ermann J, Succi MD, Zhou A, Hamilton MJ, Cao B, Korzenik JR, Glickman JN, Vemula PK, Glimcher LH, Traverso G, Langer R, Karp JM, An inflammation-targeting hydrogel for local drug delivery in inflammatory bowel disease, Sci Transl Med, 7 (2015) 300ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang M, Xu C, Liu D, Han MK, Wang L, Merlin D, Oral Delivery of Nanoparticles Loaded With Ginger Active Compound, 6-Shogaol, Attenuates Ulcerative Colitis and Promotes Wound Healing in a Murine Model of Ulcerative Colitis, J Crohns Colitis, 12 (2018) 217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yildirimer L, Thanh NT, Loizidou M, Seifalian AM, Toxicology and clinical potential of nanoparticles, Nano Today, 6 (2011) 585–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bergin IL, Witzmann FA, Nanoparticle toxicity by the gastrointestinal route: evidence and knowledge gaps, Int J Biomed Nanosci Nanotechnol, 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bouwmeester H, van der Zande M, Jepson MA, Effects of food-borne nanomaterials on gastrointestinal tissues and microbiota, Wiley Interdiscip Rev Nanomed Nanobiotechnol, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Beloqui A, Coco R, Alhouayek M, Solinis MA, Rodriguez-Gascon A, Muccioli GG, Preat V, Budesonide-loaded nanostructured lipid carriers reduce inflammation in murine DSS-induced colitis, Int J Pharm, 454 (2013) 775–783. [DOI] [PubMed] [Google Scholar]
- [17].Yang C, Zhang M, Merlin D, Advances in Plant-derived Edible Nanoparticle-based lipid Nano-drug Delivery Systems as Therapeutic Nanomedicines, J Mater Chem B, 6 (2018) 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang M, Viennois E, Prasad M, Zhang Y, Wang L, Zhang Z, Han MK, Xiao B, Xu C, Srinivasan S, Merlin D, Edible ginger-derived nanoparticles: A novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer, Biomaterials, 101 (2016) 321–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brahmbhatt M, Gundala SR, Asif G, Shamsi SA, Aneja R, Ginger phytochemicals exhibit synergy to inhibit prostate cancer cell proliferation, Nutr Cancer, 65 (2013) 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Peng F, Tao Q, Wu X, Dou H, Spencer S, Mang C, Xu L, Sun L, Zhao Y, Li H, Zeng S, Liu G, Hao X, Cytotoxic, cytoprotective and antioxidant effects of isolated phenolic compounds from fresh ginger, Fitoterapia, 83 (2012) 568–585. [DOI] [PubMed] [Google Scholar]
- [21].Yeh H.-y., Chuang C.-h., Chen H.-c., Wan C.-j., Chen T.-l., Lin L.-y., Bioactive components analysis of two various gingers (Zingiber officinale Roscoe) and antioxidant effect of ginger extracts, LWT - Food Science and Technology, 55 (2014) 329–334. [Google Scholar]
- [22].Prasad S, Tyagi AK, Ginger and its constituents: role in prevention and treatment of gastrointestinal cancer, Gastroenterol Res Pract, 2015 (2015) 142979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang M, Xiao B, Wang H, Han MK, Zhang Z, Viennois E, Xu C, Merlin D, Edible Ginger-derived Nano-lipids Loaded with Doxorubicin as a Novel Drug-delivery Approach for Colon Cancer Therapy, Mol Ther, 24 (2016) 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Merlin MZXWMKHJFCD, Oral administration of ginger-derived nanolipids loaded with siRNA as a novel approach for efficient siRNA drug delivery to treat ulcerative colitis, Nanomedicine, (2017) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ha SK, Moon E, Ju MS, Kim DH, Ryu JH, Oh MS, Kim SY, 6-Shogaol, a ginger product, modulates neuroinflammation: a new approach to neuroprotection, Neuropharmacology, 63 (2012) 211–223. [DOI] [PubMed] [Google Scholar]
- [26].Saha A, Blando J, Silver E, Beltran L, Sessler J, DiGiovanni J, 6-Shogaol from dried ginger inhibits growth of prostate cancer cells both in vitro and in vivo through inhibition of STAT3 and NF-kappaB signaling, Cancer Prev Res (Phila), 7 (2014) 627–638. [DOI] [PubMed] [Google Scholar]
- [27].Moon M, Kim HG, Choi JG, Oh H, Lee PK, Ha SK, Kim SY, Park Y, Huh Y, Oh MS, 6-Shogaol, an active constituent of ginger, attenuates neuroinflammation and cognitive deficits in animal models of dementia, Biochem Biophys Res Commun, 449 (2014) 8–13. [DOI] [PubMed] [Google Scholar]
- [28].Zick SM, Djuric Z, Ruffin MT, Litzinger AJ, Normolle DP, Alrawi S, Feng MR, Brenner DE, Pharmacokinetics of 6-gingerol, 8-gingerol, 10-gingerol, and 6-shogaol and conjugate metabolites in healthy human subjects, Cancer Epidemiol Biomarkers Prev, 17 (2008) 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Asami A, Shimada T, Mizuhara Y, Asano T, Takeda S, Aburada T, Miyamoto K, Aburada M, Pharmacokinetics of [6]-shogaol, a pungent ingredient of Zingiber officinale Roscoe (Part I), J Nat Med, 64 (2010) 281–287. [DOI] [PubMed] [Google Scholar]
- [30].Dyer EGBWJ, A RAPID METHOD OF TOTAL LIPID EXTRACTION AND PURIFICATION, Canadian Journal of Biochemistry and Physiology, 37 (1959) 7. [DOI] [PubMed] [Google Scholar]
- [31].Mircioiu C, Voicu V, Anuta V, Tudose A, Celia C, Paolino D, Fresta M, Sandulovici R, Mircioiu I, Mathematical Modeling of Release Kinetics from Supramolecular Drug Delivery Systems, Pharmaceutics, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang Y, Huo M, Zhou J, Xie S, PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel, Comput Methods Programs Biomed, 99 (2010) 306–314. [DOI] [PubMed] [Google Scholar]
- [33].Li LL, Cui Y, Guo XH, Ma K, Tian P, Feng J, Wang JM, Pharmacokinetics and Tissue Distribution of Gingerols and Shogaols from Ginger (Zingiber officinale Rosc.) in Rats by UPLC(−)Q-Exactive(−)HRMS, Molecules, 24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nikkhah Bodagh M, Maleki I, Hekmatdoost A, Ginger in gastrointestinal disorders: A systematic review of clinical trials, Food Sci Nutr, 7 (2019) 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dugasani S, Pichika MR, Nadarajah VD, Balijepalli MK, Tandra S, Korlakunta JN, Comparative antioxidant and anti-inflammatory effects of [6]-gingerol, [8]-gingerol, [10]-gingerol and [6]-shogaol, J Ethnopharmacol, 127 (2010) 515–520. [DOI] [PubMed] [Google Scholar]
- [36].Shim S, Kim S, Choi DS, Kwon YB, Kwon J, Anti-inflammatory effects of [6]-shogaol: potential roles of HDAC inhibition and HSP70 induction, Food Chem Toxicol, 49 (2011) 2734–2740. [DOI] [PubMed] [Google Scholar]
- [37].Nounou MM, El-Khordagui LK, Khalafallah NA, Khalil SA, In vitro release of hydrophilic and hydrophobic drugs from liposomal dispersions and gels, Acta Pharm, 56 (2006) 311–324. [PubMed] [Google Scholar]
- [38].Jain A, Jain SK, In Vitro Release Kinetics Model Fitting of Liposomes: An Insight, Chem Phys Lipids, (2016). [DOI] [PubMed] [Google Scholar]
- [39].Wu IY, Bala S, Skalko-Basnet N, di Cagno MP, Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes, Eur J Pharm Sci, 138 (2019) 105026. [DOI] [PubMed] [Google Scholar]
- [40].Lee JH, Yeo Y, Controlled Drug Release from Pharmaceutical Nanocarriers, Chem Eng Sci, 125 (2015) 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M, Dextran sulfate sodium (DSS)-induced colitis in mice, Curr Protoc Immunol, 104 (2014) Unit 15 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kiesler P, Fuss IJ, Strober W, Experimental Models of Inflammatory Bowel Diseases, Cell Mol Gastroenterol Hepatol, 1 (2015) 154–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M, Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation, PLoS One, 7 (2012) e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen H, Soroka DN, Zhu Y, Hu Y, Chen X, Sang S, Cysteine-conjugated metabolite of ginger component [6]-shogaol serves as a carrier of [6]-shogaol in cancer cells and in mice, Chem Res Toxicol, 26 (2013) 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen H, Lv L, Soroka D, Warin RF, Parks TA, Hu Y, Zhu Y, Chen X, Sang S, Metabolism of [6]-shogaol in mice and in cancer cells, Drug Metab Dispos, 40 (2012) 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhu Y, Warin RF, Soroka DN, Chen H, Sang S, Metabolites of ginger component [6]-shogaol remain bioactive in cancer cells and have low toxicity in normal cells: chemical synthesis and biological evaluation, PLoS One, 8 (2013) e54677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang M, Viennois E, Xu C, Merlin D, Plant derived edible nanoparticles as a new therapeutic approach against diseases, Tissue Barriers, 4 (2016) e1134415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].The dose makes the poison, Nat Nanotechnol, 6 (2011) 329. [DOI] [PubMed] [Google Scholar]
- [49].Merlin-Zhang O, Jung J-S, Viennois E, In vitro Intestinal Epithelial Wound-healing Assays Using Electric Cell-Substrate Impedance Sensing Instrument, Bio-Protocol, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Laroui H, Wilson DS, Dalmasso G, Salaita K, Murthy N, Sitaraman SV, Merlin D, Nanomedicine in GI, Am J Physiol Gastrointest Liver Physiol, 300 (2011) G371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Harvey AL, Edrada-Ebel R, Quinn RJ, The re-emergence of natural products for drug discovery in the genomics era, Nat Rev Drug Discov, 14 (2015) 111–129. [DOI] [PubMed] [Google Scholar]
- [52].Newman DJ, Cragg GM, Natural Products as Sources of New Drugs from 1981 to 2014, J Nat Prod, 79 (2016) 629–661. [DOI] [PubMed] [Google Scholar]
- [53].Wei T, Liu J, Ma H, Cheng Q, Huang Y, Zhao J, Huo S, Xue X, Liang Z, Liang XJ, Functionalized nanoscale micelles improve drug delivery for cancer therapy in vitro and in vivo, Nano Lett, 13 (2013) 2528–2534. [DOI] [PubMed] [Google Scholar]
- [54].Wang Q, Zhuang X, Mu J, Deng ZB, Jiang H, Zhang L, Xiang X, Wang B, Yan J, Miller D, Zhang HG, Delivery of therapeutic agents by nanoparticles made of grapefruit-derived lipids, Nat Commun, 4 (2013) 1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Singh R, Lillard JW Jr., Nanoparticle-based targeted drug delivery, Exp Mol Pathol, 86 (2009) 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Seiwert D, Witt H, Janshoff A, Paulsen H, The non-bilayer lipid MGDG stabilizes the major light-harvesting complex (LHCII) against unfolding, Sci Rep, 7 (2017) 5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Klimas R, Mikus G, Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: a quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide, Br J Anaesth, 113 (2014) 935–944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.