

Abstract
The Hippo pathway regulates cell proliferation and organ size through control of the transcriptional regulators YAP (yes-associated protein) and TAZ. Upon extracellular stimuli such as cell-cell contact, the pathway negatively regulates YAP through cytoplasmic sequestration. Under conditions of low cell density, YAP is nuclear and associates with enhancer regions and gene promoters. YAP is mainly described as a transcriptional activator of genes involved in cell proliferation and survival. Using a genome-wide approach, we show here that, in addition to its known function as a transcriptional activator, YAP functions as a transcriptional repressor by interacting with the multifunctional transcription factor Yin Yang 1 (YY1) and Polycomb repressive complex (PRC2) member EZH2. YAP co-localized with YY1 and EZH2 on the genome to transcriptionally repress a broad network of genes mediating a host of cellular functions, including repression of the cell-cycle kinase inhibitor p27, whose role is to functionally promote contact inhibition. This work unveils a broad and underappreciated aspect of YAP nuclear function as a transcriptional repressor and highlights how loss of contact inhibition in cancer is mediated in part through YAP repressive function.
Introduction
The Hippo-YAP pathway is a central regulator of cell fate and proliferation and is tightly regulated by mechanical cues such as tension, pressure and contact with the extracellular matrix and other cells (1). At the core of the pathway are the transcriptional co-regulators YAP and TAZ, which bind to gene promoters and enhancers through interaction with transcription factors such as the TEA-domain proteins (TEADs) and others (2, 3). YAP localization depends on cellular density, where under low cell density conditions YAP localizes to the nucleus and modulates the transcription of genes involved in cell growth and survival (4). Increased YAP activity and nuclear localization is commonly observed in a multitude of cancers including schwannoma and cancers of the liver, colon, ovarian, lung and prostate (5, 6).
YAP has previously been shown to repress the expression of mesendoderm lineage-specific genes in human embryonic stem cells (7). Additionally, YAP facilitates the recruitment of the NuRD complex to deacetylate histones and repress the expression of target genes (8). To explore the role of YAP as a transcriptional regulator, we investigated the genomic localization of YAP at low cell density in human Schwann cells. These were chosen due to the critical role YAP plays in promotion of cellular transformation and tumorigenesis, subsequent to loss of the NF2 tumor suppressor gene, which is an upstream effector of the Hippo pathway (9–12). These efforts led to identification of a transcriptional repressor function for YAP, through interaction with the multifunctional transcription factor Yin-Yang 1 (YY1) and EZH2, a member of the Polycomb repressive complex 2 (PRC2). This work unveils a broad and underappreciated aspect of YAP nuclear function and highlights how loss of contact inhibition in cancer is partly mediated through YAP’s repressive function.
Materials and methods
Human Schwann Cells-
Human Schwann cells (hSC2λ) cells were obtained from the laboratory of Dr. Margaret Wallace (13). The cells were authenticated by short tandem repeat (STR) DNA profiling (DDC Medical). Cells were maintained in low glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Atlas Biologicals) and antibiotics (100 units/ml penicillin and 100 μg/ml Streptomycin) (Gibco), at 37°C in a humidified atmosphere of 5% CO2 (v/v). Cells were tested every 3 months for mycoplasma and confirmed free of contamination.
Transfections-
Transfections were performed using an Amaxa Nucleofector with the Amaxa Cell Line Nucleofector Kit V. Lentiviral infection of hSC2λ was performed according to standard protocols. Briefly, lentvirus was prepared in HEK293T cells that were co-transfected with packaging plasmids VSVG, Δ8.2, and GIPZ YY1 shRNA gene set. Supernatant was collected 48 hr and 72 hr after transfection, and cells were infected with 6 mL of viral supernatant containing polybrene (8 μg/mL). After 48 hr, transduced cells were selected with puromycin (0.25 μg/mL) and this selection maintained for 72 hr.
Plasmids and siRNA/shRNA-
The pCMV-Flag-YAP-5SA (#27371), pCMV-Flag-YAP-S127A (#27370), pCellFree_G03 YY1 (#67082) expression plasmids were purchased from Addgene. The siGENOME Human YY1 (7528) siRNA set (MU-011796-02-0002) was purchased from GE Healthcare Dharmacon and its siGENOME non-targeting control (D-001210-01-05). The siYAP Flexitube siRNA (1027418) was purchased from Qiagen. The “AllStars” negative control siRNA (SI03650318) was purchased from Qiagen. Expression plasmids for YY1 shRNA (V2LHS_219592, V2LHS_172065, V3LHS_412955), were purchased from GE Healthcare Dharmacon. All siRNAs were used at a final concentration of 20nM.
Antibodies-
Anti-YAP antibody ChIP (Abcam, ab52771), Anti-YY1 antibody-ChIP Grade (Abcam, ab38422). Anti-EZH2 ChIP (Active motif, #39901). Anti-Lamin A/C (sc-6215 (N-18), 1:500). Anti-tubulin (T5168) anti-actin (A4700) and anti-vinculin (V4505) were from Sigma. Anti-YAP Cell Signaling (#4912). Anti-p27 Kip1 antibody (Cell signaling, 2552S). Anti-Flag (Sigma, F1804–200UG). Anti-His (Abcam, #18184). Anti-TAZ (D3I6D) (Cell signaling, 70148).
Sub-cellular fractionation-
This was carried out as previously described (14). Briefly, cells were incubated on ice for 20’ in ice-cold cytoplasmic buffer (20 mM Tris-HCl pH 7.4, 150 mM KCl, 1.5 mM MgCl2, 1 mM PMSF, 1 mM DTT, 0.5%Nonidet P-40, protease inhibitor mixture), centrifuged at 4000g for 5’ at 4°C. Supernatant was kept for the cytosolic and plasma membrane fractions. The pellet was washed 5X with nuclear washing buffer (10 mM HEPES pH7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M Sucrose, and complete protease inhibitor mixture), lysed on ice for 20’ in 400 μl of RIPA (200 mM NaCl, 50mM TRIS HCl ph=7.4, 1mM EDTA, 1% NP40, 0.25% DOC) buffer and centrifuged at 13,000g for 10’ at 4°C. Supernatant was kept as nuclear fraction. To separate the cytosolic and plasma membrane fractions the supernatant was spun at 200,000g for 30’ at 4°C and the resultant supernatant centrifuged again at 13,000g for 5’ at 4°C and kept as the cytosolic fraction. The pellet was washed in 500 μl lysis buffer (50 mM Tris pH 7.4, 1 mM EDTA, 2.5 mm MgCl2, 150 mM NaCl, and complete protease inhibitor mixture) and re-sedimented at 200,000g for 30’ at 4°C. The pellet was then resuspended in ice-cold IP buffer (1 M Tris-HCl pH 7.4, 4 M NaCl, 10% (w/v) Triton X-100, and complete protease inhibitor mixture), incubated on a rocker for 30’ at 4°C, and cleared by centrifugation at 13,000g for 15’ at 4°C. The supernatant was kept as the plasma membrane fraction.
Chromatin immunoprecipitation (ChIP) and Quantitative real-time PCR analysis-
20 million cells were fixed with 1% formaldehyde for 10’ at R.T. Fixation was halted with 125 mM glycine for 5’ at RT. Fixed cells were washed 2x with cold PBS. Cell pellets were then resuspended in ChIP lysis buffer and chromatin was sheared with Misonix S-3000 bath sonicator for 15min sonication at 280W, 30” ON and 30” OFF to obtain 0.3–0.5 kb DNA fragments. Antibody (5μg) and Dynabeads Protein A were added to the cell lysate and incubated overnight at 4°C. Beads were washed with buffer 1 (150 mM NaCL, 20 mM TrisCl pH 8.0, 5 mM EDTA, 65% w/v sucrose, 10% Triton-X-100, 20% SDS) and then washed with TE buffer. DNA was eluted by resuspending the beads in TE/1%SDS. ChIP DNA and Input were treated with RNase A (5 μg) for 1 hr at 37°C. Proteinase K (0.5 mg/mL) was added and incubated overnight at 65°C to reverse crosslinking. DNA was then purified in phenol:chloroform and resuspended in a 30 μL of elution buffer. DNA was used for real time-PCR using SYBR Green PCR kit. A standard dilution curve was obtained for each Input and 1μL of ChIP DNA was used in each PCR reaction. Melt curves were analyzed to confirm specificity of the amplified target.
Fly RNA extraction-
RNA from cells was extracted using the Qiagen RNeasy kit. RNA from Drosophila heads was extracted using the RNAaqueous-Micro Total RNA Isolation kit. cDNA was made using the SuperScript III kit (Life Technologies). qPCR was performed with SYBR Green (Applied Biosystems). Relative gene expression was calculated with the 2−ΔΔCT method. Primer sequences detail in Table S1.
ChIP–Seq library preparation and sequencing-
The ChIP-Seq libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (Cat No. E7645S) from New England Biolabs Inc. (Ipswich, MA). 1ng of fragmented DNA was used as input. The following steps were followed for library preparation: End repair, 5’ phosphorylation, dA-tailing, adapter ligation, U excision, cleanup of adaptor-ligated DNA without size selection, and library amplification. 12 to 13 cycles followed by clean-up with 1 X Agencourt AMPure XP. The quality of the libraries was assessed using the Agilent High Sensitivity DNA kit (Cat No. 5067–4626) on the Agilent 2100 Bioanalyzer (Agilent Technologies) and were quantified using the NEBNext Library Quant kit for Illumina (Cat No. E7630S) by New England Biolabs Inc. The libraries were sequenced using the NextSeq 500 High Output v2 kit (75 cycles) (Cat No. FC-404–2005) on the NextSeq 500 platform from Illumina (San Diego, CA). IGV was used to generate browser tracks (15).
Luciferase assay-
CDKN1B promoter region (From −80 to 960 relative to the TSS) was cloned in place of the PGK promoter into the pmirGLO- Dual-Luciferase plasmid (E1330 Promega). hSC2λ cells were transfected and seeded in 96-well plates. For the TEAD and YY1 mutations, the binding sites were respectively mutated from CCAT to TGTA for YY1 and from CTTC to AGGA for TEAD. On the following day, luciferase activity was measured with Dual Luciferase Assay System (Promega; #E1910) according to manufacturer’s instructions. Firefly luciferase signal was normalized with the Renilla luciferase signal.
Hydrodynamic Tail Vein Injection-
DNA (10ug/mL) was mixed in saline solution (10% vol/body weight) (2). The tail was placed under a heat lamp for 20” to increase the vascular volume and using 27-gauge needles, the DNA solution was injected at approximately 20 mL/min.
Fly Strains-
The Pho RNAi strain (#110466) and its background control (#60100) were obtained from the Vienna Drosophila RNAi Center. The RNAi line is predicted to have no off targets (RNAi-phiC31 construct and insertion data submitted by the Vienna Drosophila RNAi Center). The UAS driven yorkieS168A (BDSC 28818 UAS-yki-S168A) was obtained from the Bloomington Drosophila Stock Center. The lines were recombined with GMR-Gal4 to mediate expression in the eye (16).
CRISPR Cell Line-
The PX459 plasmid was purchased from Addgene (#62988). Guide (TCCGGACCCGGGCAACCG) targeting the first exon of YAP was cloned into PX459. hSC2λ cells were transfected with the plasmid and treated with puromycin (0.25ug/mL) for 48h. Single clones were selected, expanded and subjected to western blot analysis. Cleavage was assessed by DNA sequencing.
Proximity Ligation Assay-
Proximity ligation assay (PLA) was carried out using the Duolink In Situ Red Start Kit Mouse/Rabbit (Sigma, DUO92101) per manufacturer’s instructions. Cells were fixed in 4% PFA for 20’ at RT, washed 3 X 5’ in PBS, RT. Cells were then permeabilized in 0.3% triton-X for 3’ and blocked in blocking buffer for 2 hours at 37°C. Primary antibodies (YAP = CST-12395, YY1= CST-63227, EZH2= CST-5246) were used at 1:600. Images were taken on an Olympus FV3000RS confocal scanning microscope. DAPI was visualized at 405 nm, and Duolink red visualized at 594 nm.
ChIP-seq data analysis-
Adapter sequences and low quality read ends were trimmed using cutadapt v1.8.1 (17). For processing the ChIP-seq samples, the reads were aligned to the hg19 genome build using bowtie2 v2.2.9 (18). Post-alignment filtering was performed according to the AQUAS pipeline (github.com/kundajelab/chipseq_pipeline). Peaks were called using macs2 v2.1.1.20160309 using the shift size values calculated from the run_spp.R script from the SPP peak caller (19, 20). High-quality peaks were then identified using the idr1 pipeline, selecting peaks with an idr score of less than or equal to 0.02. HOMER v4.9 was used for peak annotation, GO analysis, motif identification, and binding heatmaps (21).
Animal experiments-
All animal experiments complied with NIH guidelines and were approved by The Scripps Research IACUC. NOD/SCID mice (6–8 weeks old) were used for the hydrodynamic tail vein injection. Luciferase labeled SC4 cells were resuspended in PBS at a concentration of 5 × 105 cells per mL. 6-week old NSG mice (Jackson Labs Stock #005557) were anesthetized with Isoflurane. 2.5 × 105 cells were injected subcutaneously in the right hind flank. Tumor growth was monitored using IVIS 200, drug treatment was started when tumors reached a threshold value. Mice were treated with EPZ005687 at dose of 10 mg/kg/day. EPZ005687 was resuspended in DMSO at 2mg/ml. Control animals received DMSO without drug. EPZ005687 was injected daily via intraperitoneal injection. Mice were sacrificed after 17 days of treatment (once control animals had tumor size that required euthanasia according to IUCAC approved protocol). Tumors were isolated weighed at the conclusion, as described (22).
Statistical analysis-
Statistical analysis of data was performed using GraphPad Prism (version 6). Individual statistical methods are described in the respective figure legends. Unpaired student’s t-test was calculated to determine the significance of the results and indicate the two-tailed P values. Unless otherwise noted, mean and standard deviation was used to assess the significance.
Data and software availability-
ChIP-seq data is stored in the Gene Expression Omnibus under accession number GSE112932.
Results
YAP binds at the promoter of p27
To understand the transcriptional network regulated by YAP, we employed human Schwann cells (hSC2λ), grown at low density, when YAP is nuclear (Figure S1A). We performed Chromatin Immunoprecipitation followed by next-generation sequencing (ChIP-seq) using an anti-YAP antibody and identified a total of 7019 peaks, including peaks at the promoters of previously identified YAP targets genes (CTGF, TEAD1, AMOTL2, ANKRD1) (Figure 1A; Figure S1B; Table S2). YAP peaks were located mainly in intron and intergenic regions and, to a lesser extent, at gene promoters (7.72%), which is in accordance with previously reported ChIP-seq data (23). Motif analysis at YAP peaks reveals that TEAD-binding consensus sequences are enriched in 57.91% of YAP peaks (Figure 1B; Table S3), in proximity to the summit of peaks (Figure 1C). Gene ontology analysis indicated that YAP is involved in structure morphogenesis, cell communication and signaling (Table S4). Among the peaks, we identified YAP binding to the promoter of the CDKN1B gene (Figure 1D), which codes for the cyclin dependent kinase inhibitor p27 (p27Kip1). Analysis of YAP ChIP-seq datasets in a variety of cell lines show enrichment of YAP within the promoter or enhancer region of p27 (23–25). We confirmed YAP binding by ChIP-qPCR and observed a significant enrichment of YAP on the CDKN1B promoter compared to an IgG control (Figure 1E and Figure S1C). We also observed enrichment of TAZ at the promoter of CDKN1B (Figure S1D). Previous studies indicate p27 mediates contact inhibition of cell proliferation by binding to cyclin-CDK complexes and preventing cells from progressing into S phase (26, 27). Therefore, we set out to further investigate the mechanism by which YAP regulates p27.
Figure 1. YAP chromatin-wide genomic distribution in Human Schwann Cells.

(A) Distribution of YAP ChIP peaks across the genome relative to gene bodies. (B) A de novo motif analysis within YAP peaks identifies a TEAD consensus sequence as the highest enriched motif. (C) Distribution of the TEAD motifs in the YAP peaks, shown as distance from peak center. (D) Gene browser tracks (green) of the YAP ChIPs at p27. Black bars underneath the track indicate the extent of the YAP peaks. Fold enrichment from 0–24 relative to input. (E) Assessment of p27 promoter enrichment by YAP ChIP-qPCR. IgG used as negative control (n=3; **** P<0.0001, two-tailed Student’s t test; error bars, SD). See also Figure S1.
Regulation of p27 expression by cell-density is YAP dependent
The mRNA and protein levels of p27 are reported to be elevated with increased cell density and cell-cell contact (28, 29), which we confirmed by qRT-PCR and Western-blot analysis in hSC2λ cells (Figure 2A). We also confirmed that the YAP target gene CTGF is upregulated at low cell density (Figure 2A). To assess the role of YAP in regulating p27 we employed hSC2λ cells in which YAP was inactivated by CRISPR mediated editing (hSC2λYAP−/−) or knocked-down by siRNA (hSC2λ-siYAP) with an efficiency of more than 80% (Figure S2A and S2B). In hSC2λYAP−/− cells grown at low density, we observed a 2.5-fold upregulation of p27 mRNA (Figure 2B). Similarly, knockdown of YAP by siRNA led to an upregulation of p27 at both the protein and mRNA levels (Figure 2C). Conversely, to assess if forced expression at high cell density of a constitutively active YAP would downregulate endogenous p27, we transfected hSC2λ cells with vector expressing a constitutively active YAP (YAP5SA) cDNA (30). As expected, we observed upregulation of the known YAP target gene CTGF and downregulation of p27 at high cell density, both at the mRNA and protein levels (Figure 2D). We also assessed the effect of TAZ overexpression by transfecting hSC2λ cells with vector expressing a constitutively active TAZ (TAZS89A) and similarly observed downregulation of p27 at the mRNA and protein levels (Figure S2C).
Figure 2. Regulation of p27 expression by cell-density is YAP dependent.

(A) CTGF and p27 mRNA, measured by qRT-PCR or p27 protein, assessed by Western blotting in hSC2λ grown at low and high densities. (B) qRT-PCR analysis of p27 mRNA and western blot analysis of protein from wildtype (WT) and YAP knockout hSC2λ. (C) qRT-PCR analysis of p27 mRNA and western blot analysis of protein from hSC2λ cells grown at low density and transfected with small interfering RNA (siRNA) control (siCtrl) or siRNA against YAP (siYAP). For mRNA, transcript levels were normalized to ACTIN (n=3, ** P= 0.0067, *** P=0.0007, two-tailed Student’s t test; error bars=SD). For western blotting GAPDH was used as a loading control. (D) Levels of CTGF and p27 mRNA were assessed by qRT-PCR in hSC2λ cells grown at high cell density and transfected with control or YAP-5SA plasmid. p27 and YAP protein assessed by Western blotting. (E) hSC2λ overexpressing a version of WT YAP or YAP-5SA mutant were implanted into the sciatic nerve of NOD/SCID mice. Following tumor formation, tumors were resected, proteins extracted and p27 protein levels assessed by Western-blotting (n=2 for each group). Vinculin was used as a loading control. (F) Analysis of dacapo mRNA in D. melanogaster Canton-S (control) or GMR-yorkie S168A. RNA from fly heads qRT-PCR for dacapo transcript levels. Representative images from fly heads from GMR/CS and GMR/ykiS168A. (mRNA transcript levels were normalized to Actin or act42a mRNA; n=3; **** P<0.0001, two-tailed Student’s t test; error bars, SD). Vinculin and tubulin are loading controls for protein. See also Figure S2.
To assess whether YAP represses p27 expression in vivo, we introduced expression vectors for wildtype YAP, constitutively active YAP (YAP5SA and YAPS127A) or control plasmid into the liver of mice by hydrodynamic tail vein injection. The levels of p27 were assessed two weeks later by qRT-PCR and a significant downregulation of p27 mRNA was observed in the presence of wildtype or activated YAP (Figure S2D). We confirmed the expression of the exogenous YAP qRT-PCR using primers against the FLAG-tag (Figure S2E). In another approach, we implanted hSC2λ cells that stably expressed either a YAP-5SA or WT YAP into the sciatic nerve of NOD/SCID mice and allowed tumors to develop. Once tumors were detected they were harvested and analyzed for the expression of p27. As expected, levels of p27 proteins were significantly reduced in the YAP-5SA tumors compared to the YAP WT control (Figure 2E). Finally, as another in vivo validation, we used the fruit fly, Drosophila melanogaster, in which the core of the Hippo pathway is highly conserved (31–33). The expression of a constitutively active form of yorkie (ykiS168A), the fly homologue of YAP, in the eye using a GMR eye specific driver, led to a significant reduction in dacapo (fly homologue of p27) mRNA levels (Figure 2F). Overall, these results indicate that YAP regulates p27 at the transcriptional level, in cultured cells and in vivo.
YY1 is required for YAP-mediated repression of p27
To identify the mechanisms mediating p27 repression by YAP, we analyzed motif distribution at the promoter of p27 and identified a Yin-Yang 1 (YY1) binding motif adjacent to the TEAD binding motif (Figure S3A). YY1 is a multifunctional zinc-finger transcription factor of the Polycomb Group protein (PcG) family that bind promoters and enhancers of various cellular and viral genes (34, 35). YY1 plays a key role in growth and differentiation and can act both as an activator and as a repressor, depending on its binding partners (34, 35). To assess whether YAP repression of p27 requires YY1, we generated a stable knockdown of YY1 in hSC2λ cells using short hairpin RNA (hSC2λshYY1). We confirmed the knockdown of YY1 by western blot analysis (Figure S3B). In hSC2λshYY1 cells, the overexpression of activated YAP failed to repress p27 both at the transcriptional and protein levels (Figure 3A and S3C). Similarly, transient knockdown of YY1 using small inhibitory RNA (siRNA) showed similar results (Figure S3D). This holds true for TAZ, where overexpression in cells transiently knockdown for YY1 using siRNA showed similar results (Figure S3E). To test whether YY1 mediates the repressive function of YAP on p27 in vivo, we employed the GMR-YkiS168A flies and crossed them to flies that carry a dsRNA construct KK110466 targeting pho (the fly homologue of YY1) (36) under conditional UAS control. This cross results in the expression of YorkieS168A in combination with downregulation of pho, specifically in the eye. As expected, the GMR-YkiS168A line shows downregulation of dacapo. While the knockdown of pho alone does not affect dacapo expression and results in a normal eye phenotype, dacapo levels were significantly rescued in the YkiS168A/pho RNAi line (Figure 3B; Figure S3F). This provides in vivo support for YY1 being required, at least in part, for YAP mediated repression
Figure 3. YY1 is required for YAP-mediated repression of p27.
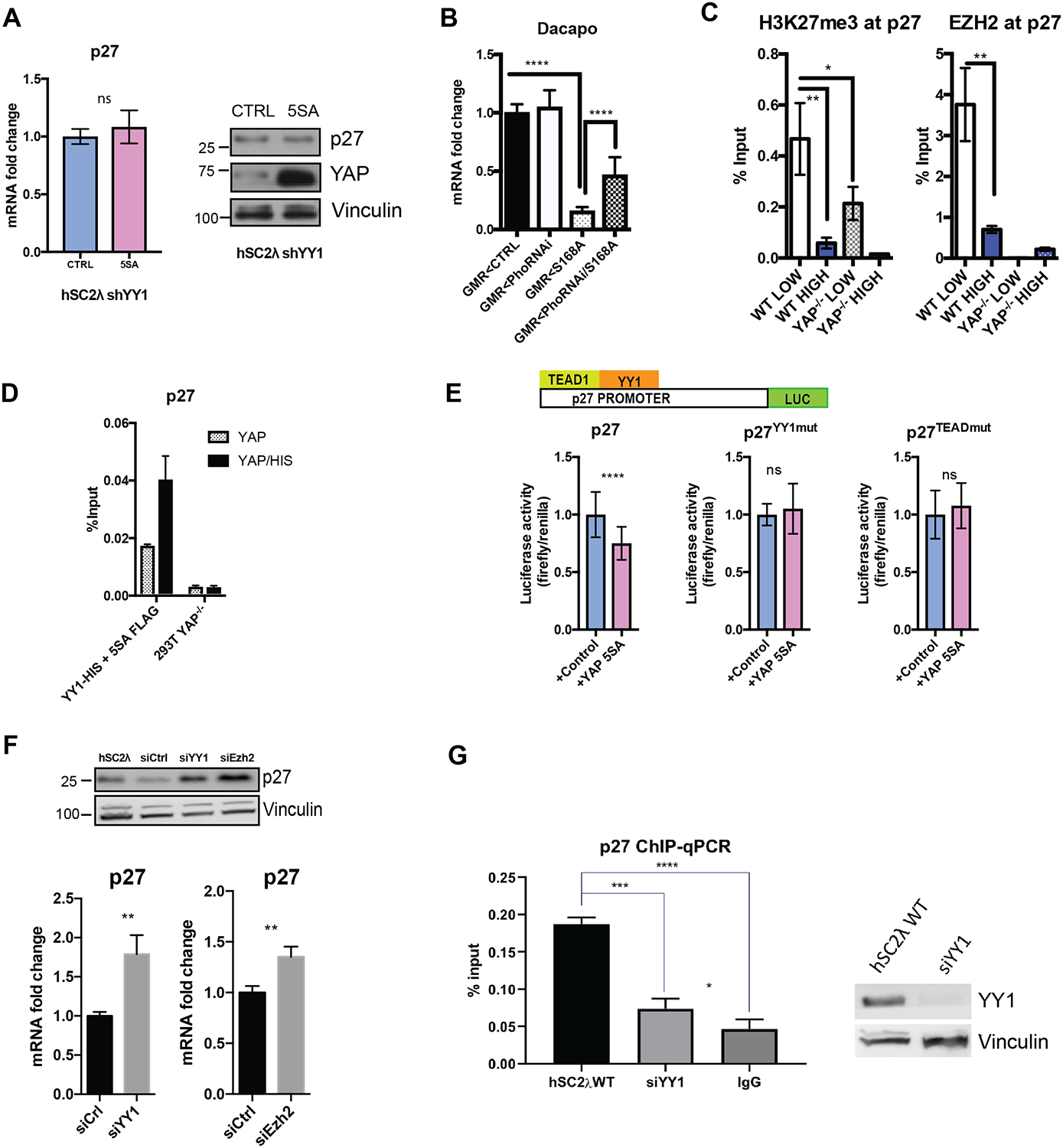
(A) p27 mRNA was measured by qRT-PCR in shYY1 hSC2λ cells transfected with YAP-5SA (5SA) or control (CTRL) plasmids. Proteins levels were assessed by Western blotting. (B) Dacapo mRNA levels were assessed in flies by qRT-PCR (transcript levels normalized to act42a; n=2 vials of 4 flies each; ****=P<0.0001, two-tailed Student’s t test; error bars=SD). GMR<CTRL=control, GMR<PhoRNAi=Pho knockdown, GMR<S168A=active Yorkie, GMR< PhoRNAi/S168A = Pho knockdown/active Yorkie. (C) Enrichment EZH2 with an anti-EZH2 antibody was assessed by ChIP-qPCR in hSC2λ WT or hSC2λ YAP−/−, at high or low cell densities at CDKN1B promoter. IgG antibody used as a control. (n=3; **P=0.0041, two-tailed Student’s t test; error bars=SD). (D) Enrichment for YY1 at the p27 promoter was assessed by sequential ChIP-qPCR. 293T YAP−/− cells were transfected with vectors for YY1-HIS and 5SA-FLAG and extracts were IP’ed with anti-FLAG (gray) followed by anti-HIS (black). (Levels shown as percentage of input, n=3). (E) hSC2λ YAP−/− cells were transfected with luciferase reporter p27 promoter (p27=wildtype, p27YY1mut=mutated YY1 binding motif, p27TEADmut=mutated TEAD binding motif) with a control or YAP-5SA plasmid. Luciferase intensity was normalized to Renilla luciferase activity. (n=3, **** P<0.0001, two-tailed Student’s t test; error bars=SD). (F) p27 protein and mRNA levels were assessed by western blot and qRT-PCR in hSC2λ cells knockdown for YY1 (siYY1) or EZH2 (siEzh2). For qRT-PCR, transcript level were normalized to actin; n=3;**P=0.0054, **P=0.0068. For western blot, vinculin was used as a loading control. (G) Assessment of p27 promoter enrichment by YAP ChIP-qPCR, in wildtype hSC2λ cells or with knockdown for YY1 (siYY1). IgG used as negative control. Western blot shows knockdown efficiency of YY1; vinculin used as loading control (n=3; **** P<0.0001, two-tailed Student’s t test; error bars, SD). See also Figure S3.
YY1 was shown to be required for the recruitment of Enhancer of zeste homologue 2 (EZH2) to chromatin in the vicinity of muscle-specific genes (37). EZH2 is the catalytic component of the PRC2 complex and silences gene expression through trimethylation of histone H3 lysine 27 (H3K27me3) (38). We performed ChIP-qPCR for EZH2 and H3K27me3 in hSC2λ cells and observed enrichment at the p27 promoter at low cell densities (Figure 3C). This enrichment was significantly reduced in hSC2λ YAP−/− cells grown at low density, suggesting that EZH2 recruitment requires YAP (Figure 3C). To test whether YAP and YY1 can co-occupy the p27 promoter, we carried out a sequential ChIP-qPCR in 293T cells in which YAP is knocked out. Cells were transfected with either FLAG-YAP5SA or a combination of 6X(HIS)-YY1 and FLAG-YAP5SA and immunoprecipitated for YAP5SA (FLAG) alone or YAP5SA (FLAG) followed by YY1 (HIS). We observed enrichment for YY1 specifically in the presence of YAP5SA at p27 promoter (Figure 3D). To test whether YY1 is required for YAP-mediated repression of p27, we cloned the promoter and first exon of p27 into a reporter plasmid upstream of a firefly luciferase cassette. Upon YAP5SA expression, we observed a decrease in luciferase signal compared to the control. However, when mutating the YY1 DNA binding site (CCAT→TGTA) in the cloned promoter sequence, we no longer observe this decrease in signal (Figure 3E). Similarly, mutation of the TEAD binding site, significantly diminished the ability of YAP5SA to repress luciferase activity (Figure 3E). Since YAP knockdown leads to upregulation of p27, we sought to assess how downregulating YY1 and EZH2 would affect p27 levels. We performed a knockdown of YY1 and EZH2 using siRNA and observed upregulation of p27 at the transcriptional and protein levels suggesting that in addition to YAP, YY1 and EZH2 are both required for p27 mediated repression (Figure 3F). Finally, to determine whether YY1 is required for YAP recruitment to the p27 promoter we assessed the enrichment for YAP at the promoter by ChIP-qPCR in the presence or absence of YY1. This analysis indicates that YY1 is required for the binding of YAP to the p27 promoter (Figure 3G).
To assess whether cells overexpressing YAP exhibit loss of contact inhibition, we performed cell-counting assays of hSC2λ cells stably expressing the activated YAP-5SA allele and observed that the YAP-5SA expressing cells have a growth advantage and continue to proliferate after the control expressing cells have reached a plateau (Figure S3G). Moreover, cell cycle analysis of the cells at high densities demonstrates a higher fraction of YAP5SA expressing cells in S-phase, compared to the control cells (Figure S3H). In addition, we observed enrichment of YY1, YAP and EZH2 at the promoter of CDKN1B in the YAP-5SA cell line, at high cell density (Figure S3I). These findings suggest that forced expression of YAP at high cell density can lead to the recruitment of YY1 and EZH2 to the CDKN1B promoter and override contact inhibition due to high cell density.
Genomic co-localization of YAP, YY1 and EZH2
To further understand YY1/YAP/EZH2 genome-wide distribution in human Schwann cells, we performed additional ChIP-seq experiments for YY1 and EZH2. We observed that all three factors co-localize at the p27 promoter (Figure 4A). To assess if YY1, YAP and EZH2 can form a complex we performed immunoprecipitation studies looking at endogenous levels of expression or in cells transfected with expression plasmids for all 3 proteins. These studies demonstrate that YY1/YAP/EZH2 can be co-immunoprecipitated using antibodies against any of the three, when overexpressed (Figure 4B–C) and at endogenous levels of expression (Figure 4D and Figure S4A). To further validate the interaction and close proximity between YAP, YY1 and EZH2, we performed a proximity ligation assay (PLA) in non-transfected hSC2λ cells. This analysis demonstrates the colocalization and close proximity of YAP-YY1 and YAP-EZH2 in the nuclei of these cells (Figure 4E).
Figure 4. YAP co-localizes and forms a complex with YY1 and EZH2.

(A) Gene browser view of the YAP (green), YY1 (blue), and EZH2 (red) ChIP libraries across the p27 promoter. Bars underneath the browser tracks indicate the span of identified peaks. (B-C) Analysis of YAP, YY1 And EZH2 interaction by Immunoprecipitation. hSC2λ were transfected with expression plasmids for all 3 proteins. Immunoprecipitation was performed using anti-YAP antibody (B) or with anti-His and anti-HA antibodies (C) as indicated. (D) Analysis of endogenous YAP, YY1 And EZH2 interaction by Immunoprecipitation. Controls were carried out using non-specific matched IgG antibodies. Additional controls using YAP−/− cells are presented in Supplementary Figure S4A (E) Proximity ligation analysis (PLA) of YAP and YY1 (TOP) or YAP and EZH2 (BOTTOM in hSC2λ cells. Cells were probed with primary antibodies recognizing YAP and YY1 or YAP and EZH2, respectively, and interactions were visualized with the Duolink In Situ Red kit (Sigma). Red dots indicate close proximity. Nuclei were visualized with DAPI stain, shown in blue. Scale bars = 50μM. YY1 or EZH2 primary antibodies alone were used as controls. Representative images are shown.
Across the genome, we observed a significant co-localization of YAP, YY1 and EZH2 (Figure 5A). The overall number of peaks identified for each mark were YAP=12480, YY1=9455, EZH2=6928. Pairwise affinity analysis of the common peaks shared between YAP and YY1 reveal a total of 8560 (64%). Of these shared YAP-YY1 peaks, 6813 (80%) were shared with EZH2 (Figure 5B). Motif analysis for triple-bound peaks revealed enrichment for TEAD1 and YY1 motif at bound sites across datasets (Figure 5C). When comparing the predicted motifs enrichment in YAP peaks versus YAP/YY1/EZH2 peaks, there is no clear enrichment for binding motifs for known transcriptional activators in the YAP peaks alone (Tables S3 and S5). Additionally, read densities for YAP and YY1 chromatin-binding show a strong overlap (Figure 5D). From the triple-bound peaks, we identified several cyclin-dependent kinase inhibitors (p57, p15, p21) as potential targets for YAP-mediated repression (Table S6). All of these targets have a YY1 motif adjacent to a TEAD motif at their respective promoters (Table S7). Overexpression of YAP in hSC2λ cells led to a significant repression of these targets (Figure 5E, TOP). The overexpression of TAZ in hSC2λ cells led similar results (Figure S5A). Significantly, in the context of downregulated YY1, the overexpression of YAP failed to repress mRNAs levels, suggesting that YY1 is required for YAP mediated repression of these targets (Figure 5E, BOTTOM). In addition, we observed a loss of H3K27me3 marks at the promoter of these genes upon YAP knockout (Figure 5F). We analyzed H3K27me3 marks at the promotors of additional genes bound by YAP/YY1/EZH2 and 3 out of 9 genes analyzed displayed a YAP-dependent H3K27me3 expression profile, suggesting that additional mechanisms can mediate the repression at other loci (Figure S5B). One possibility, given the similar repressive role for TAZ, is that TAZ may mediate the repressive function at these loci. These results suggest that YAP-mediated transcriptional repression, in conjunction with YY1, plays a broad and significant role in regulation of the cell cycle. A KEGG pathway analysis on YAP-bound genes further supports this by identifying cell cycle associated genes as significantly enriched in the list of differentially bound promoters (Figure S5C).
Figure 5. YAP co-localizes with YY1 and EZH2 to regulate cell cycle genes.

(A) Heatmaps of the common YAP-YY1-EZH2 peaks where each horizontal row is an individual locus and the vertical columns representing 25 bp blocks of the DNA surrounding the peak center ± 2Kb. The intensity of the color reflects the normalized read density of the ChIP samples. Rows are sorted by the intensity of the YAP peaks. (B) Pairwise affinity analysis of the YAP & YY1 peaks (TOP) and the common YAP/YY1 & EZH2 peaks (BOTTOM). (C) Motif analysis at YY1 and triple-bound peaks. (D) Read density of the YAP and YY1 ChIPs ± 2Kb from the peak centers. (E) p57, p15, p21 mRNA levels were measured by qRT-PCR in hSC2λ cells and hSC2λ shYY1 cells transfected with expression plasmids for YAP-5SA or control (CTRL) plasmids (n=3, **** P<0.0001, two-tailed Student’s t test; error bars=SD). (F) H3K27me3 ChIP-qPCR was performed in hSC2λ cells at low and high cell densities in the presence or absence of YAP (n=3, **** P<0.0001, *** P<0.0001, **P=0.0076 two-tailed Student’s t test; error bars=SD).
Discussion
The Hippo pathway has been shown to control cell proliferation and organ size through regulation of the transcriptional regulator YAP. Disruption of the Hippo pathway leads to YAP nuclear localization and is associated with loss of contact inhibition and increased proliferation and tumorigenesis (39). However, the precise molecular mechanisms by which YAP mediates contact inhibition remain unknown. YAP’s role as a transcriptional coactivator has been widely studied through its ability to activate the transcription of several genes including connective tissue growth factor (CTGF), epidermal growth factor receptor ligand amphiregulin (AREG) and ankyrin repeat domain 1 (ANKRD1) (40–42). YAP’s role as an activator may not fully account for the overgrowth phenotype observed in cancer suggesting that it possesses additional nuclear functions. Indeed, a limited number of studies have shown YAP can associate with the NuRD complex and transcriptionally repress differentiation markers to maintain pluripotency and repress tumor suppressor genes to promote cell growth and survival in MCF10A cells (7, 8). Our study identifies a previously unknown mechanism by which YAP can act as a transcriptional repressor through association with YY1 and the PRC2 complex and the addition of repressive H3K27me3 marks.
Our initial studies focused on CDKN1B, which codes for p27, and is a major regulator of the cell cycle that functions to prevent cells from entering S-phase (43) and is a regulator of Schwann cell differentiation (44, 45). Additionally, several studies report that p27 expression is downregulated in vestibular schwannomas (46, 47). We find that YAP represses p27 through the recruitment of YY1 and the PRC2 complex (see model in Figure 6). In addition, we show that loss of YY1 in vivo can rescue YAP-mediated repression of p27. Loss of contact inhibition of proliferation is one of the main hallmarks of cancer and is associated with decreased level of p27 and increased cell proliferation (48). Our findings that expression of active YAP can overcome contact inhibition in human Schwann cells and repress p27 expression identify a potential molecular link between the disrupted Hippo pathway, amplification of YAP in cancer and loss of contact inhibition.
Figure 6.
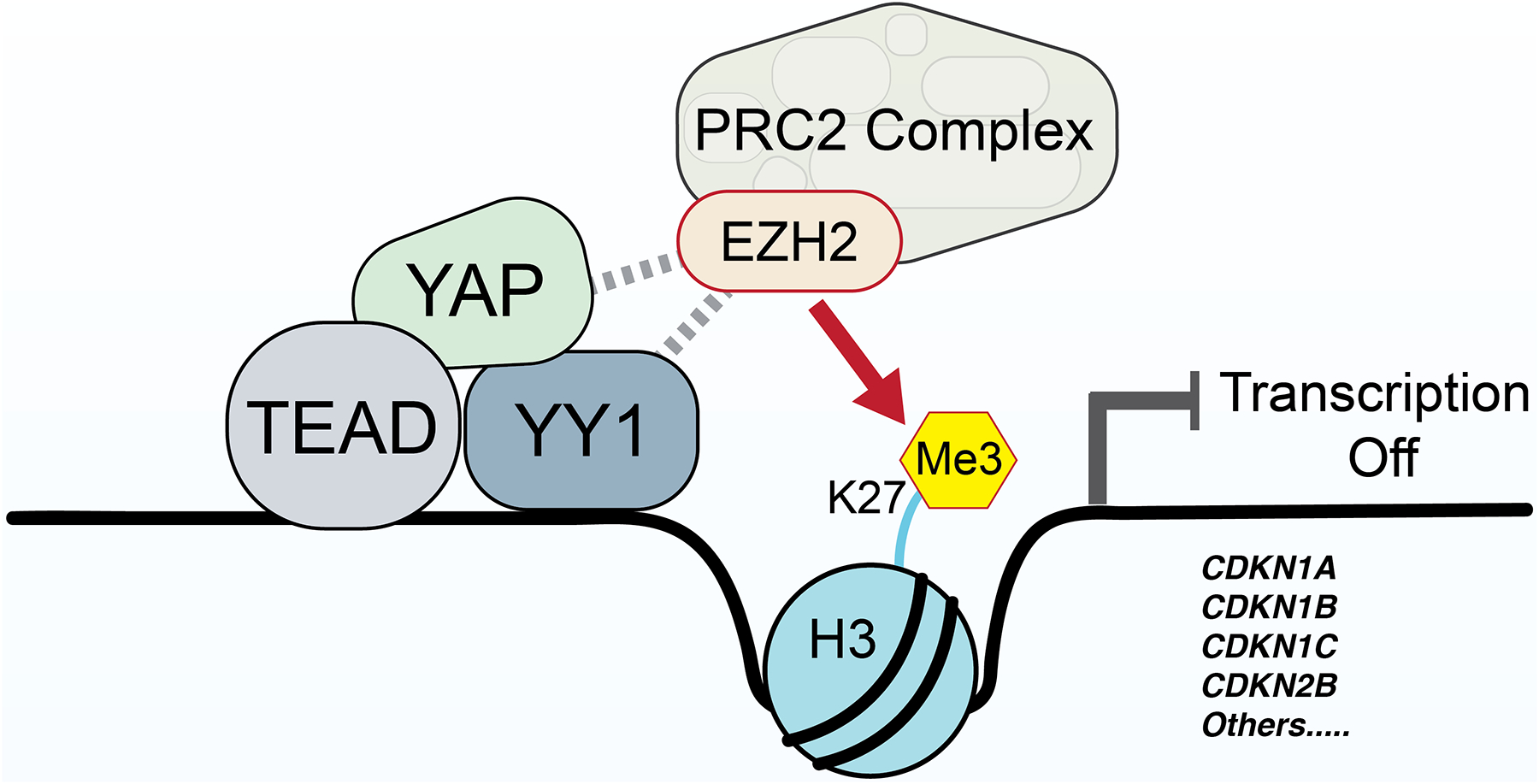
Proposed model for YAP function in inhibition of CDKN1B and other CDKI genes.
In addition to p27, our analysis identified additional targets of YAP/YY1-mediated repression. Since YAP can regulate thousands of targets, as an activator or repressor, we believe it is unlikely that the effects of YAP are mediated through a single target, or perhaps a single gene family. Rather, it is likely that the effects are mediated through hosts of genes, both up and down regulated. To dissect the role of these different YAP target genes will likely require tools that isolate YAP’s effects on individual or groups of selected targets. Further studies are thus required to elucidate the extent of YAP’s activating versus repressive functions.
Previous reports indicate that the deacetylation of H3K27 by the NuRD complex facilitates the recruitment of the PRC2 complex for gene repression (49, 50), this suggests that YAP may first associate with the NuRD complex, which can then lead to recruitment of the PRC2 complex and gene repression. Further studies are needed to determine whether such mechanisms are at play in the regulation of CDKN1B and the other CDKIs by YAP and YY1. Undoubtedly, the function of YAP in mediating cell growth is broad and involves multiple mechanisms and downstream targets. Our findings suggest that transcriptional repression represents a major mechanism through which YAP mediates transcriptional regulation of critical cellular behaviors.
Supplementary Material
Acknowledgements
We thank Dr. Ursula Ehmer (Roman-Herzog-Krebszentrum- Comprehensive Cancer Center) for sharing vectors used in the tail vein injections. Library preparation and sequencing were performed at The Scripps Research Institute Florida Genomics Core.
Funding
The work was supported by R01NS077952 (NINDS/NIH) and R01CA124495 (NCI/NIH) to J.K., R01AG045036 to W.W.J. (from the NIA/NIH). M.K is supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103620. M.E.P. is supported by R01AI095634 (NIAID/NIH) and the Frenchman’s Creek Women for Cancer Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no competing interests.
References
- 1.Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nature reviews Molecular cell biology. 2017;18(12):758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ota M, Sasaki H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development. 2008;135(24):4059–69. [DOI] [PubMed] [Google Scholar]
- 3.Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Developmental cell. 2008;14(3):388–98. [DOI] [PubMed] [Google Scholar]
- 4.Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23(7):785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boin A, Couvelard A, Couderc C, Brito I, Filipescu D, Kalamarides M, et al. Proteomic screening identifies a YAP-driven signaling network linked to tumor cell proliferation in human schwannomas. Neuro-oncology. 2014;16(9):1196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, et al. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39(11):1582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, et al. Switch enhancers interpret TGF-beta and Hippo signaling to control cell fate in human embryonic stem cells. Cell reports. 2013;5(6):1611–24. [DOI] [PubMed] [Google Scholar]
- 8.Kim M, Kim T, Johnson RL, Lim DS. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015;11(2):270–82. [DOI] [PubMed] [Google Scholar]
- 9.Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19(1):27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8(1):27–36. [DOI] [PubMed] [Google Scholar]
- 11.Guerrant W, Kota S, Troutman S, Mandati V, Fallahi M, Stemmer-Rachamimov A, et al. YAP Mediates Tumorigenesis in Neurofibromatosis Type 2 by Promoting Cell Survival and Proliferation through a COX-2-EGFR Signaling Axis. Cancer research. 2016;76(12):3507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu LMN, Deng Y, Wang J, Zhao C, Wang J, Rao R, et al. Programming of Schwann Cells by Lats1/2-TAZ/YAP Signaling Drives Malignant Peripheral Nerve Sheath Tumorigenesis. Cancer Cell. 2018;33(2):292–308 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Chang LJ, Neubauer DR, Muir DF, Wallace MR. Immortalization of human normal and NF1 neurofibroma Schwann cells. Lab Invest. 2016;96(10):1105–15. [DOI] [PubMed] [Google Scholar]
- 14.Moleirinho S, Hoxha S, Mandati V, Curtale G, Troutman S, Ehmer U, et al. Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology. 2011;29(1):24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hay BA, Maile R, Rubin GM. P element insertion-dependent gene activation in the Drosophila eye. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(10):5195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011. 2011;17(1). [Google Scholar]
- 18.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome biology. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature biotechnology. 2008;26(12):1351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38(4):576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fera D, Schultz DC, Hodawadekar S, Reichman M, Donover PS, Melvin J, et al. Identification and characterization of small molecule antagonists of pRb inactivation by viral oncoproteins. Chem Biol. 2012;19(4):518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17(9):1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang W, Kim T, Koo JS, Kim SK, Lim DS. Mechanical cue-induced YAP instructs Skp2-dependent cell cycle exit and oncogenic signaling. The EMBO journal. 2017;36(17):2510–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Estaras C, Hsu HT, Huang L, Jones KA. YAP repression of the WNT3 gene controls hESC differentiation along the cardiac mesoderm lineage. Genes & development. 2017;31(22):2250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levenberg S, Yarden A, Kam Z, Geiger B. p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene. 1999;18(4):869–76. [DOI] [PubMed] [Google Scholar]
- 27.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8(1):9–22. [DOI] [PubMed] [Google Scholar]
- 28.Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(24):8832–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Motti ML, Califano D, Baldassarre G, Celetti A, Merolla F, Forzati F, et al. Reduced E-cadherin expression contributes to the loss of p27(kip1)-mediated mechanism of contact inhibition in thyroid anaplastic carcinomas. Carcinogenesis. 2005;26(6):1021–34. [DOI] [PubMed] [Google Scholar]
- 30.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114(4):457–67. [DOI] [PubMed] [Google Scholar]
- 32.Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat Rev Cancer. 2007;7(3):182–91. [DOI] [PubMed] [Google Scholar]
- 33.Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21(8):886–97. [DOI] [PubMed] [Google Scholar]
- 34.Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1. Biochimica et biophysica acta. 1997;1332(2):F49–66. [DOI] [PubMed] [Google Scholar]
- 35.Thomas MJ, Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236(2):197–208. [DOI] [PubMed] [Google Scholar]
- 36.Brown JL, Mucci D, Whiteley M, Dirksen ML, Kassis JA. The Drosophila Polycomb group gene pleiohomeotic encodes a DNA binding protein with homology to the transcription factor YY1. Molecular cell. 1998;1(7):1057–64. [DOI] [PubMed] [Google Scholar]
- 37.Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes & development. 2004;18(21):2627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer cell. 2008;13(3):188–92. [DOI] [PubMed] [Google Scholar]
- 40.Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes & development. 2008;22(14):1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, et al. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nature cell biology. 2009;11(12):1444–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS genetics. 2015;11(8):e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, et al. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372(6506):570–3. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Yang H, Liu Y, Huan W, Zhang S, Wu G, et al. The cyclin-dependent kinase inhibitor p27(Kip1) is a positive regulator of Schwann cell differentiation in vitro. Journal of molecular neuroscience : MN. 2011;45(2):277–83. [DOI] [PubMed] [Google Scholar]
- 45.Porrello E, Rivellini C, Dina G, Triolo D, Del Carro U, Ungaro D, et al. Jab1 regulates Schwann cell proliferation and axonal sorting through p27. J Exp Med. 2014;211(1):29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seol HJ, Jung HW, Park SH, Hwang SK, Kim DG, Paek SH, et al. Aggressive vestibular schwannomas showing postoperative rapid growth - their association with decreased p27 expression. Journal of neuro-oncology. 2005;75(2):203–7. [DOI] [PubMed] [Google Scholar]
- 47.Lasak JM, Welling DB, Akhmametyeva EM, Salloum M, Chang LS. Retinoblastoma-cyclin-dependent kinase pathway deregulation in vestibular schwannomas. The Laryngoscope. 2002;112(9):1555–61. [DOI] [PubMed] [Google Scholar]
- 48.Fuse T, Tanikawa M, Nakanishi M, Ikeda K, Tada T, Inagaki H, et al. p27Kip1 expression by contact inhibition as a prognostic index of human glioma. J Neurochem. 2000;74(4):1393–9. [DOI] [PubMed] [Google Scholar]
- 49.Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, et al. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. The EMBO journal. 2012;31(3):593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morey L, Brenner C, Fazi F, Villa R, Gutierrez A, Buschbeck M, et al. MBD3, a component of the NuRD complex, facilitates chromatin alteration and deposition of epigenetic marks. Mol Cell Biol. 2008;28(19):5912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.