

Abstract
Trichloroethylene (TCE) is a widespread environmental contaminant following decades of use as an industrial solvent, improper disposal, and remediation challenges. Consequently, TCE exposure continues to constitute a risk to human health. Despite epidemiological evidence associating exposure with adverse birth outcomes, the effects of TCE and its metabolite S-(1, 2-dichlorovinyl)-L-cysteine (DCVC) on the placenta remain undetermined. Flexible and efficient macronutrient and energy metabolism pathway utilization is essential for placental cell physiological adaptability. Because DCVC is known to compromise cellular energy status and disrupt energy metabolism in renal proximal tubular cells, this study investigated the effects of DCVC on cellular energy status and energy metabolism pathways in placental cells. Human extravillous trophoblast cells, HTR-8/SVneo, were exposed to 5–20 μM DCVC for 6 or 12 h. After establishing concentration and exposure duration thresholds for DCVC-induced cytotoxicity, targeted metabolomics was used to evaluate overall energy status and metabolite concentrations from energy metabolism pathways. The data revealed glucose metabolism perturbations including a time-dependent accumulation of glucose-6-phosphate+frutose-6-phosphate (G6P+F6P) as well as independent shunting of glucose intermediates that diminished with time, with modest energy status decline but in the absence of significant changes in ATP concentrations. Furthermore, metabolic profiling suggested that DCVC stimulated compensatory utilization of glycerol, lipid, and amino acid metabolism to provide intermediate substrates entering downstream in the glycolytic pathway or the tricarboxylic acid cycle. Lastly, amino acid deprivation increased susceptibility to DCVC-induced cytotoxicity. Taken together, these results suggest that DCVC caused metabolic perturbations necessitating adaptations in macronutrient and energy metabolism pathway utilization to maintain adequate ATP levels.
Introduction
TCE is a volatile organic compound that originates exclusively from human activities. Originally commercialized in the 1920s as an oral anesthetic, its potent solvent properties were quickly discovered and harnessed, propelling TCE to become one of the most commonly used industrial and dry-cleaning solvents by midcentury.1 Today, over 80% of TCE production is utilized as a chemical intermediate in closed-system refrigerant manufacturing processes, specifically HFC-134a, a refrigerant used in car air conditioning systems.2,3 The other primary use of TCE is as a vaporized metal degreaser, although the Environmental Protection Agency (EPA) has proposed to ban this use as recently as 2017.1,4,5 As a result of its long-standing pervasive usage and its chemical properties, TCE is a ubiquitous persistent environmental contaminant found in soil, air, and water.4 TCE is currently ranked as number 16 on the U.S. Agency for Toxic Substances and Disease Registry’s Substance Priority List. Despite its detection in 1055 of 1750 current and former EPA-designated National Priorities List Superfund sites, TCE continues to makes its way into the environment with approximately 1.9 million pounds of TCE released in 2015.3,6−8 Because industrial usage and persistent environmental contamination of TCE continue, exposure through inhalation, ingestion, and skin contact remains a potential threat to human health.
TCE has as a long history of study as a potential organ-specific toxicant.1 In particular, there is extensive literature on TCE toxicity to the liver and kidney, and TCE was recently reclassified by the National Toxicology Program (NTP) and International Agency for Research on Cancer (IARC) as a known human carcinogen based on evidence that it causes kidney cancer in humans.2,9,10 In addition to recognition as a renal and liver toxicant,4 TCE has been implicated in adverse pregnancy outcomes. Although an early study found no association between maternal TCE exposure and low birth weight,11 more recent studies reported positive associations between maternal TCE exposure and low birth weight and preterm birth.12,13
The precise underlying mechanisms of adverse birth outcomes such as low birth weight and preterm birth remain largely unresolved despite considerable scientific scrutiny.14−16 Because the placenta is critical for support and protection of the fetus during pregnancy, placental disruption may play a key role in the development of adverse birth outcomes.17 For example, there is evidence that placental insufficiency, with deficient nutrient and metabolic waste exchange between mother and fetus, may contribute to early parturition.16,18 Similarly, recent epidemiology studies found significant associations between pre-eclampsia and increased risk of preterm birth or low birth weight.19,20
As a metabolically active organ in direct contact with maternal circulation, the placenta is a likely target organ for toxicity.21 Because of high blood volume and a large surface area in contact with maternal blood, the placenta is readily exposed to maternal blood-circulating TCE and its metabolites.22,23 Moreover, the placenta expresses many enzymes required for TCE biotransformation including cytochrome P450, glutathione-S-transferase, and beta-lyase.24,25 The presence of these enzymes greatly increases the risk of tissue-generated harmful TCE metabolites.
The weight of evidence indicates that biotransformation is required for TCE-mediated cytotoxicity.26 Although many prior toxicology studies focused on the TCE cytochrome P450 oxidation pathway’s metabolites trichloroacetate (TCA) and dichloroacetate (DCA), TCE renal toxicity is attributed to metabolites of the glutathione pathway.26 Moreover, in human placental cells in vitro, the TCE glutathione pathway derived metabolite S-(1,2-dichlorovinyl)-L-cysteine (DCVC) is cytotoxic27 at concentrations of TCA and DCA that fail to elicit cytotoxicity (unpublished). Furthermore, DCVC potently inhibits pathogen-stimulated cytokine release from human extraplacental membranes in culture, whereas similar and higher concentrations of TCA have no significant effect.28
Placental cells have sizable energy requirements for carrying out biological processes such as tissue remodeling, nutrient and waste transport, steroid hormone synthesis, and maintenance of homeostasis amid changing environmental conditions.29−31 In addition, ATP is required for general cell processes such as active transport, transcription, and translation.32,33 ATP is generated anaerobically by macronutrients through metabolism of glucose, lipids, and proteins through glycolysis, as well as aerobically through oxidative phosphorylation. Because of the need to maintain adequate ATP, flexible and efficient utilization of macronutrients and energy metabolism pathways is essential. DCVC causes cytotoxicity in kidney proximal tubular cells mediated by reactive oxygen species and mitochondrial dysfunction.34−40 Specifically, Lash et al. demonstrated that DCVC causes a decrease in the total adenine nucleotide pool, compromised cellular energy status, and tricarboxylic acid cycle perturbations in kidney cells.39,41 Moreover, we recently reported that DCVC causes mitochondrial perturbations, elevated reactive oxygen species generation, lipid peroxidation, and mitochondrial-mediated apoptosis in vitro utilizing a human extravillous trophoblast cell line.27,42,43 Together, the evidence suggests that cellular processes and structures responsible for generating ATP may be impacted by DCVC exposure.
On the basis of the high energy requirements of the placenta and previous studies that identified DCVC impacts on cellular energy status and ATP stability, this study investigated the effects of DCVC on cellular energy status and energy metabolism pathways in placental cells. The human extravillous trophoblast cell line HTR-8/SVneo was utilized as a model here because previous reports showed that these cells exhibited mitochondrial activity similar to other cell lines and primary trophoblasts.44−46
Experimental Procedures
Chemicals and Reagents
S-(1,2-Dichlorovinyl)-L-cysteine (DCVC), a trichloroethylene glutathione conjugation pathway metabolite, was synthesized in powder form by the University of Michigan Medicinal Chemistry Core as previously described.47 High-performance liquid chromatography (HPLC) analysis was used to determine purity (98.7%). A stock solution of 1 mM DCVC was prepared by dissolving DCVC in phosphate buffered saline and stored in small aliquots at −20 °C to minimize freeze/thaw cycles. The chemical purity of the DCVC stock solution was confirmed periodically by nuclear magnetic resonance (NMR).
RPMI 1640 culture medium with l-glutamine and without phenol red, 10 000 U/mL penicillin/10 000 μg/mL streptomycin (P/S) solution, and fetal bovine serum (FBS) were purchased from Gibco, a division of Thermo Fisher Scientific (Waltham, MA, USA). Phosphate buffered saline (PBS), Hank’s Balanced Salt Solution (HBSS), and 0.25% trypsin were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). Dimethyl sulfoxide (DMSO) was purchased from Torcis Biosciences (Bristol, UK). Dulbecco’s modified Eagle media (DMEM) with and without amino acids were purchased from US Biological Life Sciences (Salem, MA, USA).
Cell Culture and Treatment
The HTR-8/SVneo cells were obtained from Dr. Charles H. Graham (Queen’s University, Kingston, Ontario, Canada). This cell line was derived from first-trimester human placentae and immortalized with simian virus 40 large T antigen.48 The cell line expresses markers of an extravillous trophoblast phenotype and has a female genotype. HTR-8/SVneo cells were cultured as previously described.27,49 Briefly, cells were cultured between passages 78–87 in RPMI 1640 medium supplemented with 10% FBS and 1% P/S at 37 °C in a 5% CO2 humidified incubator. Cells were sustained in RPMI 1640 growth medium with 10% FBS and 1% P/S prior to and during experiments to ensure optimal cell growth as previously described.48 Cells were grown to 70–90% confluence for 24 h after subculture prior to beginning each experiment.
Just before each experiment, a DCVC stock solution aliquot was quickly thawed in a 37 °C water bath and then diluted in RPMI 1640 medium with 10% FBS and 1% P/S to final exposure concentrations of 5–20 μM DCVC. The DCVC concentrations selected for study include plausible metabolite concentrations in human blood with occupational exposure to the parent compound, trichloroethylene.50 Additionally, we selected concentrations previously determined to lack overt cytotoxicity in HTR-8/SVneo cells at the times points used in the present study.27
Cell Line Validation
Genomic DNA was extracted from HTR-8/SVneo cellsQIAamp DNA Mini Kit (Qiagen; Hilden, Germany). Microsatellite genotyping was performed using AmpFLSTR Identifier Plus PCR Amplification Kit run on a 3730XL Genetic Analyzer (Applied Biosystems; Waltham, MA, USA). The short tandem repeat profile generated for our cells was compared to the short tandem repeat profile for HTR-8/SVneo (ATCC CRL-3271) published by American Type Culture Collection (Manassas, VA, USA).51 The short tandem repeat profile was a match: CSF1PO, 12; D13S317, 9, 12; D16S539, 13; D5S818, 12; D7S820, 12; TH01, 6, 9.3; vWA, 13, 18; TPOX, 8; amelogenin gender determination marker, X.51
Measurement of Cytotoxicity
Cytotoxicity was measured with the MultiTox-Glo Multiplex Cytotoxicity Kit performed according to the manufacturer’s protocol (Promega; Madison, WI), which sequentially measured the relative number of live and dead cells in a single assay. Live cells were measured with the cell-permanent fluorescent compound glycyl-phenylalanyl-aminofluorocoumarin (GF-AFC), and dead cells were measured with the luminescent cell-impermeable compound alanyl–alanyl-phenylalanyl-aminoluciferin (AAF-Glo). Briefly, HTR-8/SVneo cells were seeded at 10 000 cells per well in a white, clear-bottom 96-well plate and incubated for 24 h. Cells were treated with medium alone (control) or DCVC (5, 10, or 20 μM) for 12, 24, or 48 h. Following exposure, GF-AFC was added directly to the media and incubated for 1 h. Fluorescence signal (emission, 400 nm; excitation, 505 nm) for viable cells was measured with a SpectraMax M2e Multi-Mode Microplate Reader (Molecular Devices). After viability quantification, AAF-Glo was added to the media and incubated at room temperature for 10 min. Luminescence signal for dead cells was quantified using the Glomax Multi Plus detection system (Promega). To normalize the data, the relative live-to-dead cell ratios were calculated by dividing the average live cell fluorescence signal by the average dead cell luminescence signal per treatment group for each time point. Camptothecin (4 μM), a toxic compound that targets the enzyme topoisomerase I and causes double-strand DNA breaks during replication,52,53 was included as a positive control at each time point.
Bicinchoninic Acid (BCA) Assay
To normalize metabolomics and western blot experiments, total protein concentration per well was measured calorimetrically using the Pierce Bicinchoninic Acid (BCA) Assay Kit (Thermo Fisher Scientific) performed according to the manufacturer’s recommended protocol. Briefly, cells were lysed with RIPA lysis buffer containing a protease inhibitor cocktail. Cell lysates from each sample (10 μL) were transferred to a 96-well clear-bottomed plate, combined with working buffer (200 μL), and incubated for 30 min at 37 °C. Following incubation, protein concentrations were determined with a SpectraMax M2e Multi-Mode Microplate Reader (OD = 562 nm) (Molecular Devices) based on comparison to a bovine serum albumin-derived standard curve ranging between 0.0625 and 2 mg/mL.
Quantification of Targeted Metabolomics
Targeted metabolomics was used to quantify a panel of energy metabolism pathway-related metabolites and amino acids. HTR-8/SVneo cells were seeded at a density of 1.6 million cells per plate in tissue culture-treated 100 mm dishes and allowed to adhere and acclimate for 48 h. Cells were then treated with medium alone (control) or DCVC (20 μM) for 6 or 12 h. Following the exposure period, cell culture medium was aspirated, and then cells were briefly washed with 0.15 M ammonium acetate to remove excess medium, rapidly flash frozen with liquid nitrogen, and stored at −80 °C. Samples were transported on ice to the University of Michigan Metabolomics Core for further processing.
Samples were prepared as previously described by Lorenz et al.54 Briefly, cells were treated with an extraction solvent containing isotope-labeled internal standards. The extraction solvent contained a mixture of chloroform, water, and methanol in a ratio of 1:1:8. Cells were lysed and scraped to create a homogenized mixture containing extracted metabolites. Metabolites were detected with reverse-phase liquid chromatography–mass spectrometry (LC–MS) on an Agilent system consisting of a 1290 UPLC coupled with a 6520 quadrupole-time-of-flight (QTOF) mass spectrometer operated in ESI- mode (Agilent Technologies, Santa Clara, CA, USA). Data were processed using MassHunter Quantitative analysis version B.07.00. Glycolytic, tricarboxylic acid, and pentose phosphate pathway metabolites were normalized to the nearest isotope-labeled internal standard and quantitated using two replicated injections of five standards to create a highly accurate linear calibration curve. Glycolytic, tricarboxylic acid, and pentose phosphate pathway metabolites were reported with specific intracellular concentrations normalized to total protein mass per dish, as measured with the BCA assay. Amino acids, lipids, and other metabolites in the analysis were normalized to the nearest internal standard, and the peak areas were used for differential analysis between groups. These metabolites were reported as relative response (RR) normalized to total protein mass per samples.54 For each time point, five independent experiments were conducted.
Evaluation of Amino Acid Deprivation Effects on DCVC-Induced Cytotoxicity
The effect of amino acid deprivation on DCVC-induced cytotoxicity was evaluated by culturing cells in different proportions of amino acid-free or amino acid-containing media with and without 20 μM DCVC. Cells were seeded into wells of a white, clear-bottom 96-well plate in complete RPMI 1640 medium at a density of 10 000 cells per well. After 24 h of incubation, the medium was changed to a mixture of complete DMEM medium containing the following proportions of amino acid-free DMEM medium: 0%, 20%, 40%, 60%, 80%, and 100%. Cytotoxicity was measured with the MultiTox-Glo Multiplex Cytotoxicity Kit (Promega), as previously described. To normalize the data, the relative live-to-dead cell ratios were calculated by dividing the live cell fluorescence signal by the dead cell luminescence signal and averaged within each treatment group for each amino acid deprivation condition. There were three independent experiments, with three replicates per DCVC and amino acid deprivation conditions in each experiment.
Immunoblotting Protein Analysis
HTR-8/SVneo cells were seeded at a density of 400 000 cells per well in a clear six-well culture plate and allowed to attach for 24 h. Following the acclimation period, cells were treated with medium alone (control) or DCVC (20 μM) for 12 h. Cells were lysed in RIPA lysis buffer and centrifuged at 14 000 rpm for 10 min at 4 °C. Lysates were heated with loading buffer at 85 °C for 3 min. Samples were loaded into commercially available Novex 4–20% Tris-Glycine Mini Gel cassettes (Invitrogen), and proteins were separated by polyacrylamide gel electrophoresis run at 125 V for 1.5 h. SeeBlue Plus2 prestained protein standard (Invitrogen) was included as a visual molecular weight reference. Following separation, proteins were transferred onto nitrocellulose membranes at 75 V for 4 h at a temperature of 4 °C. Membranes were probed using antibodies raised against small neutral amino acid transporter type 2 (ASCT2/SLC1A5) [molecular weight 75; catalog no. 8057; Cell Signaling Technologies; Research Resource Identifier (RRID): AB_10891440], large amino acid transporter heavy subunit (4F2hc/SLC3A2) antibody (molecular weight 75–120; catalog no. 13180; Cell Signaling Technologies; RRID: AB_2687475), L-type neutral amino acid transporter 1 (LAT1/SLC7A5) antibody (molecular weight 39; catalog no. 5347; Cell Signaling Technologies; RRID: AB_10695104), and AMP-activated protein kinase alpha subunit-phosphorylated (p-AMPKα) or nonphosphorylated (AMPKα) antibodies [molecular weight 62; catalog nos. 2535 or 2793; Cell Signaling Technologies; Research Resource Identifier (RRID): AB_10705605 or AB_915794]. Antibody complexes were detected by Alexa Fluor antimouse and antirabbit fluorescent-conjugated antibodies (Invitrogen) and visualized using an Odyssey CLx image scanner (Li-Cor Biosciences; Lincoln, NE, USA). Blots were quantified using Image Studio software version 5.2 and normalized to Revert Total Protein Stain (Li-Cor Biosciences). Three to four independent experiments were performed, and each experiment was performed in triplicate.
PFK Activity Enzyme Assay
Phosphofructokinase 1 (PFK1) activity was measured in cells treated for 12 h with medium alone (control) or 20 μM DCVC. PFK1 activity was measured with a commercially available colorimetric activity assay. The assay was performed according to the manufacturer’s protocol (Sigma-Aldrich). Briefly, following DCVC exposure, reaction mixture (composed of PFK1 assay buffer, PFK1 enzyme mix, PFK1 enzyme developer, ATP and PFK1 substrate) was added to each well containing NADH standards, samples, or sample blanks. After an initial 5 min incubation at 37 °C, the first absorbance measurement was recorded. Subsequent measurements were taken every 7 min until the value of the most active sample was greater than the value of the highest standard (28 to 35 min). Calorimetric absorbance (OD = 450 nm) was measured with a SpectraMax M2e Multi-Mode Microplate Reader (Molecular Devices). PFK1 activity was calculated using the following formula: PFK1 activity = [(amount of NADH generated between TInitial and Tfinal as determined by standard curve) × (sample dilution factor)]/[(reaction time) × (sample volume)]. Three independent experiments were performed, and each experiment was performed in triplicate.
Statistical Analysis
All experiments were performed independently and repeated three to five times. When applicable, technical replicates were averaged within each experiment. These data were analyzed using Student’s t-tests or one-way or two-way analysis of variance (ANOVA followed by Tukey’s post hoc test for comparison of means (GraphPad Prism version 7; GraphPad Software Inc.; San Diego, CA, USA). Experiments containing different time points and treatment were analyzed with two-way ANOVA. Experiments only containing one time point and two treatment groups including western blots and PFK activity assay were analyzed using Student’s t-tests. Metabolomics data were log2 transformed due to non-normal Gaussian distribution. After transformation, the data were normally distributed prior to statistical analysis using two-way ANOVA. Data are expressed as means ± SEM. N = Number of independent experiments. P < 0.05 was considered statistically significant in all experiments.
Results
DCVC Cytotoxicity
Because the objective of this study was to investigate energy metabolism under conditions that were not lethal to cells, we measured cytotoxicity in HTR-8/SVneo cells at lower concentrations (5–20 μM DCVC) and for a larger range of time points (12–48 h) than previously reported by Hassan et al.27 DCVC induced cytotoxicity in time- and concentration-dependent manners after 24 and 48 h exposure but not 12 h (ANOVA interaction effect, P < 0.0001, Figure 1). After 24 h of exposure, only 20 μM DCVC decreased the live-to-dead cell ratio significantly by 51% (P = 0.002). However, after 48 h of exposure, both 10 and 20 μM DCVC reduced the live-to-dead cell ratio by 55% and 67%, respectively (P < 0.0008). These experiments validated previous findings that exposure to 20 μM DCVC for 24 h is cytotoxic to HTR-8/SVneo cells27 while establishing an exposure duration threshold of 48 h for cytotoxicity with 10 μM DCVC.
Figure 1.

DCVC cytotoxicity. HTR-8/SVneo cells were treated for 12, 24, or 48 h with medium alone (control), or with 5, 10, or 20 μM DCVC. The MultiTox-Glo Multiplex Cytotoxicity Kit (Promega) was used to measure the relative number of live and dead cells within a single well as described in the Experimental Procedures. Graphical representation shows live-to-dead cell ratios as percent control within each time point. Bars represent means ± SEM. Data were analyzed by two-way ANOVA (interaction between time and treatment, P < 0.0001) with post hoc Tukey multiple comparisons. Pound sign indicates significant difference compared to same treatment at all earlier time points: #P < 0.0001. At symbol indicates significant difference compared to same treatment at 12 h time point: @P < 0.03. Asterisk indicates significant difference compared to medium alone (control) within same time point: *P = 0.0008. Plus sign indicates significant difference compared to control and 5 μM DCVC within same time point: +P < 0.02. N = 3 independent experiments for each time point, with three replicates per treatment in each experiment. Camptothecin (4 μM) was included as a positive control and decreased the live-to-dead cell ratio by 55.6% ± 2.17% at 12 h, 80.68% ± 0.531% at 24 h, and 32.89% ± 0.039%% at 48 h.
DCVC-Induced Changes in Cellular Energy Status Indicators
We focused our investigation on cellular energy metabolism because DCVC was previously shown to deplete ATP concentrations and compromise cellular energy status in renal proximal tubular cells.39 The overall cellular energy status describes a cell’s ability to maintain adequate ATP levels.55,56 To evaluate the effect of DCVC on the overall energy status of treated HTR-8/SVneo cells, we first used targeted metabolomics to measure concentrations of key energy metabolites. Then we analyzed the ratios of key energy metabolite couples including adenylate and guanylate nucleotides, electron donors/acceptors, and a phosphate group donor/acceptor.
Intracellular Concentrations of Key Energy Metabolites
Treatment with DCVC-induced changes in intracellular concentrations of key energy metabolites, as shown in Figure 2A. Regarding effects on adenylate and guanylate nucleotides, the primary energy drivers of physiological processes in cells, 20 μM DCVC significantly increased AMP, ADP, and GMP intracellular concentrations by at least 1.4-fold after 6 and 12 h exposures, whereas GDP and GTP concentrations increased significantly only after 12 h, compared to time-matched controls (Figure 2Ai; P < 0.05). ATP concentrations did not change significantly despite changes in concentrations of other adenylate nucleotides. Phosphocreatine, a phosphate donor critical for rapid regeneration of ATP during high energy expenditure, decreased 1.6-fold after 12 h (P = 0.0002) but was not significantly changed after 6 h (Figure 2Aii). Concomitantly, creatine byproduct concentrations increased nearly two-fold at both 6 and 12 h (Figure 2Aii; P < 0.01). Lastly, NADH, an electron carrier that shuttles electrons from different pathways to the electron transport chain, increased 2.2-fold after 6 h (P = 0.0007) but was not significantly increased after 12 h exposure compared to time-matched controls (Figure 2Aiii). The NAD+ concentrations did not significantly change at either the 6 or 12 h time point.
Figure 2.

DCVC-induced changes in key cellular energy status indictors. Targeted metabolomics analysis was used to measure concentrations of energy status metabolites in HTR-8/SVneo cells treated with medium alone (control) or 20 μM DCVC for 6 or 12 h. (A) Graphical representations of concentrations of: (i) adenylate and guanylate nucleotides, (ii) phosphate donor and product phosphocreatine and creatine, and (iii) electron transporters NAD+ and NADH. Boxes represent first quartile, median, and third quartile; whiskers represent minimum and maximum concentrations. (B) Graphical representations of energy metabolite ratios derived from metabolite concentrations: (i) ATP:AMP, (ii) ATP:ADP, (iii) phosphocreatine:creatine, and (iv) NADH:NAD+. Bars represent ratio means ± SEM. All data were analyzed by two-way ANOVA (interaction between time and treatment varied by metabolite, P < 0.05) with post hoc Tukey multiple comparisons. Asterisks indicate significant differences compared to medium alone (control): *P < 0.0419, **P < 0.0097, ***P < 0.001. Pound signs indicates significant differences compared to same treatment at all earlier time points: #P = 0.0116, ##P = 0.0026, ###P < 0.001. N = 5 independent experiments for each time point. (C) AMPK signaling pathway was evaluated with western blotting analysis and normalized to total protein. (i) Graphical representation of p-AMPKα:p-AMPKα ratio and (ii) representative western blotting images. Bars represent means ± SEM. Data were analyzed with student t tests. N = 3 independent experiment, with three replicates per treatment in each experiment.
Ratios of Key Energy Metabolite Couples
Ratios of key energy metabolite couples are shown in Figure 2B, calculated from the concentrations reported in Figure 2A. Because these energy metabolites are normally maintained in narrow nonequilibrium concentrations for optimal physiologic function, changes in ratios of critical metabolite couples may indicate compromise of the intracellular homeostasis that maintain the concentrations.56,57 The ATP:AMP ratio decreased 72% and 62% after 6 and 12 h exposure to DCVC, respectively, whereas the ATP:ADP ratio decreased by 44% only after 12 h (Figure 2Bi,Bii; P < 0.01). After 6 and 12 h DCVC exposure, the phosphocreatine:creatine ratio decreased 55% and 65%, respectively (Figures 2Biii; P < 0.0001). On the other hand, the NADH:NAD+ ratio, demonstrating the opposite pattern, increased by 128% after 6 h but did not change significantly after 12 h (Figures 2Biv; P = 0.0084). These fluctuating energy metabolite ratios indicate that short-duration DCVC treatment caused an overall mild decline in cellular energy status and a buildup of NADH.
AMP-Kinase Phosphorylation
Because a decreased ATP:AMP ratio could reflect activation of the energy stress response AMP-kinase (AMPK) signaling pathway,56 we measured levels of phosphorylated (activated) and total AMPKα using western blot analyses (Figure 2C). We observed no significant treatment-related changes when comparing the ratio of p-AMPKα to AMPKα protein levels between nontreated control and DCVC-treated cells, suggesting that the increase in the AMP/ATP ratio was not sufficient to promote AMPK phosphorylation.
Profiling DCVC Effect on Energy Metabolism Pathway Utilization
DCVC has previously been shown to alter concentrations of tricarboxylic acid cycle intermediates in renal proximal tubular cells,41 prompting us to further investigate DCVC-induced effects on specific energy metabolism pathways and related pathways using targeted metabolomics to quantify a panel of metabolites unique to each pathway. Indeed, nine pathways had one or more metabolite concentrations significantly altered by exposure to 20 μM DCVC at either the 6 h or 12 h time points. An overview of DCVC-induced changes in energy metabolism pathways is summarized in Figure 3A, with affected pathways in dark gray boxes and notable impacted metabolites outlined in light gray rectangles. The outlined metabolite concentrations are individually displayed in Figure 3B for each pathway.
Figure 3.


Effects of DCVC on energy metabolism pathways. HTR-8/SVneo cells were treated with medium alone (control) or 20 μM DCVC for 6 or 12 h. Targeted metabolomics analysis was used to measure a panel of intracellular metabolites unique to specific energy metabolism pathways. (A) Overview of DCVC-induced changes in integrated energy metabolism pathways. Blue arrows indicate pathway directionality. Metabolite names in red indicate altered concentrations between treatment groups within same time point (P < 0.05). Purple and pink arrows indicate direction of change in concentrations within 6 or 12 h time points, respectively. Green star symbols indicate altered concentrations between time points within same treatment group (P < 0.05). All other symbols are indicated in figure legend. (B) Graphical representations of selected metabolite concentrations grouped by energy metabolic pathway. Background color indicates corresponding pathway on integrated overview in panel A. Pathways represented include: (i) glucose metabolism, (ii) pentose phosphate pathway, (iii) purine pathways, (vi) hexosamine biosynthesis pathway, (v) glycolysis, (vi) TCA cycle pathway, (vii) glycerol metabolism pathway, (viii) β-oxidation pathway, and (ix) amino acid metabolism pathways. Within each graph, boxes represent first quartile, median, and third quartile; whiskers represent minimum and maximum. All data were log2 transformed prior to statistical analysis to achieve normal Gaussian distribution. Data were analyzed by two-way ANOVA (interaction between time and treatment varied depending on metabolite, P < 0.05) with post hoc Tukey multiple comparisons. Asterisks indicate significant differences compared to medium alone (control): *P < 0.05, **P < 0.01, ***P < 0.001. Pound signs indicate significant differences compared to same treatment at all earlier time points: #P < 0.05, ##P < 0.01, ###P < 0.001. N = 5 independent experiments for each time point. (C) Graphical representation of phosphofructokinase 1 activity after 12 h DCVC treatment. PFK1 activity was measured with a commercially available enzyme activity assay kit (Sigma-Aldrich). Bars represent means ± SEM. Data were analyzed with student t test. N = 3 independent experiments, with three replicates per treatment in each experiment.
Glucose Metabolism Pathway
DCVC had no significant effect on glucose concentrations compared to nontreated controls at either 6 or 12 h: however, glucose concentrations decreased from 6 to 12 h regardless of treatment (Figure 3Bi; P < 0.004). Once glucose enters the cell via GLUT membrane transporters, it undergoes reversible phosphorylation yielding glucose-6-phosphate (G6P), which is further isomerized to fructose-6-phosphate (F6P).58 Although our analysis was unable to differentiate between G6P and F6P, DCVC treatment increased G6P+F6P metabolites 1.4- and 2.4-fold at 6 and 12 h, respectively, compared to time-matched controls (Figure 3Bi; P ≤ 0.03). These results demonstrate that 20 μM DCVC treatment caused a substantial time-dependent accumulation of F6P+G6P, indicating possible perturbation of downstream glucose catabolism.
Phosphofructokinase 1 Activity
Because a time-dependent accumulation of F6P+G6P was observed, we measured DCVC effects on activity of the enzyme phosphofructokinase 1 (PFK1), which catalyzes the rate-limiting phosphorylation of F6P to frucose-1,6-bisphosphate. We observed no significant change in activity after 12 h of treatment with 20 μM DCVC, compared to time-matched controls (Figure 3C). These results indicate that F6P+G6P accumulation was not caused by reduced enzymatic activity.
Pentose Phosphate, Hexosamine, and Purine Nucleotide Pathways
Notably, the metabolic fate of the accumulated F6P+G6P is a critical branching point for glucose metabolism: although most molecules proceed through glycolysis, a small quantity shunt into the pentose phosphate pathway (PPP) and downstream purine nucleotide pathways (PNP), or into the hexosamine biosynthesis pathway (HBP) (Figure 3B). Treatment with 20 μM DCVC significantly increased metabolite concentrations in each of these pathways: PPP five-carbon sugars (R5P+X5P) increased after 6 h but not 12 h (Figure 3Bii; P = 0.0004); PNP metabolites inosine and inosine monophosphate increased after 6 and 12 h, albeit a smaller increase after 12 h (Figure 3Biii; P < 0.04); and HBP metabolites N-acetyl-glucosamine-1-phosphate (NAcG1P) and uridine diphosphate N-acetyl-glucosamine increased after 6 h or both time points, respectively, compared to time-matched controls (Figure 3Biv; P < 0.03). These elevated metabolite concentrations suggest that 20 μM DCVC treatment increased F6P+G6P shunting from the glycolytic pathway after 6 h but not 12 h.
Glycolytic Pathway
Despite increased shunting, the vast majority of F6P+G6P molecules are further phosphorylated into frucose-1,6-bisphosphate (F1,6bP) during the first committed and most heavily regulated glycolytic step. F1BP then splits into two molecules, glyceraldehyde-3-phosphate (Ga3P) and dihydroxyacetone phosphate (DHAP), which rapidly interconvert. Although F1,6bP concentrations did not change in 20 μM DCVC-exposed cells, Ga3P and DHAP concentrations increased by 2.5- and 2.8-fold after 6 h, and 1.8- and 2-fold after 12 h, respectively, compared with time-matched control cells (Figure 3v; P < 0.0003). The remaining downstream glycolytic metabolites, including 2-phosphoglycerate+3-phosphoglycerate (2PG+3PG) and phosphoenolpyruvate (PEP), were not significantly altered by DCVC treatment. However, lactate, the terminal anaerobic glycolytic metabolite, increased by 1.4-fold with 20 μM DCVC after 6 h but not 12 h, compared to time-matched controls (Figure 3Bv; P = 0.04). These results demonstrate that 20 μM DCVC elevated Ga3P and DHAP intracellular concentrations, indicating a potential DCVC-induced compensatory mechanism occurring in upstream glycolysis.
Tricarboxylic Acid Cycle
Under aerobic conditions, the final step of the glycolytic pathway converts pyruvate to acetyl CoA in preparation for entrance into tricarboxylic acid cycle (TCA). Treatment with 20 μM DCVC for 6 and 12 h increased acetyl CoA concentrations approximately 2.4-fold compared to time-matched controls (Figure 3Bvi; P = 0.01). Of the TCA metabolites measured in our panel, only malate had elevated concentrations with DCVC treatment after 12 h, compared to time-matched controls (Figure 3Bvi; P = 0.01). DCVC had no significant impact on the other TCA metabolites, citrate+isocitrate and succinate. In comparison to the other energy metabolic pathways examined, the TCA cycle appeared largely unaffected by DCVC treatment under our experimental conditions.
Alternative Bioenergetics Fuel Sources
Glycerol Metabolism Pathway
Because DCVC increased Ga3P and DHAP concentrations along with G6P+F6P accumulations in the absence of changes in glycolytic downstream metabolites, we hypothesized that there may be alternative macronutrient source(s) providing glycolytic intermediates. Importantly, DHAP is also a downstream metabolite in the glycerol metabolism pathway (Figure 3Bvii), providing a crucial entry point at which glycerol derived from lipid molecules may enter the glycolytic pathway midstream. Indeed, concentrations of glycerol-3-phosphate (GL3P), an upstream metabolite of DHAP in the glycerol catabolism pathway, increased 1.5-fold in DCVC-exposed cells at 6 and 12 h compared to time-matched controls (Figure 3Bvii P < 0.0001). Moreover, free oleic fatty acid concentrations, possibly derived extracellularly or from β-oxidation, were increased four-fold with DCVC treatment for 6 and 12 h, compared with time-matched controls (Figure 3Bviii; P < 0.0001). The DCVC-stimulated increases of intracellular GL3P, oleate, and acetyl-coA concentrations may reflect the use of lipids as an alternative fuel source.
Amino Acid Metabolism and Transport
Under certain conditions, amino acids can enter the glycolytic pathway as pyruvate, acetyl-CoA, or other TCA intermediates. Alanine, glutamine, and glutamate are among the most important amino acids for energy metabolism. Alanine recycles the carbon skeletons from metabolized branched amino acids into glucose via the Cahill cycle and interconverts with pyruvate.59−61 Glutamine and glutamate enter the TCA cycle via interconversion with each other and to the TCA intermediate α-ketoglutarate.62 Treatment with 20 μM DCVC increased intracellular concentrations of multiple amino acids (Figure 3Bix). Alanine concentrations were significantly elevated after 6 and 12 h, whereas glutamine and glutamate concentrations were only elevated after 6 h treatment with DCVC, compared to time-matched controls (Figure 3Bix; P < 0.05). Among essential amino acids, DCVC treatment increased tryptophan concentrations by nearly four-fold after 6 and 12 h, whereas leucine+isoleucine concentrations increased 1.4-fold at 6 h only, and histidine increased 1.6-fold at 12 h only, compared to time-matched controls (Figure 3Bix; P < 0.04). These results suggest that DCVC may stimulate uptake and utilization of amino acids as compensatory macronutrient sources.
Membrane transporters involved in cellular uptake of extracellular free amino acids were investigated as potential contributors to the increased intracellular concentrations of energy-relevant amino acids. Western blotting revealed that treatment with 20 μM DCVC for 12 h stimulated a 1.7-fold increased abundance of the alanine, serine, cysteine transporter 2 (ASCT2/SLC1A5) protein, an amino acid transporter responsible for transporting small neutral amino acids (Figure 4A; P = 0.047 compared to time-matched control). In contrast, no statistically significant differences were detected in protein expression of the 4F2 cell-surface antigen heavy chain (4F2hc/SLC3A2) and L-type amino acid transporter 1 (LAT1/SLC7A5) proteins that form a heterodimer transporter of large neutral amino acids, despite apparent trends toward DCVC-stimulated increases (Figure 4B,C). The finding of increased ASCT2/SLC1A5 abundance suggests that increased amino acid transport may contribute to the observed DCVC-induced increases in amino acid concentrations.
Figure 4.

DCVC-induced changes in amino acid transporter levels. Energy-relevant amino acid transporter levels in HTR-8/SVneo cells treated with medium alone (control) or 20 μM DCVC for 12 h were evaluated with western blotting analysis and normalized to total protein. (A) Small neutral amino acid transporter ASCT2/SLC1A5. (B) Large amino acid transporter heavy subunit 4F2hc/SLC3A2. (C) Large neutral amino acid transporter LAT1/SLC7A5. (D) Representative western blot images. Bars represent means ± SEM as percent control. Data were analyzed with student t tests. Asterisks indicate significant difference compared to medium alone (control): * P = 0.0474. N = 3 independent experiments, with three replicates per treatment in each experiment.
Exacerbation of DCVC-Stimulated Cytotoxicity by Amino Acid Deprivation
To further establish the role of amino acid utilization as a compensatory biofuel source, HTR-8/SVneo cells were cultured with different concentrations of amino-acid-free medium in the absence and presence of 20 μM DCVC for 12 h. The live-to-dead cell ratio of cell treated with DCVC compared to nontreated controls decreased inversely with increasing proportions of amino acid-free medium. For cells cultured in 100% amino acid-free medium, DCVC treatment decreased the live-to-dead cell ratio significantly by 91% compared to nontreated cells (Figure 5; P < 0.0001). For cells cultured in 80% amino acid-free medium, DCVC treatment decreased the live-to-dead cell ratio significantly by 53% compared to nontreated cells (Figure 5; P < 0.0001). However, cells cultured in proportions of amino acid-free medium at 60% or less, did not show significant differences in the live-to-dead cell ratio between DCVC-treated and nontreated cells. These data demonstrate that amino acid deprivation exacerbated DCVC-stimulated cytotoxicity, revealing a substantial role for amino acids in HTR-8/SVneo adaptation and survival.
Figure 5.

Effects of amino acid deprivation on DCVC-induced cytotoxicity. HTR-8/SVneo cells were cultured in DMEM containing the following proportions of amino acid-free medium: 0%, 20%, 40%, 60%, 80%, and 100%, mixed with respective proportions of amino acid-containing DMEM medium. Within each culture condition, cells were also treated in triplicate with medium alone (control) or 20 μM DCVC for 12 h. The MultiTox-Glo Multiplex Cytotoxicity Kit (Promega) was used to measure live and dead cells, as previously described in the Experimental Procedures. Graphical representation shows live-to-dead cell ratios as a percentage of nontreated controls within each respective cell culture condition group. Bars represent means ± SEM. Data were analyzed by two-way ANOVA (interaction between cell culture conditions and DCVC treatment, P < 0.0001) with post hoc Tukey multiple comparisons. Asterisks indicate significant differences compared to medium alone (control) within respective cell culture conditions groups: *P < 0.0002. Dollar symbol indicates significant difference compared to same DCVC treatment cultured with amino acid-free medium: $P = 0.0137. N = 3 independent experiments, with three replicates per treatment and cell culture condition groups in each experiment.
Energy Pathway Metabolite Ratios
To confirm DCVC-induced alterations to energy metabolism pathways, we calculated metabolite ratios to determine metabolite flux in glucose metabolism, upstream glycolysis, and other connected pathways. The metabolite ratios are graphically displayed in the context of the respective pathways they represent in Figure 6.
Figure 6.

Energy pathway metabolite ratios. Graphical representations of energy metabolite ratio calculated from metabolite concentrations representing upstream glycolysis, pentose phosphate pathway, and glycerol metabolism pathway. (A) Glucose:G6P+F6P ratio, (B) G6P+F6P:R5P+X5P ratio, (C) G6P+F6P:R5P+X5P ratio, (D) G6P+F6P:NAcG1P ratio, (E) F1BP:Ga3P+DHAP ratio, (F) Ga3P+DHAP:2G+3PG ratio, (G) Ga3P+DHAP:GL3P ratio. Metabolite names in dark red indicate metabolites with noteworthy DCVC-induced fluctuations. Boxes within each graph represent first quartile, median, and third quartile; whiskers represent minimum and maximum. All data were log2 transformed prior to statistical analysis to achieve normal Gaussian distribution. Data were analyzed by two-way ANOVA (interaction between time and treatment varied depending on metabolite, P < 0.05) with post hoc Tukey multiple comparisons. Asterisks indicate significant differences compared to medium alone (control): *P < 0.05, **P < 0.01, ***P < 0.001. Pound signs indicate significant differences compared to same treatment at all earlier time points: #P < 0.05, ##P < 0.01, ###P < 0.001.
G6P+F6P Time-Dependent Accumulation
DCVC treatment for 12 h increased the glucose:G6P+F6P ratio compared to time-matched controls (Figure 6A; P = 0.01). Concomitantly, the downstream G6P+F6P:F1BP ratio decreased at 6 and 12 h (Figure 6B; P = 0.0003). These ratio changes indicate metabolite flux favoring G6P+F6P, consistent with the finding that DCVC increased G6P+F6P intracellular concentration.
Pentose Phosphate and Hexosamine Biosynthesis Shunts
Following 12 h of exposure to 20 μM DCVC, the G6P+F6P:R5P+X5P ratio, representing the PPP, increased significantly (Figure 6C; P = 0.005), whereas the G6P+F6P:NAcG1P ratio, representing the HBP, did not significantly change compared to time-matched controls. Following 6 h of exposure to 20 μM DCVC, neither ratio was significantly altered, despite an apparent decreasing trend for both ratios, compared to time-matched controls.
These results may indicate potential DCVC-stimulated metabolic fluctuations favoring the PPP and HBP after 6 h, whereas the evidence indicates a reverse trend after 12 h with metabolic fluctuations favoring the glycolytic pathway.
Alternative Bioenergetics Fuel Sources via Glycerol Metabolism
The calculated metabolite ratio for F1BP:Ga3P+DHAP decreased significantly with 20 μM DCVC treatment after 6 and 12 h, whereas the Ga3P+DHAP:2PG+3PG ratio increased only after 6 h, compared to time-match controls (Figure 6E,F; P < 0.002). These results indicate DCVC-stimulated metabolic fluctuations favoring Ga3P and DHAP formation after 6 h that eased slightly after 12 h. Furthermore, 20 μM DCVC treatment increased the metabolite ratio of Ga3P+DAHP:GL3P after 6 h but not 12 h, compared to time-matched controls (Figure 6G; P = 0.0008). Importantly, this latter ratio indicates DCVC-induced metabolite fluctuation toward the glycolytic pathway, especially after 6 h, consistent with glycerol metabolism intermediates entering glycolysis.
Discussion
Epidemiologic evidence has associated maternal TCE exposure with increased risk of adverse birth outcomes. However, the effects of TCE and its metabolite S-(1, 2-dichlorovinyl)-L-cysteine (DCVC) on the placenta remain largely undetermined. Because of previous evidence of DCVC-induced energy perturbations in kidney cells,39,41 and the importance of maintaining sufficient ATP levels for placental cell function,30 this study investigated the effects of nonlethal DCVC concentrations on cellular energy status, macronutrients, and energy metabolism in placental cells. Here, we report DCVC-stimulated changes in energy metabolism pathway and macronutrient utilization in HTR-8/SVneo trophoblast cells.
To our knowledge, the current study is the first to use targeted metabolomics to evaluate DCVC-induced changes in cellular energy metabolism for any cell type, although a previous study used a metabolomics approach to investigate TCE-induced changes to the metabolome in the context of embryogenesis.63 In agreement our results, the latter study found increased histidine, glutamine, isoleucine, and decreased phosphocreatine concentrations in Japanese medaka fish exposed to 0–175 mg/L of TCE during embryogenesis: however, contrary to our results, ATP and glutamate and ATP concentrations decreased.63 Although the latter study of fish embryogenesis involved TCE exposure and not specifically DCVC, it is noteworthy that important energy-related metabolite concentrations were affected in the context embryogenesis, suggesting that TCE exposure may affect energy metabolism related to fetal development or pregnancy.
DCVC-Induced Changes to Cellular Energy Status
The cellular energy status, indicated by ratios comparing intracellular concentrations of key energy metabolite couples, describes cells’ ability to meet and maintain sufficient ATP levels to carry out energy-dependent processes.55,56 Our targeted metabolomics analysis revealed many DCVC-induced changes to intracellular concentrations of energy-relevant metabolites and corresponding ratios, indicating an apparent decline in cellular energy status, similar to a prior study conducted in rat kidney cells which used DCVC concentrations an order of magnitude higher than those used here.41 Importantly, we observed elevated ADP and AMP concentrations along with depressed ATP:ADP and ATP:AMP ratios compared to controls 6 and 12 h post DCVC exposure. However, despite these findings, we observed no overall changes to ATP intracellular concentrations after 6 or 12 h, nor activation of the AMPK energy stress pathway after 12 h of exposure. Taken together, these results suggest that despite the apparent cellular energy status decline, adaptive changes in energy production were sufficient to maintain ATP levels in HTR-8/SVneo cells under our exposure conditions.
In addition to cellular energy status, ratios of energy metabolite couples may serve as predictors or regulators of metabolic activity.55,56 During pregnancy, the ratio of ATP:ADP in placental tissue does not change in the first, second, or third-trimester under normal circumstances.64 However, a decreased ATP:ADP ratio was observed in primary cultures of cytotrophoblasts isolated from placentae of term pregnancies complicated with intrauterine growth restricted (IUGR) compared to normal term placentae.65 Moreover, the decreased ratio in the latter study corresponded with higher susceptibility to spontaneous apoptosis in the cytotrophoblasts,65 a defining characteristic of abnormal placental development.66 A study by Crocker et al. provides an example in which the ATP:ADP ratio not only is an indicator of energy status but also appears to be a metabolic regulator or predictor of cell fate.55,56 Although not directly tested, we suggest that the DCVC-induced changes in energy status ratios reported here may serve as early indicators and subsequent regulators of cell fate associated abnormal placental growth, as observed by others in IUGR.67
DCVC-Induced Changes to Macronutrients and Energy Metabolism Pathways
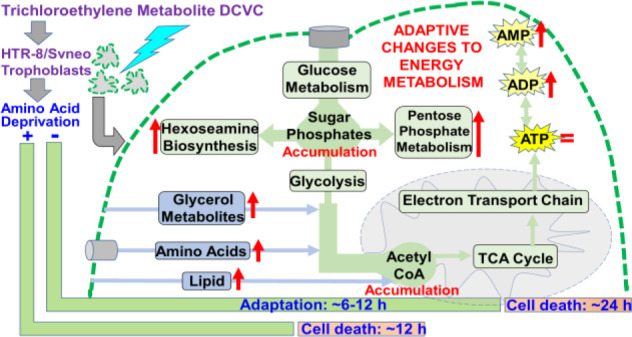
Cellular energy metabolism pathways are tightly regulated and highly interconnected, enabling cells to precisely adjust macronutrient and metabolism pathway utilization for optimal ATP synthesis.29,30 This facilitates crucial plasticity necessary to meet the energy demands of the placenta amidst constantly changing environmental conditions.30,68 In normal early pregnancies, anaerobic glycolysis fueled by glucose derived from maternal glandular histiotroph is the preferred source of energy over oxidative phosphorylation as outlined in Figure 7A.23,69−71 In the present study, we demonstrated DCVC-induced changes in intracellular metabolite concentrations from nine interconnected metabolism pathways. On the basis of these concentrations altered by DCVC exposure and corresponding metabolite flux ratios, we present an integrated framework proposing how DCVC may alter bioenergetic fuel sources and pathway utilization following 6 and 12 h of exposure as summarized in Figure 7B. A caveat to our steady state metabolomics approach is that we did not directly measure flux of metabolites through these pathways but are predicting the flux based on buildups at key regulatory points.
Figure 7.

Proposed DCVC-induced energy metabolism alterations HTR-8/SVneo cells. (A) Overview of normal cellular energy metabolism pathways in first-trimester placental cells. First-trimester placental cells survive in a low-oxygen environment. As a result, these cells prefer glycolysis fueled by glucose as their primary source of energy over oxidative phosphorylation. Despite this preference, the cells are capable of using other macronutrient metabolism pathways to fuel oxidative phosphorylation for additional ATP synthesis. (B) Summary of proposed DCVC-induced energy metabolism alterations in HTR-8/SVneo cells. 1: Following glucose phosphorylation, G6P+F6P accumulated in a time-dependent manner. Conversely, PPP and HBP shunting of G6P+F6P was elevated at 6 h and diminished with time, suggesting two independent processes. 2: At 6 and 12 h, alternative bioenergentic fuel sources and pathways including amino acid, lipid, and glycerol metabolism provided intermediates that enter glycolysis downstream of the G6P+F6P accumulation or enter the TCA cycle as acetyl CoA. Additionally, Ga3P and DHAP concentrations were elevated at both time points, suggesting another possible glycolytic perturbation. 3: Acetyl-CoA concentrations were increased at both time points, but TCA cycle metabolites were largely unchanged, indicating that DCVC likely does not directly affect the TCA cycle. 4: Although ATP levels are sustained, adenylate nucleotide ratios shifted down, and ADP and AMP concentrations increased.
During glucose metabolism, we observed time-dependent elevated G6P+F6P concentrations in DCVC-treated cells and DCVC-stimulated ratio shifts for metabolites upstream (glucose:G6P+F6P), and downstream (G6P+F6P:F1BP), of G6P+F6P; both heavily favoring metabolite flux toward G6P+F6P. Together, these results indicated a time-progressive accumulation of G6P+F6P metabolites resulting from DCVC exposure between 6 and 12 h. Furthermore, we established that the time-progressive G6P+F6P accumulation was not caused by a change in PFK1 activity, the enzyme that converts F6P to F1BP. Considering that PFK1 is the most regulated glycolytic enzyme,72 there may be DCVC-induced changes to other regulatory mechanisms. For example, not only is F6P converted to F1BP via PFK1 but F6P is simultaneously converted by the enzyme PFK2 to fructose-2,6 bisphosphate (F2BP) for the purpose of feed-forward up-regulation of PFK1 activity.73 As a result of this complex regulation, determining the exact target of DCVC requires further investigation. Nevertheless, the time-progressive G6P+F6P accumulation represents a significant DCVC-induced disruption at a key regulatory point for multiple energy metabolism pathways.
Shunting of glucose metabolism intermediates was also detected, as indicated by elevated concentrations of R5P+X5P and NAcG1P from the PPP and HBP, respectively, following 6 h of DCVC exposure but not 12 h. Moreover, DCVC treatment stimulated metabolite ratios that shifted toward the PPP and HBP after 6 h, but not 12 h. Together, these results indicated DCVC-induced elevated G6P+F6P shunting away from glycolysis in favor of the PPP and HBP, a pattern that peaked at 6 h and diminished with time. Because G6P+F6P shunting toward the PPP and HBP decreased with time, whereas the G6P+F6P accumulation increased with time, these observations suggest opposing G6P+F6P metabolite fluctuations. Although not directly tested, diminished G6P+F6P shunting may have exacerbated the G6P+F6P accumulation between 6 and 12 h of DCVC exposure. Regardless, the opposing nature of these metabolite flux patterns likely suggests independent processes. However, the precise relationship between these observations requires further experimental clarification outside of the scope this study. Despite the need for further investigation, the results presented here offer compelling evidence of DCVC-induced disruptions centered around G6P+F6P.
Interestingly, DCVC treatment revealed likely compensatory use of alternative macronutrient fuel sources based on evidence that DCVC treatment for 6 and 12 h increased intracellular concentrations of glycerol metabolism intermediates, lipids, and energy-relevant amino acids, accompanied by changes in metabolite ratios favoring catabolic flux patterns. Furthermore, DCVC treatment also elevated Ga3P+DHAP and acetyl CoA intracellular concentrations. The latter metabolite changes frequently occur in association with increased utilization of alternative macronutrient fuel sources because Ga3P+DHAP and acetyl CoA are entry points into energy metabolism for glycerol intermediates, some amino acids, and some lipids.74,75 Additionally, because the lower glycolytic pathway and TCA cycle downstream of these entry points were largely unaffected by DCVC treatment, this pattern suggests a successful compensatory mechanism, likely related to increased energy demand or the previously described changes in glucose metabolism following treatment with DCVC. Increased utilization of alternative macronutrient fuel sources appears to be an important adaptation mechanism by which the cells maintain enough energy production, even when challenged by DCVC treatment.
DCVC treatment for 6 or 12 h increased some intracellular amino acid concentrations and amino acid transporter levels. This suggested a role for amino acids in metabolic compensation, also supported by our demonstration that amino acid deprivation in cell culture media profoundly increased cytotoxicity in DCVC-treated HTR-8/SVneo cells compared to cells cultured in amino acid-replete media. These results not only confirm the obligatory role of amino acids in successful compensatory mechanisms but also potentially reveal a direct relationship between amino acid availability and DCVC exposure, in which amino acid deprivation increases susceptibility to DCVC-induced cell death. There are many factors that may result in placental amino acid deficiency in pregnant women such as maternal protein malnutrition, malabsorption, genetic factors, or metabolic diseases.76,77 These data indicate that amino acid deficiency during pregnancy could interact with environmental TCE exposure to exacerbate structural abnormalities and placental dysfunction, increasing the risk of adverse birth outcomes,78 though further studies are needed to link this cellular phenotype with human outcomes.
DCVC treatment did not change TCA cycle concentrations of the metabolites included in our panel except for a slightly elevated malate concentration following 12 h of exposure. This finding contrasts with a study by Lash and Anders41 that showed substantial increases in succinate and isocitrate concentrations in rat renal proximal tubular cells. Possible explanations for the different study findings may include the different cell types (placenta vs kidney) or DCVC concentrations (20 μM vs 1 mM) used in the present study compared to the study by Lash and Anders.41 The Lash and Anders study further described DCVC inhibition of corresponding enzymes succinate dehydrogenase and isocitrate dehydrogenase.41 Because no substantial changes in these metabolites were observed in the HTR-8/SVneo cells, the effect of DCVC on specific TCA cycle enzymes was not further investigated in the present study. In addition, prior studies suggest that DCVC metabolites may inhibit thiol-containing enzymes by acting as electrophiles in a physical interaction with the sulfide groups of these enzymes, but we did not explore such possibilities in the present study.41,79,80 Regardless, our results suggest that TCA cycle metabolites were likely not the primary target of DCVC-induced changes in energy metabolism in HTR-8/SVneo cells at the concentrations and exposure durations tested here.
S-(1,2-Dichlorovinyl)-L-cysteine (DCVC) Concentrations
Because the aim of the current study was to investigate the effects of nonlethal DCVC concentrations on energy in HTR-8/SVneo cells, we reaffirmed previous findings indicating that 24 h exposure to 20 μM DCVC or higher caused cell death, whereas 12 h exposure to 20 μM DCVC or lower was not lethal to the cells.27,42 Furthermore, we established an exposure duration threshold for detectable cell death at the lower concentration of 10 μM DCVC following 48 h of exposure. Contrary to the current study, previous studies investigating energy or energy metabolism responses to DCVC treatment in different cell types mostly used cytotoxic concentrations up to 1 mM, an order of magnitude higher than concentrations used here.36,39,41 Thus, the findings reported here contribute novel evidence of cellular energy responses to nonlethal DCVC concentrations.
The DCVC concentrations used in the present study are relevant to human occupational exposures based on levels of TCE that workers may encounter in an occupational setting. The Occupational Safety and Health Administration (OSHA) Permissible Exposure Limit (PEL) is 100 ppm averaged over an 8 h work day.3 In one study, female volunteers exposed to the PEL of TCE for 4 h by inhalation had an average peak blood concentration of 13.4 μM for the metabolic precursor to DCVC, S-(1,2-dichlorovinyl) glutathione.50 This peak blood concentration is included within the range of concentrations used in our study of 5–20 μM DCVC. Moreover, another study detected volatilized TCE concentrations up to 229 ppm, more than twice the PEL, in 80 exposed workers (29% women) wearing personal aerosolized monitoring devices,81 demonstrating the exposure levels above the PEL are possible. Thus, our study contributes valuable evidence of the effects of plausible DCVC blood concentrations from occupational TCE exposure in human placental cells.
HTR-8/SVneo Cells
Because of the lack of comparable animal models and limited availability of early human placental tissues, HTR-8/SVneo cells, a widely used immortalized trophoblast cell line originally derived from noncancerous first-trimester placental tissue, were used here to model extravillous trophoblasts. Extensive characterization by multiple research groups has collectively demonstrated that these cells display a combination of distinctive extravillous trophoblast molecular markers and functional attributes.82 For example, HTR-8/SVneo cells express a combination of cytokeratin 7 (CK7), histocompatibility antigen, class I, G (HLA-G) (when grown on Matrigel), and α5β1 integrin dimers.83−87 Moreover, the cells display a mesenchymal phenotype based on proteomics88 and stress-induced altered invasion capabilities.85,89,90 Two recent studies reported that HTR-8/SVneo cells contain mixed populations of cells;91,92 however, neither of these studies used any validation methods to verify the identity of their cell lines nor did they obtain their cells from the originator Dr. Charles Graham.48 In contrast, we obtained HTR-8/SVneo cells directly from Dr. Charles Graham’s laboratory, and we authenticated the identity of the cells using short tandem repeat profiling. Of specific relevance to the current study, prior reports found that trophoblast cell lines, including HTR-8/SVneo cells45 and BeWo cells,46 as well as primary trophoblasts,44 maintain typical mitochondrial activity as assessed by oxygen consumption rate measured with the Seahorse XF analyzer. The prior study from our laboratory43 was performed under experimental conditions that match those used in the current study.
Despite the benefits of using a cell line, there are also drawbacks. The immortalization processes changes the cells to allow them to continue proliferating in culture conditions for a longer time period than primary cells.48 Furthermore, in vitro cell culture conditions may contribute to genetic and epigenetic differences observed between HTR-8/SVneo cells and freshly isolated extravillous trophoblasts.93,94 Additionally, because in vitro experiments do not reflect the complicated in vivo dynamics within the fetal-uteroplacental unit, further studies in primary extravillous trophoblasts, villous explants, and other models are needed to confirm our results.
Summary and Conclusion
In summary, we present evidence detailing alterations to macronutrient and energy pathway metabolites in HTR-8/SVneo cells exposed to DCVC at relatively low concentrations that are relevant to human occupational TCE exposure. Taken together, the results presented here suggest that DCVC caused metabolic perturbations necessitating adaptations in macronutrient and energy metabolism pathway utilization, while successfully maintaining sufficient ATP concentrations. Our findings demonstrate the biological plausibility of DCVC-induced placental injury and provide new insights into the toxicological mechanisms of action of TCE and its metabolite DCVC.
Acknowledgments
We thank Maureen Kachman, Charles Evans, Angela Wiggins, and Kari Bonds of the University of Michigan’s Metabolomics Core for their assistance with LC–MS targeted metabolomics quantification. We also thank Maureen Sartor and Alla Karnovsky of the Omics and Bioinformatics Core of the Michigan Center on Lifestage Environmental Exposures and Disease for assistance with metabolomics statistical analyses. Additionally, we thank Larisa Yeomans of the University of Michigan Biochemical Nuclear Magnetic Resonance Core for verifying the chemical structure of DCVC. Finally, we gratefully acknowledge Anthony Su, Faith Bjork, JeAnna Redd, and Molly Mulcahy for assistance with learning new laboratory techniques and for helpful scientific discussions, and Monica Smolinski for assistance with BCA assays.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.9b00356.
Raw intracellular concentrations of 75 targeted metabolites detected with LC–MS, isolated from HTR-8/SVneo cells exposed to 20 μM DCVC for 6 or 12 h and normalized to total protein per sample (XLSX)
This work was supported by the National Institute of Environmental Health Sciences, National Institutes of Health (Grant Nos. P42 ES017198, P30 ES017885, and T32 ES007062), the National Institute of Diabetes and Digestive Kidney Diseases (Grant Nos. R01 DK107535 and P30 DK089503), and the University of Michigan. Additional fellowship support for SMH was from the Michigan Institute for Clinical and Health Research funded by the National Center for Advancing Translational Sciences (NCATS), NIH (UL1TR002240). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS, NIH, or the University of Michigan.
The authors declare no competing financial interest.
Supplementary Material
References
- Waters E. M.; Gerstner H. B.; Huff J. E. (1977) Trichloroethylene. I. An overview. J. Toxicol. Environ. Health 2, 671–707. 10.1080/15287397709529469. [DOI] [PubMed] [Google Scholar]
- NTP . (2015) National Toxicology Program Monograph on Trichloroethylene, in Reports on Carcinogens, National Toxicology Program. [Google Scholar]
- ATSDR . (2016) Public Health Statement on Trichloroethylene, Agency for Toxic Substances and Disease Registry. [Google Scholar]
- Chiu W. A.; Jinot J.; Scott C. S.; Makris S. L.; Cooper G. S.; Dzubow R. C.; Bale A. S.; Evans M. V.; Guyton K. Z.; Keshava N.; Lipscomb J. C.; Barone S. Jr.; Fox J. F.; Gwinn M. R.; Schaum J.; Caldwell J. C. (2013) Human health effects of trichloroethylene: key findings and scientific issues. Environ. Health Perspect. 121, 303–311. 10.1289/ehp.1205879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA . (2017) Trichloroethylene (TCE); Regulation of Use in Vapor Degreasing under TSCA §6(a) (RIN 2070-AK11) (Agency E. P., Ed.) p 119, Federal Regis. [Google Scholar]
- EPA . (2018) National Priorities List, Environmental Protection Agency.
- ATSDR . (2017) 2017 Substance Priority List, U.S. Agency for Toxic Substances and Disease Registry. [Google Scholar]
- EPA . (2017) TRI Explorer: Release Trends Report, Environmental Protection Agency. [Google Scholar]
- Guha N.; Loomis D.; Grosse Y.; Lauby-Secretan B.; El Ghissassi F.; Bouvard V.; Benbrahim-Tallaa L.; Baan R.; Mattock H.; Straif K. (2012) Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. Lancet Oncol. 13, 1192–1193. 10.1016/S1470-2045(12)70485-0. [DOI] [PubMed] [Google Scholar]
- Rusyn I.; Chiu W. A.; Lash L. H.; Kromhout H.; Hansen J.; Guyton K. Z. (2014) Trichloroethylene: Mechanistic, epidemiologic and other supporting evidence of carcinogenic hazard. Pharmacol. Ther. 141, 55–68. 10.1016/j.pharmthera.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagakos S.; Wessen B.; Zelen M. (1986) An analysis of contaminated well water and health eff ects in Woburn, Massachusetts. J. Am. Stat. Assoc. 81, 583–596. 10.1080/01621459.1986.10478307. [DOI] [Google Scholar]
- Forand S. P.; Lewis-Michl E. L.; Gomez M. I. (2012) Adverse birth outcomes and maternal exposure to trichloroethylene and tetrachloroethylene through soil vapor intrusion in New York State. Environ. Health Perspect. 120, 616–621. 10.1289/ehp.1103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckart P. Z.; Bove F. J.; Maslia M. (2014) Evaluation of contaminated drinking water and preterm birth, small for gestational age, and birth weight at Marine Corps Base Camp Lejeune, North Carolina: a cross-sectional study. Environ. Health 13, 99. 10.1186/1476-069X-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R. (2008) Spontaneous preterm birth, a clinical dilemma: etiologic, pathophysiologic and genetic heterogeneities and racial disparity. Acta Obstet. Gynecol. Scand. 87, 590–600. 10.1080/00016340802005126. [DOI] [PubMed] [Google Scholar]
- March of Dimes P., and Save the Children, WHO. (2012) Born Too Soon: The Global Action Report on Preterm Birth (Howson C. P., K M., and Lawn J. E., Eds.) World Health Organization, Geneva. [Google Scholar]
- Morgan T. (2014) Placental Insufficiency Is a Leading Cause of Preterm Labor. NewReviews 15, 5618–e5525. 10.1542/neo.15-12-e518. [DOI] [Google Scholar]
- Ilekis J. V.; Tsilou E.; Fisher S.; Abrahams V. M.; Soares M. J.; Cross J. C.; Zamudio S.; Illsley N. P.; Myatt L.; Colvis C.; Costantine M. M.; Haas D. M.; Sadovsky Y.; Weiner C.; Rytting E.; Bidwell G. (2016) Placental origins of adverse pregnancy outcomes: potential molecular targets: an Executive Workshop Summary of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Am. J. Obstet. Gynecol. 215, S1–S46. 10.1016/j.ajog.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan T. K. (2016) Role of the Placenta in Preterm Birth: A Review. Am. J. Perinatol 33, 258–266. 10.1055/s-0035-1570379. [DOI] [PubMed] [Google Scholar]
- Davies E. L.; Bell J. S.; Bhattacharya S. (2016) Preeclampsia and preterm delivery: A population-based case-control study. Hypertens. Pregnancy 35, 510–519. 10.1080/10641955.2016.1190846. [DOI] [PubMed] [Google Scholar]
- Oshvandi K.; Jadidi A.; Dehvan F.; Shobeiri F.; Cheraghi F.; Sangestani G.; Moghadari Koosha B.; Takarli F.; Aghababaie S. (2018) Relationship between Pregnancy-induced Hypertension with Neonatal and Maternal Complications. International Journal of Pediatrics 6, 8587–8594. 10.22038/IJP.2018.31750.2809. [DOI] [Google Scholar]
- Goodman D. R.; James R. C.; Harbison R. D. (1982) Placental toxicology. Food Chem. Toxicol. 20, 123–128. 10.1016/S0278-6915(82)80018-5. [DOI] [PubMed] [Google Scholar]
- Laham S. (1970) Studies on placental transfer. Trichlorethylene. IMS Ind. Med. Surg 39, 46–49. [PubMed] [Google Scholar]
- Burton G. J.; Fowden A. L. (2015) The placenta: a multifaceted, transient organ. Philos. Trans. R. Soc., B 370, 20140066. 10.1098/rstb.2014.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J.; Jeong S. K.; Na K.; Lee M. J.; Lee S. H.; Lim J. S.; Cha H. J.; Cho J. Y.; Kwon J. Y.; Kim H.; Song S. Y.; Yoo J. S.; Park Y. M.; Kim H.; Hancock W. S.; Paik Y. K. (2013) Comprehensive genome-wide proteomic analysis of human placental tissue for the Chromosome-Centric Human Proteome Project. J. Proteome Res. 12, 2458–2466. 10.1021/pr301040g. [DOI] [PubMed] [Google Scholar]
- Myllynen P.; Pasanen M.; Pelkonen O. (2005) Human placenta: a human organ for developmental toxicology research and biomonitoring. Placenta 26, 361–371. 10.1016/j.placenta.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Lash L. H.; Chiu W. A.; Guyton K. Z.; Rusyn I. (2014) Trichloroethylene biotransformation and its role in mutagenicity, carcinogenicity and target organ toxicity. Mutat. Res., Rev. Mutat. Res. 762, 22–36. 10.1016/j.mrrev.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan I.; Kumar A. M.; Park H. R.; Lash L. H.; Loch-Caruso R. (2016) Reactive Oxygen Stimulation of Interleukin-6 Release in the Human Trophoblast Cell Line HTR-8/SVneo by the Trichlorethylene Metabolite S-(1,2-Dichloro)-L-Cysteine. Biol. Reprod. 95, 66. 10.1095/biolreprod.116.139261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldenow E.; Hassan I.; Chames M. C.; Xi C.; Loch-Caruso R. (2015) The trichloroethylene metabolite S-(1,2-dichlorovinyl)-L-cysteine but not trichloroacetate inhibits pathogen-stimulated TNF-alpha in human extraplacental membranes in vitro. Reprod. Toxicol. 52, 1–6. 10.1016/j.reprotox.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A. J. (2012) Oxygen delivery and fetal-placental growth: beyond a question of supply and demand?. Placenta 33 (Suppl 2), e16–22. 10.1016/j.placenta.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Vaughan O. R.; Fowden A. L. (2016) Placental metabolism: substrate requirements and the response to stress. Reprod Domest Anim 51 (Suppl 2), 25–35. 10.1111/rda.12797. [DOI] [PubMed] [Google Scholar]
- Mando C.; De Palma C.; Stampalija T.; Anelli G. M.; Figus M.; Novielli C.; Parisi F.; Clementi E.; Ferrazzi E.; Cetin I. (2014) Placental mitochondrial content and function in intrauterine growth restriction and preeclampsia. American journal of physiology. Endocrinology and metabolism 306, E404–413. 10.1152/ajpendo.00426.2013. [DOI] [PubMed] [Google Scholar]
- Lager S.; Powell T. L. (2012) Regulation of nutrient transport across the placenta. J. Pregnancy 2012, 179827. 10.1155/2012/179827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher G. J.; Kelley L. K.; Smith C. H. (1987) ATP-dependent calcium transport across basal plasma membranes of human placental trophoblast. Am. J. Physiol. 252, C38–46. 10.1152/ajpcell.1987.252.1.C38. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Cai J.; Anders M. W.; Stevens J. L.; Jones D. P. (2001) Role of mitochondrial dysfunction in S-(1,2-dichlorovinyl)-L-cysteine-induced apoptosis. Toxicol. Appl. Pharmacol. 170, 172–180. 10.1006/taap.2000.9107. [DOI] [PubMed] [Google Scholar]
- Xu F.; Papanayotou I.; Putt D. A.; Wang J.; Lash L. H. (2008) Role of mitochondrial dysfunction in cellular responses to S-(1,2-dichlorovinyl)-L-cysteine in primary cultures of human proximal tubular cells. Biochem. Pharmacol. 76, 552–567. 10.1016/j.bcp.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Water B.; Zoeteweij J. P.; de Bont H. J.; Nagelkerke J. F. (1995) Inhibition of succinate:ubiquinone reductase and decrease of ubiquinol in nephrotoxic cysteine S-conjugate-induced oxidative cell injury. Molecular pharmacology 48, 928–937. [PubMed] [Google Scholar]
- van de Water B.; Zoeteweij J. P.; de Bont H. J.; Mulder G. J.; Nagelkerke J. F. (1994) Role of mitochondrial Ca2+ in the oxidative stress-induced dissipation of the mitochondrial membrane potential. Studies in isolated proximal tubular cells using the nephrotoxin 1,2-dichlorovinyl-L-cysteine. J. Biol. Chem. 269, 14546–14552. [PubMed] [Google Scholar]
- Lash L. H.; Putt D. A.; Hueni S. E.; Krause R. J.; Elfarra A. A. (2003) Roles of necrosis, Apoptosis, and mitochondrial dysfunction in S-(1,2-dichlorovinyl)-L-cysteine sulfoxide-induced cytotoxicity in primary cultures of human renal proximal tubular cells. J. Pharmacol. Exp. Ther. 305, 1163–1172. 10.1124/jpet.102.046185. [DOI] [PubMed] [Google Scholar]
- Lash L. H.; Anders M. W. (1986) Cytotoxicity of S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine in isolated rat kidney cells. J. Biol. Chem. 261, 13076–13081. [PubMed] [Google Scholar]
- Chen Q.; Jones T. W.; Brown P. C.; Stevens J. L. (1990) The mechanism of cysteine conjugate cytotoxicity in renal epithelial cells. Covalent binding leads to thiol depletion and lipid peroxidation. J. Biol. Chem. 265, 21603–21611. [PubMed] [Google Scholar]
- Lash L. H.; Anders M. W. (1987) Mechanism of S-(1,2-dichlorovinyl)-L-cysteine- and S-(1,2-dichlorovinyl)-L-homocysteine-induced renal mitochondrial toxicity. Molecular pharmacology 32, 549–556. [PubMed] [Google Scholar]
- Elkin E. R.; Harris S. M.; Loch-Caruso R. (2018) Trichloroethylene metabolite S-(1,2-dichlorovinyl)-L-cysteine induces lipid peroxidation-associated apoptosis via the intrinsic and extrinsic apoptosis pathways in a first-trimester placental cell line. Toxicol. Appl. Pharmacol. 338, 30–42. 10.1016/j.taap.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkin E. R.; Bridges D.; Loch-Caruso R. (2019) The trichloroethylene metabolite S-(1,2-dichlorovinyl)-L-cysteine induces progressive mitochondrial dysfunction in HTR-8/SVneo trophoblasts. Toxicology 427, 152283. 10.1016/j.tox.2019.152283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario F. J.; Gupta M. B.; Myatt L.; Powell T. L.; Glenn J. P.; Cox L.; Jansson T. (2019) Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci. Rep. 9, 246. 10.1038/s41598-018-36265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Aranguren L. C.; Espinosa-Gonzalez C. T.; Gonzalez-Ortiz L. M.; Sanabria-Barrera S. M.; Riano-Medina C. E.; Nunez A. F.; Ahmed A.; Vasquez-Vivar J.; Lopez M. (2018) Soluble Fms-Like Tyrosine Kinase-1 Alters Cellular Metabolism and Mitochondrial Bioenergetics in Preeclampsia. Front. Physiol. 9, 83. 10.3389/fphys.2018.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lojpur T.; Easton Z.; Raez-Villanueva S.; Laviolette S.; Holloway A. C.; Hardy D. B. (2019) Delta9-Tetrahydrocannabinol leads to endoplasmic reticulum stress and mitochondrial dysfunction in human BeWo trophoblasts. Reprod. Toxicol. 87, 21–31. 10.1016/j.reprotox.2019.04.008. [DOI] [PubMed] [Google Scholar]
- McKinney L. L.; Picken J. C. Jr.; Weakley F. B.; Eldridge A. C.; Campbell R. E.; Cowan J. C.; Biester H. E. (1959) Possible Toxic Factor of Trichloroethylene-extracted Soybean Oil Meal3. J. Am. Chem. Soc. 81, 909–915. 10.1021/ja01513a039. [DOI] [Google Scholar]
- Graham C. H.; Hawley T. S.; Hawley R. G.; MacDougall J. R.; Kerbel R. S.; Khoo N.; Lala P. K. (1993) Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp. Cell Res. 206, 204–211. 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- Tetz L. M.; Cheng A. A.; Korte C. S.; Giese R. W.; Wang P.; Harris C.; Meeker J. D.; Loch-Caruso R. (2013) Mono-2-ethylhexyl phthalate induces oxidative stress responses in human placental cells in vitro. Toxicol. Appl. Pharmacol. 268, 47–54. 10.1016/j.taap.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash L. H.; Putt D. A.; Brashear W. T.; Abbas R.; Parker J. C.; Fisher J. W. (1999) Identification of S-(1,2-dichlorovinyl)glutathione in the blood of human volunteers exposed to trichloroethylene. J. Toxicol. Environ. Health, Part A 56, 1–21. 10.1080/009841099158204. [DOI] [PubMed] [Google Scholar]
- ATCC . (2015) American Type Culture Collection Product Sheet: HTR8/SVneo (ATCC® CRL3271), American Type Culture Collection. [Google Scholar]
- Hsiang Y. H.; Hertzberg R.; Hecht S.; Liu L. F. (1985) Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 260, 14873–14878. [PubMed] [Google Scholar]
- Ryan A. J.; Squires S.; Strutt H. L.; Johnson R. T. (1991) Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res. 19, 3295–3300. 10.1093/nar/19.12.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M. A.; Burant C. F.; Kennedy R. T. (2011) Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem. 83, 3406–3414. 10.1021/ac103313x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantama M.; Yellen G. (2014) Imaging changes in the cytosolic ATP-to-ADP ratio. Methods Enzymol. 547, 355–371. 10.1016/B978-0-12-801415-8.00017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie D. G. (2003) Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144, 5179–5183. 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- Guimaraes-Ferreira L. (2014) Role of the phosphocreatine system on energetic homeostasis in skeletal and cardiac muscles. Einstein (Sao Paulo) 12, 126–131. 10.1590/S1679-45082014RB2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. T.; Kato T. (1985) Liver-specific glucose-6-phosphatase is not present in human placenta. J. Inherited Metab. Dis. 8, 92–94. 10.1007/BF01801675. [DOI] [PubMed] [Google Scholar]
- Holecek M. (2018) Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. 15, 33. 10.1186/s12986-018-0271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P.; Pozefsky T.; Marliss E.; Cahill G. F. Jr. (1970) Alanine: key role in gluconeogenesis. Science 167, 1003–1004. 10.1126/science.167.3920.1003. [DOI] [PubMed] [Google Scholar]
- Mallet L. E.; Exton J. H.; Park C. R. (1969) Control of gluconeogenesis from amino acids in the perfused rat liver. J. Biol. Chem. 244, 5713–5723. [PubMed] [Google Scholar]
- Stumvoll M.; Perriello G.; Meyer C.; Gerich J. (1999) Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 55, 778–792. 10.1046/j.1523-1755.1999.055003778.x. [DOI] [PubMed] [Google Scholar]
- Viant M. R.; Bundy J. G.; Pincetich C. R.; de Ropp J. S.; Tjeerdema R. S. (2005) NMR-derived developmental metabolic trajectories: an approach for visualizing the toxic actions of trichloroethylene during embryogenesis. Metabolomics 1, 149–158. 10.1007/s11306-005-4429-2. [DOI] [Google Scholar]
- Cindrova-Davies T.; van Patot M. T.; Gardner L.; Jauniaux E.; Burton G. J.; Charnock-Jones D. S. (2015) Energy status and HIF signalling in chorionic villi show no evidence of hypoxic stress during human early placental development. Mol. Hum. Reprod. 21, 296–308. 10.1093/molehr/gau105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker I. P.; Cooper S.; Ong S. C.; Baker P. N. (2003) Differences in apoptotic susceptibility of cytotrophoblasts and syncytiotrophoblasts in normal pregnancy to those complicated with preeclampsia and intrauterine growth restriction. Am. J. Pathol. 162, 637–643. 10.1016/S0002-9440(10)63857-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp A. N.; Heazell A. E.; Crocker I. P.; Mor G. (2010) Placental apoptosis in health and disease. Am. J. Reprod. Immunol. 64, 159–169. 10.1111/j.1600-0897.2010.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aardema M. W.; Oosterhof H.; Timmer A.; van Rooy I.; Aarnoudse J. G. (2001) Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta 22, 405–411. 10.1053/plac.2001.0676. [DOI] [PubMed] [Google Scholar]
- Illsley N. P.; Caniggia I.; Zamudio S. (2010) Placental metabolic reprogramming: do changes in the mix of energy-generating substrates modulate fetal growth?. Int. J. Dev. Biol. 54, 409–419. 10.1387/ijdb.082798ni. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton G. J.; Watson A. L.; Hempstock J.; Skepper J. N.; Jauniaux E. (2002) Uterine glands provide histiotrophic nutrition for the human fetus during the first trimester of pregnancy. J. Clin. Endocrinol. Metab. 87, 2954–2959. 10.1210/jcem.87.6.8563. [DOI] [PubMed] [Google Scholar]
- James J. L.; Stone P. R.; Chamley L. W. (2006) The regulation of trophoblast differentiation by oxygen in the first trimester of pregnancy. Hum. Reprod. Update 12, 137–144. 10.1093/humupd/dmi043. [DOI] [PubMed] [Google Scholar]
- Hoang V. M.; Foulk R.; Clauser K.; Burlingame A.; Gibson B. W.; Fisher S. J. (2001) Functional proteomics: examining the effects of hypoxia on the cytotrophoblast protein repertoire. Biochemistry 40, 4077–4086. 10.1021/bi0023910. [DOI] [PubMed] [Google Scholar]
- Mor I.; Cheung E. C.; Vousden K. H. (2011) Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harbor Symp. Quant. Biol. 76, 211–216. 10.1101/sqb.2011.76.010868. [DOI] [PubMed] [Google Scholar]
- Berg J. M., Tymoczko J. L., Gatto J. G., and Stryer L. (2015) Biochemistry, 8th ed., W. H. Freeman, Stuttgart, Germany. [Google Scholar]
- Lipmann F. (1940) A phosphorylated oxidation product of pyruvic acid. J. Biol. Chem. 134, 4630464. [Google Scholar]
- Pietrocola F.; Galluzzi L.; Bravo-San Pedro J. M.; Madeo F.; Kroemer G. (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821. 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Dunford L. J.; Sinclair K. D.; Kwong W. Y.; Sturrock C.; Clifford B. L.; Giles T. C.; Gardner D. S. (2014) Maternal protein-energy malnutrition during early pregnancy in sheep impacts the fetal ornithine cycle to reduce fetal kidney microvascular development. FASEB J. 28, 4880–4892. 10.1096/fj.14-255364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S.; Patro N.; Patro I. K. (2018) Maternal Protein Malnutrition: Current and Future Perspectives of Spirulina Supplementation in Neuroprotection. Front. Neurosci. 12, 966. 10.3389/fnins.2018.00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordas K.; Lonnerdal B.; Stoltzfus R. J. (2007) Interactions between nutrition and environmental exposures: effects on health outcomes in women and children. J. Nutr. 137, 2794–2797. 10.1093/jn/137.12.2794. [DOI] [PubMed] [Google Scholar]
- Stonard M. D.; Parker V. H. (1971) 2-Oxoacid dehydrogenases of rat liver mitochondria as the site of action of S-(1,2 dichlorovinyl)-L-cysteine and S-(1,2-dichlorovinyl)-3-mercaptopropionic acid. Biochem. Pharmacol. 20, 2417–2427. 10.1016/0006-2952(71)90242-5. [DOI] [PubMed] [Google Scholar]
- van de Water B.; Zoeteweij J. P.; Nagelkerke J. F. (1996) Alkylation-induced oxidative cell injury of renal proximal tubular cells: involvement of glutathione redox-cycle inhibition. Arch. Biochem. Biophys. 327, 71–80. 10.1006/abbi.1996.0094. [DOI] [PubMed] [Google Scholar]
- Walker D. I.; Uppal K.; Zhang L.; Vermeulen R.; Smith M.; Hu W.; Purdue M. P.; Tang X.; Reiss B.; Kim S.; Li L.; Huang H.; Pennell K. D.; Jones D. P.; Rothman N.; Lan Q. (2016) High-resolution metabolomics of occupational exposure to trichloroethylene. Int. J. Epidemiol 45, 1517–1527. 10.1093/ije/dyw218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan N. J.; Paiva P.; Dimitriadis E.; Salamonsen L. A. (2010) Models for study of human embryo implantation: choice of cell lines?. Biol. Reprod. 82, 235–245. 10.1095/biolreprod.109.077800. [DOI] [PubMed] [Google Scholar]
- Irving J. A.; Lysiak J. J.; Graham C. H.; Hearn S.; Han V. K.; Lala P. K. (1995) Characteristics of trophoblast cells migrating from first trimester chorionic villus explants and propagated in culture. Placenta 16, 413–433. 10.1016/0143-4004(95)90100-0. [DOI] [PubMed] [Google Scholar]
- Khan G. A.; Girish G. V.; Lala N.; Di Guglielmo G. M.; Lala P. K. (2011) Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol. Endocrinol. 25, 1431–1443. 10.1210/me.2010-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilburn B. A.; Wang J.; Duniec-Dmuchowski Z. M.; Leach R. E.; Romero R.; Armant D. R. (2000) Extracellular matrix composition and hypoxia regulate the expression of HLA-G and integrins in a human trophoblast cell line. Biol. Reprod. 62, 739–747. 10.1095/biolreprod62.3.739. [DOI] [PubMed] [Google Scholar]
- Damsky C. H.; Librach C.; Lim K. H.; Fitzgerald M. L.; McMaster M. T.; Janatpour M.; Zhou Y.; Logan S. K.; Fisher S. J. (1994) Integrin switching regulates normal trophoblast invasion. Development 120, 3657–3666. [DOI] [PubMed] [Google Scholar]
- Zdravkovic M.; Aboagye-Mathiesen G.; Guimond M. J.; Hager H.; Ebbesen P.; Lala P. K. (1999) Susceptibility of MHC class I expressing extravillous trophoblast cell lines to killing by natural killer cells. Placenta 20, 431–440. 10.1053/plac.1999.0393. [DOI] [PubMed] [Google Scholar]
- Szklanna P. B.; Wynne K.; Nolan M.; Egan K.; Ainle F. N.; Maguire P. B. (2017) Comparative proteomic analysis of trophoblast cell models reveals their differential phenotypes, potential uses and limitations. Proteomics 17, 1700037–1700042. 10.1002/pmic.201700037. [DOI] [PubMed] [Google Scholar]
- Graham C. H.; Connelly I.; MacDougall J. R.; Kerbel R. S.; Stetler-Stevenson W. G.; Lala P. K. (1994) Resistance of malignant trophoblast cells to both the anti-proliferative and anti-invasive effects of transforming growth factor-beta. Exp. Cell Res. 214, 93–99. 10.1006/excr.1994.1237. [DOI] [PubMed] [Google Scholar]
- Liu L.; Wang Y.; Shen C.; He J.; Liu X.; Ding Y.; Gao R.; Chen X. (2016) Benzo(a)pyrene inhibits migration and invasion of extravillous trophoblast HTR-8/SVneo cells via activation of the ERK and JNK pathway. J. Appl. Toxicol. 36, 946–955. 10.1002/jat.3227. [DOI] [PubMed] [Google Scholar]
- Takao T.; Asanoma K.; Kato K.; Fukushima K.; Tsunematsu R.; Hirakawa T.; Matsumura S.; Seki H.; Takeda S.; Wake N. (2011) Isolation and characterization of human trophoblast side-population (SP) cells in primary villous cytotrophoblasts and HTR-8/SVneo cell line. PLoS One 6, e21990. 10.1371/journal.pone.0021990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou-Kheir W.; Barrak J.; Hadadeh O.; Daoud G. (2017) HTR-8/SVneo cell line contains a mixed population of cells. Placenta 50, 1–7. 10.1016/j.placenta.2016.12.007. [DOI] [PubMed] [Google Scholar]
- Novakovic B.; Gordon L.; Wong N. C.; Moffett A.; Manuelpillai U.; Craig J. M.; Sharkey A.; Saffery R. (2011) Wide-ranging DNA methylation differences of primary trophoblast cell populations and derived cell lines: implications and opportunities for understanding trophoblast function. Mol. Hum. Reprod. 17, 344–353. 10.1093/molehr/gar005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilban M.; Tauber S.; Haslinger P.; Pollheimer J.; Saleh L.; Pehamberger H.; Wagner O.; Knofler M. (2010) Trophoblast invasion: assessment of cellular models using gene expression signatures. Placenta 31, 989–996. 10.1016/j.placenta.2010.08.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.