

Abstract
Purpose:
Malignant peripheral nerve sheath tumors (MPNSTs) are deadly sarcomas that lack effective therapies. In most MPNSTs, the retinoblastoma (RB1) tumor suppressor is disabled by hyperactivation of cyclin dependent kinases (CDKs), commonly through loss of CDK inhibitory proteins such as p27(Kip1). RABL6A is an inhibitor of RB1 whose role in MPNSTs is unknown. To gain insight into MPNST development and establish new treatment options, we investigated RABL6A-RB1 signaling and CDK inhibitor-based therapy in MPNSTs.
Experimental Design:
We examined patient-matched MPNSTs and precursor lesions by RNA-Seq and IHC. Molecular and biological effects of silencing RABL6A and/or p27 in MPNST lines and normal human Schwann cells were determined. Tumor suppressive effects of CDK inhibitors were measured in MPNST cells and orthotopic tumors.
Results:
RABL6A was dramatically upregulated in human MPNSTs compared to precursor lesions, which correlated inversely with p27 levels. Silencing RABL6A caused MPNST cell death and G1 arrest that coincided with p27 upregulation, CDK downregulation and RB1 activation. The growth suppressive effects of RABL6A loss, and its regulation of RB1, were largely rescued by p27 depletion. Importantly, reactivation of RB1 using a CDK4/6 inhibitor (palbociclib) killed MPNST cells in vitro in a RABL6A-dependent manner and suppressed MPNST growth in vivo. Low-dose combination of drugs targeting multiple RB1 kinases (CDK4/6, CDK2) had enhanced anti-tumorigenic activity associated with potential MPNST cell re-differentiation.
Conclusions:
RABL6A is a new driver of MPNST pathogenesis that acts in part through p27-RB1 inactivation. Our results suggest RB1 targeted therapy with multiple pathway drugs may effectively treat MPNSTs.
Keywords: MPNST, RABL6A, p27Kip1, RB1, CDK inhibitors
Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive sarcomas that develop in the connective tissue surrounding nerves. These tumors arise sporadically (in ~50% of cases) or in patients with the cancer predisposition syndrome, neurofibromatosis type I (NF1), through transformation of plexiform neurofibromas (PNFs) (1, 2). NF1 patients are at higher risk of MPNST development than the general population, and MPNSTs are the leading cause of NF1 patient death (3). The 5-year survival rate for MPNST patients is 20–35% and significantly worsens with non-resectable or metastatic tumors (4, 5). The only potential cure for localized MPNST is complete surgical resection; however, many tumors are non-resectable or cannot be fully resected due to their location and/or large size. Additionally, treatment with conventional radiation and chemotherapy for inoperable tumors does not reduce mortality. Current treatment with ifosfamide and doxorubicin combination has high toxicity with little benefit (6–10). Thus, greater understanding of the key molecular events driving MPNST formation is critically needed to guide therapy and identify potential targets for new anticancer strategies.
The retinoblastoma (RB1) protein is one of the most important tumor suppressors in human cancers (11–13), and its inactivation is a major event in the pathogenesis of MPNST (14, 15). RB1 blocks G1-S phase transition in cells by sequestering E2F transcription factors necessary for S phase entry (16). It is inactivated through phosphorylation by cyclin-dependent kinases (CDKs), which results in the release of E2F and subsequent transcription of E2F regulated genes required for DNA replication (16, 17). Endogenous inhibitors of the CDKs, such as p16INK4a and p27(Kip1), enforce RB1-mediated cell cycle arrest by blocking RB1 phosphorylation. Besides its well-established antiproliferative activity, RB1 can also promote apoptosis (18, 19). In most MPNSTs, the RB1 gene remains wild-type and inactivation occurs through aberrant CDK activity, largely due to loss of p16INK4a and p27(Kip1) (15, 20). Inactivation of CDKN2A, the locus encoding p16INK4a, is the hallmark event associated with PNF-to-MPNST malignant transformation in NF1 patients. In fact, loss of CDKN2A and the adjacent CDKN2B gene (which encodes a related p15INK4b protein) is the only known molecular change that defines the transitional lesion between PNFs and MPNSTs, called atypical neurofibromatosis neoplasm of unidentified biologic potential (ANNUBP) (21). This suggests pharmacological inhibition of hyperactive CDKs may be effective against MPNSTs and possibly NF1-associated, pre-malignant lesions.
RABL6A (also called Parf, RBEL1, c9orf86), a recently discovered RAB-like GTPase, is implicated in promoting the pathogenesis of multiple human cancers, including breast and pancreatic (both adenocarcinoma and neuroendocrine) tumors (22–26). RABL6A acts through multiple mechanisms that are only partly defined to control tumor cell proliferation and survival (22, 26–29). For example, RABL6A promotes ERK signaling (26, 27), activates AKT by inhibiting tumor suppressive protein phosphatase 2A (PP2A) (23), and inhibits p53 by enhancing its degradation via Mdm2-mediated ubiquitination (28). RABL6A also negatively regulates RB1 in pancreatic neuroendocrine tumor and osteosarcoma cells by promoting its phosphorylation (22, 24). Because disruptions in the RB1 pathway are key to MPNST development, this study sought to define the role of RABL6A in MPNST pathogenesis.
Here, we show that RABL6A protein expression and signaling is significantly upregulated in MPNSTs versus matched, benign neurofibromas (NFs) from the same NF1 patients. Intermediate levels of RABL6A exist in ANNUBPs, the precursors to MPNSTs. Cell-based analyses revealed RABL6A is necessary for MPNST cell survival and proliferation. RABL6A negatively regulates p27 expression in MPNST cells, which causes increased phosphorylation of RB1 at CDK4/6-targeted sites, thereby inactivating RB1. Depletion of p27 attenuates the molecular and biological phenotypes caused by RABL6A loss. Importantly, pharmacological inhibition of CDK4/6 kills MPNST cells in a RABL6A-dependent manner and blocks orthotopic tumor growth in vivo. Combination therapy with lowered doses of multiple CDK inhibitors has a more pronounced suppressive effect against MPNST cells and tumors than CDK monotherapy. Together, these studies define a new role for the RABL6A-p27-RB1 pathway in MPNST pathogenesis and highlight the potential of RB1 targeted therapy to combat this deadly cancer.
Materials and Methods
Tissue microarray.
A total of 12 paired neurofibromas and MPNSTs (i.e., matched tumors arising in the same patient), 1 unpaired neurofibroma and 2 unpaired MPNSTs were obtained from the University of Iowa Department of Pathology with previous approval from the Institutional Review Board (IRB ID# 201507708). Upon further review, 3 ANNUBPs were identified. Neurofibromas, ANNUBPs and MPNST utilized in the array were reviewed by multiple pathologists (BWD and MRT) and classified according to recent consensus criteria (21). Peripheral nerve was included as control tissue. The TMA was constructed by arraying the neoplasms in duplicate consisting of 1.0-mm cores taken from formalin fixed paraffin embedded tissue and assembled using a MTA-1 tissue arrayer from Beecher Instruments (Sun Prarie, WI). Immunohistochemistry for RABL6A and p27 was evaluated and expressed semi-quantitatively as 3 (strong expression), 2 (intermediate expression), 1 (weak expression), or 0 (no expression). The percentage of cells positive were recorded for each core. H-score was calculated as the product of the expression score multiplied by % cells positive.
RNA-Seq.
Nucleic acid was extracted from formalin fixed, paraffin embedded (FFPE) tissue cores taken adjacent to those used to construct the tissue microarray. Total RNA was extracted using the RNeasy FFPE Kit (Qiagen, Valencia, CA) and its quality assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-Seq was performed at the University of Iowa Institute of Human Genetics, Genomics Division (Iowa City, IA) using the Agilent SureSelect RNA Direct protocol with the SureSelect Human All Exon V6 + COSMIC enrichment library. Sequencing was performed on the Illumina HiSeq 4000 genome sequencer (Illumina, San Diego, CA) using a 150 bp paired-end sequencing-by-synthesis chemistry.
To identify differentially expressed genes in malignant versus benign tumors, RNA-Seq reads were processed with the pseudoalignment tool Kallisto and differential expression was analyzed with the statistical R package Sleuth (30). Default settings were used with the exception that fold change values were calculated using a log2 and not natural log transformation. Samples with less than 10 million mapping reads were excluded from further analysis. Ingenuity Pathway Analysis (IPA) was used to identify functionally related pathways differentially regulated between paired patient tumors. IPA, specifically the analysis module named “canonical pathways” was used to identify signaling pathways that were enriched for within the overall set of genes differentially expressed between paired patient PNFs versus atypicals and MPNSTs. For this pathway analysis, only genes that exhibited a p-value < 0.05 and q-value of < 0.2 in the differential expression analysis were used. This pathway analysis found 83 pathways that were significant after multiple testing correction using the Benjamini-Hochberg procedure. Of these 83 pathways only 54 had non-zero activity z-scores. From these 54 pathways we chose 21 pathways that exhibited high magnitude z-scores (i.e., the most activated or inhibited pathways) and the most significant enrichment. To simplify the figure display, those 21 pathways were consolidated into five broader “super” pathways from which we selected a subset of genes that were the most differentially expressed between the tumor types (fold change of > 1.5 or < −1.5). Morpheus software used to generate the heatmaps (https://software.broadinstitute.org/morpheus). The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (31) and are accessible through GEO series accession number GSE145064 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145064).
Cell culture.
Three well characterized human MPNST cell lines were used for these studies: the sporadic human MPNST cell line STS26T (referred to as ‘26T’; NF1 wild-type; Cellosaurus RRID:CVCL_8917), NF1-associated human MPNST cell line S462 (Cellosaurus RRID:CVCL_1Y70), and NF1-associated human MPNST cell line sNF96.2 (ATCC CRL-2884). In addition, two novel patient-derived cultures (named MT1 and MT2) were isolated from distinct metastatic MPNST lesions in a single NF1 patient by the Tanas laboratory. MT1 and MT2 were passaged extensively in culture until fibroblasts were depleted by senescence; cells were then karyotyped to establish genomic similarity to the original lesions. All MPNST cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 4mM glutamine, and 100μg/mL penicillin-streptomycin. Primary human Schwann cells (NHSC) isolated from human spinal nerve were purchased from ScienCell Research Laboratories (Cat. # 1700) and maintained in Schwann Cell Medium (Cat. #1701), Growth Supplement (Cat. #1752) and 5% FBS. Cells were routinely tested for mycoplasma contamination (LONZA MycoAlert Mycoplasma Detection Kit) and found to be negative (most recent date of testing for 26T and sNF96.2 was 02/06/2020 and 07/02/2018 for S462), except 26T, which occasionally tested in the low positive range and was treated with Plasmocin (Invivogen). Consequently, most experiments utilized S462 and sNF96.2 cells. Cryopreserved stocks of each cell line were made at low passage numbers (less than 3–6 passages after receipt) and cells were used in experiments for a maximum of 8–10 passages (roughly 4 weeks) after being thawed.
RNA interference, virus production, and infection.
Human RABL6A shRNAs in the pLKO.1 lentiviral vector (Open Biosystems, Huntsville, AL) have been described (26). Lentiviruses encoding RABL6A shRNAs (KD1 and KD2), p27 shRNA, and empty vector (CON) were produced by lipofection of viral constructs in HEK293T cells and infected into target cells using established methods (22, 26). Transduced cells were harvested 3 to 5 days after infection, depending on the assay. For combined silencing of RABL6A and p27, cells were sequentially infected with p27 shRNA viruses followed by RABL6A shRNA viruses (22).
Cell proliferation and survival assays.
Cell number and viability were quantified by trypan blue exclusion (1:1 v/v) and manual counting using a hemocytometer. Relative cell numbers were quantified using alamarBlue® (ThermoFisher Scientific) or Cell-Quant (Genecopoeia) assays in a 96-well format per manufacturer’s instructions. To measure DNA synthesis, cells were plated onto chamber slides and the next day incubated with 10μM 5-ethynyl-2’-deoxyuridine (EdU) for 5 to 8hrs. EdU incorporation was detected by staining with Andy Fluor 488 dye (Genecopoeia Cat. #A003). 4’, 6’-diamidino-2-phenylinodole (DAPI) was used to detect nuclei. Confocal microscopy (Zeiss LSM 710, Germany) was performed to capture fluorescence images and quantify EdU positivity (100 or more cells per sample counted) from 3 or more independent experiments. DNA content was measured by flow cytometry after propidium iodide staining and analyzed using MOD Fit LT software (Verity Software House).
Antibodies.
For western blotting, antibodies were used according to supplier guidelines (Supplemental Table 1). Antibodies included those specific for p-RB1-S807/811 (no. 8516), RB1 (no. 9309), p27 (no. 3686), and cleaved PARP (no. 5625) from Cell Signaling Technology; GAPDH (no. Ab8245) from Abcam; vinculin (no. V9131), gamma-tubulin (no. T6557), and HRP-coupled secondary antibodies (nos. NA934 and NA935) from Sigma; and CDK2 (no. sc-6248) and beta-actin (no. sc-8432) from Santa Cruz. RABL6A antibodies (monoclonal and polyclonal) were produced in the Quelle laboratory (26, 32).
Western blotting.
Cellular proteins were isolated by lysis in SDS-PAGE loading buffer and identical cell equivalents electrophoresed through polyacrylamide gels. Frozen tumor pieces were pulverized in LN2 using a mortar and pestle and lysed for 30min on ice with RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) containing 1 mM NaF, protease and phosphatase inhibitor cocktails (Sigma, P-8340 & P-0044) and 30 μM phenylmethylsulfonyl fluoride (PMSF). Protein concentrations of extracts were determined by BCA protein assay (ThermoFisher, Cat. # 23228). Proteins were transferred from gels onto PVDF membranes (Millipore), which were blocked with 5% non-fat milk or 5% BSA in TBST (Tris-buffered saline containing Tween-20) depending on the antibody (detailed blotting conditions in Supplemental Table 1). Proteins were detected using HRP-conjugated secondary antibodies and enhanced chemiluminescence (ECL, Amersham, Buckinghamshire, UK). Densitometry quantification was performed using ImageJ (NIH).
Immunofluorescence.
Following infection, cells were plated on poly-D-lysine coated chamber slides (Corning Ref 354632) and incubated overnight. Slides were washed once with PBS, fixed for 15min at room temperature with 16% formaldehyde in PBS, washed 3X with PBS and blocked (PBS with 5% FBS, 0.3% Triton X-100) for 1hr at room temperature. Slides were rinsed 3X with PBS and incubated at 4C overnight in primary antibody solution (PBS with 1% BSA, 0.3% Triton X-100) using Cell Signaling Technology antibodies to p-RB1 S807/811 (no. 8516, 1:1600) or p27 (no. 3686, 1:800). Slides were then washed with PBS and incubated in secondary antibody solution (Anti-Rabbit IgG, Alexa Fluor 488 and 647) for 1hr at room temperature. DAPI was used to detect nuclei. 100 or more cells per sample were counted from 3 or more experiments to quantify results.
Drug response assays.
Palbociclib (CDK4/6 inhibitor, no. S1116, dissolved in sterile ddH20) and Dinaciclib (CDK2 inhibitor, no. S2768, dissolved in DMSO) were purchased from Selleckchem (Houston, TX, USA) and stock solutions stored at −80C. MPNST cell lines were seeded at 1,000 cell per well in 96-well flat-bottom dishes. Varying concentrations of the drugs were added the next day and cells exposed for 3 days. Each condition was performed in triplicate. Samples were assayed for relative cell number using alamarBlueR (Thermo Fisher Scientific, DAL1025) or Cell-Quant ™ (GeneCopoeia, A014) cell proliferation assays. Results were quantified using a fluorescence microplate reader by measuring absorbance at 540/570nm for alamarBlueR and 480/530nm for Cell-Quant ™. For combination studies, percent inhibition was calculated and analyzed using SynergyFinder (https://synergyfinder.fimm.fi/synergy/) software. For molecular analyses, cells in 10cm dishes were treated for 2–3 days at 37C, 5% CO2 and evaluated for viability (trypan blue exclusion), DNA content (flow cytometry), and protein status (western blotting).
Colony Formation Assays.
S462 and sNF96.2 MPNST cell lines were seeded in 6-well plates at 150 and 300c/well or 1000 and 2000c/well, respectively. Each drug concentration was performed in triplicate. Drug concentrations used for S462 assays were: 0.8μM and 1.5μM palbociclib, 4–6nM dinaciclib, 0.8μM palbociclib + 4–6nM dinaciclib, and 1.5μM palbociclib + 4–6nM dinaciclib. Drug concentrations used for sNF96.2 assays were: 62.5nM and 125nM palbociclib, 2–3nM dinaciclib, 62.5nM palbociclib + 2–3nM dinaciclib, and 125nM palbociclib + 2–3nM dinaciclib. Media plus drug was changed twice per week for 12 – 17 days of treatment. Colonies were washed, fixed, Giemsa stained, and images taken with an Oxford Optronix GelCount™ imager. Percent area covered was calculated using ImageJ.
Mouse orthotopic xenografts and treatment studies.
Mice were housed in the University of Iowa Animal Care barrier facility in rooms with a 12-hour light-dark cycle and free access to water and food. For tumor studies, the left hind leg of eight-week-old immunodeficient (NOD scid gamma) male and female mice was surgically opened and the sciatic nerve was injected with sNF96.2 or S462 cells (5×105c) suspended in 10uL DMEM. Tumor volume, assessed by daily caliper measurements, was determined using the formula: (length × width × thickness × π) / 6. When tumor volumes reached an average of 200mm3, mice were randomized into treatment groups. For monotherapy studies, mice were treated daily by oral gavage with vehicle control (ddH20) or palbociclib (50mg/kg). For the palbociclib + dinaciclib combination study, mice with sNF96.2 tumors were randomized into four treatment groups (vehicle, palbociclib, dinaciclib, combination). Mice were treated daily by oral gavage with vehicle control (ddH20) or palbociclib (25mg/kg) or by intraperitoneal injection of dinaciclib dissolved in 20% w/v 2-hydroxypropyl-beta-cyclodextrin (HPBCD) (16mg/kg). Mouse body weights were recorded 3 times per week. Animals were observed for signs of toxicity (weight loss, ruffled fur, immobility, and abdominal rigidity). Due to toxicity from dinaciclib treatment, dosage was reduced to 8mg/kg after one week of treatment. Mice were sacrificed when the first mouse tumor reached 2000mm3. Tumor tissues, harvested 2hr post-final dose of treatment, were weighed and split for processing by fixation in 10% neutral buffered formalin or flash frozen in LN2.
Histopathological analysis.
All immunohistochemistry procedures were performed in a consistent manner to limit tissue staining factors that could create bias for interpretation of immunostaining (33). Formalin (10% neutral buffered formalin) fixed tumors were routinely processed, paraffin embedded, sectioned (~4 μm) onto glass slides, and hydrated through series of xylene and alcohol baths. Antigen retrieval and application of immuno reagents are summarized in Supplemental Table 1. 3,3′-diaminobenzidine (DAB, brown staining) was used as the chromogen, and Harris hematoxylin (basophilic staining) was used as the counterstain. Sections were dehydrated through a series of alcohol and xylene baths, and coverslipped. Immunostaining for RABL6A, p27, Ki-67, phospho-RB1 S807/811, and S100B was conducted utilizing validated protocols (34, 35). Slides were reviewed by a sarcoma pathologist (Dr. Munir Tanas).
Statistics.
Western data were imaged by scanning densitometry and quantified by ImageJ (NIH). Values for phosphoproteins were normalized to expression of the total protein while values for all other proteins were normalized to loading control. Differences in levels of protein phosphorylation or expression were displayed as fold change relative to untreated counterparts or vector controls. Quantified data were presented as the mean +/− SD or SEM, as indicated. All P values, unless otherwise specified, were obtained by Student’s t-test and adjusted for multiple comparisons using Dunnett’s test or Bonferroni’s method, as indicated. Overall differences between curves were assessed using generalized linear regressions. An adjusted P value less than 0.05 was considered statistically significant.
Study approval.
The study of patient tumors was performed after approval by the University of Iowa Hospitals and Clinics institutional review board (IRB ID# 201507708). Studies were conducted in accordance with ethical guidelines in the Belmont Report. Written consent was not obtained from subjects for this retrospective study using residual specimens as it was not required for IRB approval. All mouse studies were conducted according to protocols approved by the University of Iowa Institutional Animal Care and Use Committee (protocol #7112074: Targeting RB1 Pathway in Sarcomas). All efforts were made to minimize animal suffering.
Results
Upregulation and activation of RABL6A during the PNF to MPNST transition in NF1 patient specimens
About one-third of NF1 patients develop benign plexiform neurofibromas (PNFs), which cause significant morbidity and have a high risk for transformation into MPNSTs (36). The step-wise progression to malignancy in these patients involves an intermediate neoplasm called ANNUBP, providing a unique system to study molecular alterations promoting PNF to ANNUBP to MPNST transformation. We generated a tissue microarray (TMA) of 32 patient-matched NF/PNF and MPNST pairs that included 3 ANNUBPs from 12 NF1 patients cared for at the Iowa NF Clinic. Two different tumor cores were arrayed per specimen and normal peripheral nerve was included as control. For some patients, multiple tumors were arrayed and for several the MPNST arose from the paired PNF. Immunohistochemical (IHC) staining of Ki-67 verified increased proliferation in MPNSTs compared to precursor lesions (NF mean: 0.27, ANNUBP mean: 0, MPNST mean: 24.8) (Supplemental Figure 1A and 1B).
IHC analysis of RABL6A showed differential expression in the three groups, where NFs displayed the lowest expression (mean: 16.5) followed by intermediate levels in ANNUBPs (mean: 41.2) and dramatically higher expression in MPNSTs (152.6) (Figure 1, A-C). The induction of RABL6A coincided with loss of nuclear p27 (Figure 1, D and E), an event associated with poor survival in MPNST patients (15, 37). Down-regulation of p27 during the NF-to-MPNST transition can be caused by reduced transcription as well as mis-localization of p27 from the nucleus to the cytoplasm (15, 38). MPNSTs as well as ANNUBPs in our TMA mostly displayed an absence of p27 staining (MPNST mean: 2.0; ANNUBP mean: 2.3; NF mean: 65.1) with little evidence of protein mis-localization (Figure 1D).
Figure 1. Upregulation and activation of RABL6A during the PNF to MPNST transition in NF1 patient specimens.
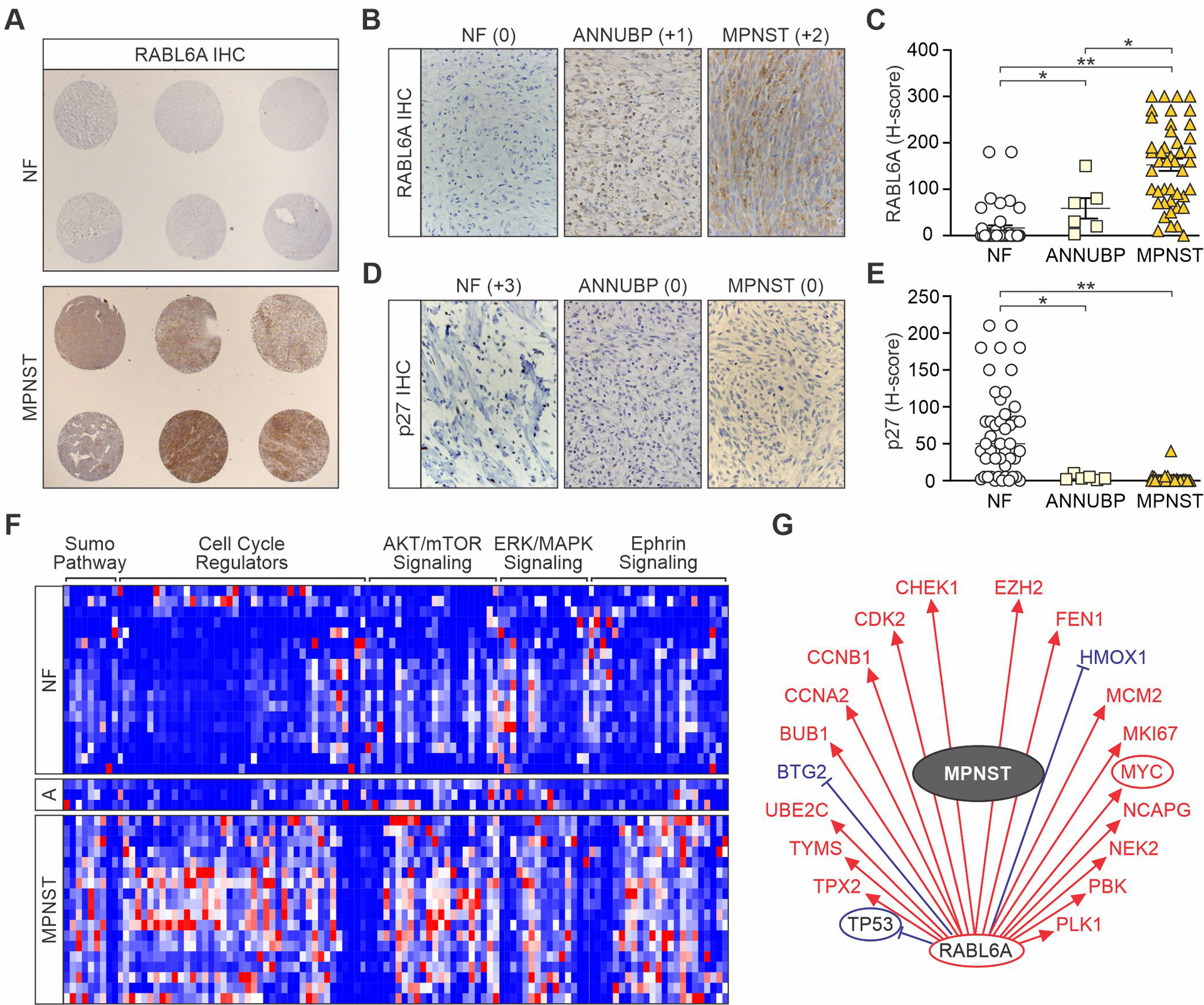
(A) Immunohistochemical (IHC) staining of RABL6A in tissue microarrays (TMAs) of patient-matched NF (mainly plexiform NFs) and MPNSTs. Images at 10X magnification. Higher magnification images (400X) of RABL6A (B) and p27 (D) IHC staining of NF, ANNUBP and MPNST cores from the TMA. Stain intensity indicated in parentheses. H-scores, representing the stain intensity × percent of cells stained, of RABL6A (C) and p27 (E) in NF, ANNUBP and MPNST samples. Error bars, SEM. P-value, Student’s t-test. *, p<0.05; **, p<0.01. (F) Heat-map of RNA-Seq data with hierarchical clustering of a subset of functionally related differentially expressed genes (n=110) between paired patient NFs, ANNUBPs (designated A) and MPNSTs. Genes were categorized by IPA software. Red, relatively increased expression; blue, relatively decreased expression. Genes within each group are listed in Supplemental Table 2. (G) An MPNST gene activity wheel, depicting select genes associated with RABL6A expression, shows MPNSTs have an activated ‘RABL6A signature’ of gene expression (22, 23). Transcript levels of the indicated genes in MPNSTs: decreased (blue), increased (red), unchanged (black). Circled genes reflect pathway activation (red) or inhibition (blue). Expression of RABL6A is associated with increased (red arrows) or decreased (blue bars) mRNA levels of the displayed genes.
RNA sequencing (RNA-seq) was performed on the TMA samples to identify genes and pathways differentially regulated in MPNSTs relative to benign lesions from the same patients. A total of 1326 genes were significantly altered (>1.5 or <−1.5-fold change, q<0.20) in MPNSTs (Supplemental Figure 1C, Supplemental Table 2). In agreement with prior MPNST profiling studies, MPNSTs displayed upregulation of TWIST1, a pro-tumorigenic neural stem cell marker that inhibits p53 and RB1 signaling (39, 40), as well as increased expression of oncogenic mitotic kinase genes, such as AURKA, BUB1, and NEK2 (41–43). ANNUBPs and MPNSTs also displayed the expected downregulation of p16INK4a transcripts whereas RABL6 mRNA levels were statistically indistinguishable in the benign and malignant tumors (Supplemental Figure 1D and 1E).
Ingenuity Pathway Analysis (IPA) software classified the MPNST gene set into broad functional categories that were significantly enriched for within those tumors. One hundred and ten genes from a subset of those pathways that exhibited the highest magnitude z-scores and greatest enrichment are shown in Figure 1F. Activation of Ras effector pathways (ERK/MAPK, AKT/mTOR) and cell cycle promoting pathways featured prominently among the MPNST gene sets as well as inhibition of G1/S, G2/M and DNA damage checkpoint signaling (Figure 1F, Supplemental Table 2). ANNUBPs exhibited an intermediate gene expression profile with greater similarity to PNFs for genes in cell cycle, ERK/MAPK and sumoylation pathways while more closely resembling MPNSTs for AKT/mTOR and ephrin signaling genes. Strikingly, while the RABL6 transcript is not increased in MPNSTs, IPA revealed that the RABL6A pathway is activated in MPNSTs (Figure 1G) consistent with elevated RABL6A protein levels. Many genes within the known ‘RABL6A signature’ (22, 23) overlap significantly with dysregulated MPNST genes, particularly those associated with aberrant cell cycle progression. The stark protein upregulation and activation of RABL6A in malignant lesions strongly implied a role for RABL6A in promoting MPNST pathogenesis.
RABL6A is required for MPNST cell survival and proliferation
Examination of sporadic and NF1-associated MPNST cell lines revealed increased RABL6A protein expression in MPNSTs compared to normal human Schwann cells (NHSCs), the cell of origin for these tumors (Figure 2A) (41). To investigate the potential oncogenic role of RABL6A in MPNST cells, endogenous RABL6A was silenced in NHSCs and MPNST cell lines via RNA interference with two different short hairpin RNAs (KD1 and KD2), as described (22, 26, 29). Studies were performed in S462 and 26T cells, which express the highest levels of RABL6A and are two of the most studied MPNST cell lines (44), as well as the NF1-associated MPNST cell line, sNF96.2 (45, 46). Knockdown of RABL6A was verified by western blotting (Figure 2B; Supplemental Figure 2A) and in all three MPNST lines caused significant cell death relative to empty vector control, whereas no death was caused by RABL6A loss in NHSCs (Figure 2C). The cell death induced by RABL6A silencing coincided with induction of cleaved PARP, a marker of apoptosis, in 26T and S462 MPNST cells (less so in sNF96.2) (Figure 2B). Thus, unlike non-transformed Schwann cells, MPNST cells depend on RABL6A for survival.
Figure 2. RABL6A is required for MPNST cell survival and proliferation.
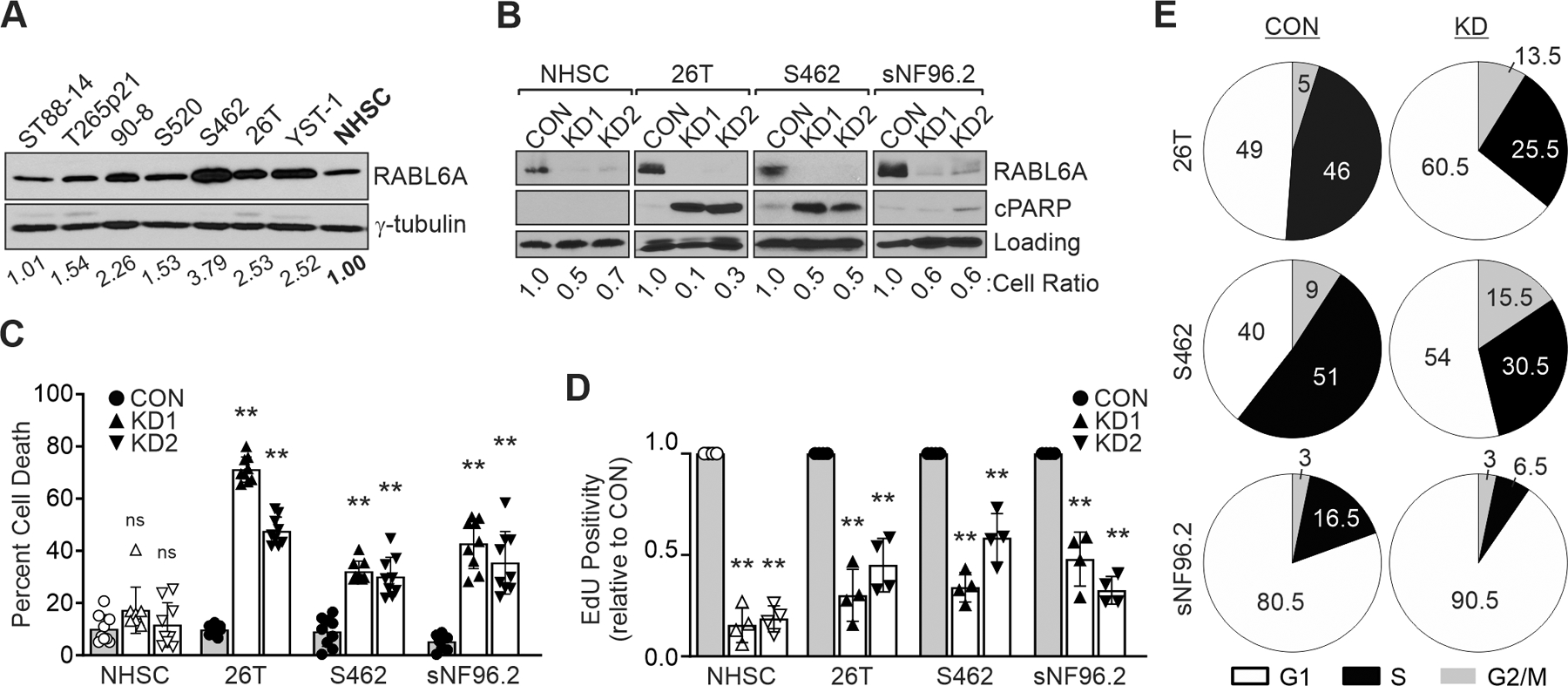
(A) Western blots show elevated RABL6A levels in both NF1-associated (T265p21, 90–8, S520 and S462) and sporadic (STS26T [26T]c and YST-1) MPNST cell lines versus precursor, primary normal human Schwann cells (NHSC). (B) Representative westerns show that knockdown of RABL6A (KD1 and KD2) causes induction of cleaved PARP, a marker of apoptosis, in MPNST cells but not in NHSC. Loading control, GAPDH or γ-tubulin. Cell number relative to CON (cell ratio) for each cell line is shown. CON, empty vector control. (C) Quantification of cell death, as measured by trypan blue exclusion, shows RABL6A knockdown induces the death of MPNST cells but not NHSC. P-value, Student’s t-test with Dunnett’s correction for KD versus CON; **, p<0.01. ns, not significant. (D) RABL6A loss in both NHSC and MPNST cells causes reduced DNA synthesis, as measured by EdU positivity relative to CON cells (set at 1.0). **, P<0.001 for all samples compared to controls for each cell line. (E) Pie chart schematics of representative DNA content analyses show RABL6A loss causes G1 accumulation and decreased S phase. Data in B – E are from three of more independent experiments. C, D: Error bars, SD from mean.
Prior studies have shown RABL6A is required for normal and tumor cell proliferation (22, 26–28). In both NHSC and MPNST cells, RABL6A silencing caused a significant reduction in cell number (Figure 2B, see cell ratios). For RABL6A depleted NHSCs, decreased cell numbers in the absence of cell death (Figures 2B and 2C) suggest RABL6A loss in non-transformed cells causes cell cycle arrest, which was confirmed by reduced DNA replication, as measured by reduced EdU incorporation (Figure 2D). MPNST cells lacking RABL6A likewise displayed decreased DNA synthesis (Figure 2D) and their impaired proliferation was verified by flow cytometric analyses showing an accumulation of RABL6A-depleted cells in G1 phase coincident with fewer cells in S phase (Figure 2E; Supplemental Figure 3). Together, these data demonstrate a critical role for RABL6A in MPNST cell survival and proliferation whereas NHSCs selectively require RABL6A for cell cycle progression.
RABL6A promotes MPNST pathogenesis through inactivation of the p27-RB1 pathway
In pancreatic neuroendocrine tumor cells, RABL6A promotes G1 to S phase progression through multiple mechanisms that include inactivation of the RB1 tumor suppressor (22, 23). Given the G1 phase arrest caused by RABL6A downregulation in MPNST cells, we examined the effect of RABL6A loss on RB1 signaling. Knockdown of RABL6A led to significant increases in expression of the CDK inhibitor and RB1 activator, p27, in all three MPNST cell lines as well as in NHSCs (Figure 3, A and B; Supplemental Figure 2A). The marked rise in p27 protein levels in RABL6A depleted cells coincided with moderate to little increase in p27 mRNA expression (Supplemental Figure 3D), suggesting RABL6A may regulate p27 through both transcriptional and post-transcriptional mechanisms.
Figure 3. RABL6A inactivates the RB1 tumor suppressor pathway in MPNST cells.
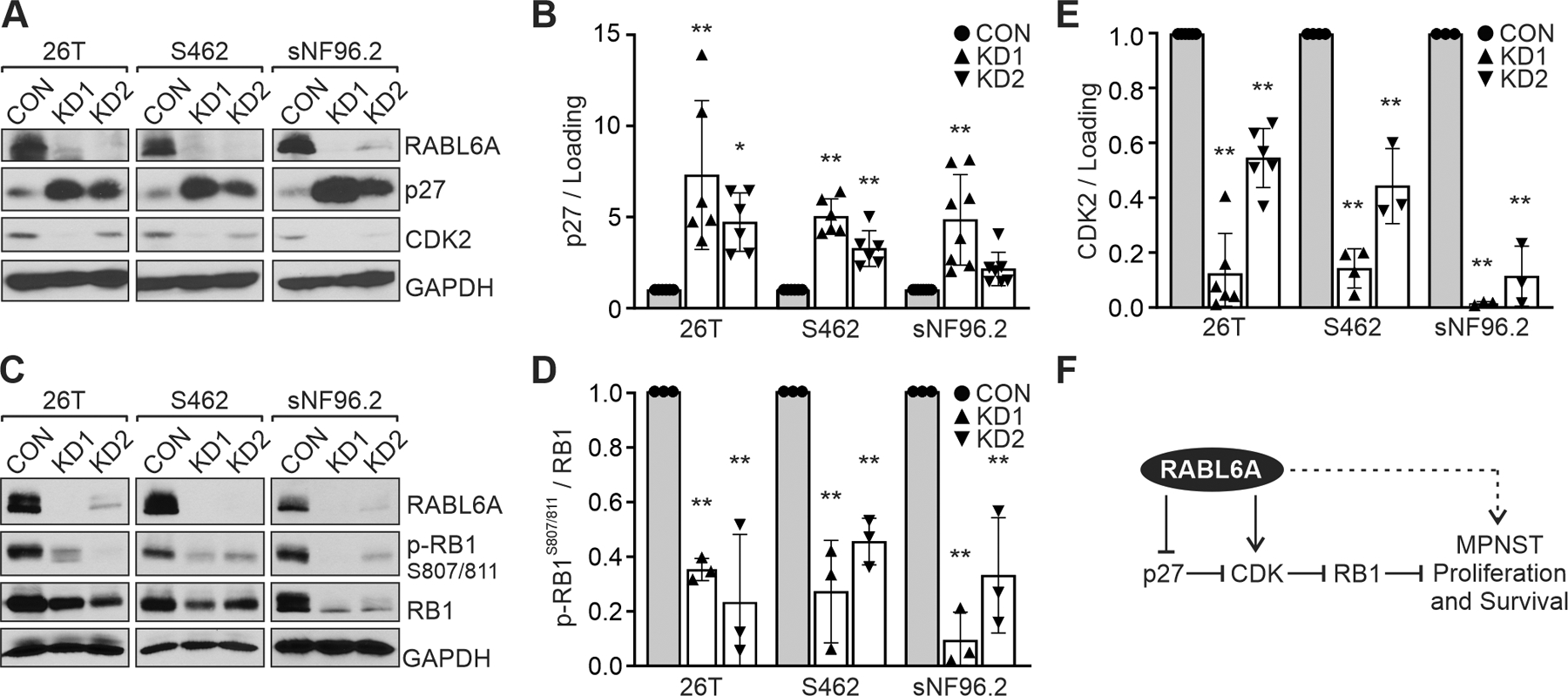
(A) Representative westerns of MPNST cell lines expressing empty vector (CON) versus RABL6A shRNAs (KD1 and KD2). RABL6A loss causes p27 upregulation and CDK2 downregulation. (B) ImageJ quantification of western data from three or more experiments showing fold increase in p27 expression following RABL6A knockdown. (C) Representative westerns show loss of RABL6A in MPNST cells causes reduced phosphorylation of RB1 at CDK4/6 targeted sites (S807/811). (D, E) ImageJ quantification of western data from three or more experiments showing reduced levels of phospho-RB1 (D) and CDK2 (E) in RABL6A knockdown cells relative to CON cells (set at 1.0). (F) Proposed model of the RABL6A-p27-RB1 pathway and how it contributes to MPNST cell proliferation and survival. Dashed arrow indicates RABL6A may also act through RB1-independent mechanisms to promote MPNST pathogenesis. Data in B, D and E are normalized to loading control or total RB1, as indicated. Error bars, SD from mean. P-value, Student’s t-test with Dunnett’s correction for each KD versus the CON (set at 1.0) for each cell line. *, p<0.05; **, p<0.01.
Consistent with the rise in p27 protein levels, loss of RABL6A led to decreased phosphorylation of RB1 at CDK4/6 targeted sites (S807/811) (Figure 3, C and D; Supplemental Figure 2, A and B). Hypo-phosphorylated RB1 halts G1-S phase progression by sequestering and repressing E2F transcription factors (47). A key transcriptional target of E2F1 is CDK2, which is required for G1 exit and S phase entry (16). Quantitative RT-PCR and western analyses verified decreased expression of CDK2 mRNA and protein in cells lacking RABL6A (Supplemental Figure 3E; Figure 3, A and E). These data support a role for RABL6A in negatively regulating the p27-RB1 pathway and thereby promoting MPNST cell proliferation (Figure 3F).
Downregulation of the p27 tumor suppressor has potential clinical importance in MPNST patients as its cytoplasmic mis-localization connotes worse outcome (15). Thus, we silenced p27 to test its importance in the RABL6A knockdown phenotype. S462 MPNST cells, as well as 26T and sNF96.2 MPNST cells, were sequentially infected with p27 shRNA (shp27) or empty vector (EV) viruses followed by CON or RABL6A-targeted KD1 and KD2 lentiviruses. Depleting RABL6A alone caused substantial cell death and reduction in cell number which was rescued by concomitant loss of p27 (Figure 4A; Supplemental Figure 4, A and F). Western analyses confirmed effective knockdown of both RABL6A and p27 (Figure 4B; Supplemental Figure 4, D and E). Individual loss of RABL6A caused upregulation of p27 and decreased RB1 phosphorylation, as expected, while dual inactivation of p27 and RABL6A significantly restored phosphorylation of RB1 (Figure 4, B and C; Supplemental Figure 4, C and H). Likewise, p27 loss reinstated DNA synthesis in RABL6A knockdown cells (Figure 4, D and E; Supplemental Figure 4, B and G). Similar results were observed in NHSCs (Supplemental Figure 2, C-F), demonstrating RABL6A regulates normal cell proliferation through the p27-RB1 axis. Together, these data establish that RABL6A promotes MPNST cell survival and cell cycle progression via p27 downregulation and consequent phosphorylation (inactivation) of RB1.
Figure 4. RABL6A promotes MPNST cell proliferation and survival via p27-RB1 inactivation.
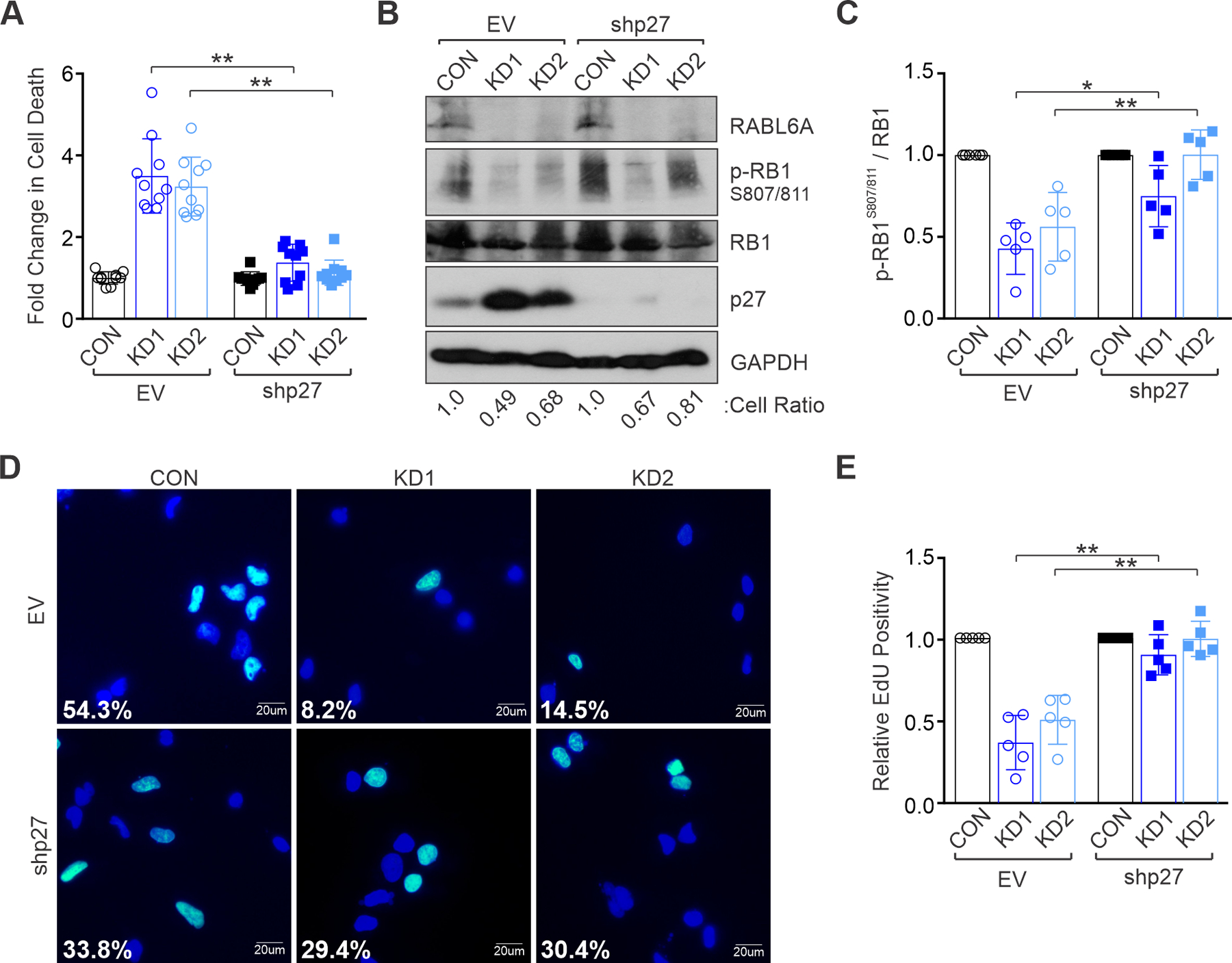
S462 MPNST cells were transduced with empty vector (EV) or p27 shRNA viruses and subsequently infected with control (CON) or RABL6A shRNAs (KD1 and KD2). (A) Quantification of the fold change in cell death after RABL6A knockdown shows that p27 inactivation rescues cell death caused by RABL6A loss. (B) Representative westerns show partial (KD1) to full (KD2) restoration of RB1 phosphorylation (p-RB1) upon co-depletion of p27 with RABL6A. Below, cell ratio (relative to control cells) for the displayed experiment. (C) ImageJ quantification of western data from three or more experiments showing partial to full restoration of phospho-RB1 when p27 is depleted in tandem with RABL6A. (D) Representative images of EdU staining show p27 inactivation rescues DNA synthesis in RABL6A depleted cells. Percent EdU positive cells (cyan colored nuclei versus dark blue nuclei for EdU negative cells) are denoted. (E) Quantification of EdU positivity from three independent experiments. Data in A, C, and E are from three of more independent experiments. Error bars, SD from mean. P-value, Student’s t-test with Bonferroni’s correction. *, p<0.05; **, p<0.01.
The CDK4/6 inhibitor, palbociclib, suppresses MPNST cell proliferation and viability in a RABL6A-dependent manner
In most MPNSTs, the RB1 tumor suppressor remains wild-type and is inactivated through loss of CDK inhibitors and consequent CDK hyperactivation (14, 15). Therefore, pharmacological inhibition of the CDKs may prove efficacious against MPNSTs. Palbociclib, an orally active, small molecule inhibitor of CDK4/6, is currently FDA approved for hormone receptor (HR) positive and human epidermal growth factor receptor 2 (HER2) negative advanced breast cancer in combination with letrozole (an aromatase inhibitor that downregulates cyclin D1, the partner of CDK4/6) (48–50).
MPNST lines treated with increasing concentrations of palbociclib were sensitive to the drug with IC50s between 0.4 to 4.27 μM (Figure 5A). Those results are similar to or better than IC50s reported for other patient derived sarcoma lines including two MPNSTs (51). It is established that palbociclib induces RB1-dependent G1 arrest and senescence in cells even while reducing total levels of RB1 (51–53). The drug can also induce apoptosis, which is heightened in cells with elevated CDK4/6 activity (51). As shown in Figure 5B, palbociclib caused significant death in MPNST cells concomitant with RB1 activation (i.e., reduced phosphorylation at S807/811). Essentially no effect of palbociclib was seen on p27 expression in the MPNST cells (Figure 5B), as anticipated. Interestingly, palbociclib variably reduced RABL6A detection depending on the cell line. Since RABL6A levels appear unaffected by palbociclib when probed using a different antibody to the protein (data not shown), the altered detection may reflect changes in RABL6A phosphorylation or other post-translational modification that impairs recognition by the polyclonal antibody.
Figure 5. MPNST cells are inhibited by the CDK4/6 inhibitor, palbociclib, in a RABL6A-dependent manner.
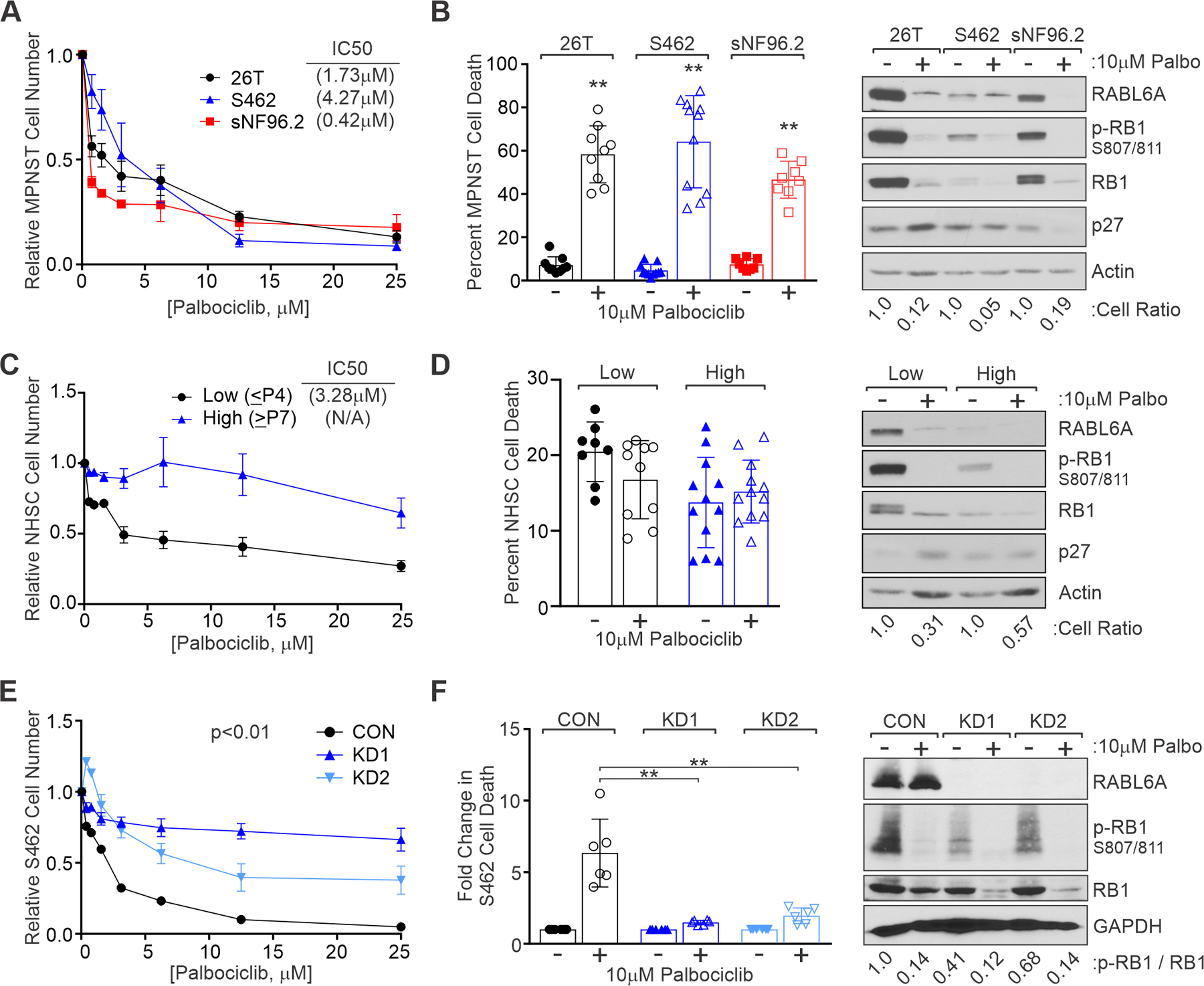
(A) MPNST cells were treated for 3 days with varying concentrations of palbociclib and relative cell number assessed by CellQuant. IC50 values indicated at top right. (B) Left, MPNST cells were treated for 2 days with 10 μM palbociclib and viability assessed by trypan blue exclusion. Right, Representative westerns showing reduced p-RB1 at CDK4/6 sites after drug treatment. Below, cell ratios relative to untreated controls show palbociclib reduces MPNST cell number. (C) Relative number of primary human NHSCs after palbociclib treatment for 3 days, as measured by AlamarBlue, in low passage (≤ P4) or high passage (≥ P7) cells. IC50 value for low passage NHSC at top right. (D) Left, Percent cell death in low versus high passage NHSCs showing no induction of death by palbociclib. Right, Representative westerns verifying reduced p-RB1 S807/811 in drug treated cells. (E) S462 CON and RABL6A depleted cells (KD1 and KD2) were treated with palbociclib at varying concentrations for 3 days and relative cell number assessed by AlamarBlue. Loss of RABL6A reduces S462 cell sensitivity to palbociclib. (F) Left, Fold change in cell death reveals cells lacking RABL6A undergo reduced drug induced death compared to CON cells. Right, Representative westerns show that palbociclib effectively reduces p-RB1 807/811 in CON and KD cells. Below, ratio of p-RB1 to total RB1. Data in A-F obtained from three or more experiments. Error bars, SD from mean. B, D, F: P-value, Student’s t-test (F: with Dunnett’s correction); **, p<0.01. E: P-values determined by a generalized linear model to assess the difference between the curves.
We next evaluated palbociclib against NHSCs to gauge potential adverse effects that might occur during patient treatment. NHSCs are primarily senescent in humans unless subjected to injury (54–56); therefore, we tested palbociclib against actively proliferating, low passage (≤ P4) and largely senescent, higher passage (≥ P7) cells. Palbociclib had little activity against later-passage NHSCs, which more accurately represent the vast majority of non-proliferating Schwann cells in humans (Figure 5C). Importantly, palbociclib induces arrest (not death) in NHSCs (Figure 5D, left) that mirrors effective activation of RB1 due to reduced phosphorylation (Figure 5D, right). To further assess the utility of palbociclib treatment, we tested its efficacy against two patient-derived cultures (designated MT1 and MT2) isolated from separate metastatic lesions in a single patient. MT1 and MT2 displayed similar sensitivity to palbociclib (IC50s of 12.7 μM and 14.5 μM, respectively) (Supplemental Figure 5A) as other patient-derived MPNST cultures (51). Together, these data support the use of palbociclib against MPNSTs while demonstrating no adverse effects besides arrest in normal cells, in agreement with its excellent safety profile in people.
Because RABL6A negatively regulates the RB1 pathway, we tested if palbociclib inhibited MPNST cell growth in a RABL6A-dependent manner. S462 control and RABL6A knockdown cells were treated with increasing concentrations of palbociclib and relative cell number was quantified. RABL6A-depleted cells were less sensitive to palbociclib than control cells (Figure 5E), which correlated with reduced drug-induced cell death (Figure 5F, left. Comparable results were obtained in 26T and sNF96.2 MPNST cells (Supplemental Figure 6). RABL6A knockdown cells still retained partial response to palbociclib (Figure 5E), likely due to the further decrease in RB1 phosphorylation at S807/811 following drug treatment (Figure 5F, right). These data demonstrate that MPNST cell response to the CDK4/6 inhibitor, palbociclib, is partially dependent on RABL6A expression. The finding that RABL6A levels influence cellular sensitivity to palbociclib supports the conclusion that CDK4/6 is a biologically relevant downstream effector of RABL6A.
CDK4/6 inhibition significantly reduces MPNST growth in vivo
To mimic de novo MPNST formation, we generated orthotopic xenografts using sNF96.2 cells injected into the sciatic nerve of immunocompromised NSG mice (45, 57). Tumors were grown to approximately 200 mm3 in size and then treated by daily oral gavage with vehicle control (water) or 25 mg/kg palbociclib. Treatment with palbociclib significantly reduced tumor growth compared to vehicle control (Figure 6, A and B). Similarly, effective inhibition of tumor growth was observed in S462 orthotopic xenografts treated with palbociclib (Supplemental Figure 7, A and B). In both cases, however, some tumors began re-growing near the end of drug treatment, indicative of acquired tumor resistance. The drug was well tolerated by animals with no behavioral abnormalities or significant reduction in body weight seen during the treatment period (Supplemental Figure 7, E and F).
Figure 6. CDK4/6 inhibition significantly reduces MPNST growth in vivo.
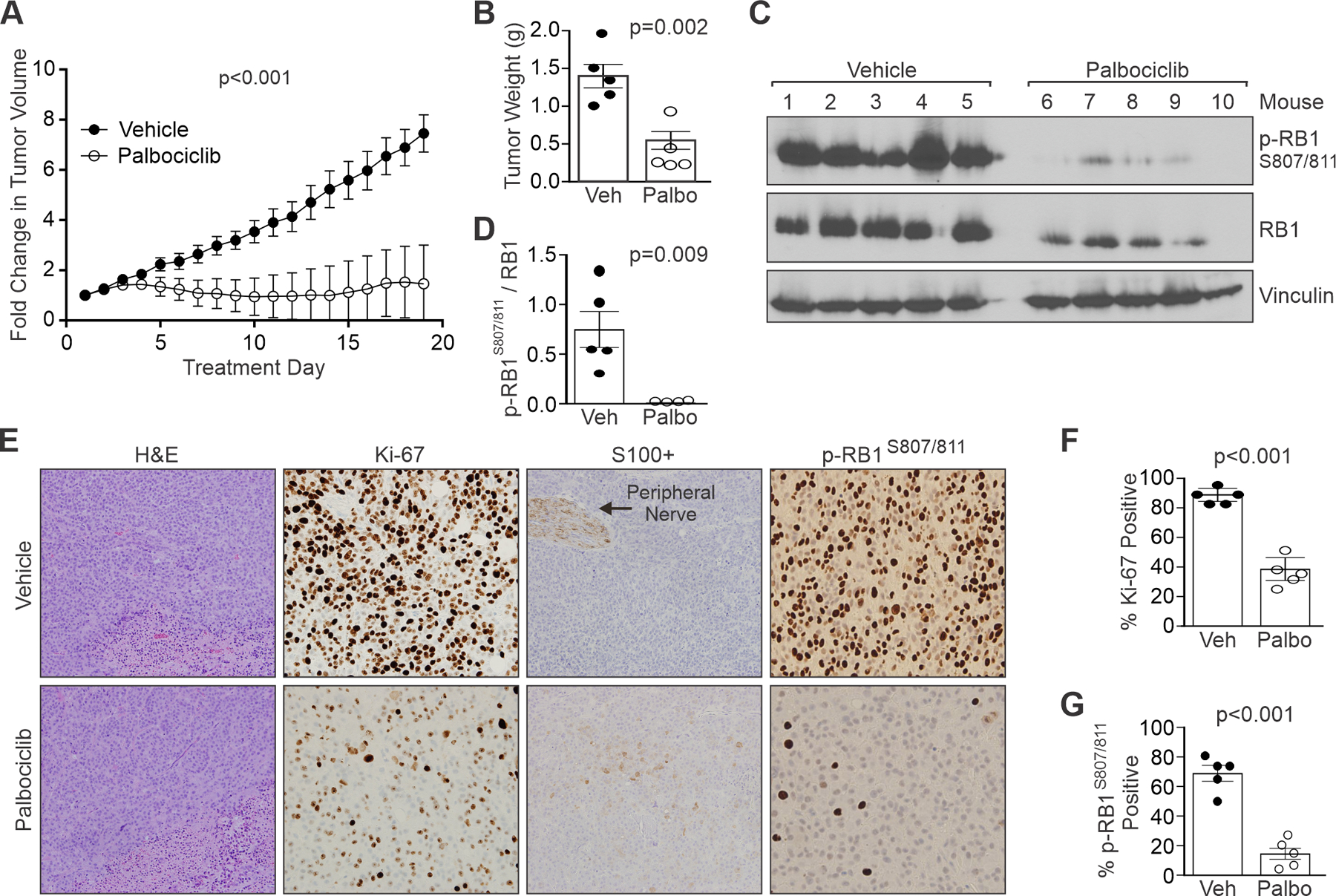
Orthotopic xenograft MPNSTs of sNF96.2 cells were generated in NSG mice. (A) Fold change in tumor volume over time in vehicle versus palbociclib treated mice. Palbociclib (25 mg/kg) or vehicle (water) was administered by oral gavage daily. Five mice per group. (B) Comparison of tumor weights after excision show palbociclib (Palbo) significantly reduces tumor weight compared to vehicle (Veh). (C) Western analyses of excised tumors show decreased phosphorylation of RB1 at CDK4/6 targeted sites (S807/811). (D) ImageJ quantification of the ratio of phospho-RB1 S807/811 to total RB1 in tumor samples. (E) H&E and IHC staining for Ki-67, S100+, and phospho-RB1 S807/811 in tumor samples from vehicle and palbociclib treated mice. Arrow, positive control peripheral nerve for S100 positivity. H&E images at 400X magnification, and IHC images at 200X magnification. (F) Ki-67 positivity in Veh or Palbo treated tumors. (G) phospho-RB1 S807/811 positivity in Veh or Palbo treated tumors. A, F, G: Error bars, SEM. B, D: Error bars, SD from the mean. A: P-value determined by a generalized linear model to assess the difference between the curves. B, D, F, G: P-value, Student’s t-test.
Excised tumors were evaluated by western blotting and IHC staining. RB1 phosphorylation at S807/811 was markedly decreased in palbociclib treated tumors versus controls in both western (Figure 6, C and D) and IHC (Figure 6, E and G; Supplemental Figure 7, C and D) tumor analyses. Expression of the proliferation marker, Ki-67, showed a significant decrease in its magnitude per cell as well as the percentage of positive cells following palbociclib treatment (Figure 6, E and F; Supplemental Figure 7C), reflecting G1 phase arrest (58, 59). While reduced Ki-67 expression and positivity were seen in palbociclib treated S462 xenografts, results were more variable with focal intratumoral regions of high Ki-67 (Supplemental Figure 7C). Palbociclib monotherapy in vivo did not cause tumor cell death, as measured by a lack of cleaved caspase-3 expression (negative data not shown). Interestingly, expression of S100B, a Schwann cell lineage marker that is lost in MPNSTs, was modestly induced by palbociclib treatment in mice with sNF96.2 tumors (Figure 6E) but not those with S462 tumors (Supplemental Figure 7C). Together, these data demonstrate the efficacy of CDK4/6 targeted drugs for MPNST treatment.
Combination CDK inhibitor therapy enhances RB1 activation and MPNST suppression
A major limitation of cancer therapy is drug resistance, and with CDK4/6 inhibitors acquired tumor resistance is often mediated by increased CDK2 activity (50, 53, 60). RB1 inactivation by CDKs is a sequential process in which cyclin D-CDK4/6 kinases prime RB1 for additional phosphorylation by cyclin E/A-CDK2 at different S/T sites, with both required for full RB1 inactivation (16, 17). CDK2 is also capable of compensating for the absence of CDK4/6 (61, 62). Given the high levels of activated CDK2 and CDK4/6 in MPNSTs (consistent with the 12–13-fold increase in their mRNA levels in patient MPNSTs, Supplemental Table 2), we hypothesized that dual therapy targeting both kinases would be superior to monotherapy with each inhibitor alone. No drugs specifically targeting CDK2 exist, but some display better selectivity for CDKs than other kinases. Dinaciclib, a potent inhibitor of CDK2 with nearly equal activity against CDKs 1, 5 and 9, has shown promising anti-cancer activity in the clinic (53, 63, 64).
Dose response curves revealed all three MPNST cell lines were highly sensitive to dinaciclib with IC50s in the low nM range (Figure 7A). The Bliss independence method (65) was used to evaluate synergism between varying concentrations of palbociclib and dinaciclib in sNF96.2 cells, with the highest synergy attained at low doses for each drug in cell proliferation assays (Figure 7B). Significant synergism between the drugs was also achieved in clonogenic survival assays where each drug alone had only moderate activity (Figure 7C). Palbociclib and dinaciclib were likewise synergistic in S462 cells (Supplemental Figure 8) and patient-derived MT1 and MT2 cultures (Supplemental Figure 5C).
Figure 7. Combination therapy with multiple CDK inhibitors enhances MPNST suppression in vivo.
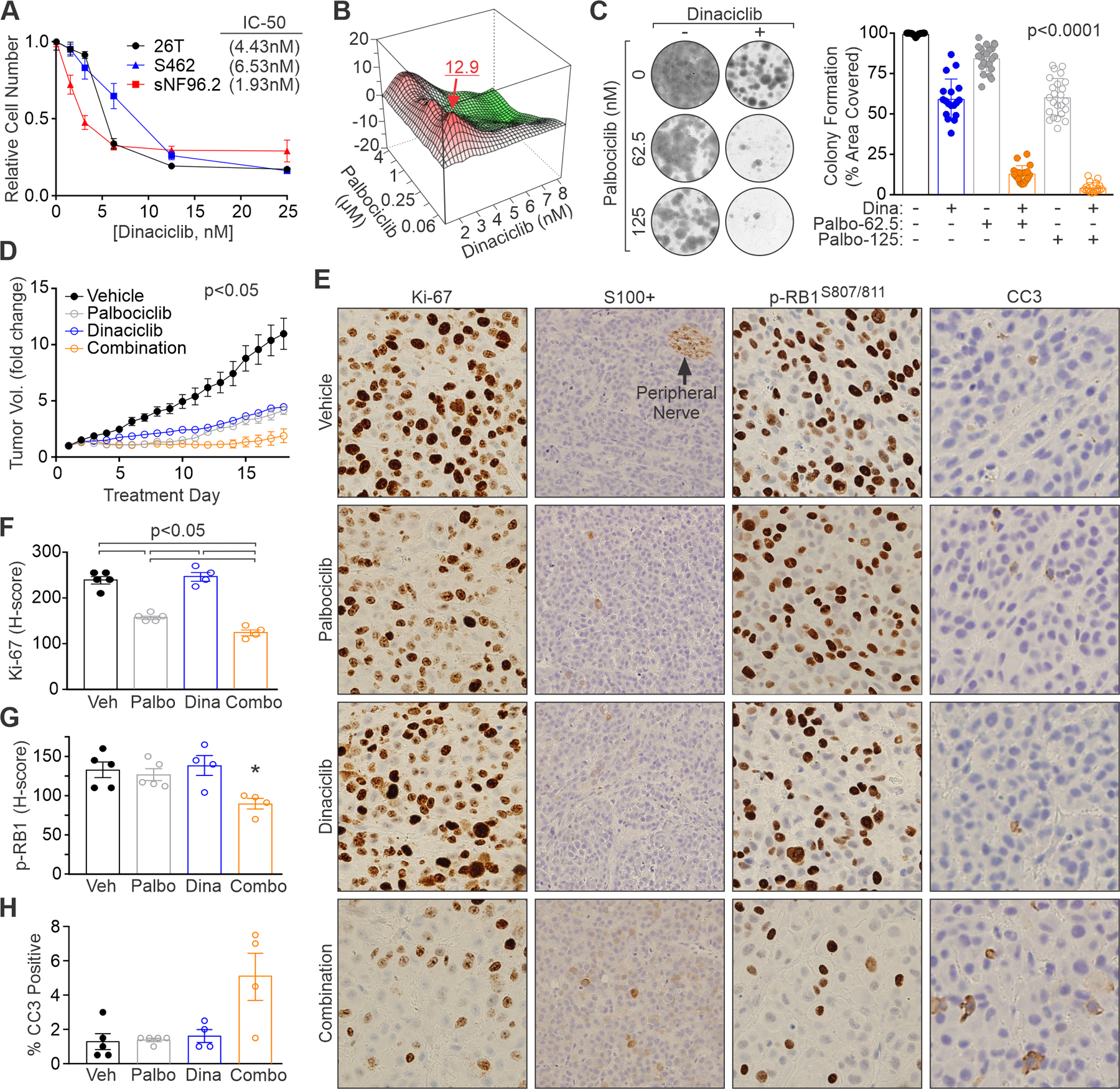
(A) MPNST cells were treated for 3 days with the indicated concentrations of dinaciclib and relative cell number quantified by AlamarBlue. IC50 values are shown. (B) Contour plot of interaction index (Bliss independence model) for the combination of low doses of CDK4/6 (palbociclib) and CDK2 (dinaciclib) inhibitors in sNF96.2 cells. Red, synergy; green, antagonism. (C) Colony formation assays in sNF96.2 cells show synergism between low concentrations of dinaciclib (2 or 3 nM) and palbociclib. Left, representative images. Right, quantification of percentage area covered by cells from 3 or more experiments. P<0.0001 for all comparisons except Dina vs Palbo-125. (D) Fold change in tumor volume for sNF96.2 orthotopic tumors treated daily with vehicle (water), palbociclib (0.5 mg/dose), dinaciclib (0.16 mg/dose) or the combination. P<0.05 for all comparisons except Dina vs Combo and Dina vs Palbo, which were non-significant. (E) IHC staining for Ki-67, S100+, and phospho-RB1 S807/811 in sNF96.2 tumor samples after treatment. H&E images at 400X magnification. IHC images at 200X magnification. Quantification of (F) Ki-67, (G) phospho-RB1 S807/811, and (H) CC3 in treated tumor samples. G: *, p<0.05 for Combo vs Veh and Combo vs Dina, other comparisons not significant. H: no comparisons significant. A, C: Error bars, SD from the mean. D, F, G, H: Error bars, SEM. C, F, G, H: P-value, Student’s t-test with Bonferroni correction. D: P-values determined by a generalized linear model to assess the difference between the curves.
The in vivo efficacy of combined CDK therapy was tested against sNF96.2 orthotopic xenografts. We lowered the dose of palbociclib relative to the monotherapy study in Figure 6, and also used a lower dose of dinaciclib compared to prior work (66). The combination of palbociclib and dinaciclib restricted tumor growth better than either drug alone (Figure 7D). Minimal weight loss in the mice (5–10% body weight) was observed but could not be attributed to drug since the same weight change occurred in vehicle control animals (Supplemental Figure 7G). However, mice treated with dinaciclib or dinaciclib plus palbociclib suffered from diarrhea. A reduction in dinaciclib dose after 1-week of therapy was better tolerated.
IHC staining of the tumors revealed enhanced anti-proliferative activity (i.e., reduced Ki-67 positivity and levels) of the combination relative to single drug-treated tumors (Figure 7, E and F). Combining palbociclib and dinaciclib also caused the greatest accumulation of active, hypo-phosphorylated RB1 (Figure 7, E and G). These changes correlated with a trend toward increased apoptosis (cleaved caspase-3 levels) in the dual-treated tumors although it was not statistically significant (Figure 7, E and H). S100B expression, undetectable in vehicle control tumors, was seen in only a few scattered cells in tumors exposed to low dose dinaciclib or palbociclib alone (Figure 7E). By comparison, treatment with both CDK inhibitors induced regional areas of low-level S100 expression across the tumors with higher levels (like those seen in peripheral nerve) visible in a small percentage of cells. These data demonstrate increased tumor suppressive activity of multi-CDK inhibitor therapy against MPNSTs.
Discussion
MPNST patients lack effective therapies, necessitating novel, more targeted treatments. RB1 signaling is often disrupted in MPNSTs through loss of CDK inhibitory proteins, such as p27, and increased CDK2 and CDK4/6 activity (15, 21, 67). Here, we identify RABL6A as a new driver of MPNST pathogenesis and show it acts by inhibiting two powerful suppressors of the disease, p27 and RB1. Our findings suggest that RB1 reactivating drugs are effective options for MPNST treatment and that combination therapies targeting multiple CDKs exert more complete responses which may ultimately reduce drug resistance in tumors.
A key conclusion of this work is that MPNSTs are addicted to RABL6A for their survival whereas non-transformed Schwann cells remain viable in its absence. The proliferation of both cell types, however, depends on RABL6A. These findings reinforce prior evidence that RABL6A is oncogenic in other human cancers, such as pancreatic and breast adenocarcinomas (25, 26), pancreatic neuroendocrine tumors (22, 23), and osteosarcomas (24). MPNSTs are now the second type of sarcoma displaying reliance on RABL6A. There are more than fifty different types of soft tissue sarcoma, essentially all of which are rare tumors that lack meaningful biomarkers and desperately need effective treatments (68). Future studies of RABL6A across a broad range of sarcoma types will reveal if it is a general driver of sarcomagenesis and if its activity represents a valuable target for sarcoma therapy. RABL6A itself may be druggable since it has intrinsic GTPase activity (69), but GTPase inhibitors have been notoriously unsuccessful in the clinic; thus, strategies targeting regulators or downstream effectors of RABL6A likely have higher potential of success for therapy.
Several observations establish that RABL6A-mediated inactivation of the p27-RB1 tumor suppressor pathway is central to its oncogenic activity in MPNSTs. Silencing RABL6A in three different MPNST cell lines caused upregulation of p27 and activation of RB1 coincident with cellular arrest and death. Conversely, silencing p27 in RABL6A depleted cells restored RB1 phosphorylation, cellular viability and cell cycle progression into S phase where levels of DNA synthesis paralleled those of control cells. Thus, the RABL6A knockdown phenotype is dependent on p27 induction, demonstrating that RABL6A is an essential negative regulator of p27 in MPNST. In agreement, RABL6A is strikingly upregulated in patient MPNSTs relative to patient-matched, benign precursor lesions and this correlated inversely with p27 protein levels. Earlier work found that p27 inactivation in MPNSTs confers reduced survival (15), predicting that elevated RABL6A in MPNSTs may likewise connote worse outcomes. RABL6A expression is a biomarker of poor survival in breast and pancreatic cancers (25, 26), but whether that is also true for MPNSTs will need to be tested in a larger cohort of patient specimens than examined here. Excitingly, our findings suggest loss of p27 and moderate upregulation of RABL6A protein represent new features of ANNUBPs, the precursors to MPNSTs whose molecular definition is still unfolding (21).
Mechanisms underlying RABL6A inhibition of p27 remain to be defined although our results predict both transcriptional and post-transcriptional events are involved. Inactivation of p27 is common in aggressive human cancers (70) and occurs through gene mutation, transcriptional silencing, and/or dysregulation of the protein through phosphorylation changes that dictate cytoplasmic mislocalization, ubiquitination and proteasomal degradation (71). In MPNSTs, the CDKN1B gene encoding p27 generally remains intact but it can be downregulated via MEIS1 overexpression and ID1-mediated transcriptional suppression (38) or by nuclear exclusion (15). The latter ultimately leads to its degradation (71). Following RABL6A depletion, we observed little to moderate upregulation of p27 mRNA coincident with a robust rise in p27 protein levels that surpassed the fold induction of its mRNA. While it is possible RABL6A affects MEIS1-ID1 activity, RNA-Seq did not detect p27 mRNA downregulation in MPNSTs compared to patient-matched NFs/PNFs, suggesting post-translational regulation of p27 protein in patient tumors may predominate. Phosphorylation of p27 at S10 is mediated by AKT, KIS, CDK5 and CDK16 kinases whereas T187 phosphorylation is mediated by CDK2 (71, 72). RABL6A is essential for AKT activation in pancreatic neuroendocrine tumors (23) and here we showed it promotes CDK2 expression, supporting the prospect that RABL6A may destabilize p27 protein by enhancing its phosphorylation at AKT and/or CDK2 specific sites.
A common chemotherapy for MPNST patients is combination of ifosfamide and doxorubicin (44, 73), a treatment with high toxicity and limited efficacy. RB1 targeted therapy using CDK inhibitors has not been used for MPNST treatment, but our data along with prior evidence of elevated CDK2 and CDK4/6 activity in patient tumors support its use for MPNST treatment. All MPNST cell lines and patient-derived cells tested were sensitive to palbociclib (albeit with IC50s in the low micromolar range), in keeping with a recent sarcoma study (which included an MPNST patient-derived xenograft cell line) showing palbociclib was most active against tumors with elevated CDK4 (51). Nearly all patients with advanced or metastatic liposarcoma have CDK4 amplification, and a phase 2 trial of palbociclib in those patients (NCT01209598) was encouraging with favorable progression-free survival and occasional tumor response (74). Currently, a phase 2 multi-center trial of palbociclib monotherapy in advanced sarcomas with CDK4 overexpression is being conducted in Spain (NCT03242382), and CDK4/6 inhibitors are considered high priority agents that warrant testing in metastatic / relapsed Ewing sarcoma (75). In our MPNST analyses, palbociclib efficacy depended on RABL6A expression, implying that high RABL6A levels and/or activity in MPNSTs (and possibly other tumors) may predict which patients will respond better to CDK4/6 targeted therapy.
Development of drug resistance to monotherapy, especially with drugs that are cytostatic in vivo (like palbociclib and dinaciclib), is a significant clinical problem driving the use of rational combination therapies. In both sNF96.2 and S462 tumor models, palbociclib caused a pronounced G1 arrest but emerging resistance (regrowth of arrested tumors or presence of high Ki-67 regions) was evident near the end of treatment. Several mechanisms of acquired resistance to CDK4/6 inhibitor therapy have been described, including cyclin D upregulation, amplification of cyclin E and/or increased expression/activity of CDK2 (reviewed in (50, 53, 60). Hyperactivation of CDK5 might also confer resistance to CDK4/6 inhibitors since it can inactivate RB1 via S807/811 phosphorylation (76). Our study showed synergistic anti-tumor effects of pairing palbociclib with the CDK2 inhibitor, dinaciclib. However, since dinaciclib also targets CDKs 1, 5 and 9 (53), it remains to be seen if suppression of CDK2 activity or the other kinases mediated the synergy with palbociclib. The possibility that other CDKs inhibited by dinaciclib could be important targets in MPNSTs is exciting since it may expand the repertoire of potential therapeutics to battle this disease.
Intriguingly, sNF96.2 tumors treated with CDK inhibitors promoted low level expression of S100, an established marker of precursor Schwann cells that is lost during malignant transformation and completely absent from dedifferentiated, high grade MPNSTs (44). S100 expression was greatest in tumors treated with the combination of palbociclib plus dinaciclib compared to either drug alone, which correlated with increased RB1 activation. This suggests that effective RB1 activation through CDK inhibition may cause MPNSTs to adopt a pre-malignant phenotype. The ability of CDK2 and CDK4/6 inhibitors to promote tumor reversion to a less aggressive, more targetable tumor type has not to our knowledge been described. The effect was not universal since a second MPNST cell line (S462) displayed no induction of S100 expression following palbociclib treatment alone in vivo. Further investigation is needed to better understand how inhibition of RB1-directed CDKs may potentially promote MPNST cell re-differentiation and to identify additional drugs that may enhance it further. Other chemotherapeutic combinations that promote tumor re-differentiation have been used to more effectively treat acute promyelocytic leukemia and neuroblastoma (77).
In conclusion, our findings justify and highlight the potential of RB1 targeted therapy for MPNST treatment. Drug combinations that inhibit multiple, RB1-directed CDKs had greater anti-tumor activity against MPNSTs than monotherapy with each CDK inhibitor alone. Dinaciclib, however, with its less specific anti-CDK activity caused diarrhea in the mice. Although generally well tolerated in patients, adverse events associated with dinaciclib treatment include orthostatic hypotension, elevated uric acid, cytopenia and tumor lysis syndrome (53). Strategic dosing would be critical for successful therapy with combinations involving dinaciclib. Alternative agents that indirectly modulate RB1 signaling via effects on cyclin D/E and p27 transcription, such as Ras-MEK-ERK and PI3K-AKT-mTOR inhibitors, have shown substantial synergy and therapeutic efficacy with CDK4/6 inhibitors against other cancers (50, 60). A logical option for MPNST therapy would be combination of palbociclib with a MEK inhibitor since MPNSTs are Ras-driven (78) and the MEK inhibitor, selumetinib, has had remarkable success treating plexiform neurofibromas in NF1 patients (79–81). Moreover, this drug combination was found to activate a senescence-associated secretory phenotype which promotes NK cell-mediated cytotoxicity and prolonged tumor control in lung cancer models with mutant Ras (82). With so many advances being made in targeted combination therapies for common cancers, there is no better time to leverage these findings to improve the treatments and outcomes for rare malignancies like MPNSTs.
Supplementary Material
Translational Relevance:
RB1 is one of the most commonly inactivated tumor suppressors in human cancers. It can be reactivated in tumors using specific inhibitors of CDK4/6 kinases, such as palbociclib, which is approved for metastatic breast cancer and in advanced clinical trials for many other cancers. However, tumor resistance to CDK4/6 inhibitor monotherapy arises due to CDK2 hyperactivation, consequently combination therapies with other targeted agents holds greater promise. Currently, there are no effective therapies for MPNSTs, yet RB1 pathway mutations that hyperactivate CDK4/6 and CDK2 kinases are defining events in MPNST pathogenesis. Our study identifies RABL6A as an essential driver of MPNSTs that negatively regulates the RB1 pathway and sensitizes tumor cells to CDK4/6 inhibitors. Moreover, low-dose combinations of CDK4/6 and CDK2 inhibitors display enhanced tumor suppressive activity against MPNSTs. Our findings suggest RB1 targeted combination therapies may effectively treat MPNSTs as well as other RABL6A-driven tumors and reduce tumor resistance.
Acknowledgements
We thank Drs. Nancy Ratner and David Gordon for generously sharing cells and drug testing expertise, respectively. We are grateful to personnel in the core facilities (Flow Cytometry, Central Microscopy, Genomics and Biostatistics) at the University of Iowa College of Medicine and Holden Comprehensive Cancer Center for their assistance, particularly Tim Ginader and Sarah Bell in Biostatistics. We thank Colleen Fullenkamp from the Tanas laboratory for generating MT1 and MT2 cultures, and Drs. Agshin Taghiyev and Richard Van Rheeden in the Darbro laboratory for assistance with TMA analyses. We also thank colleagues within the NF1 Synodos Consortium and Iowa Sarcoma Research Group, particularly Drs. Peggy Wallace, Varun Monga and Mohammed Milhem, for their constructive thoughts during this study. This work was supported by a Children’s Tumor Foundation (CTF) Synodos for Neurofibromatosis-1 grant (JMW and DKM; Projects 6 and 7, DEQ and BWD), University of Iowa Sarcoma Multidisciplinary Oncology Group pilot award (DEQ), Holden Comprehensive Cancer Center Mezhir Research Award (DEQ with BWD, RDD, MRT), Pharmacological Sciences Training Grant 2T32 GM0677954 (JLK), Young Investigator Award from the CTF (JLK), NCI Neuroendocrine Tumor SPORE P50 CA174521 (Project 2, DEQ and BWD), and an NCI Core Grant P30 CA086862 (University of Iowa Holden Comprehensive Cancer Center).
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References
- 1.Widemann BC. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr Oncol Rep. 2009;11(4):322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, et al. Malignant peripheral nerve sheath tumors. Oncologist. 2014;19(2):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang IK, Hahn SM, Kim HS, Kim SK, Kim HS, Shin KH, et al. Outcomes of Treatment for Malignant Peripheral Nerve Sheath Tumors: Different Clinical Features Associated with Neurofibromatosis Type 1. Cancer Res Treat. 2017;49(3):717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroep JR, Ouali M, Gelderblom H, Le Cesne A, Dekker TJ, Van Glabbeke M, et al. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: an EORTC soft tissue and bone sarcoma group study. Ann Oncol. 2011;22(1):207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Judson I, Verweij J, Gelderblom H, Hartmann JT, Schoffski P, Blay JY, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415–23. [DOI] [PubMed] [Google Scholar]
- 6.Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014–22. [DOI] [PubMed] [Google Scholar]
- 7.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, and Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reilly KM, Kim A, Blakely J, Ferner RE, Gutmann DH, Legius E, et al. Neurofibromatosis Type 1-Associated MPNST State of the Science: Outlining a Research Agenda for the Future. J Natl Cancer Inst. 2017;109(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaushal A, and Citrin D. The role of radiation therapy in the management of sarcomas. Surg Clin North Am. 2008;88(3):629–46, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zehou O, Fabre E, Zelek L, Sbidian E, Ortonne N, Banu E, et al. Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1: a 10-year institutional review. Orphanet J Rare Dis. 2013;8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Fiore R, D’Anneo A, Tesoriere G, and Vento R. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol. 2013;228(8):1676–87. [DOI] [PubMed] [Google Scholar]
- 12.Sherr CJ. Principles of tumor suppression. Cell. 2004;116(2):235–46. [DOI] [PubMed] [Google Scholar]
- 13.Mittnacht S The retinoblastoma protein--from bench to bedside. Eur J Cell Biol. 2005;84(2–3):97–107. [DOI] [PubMed] [Google Scholar]
- 14.Berner JM, Sorlie T, Mertens F, Henriksen J, Saeter G, Mandahl N, et al. Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: studies of CDKN2A and other genes of the pRB pathway. Genes Chromosomes Cancer. 1999;26(2):151–60. [PubMed] [Google Scholar]
- 15.Kourea HP, Cordon-Cardo C, Dudas M, Leung D, and Woodruff JM. Expression of p27(kip) and other cell cycle regulators in malignant peripheral nerve sheath tumors and neurofibromas: the emerging role of p27(kip) in malignant transformation of neurofibromas. Am J Pathol. 1999;155(6):1885–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harbour JW, and Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14(19):2393–409. [DOI] [PubMed] [Google Scholar]
- 17.Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38(1):12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15(3):184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, et al. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 2013;27(9):1003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miettinen MM, Antonescu CR, Fletcher CDM, Kim A, Lazar AJ, Quezado MM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagen J, Muniz VP, Falls KC, Reed SM, Taghiyev AF, Quelle FW, et al. RABL6A promotes G1-S phase progression and pancreatic neuroendocrine tumor cell proliferation in an Rb1-dependent manner. Cancer Res. 2014;74(22):6661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umesalma S, Kaemmer CA, Kohlmeyer JL, Letney B, Schab AM, Reilly JA, et al. RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth. J Clin Invest. 2019;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang H, Ji F, Sun J, Xie Y, Xu Y, and Yue H. RBEL1 is required for osteosarcoma cell proliferation via inhibiting retinoblastoma 1. Mol Med Rep. 2016;13(2):1275–80. [DOI] [PubMed] [Google Scholar]
- 25.Li YY, Fu S, Wang XP, Wang HY, Zeng MS, and Shao JY. Down-regulation of c9orf86 in human breast cancer cells inhibits cell proliferation, invasion and tumor growth and correlates with survival of breast cancer patients. PLoS One. 2013;8(8):e71764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muniz VP, Askeland RW, Zhang X, Reed SM, Tompkins VS, Hagen J, et al. RABL6A Promotes Oxaliplatin Resistance in Tumor Cells and Is a New Marker of Survival for Resected Pancreatic Ductal Adenocarcinoma Patients. Genes Cancer. 2013;4(7–8):273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montalbano J, Lui K, Sheikh MS, and Huang Y. Identification and characterization of RBEL1 subfamily of GTPases in the Ras superfamily involved in cell growth regulation. J Biol Chem. 2009;284(27):18129–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lui K, An J, Montalbano J, Shi J, Corcoran C, He Q, et al. Negative regulation of p53 by Ras superfamily protein RBEL1A. J Cell Sci. 2013;126(Pt 11):2436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, Hagen J, Muniz VP, Smith T, Coombs GS, Eischen CM, et al. RABL6A, a novel RAB-like protein, controls centrosome amplification and chromosome instability in primary fibroblasts. PLoS One. 2013;8(11):e80228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods. 2017;14(7):687–90. [DOI] [PubMed] [Google Scholar]
- 31.Edgar R, Domrachev M, and Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tompkins V, Hagen J, Zediak VP, and Quelle DE. Identification of novel ARF binding proteins by two-hybrid screening. Cell Cycle. 2006;5(6):641–6. [PubMed] [Google Scholar]
- 33.Meyerholz DK, and Beck AP. Principles and approaches for reproducible scoring of tissue stains in research. Lab Invest. 2018;98(7):844–55. [DOI] [PubMed] [Google Scholar]
- 34.White KA, Swier VJ, Cain JT, Kohlmeyer JL, Meyerholz DK, Tanas MR, et al. A porcine model of neurofibromatosis type 1 that mimics the human disease. JCI Insight. 2018;3(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyerholz DK, Ofori-Amanfo GK, Leidinger MR, Goeken JA, Khanna R, Sieren JC, et al. Immunohistochemical Markers for Prospective Studies in Neurofibromatosis-1 Porcine Models. J Histochem Cytochem. 2017;65(10):607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, and Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. 1999;155(6):1879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brekke HR, Kolberg M, Skotheim RI, Hall KS, Bjerkehagen B, Risberg B, et al. Identification of p53 as a strong predictor of survival for patients with malignant peripheral nerve sheath tumors. Neuro Oncol. 2009;11(5):514–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel AV, Chaney KE, Choi K, Largaespada DA, Kumar AR, and Ratner N. An ShRNA Screen Identifies MEIS1 as a Driver of Malignant Peripheral Nerve Sheath Tumors. EBioMedicine. 2016;9:110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14(1):79–89. [DOI] [PubMed] [Google Scholar]
- 40.Zhao Z, Rahman MA, Chen ZG, and Shin DM. Multiple biological functions of Twist1 in various cancers. Oncotarget. 2017;8(12):20380–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, et al. Large-scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006;66(5):2584–91. [DOI] [PubMed] [Google Scholar]
- 42.Subramanian S, Thayanithy V, West RB, Lee CH, Beck AH, Zhu S, et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J Pathol. 2010;220(1):58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stricker TP, Henriksen KJ, Tonsgard JH, Montag AG, Krausz TN, and Pytel P. Expression profiling of 519 kinase genes in matched malignant peripheral nerve sheath tumor/plexiform neurofibroma samples is discriminatory and identifies mitotic regulators BUB1B, PBK and NEK2 as overexpressed with transformation. Mod Pathol. 2013;26(7):930–43. [DOI] [PubMed] [Google Scholar]
- 44.Kim A, Stewart DR, Reilly KM, Viskochil D, Miettinen MM, and Widemann BC. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma. 2017;2017:7429697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perrin GQ, Li H, Fishbein L, Thomson SA, Hwang MS, Scarborough MT, et al. An orthotopic xenograft model of intraneural NF1 MPNST suggests a potential association between steroid hormones and tumor cell proliferation. Lab Invest. 2007;87(11):1092–102. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, Yin B, Wang B, Ma Z, Liu W, and Lv G. MicroRNA-210 promotes proliferation and invasion of peripheral nerve sheath tumor cells targeting EFNA3. Oncol Res. 2013;21(3):145–54. [DOI] [PubMed] [Google Scholar]
- 47.Henley SA, and Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lynce F, Shajahan-Haq AN, and Swain SM. CDK4/6 inhibitors in breast cancer therapy: Current practice and future opportunities. Pharmacol Ther. 2018;191:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walker AJ, Wedam S, Amiri-Kordestani L, Bloomquist E, Tang S, Sridhara R, et al. FDA Approval of Palbociclib in Combination with Fulvestrant for the Treatment of Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin Cancer Res. 2016;22(20):4968–72. [DOI] [PubMed] [Google Scholar]
- 50.Sherr CJ, Beach D, and Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016;6(4):353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez M, Munoz-Galvan S, Jimenez-Garcia MP, Marin JJ, and Carnero A. Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget. 2015;6(38):40557–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade MA, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Molecular Cancer Therapeutics. 2004;3(11):1427–37. [PubMed] [Google Scholar]
- 53.Whittaker SR, Mallinger A, Workman P, and Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017;173:83–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levi AD, Bunge RP, Lofgren JA, Meima L, Hefti F, Nikolics K, et al. The influence of heregulins on human Schwann cell proliferation. J Neurosci. 1995;15(2):1329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asbury AK. Schwann cell proliferation in developing mouse sciatic nerve. A radioautographic study. J Cell Biol. 1967;34(3):735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mirsky R, and Jessen KR. Schwann cell development, differentiation and myelination. Curr Opin Neurobiol. 1996;6(1):89–96. [DOI] [PubMed] [Google Scholar]
- 57.Turk AN, Byer SJ, Zinn KR, and Carroll SL. Orthotopic xenografting of human luciferase-tagged malignant peripheral nerve sheath tumor cells for in vivo testing of candidate therapeutic agents. J Vis Exp. 2011(49). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sobecki M, Mrouj K, Colinge J, Gerbe F, Jay P, Krasinska L, et al. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017;77(10):2722–34. [DOI] [PubMed] [Google Scholar]
- 59.Miller I, Min M, Yang C, Tian C, Gookin S, Carter D, et al. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018;24(5):1105–12 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Leary B, Finn RS, and Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–30. [DOI] [PubMed] [Google Scholar]
- 61.Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118(4):477–91. [DOI] [PubMed] [Google Scholar]
- 62.Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118(4):493–504. [DOI] [PubMed] [Google Scholar]
- 63.Ghia P, Scarfo L, Perez S, Pathiraja K, Derosier M, Small K, et al. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood. 2017;129(13):1876–8. [DOI] [PubMed] [Google Scholar]
- 64.Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood. 2015;125(3):443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, and Yang H. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen. 2014;19(5):817–21. [DOI] [PubMed] [Google Scholar]
- 66.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9(8):2344–53. [DOI] [PubMed] [Google Scholar]
- 67.Zhou H, Coffin CM, Perkins SL, Tripp SR, Liew M, and Viskochil DH. Malignant peripheral nerve sheath tumor: a comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am J Surg Pathol. 2003;27(10):1337–45. [DOI] [PubMed] [Google Scholar]
- 68.Katz D, Palmerini E, and Pollack SM. More Than 50 Subtypes of Soft Tissue Sarcoma: Paving the Path for Histology-Driven Treatments. Am Soc Clin Oncol Educ Book. 2018;38:925–38. [DOI] [PubMed] [Google Scholar]
- 69.Montalbano J, Jin W, Sheikh MS, and Huang Y. RBEL1 is a novel gene that encodes a nucleocytoplasmic Ras superfamily GTP-binding protein and is overexpressed in breast cancer. J Biol Chem. 2007;282(52):37640–9. [DOI] [PubMed] [Google Scholar]
- 70.Lee J, and Kim SS. The function of p27 KIP1 during tumor development. Exp Mol Med. 2009;41(11):765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hnit SS, Xie C, Yao M, Holst J, Bensoussan A, De Souza P, et al. p27(Kip1) signaling: Transcriptional and post-translational regulation. Int J Biochem Cell Biol. 2015;68:9–14. [DOI] [PubMed] [Google Scholar]
- 72.Yanagi T, Tachikawa K, Wilkie-Grantham R, Hishiki A, Nagai K, Toyonaga E, et al. Lipid Nanoparticle-mediated siRNA Transfer Against PCTAIRE1/PCTK1/Cdk16 Inhibits In Vivo Cancer Growth. Mol Ther Nucleic Acids. 2016;5(6):e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Higham CS, Steinberg SM, Dombi E, Perry A, Helman LJ, Schuetze SM, et al. SARC006: Phase II Trial of Chemotherapy in Sporadic and Neurofibromatosis Type 1 Associated Chemotherapy-Naive Malignant Peripheral Nerve Sheath Tumors. Sarcoma. 2017;2017:8685638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dickson MA, Schwartz GK, Keohan ML, D’Angelo SP, Gounder MM, Chi P, et al. Progression-Free Survival Among Patients With Well-Differentiated or Dedifferentiated Liposarcoma Treated With CDK4 Inhibitor Palbociclib: A Phase 2 Clinical Trial. JAMA Oncol. 2016;2(7):937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bailey K, Cost C, Davis I, Glade-Bender J, Grohar P, Houghton P, et al. Emerging novel agents for patients with advanced Ewing sarcoma: a report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Res. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pozo K, Castro-Rivera E, Tan CF, Plattner F, Schwach G, Siegl V, et al. The Role of Cdk5 in Neuroendocrine Thyroid Cancer. Cancer Cell. 2013;24(4):499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de The H Differentiation therapy revisited. Nat Rev Cancer. 2018;18(2):117–27. [DOI] [PubMed] [Google Scholar]
- 78.Ratner N, and Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15(5):290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. New England Journal of Medicine. 2016;375(26):2550–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jessen WJ, Miller SJ, Jousma E, Wu JQ, Rizvi TA, Brundage ME, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. Journal of Clinical Investigation. 2013;123(1):340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jousma E, Rizvi TA, Wu JQ, Janhofer D, Dombi E, Dunn RS, et al. Preclinical assessments of the MEK inhibitor PD-0325901 in a mouse model of Neurofibromatosis type 1. Pediatric Blood & Cancer. 2015;62(10):1709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018;362(6421):1416-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.