

Abstract
Background:
Tumor genomic features have been of particular interest because of their potential impact on the tumor immune microenvironment and response to immunotherapy. Due to the substantial heterogeneity, an integrative approach incorporating diverse molecular features is needed to characterize immunological features underlying primary resistance to immunotherapy and for the establishment of novel predictive biomarkers.
Methods:
We developed a pan-cancer deep machine-learning model integrating tumor mutation burden, microsatellite instability and somatic copy number alterations to classify tumors of different types into different genomic clusters, assessed the immune microenvironment in each genomic cluster and the association of each genomic cluster with response to immunotherapy.
Results:
Our model grouped 8,646 tumors of 29 cancer types from the Cancer Genome Atlas into four genomic clusters. Analysis of RNA-sequencing data revealed distinct immune microenvironment in tumors of each genomic class. Furthermore, applying this model to tumors from two melanoma immunotherapy clinical cohorts demonstrated that patients with melanoma of different genomic classes achieved different benefit from immunotherapy. Interestingly, tumors in cluster 4 demonstrated a cold immune microenvironment and lack of benefit from immunotherapy despite high microsatellite instability burden.
Conclusion:
Our study provides a proof-for-principle that deep learning modeling may have the potential to discover intrinsic statistical cross-modality correlations of multifactorial input data to dissect the molecular mechanisms underlying primary resistance to immunotherapy, which likely involves multiple factors from both the tumor and host at different molecular levels.
Keywords: Deep learning, cancer genomics, immunogenomics, immune checkpoint blockade
Statement of translational relevance
In this study, we developed a deep machine-learning model integrating tumor mutation burden, microsatellite instability and somatic copy number alterations that classified 8,646 tumors of 29 cancer types from the Cancer Genome Atlas into four distinct genomic clusters. De-convolution of RNA-sequencing data revealed unique immune microenvironment associated with each genomic cluster. Applying this model to exome sequencing data from two immunotherapy clinical cohorts demonstrated that patients with tumors of different genomic clusters had significantly different benefit from immunotherapy. This study provided proof-for-principle evidence that deep learning modeling may have the potential to discover cross-modality correlations of multifactorial input data to delineate the interplay between tumor and host factors by integrating genomic, epigenetic, transcriptomic, proteomic, microbiomic etc. datasets to dissect the mechanisms underlying resistance to immunotherapy and to establish novel predictive markers to precisely select patients who will benefit from immunotherapy.
Introduction
Immune checkpoint blockade (ICB) therapy targeting the cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and programmed death-1 (PD-1) pathways has demonstrated unprecedented rates of durable clinical benefit in patients with various cancers(1–6). However, response rates to single agent ICB tend to be limited to 20-30% of patients across various cancer types(3,4,6–9). Although dual ICB (α-PD-1 + α-CTLA4) and combination of ICB with chemotherapy have achieved promising responses in metastatic melanoma(10) and non-small cell lung cancer (NSCLC) patients(11), primary resistance is still observed in ~50% of patients and combination approaches have not yet been proven effective in many other tumor types. Currently, there are no optimal biomarkers to predict response to ICB. PD-L1 is approved as a predictive marker for ICB therapy in certain cancer types, but robust responses in patients with low PD-L1 argue against the value of PD-L1 as an exclusionary predictive biomarker(12,13). Understanding ICB resistance mechanisms and establishing reliable predictive biomarkers, particularly those which can be applied to multiple cancer types presents a current unmet clinical and scientific need.
Common mechanisms utilized by different cancers to evade the host’s anti-tumor immune response include selection of cancer cells with specific molecular aberrations that the host immune system cannot recognize or destroy and induction of an immunosuppressive tumor microenvironment. The molecular profiling of cancers treated with ICB has enabled us to identify several molecular features associated with response or resistance to ICB across cancer types, including expression of T cell signaling pathway genes such as IFN-γ(14), total mutation/neoantigen burden(15–17), microsatellite instability(5), infiltration of tumor-specific cytotoxic T cells(18), and somatic copy number alterations (SCNAs)(19). Cancer genomic features have been the focus of intense scrutiny with regards to their impact on ICB resistance as well as their potential as predictive biomarkers. However, due to the substantial heterogeneity between cancer types and patients, none of these features is exclusionary. For example, high tumor mutation burden (TMB) was reported to contribute to favorable response to ICB in patients with melanoma and NSCLC(15–17), which are generally on the higher end of the TMB spectrum. On the other hand, a relatively high response rate was also observed in patients with kidney cancer, which has a lower TMB(20), suggesting other molecular features are likely involved. Therefore, an integrative approach incorporating complementary molecular features is warranted to understand the unifying molecular mechanisms underlying resistance to ICB and for the identification of subgroups of patients with unique molecular features and immune microenvironments that may impact clinical benefit from ICB.
Through the use of large-scale molecular profiling techniques over the past decade, we have compiled an overwhelming amount of complex data on cancer genomics that are beyond the capabilities of traditional research approaches. Deep learning algorithms allow efficient extraction of complex data with multiple levels of abstraction and have recently proven remarkably powerful in many fields ranging from visual object recognition(21,22), speech recognition(23), and medical diagnosis(24), to genomics(25,26). Importantly, deep learning approaches also present advantages for integrative analysis of multiple types of data over traditional machine learning approaches(27,28). In this study, we applied a multifactorial deep learning framework to whole exome sequencing (WES) data derived from large pan-cancer cohorts as well as clinical cohorts treated with ICB to determine the common genomic basis for ICB-resistance.
Materials and methods
TCGA and clinical trial data
To build a multifactorial deep learning model, we integrated genomic data consisting of 8,646 the Cancer Genome Atlas (TCGA) samples across 29 tumor types (Table S1). Gene expression data, clinical data, and mutation annotation format (MAF) files for somatic mutations were obtained from GDAC Firehose Standard Data (29) (stddata_20160128 stddata Run). GISTIC2 (30) analysis of copy number data from Affymetrix SNP 6.0 arrays was obtained from GDAC Firehose Standard Analysis (31) (analyses_20150821 analyses Run). For detecting Microsatellite instability (MSI), paired tumor-normal WES data were downloaded from dbGaP and Genomic Data Commons(GDC) Legacy Archive whose data were generated based on GRCh37 reference genome(32). The data of cohorts from Van Allen’s study (16) (108 patients) and Snyder’s study (17) (64 patients) were downloaded from dbGaP (33) (accession no. phs000452.v2.p1) and dbGap (accession no. phs001041.v1.p1), respectively. All chromosome coordinates for the TCGA data and trial data in all analyses of this study were based on the GRCh37.
Genomic data processing for model
To represent the TCGA samples using genomic data, we first selected a set of genomic alteration features from several different types of genomic data, including MSI, SCNA and modified TMB (mTMB) (defined as the total number of unique genes with mutations). Variation in these features has been shown to be associated with sensitivity to immune checkpoint blockade therapy (5,15–17,19) and we anticipated that their integration would allow stratification of differential clinical phenotypes.
SCNA data processing:
the SCNA genomic features were defined as the recurrent regions of copy number change determined by algorithm GISTIC2 (30). SCNA processing methods from a previous study(34) were used to determine SCNA features and their binary statuses in each sample. The details of these methods are as follows: The peak regions of the GISTIC results for all tumor types were extracted as SCNA features. For peak regions with the same genes, only one peak region was kept. To determine SCNA events, the discrete copy number calls provided by GISTIC were used: −2, homozygous deletion; −1, heterozygous loss; 0, diploid; 1, one copy gain; 2, high-level amplification or multiple-copy gain. The copy number of the peak region was defined as altered when more than 50% of genes in an amplification or deletion peak region were high-level amplifications (2) or homozygous losses (−2). To generate the SCNA by sample binary matrix, for each tumor sample we computed whether the peak region was altered for each SCNA feature.
MSI data processing:
MSI was identified at the microsatellite genomic locus level using MSIsensor (35). MSI events were detected using MSIsensor (35) (v1.0) with paired tumor-normal WES data. MSIsensor first scans the whole reference genome to find all microsatellite sites, then assesses whether each of the microsatellite sites was mutated or not by comparing the length distribution of microsatellites in paired tumor-normal sequencing data using a chi-square goodness of fit test. The default parameters were used in MSIsensor. We treated a microsatellite site as altered if the microsatellite sites with somatic mutations tested by MSIsensor had an FDR value less than 0.05. The genomic features of each microsatellite site were annotated by ANNOVAR (36) (v2.4). We limited our analysis to the microsatellite sites located in gene regions as they were more likely to alter gene functions. These microsatellite sites were used to construct the binary microsatellite feature by sample matrix based on the somatic mutation status of each microsatellite site of each sample. We then calculated the alteration frequency among all samples for each microsatellite site. Only the 5,000 most frequently altered microsatellite sites were selected as MSI features for modeling and kept for downstream analysis. The variants at these microsatellite loci accounted for 60% detected MSI alterations of 110k microsatellite loci.
mTMB data processing:
We defined mTMB as the total number of unique mutated genes in each sample. We considered a gene mutated when at least one mutation occurred in this gene. To remove mutations with little or no functional relevance, only 7 kinds of mutations from MAF files were considered in this study, including Frame_Shift_Del, Frame_Shift_Ins, Nonsense_Mutation, Splice_Site, Translation_Start_Site, Nonstop_Mutation, and Missense_Mutation. For mTMB features, we selected frequently mutated genes gathered from i) top 5,000 most frequently mutated genes directly extracted from TCGA MAF data and ii) significantly mutated genes derived by identification of driver gene studies(37).
In total, 11,385 genomic alteration features were constructed including 558 copy number gains, 729 copy number losses, 5,000 altered microsatellite loci and 5,098 genes. We then characterized the SCNA, MSI and mTMB features in each tumor sample in a binary fashion indicating whether a genomic alteration occurred or not in each tumor sample. This resulted in three binary features by sample matrices constructed from SCNA, MSI and mTMB, respectively, where 1 represented the presence of a genomic alteration and 0 stands for the absence of a genomic alteration in the matrix.
Deep learning model
MultiModal Network:
To capture both internal and cross-modality correlation among multi-genomic features across the pan-cancer sample space, we developed a deep learning model by extending a multifactorial network from previous studies (27,28) (Fig. 1a). The model contains two stages: i) Construction of modality-specific deep belief networks (DBN) to extract features of multifactorial inputs and ii) Application of deep AutoEncoders(DAEs) to perform stratification analysis. In the first stage, each DBN takes one type of mTMB, SCNA, and MSI genomic alteration matrix dataset as input to extract its high levels of presentation. In the second stage, the model combines the hidden units that represent high level features of multimodal datasets to generate common hidden units, then feeds them to the DAEs. Fig. S1a shows the detailed structure of the MultiModal Network, which takes MSI, SCNA and mTMB data as inputs.
Figure 1.
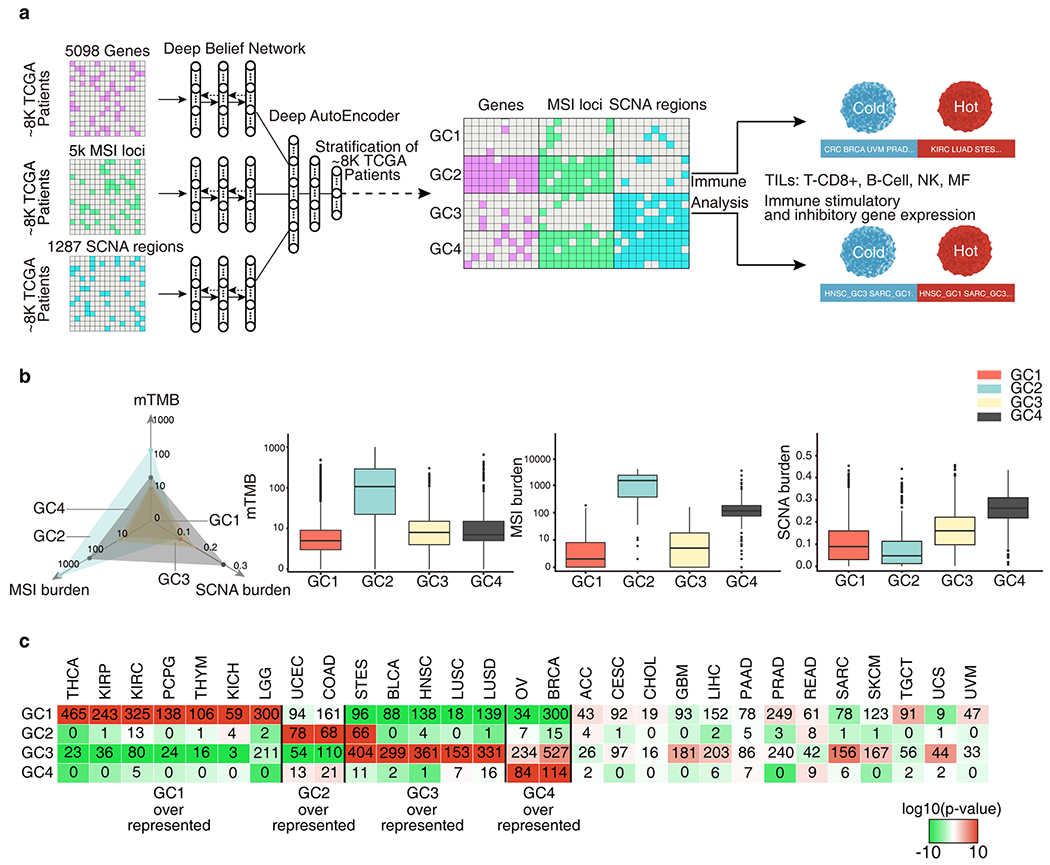
A multifactorial deep learning model reveals novel cancer subtypes and cancer families in terms of intrinsic relationships between the genomic alterations. a, Genomic features of 8,646 TCGA samples across 29 cancer types were extracted from frequently altered somatic mutation genes, microsatellite instability (MSI) loci and somatic copy number alteration (SCNA) regions. A deep learning model was constructed first extracting the high-level presentations of each kind of genomic features using deep belief networks (DBN), then perform deep AutoEncoders (DAEs) on the combined presentations for stratification. Four genomic classes(GCs) were obtained by the stratification. Cancers and cancer subtypes in GCs were identified with immune cold and immune hot microenvironment by the immune gene expression and Tumor-infiltrating lymphocytes (TILs) analysis. b, The mTMB, MSI burden, SCNA burden of each GC. The axis of TMB and MSI burden were scaled by a factor of log10. c, Number of cases in each GC across tumor types. Colors in boxes represent the log10(p-value) calculated from a hypergeometric test comparing the fraction of samples of a given cancer type in a GC to the fraction of samples that are in that GC overall. The red color indicate levels of enrichment while the green color and negative numbers indicate levels of depletion. Samples exceeding an enrichment threshold of 10 are grouped together.
Feature extraction:
DBN, which can be formed by a stacking Restricted Boltzmann Machine(RBM) (38) as showed in Fig. S1b, is a graphical model which learns to extract hierarchical representation of the input data. DBN can be trained in a greedy manner layer by layer (39). In this paper, three DBNs were trained with mTMB, MSI and SCNA datasets independently to perform feature extraction.
RBM:
RBM is a two layer and bi-directionally connected neural network where each layer is a vector of stochastic processing units (Fig. S1c). The first layer is an input layer where dimensionality is identical with the dimensionality of the input. The second layer is a hidden layer which detects high level features. The joint configuration (v, h) of the visible layer and hidden layer in RBM have an energy function (40):
| (1) |
Where n and m are the dimensionalities of the input layer and hidden layer respectively, vi, i=1,2,…,n is the variable of the input layer, hj, j=1,2,…,n is the variable of the hidden layer, b and c are bias parameters, w stands for weight parameter between the input layer and hidden layer. These parameters are learned during training.
Because of the specific structure of RBMs, visible and hidden units are conditionally independent on one-another. In general, RBMs use binary units, then we can get the following conditional distribution:
| (2) |
| (3) |
Where sigm(x) is a sigmoid activation function:
| (4) |
As shown in Fig. S1a, the first stage consists of two separate DBNs, each one takes one of genomic alteration datasets as an input respectively to reduce dimensionality and extract high level representation. When the number of datasets changed, the number of DBNs can be changed accordingly. The top hidden layer of the DBN is the low-dimensional features learned from the input. As independence between the DBNs, each DBN learns intra-modality relationship.
Clustering:
DAE (40) usually is used for dimension reduction, data reconstruction, data compression. Here we applied DAE for clustering features extracted from DBNs. Our DAE model consists of two symmetrical DBNs. The first network works as an encoder to encode input data to a code layer. The second network works as a decoder to recover input data from code layer. By minimizing the divergence between input data and its reconstruction, encoders and decoders can be trained together. We can initialize DAE weights by unrolling a pre-trained DBN. The whole network with 3 hidden layers is illustrated in Fig. S1d.
For a DAE with 3 hidden layers as shown in Fig. S1d, the forward of DAE can be derived:
| (5) |
| (6) |
| (7) |
| (8) |
DAE is trained by minimizing reconstruction error, which can be measured in many ways, such as traditional squared error:
| (9) |
In this study, we applied the DAE as a clustering tool. Because the hidden units are binary, we can take each configuration of all code units in code layer as a cluster. Namely, the number of derived the clusters is primarily related to number of the units of last layer (n) in DAE: it is less than or equal to 2n. For example, we can group data into 24 = 16 categories assuming that there are four units in code layer. In this stage, first, the top hidden layers from each modality-specific DBNs are combined into a common hidden layer. Next, the DAE takes the common hidden layer as inputs to perform cluster analysis. As union of all top hidden layers, the DAE can learn cross-modality correlations between the three types of input genomic datasets. In this study, we iteratively run the model for 100 times with randomly given n from 4 to 8 and found the most of clustering results (>90%) converge to 4 classes and distribution of all samples to the classes remains similar. Our model stratified all tumor samples into four genomic classes (GCs).
Immune cell infiltration and tumor purity analyses
Immune infiltration was inferred using a computational method proposed in a previous study (41). Briefly, this method uses immune cell gene expression profiles from the Immunological Genome Project (ImmGen) as references to determine weights relating to the specificity by which genes are expressed in different immune cell subtypes. The distribution of these gene weights throughout an individual patient’s ranked gene expression profile is then used to calculate each patient’s immune infiltration score for a given reference ImmGen cell type. The infiltration scores from four ImmGen reference cell types were used in this study: T.8MEM.SP.OT1.D45.LISOVA, B.FRE.BM, NK.DAP12neg.SP, and MF.LU, which represent infiltration from CD8+ T cells, B cells, NK cells, and myeloid cells, respectively. These representative profiles were chosen due to their high correlation with their respective cell type in peripheral blood mononuclear cell mixtures and low correlations with tumor purity, as described previously (41). When implementing this method, all 8,646 TCGA samples were input into the method at the same time. Tumor purity was calculated from each TCGA patient gene expression profile using the ESTIMATE algorithm (42).
Pathway enrichment analysis using GSEA
Gene Set Enrichment Analysis (GSEA) was used to identity the gene sets that are over-expressed or under-expressed in GC compared to other GCs within each tumor type. The pathways from Kyoto Encyclopedia of Genes and Genomes (KEGG), REACTOME, BioCarta pathways downloaded from MSigDB were used for enrichment analysis. The GSEA analysis consist following steps: 1) Gene expression (RNASeq_V2 raw counts) were downloaded data from TCGA; 2) EdgeR was used for differential expression analysis by comparing one GC to other GCs within the same tumor type; 3) A weighted gene rank list was generated from EdgeR results for each tumor type of each GC. The weight of each gene was defined as negative log10 of the EdgeR analysis-derived FDR multiplied by the sign of the logFC (log fold change); 4) the GSEA Preranked tool were used for enrichment analysis by import the weighted gene rank list and pathways.
Survival Analysis
Overall survival (OS) was calculated for clinical trial samples with the clinical data available the original study. Kaplan-Meier survival plots were generated with package “survival” (v2.41) in R. P values were assessed using a log-rank test.
Trial data and analysis
We constructed SCNA and MSI feature data by applying VarScan2 (43) and MSISensor (35) to the two WES datasets. The mTMB feature of mutated genes was calculated using the somatic mutation data from the clinical study(16,17).
The original SRA files of patients from Van Allen’s and Snyder’s study were downloaded from dbGap and converted to FASTQ files using SRA Toolkit (v2.8.0). The reads were then aligned to the GRCh37 reference genome using BWA(44) (v0.7.13). To fit the samples from these cohorts into our model, we constructed MSI and SCNA matrices using similar methods to these described above. For mTMB feature construction, we used the original mutation data from each source paper. As MSISensor (v1.0) was sensitive to sequencing depth, we normalized the sequencing depth of the samples in each cohort to be the same as the corresponding TCGA cancer type’s average sequencing depth. The normalization was performed by randomly sampling reads from the BAM file using Samtools(45) (v0.1.19) as the average depth of the samples in each cohort was higher than the TCGA corresponding cancer type’s depth. As we did not have access to SNP6.0 array data for these cohorts for SCNA matrix construction, we used the methods from a previous study(19) for copy number variation calling which are detailed as follows: 1) to call SCNA, the duplicated reads were removed using Picard (v2.9.0) and then 2) the ratio between the total number of reads from normal sample and the total number of reads from tumor sample was calculated for each sample for later use; 3) each pair of duplicated read-removed files were converted into mpileup format using samtools and the GRCh37 reference genome; 4)the mpileup file of each sample was input into VarScan2 (43) (v3.9.0) set to copynumber mode, with the data ratio parameter on and default parameters for the remainder; 5) the output segment files were adjusted for GC content using VarScan copyCaller mode; 6) adjusted segment files were further segmented using R and DNAcopy (46) library (v.1.44). The method smooth.SNA and segment in the DNAcopy library were used to perform the segmentation. The default parameters for smooth.CNA and the parameters min.width = 5 and splits = “sdundo” for segment were used; 7) the small segments were merged into large segments with mergeSegments.pl function provided by Varscan 2 with default parameters. Then, gene level copy number variation was determined as the mean of the copy number change of the segments within the gene. Because we determine whether SCNA features were altered or not based on the number of the significantly amplified or deleted genes in each SCNA region as described in the previous section, thresholds for determining the gene amplification and deletion were needed for this analysis. To determine these thresholds, we hypothesized that the mean number of SCNA events were similar between cohorts and the corresponding cancer type from TCGA. Then, for each cohort, we calculated the mean number of SCNA events in TCGA samples of the corresponding cancer and fit the amplification and deletion threshold to get the same number of SCNA events. The feature by sample matrices generated by MSI, SCNA and somatic mutation data were fed into the model trained by the TCGA data to predict the genomic classes of the patients from each cohort respectively.
In the Van Allen’s study, 1). Clinical benefit was defined using a composite end point of complete response or partial response to ipilimumab by RECIST criteria(47)or stable disease by RECIST criteria with OS greater than 1 year (n = 27). 2). No clinical benefit was defined as progressive disease by RECIST criteria or stable disease with OS less than 1 year (n = 73). 3). Long-term survival was defined as patients who had early tumor progression (progression-free survival <6 months) (n = 10) and achieved long-term survival (OS >2 years) after ipilimumab treatment.
In the Snyder’s study, 1). A long-term clinical benefit was defined by radiographic evidence of freedom from disease or evidence of a stable or decreased volume of disease for more than 6 months. 2). Minimal or no benefit was defined by tumor growth on every computed tomographic scan after the initiation of treatment (no benefit) or a clinical benefit lasting 6 months or less (minimal benefit).
Results
A deep-learning model for pan-cancer analysis classifies tumors into four genomic classes
In this study, we developed a multifactorial deep learning model to integrate three major genomic alteration features that have been reported to be associated with response to ICB in multiple cancer types(5,15–17,19): MSI burden (represented as the total number of MSI alterations(35)), SCNA burden (represented as the sum of the log2-transformed copy number ratio [tumor versus normal] of genomic segments normalized by segment length) and mTMB derived from 8,646 TCGA samples across 29 tumor types (Fig. 1a and Table S1). To capture both internal and cross-modality correlation among multi-genomic features across cancer samples, the deep learning model by extending a multifactorial network from previous studies(27,28). The model contains two stages: i) Construction of modality-specific DBN to extract features of multifactorial inputs consisting from mTMB, SCNA, and MSI and ii) Application of DAEs to perform stratification analysis (Fig. 1a).
As expected, mTMB was strongly correlated with traditional TMB (the total number of non-synonymous mutations), while mTMB, MSI burden and SCNA burden were weakly to moderately correlated to each other (Fig. S2) suggesting each of these 3 genomic features may have a distinct impact on the biology of tumors. Our model stratified all tumor samples into four GC (Fig. 1b and Table S2). GC1 and GC3 appeared to be genomically stable and were characterized by a low or intermediate mTMB, MSI and SCNA burden (thereafter named GC1:TlowMlowSlow or GC3:TlowMlowSmid). Conversely, GC2 and GC4 were generally genomically unstable, with high mTMB, high MSI burden and low SCNA burden in GC2 (thereafter named GC2:ThiMhiSlow) while high MSI burden, high SCNA burden and low mTMB was seen in GC4 (thereafter named GC4:TlowMhiShi). Tumors in GC2 had the highest MSI burden (median = 722), and highest mTMB (median = 105) among the four GC classes, while GC4 tumors were characterized by the highest SCNA burden (median = 0.26). Tumors in GC1 had the lowest mTMB (median = 4) and MSI burden (median = 1) while tumors in GC3, the largest cluster (48.7%, n = 4,213 samples), were intermediate for all 3 factors.
We next investigated the distribution of the 4 GCs within each individual cancer type by calculating the enrichment score of different GC (% tumors of a given cancer type clustered into each GC versus % tumors of any cancer types clustered into that particular GC) and Hypergeometric tests were used to evaluate statistical significance. As shown in Fig. 1c, THCA, KIRP, KIRC, PCPG, THYM, and KICH, LGG were over-represented in GC1:TlowMlowSlow; UCEC and COAD over-represented in GC2:ThiMhiSlow; STES, BLCA, HNSC, LUSC and LUAD over-represented in GC3:TlowMlowSmid; and OV and BRCA over-represented in GC4:TlowMhiShi. The remaining cancer types were relatively evenly distributed across all GCs (Fig. 1c).
Tumors of different GCs have a distinct tumor immune microenvironment
To investigate the association between GCs and tumor immune environment, we next deconvoluted the TCGA RNA sequencing data to infer the tumor immune microenvironment including infiltrating immune cells (CD8+ T cells, B cells, natural killer [NK] cells, and macrophages(41)), expression of 70 immune-related genes including PD1/PD-L1 (PDCD1/CD274), and GSEA immune pathway analysis (Fig. 2a and Fig. S3)(48,49) Overall, GC2:ThiMhiSlow tumors were characterized by a high level of TIL infiltration, high expression of immune genes, and up-regulated immune pathways suggesting an active or “hot” immune microenvironment. Alternately, GC4:TlowMhiShi tumors demonstrated low immune cell infiltration and low level of immune gene expression suggesting an inactive or “cold” immune microenvironment (Fig. 2a and Fig. S3). There were 3 main cancer types (COAD, STES and UCEC) with tumors clustered into GC2:ThiMhiSlow, which was characterized by high MSI and high mTMB. These findings are consistent with previous reports regarding the association between high MSI burden and active immune response in colorectal cancer(50,51), stomach cancer(52) and uterine cancer(41). In a recent pan-cancer analysis on the same TCGA cohorts, Bailey et al also demonstrated high MSI burden was associated with a “hot” immune microenvironment (53). On the other hand, GC4:TlowMhiShi tumors, exampled by OV, COAD and BRCA, demonstrated an immune cold microenvironment (Fig. 2a), despite GC4:TlowMhiShi had a considerably high level of MSI burden (Fig. 1b) and DNA mismatch repair (MMR) gene mutations (Fig. 2b and Fig. S4). Importantly, GC4:TlowMhiShi tumors were distinguished from GC2:ThiMhiSlow tumors by the high level of SCNA burden. In contrast, GC2 patients rarely exhibited SCNAs across genome. This suggests a distinct type of genomic instability in play that may be associated with different immunogenicity from GC2:ThiMhiSlow tumors. GC1:TlowMlowSlow and GC3:TlowMlowSmid are much larger and heterogeneous genomic classes with varying immune microenvironments (Fig. 2a). For HNSC, PAAD, STES, SKCM, KIRP and ACC, GC1 samples displayed hotter immune environments than GC3 samples. However, in BRCA, LGG and SARC, the opposite was observed: GC3 LGG, SARC, and BRCA samples displayed hotter immune environments than their GC1 counterparts. HNSC GC1 tumors had significantly higher levels of TIL infiltration and immune-related genes and up-expressed immune pathways, including those encoding the checkpoint proteins PD-1 and PD-L1 than GC3 tumors (Fig. 2a and Fig. 3a, b). Another interesting example was BRCA. In our analysis, TNBC was enriched in GC3:TlowMlowSmid (p = 0.05) (Fig. 3c). Immune microenvironment analysis demonstrated although BRCA was generally an immune-cold cancer type, GC3 BRCA tumors had significantly higher immune cell infiltration (CD8+ T, B and NK cells), immune gene expression, and up-regulated immune pathways compared to other BRCA tumors (Fig. 2a and Fig. S3). Of particular interest, comparison of TNBC to non-TNBC in different GCs, demonstrated that TNBC tumors had overall higher immune infiltration and the difference was even more obvious for TNBC tumors clustered into GC3 (Fig. 3d).
Figure 2.
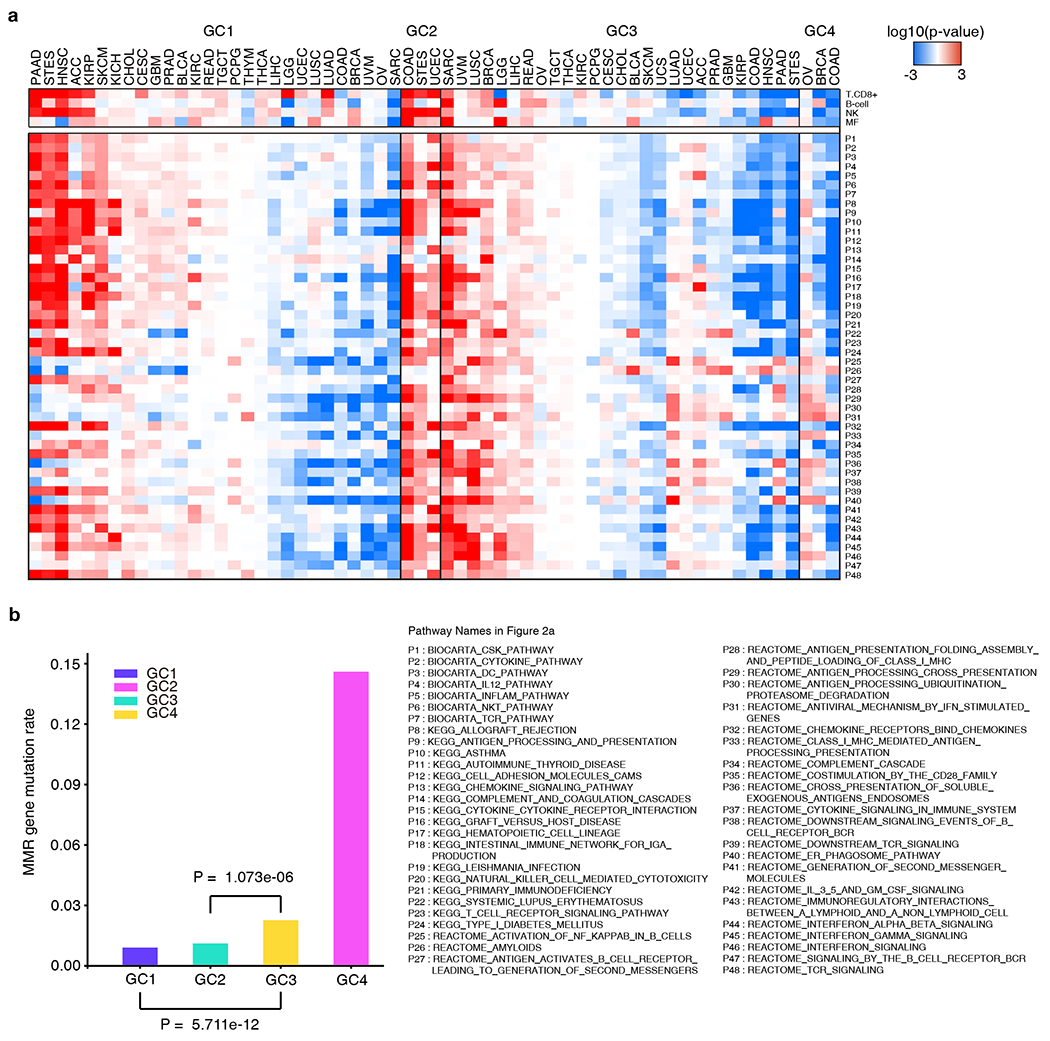
Immune profile and objective response rate (ORR) comparisons between GCs. a, Comparison of expression of immune pathways between GCs. Expression of immune pathway comparisons between samples of a given cancer type in a specific GC and samples of the same cancer type not in that GC. Color intensities in boxes represent the log10(FDR) calculated by GSEA, red color indicates high expression and blue color indicates low expression. b, Mismatch repair gene mutation frequency of each GC. The mutation frequency of the MMR genes (MLH1, MLH3, MSH2, PMS2, MSH3, MSH6, MSH4, PMS1) is calculated as the number of mutation events in a GC divided by the product of the number of MMR genes and the sample size of this GC. P value was calculated using Chi-square test comparing the mutation frequency of MMR genes in one GC to another.
Figure 3.
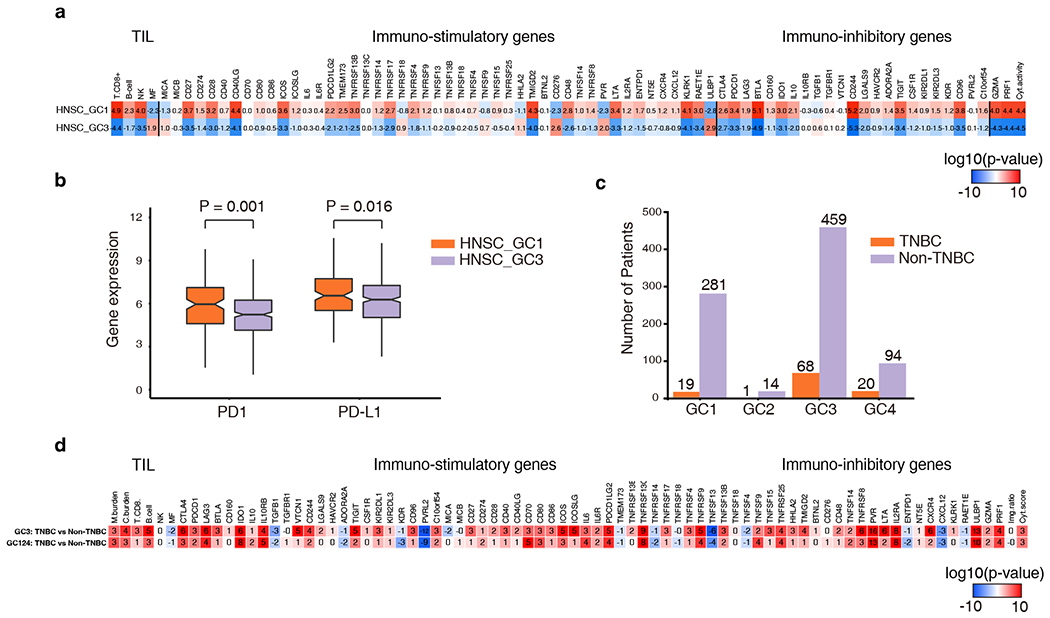
Comparisons of GC tumor immune profiles of the head and neck cancer (HNSC) and the triple negative breast cancer (TNBC). a, Immune profile difference between HNSC GC1 and HNSC GC3. Numbers in boxes represent the log10(p-value) calculated from a Wilcoxon test comparing the given immune feature between each group of samples. The higher red intensity and positive numbers indicate a more significant level of enrichment while the higher blue intensity and negative numbers indicate a more significant level of depletion. b, PD1 and PD-L1 expression of HNSC GC1 and HNSC GC3. Gene expression is measured as the log2 expected count quantified by RSEM. P value was calculated by the Wilcoxon test. c, The distribution of TNBC and non-TNBC in each GC. Fisher’s exact test was used to compare the fraction of TNBC in one GC to other GCs. TNBC GC1 was significant depleted (p <0.001), and TNBC GC3 and TNBC GC4 were significant enriched (p = 0.05, p = 0.02 respectively). d, Immune profile difference between TNBC and Non-TNBC in GC3 and other GCs. Numbers and colors are similar as these in a.
Different GC is associated with distinct biology and patient survival
Next, we explored whether tumors of different GCs have different biological and clinical behaviors. We utilized GSEA analysis to identify overall gene sets and pathways associated with each GC and revealed significantly differentially expressed pathways including cell cycle, DNA repair, metabolism etc. At pan-cancer level (Fig. S5a), G1 was associated with low expression of most above-mentioned pathways, while GC2, 3 and 4 were characterized by the high expression of nearly all the pathways. G3 tumors had the highest expression of the majority of cell cycle, DNA repair, and metabolism-related pathways. At cancer-specific level (Fig. S5b), tumors in GC2 and GC4 showed similar patterns to those at pan-cancer level and GC1 and GC3 were heterogeneous. In GC1 tumors, cell cycle, DNA repair, metabolism related pathways were under-expressed in most of the tumor types except KIPAN, CHOL, THYM, and PCPG. In GC3, cell cycle related pathways were over-expressed in SARC, BRCA UVM, LUSC, GBMLGG, ACC, LUAD, BLCA, LIHC, PRAD, PAAD, HNSC, and SKCM but under-expressed in STES, PCPG, and THYM.
Furthermore, discovered that patients with tumors of different GCs demonstrated distinct OS. As shown in Fig. 4a, patients with tumors clustered into GC2(associated with hot immune microenvironment) demonstrated longer overall survival (OS) than GC4 patients (associated with cold immune microenvironment), highlighting the prognostic importance of the immune microenvironment of tumors regardless of cancer type. In addition, GC1 patients (TlowMlowSlow) had longer OS than GC3 patients (TlowMlowSmid), suggesting the negative impact of SCNA burden on patient survival. Cancer-specific analyses also revealed the association between GCs with OS in multiple cancer types with similar trend as in pan-cancer analysis (Fig. 4b–g).
Figure 4.

Overall survival of patients from each GC. a, All patients from pan-cancers, b, ACC, c, BRCA, d, GBMLGG, e, LIHC, f, SARC, g, UCEC. Cancer types with p values < 0.05 were not shown. P value was calculated by the log-rank test.
Genomic classes are associated with response to checkpoint blockade
To further investigate whether the GCs defined by our deep learning model were associated with clinical benefit from ICB, we applied our model to WES data from a clinical cohort of metastatic melanoma treated with anti-CTLA4 (16). Patients (n = 108) were classified into GC1 (n = 54), GC2 (n = 10), GC3 (n = 35), and GC4 (n = 9). GSEA analysis from the 42 tumors with RNA-seq data available demonstrated that pathways related to cell cycle, DNA repair and metabolism were upregulated in GC3 compared to GC1, while immune-related pathways were upregulated in GC1 compared to GC3, consistent with results from the high-quality data of resected melanomas in TCGA (Fig. S6) indicating our model could be applied to data derived from small clinical specimens. Of course, this application could be affected by availability and quality of clinical specimens depending on the size and anatomic site of biopsy.
Overall, 26 of 108 patients had clinical benefit (Fig. 5a) from anti-CTLA4 treatment. Of particular interest, none of the 9 patients with GC4:TlowMhiShi tumors achieved clinical benefit (Fig. 5a and Fig. S7a–c) and their OS was significantly shorter compared to patients with other GC tumors (Fig. 5b and Fig. S7e–g). Importantly, GC4:TlowMhiShi tumors are characterized by the highest SCNA burden (Fig. S8), which has been reported to be associated with lack of response to ICB and inferior survival (19). Furthermore, comparing patients with lower levels of SCNA events (GC1 and GC2) with those with a higher degree of SCNAs (GC3 and GC4) demonstrated that patients with tumors of GC3/GC4 had significantly lower response rate (p = 0.003) (Fig. S7d) and shorter OS than patients with tumors of GC1/GC2 (p = 0.009) (Fig. S7h).
Figure 5.
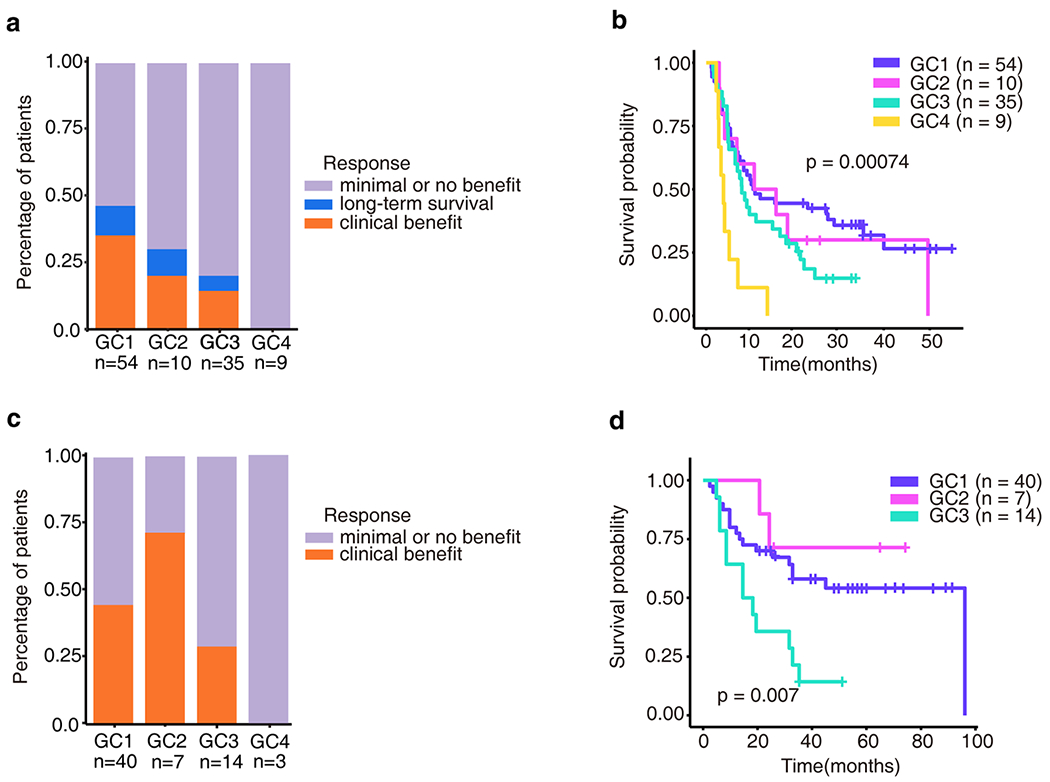
Genomic clusters are associated with clinical response from immunotherapy. a, Clinical response to immunotherapy in patients with metastatic melanoma from Van Allen’s cohort in each GC. 108 patients from Van Allen’s study were classified into four GCs. The clinical response of patients with minimal or no benefit (n = 73), long-term survival (n = 9), clinical benefit (n = 26) were defined in Van Allen’s study as described in methods. b, Overall survival of patients with metastatic melanoma from Van Allen’s cohort in each GC. P value was calculated by the log-rank test. c, Clinical response to immunotherapy in patients with metastatic melanoma from Snyder’s cohort in each GC. 64 patients from Snyder’s study were classified into four GCs. The clinical response of patients with long-term clinical benefit (n = 27), minimal or no benefit(n = 37) were defined in Snyder’s study as described in methods. d, Overall survival of patients with metastatic melanoma from Snyder’s cohort in each GC. P value was calculated by the log-rank test. There are 3 patients in GC4, not included in OS calculation.
We further tested our model on WES data from another clinical cohort of 64 patients with metastatic melanoma treated with anti-CTLA-4 (Snyder’s cohort) (17) and observed a similar trend (Fig. 5c, d). A combination of the two datasets (172 patients) further confirmed that patients with tumors clustered into GC4 had the lowest rate of clinical benefit and shortest OS compared to patients with tumors of other GCs (Fig. S9a, b). Furthermore, patients with tumors of GC1/GC2 had higher rate of clinical benefit and longer OS than patients with tumors of GC3/GC4 (p<0.0001) (Fig. S9c, d). The associations of GCs with OS remained significant after adjusting potential confounding factors in a subsequent multivariate analysis (Table S3).
Interestingly, a simple combination of TMB, MSI burden and SCNV burden was not able to predict patients’ benefit from ICB. Using median as cutoffs, we defined the genomic features reportedly associated with benefit from ICB as good markers including TMB-high (>median), MSI-high (>median) and CNV-low (<median). Accordingly, we grouped the 172 patients from the Van Allen and Snyder cohorts into group A, B, C and D with 3, 2, 1, and 0 good markers, respectively. However, no association was observed with patient’s response and survival (Fig. S10).
We further compared the predictive performance of GCs from our model to benchmark markers that have been reported to associate with clinical benefit from ICB including TMB, predicted neoantigen burden, mutations in PTEN, B2M and MMR etc. B2M mutation was associated with worse response in Van Allen’s dataset (p = 0.009) but the significance was lost when both clinical datasets were combined (Fig. S11 and Table S4). In the combined datasets, patients with MMR deficiencies showed better response rate (p = 0.027), which wasn’t observed in individual dataset (Fig. S11 and Table S4). Otherwise, none of the markers were associated with benefit from ICB with respect to response rate or OS in either cohort alone or in combination (Fig. S11, 12 and Table S4).
Discussion
ICB has revolutionized the therapeutic landscape of various cancers. However, primary resistance occurs in a considerable proportion of patients across different cancer types(3,4,6,9,54). Understanding the mechanisms underlying primary resistance to ICB across different cancers presents an unmet need. Genomic aberrations have been of particular interest to clinicians and researchers not only because of their profound impact on cancer biology, but also because of their potential as biomarkers, thanks to the fact that DNA is generally stable and technologies for DNA analysis are relatively mature. Recently, extensive studies have explored the predictive value of genomic features to immunotherapy and TMB, MSI, and SCNA have been associated with therapeutic response or resistance(5,15,19) across different cancer types. However, substantial discrepancies have been observed between different cancer types and even within the same cancer types(16,19,20) for each one of these genomic features individually as predictive biomarkers reflecting the profound heterogeneous nature of various types of cancers.
In the current study, utilizing a deep learning model, we subclassified various tumors into four distinct genomic clusters based on three genomic features: mTMB, SCNAs, and MSI. Interestingly, each cluster was associated with a unique immune landscape highlighting the complex relationship between the tumor genomic landscape and host anti-tumor immunity. Furthermore, applying this model to tumors from two clinical cohorts of metastatic melanoma treated with ICB demonstrated that different GCs were associated with distinct responses to ICB. Of particular interest, a simple combination of TMB, MSI burden and SCNV burden or using mutations of benchmark genes was not able to satisfactorily predict patient’s benefit from ICB. These results highlighted the complexity of molecular determinants of immune landscape and response to ICB and emphasized the advantage of an unsupervised approach such as that used in our model in deconvoluting complex data. Unfortunately, the current study was limited by the availability of genomic and transcriptomic data at exome level from patients treated with ICB. Therefore, these findings need to be validated in future ICB cohorts of larger sizes and different tumor types.
From a computational method standpoint, recent studies have shown the advantages of deep learning-based approaches over traditional machine learning methods in fields such as computer vision and natural language processing in terms of prediction precision and computational performance(55). In medical related areas, successful application of deep learning-based approaches have also been documented(56–58). The main goal of this multimodal deep learning model is to develop an unsupervised machine learning method capable of integrating multi-omics datasets and capturing both intrinsic statistical properties within each type of data and correlations between each type of data. Theoretically, the model is even able to integrate very diverse formats of data including image and audio(27), which is particularly useful in this era of precision oncology where multiple levels of multi-faceted data must be fused together to help clinician diagnose and tailor therapy. However, most conventional clustering methods are not designed to deal with cross-platform data. Furthermore, compared to traditional clustering methods such as K-means, the proposed methods in this study demonstrate advantages. For example, K-means based methods require the user to determine the number of clusters and to choose initial cluster centers. The clustering results are normally very sensitive to the user’s choice. Unlike K-means method, our method is a probabilistic framework, which remains stable under perturbation of initial states. Moreover, our methods do not require users to specify the number of clusters in advance. Bayesian approaches require a prior distribution (e.g., normal distribution) about latent variables, which is often not the case in the genomic data. In our method, the distribution of hidden variables is generated automatically from its conditional distribution on visible variables. Thus, our approach has more advantages than traditional unsupervised machine learning methods and is more suitable for dealing with multi-platform cancer data.
Interactions between tumor cells and the immune system are complex. Evidence has emerged that response and resistance to immunotherapy are associated with multiple tumor and host factors(59–61). However, a majority of previous studies have focused on individual factors since comprehensive analysis incorporating multiple factors is challenging by traditional approaches. As a proof for principle, our study demonstrates that multifactor deep learning is advantageous in discovering intrinsic statistical cross-modality correlations of multifactorial input data and uncovering novel molecular subtypes. With the increasing implementation of large scale profiling technologies at an affordable price applied to various biological specimens from patients with various cancers treated with immunotherapies, a tremendous amount of comprehensive data are being generated. In the foreseen future, similar models may be deployed to delineate the interplay between tumor and host factors by integrating multiple layers of omics datasets related to features such as genomics, epigenetics, gene expression, proteomics, microRNA, and microbiome to understand the molecular mechanisms underlying primary and acquired resistance to immunotherapies and to establish exclusive predictive markers to precisely select patients who will benefit from immunotherapy and avoid unnecessary toxicities.
Supplementary Material
Acknowledgements
This study was supported by the National Natural Science Foundation of China (61571202) and the Junior Thousand Talents Program of China, MD Anderson Lung cancer Moon Shot Program, MD Anderson Physician Scientist Program, Khalifa Scholar Award, Duncan Family Institute, Sabin Family Foundation, the Cancer Prevention and Research Institute of Texas Multi-Investigator Research Award grant, T.J. Martell Foundation. This work is also supported by the Cancer Prevention Research Institute of Texas (CPRIT) (RR180061 to CC) and the National Cancer Institute of the National Institutes of Health (1R21CA227996 to CC). CC is a CPRIT Scholar in Cancer Research.
Conflict of interest
J.Z. is a consultant for Geneplus-Beijing, AstraZeneca and receives honoraria from Roche, OrigiMed, Innovent and Bristol-Myers Squibb. I.I.W. receives honoraria from Roche/Genentech, Ventana, GlaxoSmithKline, Celgene, Bristol-Myers Squibb, Synta Pharmaceuticals, Boehringer Ingelheim, Medscape, Clovis, AstraZeneca, and Pfizer, and research support from Roche/Genentech, Oncoplex, and HGT. X.Y., is a current employee of Geneplus-Beijing. X.Y. and hold leadership positions and stocks of Geneplus-Beijing. A.P.F. is a consultant for Geneplus-Beijing. Z. L. is a paid speaker by Geneplus-Beijing in 2016 and 2017. The authors have no additional financial interests.
Reference
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. Massachusetts Medical Society ; 2010;363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N Engl J Med. Massachusetts Medical Society; 2015;372:2018–28. [DOI] [PubMed] [Google Scholar]
- 3.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N Engl J Med. Massachusetts Medical Society; 2015;373:1627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. Massachusetts Medical Society; 2016;375:1856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. American Association for the Advancement of Science; 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. Massachusetts Medical Society; 2015;373:1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N Engl J Med. Massachusetts Medical Society; 2015;373:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehra R, Seiwert TY, Mahipal A, Weiss J, Berger R, Eder JP, et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC): Pooled analyses after long-term follow-up in KEYNOTE-012. J Clin Oncol. American Society of Clinical Oncology; 2016;34:6012. [Google Scholar]
- 9.Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and Antitumor Activity of Anti–PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. American Society of Clinical Oncology; 2015;33:4015–22. [DOI] [PubMed] [Google Scholar]
- 10.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016. page 275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015. page 32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. Elsevier; 2016;167:397–404. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. American Association for the Advancement of Science; 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van-Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. American Association for the Advancement of Science; 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med. Massachusetts Medical Society; 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Concha-Benavente F, Gillison ML, Blumenschein GR, Harrington K, Fayette J, Colevas AD, et al. Characterization of potential predictive biomarkers of response to nivolumab in CheckMate 141 in patients with squamous cell carcinoma of the head and neck (SCCHN). J Clin Oncol. American Society of Clinical Oncology; 2017;35:6050. [Google Scholar]
- 19.Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. American Association for the Advancement of Science; 2017;355:eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Velasco G, Miao D, Voss MH, Hakimi AA, Hsieh JJ, Tannir NM, et al. Tumor Mutational Load and Immune Parameters across Metastatic Renal Cell Carcinoma Risk Groups. Cancer Immunol Res. American Association for Cancer Research; 2016;4:820–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krizhevsky A, Sutskever I, Hinton GE. Imagenet classification with deep convolutional neural networks. Adv Neural Inf Process Syst. 2012;1097–105. [Google Scholar]
- 22.Farabet C, Couprie C, Najman L, LeCun Y. Learning Hierarchical Features for Scene Labeling. IEEE Trans Pattern Anal Mach Intell. 2013;35:1915–29. [DOI] [PubMed] [Google Scholar]
- 23.Hinton G, Deng L, Yu D, Dahl G, Mohamed A, Jaitly N, et al. Deep Neural Networks for Acoustic Modeling in Speech Recognition: The Shared Views of Four Research Groups. IEEE Signal Process Mag. 2012;29:82–97. [Google Scholar]
- 24.Esteva A, Kuprel B, Novoa RA, Ko J, Swetter SM, Blau HM, et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature. Nature Publishing Group; 2017;542:115–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. NIH Public Access; 2015;347:1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods. Nature Publishing Group; 2015;12:931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ngiam J, Khosla A, Kim M, Nam J, Lee H, Ng AY. Multimodal deep learning. Proc 28th Int Conf Mach Learn ICML 2011. 2011. page 689–96. [Google Scholar]
- 28.Liang M, Li Z, Chen T, Zeng J. Integrative Data Analysis of Multi-Platform Cancer Data with a Multimodal Deep Learning Approach. IEEE/ACM Trans Comput Biol Bioinforma. IEEE Computer Society Press; 2015;12:928–37. [DOI] [PubMed] [Google Scholar]
- 29.Broad Institute TCGA Genome Data Analysis Center. Analysis-ready standardized TCGA data from Broad GDAC Firehose 2016_01_28 run. Broad Institute of MIT and Harvard; 2016. [Google Scholar]
- 30.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. BioMed Central; 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broad Institute TCGA Genome Data Analysis Center. Analysis-ready standardized TCGA data from Broad GDAC Firehose 2015_08_21 analysis. Broad Institute of MIT and Harvard; 2015. [Google Scholar]
- 32.Jensen MA, Ferretti V, Grossman RL, Staudt LM. The NCI Genomic Data Commons as an engine for precision medicine. Blood, J Am Soc Hematol. American Society of Hematology Washington, DC; 2017;130:453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The data/analyses presented in the current publication are based on the use of study data downloaded from the dbGaP web site, under dbGaP accession no. phs000980.v1.p1.and no. phs000452.v2.p1.
- 34.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. Oxford University Press; 2014;30:1015–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. Nature Publishing Group; 2013;502:333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer A, Igel C. An Introduction to Restricted Boltzmann Machines Prog Pattern Recognition, Image Anal Comput Vision, Appl. Springer, Berlin, Heidelberg; 2012. page 14–36. [Google Scholar]
- 39.Bengio Y, Lamblin P, Popovici D, Larochelle H. Greedy Layer-Wise Training of Deep Networks. Adv Neural Inf Process Syst. 2007;153–60. [Google Scholar]
- 40.Hinton GE, Salakhutdinov RR. Reducing the Dimensionality of Data with Neural Networks. Science (80- ). 2006;313:504–7. [DOI] [PubMed] [Google Scholar]
- 41.Varn FS, Wang Y, Mullins DW, Fiering S, Cheng C. Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer Res. American Association for Cancer Research; 2017;77:1271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. Nature Publishing Group; 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. Oxford University Press; 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. Oxford University Press; 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seshan VE, Olshen AB. DNAcopy: A Package for Analyzing DNA Copy Data. 2017;
- 47.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. Elsevier; 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 48.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. NIH Public Access; 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. Cell Press; 2017;18:248–62. [DOI] [PubMed] [Google Scholar]
- 50.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science (80- ). 2006;313:1960–4. [DOI] [PubMed] [Google Scholar]
- 51.Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res. 2015;75:3446–55. [DOI] [PubMed] [Google Scholar]
- 52.Li B, Severson E, Pignon J-C, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. BioMed Central; 2016;17:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. Elsevier; 2018;173:371–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N Engl J Med. Massachusetts Medical Society ; 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. Nature Publishing Group; 2015;521:436. [DOI] [PubMed] [Google Scholar]
- 56.Eraslan G, Avsec Ž, Gagneur J, Theis FJ. Deep learning: new computational modelling techniques for genomics. Nat Rev Genet. Nature Publishing Group; 2019;1. [DOI] [PubMed] [Google Scholar]
- 57.Esteva A, Robicquet A, Ramsundar B, Kuleshov V, DePristo M, Chou K, et al. A guide to deep learning in healthcare. Nat Med. Nature Publishing Group; 2019;25:24–9. [DOI] [PubMed] [Google Scholar]
- 58.Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A. A primer on deep learning in genomics. Nat Genet. Nature Publishing Group; 2019;51:12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spranger S, Sivan A, Corrales L, Gajewski TF. Tumor and Host Factors Controlling Antitumor Immunity and Efficacy of Cancer Immunotherapy. Adv Immunol. 2016. page 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017. page 707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med. 2016;375:1767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.