

Abstract
Therapies for lung cancer patients initially illicit desirable responses, but the presence of hypoxia and drug resistant cells within tumors ultimately lead to treatment failure. Disulfiram (DSF) is an FDA approved, copper chelating agent that can target oxidative metabolic frailties in cancer vs. normal cells and be repurposed as an adjuvant to cancer therapy. Clonogenic survival assays showed that DSF (50–150 nM) combined with physiological levels of Cu (15 μM CuSO4) was selectively toxic to H292 NSCLC cells versus normal human bronchial epithelial cells (HBEC). Furthermore, cancer cell toxicity was exacerbated at 1% O2, relative to 4 or 21% O2. This selective toxicity of DSF/Cu was associated with differential Cu ionophore capabilities. DSF/Cu treatment caused a >20-fold increase in cellular Cu in NSCLCs, with nearly two-fold higher Cu present in NSCLCs vs. HBECs and in cancer cells at 1% O2 vs. 21% O2. DSF toxicity was shown to be dependent on the retention of Cu as well as oxidative stress mechanisms, including the production of superoxide, peroxide, lipid peroxidation, and mitochondrial damage. DSF was also shown to selectively (relative to HBECs) enhance radiation and chemotherapy-induced NSCLC killing and reduce radiation and chemotherapy resistance in hypoxia. Finally, DSF decreased xenograft tumor growth in vivo when combined with radiation and carboplatin. These results support the hypothesis that DSF could be a promising adjuvant to enhance cancer therapy based on its apparent ability to selectively target fundamental differences in cancer cell oxidative metabolism.
Keywords: Disulfiram, ATP7B, metabolic oxidative stress, radiation oncology, hypoxia
Graphical Abstract
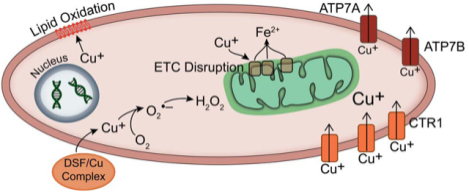
Introduction
Current therapy regimens for non-small cell lung cancer (NSCLC) include surgery, radiation, and chemotherapy. While these treatments can show excellent initial responses, tumors often become resistant, leading to 5-year survival rates of <20% (1). Factors contributing to this resistance include hypoxia and resistant populations of cancer stem cells (2).
DSF is an inhibitor of aldehyde dehydrogenase that has historically been used for the treatment of alcoholism (3). Recently, it has gained attention as a potential anti-cancer agent, causing cancer cell killing when combined with physiological levels of Cu both in vitro and in vivo (4–8). Suggested mechanisms for DSF toxicity include inhibition of proteasome activity, G2/M cell cycle arrest, induction of apoptosis, inhibition of Nrf2 expression, inhibition of TGF-beta induced epithelial-to-mesenchymal transitions, and inhibition of the p97-NPL4 pathway (3, 4, 7–10). DSF as a cancer therapeutic is well tolerated, with few toxicities at blood levels of 1.3 μM (11). Clinical trials in cancer patients suggest that dosing up to 500 mg is possible and well tolerated in combination with chemotherapy (12). The limited side effects suggest that DSF may be selectively toxic to tumor cells vs. normal tissue, but these differential effects have not been demonstrated or explored mechanistically. Furthermore, while previous studies support the use of DSF as an anti-cancer agent, the causal role of oxidative metabolism and the production of specific reactive oxygen species (ROS), such as O2•− and H2O2, was not determined.
Cu chelation by DSF is believed to reduce Cu(II) to Cu(I), which can further react with O2 to produce O2•− and subsequently H2O2 (reactions 1–3) (6, 13). This reaction also produces an intermediate compound, bitt-42+, which can decompose in buffered conditions.
| [1] |
| [2] |
| [3] |
| [4] |
Although these reactions have been hypothesized to occur within cancer cells, there is no direct evidence supporting a causal role for these reactions in tumor cell toxicity. DSF is also hypothesized to act as a copper ionophore in cancer cells, delivering increasing levels of Cu into the cell (8). With increased levels of intracellular Cu, the subsequent production of O2•− and H2O2 may allow for cancer cell toxicity, especially when reactions with metal ions produce highly cytotoxic hydroxyl radicals (HO•) (reaction 4). It has been shown that hypoxia can increase cellular uptake of Cu in both the cytoplasm and the nucleus, but the role of O2 in the anti-cancer effects of DSF is unknown (14).
Previous studies have shown that H2O2 production from ascorbate can induce radio-chemo-sensitization (15, 16). Since DSF has been proposed to disrupt redox metabolism in cancer cells, DSF and Cu may cause sensitization through similar mechanisms. Furthermore, previous studies have shown that DSF can inhibit ALDH1 activity, inhibit mammo-sphere formation, decrease radiation-induced increases in the %ALDH1+ cancer stem cells, and inhibit 4T1 cell lung metastasis in mouse models (17–19). Finally, DSF has been suggested to prolong patient survival when given with standard chemotherapies (20, 21). These data support the hypothesis that DSF may cause radio-chemo-sensitization and target cancer stem cells.
The current study fills many gaps in knowledge by showing that DSF in combination with physiologically relevant concentrations of Cu causes selective toxicity to human lung and breast cancer cells vs. normal epithelial cells that is greater in hypoxia. DSF toxicity is shown to occur through a Cu-mediated mechanism that involves O2•− and H2O2. Furthermore, Cu efflux through ATP7B plays an important role in this cancer cell toxicity. Finally, studies both in vitro and in vivo demonstrate that DSF selectively sensitizes cancer cells to radio-chemo-therapies and causes toxicity in radio-resistant and chemo-resistant hypoxic cells as well as cancer stem cells. Overall, these results support the hypothesis that DSF represents a promising adjuvant to radio-chemo-therapies that exploits fundamental differences in cancer cell redox metabolism.
Methods
Cell Culture
The NSCLC cell line NCI-H292 was obtained from ATCC. The NSCLC cell line H1299T was obtained from Dr. Bryan Allen’s lab (University of Iowa) and was derived from H1299 cells (ATCC) that were expanded in a mouse xenograft and selected for aggressive growth in animals. H1299T cells were verified to have the same genetic profile as H1299 cells through IDEXX BioResearch. H1299T-CAT cells were derived from the H1299T cell line as previously described (22). Cancer cells were cultured in RPMI media (Gibco) supplemented with 10% fetal bovine serum (Atlanta Biologicals). Normal human bronchial epithelial cells (HBEC) and normal human mammary epithelial cells (HMEC) were obtained from Cell Applications and cultured in HBEC or HMEC media (Cell Applications). All cells were cultured at 21% O2 (37°C, 5% CO2) unless otherwise noted.
Cell Treatments
20 mM disulfiram (DSF, Sigma) stock was dissolved in DMSO and further diluted to 20 μM in PBS. 10 mM CuSO4 (Sigma) stock was dissolved in water. 3–5 mM GC4419 (Galera Therapeutics) stock was dissolved in bicarbonate buffer. 100 mM buthionine sulfoximine (BSO, Sigma) stock was dissolved in PBS. 1 mg/mL auranofin (AUR, Enzo Life Science) stock was dissolved in ethanol and then further diluted 1:10 in PBS. 1 mg/mL cisplatin (Fresenius Kabi) stock was diluted 1:10 in PBS. 1 mg/mL doxycycline (Fisher) stock was dissolved in water. 10 mM bathocuproinedisulfonic acid (BCS, Sigma) stock was dissolved in water. For drug treatments, cells were plated at least 48 h prior to treatment and allowed to grow to 40–80% confluence. Cancer cell drug treatments were given in RPMI media with 10% FBS. Tumor vs. normal cell drug treatments were given in HBEC media. Final drug concentrations are reported in figure legends. Radiation in vitro was delivered using a 6,000 Ci 137Cs source (JL Shepherd and Associates, San Fernando, CA) with doses of 0–6 Gy (dose rate, ≈ 0.65 Gy/min).
Clonogenic Survival Assay
Clonogenic survival assays were performed on exponentially growing cultures as described (16). Attached and non-attached cells were collected with trypsin-EDTA immediately following treatment. Low densities (150 – 100,000) were re-plated and grown for 8 – 14 days prior to staining using Coomassie Blue. Colonies >50 cells were considered survivors and survival data were normalized to sham treated controls.
Total Cu Quantification
Following treatment, cells were washed, scraped into cold PBS, and protein was measured by Lowry assay (23). Cells were then pelleted and dissolved in nitric acid. For xenograft samples, wet tumor weights were determined, and tumors were dissolved in nitric acid. Total Cu was assessed using inductively coupled plasma mass spectrometry (ICP-MS) at the State Hygienic Laboratory (Ankeny, IA). Cu quantities were calculated as ng Cu per mg total protein. For cell culture experiments where biological replicates were analyzed by the State Hygienic Lab on separate dates, results were normalized to control, and relative Cu amounts were reported. For mouse xenograft experiments, two samples of each tumor were analyzed, and the average of these two samples was reported.
Cu-64 Influx/Efflux
Cells were plated in 6-well plates at 65,000 cells/well and allowed to attach for 24 h. At time 0, medium was replaced, and cells were treated with 150 nM DSF and 15 μM CuCl2 containing 37 kBq 64CuCl2 (University of Wisconsin Madison, Department of Radiology) at 21% or 1% O2. CuCl2 uptake was monitored by 64CuCl2 radiotracer. At varying time points, cells were removed from the incubator, medium was removed, and cells were washed three times with ice-cold PBS. Cells were lysed using 0.5 mL of 0.5 M NaOH and transferred into 10 mL of Econo-Safe™ liquid scintillation cocktail (Research Product International, Mount Prospect, IL). Radioactivity was counted for 2 min on Tri-Carb 2800TR Liquid Scintillation Analyzer (PerkinElmer, Waltham, MA) and cell numbers from untreated dishes were used to determine counts per minute (CPM) per cell.
Cell Free Measurement of Cu(I) with Neocuproine
In cell free buffer, the Cu(I) chelator, neocuproine (NC), was used to determine the production of Cu(I) from DSF as previously described by absorbance at 448 nm (24, 25). In phosphate buffer, 2 mM NC and 50–100 μM DSF were combined, and 2 mM CuSO4 was added at time 0. Absorbance at 448 nm was measured with the Tecan Infinite M200 spectrophotometer over 60 min. Spectra were then taken of the NC-Cu(I) complex, the combination of DSF and Cu in the absence of NC, and the combination of DSF and NC in the absence of Cu.
Intracellular Hydrogen Peroxide
The determination of intracellular steady-state [H2O2] was accomplished using the inhibition of catalase activity in the presence of 3-aminotriazole (3-AT) as described (26–28). Briefly, cells were infected with 100 MOI of the AdCAT adenovirus to achieve baseline catalase activity of 150 – 400 mkU/mg protein. Cells were then treated with 100 nM DSF and 15 μM CuSO4 for 1 h; then 50 mM 3-AT was added, and allowed to incubate for an additional 1 h, sampling every 15 min. 3-AT reacts in a one-to-one ratio with catalase in the presence of H2O2, which stoichiometrically inhibits catalase activity (26). Inhibition of catalase activity was monitored over time and intracellular [H2O2] was calculated using pseudo-first order kinetics: [H2O2] = k′AT/k1, where k1 = 1.7 × 107 M−1 s−1 (26–28).
Adenovirus Overexpression
Adenovirus containing plasmids for overexpression of catalase, Cu, Zn superoxide dismutase (CuZnSOD), and glutathione peroxidase 4 long form (GPx4L) were obtained from ViraQuest (North Liberty, IA). For infection, cells were grown for 48 h, 100 MOI of adenovirus was added in serum free media, and cells were incubated for at least 6 h. Medium containing FBS was then added to cells, and cells recovered for 24–48 h prior to treatment. Overexpression was confirmed in each experiment with activity assays.
Catalase Activity Assay
Catalase activity was determined from frozen pellets at room temperature as described by Beers and Sizer with the analysis of Aebi (29–31). Activity was normalized per mg cellular protein, as determined by the Lowry assay (23).
SOD Activity Assay
At the time of clonogenic survival assays, a fraction of trypsinized cells was pelleted and frozen at −20 °C. SOD activity was determined using an indirect competitive inhibition assay according to the method of Spitz and Oberley and normalized per mg protein (32).
GPx4 Activity Assay
GPx4 activity was determined as previously described (33, 34). Assay buffer consisted of 100 mM (pH 8.0) Tris/HCl, 1.5 mM NaN3 to inhibit catalase, 2.0 mM EDTA (chelator), and 0.1% Triton X-100, peroxide free (Sigma-Aldrich, surfactant). Stock solutions were as follows: 15 U/mL glutathione disulfide reductase (GR, Sigma-Aldrich), 60 mM glutathione (GSH, Sigma-Aldrich) in assay buffer, and 4 mM NADPH in 0.1% NaHCO3. At the time of clonogenic survival assays, a fraction of cells was pelleted and frozen at −20 °C. On day of analysis, pellets were resuspended in assay buffer. The buffer, GR solution, GSH solution, NADPH solution, and sample were combined in a cuvette and were incubated at 37°C for 5 min. The oxidation of NADPH was monitored at 340 nm (ε = 6,220 M−1 cm−1) for 300 s (5 min). Background oxidation of NADPH was monitored for the first 100 s, then 15 μL of ≈ 2 mM phosphatidylcholine hydroperoxide (PCOOH) solution was added into the cuvette to start the reaction. PCOOH was synthesized using L-α-phosphatidylcholine Type III/S (Sigma-Aldrich) and soybean lipoxidase Type V (Sigma-Aldrich) (35). Observed activity was normalized to the protein content of the sample as determined by the DC Protein Assay (Bio-Rad) (23).
Flow Cytometry for MitoSOX, C11-BODIPY, and JC-1
Cells were plated, incubated for 48 h, then treated with DSF/Cu for 4 h. Media was removed and replaced with fresh media, and incubated for an additional 24 h. Cells were then collected with trypsin-EDTA and stained with 5 μM C11-BODIPY in HBSS, or 2 μM MitoSOX or 1.5 μg/mL JC-1 in PBS + 2% FBS + 5 mM pyruvate for 15 min at 37°C. Stain was removed, cells were resuspended in cold PBS, and analyzed using the BD LSR flow cytometer. Hoechst negative cells were used to analyze at least 20,000 live cells/sample.
Stable ATP7B Overexpression
Empty vector (pGCsamEN) and ATP7B overexpression vector were obtained from Dr. Hartmut Schmidt (Münster University Hospital) (36, 37). Retrovirus containing these vectors were produced using GP293 cells and VSV-G helper vector. Virus was collected from GP293 cell cultures, centrifuged, and passed through 0.45 μm filters. H292 cells were grown for 24 h, exposed to virus for 24 h, and clones were selected with 750 μg/mL G418. For expansion of single clones, cells were re-plated in 150 mm dishes, with 500 cells per dish, grown for two weeks, and colonies were picked.
ATP7B Western Blot and qPCR
Cells were lysed in RIPA buffer with protease inhibitor and were analyzed by western blotting procedure using primary antibodies: ATP7B (1:100, Santa Cruz), β-tubulin (1:1000, Iowa Hybridoma Facility). Secondary antibodies: anti-mouse and anti-rabbit (1:5000, Cell Signaling). All antibodies were in 5% milk in TBST.
Cells were lysed and RNA was extracted using the Qiagen RNeasy Mini Kit and RNAse-free DNAse. cDNA was produced using the Bio-Rad iSCRIPT cDNA synthesis kit. qPCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems).
ATP7B primer sequences (5’−3’):
forward-TCCTCTGTGTCTGTGGTGCTC
reverse-ATGCGCCTGTGCCTCATAC
18s primer sequences (5’−3’):
forward-AACTTTCGATGGTAGTCGCCG
reverse-CCTTGGATGTGGTAGCCGTTT
ALDH Activity for Cancer Stem Cells
ALDH activity was measured using the Aldefluor Kit (STEMCELL Technologies), using Hoechst as a live/dead cell marker. Cells were stained per the kit instructions, analyzed by the BD LSR flow cytometer, and gates were set so negative controls for each sample (DEAB treated) were 0.1% positive. At least 10,000 live cells were analyzed for each sample. Live cell sorting was performed using the BD FACSAria cell sorter.
Mouse Xenograft Studies
Female athymic nude mice were purchased from Envigo and 106 H292 cells were injected into the right flank. When tumor volumes averaged 100–300 mm3, mice were randomized into control, DSF, radiation + carboplatin, or radiation + carboplatin + DSF groups. Radiation consisted of 3 × 6 Gy radiation doses on days 1, 3, and 5. Radiation was administered under anesthesia (ketamine) using a Pantak x-ray source operated at 200 kVp, 15 mA, with 0.35 mm Cu + 1.5 mm Al filtered x-rays at a dose rate of ≈ 1.4 Gy/min. Mice were shielded using lead coffins as described (16). Carboplatin was administered by IP in normal saline as 2 × 15 mg/kg doses on days 1 and 8. DSF in vegetable oil (100 mg/kg) was given daily by oral gavage (and continued for the entire experiment). Groups not receiving DSF received vegetable oil alone. Carboplatin and DSF were administered ≈ 3–6 h prior to irradiation. Tumor volumes were measured using Vernier calipers and mice were weighed daily until they met criteria of 1.5 cm for 2 days.
Statistical Analysis
Results were presented as mean +/− SEM, and significance was determined as p<0.05. Results were analyzed using GraphPad Prism (GraphPad Software, Inc.). Analysis of three or greater samples was done by one-way and two-way ANOVA using multiple comparisons with Tukey or Sidak post hoc tests. Student’s t-test was used when only two samples were analyzed. Radiation dose response curves were fit to the linear quadratic equation and compared using the extra sum-of-squares F test. Survival analysis of in vivo results was done by the log-rank Mantel-Cox test.
Results
DSF induces selective killing in tumor cells and in hypoxia
To demonstrate that Cu is necessary for DSF toxicity, cancer cells were treated with DSF and increasing Cu concentrations for 24 h. Only in the presence of both Cu and DSF was clonogenic cancer cell killing induced [Supp Fig S1B]. Since the serum concentration of Cu in healthy individuals is ≈ 15 μM and culture media is only 0.25 μM, 15 μM CuSO4 was added in all cell experiments (38–40). Since human serum contains ceruloplasmin and albumin that can bind Cu, 10% fetal bovine serum containing ceruloplasmin was also used in cancer cell culture media. The exact levels of these Cu binding proteins in cell culture experiments do not perfectly recapitulate levels found in human serum. However, unbound Cu in human serum averages between 0.5–1 μM (41), and results show that Cu supplementation with 1 μM is sufficient to cause cancer cell killing by DSF [Supp Fig S1B].
When normal human bronchial epithelial cells (HBECs) and H292 NSCLC cells were treated with 50–100 nM DSF (doses lower than 1.3 μM plasma levels seen with 250 mg oral dosing (11)), DSF/Cu caused selective toxicity to H292s [Fig 1A]. Similar results were obtained with comparisons of human SUM159 breast cancer cells and normal human mammary epithelial cells (HMECs) [Supp Fig S1A].
Figure 1. DSF in combination with Cu induces selective clonogenic cell killing in tumor vs. normal cells and in hypoxic cancer cells.
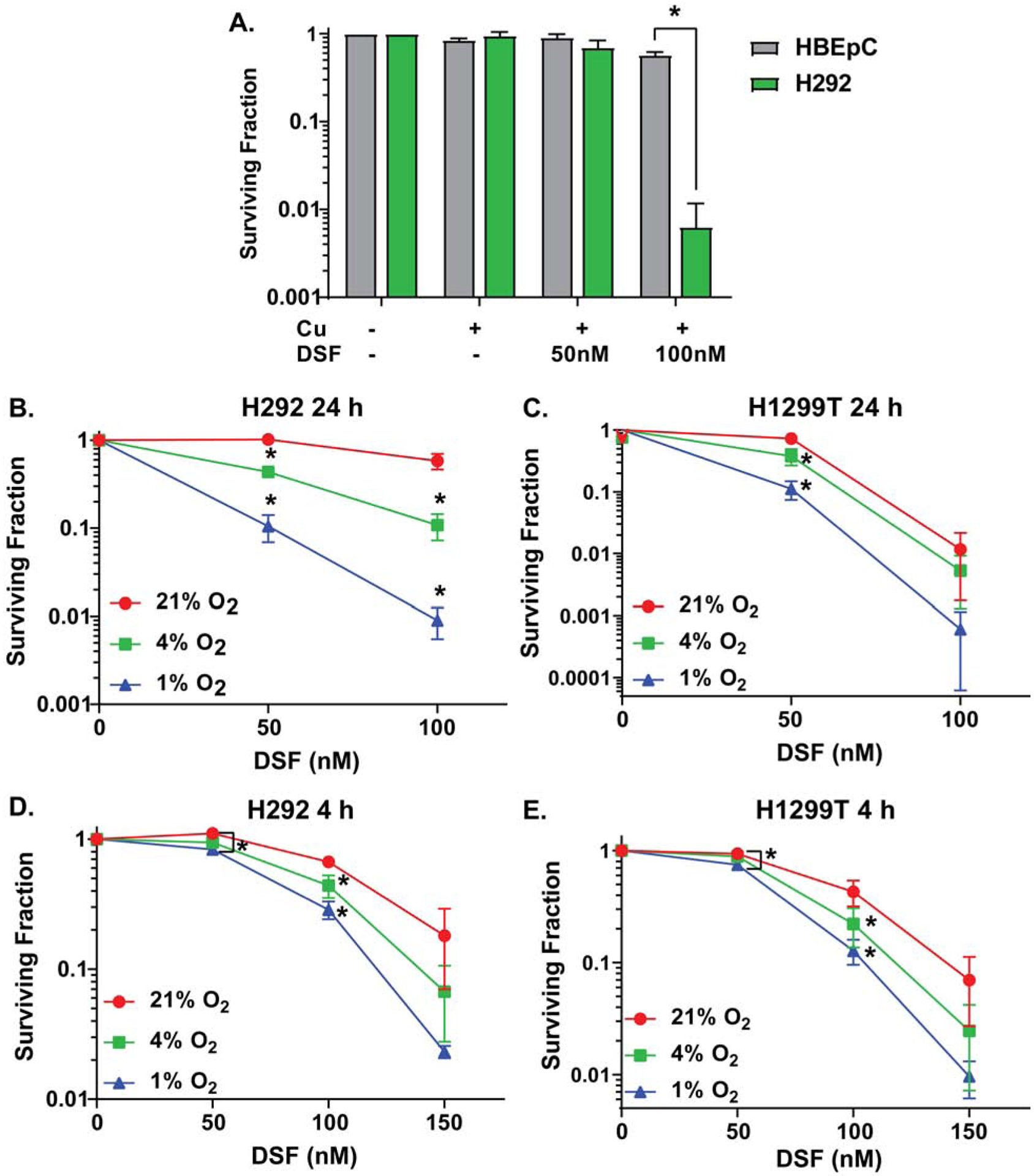
(A) Clonogenic survival of H292 and HBEC cells treated with physiological levels of drugs (15 μM CuSO4 and 50–100 nM DSF for 4 h) at a physiologically relevant O2 tension (4% O2). (B–E) Clonogenic survival of H292 (B,D) and H1299T (C,E) cells treated with 50–150 nM DSF and 15 μM CuSO4 (on all plates) for 24 and 4 h at 21%, 4%, and 1% O2. n = 3, at least 3 cloning dishes per treatment, error bars ± SEM, * is p < 0.05 compared to 21% O2 or as indicated.
Since Cu was critical to DSF-induced cancer cell killing and Cu can react with O2, it was hypothesized that O2 tension could mediate DSF/Cu cancer cell toxicity. DSF/Cu was found to be significantly more toxic to H292 and H1299T NSCLC cells at lower O2 tensions with toxicity at 1% > 4% > 21% O2 [Fig 1B–E].
DSF acts as a Cu ionophore and toxicity is dependent on redox cycling of Cu
DSF is hypothesized to act as a copper ionophore in cancer cells, delivering increased Cu concentrations into the cell (9, 42). Fig 2AB shows that DSF/Cu increased total cellular Cu in both H292 cancer cells and HBEC normal cells, but levels were significantly higher in H292s vs. HBECs and in cancer cells treated at 1% O2 vs. 21%. Experiments with 64Cu showed that the influx rate of Cu was greater in cancer cells treated at 1%, relative to 21% O2, leading to a higher retention of Cu in hypoxia after 4 h [Fig 2C].
Figure 2. DSF acts as a Cu ionophore and DSF toxicity is dependent on Cu.
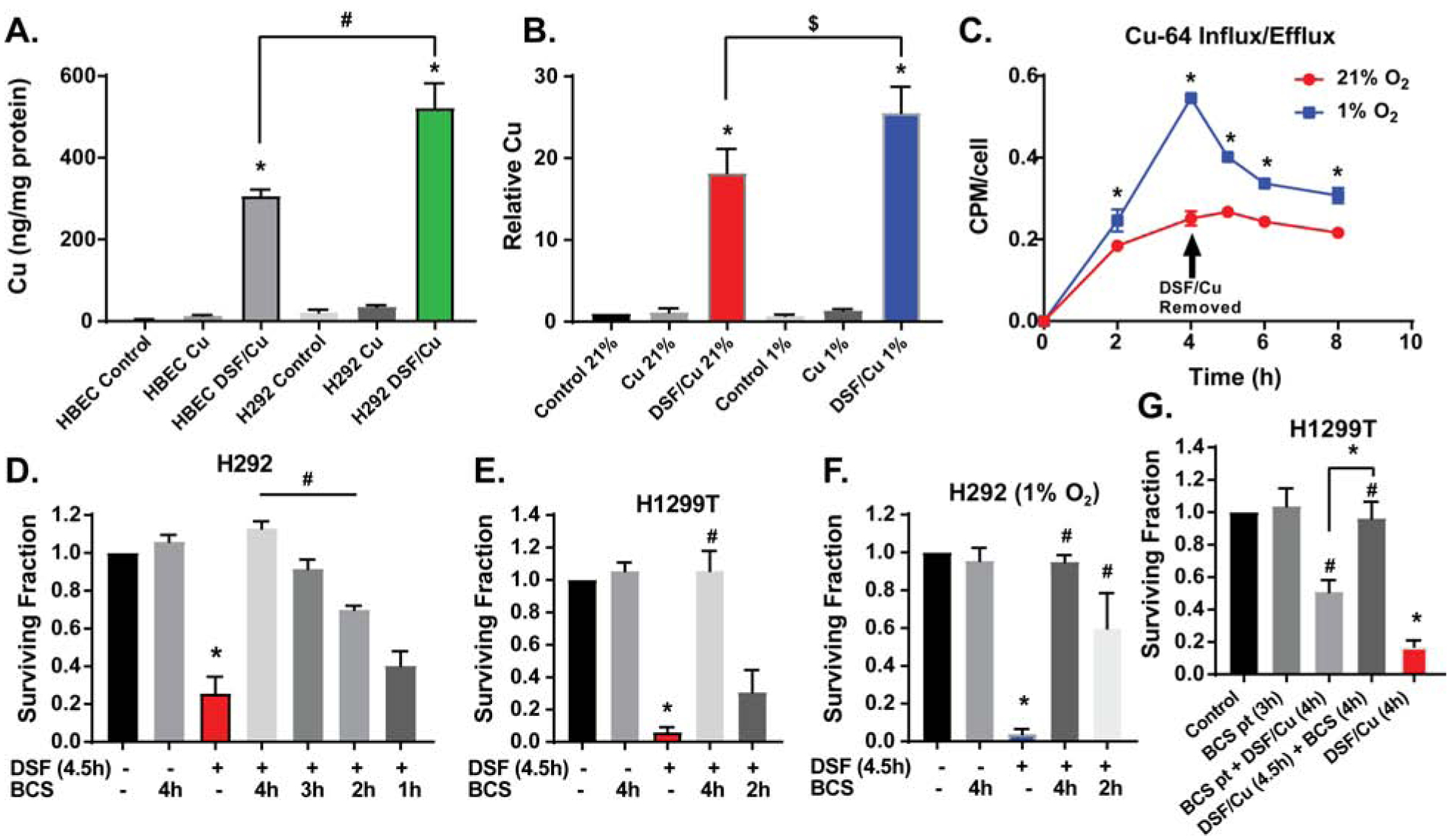
(A,B) Intracellular Cu was measured by ICP-MS following treatment of (A) HBEC and H292 cells with 100 nM DSF and 15 μM CuSO4 for 4 h at 4% O2 as well as (B) H292 cells with 150 nM DSF and 15 μM CuSO4 for 4 h at 21% and 1% O2 ($ indicates p<0.05 by paired t-test). (C) Cu influx and efflux were measured by treating H292 cells with 150 nM DSF, 15 μM CuCl2, and 37 kBq Cu64Cl2 for 4 h at 21% and 1% O2. Clonogenic survival assays were performed on (D) H292 cells and (E) H1299T cells treated with 150 nM DSF and 15 μM CuSO4 (on all plates) for 4.5 h at 21% O2 with the addition of 100 μM BCS during the last 1–4 h of treatment; (F) H292 cells treated with 150 nM DSF and 15 μM CuSO4 (on all plates) at 1% O2 for 4.5 h with the addition of 100 μM BCS during the last 2–4 h of treatment; as well as (G) H1299T cells treated with BCS for a 3 h pre-treatment, washed with PBS, then treated with 150 nM DSF and 15 μM CuSO4 (on all plates) for 4 h as well as treated with DSF/Cu for 4.5 h with the addition of 100 μM BCS for 4 h. n = 3, at least 3 cloning dishes per treatment, error bars ± SEM, * is p < 0.05 compared to control or as indicated, # is p < 0.05 compared to DSF/Cu or as indicated.
When the Cu chelator, bathocuproinedisulfonic acid (BCS), was added to cells 30 min after DSF/Cu treatment and present during 4 h of exposure, cancer cell toxicity was completely inhibited [Fig 2D,E]. However, when BCS was only present during the last 3, 2, or 1 h of treatment, toxicity was only partially inhibited. This effect was recapitulated when H292 cells were treated at 1% O2 [Fig 2F]. Consistent with BCS affecting Cu reactions inside cancer cells, 3h pretreatment (pt) with BCS [Fig 2G] followed by BCS removal also partially but significantly inhibited DSF toxicity. In contrast, when BCS was added 30 min after DSF/Cu, toxicity was completely inhibited, indicating that BCS was acting both outside and inside the cells to block Cu toxicity [Fig 2G].
BCS primarily binds Cu(I) (43), so to further evaluate the role of Cu redox cycling, the production of Cu(I) was observed in a cell free system using neocuproine (NC), a Cu(I) specific chelator that absorbs at 448 nm when bound to Cu(I) (24, 25). Results showed an accumulation of the NC-Cu(I) complex over time, suggesting that DSF reduced Cu(II) to Cu(I) [Supp Fig S2K]. The production of Cu(I) was also dependent on the dose of DSF. At the end of the reaction, spectra of the NC-Cu(I) complex in the presence and absence of DSF and Cu confirmed that the combination of DSF, NC, and Cu was necessary for the formation of the NC-Cu(I) complex [Supp Fig S2L]. These data, together with the survival data using BCS, support the hypothesis that DSF affects both the influx and redox cycling of Cu, which initiates cancer cell toxicity.
DSF/Cu toxicity is dependent on peroxide and superoxide and induces lipid oxidation and mitochondrial stress
Cu(I) can react with O2 to produce O2•− and H2O2 (6, 13). To determine if DSF affected intracellular [H2O2] a specific assay for H2O2 using 3-AT was used (26, 44). Results showed that DSF significantly increased [H2O2] in cancer cells [Fig 3A], and adenovirus-mediated overexpression of catalase in H1299T cells led to a partial but significant inhibition of DSF/Cu toxicity [Fig 3B, Supp Fig S2A]. Furthermore, when auranofin (AU) and buthionine sulfoximine (BSO) were used to inhibit the thioredoxin and glutathione-dependent peroxide-scavenging pathways in H292 cells (5, 45), the toxicity of these agents was significantly enhanced [Fig 3C, Supp Fig S2F].
Figure 3. DSF/Cu toxicity is dependent on peroxide and superoxide and induces lipid oxidation and mitochondrial oxidative stress.
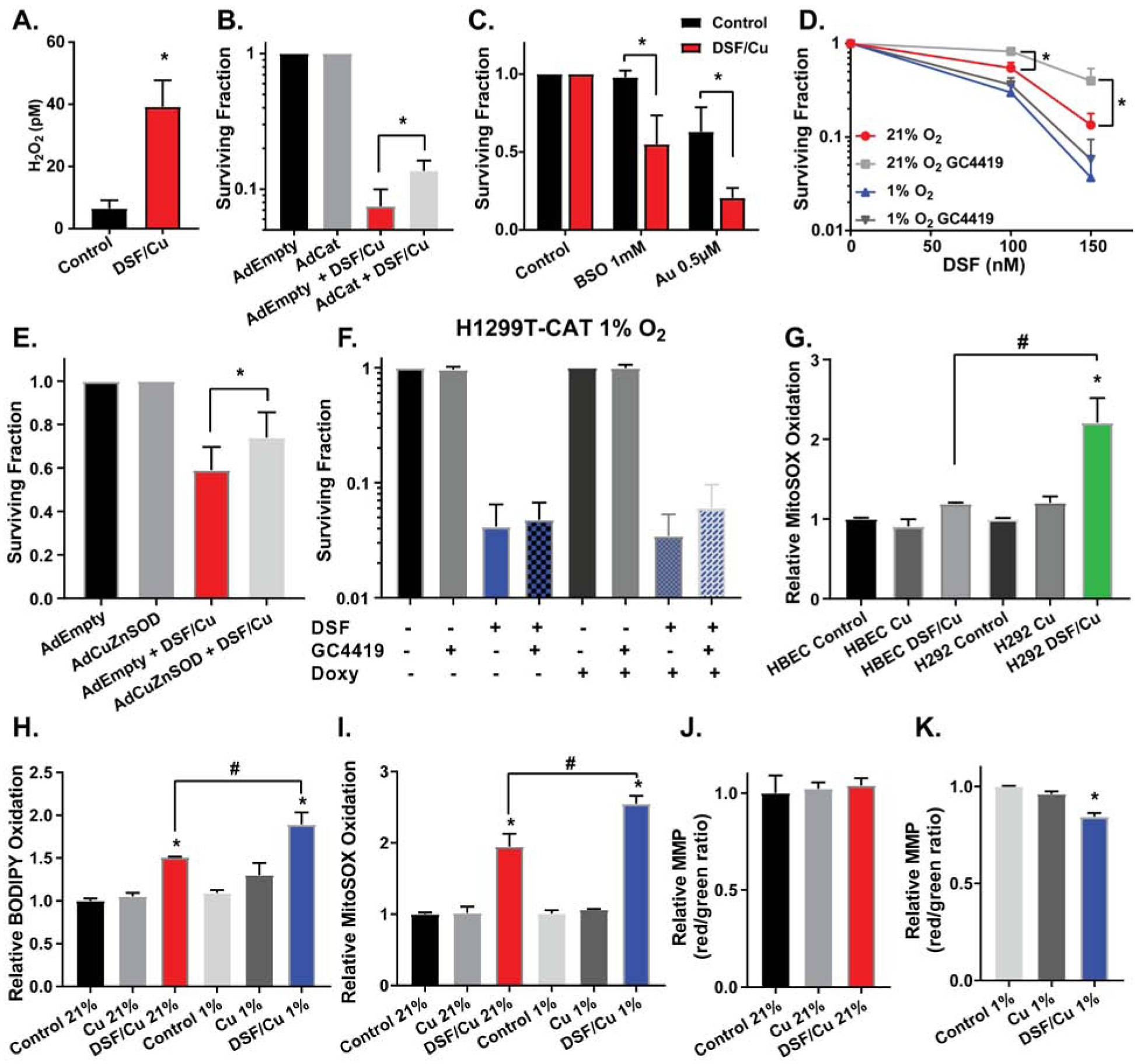
(A) 3-Aminotriazole (3-AT) mediated inhibition of catalase assay was used to determine intracellular H2O2. H1299T cells transfected with Ad-catalase were pre-treated with 15 μM CuSO4 and 100 nM DSF for 1 h and time points were collected over an additional 1 h. Clonogenic survival assays were performed on (B) H1299T cells transfected with ad-empty or ad-catalase and treated with 150 nM DSF and 15 μM CuSO4 (on all plates) for 2 h at 21% O2 (* is p < 0.05 by paired t-test), (C) H292 cells treated with 150 nM DSF, 15 μM CuSO4 (on all plates), 1 mM BSO, and 0.5 μM auranofin for 4 h at 21% O2, (D) H1299T cells transfected with ad-empty or ad-CuZnSOD and treated with 100 nM DSF and 15 μM CuSO4 (on all plates) for 2 h at 21% O2 (* is p < 0.05 by paired t-test), (E) H1299T cells treated with 100–150 nM DSF, 15 μM CuSO4 (on all plates), and 10 μM GC4419, an SOD mimetic, for 4 h at 21% and 1% O2, and (F) H1299T-CAT cells pre-treated with 0.5 μg/ml doxycycline for 48 h and then treated with 150 nM DSF, 15 μM CuSO4 (on all plates), and 10 μM GC4419 for 4 h at 1% O2. (G) MitoSOX oxidation was assessed by flow cytometry after HBEC and H292 cells were treated with 100 nM DSF and 15 μM CuSO4 for 4 h and then incubated with fresh media for an additional 24 h, all at 4% O2 (n = 4). (H) C-11 BODIPY oxidation was assessed by flow cytometry after H1299T cells were treated with 150 nM DSF and 15 μM CuSO4 for 4h at 21% and 1% O2 and then incubated with fresh media for an additional 24 h at 21% O2 (n = 4). (I) MitoSOX oxidation was assessed by flow cytometry on H1299T cells treated with 150 nM DSF and 15 μM CuSO4 for 4h at 21% and 1% O2 and then incubated with fresh media for an additional 24 h at 21% O2 (n = 4). (J,K) Mitochondrial membrane potential (MMP) was assessed with JC-1 and flow cytometry after H1299T cells were treated with 150 nM DSF and 15 μM CuSO4 for 4h at 21% and 1% O2 and then incubated with fresh media for an additional 24 h at 21% O2 (n = 4). n = 3 unless indicated otherwise, at least 3 cloning dishes per treatment, error bars ± SEM, * is p < 0.05 compared to control or as indicated, # is p < 0.05 compared to DSF/Cu 21% or as indicated.
Consistent with a role of O2•− in DSF toxicity, treatment with the SOD mimic (GC4419) and adenovirus-mediated overexpression of CuZnSOD were shown to partially inhibit DSF/Cu toxicity in H1299T [Fig 3D,E] and H292 cells [Supp Fig S2G], with more pronounced effects at 21% O2 than 1% O2. Finally, when H1299T-CAT cells were induced to overexpress catalase in the presence of GC4419 in 1% O2, there was no significant protection from DSF/Cu toxicity, supporting the hypothesis that O2•− and H2O2 play a limited role in DSF toxicity at 1% O2 [Fig 3F].
As a redox active metal ion, Cu(I) can interact with numerous critical cellular components, including membrane lipids and Fe-S clusters of cytoplasmic and mitochondrial proteins [Fig 4A] (46–49). Studies have shown that Cu(I) is more thermodynamically favorable than Cu(II) for initiating lipid peroxidation chain reactions, leading to the formation of lipid hydroperoxides (47, 50). When H1299T cells were treated with DSF/Cu for 4 h, then incubated in fresh media for 24 h, flow cytometry analysis of lipid peroxidation with C11-BODIPY showed a significant induction of lipid oxidation occurred with DSF/Cu at 21% O2 that was further increased at 1% O2 [Fig 3H]. A scavenger of membrane lipid hydroperoxides, glutathione peroxidase 4, was overexpressed in H1299T cells, partially inhibiting the increased toxicity of DSF/Cu noted in hypoxia [Supp Fig S2D,E].
Figure 4. DSF/Cu toxicity is dependent on intracellular copper retention.
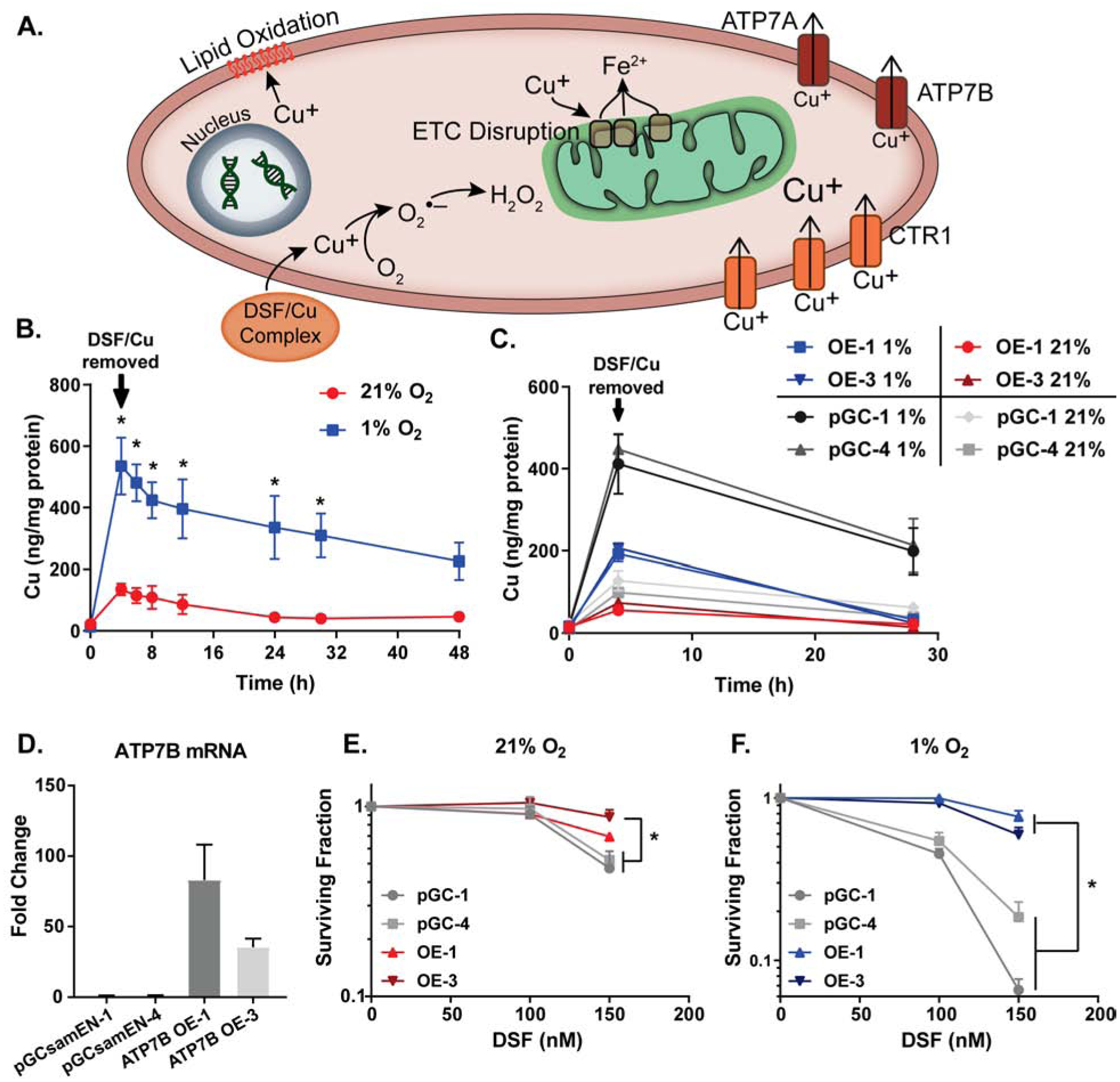
(A) Proposed mechanism of DSF/Cu toxicity. Cu is delivered to cancer cells and interacts with cellular components to cause damage and death. (B,C) Intracellular Cu was measured by ICP-MS following treatment of (B) H292 cells with 150 nM DSF and 15 μM CuSO4 for 4 h at 21% and 1% O2 followed by replacement of media and continued incubation for 48 h at 21% O2 and (C) H292-pGCsamEN-1/4 (empty vector) cells and H292-ATP7B OE-1/3 (overexpression) cells treated with 150 nM DSF and 15 μM CuSO4 for 4 h at 21% and 1% O2 followed by replacement of media and continued incubation for 24 h at 21% O2. (D) mRNA levels of ATP7B were analyzed by qPCR in pGCsamEN empty vector clones 1 and 4 as well as ATP7B overexpression clones 1 and 3 of H292 cells. (E,F) Clonogenic survival assays were performed on H292-pGCsamEN-1/4 cells and H292-ATP7B OE-1/3 cells treated with 100–150 nM DSF and 15 μM CuSO4 (on all plates) for 4 h at 21% and 1% O2. n = 3, at least 3 cloning dishes per treatment, error bars ± SEM, * is p < 0.05 compared to 21% O2 or as indicated.
Previous studies have suggested that Cu(I), but not Cu(II), is capable of disrupting Fe-S clusters, causing release of labile Fe and decreased protein activity (48, 49). Since previous studies have also shown that H2O2 and O2•− increase labile iron pools, resulting in decreased activity of Fe-S containing electron transport chain enzymes (16), mitochondrial function was assessed following DSF/Cu treatment. DSF/Cu at 1% but not 21% O2, disrupted mitochondrial membrane potential (MMP) in both H1299T and H292 cells as assessed by JC-1 staining [Fig 3J,K, Supp Fig S2I,J]. Decreased MMP in cancer cells with DSF treatment was also accompanied by increased MitoSOX oxidation, with higher levels at 1% O2 vs. 21% O2 in both H1299T and H292 cells [Fig 3I, Supp Fig S2H]. Furthermore, DSF-induced MitoSOX oxidation was not seen in normal HBECs when compared to H292s at 4% O2 [Fig 3G], suggesting that DSF-induced mitochondrial oxidative stress is greater in cancer vs. normal cells.
DSF/Cu toxicity is dependent on intracellular Cu retention
While oxidative stress mechanisms appear to play a role in the differential DSF toxicity between normoxic and hypoxic cells, the main driver of these oxidative mechanisms appears to be retention of Cu(I) [Fig 4A]. In support of this hypothesis, H292 cells treated with DSF/Cu for 4 h demonstrated highly significant increases (10-fold) in total Cu levels at 21% O2 that were further increased 4-fold at 1% O2. Furthermore, Cu levels remained significantly elevated in the cells treated at 1% O2 even 26 h after DSF/Cu was removed, while intracellular Cu returned to normal levels at 21% O2 at that time point [Fig 4B]. Most importantly, overexpression of the Cu export protein, ATP7B (mutations in this protein are responsible for Wilson disease (36)), showed decreased accumulation of Cu, a rapid return to normal Cu levels following DSF treatment, as well as significant resistance to clonogenic cell killing from DSF/Cu at both 21% and 1% O2, compared with empty vector controls [Fig 4C–F, Supp Fig S1C]. Furthermore, protection from DSF toxicity with ATP7B overexpression was more pronounced in hypoxia, relative to normoxia [Fig 4E,F].
DSF/Cu enhances radiation and chemotherapy responses in cancer but not normal cells
H1299T and H292 cells demonstrated significant radio-sensitization when treated for 4h with DSF at 21% O2 prior to irradiation [Fig 5AB]. Furthermore, DSF/Cu inhibited radioresistance in H292 cells treated with DSF/Cu for 4 h prior to irradiation at either 21% or 1% O2 [Fig 5C]. Similar results in hypoxia were observed in H1299T cells, especially at higher doses of radiation [Supp Fig S3I]. When H292 cells were treated with DSF/Cu and cisplatin for 4 h at 21% O2, significant chemo-sensitization was noted [Fig 5D] with similar trends observed in H1299T cells that did not reach statistical significance (p=0.24) [Supp Fig S3G]. DSF/Cu and cisplatin also induced chemo-sensitization in hypoxia when H1299T cells were treated for 4h at 21% vs. 1% O2 [Fig 5E], with H292s also trending towards significance (p=0.16) [Supp Fig S3H].
Figure 5. DSF/Cu enhances radiation and chemotherapy toxicity in tumor cells but not normal cells.
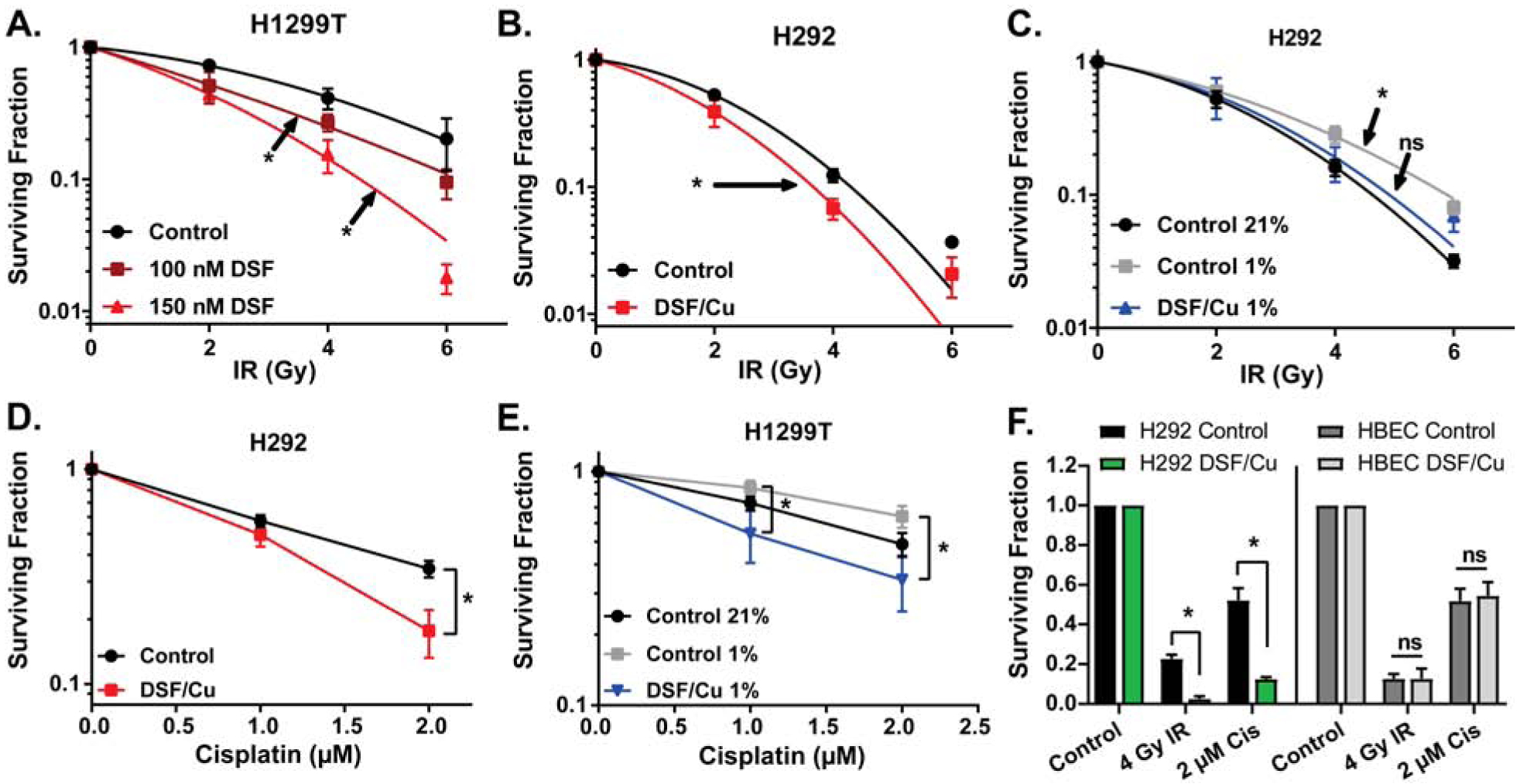
Clonogenic survival assays were performed on (A) H1299T cells treated with 100–150 nM DSF and 15 μM CuSO4 (on all plates) for 4 h at 21% O2 prior to irradiation with 0–6 Gy; (B) H292 cells treated with 150 nM DSF and 15 μM CuSO4 (on all plates) for 4 h at 21% O2 prior to irradiation with 0–6 Gy; (C) H292 cells treated with 100 nM DSF and 15 μM CuSO4 (on all plates) for 4 h prior to irradiation with 0–6 Gy at 21% O2 and 1 % O2 (n = 4–5); (D) H292 cells treated with 150 nM DSF, 15 μM CuSO4 (on all plates), and 1–2 μM cisplatin for 4 h at 21% O2 (n = 4); (E) H1299T cells treated with 150 nM DSF, 15 μM CuSO4 (on all plates), and 1–2 μM cisplatin for 4 h at 21% O2 and 1% O2 (n = 5); (F) H292 and HBEC cells treated with 100 nM DSF, 15 μM CuSO4 (on all plates), and 2 μM cisplatin for 4 h at 4% O2 prior to irradiation with 4 Gy. n = 3 unless otherwise noted, at least 3 cloning dishes per treatment, error bars ± SEM, * indicates p < 0.05 compared to control or as indicated.
Studies have shown that radiation can result in the selection of radio-resistant cancer stem cell populations (51). When H292 cells were treated with DSF/Cu for 4 h prior to irradiation at 21% O2 and then treated with DSF/Cu for an additional 24 h, DSF/Cu inhibited the increase in the %ALDH+ cells as a marker for cancer stem cells [Supp Fig S3A]. This decrease in the %ALDH+ cells was accompanied by significant decreases in clonogenic survival [Supp Fig S3D]. When H292 cells were live sorted into ALDH+ and ALDH− populations, plated, and allowed to attach for 2–3 days, before treatment with DSF/Cu for 4 h followed by irradiation at 21% O2, both ALDH+ and ALDH− cells were sensitive to DSF/Cu-induced clonogenic cell killing, and DSF/Cu-induced radio-sensitization was noted in ALDH+ cells as well as trending towards significant in ALDH− cells (p=0.27) [Supp Fig S3BC]. These results demonstrate that in contrast to radiation alone, ALDH+ are not relatively resistant to radiation with DSF/Cu.
When H292 and HBEC cells were treated in the same experiment with combinations of radiation and chemotherapy, DSF/Cu was able to significantly sensitize cancer cells to radiation and cisplatin but caused no sensitization in normal HBEC cells [Fig 5F]. Furthermore, the toxicities of DSF/Cu and radiation or cisplatin were clearly additive in the cancer cells but not in normal cells [Supp Fig S3E,F].
DSF with standard of care therapies inhibits tumor cell growth in vivo
H292 cells were injected into female athymic nude mice, and mice were randomized into control, DSF (100 mg/kg daily), radiation (3 × 6 Gy) + carboplatin (2 × 15 mg/kg; R/C), and R/C + DSF groups. Results of in vivo studies showed that DSF treatment alone was unable to significantly alter tumor volume or survival [Fig 6A,B], while DSF + R/C resulted in a significant reduction in tumor volume, compared to R/C alone [Fig 6A]. This reduction in tumor volume was associated with an increase in survival as well, which trended (p=0.057) towards significance [Fig 6B]. Mouse weights demonstrated that R/C treatment alone led to substantial weight loss, but the addition of DSF significantly inhibited this weight loss [Fig 6C]. When tumors were collected at the time tumor size reached criteria, DSF treatment was shown to cause a significant increase in Cu levels [Fig 6D].
Figure 6. DSF with standard of care therapies inhibits tumor cell growth in vivo.
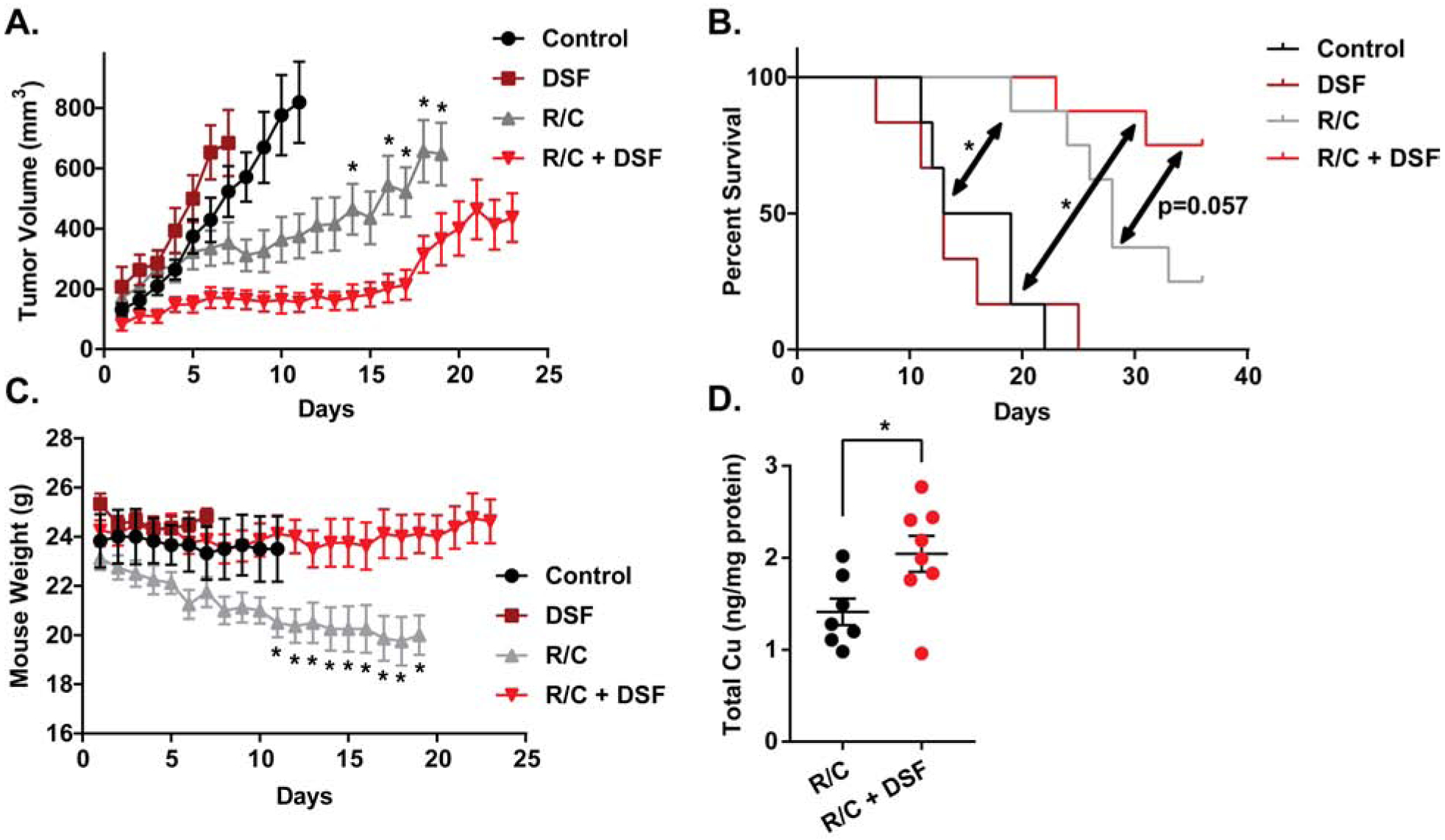
H292 xenografts were treated with radiation and chemotherapy (R/C, 15 mg/kg carboplatin on days 1 and 8, 6 Gy IR on days 1, 3, and 5) and 100 mg/kg DSF daily. (A) Tumor volume, (B) mouse weights, and (C) survival were measured. (D) Total Cu was measured by ICP-MS on H292 xenografts treated with R/C and R/C+DSF. Error bars ± SEM, * indicates p < 0.05 compared to R/C+DSF or as noted.
Discussion
The current study shows that DSF in combination with physiologically relevant concentrations of Cu causes selective toxicity to lung cancer cells through a Cu-mediated mechanism and that O2•− and H2O2 as well as Cu retention play important roles in this toxicity. The exact roles of each of these reactive substances vary significantly based on oxygen tension. When oxygen is limited, the effects of O2•− and H2O2 are limited, and the damage caused by Cu(I) directly appears more substantial. Cu(I) interacts with lipids to cause increased lipid oxidation at hypoxia. It also interacts with Fe-S clusters to cause mitochondrial dysfunction and disruption of electron transport, leading to decreased mitochondrial membrane potential and increased mitochondrial O2•− production in low O2. The differential effects of oxidative damage at varying oxygen tensions are also important factors in the toxicities of DSF/Cu in tumor and normal cells. Mitochondrial dysfunction and Cu uptake and retention appear to be less pronounced in normal vs. cancer cells, suggesting that reduced intracellular Cu levels ultimately lead to reduced redox reactions of Cu and lower levels of oxidative stress in normal cells. Thus, our data supports the hypothesis that differences in the redox biochemistry of Cu in cancer vs. normal cells significantly contributes to the differential effects of DSF in these cell populations.
Similar to differences in tumor and normal cells, the ultimate cause of the differential levels of oxidative damage between cells at hypoxia and normoxia appears to be driven by the increase uptake and retention of intracellular Cu and the downstream redox reactions that occur from this Cu. In tumor cells and hypoxic cells, the levels of Cu were higher after only 4 h of DSF/Cu treatment. Additionally, Cu levels were higher in mouse xenografts treated with DSF. Since DSF/Cu toxicity is dependent on Cu and its reactions with oxygen, lipids, and Fe-S clusters, increased intracellular Cu levels in cancer cells appear to cause greater redox cycling of the metal and greater damage to the cell. While the cause of the increased Cu levels with DSF treatment is not fully understood, 64Cu uptake data as well as ICP-MS data support the conclusion that hypoxia can increase the influx and decrease the efflux of Cu, causing a greater accumulation of Cu. These alterations may be due to differences in the levels and functions of Cu trafficking proteins, such as Ctr1 (import), Atox1 (intracellular trafficking), and ATP7A/B (export). The current study shows that overexpressing ATP7B leads to both a decreased accumulation and more rapid efflux of Cu, leading to diminished effects of DSF on cancer cell clonogenic cell killing. These results support a critical role for Cu metabolism in the anti-cancer effects of DSF in NSCLC.
Given the importance of Cu and Cu metabolism on DSF cell killing in this study, it is hypothesized that reactions of Cu may also contribute to the mechanisms of DSF cell killing described in other literature. DSF/Cu is considered an inhibitor of proteasomes, and one study has actually shown that Cu itself is capable of inhibiting proteasomes, with Cu(I) being more effective than Cu(II) (52). Other studies have shown that the antioxidant compound n-acetylcysteine (NAC) can inhibit the DSF/Cu-induced activation of cJun, p38, and Nrf2, suggesting that oxidative stress is responsible for downstream signaling effects of DSF/Cu (10, 53). While studies have suggested multiple other mechanisms of DSF cell killing, results from this study and those referenced above highlight the overall impact that redox reactions of Cu play on the differential toxicity between normal cells and cancer cells at normoxia as well as hypoxia.
Finally, in our studies, we were able to show that DSF enhances radiation and chemotherapy toxicity in cancer cells at both normoxia and hypoxia while not causing additional toxicity to normal cells. Furthermore, DSF/Cu was able to inhibit the percent increase of ALDH+ cancer stem cells following radiation, while retaining the ability to clonogenically inactive both ALDH+ and ALDH− cells. Since hypoxic cells and cancer stem cells are both thought to contribute to treatment failure, DSF may provide a significant non-toxic adjuvant in cancer therapy that could allow for increased survival and decreased progression of disease. To support this hypothesis, in vivo studies showed enhanced efficacy when DSF was combined with radio-chemo-therapy in a NSCLC xenograft model (H292 cells). Furthermore, DSF also appeared to protect mice from weight loss that was caused by treatment with radio-chemo-therapy. Given that DSF is already FDA-approved, these studies provide substantial evidence to suggest that DSF could be advanced to clinical trials in the setting of NSCLC.
Supplementary Material
Highlights.
Disulfiram causes selective toxicity in cancer cells vs. normal cells.
Disulfiram toxicity is enhanced at hypoxia.
Disulfiram causes selective radio-chemo-sensitization in cancer cells and hypoxia.
O2•−, H2O2, lipid oxidation, and Cu retention/efflux (via ATP7B) govern toxicity.
Acknowledgements
The authors thank the Radiation and Free Radical Research Core in the Holden Comprehensive Cancer Center as well as Amanda Kalen and Dr. Michael McCormick for technical assistance. The authors thank Gareth Smith for his assistance with developing high-quality figure graphics. The authors thank the Flow Cytometry Core in the Holden Comprehensive Cancer Center for assistance with MitoSOX, C11-BODIPY, and JC-1 assays. This work was supported by NIH F30CA213817, R01CA182804, T32GM007337, T32CA078586, P30CA086862, P01CA217797, P50CA174521 as well as the Iowa Center for Research by Undergraduates and the Biomedical Scholars Summer Undergraduate Research Program. We also would like to thank Dr. Dennis Riley from Galera Therapeutics, Inc., for supplying the GC4419 SOD mimetic.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts: Douglas R. Spitz PhD and Bryan G. Allen MD PhD have a sponsored research agreement with Galera Therapeutics, Inc. at the University of Iowa and are unpaid scientific consultants for Galera Therapeutics.
References
- 1.Cancer Stat Facts: Lung and Bronchus Cancer NCI: Surveillance, Epidemiology, and End Result Program [cited 2020 Jan 22]. Available from: https://seer.cancer.gov/statfacts/html/lungb.html.
- 2.Walsh JC, Lebedev A, Aten E, Madsen K, Marciano L, Kolb HC. The Clinical Importance of Assessing Tumor Hypoxia: Relationship of Tumor Hypoxia to Prognosis and Therapeutic Opportunities. Antioxidants & Redox Signaling. 2014;21(10):1516–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duan L, Shen H, Zhao G, Yang R, Cai X, Zhang L, Jin C, Huang Y. Inhibitory effect of Disulfiram/copper complex on non-small cell lung cancer cells. Biochemical and Biophysical Research Communications. 2014;446(4):1010–6. [DOI] [PubMed] [Google Scholar]
- 4.Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a Clinically Used Anti-Alcoholism Drug and Copper-Binding Agent, Induces Apoptotic Cell Death in Breast Cancer Cultures and Xenografts via Inhibition of the Proteasome Activity. Cancer Research. 2006;66(21):10425–33. [DOI] [PubMed] [Google Scholar]
- 5.Papaioannou M, Mylonas I, Kast RE, Brüning A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin-mediated thioredoxin inhibition. Oncoscience. 2014;1(1):21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupte A, Mumper RJ. An investigation into copper catalyzed D-penicillamine oxidation and subsequent hydrogen peroxide generation. Journal of inorganic biochemistry. 2007;101(4):594–602. [DOI] [PubMed] [Google Scholar]
- 7.Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, Ozdian T, Bartkova J, Turi Z, Moudry P, Kraus M, Michalova M, Vaclavkova J, Dzubak P, Vrobel I, Pouckova P, Sedlacek J, Miklovicova A, Kutt A, Li J, Mattova J, Driessen C, Dou QP, Olsen J, Hajduch M, Cvek B, Deshaies RJ, Bartek J. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han D, Wu G, Chang C, Zhu F, Xiao Y, Li Q, Zhang T, Zhang L. Disulfiram inhibits TGF-beta-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-kappaB/Snail pathway. Oncotarget. 2015;6(38):40907–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allensworth JL, Evans MK, Bertucci F, Aldrich AJ, Festa RA, Finetti P, Ueno NT, Safi R, McDonnell DP, Thiele DJ, Van Laere S, Devi GR. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Molecular oncology. 2015;9(6):1155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zha J, Chen F, Dong H, Shi P, Yao Y, Zhang Y, Li R, Wang S, Li P, Wang W, Xu B. Disulfiram targeting lymphoid malignant cell lines via ROS-JNK activation as well as Nrf2 and NF-kB pathway inhibition. Journal of Translational Medicine. 2014;12:163-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faiman MD, Jensen JC, Lacoursiere RB. Elimination kinetics of disulfiram in alcoholics after single and repeated doses. Clinical Pharmacology & Therapeutics. 1984;36(4):520–6. [DOI] [PubMed] [Google Scholar]
- 12.Huang J, Campian JL, Gujar AD, Tran DD, Lockhart AC, DeWees TA, Tsien CI, Kim AH. A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy. Journal of neuro-oncology. 2016;128(2):259–66. [DOI] [PubMed] [Google Scholar]
- 13.Lewis DJ, Deshmukh P, Tedstone AA, Tuna F, O’Brien P. On the interaction of copper(II) with disulfiram. Chemical communications (Cambridge, England). 2014;50(87):13334–7. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Ge Y, Kang YJ. Featured Article: Effect of copper on nuclear translocation of copper chaperone for superoxide dismutase-1. Experimental Biology and Medicine. 2016;241(14):1483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du J, Cieslak JA, Welsh JL, Sibenaller ZA, Allen BG, Wagner BA, Kalen AL, Doskey CM, Strother RK, Button AM, Mott SL, Smith B, Tsai S, Mezhir J, Goswami PC, Spitz DR, Buettner GR, Cullen JJ. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer research. 2015;75(16):3314–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, Sandhu S, Carlisle TL, Smith MC, Abu Hejleh T, Berg DJ, Zhang J, Keech J, Parekh KR, Bhatia S, Monga V, Bodeker KL, Ahmann L, Vollstedt S, Brown H, Shanahan Kauffman EP, Schall ME, Hohl RJ, Clamon GH, Greenlee JD, Howard MA, Shultz MK, Smith BJ, Riley DP, Domann FE, Cullen JJ, Buettner GR, Buatti JM, Spitz DR, Allen BG. O2- and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer cell. 2017;31(4):487–500.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Li W, Patel SS, Cong J, Zhang N, Sabbatino F, Liu X, Qi Y, Huang P, Lee H, Taghian A, Li J-J, DeLeo AB, Ferrone S, Epperly MW, Ferrone CR, Ly A, Brachtel EF, Wang X. Blocking the formation of radiation–induced breast cancer stem cells. Oncotarget. 2014;5(11):3743–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Wang L, Cui W, Yuan X, Lin L, Cao Q, Wang N, Li Y, Guo W, Zhang X, Wu C, Yang J. Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells. Oncotarget. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JY, Cho Y, Oh E, Lee N, An H, Sung D, Cho T-M, Seo JH. Disulfiram targets cancer stem-like properties and the HER2/Akt signaling pathway in HER2-positive breast cancer. Cancer Letters. 2016;379(1):39–48. [DOI] [PubMed] [Google Scholar]
- 20.Nechushtan H, Hamamreh Y, Nidal S, Gotfried M, Baron A, Shalev YI, Nisman B, Peretz T, Peylan-Ramu N. A Phase IIb Trial Assessing the Addition of Disulfiram to Chemotherapy for the Treatment of Metastatic Non-Small Cell Lung Cancer. The Oncologist. 2015;20(4):366–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lun X, Wells JC, Grinshtein N, King JC, Hao X, Dang NH, Wang X, Aman A, Uehling D, Datti A, Wrana JL, Easaw JC, Luchman A, Weiss S, Cairncross JG, Kaplan DR, Robbins SM, Senger DL. Disulfiram when Combined with Copper Enhances the Therapeutic Effects of Temozolomide for the Treatment of Glioblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(15):3860–75. [DOI] [PubMed] [Google Scholar]
- 22.Heer CD, Davis AB, Riffe DB, Wagner BA, Falls KC, Allen BG, Buettner GR, Beardsley RA, Riley DP, Spitz DR. Superoxide Dismutase Mimetic GC4419 Enhances the Oxidation of Pharmacological Ascorbate and Its Anticancer Effects in an H2O2-Dependent Manner. Antioxidants (Basel, Switzerland). 2018;7(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The Journal of biological chemistry. 1951;193(1):265–75. [PubMed] [Google Scholar]
- 24.Tutem E, Apak R, Baykut F. Spectrophotometric determination of trace amounts of copper(I) and reducing agents with neocuproine in the presence of copper(II). Analyst. 1991;116(1):89–94. [Google Scholar]
- 25.Gouda AA, Amin AS. Copper(II)–neocuproine reagent for spectrophotometric determination of captopril in pure form and pharmaceutical formulations. Arabian Journal of Chemistry. 2010;3(3):159–65. [Google Scholar]
- 26.Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, Buettner GR, Venkataraman S, Mackey MA, Flanagan SW, Oberley LW, Spitz DR. Mitochondrial and H2O2 Mediate Glucose Deprivation-induced Stress in Human Cancer Cells. Journal of Biological Chemistry. 2005;280(6):4254–63. [DOI] [PubMed] [Google Scholar]
- 27.Sciegienka SJ, Solst SR, Falls KC, Schoenfeld JD, Klinger AR, Ross NL, Rodman SN, Spitz DR, Fath MA. D-penicillamine combined with inhibitors of hydroperoxide metabolism enhances lung and breast cancer cell responses to radiation and carboplatin via H2O2-mediated oxidative stress. Free radical biology & medicine. 2017;108:354–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Royall JA, Gwin PD, Parks DA, Freeman BA. Responses of vascular endothelial oxidant metabolism to lipopolysaccharide and tumor necrosis factor-alpha. Archives of biochemistry and biophysics. 1992;294(2):686–94. [DOI] [PubMed] [Google Scholar]
- 29.Beers RF Jr., Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. The Journal of biological chemistry. 1952;195(1):133–40. [PubMed] [Google Scholar]
- 30.Aebi H Catalase in vitro. Methods in enzymology. 1984;105:121–6. [DOI] [PubMed] [Google Scholar]
- 31.Spitz DR, Elwell JH, Sun Y, Oberley LW, Oberley TD, Sullivan SJ, Roberts RJ. Oxygen toxicity in control and H2O2-resistant Chinese hamster fibroblast cell lines. Archives of biochemistry and biophysics. 1990;279(2):249–60. [DOI] [PubMed] [Google Scholar]
- 32.Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Analytical biochemistry. 1989;179(1):8–18. [DOI] [PubMed] [Google Scholar]
- 33.Maiorino M, Gregolin C, Ursini F. [47] Phospholipid hydroperoxide glutathione peroxidase. Methods in enzymology: Academic Press; 1990. p. 448–57. [DOI] [PubMed] [Google Scholar]
- 34.Wang HP, Qian SY, Schafer FQ, Domann FE, Oberley LW, Buettner GR. Phospholipid hydroperoxide glutathione peroxidase protects against singlet oxygen-induced cell damage of photodynamic therapy. Free radical biology & medicine. 2001;30(8):825–35. [DOI] [PubMed] [Google Scholar]
- 35.Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochimica et biophysica acta. 1985;839(1):62–70. [DOI] [PubMed] [Google Scholar]
- 36.Guttmann S, Bernick F, Naorniakowska M, Michgehl U, Groba SR, Socha P, Zibert A, Schmidt HH. Functional Characterization of Novel ATP7B Variants for Diagnosis of Wilson Disease. Frontiers in pediatrics. 2018;6:106-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandhok G, Horvath J, Aggarwal A, Bhatt M, Zibert A, Schmidt HH. Functional analysis and drug response to zinc and D-penicillamine in stable ATP7B mutant hepatic cell lines. World journal of gastroenterology. 2016;22(16):4109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zowczak M, Iskra M, Torliński L, Cofta S. Analysis of serum copper and zinc concentrations in cancer patients. Biological Trace Element Research.82(1):1–8. [DOI] [PubMed] [Google Scholar]
- 39.Glassman AB, Rydzewski RS, Bennett CE. Trace metal levels in commercially prepared tissue culture media. Tissue & cell. 1980;12(4):613–7. [DOI] [PubMed] [Google Scholar]
- 40.Golovine K, Uzzo RG, Makhov P, Crispen PL, Kunkle D, Kolenko VM. Depletion of intracellular zinc increases expression of tumorigenic cytokines VEGF, IL-6 and IL-8 in prostate cancer cells via NF-κB dependent pathway. The Prostate. 2008;68(13):1443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMillin GA, Travis JJ, Hunt JW. Direct Measurement of Free Copper in Serum or Plasma Ultrafiltrate. American Journal of Clinical Pathology. 2009;131(2):160–5. [DOI] [PubMed] [Google Scholar]
- 42.Tardito S, Bassanetti I, Bignardi C, Elviri L, Tegoni M, Mucchino C, Bussolati O, Franchi-Gazzola R, Marchiò L. Copper Binding Agents Acting as Copper Ionophores Lead to Caspase Inhibition and Paraptotic Cell Death in Human Cancer Cells. Journal of the American Chemical Society. 2011;133(16):6235–42. [DOI] [PubMed] [Google Scholar]
- 43.Chen D, Darabedian N, Li Z, Kai T, Jiang D, Zhou F. An improved Bathocuproine assay for accurate valence identification and quantification of copper bound by biomolecules. Analytical biochemistry. 2016;497:27–35. [DOI] [PubMed] [Google Scholar]
- 44.Wagner BA, Evig CB, Reszka KJ, Buettner GR, Burns CP. Doxorubicin increases intracellular hydrogen peroxide in PC3 prostate cancer cells. Archives of biochemistry and biophysics. 2005;440(2):181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hatori Y, Lutsenko S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants. 2016;5(3):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maiorino M, Zamburlini A, Roveri A, Ursini F. Copper-induced lipid peroxidation in liposomes, micelles, and LDL: Which is the role of vitamin E? Free Radical Biology and Medicine. 1995;18(1):67–74. [DOI] [PubMed] [Google Scholar]
- 48.Macomber L, Imlay JA. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(20):8344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsvetkov P, Detappe A, Cai K, Keys HR, Brune Z, Ying W, Thiru P, Reidy M, Kugener G, Rossen J, Kocak M, Kory N, Tsherniak A, Santagata S, Whitesell L, Ghobrial IM, Markley JL, Lindquist S, Golub TR. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nature chemical biology. 2019;15(7):681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burkitt MJ. A Critical Overview of the Chemistry of Copper-Dependent Low Density Lipoprotein Oxidation: Roles of Lipid Hydroperoxides, α-Tocopherol, Thiols, and Ceruloplasmin. Archives of biochemistry and biophysics. 2001;394(1):117–35. [DOI] [PubMed] [Google Scholar]
- 51.Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem cells (Dayton, Ohio). 2012;30(5):833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao YAN, Chen DI, Zhang XIA, Cui Q, Fan Y, Bi C, Dou QP. Molecular study on copper-mediated tumor proteasome inhibition and cell death. International journal of oncology. 2010;37(1):81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL, Wang W. Disulfiram modulated ROS-MAPK and NFkappaB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011;104(10):1564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.