

Abstract
The MAPK pathways are an enduring area of interest due to their essential roles in cell processes. Increased expression and activity can lead to a multitude of diseases, sparking research efforts in developing inhibitors against these kinases. Though great strides have been made in developing MEK1/2 inhibitors, there is a notable lack of chemical probes for MEK3–7, given their central role in stimuli response, cell growth, and development. This review summarizes the progress that has been made on developing small molecule probes for MEK3–7, the specific disease states in which they have been studied, and their potential to become novel therapeutics.
Keywords: Kinase, small molecule inhibitors, mitogen-activated protein kinases, drug discovery, structure-activity relationships
Graphical Abstract
1. Introduction
Mitogen-activated protein kinase (MAPK) pathways are highly conserved cascades that interface extracellular stimuli with the molecular machinery necessary for multiple cell processes.1–3 The various classes of MAP kinases are separated into three tiers, with MAP3K phosphorylating and activating downstream MAP2K (also known as MEK), which subsequently phosphorylates and activates MAPK (Figure 1A). Since the discovery of the first MAPK pathway, known canonically as the ‘classical MAPK pathway’, and its associated implications in various diseases, several enzymes along the pathway have become targets for novel therapeutics. MEK1 and MEK2 in particular have been extensively researched, with small molecules being heavily studied in clinical settings.4–7 This focused research has resulted in significant success to date, including three FDA-approved drugs for use individually or in combination with other therapeutics as cancer therapy (1–3).7–10 However, there has been less effort on developing small molecule inhibitors of the rest of the MEK family. Given that the MAPK pathways as a whole are involved in essential cellular processes, it comes as no surprise that MEK3–7 are also implicated in many disease states.1–2 The progress made thus far has begun to pave the way for additional advances in probe development and understanding of the MAPK pathways. In this review, we document the recent advances in the development of small molecule inhibitors against ‘non-classical’ MEKs and highlight their future potential to aid in elucidating the role of these kinases in prominent and relevant global health issues and diseases.
Figure 1.
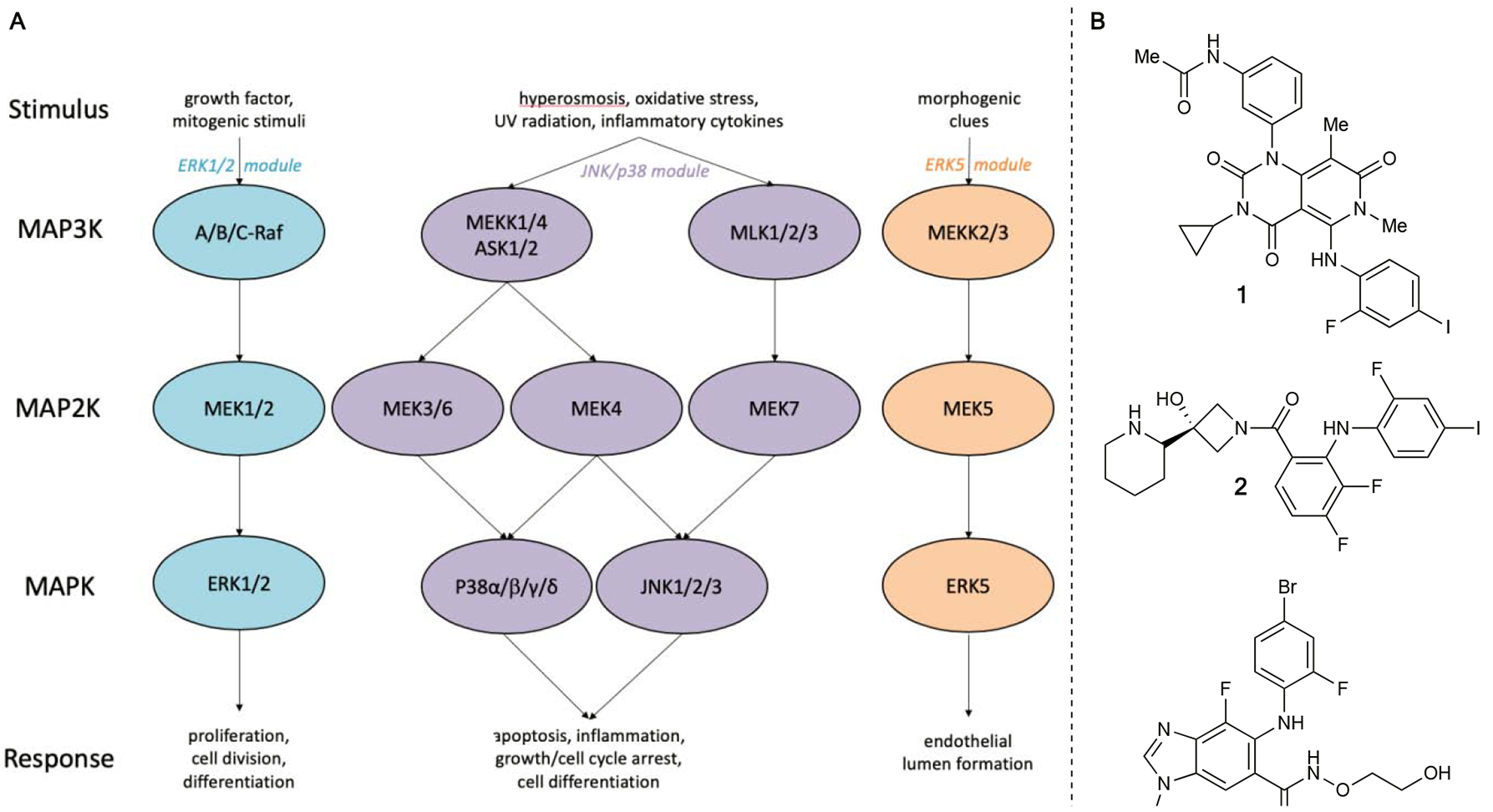
A) Simplified overview of the MAPK Pathway B) FDA-approved MEK inhibitors
2. MEK3/6
Two of the three MEKs within the p38 pathway, MEK3 and MEK6, are involved in cell cycle arrest and have been implicated in inflammatory diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease11–16, as well as various cancers17–19, Alzheimer’s20–21 and cardiovascular dysfunction22–23. Despite the myriad of disease states related to MEK3/6, few small molecule probes have been developed thus far, limiting further investigation of their roles in each disease.
3-aminopicolinamide and imidazolopyridine inhibitors:
Using a fragment-based drug discovery strategy, Swann and coworkers discovered picolinamide- and imidazolopyridine-derived compounds 4 and 5, both with IC50 values of 4 nM against MEK3.24 5 in particular contains a unique isothiazole ring not commonly seen in small molecule inhibitors. Subsequent selectivity panels of these compounds against MAPK kinases downstream of MEK3 as well as MEK1/2 showed high preference for MEK3 and MEK6. 4 had less than 9-fold selectivity for MEK3 over MEK6 while maintaining >100-fold selectivity over all other kinases in the panel. Conversely, 5 had 12-fold selectivity for MEK3 over MEK6, but did not fully avoid all off-target binding against the other kinases in the panel.
Notably, the selectivity of 5 for MEK3 over MEK1 dropped to 86-fold, while selectivity over TAK1 dropped to 23-fold. However, subjection of both compounds to human leukemic monocyte U937 cells revealed attenuation of potency. Quantification using an enzyme-linked immunosorbent assay (ELISA) revealed dose-dependent inhibitory activity, with IC50 values of 2.7 μM and 0.73 μM for 4 and 5, respectively. This study represents the development of the first potent and selective MEK3/6 inhibitors and the beginnings of a potential highly potent and solely selective inhibitor of MEK3.
Gossypetin:
The flavonoid gossypetin (6) is isolated from the flowers and calyx of Hibiscus sabdariffa (roselle), and has been shown to have potential antioxidant, antibacterial, and anticancer activity.25–28 Gossypetin was first discovered by Kim and coworkers due to its selective inhibition of p38 phosphorylation in esophageal cancer cells.29 Subsequent in vitro assays by the authors confirmed that gossypetin inhibits MEK3/6 and not AKT1 (protein kinase B), extracellular signal-regulated kinase-2 (ERK2), p38, or epidermal growth factor receptor (EGFR). Cellular studies on human esophageal squamous cell carcinoma (ESCC) lines (KYSE30, KYSE410, KYSE450, KYSE510) revealed additionally consequential effects on the cell cycle, growth, and apoptosis. Gossypetin was found to inhibit growth of ESCC lines in a dose-dependent manner, inhibiting all growth at 10 μM after 48 hours of incubation. Additionally, high ratios of dead to live cells were observed with gossypetin-treated cultures. Further investigation revealed that pro-apoptotic markers (cleaved caspase 3, 7, BAX, and cytochrome c) were strongly induced, while anti-apoptotic markers (BCL2 and BCL-XL) were reduced at increasing doses, suggesting direct influence of gossypetin on apoptosis. Administration of gossypetin upon synchronization and release of the cell cycle showed a reduced S Phase and induced G2 phase cell cycle arrest after 48 hours of incubation. Additionally, cell cycle marker protein CDKN1B showed increased expression upon treatment.
Following these in-depth cellular studies, the authors investigated the effects of gossypetin in vivo. Employing patient-derived xenograft (PDX) models, patient-derived esophageal tumor tissues were inoculated into immunodeficient mice. After oral administration of gossypetin five times a week over the course of 21 days, tumor growth was decreased by over 60%. Additionally, expression of tumor proliferation marker Ki67 was significantly decreased, and there were no toxic effects on other tissues. However, gossypetin affects a number of biological markers other than MEK3/6, as shown in this report, increasing the chances that gossypetin may have other off-target effects not studied here, directly or through multiple potential metabolites or conjugates that are often formed in vivo by such polyphenolic species. Nevertheless, this study further elucidated the role of MEK3/6 in ESCC and added validation to MEK3/6 as a therapeutic target for this disease state.
Ethacrynic acid:
Interaction determination using unpurified proteins (IDUP) is a method developed by Liu and coworkers to find small molecule-protein interactions by amplifying DNA sequences that encode for target and DNA-linked ligand pairs.30 As proof-of-concept, by using a library of DNA-linked small molecules against a mixture of proteins in unpurified lysate, they discovered that ethacrynic acid (7) was a MEK6 inhibitor.31 Ethacrynic acid was confirmed as a hit and evaluated to have an IC50 value of 4.5 μM in subsequent in vitro assays. Additionally, since ethacrynic acid contains a conjugate acceptor, the possibility of it forming a covalent adduct with the C38 residue was investigated. Non-electrophilic analogs of ethacrynic acid showed >30-fold decrease in potency, and a C38A mutation resulted in a 3.3-fold loss in potency. Mass spectrometry analysis confirmed covalent engagement. Through the IDUP method and an additional in vitro MEK family screening panel, ethacrynic acid was determined to be about 3-fold more selective for MEK6 over both MEK3 and MEK4, 9-fold more selective over MEK2, and 12-fold more selective over MEK1. The ability of ethacrynic acid to inhibit MEK6 selectively over MEK5 or MEK7 was not evaluated.
Given that the inhibition of MEK6 is unrelated to the current uses of ethacrynic acid,32 further development of ethacrynic acid or derivatives could provide a more potent and selective MEK6 probe to investigate the role of MEK6 in various diseases. Additionally, the site of alkylation is Cys38, a non-active site location, and is unique among the MEK family. Little is known about the function of that protein area, and a selective probe developed from ethacrynic acid could be used to investigate not only the role of MEK6, but the individual functions of specific regions within the structure of MEK6. In addition to the application of IDUP, this study provides the first example of a modestly selective MEK6 inhibitor.
3. MEK4/MEK7
As part of the stress-activated JNK/p38 signaling pathway, MEK4 and MEK7 have attracted great interest due to the myriad of diseases associated with them, with cancer being particularly prevalent.33 MEK7 exclusively activates JNK, while MEK4 activates both JNK and p38.1–3, 6, 16, 33 MEK4 and MEK7 have also been implicated in heart disease as well as neurological damage when upregulated in certain cases.34–37
7, 3’, 4’-Trihydroxyisoflavone (THIF):
Daidzein (8) is a naturally occurring isoflavone found in soy foods and other various beans, legumes and sprouts.38 Dong and coworkers investigated the effects of THIF (9), a major metabolite of daidzein, on ultraviolet B (UVB)-induced skin cancer.39 First, daidzein and THIF were tested against COX-2 activity in JB6 P+ (murine skin) cells. Previous studies have shown that COX-2, an enzyme that normally mediates conversion from arachidonic acid to prostaglandin, is overexpressed in skin upon UVB irradiation, and is therefore predicted to play a role in propagating skin cancer.40 However, while THIF showed activity against COX-2, daidzein was ineffective, even up to 60 μM. Daidzein and THIF were then tested against NF-κb since COX-2 is regulated by NF-κb.41 Again, THIF was observed to be more effective. Additionally, in murine models, it has been demonstrated that UVB activates the p38 and JNK pathways, leading to the development of skin cancer.42 The authors used JB6 P+ cells to investigate the effects THIF on these pathways. It was found that JNK and p38 phosphorylation were inhibited, but ERK, MEK3, MEK5, MEK6, and Cot phosphorylation were not affected and further investigation into upstream kinases revealed ATP-competitive inhibition of MEK4 and Cot. To confirm that this inhibition also occurs in vivo, mice with UVB-induced skin cancer were topically treated with THIF. COX-2 expression and phosphorylation of p38, JNK, and NF-κb were attenuated post-treatment. Additionally, daidzein remained ineffective against the cancer, and MEK4 and Cot inhibition was confirmed. This report highlights the potential use of MEK4 inhibitors in further studying the effects of UVB-induced skin cancer.
HWY336:
Protoberberine alkaloids have exhibited activity against human cancer cell lines as well as anti-tumor activity in vivo.43–44 Based on this precedent, Song and coworkers investigated the effects of a protoberberine alkaloid derivative against MEK4 and MEK7 and screened a library of derivatives based on this core against a panel of kinases in the MEK and MAPK families.45
HWY336 (10) was identified as a selective inhibitor of MEK4 and MEK7 using an in vitro kinase assay, and evaluated to have an IC50 value of 6 μM against MEK4 and 10 μM against MEK7. To validate this result, sorbitol-treated human embryonic kidney 293T (HEK293T) cells were treated with HWY336 at various doses. After incubation for 3 hours, treatment with 9 μM HWY336 led to reduced phospho-JNK (p-JNK), while treatment at 12 μM resulted in the complete disappearance of p-JNK. These results, coupled with binding affinity and wash experiments suggesting that HWY336 binds directly to MEK4, indicates that HWY336 is both a MEK4 and MEK7 inhibitor. Further experiments showing phosphorylation inhibition of MEK4 could indicate additional upstream inhibition, but selectivity for MEK4 or MEK7 over related kinases was not tested. While not singularly selective, this report was the first to develop a class of compounds for inhibition of MEK4/MEK7. The existing activity of HWY336 hinted at the potential for developing selective compounds against MEK4 or MEK7.
Genistein:
Genistein (11) is an isoflavone that was originally isolated from soybeans.46 Consumption of foods with high levels of genistein has been previously associated with decreased prostate cancer metastasis and mortality from prostate cancer.47–48 Genistein has also been shown to inhibit matrix metalloproteinase-2 (MMP-2) expression and cell invasion.49–50 In human prostate cancer cells, Bergan and coworkers previously established that genistein treatment blocks activation of p38, as well as MAP kinase-activated protein kinase 2 (MAPKAPK2) and heat shock protein 27 (HSP27), which are all downstream of the p38 pathway.49, 51 However, the exact pharmacological mechanism of genistein was unknown. In further investigations with prostate cancer cells, Bergan and coworkers found that genistein directly inhibited MEK4 with an IC50 value of 400 nM.52 Additionally, a structurally-activated form of MEK4 (MEK4-EE) exhibited greatly increased p38 phosphorylation and MMP-2 expression. However, genistein treatment did not affect levels of phospho-MEK4 (p-MEK4). Cells lacking MEK4 expression did not respond to genistein treatment, supporting the hypothesis that genistein directly inhibits MEK4. It is worth noting that activity against MEK3 was not investigated, and therefore the mode of action of genistein has not been localized to one pathway. Additionally, Xu and coworkers identified that genistein affects the MEK5/ERK5 pathway in MBA-MB-231 breast cancer cells through induction of apoptosis.53 However, based on Phase II clinical trials conducted with genistein in this report as well as subsequent studies on long-term consumption of genistein, MEK4 appears to be a promising pharmaceutical target for metastatic prostate cancer.54
3-Arylindazoles:
Leveraging a 50,000-compound library screen, our laboratory identified 3-arylindazole 12 as a novel inhibitor of MEK4 with an IC50 value of 190 nM.55 12 was first vetted for aggregation and then subjected to a TR-FRET assay to determine if 12 was bound to the active site of MEK4. Upon confirmation that 12 was indeed binding to the active site, an ATP titration was performed to confirm the binding mode of 12, where it was observed that the potency of the inhibitor decreased with increasing ATP concentration. Fluorescence thermal shift (FTS) assays determined that 12 increased the melting point of three different constructs of MEK4 (full length, inactive kinase domain, and MEK4-EE phosphomimetic mutant), indicating that 12 stabilized MEK4 with no preference for the active or inactive form. Further SAR studies were then conducted on 12 using the functional and selectivity-profiling platform previously developed by our laboratory.56–57 13 was discovered in the SAR study with an IC50 value of 41 nM. Once it was determined that 13 was selective for MEK4 over the other MEK family members, 12 and 13 were both subjected to a kinome-wide screening panel (including the seven MEK family members). The original hit 12 was found to inhibit 5 out of the 57 kinases tested at >35% inhibition. The SAR-discovered lead, 13, was found to inhibit 12 out of the 57 kinases. However, the authors note that both compounds were tested at 10 μM and fully inhibited MEK4. Since 13 is 3- to 4-fold more potent than 12, it is possible that 13 would more selective at a concentration that gives equal MEK4 inhibition. 13 is the first report of a potent and selective MEK4 inhibitor and holds potential for further modifications that could improve its performance in cells and in vivo.
Prenylated quinolinecarboxylic acids (PQA):
Prior to discovering PQAs, Homma and coworkers reported Ppc-1 (14) as a mitochondrial oxygen consumption enhancer.58 The authors noticed that Ppc-1 also exhibited neuroprotectant properties against glutamate-induced cell death in hippocampal cultures, so other PQA derivatives were tested against hippocampal cultures. PQA-11 (15) was identified as the most potent neuroprotectant of the derivatives tested, with an IC50 value of 127 nM.59 Because JNK and MEK4 are involved in the cell death pathway through activation of caspase-3,34, 60 the phosphorylation state of these enzymes post-treatment with PQA-11 was examined. Human neuroblastoma SH-SY5Y cells were treated with N-methyl-4-phenylpyridium (MPP+) to activate the pathway. When treated with PQA-11, phosphorylation of MEK4 and JNK, were suppressed, and cell death was attenuated. Similar results were seen when the SH-SY5Y cells were instead treated with aggregated Aβ-peptides, a different inducer of neuron death. To further study inhibition of the pathway by PQA-11, PQA-11 binding was probed by the quartz crystal microbalance (QCM) method. PQA-11 was immobilized on a gold surface, and when inactive MEK4 was added, the frequency of gold self-oscillation decreased in a dose-dependent manner. No response was seen with active MEK4 or an upstream kinase, LRRK2, confirming PQA-11 binding with inactive MEK4. Knockdown of MEK4 also revealed suppression of caspase-3. Lastly, PQA-11 was administered to mice previously treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Using tyrosine hydroxylase-positive neurons as a marker for dopaminergic neurons, the number of neurons observed was reduced with MPTP treatment and restored with subsequent PQA-11 treatment for two weeks. This report brings to light the effects of MEK4 inhibition on neurons, which could be further explored in the context of neurodegenerative diseases.
Paeoniflorin:
Xu and coworkers reported paeoniflorin (16), the active ingredient of the natural herbal medicine Paeonia lactiflora Pall61, as a neuroprotectant against tributyltin chloride-induced apoptosis in hypothalamic neurons.62 Treatment with tributyltin alone resulted in a 43% increase in apoptosis. Conversely, pre-incubation of cells with paeoniflorin followed by treatment with tributyltin saw a 22–34% dose-dependent decrease in apoptosis from the initial increase. During these experiments, there was also a marked dose-dependent decrease in MMP levels. The authors then examined the neuroprotective mechanism, where they saw that tributyltin chloride treatment significantly increases levels of p-JNK. Pre-incubation with paeoniflorin followed by the same tributyltin chloride treatment significantly decreased p-JNK levels from the initial increase. While this report does not confirm engagement of paeoniflorin with MEK4, it does additionally highlight the role of MEK4 and the proceeding pathway in neurodegenerative states.
Growth Arrest and DNA damage-inducible 45β (GADD45β-I):
GADD45β-I is a small length peptide that was designed by Borsello and coworkers by optimizing the minimal essential domains of interaction with MEK7.63 Based on the interacting domains of GADD45β and MEK7,64 GADD45β-I includes a transactivator of transcription (TAT) peptide for membrane permeability, the crucial portions of the GADD45β peptide, and a spacer peptide to improve solubility. Overactivation of N-methyl-D-aspartic acid (NMDA) receptors causes excitotoxity, which in turn activates the JNK pathway.65 NMDA and GADD45β-I treatments did not impact MEK4 activity, but the authors demonstrated that GADD45β-I suppresses NMDA activation of MEK7. GADD45β-I exhibited no toxicity against cultured neurons, but fully protected them from NMDA-induced excitotoxicity at 5 μM. After pre-treatment with NMDA for 12 hours, 56% of the neurons were protected. GADD45β-I was then administered in two in vivo stroke-inducing models. In the middle cerebral artery occlusion (MCAo) model, MEK7 was activated after the stroke was induced. Pre-treatment with GADD45β-I prior to the induced stroke resulted in a 43% reduction in tissue death. Six- and twelve-hour post-treatments produced 42% and 8% decreases in tissue death respectively, indicating that twelve hours is beyond the therapeutic window of GADD45β-I. Additionally, the significant decrease in tissue death was maintained for a week following the induced stroke, where pre-treatment with GADD45β-I reduced the final tissue death volume by about half. This data was reproduced in the thromboembolic model. To probe the mechanism of action further, the levels of phosphorylated MEK7 (p-MEK7) and p-JNK were analyzed. MCAo increased levels of p-MEK7 and p-JNK, while GADD45β-I treatment showed a decrease in p-MEK7 and p-JNK, which was maintained six hours after the induced stroke. MEK4 basal activity was modestly increased upon treatment with GADD45β-I, further indicating that GADD45β-I affects the MEK7 pathway without disturbing MEK4 activity.
In addition to reducing stroke-related tissue death, Liu and coworkers investigated the effect of GADD45β-I treatment on endoplasmic reticulum (ER) stress-induced cytotoxicity in neuronal HT22 cells.66 Tunicamycin (TM) treatments were used to induce ER stress, and cell viability was significantly reduced 24 hours after treatment. This was partially prevented by 5 μM GADD45β-I treatment. Using a fluorescent ER probe, they were able to observe increased ER cytotoxicity after TM treatment, and decreased ER cytotoxicity upon a follow-up treatment of GADD45β-I. These two reports together demonstrate the importance of MEK7 inhibition in alleviating stress-related neurocytotoxicity.
5Z-7-Oxozeaenol (5Z7O):
Upon successful co-crystallization and determination of the inhibitor-protein complex, Kinoshita and coworkers observed 5Z7O (17) covalently bound to MEK7.67 Further investigation of the mechanism of action of 5Z70 revealed that the electrophilic moiety on 5Z7O was required for potency. Additionally, through mutation experiments, it was discovered that 5Z7O was covalently bound to Cys218, a cysteine that is uniquely present in MEK7. However, 5Z7O has an IC50 value of 1.3 μM against MEK7 in vitro, and is not selective over MEK1, ERK2, and TAK1. These findings showcase the potential of 5Z7O derivatives to serve as highly potent and selective MEK7 inhibitors in addition to providing a clearer picture of other non-covalent interactions within the binding pocket.
Indazole-based arylacylamides:
Following an in silico screen of nearly 118,000 acrylamide compounds against MEK7 using their developed software DOCKovalent68, London and coworkers selected the ten best compounds for synthesis. Of the ten compounds, MKK7-COV-1 (18) was the most potent at 11 nM.69 Through a series of substitutions, analogs MKK7-COV-2 through 4 (19–21) were discovered to have IC50 values of 6, 5, and 14 nM respectively. Time course studies additionally showed extremely fast binding kinetics of 20 at various concentrations. Optimized compounds 19 and 20 were then subjected to studies in HEK293 cells. After activation of the pathway by sorbitol, the optimized compounds were found to have EC50 values of 2.1 and 1.3 μM respectively for reducing levels of p-JNK. Biological replicates in 3T3 and Beas2B cells showed similar results. At higher concentrations of 10 μM, varied responses were seen in levels of phospho-ERK (p-ERK) and phospho-p38 (p-p38). Higher concentrations of 20 resulted complete inhibition of p-p38, while higher concentrations of 19 resulted in lowered levels of p-ERK in addition to complete inhibition of p-p38. On-target activity was validated through MEK7 and MEK4/7 double knockouts, where treatment with 18–21 resulted in no change. Additionally, kinome-wide selectivity (panel of 76 kinases) was assessed for 19 and 20 at a concentration of 1 μM. 19 inhibited aurora kinase B, LRRK2, JAK3 and MEK4 at >75% inhibition, while 20 inhibited aurora kinase B, LRRK2, FTL3 and MEK4 at >75% inhibition. Additionally, both lead compounds were weak inhibitors of MEK4 at concentrations of 4.1 and 7.8 –M. Follow-up in vitro assays showed that 21, while having a slightly lower potency against MEK7, showed no detectable inhibition of MEK4 up to 10 –M. Proteome-wide selectivity was also assessed by labeling MEK7 with slightly modified inhibitor 22 outfitted with an alkyne. With this modified inhibitor, the authors were able to selectively label MEK7 at a concentration of 2 μM. Higher concentrations started to show off-target effects, but MEK7 was still the predominant target at 5 μM. A co-crystal structure of MEK7 with 20 confirmed the covalent binding mechanism. These covalent inhibitors represent the first reported selective and potent MEK7 inhibitors, which opens up many possibilities for further application. These probes can be utilized to further elucidate the functions of MEK7 and the downstream kinases, as well as in in vivo applications to examine the role of MEK7 in implicated disease states.
Pyrazolopyrimidine-based acrylamides:
Inspired by a previously discovered epidermal growth factor receptor (EGFR) inhibitor (23) with moderate activity against MEK7,70 Rauh and coworkers reported a class of pyrazolopyrimidine covalent inhibitors against MEK7.71 After some derivatization work, 24 was discovered, with an IC50 value of 10 nM. The modification made to access 24 also greatly increased selectivity for MEK7 over EGFR compared to the other explored inhibitors. Additionally, at a concentration of 1 μM, 24 was exceptionally selective against a panel of kinases, only showing over 50% inhibition for seven out of 320 kinases. Covalent engagement was confirmed by mass spectrometry studies as well as X-ray crystallography. At a concentration of 1 μM in dorsal root ganglion (DRG) cells, formation of p-JNK was completely inhibited. This report continues to build upon developing highly potent and selective MEK7 covalent inhibitors, taking advantage of the cysteine within the active site that is unique within the MEK family.
1,4-napthoquinones:
Prior to exploration of the 1,4-napthoquinone MEK7 inhibitors, Cdc25B inhibitor NSC95397 (25) was also observed to be a weak MEK7 inhibitor.72 Based on the scaffold of 25, Quinn and coworkers designed a small molecule library of both natural and synthetic compounds to be tested against MEK7.73 All natural derivatives were weak inhibitors or inactive against MEK7, while synthetic compounds 26–28 were the most active inhibitors of the library. The most potent compound was 26 with an IC50 value of 230 nM- however, with the exception of 28, all other compounds that were active against MEK7 were also equally potent against Cdc25A and Cdc25B. Compound 28, while less effective with an IC50 of 1.9 μM, was 17-fold more selective over Cdc25A and 12-fold more selective over Cdc25B. Additionally, compounds 26–28 had low to no activity against MEK4 and displayed moderate to strong cytotoxic activity against a cancer cell line panel. While these compounds hit both isoforms of Cdc25 as well as MEK7, they are selective over MEK4 and appear to be potent against cancer cells. This report showcases a new class of compounds that have excellent potential to be developed into potent and selective cell permeable MEK7 inhibitors, as well the anti-cancer properties that these compounds can have.
4. MEK5
MEK5 has been demonstrated to be the sole activator of ERK5.1, 3, 6, 16, 33 However, this selective substrate activation does not preclude its role in various diseases. MEK5 has been implicated in cardiovascular development,74–75 neuronal survival,76–77 and a myriad of cancers.78–80 Of particular interest is the elevated expression of MEK5 present in a large portion of breast cancer tumors analyzed.81 Like MEK3/6, only a few MEK5 inhibitors have been developed to date.
BIX02188 and BIX02189:
In 2008, Snow and coworkers reported the identification of the first MEK5 inhibitors.82 Indolinone-6-carboxamide compounds BIX02188 (29) and BIX02189 (30) were found to inhibit MEK5 with IC50 values of 4.3 nM and 1.5 nM, respectively. The compounds were subsequently screened against a panel of closely related kinases (MEK1, MEK2, ERK1, ERK5, JNK2, TGFβR1, EGFR, STK16). Both compounds were extremely selective for MEK5 over all other kinases, with the exception of ERK5 and TGFβR1. BIX02189 maintained 810-fold selectivity over ERK5 and 1800-fold selectivity over TGFβR1, while BIX02189 maintained approximately 60-fold selectivity over ERK5 and 580-fold selectivity over TGFβR1. In a more extensive kinase panel study, both inhibitors showed greater than 100-fold selectivity for MEK5 for 85 out of the 87 kinases in the panel at a concentration of 3 or 10 μM. To investigate the downstream effects of MEK5 inhibition on ERK5 phosphorylation, HeLa cells were incubated with inhibitor for 90 minutes, followed by MEK5/ERK5 pathway activation by sorbitol treatment. Immunoblotting of the resulting cell lysate showed dose-dependent inhibition of phospho-ERK5. At 10 μM, neither BIX02188 nor BIX02189 affected phosphorylation of ERK1/2, JNK1/2, or p38. However, inhibition of transcriptional activation of MEF2C, a downstream MEK5/ERK5 substrate, was observed. Further investigation of these compounds using a luciferase-based activity assay showed single digit or sub-micromolar IC50 values in both HeLa and HEK293 cells, highlighting the potential of these inhibitors as effective chemical probes. Rönnstrand and coworkers employed BIX02188 to determine the role of MEK5 in leukemia development and discovered that MEK5 may represent an alternative target against Fms-like tyrosine kinase-3 internal tandem duplication (FLT3-ITD) positive leukemia.83
Benzimidazole-based inhibitors:
While attempting to develop CDK5 inhibitors, Cavanaugh and coworkers screened their compound library for activity against MEK5, since CDK5 and MEK5 are both serine/threonine kinases.80, 84 After preliminary testing, their novel benzimidazole scaffold was instead used as a screening platform for developing novel MEK5 inhibitors. Compounds were screened by western blot analysis of MEK5-mediated ERK5 phosphorylation in HEK293 cells. The MEK5/ERK5 pathway was activated by a 30-minute treatment with epidermal growth factor (EGF), then treated with inhibitor at a final concentration of 10 μM. In their first study, they identified 31 as a novel MEK5 inhibitor, which was inactive against CDK5.85 However, they found only slight selectivity for ERK5 phosphorylation over ERK1/2 phosphorylation. A subsequent SAR study expanded on 31 and identified four more novel MEK5 inhibitors.86 32–35 displayed interesting activity, showing either moderate inhibition of ERK5 phosphorylation with high selectivity over ERK1/2 phosphorylation, or activation of ERK1/2 phosphorylation. It is worth noting that these compounds did not inhibit MEK1/2. Additionally, the low-affinity of these compounds to the ATP-site of MEK5 suggests another mode of action. It is unclear whether or not these compounds exhibit off-target effects, but preliminary NCI 60-cell line screens show selective growth inhibition of the MCF-7 cell line. Further studies with these chemical probes could deconvolute the role of MEK5 and ERK5 phosphorylation inhibition in selectively stopping MCF-7 growth, in addition to roles that MEK5 may play in other disease states.
Anthranilic acid-based inhibitors:
Continuing their exploration of MEK5 inhibitors, Cavanaugh and coworkers examined anthranilic acid-based inhibitors, drawing inspiration from MEK1/2 inhibitor PD325901.87–88 Starting with a N,N-diphenylaniline core, a homology model of MEK5 was used to examine potential variations of the scaffold. Four main areas of the chosen scaffold were altered to tune for MEK5 selectivity and potency. The resulting 24 compounds were screened at an initial concentration of 10 μM against EGF triple-negative breast cancer MDA-MB-231 cells. The most potent and selective of the compounds against MEK5 was 36, invoking an 82% decrease in pERK5 while promoting a moderate increase (~20%) in phospho-ERK1/2 (p-ERK1/2) normalized against DMSO treatment. To further validate MEK5 as a target for stopping tumor growth, severe combined immune deficient (SCID), beige, female mice with MDA-MB-231/matrigel xenografts were treated with compound 37, one of their first compounds to display significant MEK5 inhibition. With treatment over 31 days, tumor volume was significantly lower than that of the DMSO treatment, suggesting that the role of MEK5 in tumorigenesis is significant and that these anthranilic acid-based inhibitors could be used to further probe the role of MEK5 in propagating triple-negative breast cancer.
5. Conclusions
Research and activity around the ‘non-classical’ MEKs has grown extensively in the past several years. Natural products, in silico modeling, existing drug molecule scaffolds, fragment libraries, and high throughput screens have all aided in the advancement of many of the reported compounds above. However, there are still advances yet to be made, especially given that there are inherent challenges when targeting MEKs, such as cellular potency and selectivity.
Surveying the MEK family, it is readily apparent that each MEK family member is involved in various high impact and prevalent diseases. Cancers, neurological diseases, inflammation, and cardiovascular diseases are all affected by the ‘non-classical’ MEKs. Some studies have even built precedence for developing a combination therapeutic using existing MEK1/2 inhibitors with a ‘non-classical’ MEK inhibitor.89 This strategy would of course necessitate further progress towards developing ‘non-classical’ inhibitors, but could indeed be a promising avenue to develop the next novel treatment. However, it is more pressing to first develop highly selective ‘non-classical’ MEK inhibitors. With more selective and potent inhibitors, the scientific community would be able to gain a better understanding of each MEK pathway, both independently and synergistically. Each new compound discovery aids in unraveling the role of these MEKs in cellular processes as well as their respective disease states. With this better understanding comes more efficient and effective inhibitor development, in turn greatly increasing the chances that a novel therapeutic agent targeting ‘non-classical’ MEKs will become a reality.
Figure 2.
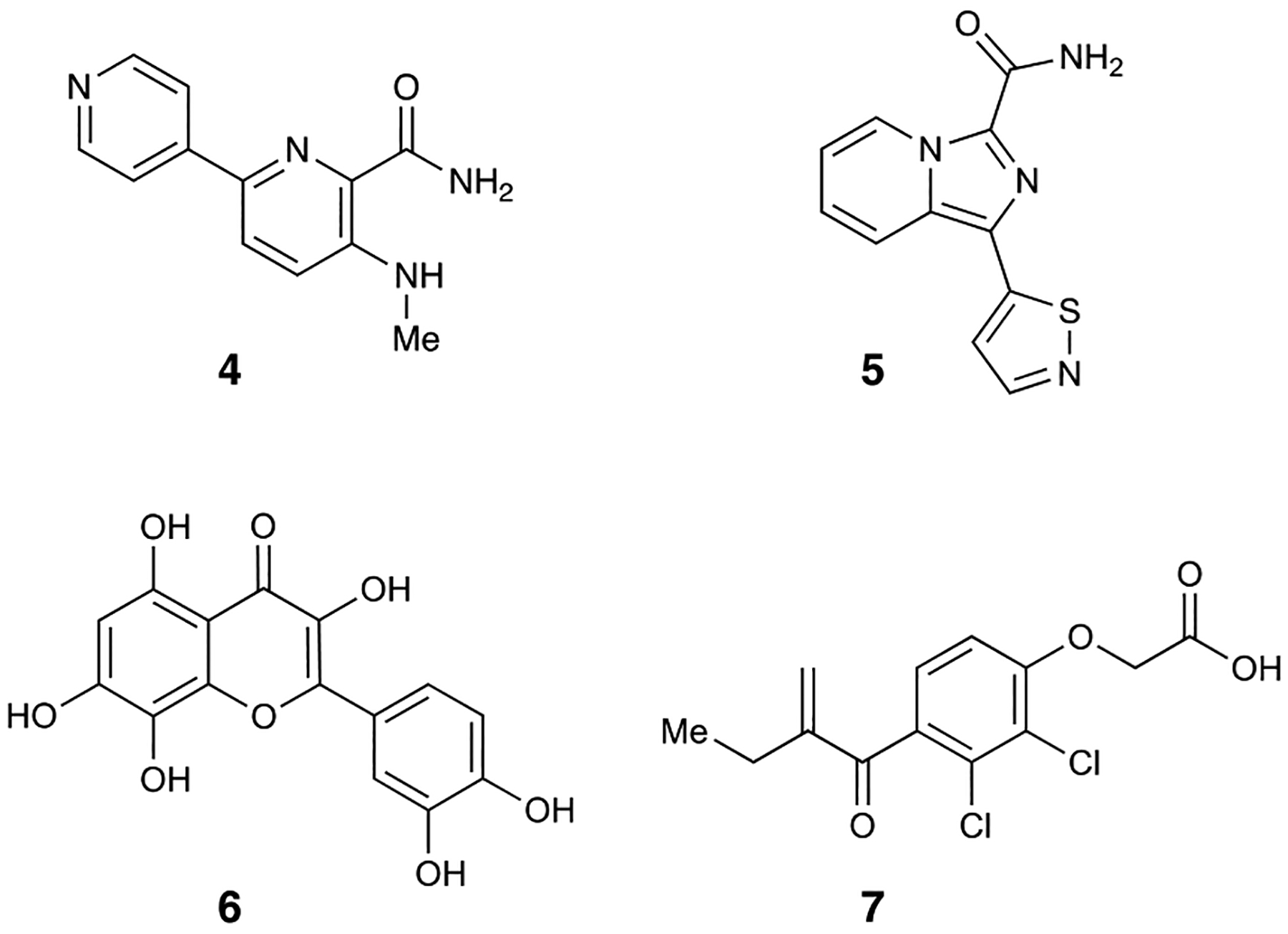
MEK3/6 inhibitors
Figure 3.
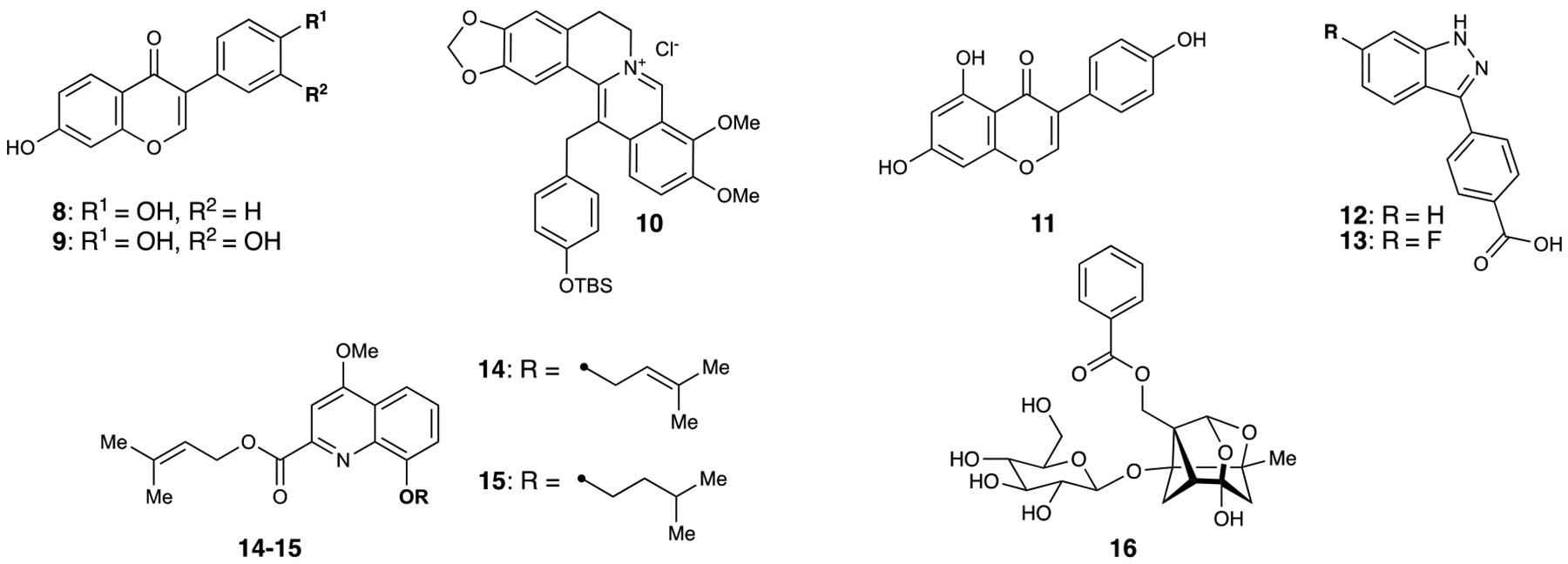
MEK4 inhibitors
Figure 4.
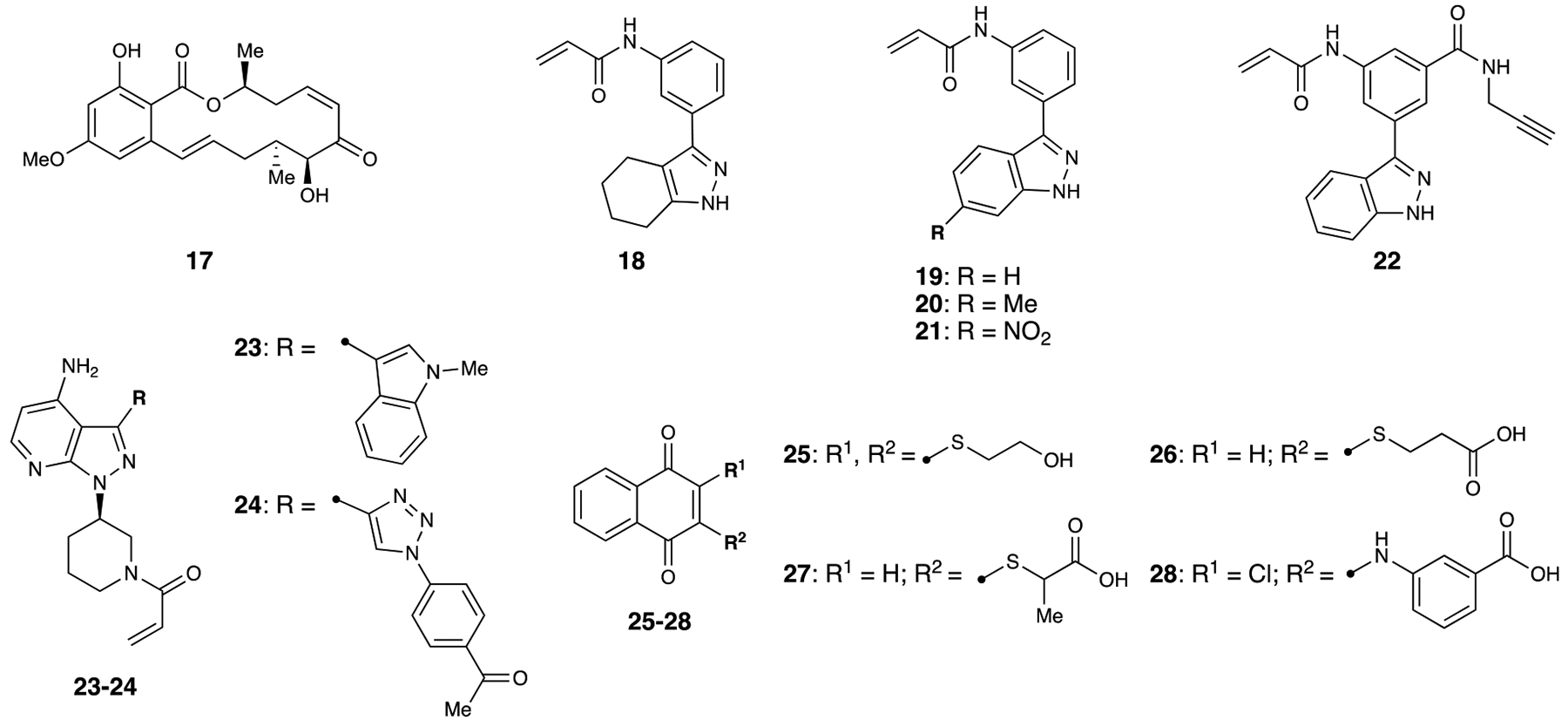
MEK7 inhibitors
Figure 5.
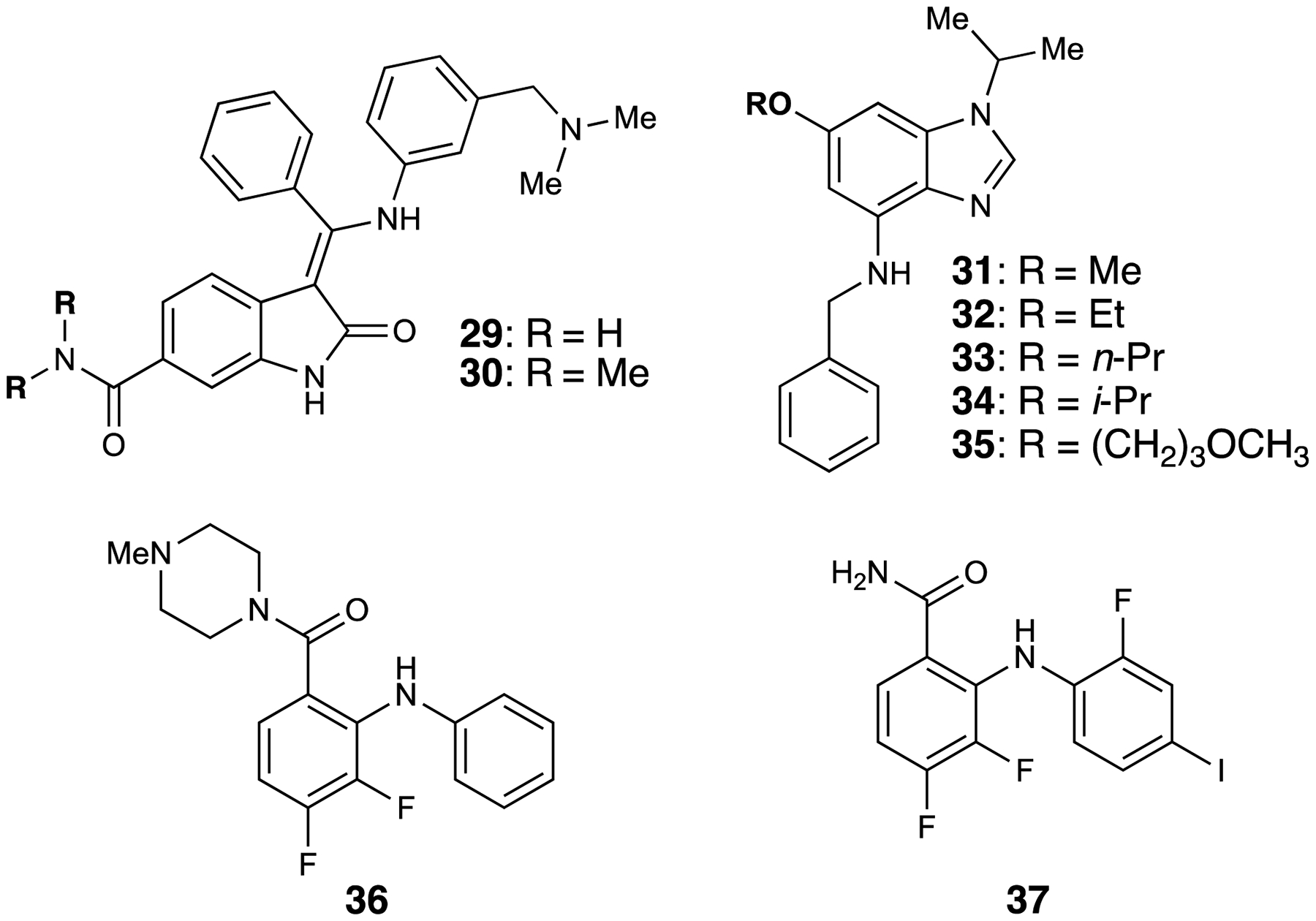
MEK5 inhibitors
Acknowledgments
Funding for these efforts has been provided by the National Institutes of Health, National Cancer Institute (NCI; R01 CA188015) and Northwestern. AJK is supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T32GM105538. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank Meghan Orr (NU), Dalton Kim (NU), and David Burke (NU) for manuscript proofreading.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang W; Liu HT, MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9. [DOI] [PubMed] [Google Scholar]
- 2.Raman M; Chen W; Cobb MH, Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100. [DOI] [PubMed] [Google Scholar]
- 3.Chang L; Karin M, Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37. [DOI] [PubMed] [Google Scholar]
- 4.Akinleye A; Furqan M; Mukhi N; Ravella P; Liu D, MEK and the inhibitors: from bench to bedside. J. Hematol. Oncol 2013, 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng Y; Tian H, Current Development Status of MEK inhibitors. Molecules 2017, 22, 1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burotto M; Chiou VL; Lee J-M; Kohn EC, The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez JN; Wang T; Cohen MS, BRAF and MEK Inhibitors: Use and Resistance in BRAF-Mutated Cancers. Drugs 2018, 78, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falchook GS; Lewis KD; Infante JR; Gordon MS; Vogelzang NJ; DeMarini DJ; Sun P; Moy C; Szabo SA; Roadcap LT; Peddareddigari VGR; Lebowitz PF; Le NT; Burris HA; Messersmith WA; O’Dwyer PJ; Kim KB; Flaherty K; Bendell JC; Gonzalez R; Kurzrock R; Fecher LA, Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribas A; Gonzalez R; Pavlick A; Hamid O; Gajewski TF; Daud A; Flaherty L; Logan T; Chmielowski B; Lewis K; Kee D; Boasberg P; Yin M; Chan I; Musib L; Choong N; Puzanov I; McArthur GA, Combination of vemurafenib and cobimetinib in patients with advanced BRAFV600-mutated melanoma: a phase 1b study. Lancet Oncol. 2014, 15, 954. [DOI] [PubMed] [Google Scholar]
- 10.Cho M; Gong J; Frankel P; Synold TW; Lim D; Chung V; Chao J; Li D; Chen Y; Sentovich S; Melstrom K; Singh G; Luevanos E; Fakih M, A phase I clinical trial of binimetinib in combination with FOLFOX in patients with advanced metastatic colorectal cancer who failed prior standard therapy. Oncotarget 2017, 8, 79750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar S; Boehm J; Lee JC, p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Disc 2003, 2, 717. [DOI] [PubMed] [Google Scholar]
- 12.Lee JC; Kumar S; Griswold DE; Underwood DC; Votta BJ; Adams JL, Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology 2000, 47, 185. [DOI] [PubMed] [Google Scholar]
- 13.Meng H; Gao Y; Kang YF; Zhao YP; Yang GJ; Wang Y; Cao Y; Gan YH; Xie QF, Molecular Changes Involving MEK3–p38 MAPK Activation in Chronic Masticatory Myalgia. J. Dent. Res 2016, 95, 1169. [DOI] [PubMed] [Google Scholar]
- 14.Chabaud-Riou M; Firestein GS, Expression and Activation of Mitogen-Activated Protein Kinase Kinases-3 and −6 in Rheumatoid Arthritis. Am. J. Pathol 2004, 164, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshizawa T; Hammaker D; Boyle DL; Corr M; Flavell R; Davis R; Schett G; Firestein GS, Role of MAPK Kinase 6 in Arthritis: Distinct Mechanism of Action in Inflammation and Cytokine Expression. J. Immunol 2009, 183, 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frémin C; Meloche S, From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J. Hematol. Oncol 2010, 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z; Fu J; Li N; Shen L, Quantitative proteome analysis identifies MAP2K6 as potential regulator of LIFR-induced radioresistance in nasopharyngeal carcinoma cells. Biochem. Biophys. Res. Commun 2018, 505, 274. [DOI] [PubMed] [Google Scholar]
- 18.Li Z; Li N; Shen L, MAP2K6 is associated with radiation resistance and adverse prognosis for locally advanced nasopharyngeal carcinoma patients. Cancer Manag. Res 2018, 10, 6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin S; Liu K; Zhang Y; Jiang M; Lu R; Folts CJ; Gao X; Noble MD; Zhao T; Zhou Z; Lan X; Que J, Pharmacological targeting of p38 MAP-Kinase 6 (MAP2K6) inhibits the growth of esophageal adenocarcinoma. Cellular Signalling 2018, 51, 222. [DOI] [PubMed] [Google Scholar]
- 20.Lemche E, Early Life Stress and Epigenetics in Late-onset Alzheimer’s Dementia: A Systematic Review. Curr. Genomics 2018, 19, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munoz L; Ammit AJ, Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561. [DOI] [PubMed] [Google Scholar]
- 22.Liao P; Georgakopoulos D; Kovacs A; Zheng M; Lerner D; Pu H; Saffitz J; Chien K; Xiao R-P; Kass DA; Wang Y, The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci 2001, 98, 12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asrih M; Mach F; Nencioni A; Dallegri F; Quercioli A; Montecucco F, Role of Mitogen-Activated Protein Kinase Pathways in Multifactorial Adverse Cardiac Remodeling Associated with Metabolic Syndrome. Mediators Inflamm. 2013, 2013, 367245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams M; Kobayashi T; Lawson JD; Saitoh M; Shimokawa K; Bigi SV; Hixon MS; Smith CR; Tatamiya T; Goto M; Russo J; Grimshaw CE; Swann S, Fragment-based drug discovery of potent and selective MKK3/6 inhibitors. Bioorg. Med. Chem. Lett 2016, 26, 1086. [DOI] [PubMed] [Google Scholar]
- 25.Khan A; Manna K; Das DK; Kesh SB; Sinha M; Das U; Biswas S; Sengupta A; Sikder K; Datta S; Ghosh M; Chakrabarty A; Banerji A; Dey S, Gossypetin ameliorates ionizing radiation-induced oxidative stress in mice liver—a molecular approach. Free Radical Res. 2015, 49, 1173. [DOI] [PubMed] [Google Scholar]
- 26.Lin H-H, In Vitro and in Vivo Atheroprotective Effects of Gossypetin against Endothelial Cell Injury by Induction of Autophagy. Chem. Res. Toxicol 2015, 28, 202. [DOI] [PubMed] [Google Scholar]
- 27.Khan A; Manna K; Bose C; Sinha M; Das DK; Kesh SB; Chakrabarty A; Banerji A; Dey S, Gossypetin, a naturally occurring hexahydroxy flavone, ameliorates gamma radiation-mediated DNA damage. Int. J. Radiat Biol 2013, 89, 965. [DOI] [PubMed] [Google Scholar]
- 28.Lee M-S; Tsai C-W; Wang C-P; Chen J-H; Lin H-H, Anti-prostate cancer potential of gossypetin via inducing apoptotic and autophagic cell death. Mol. Carcinog 2017, 56, 2578. [DOI] [PubMed] [Google Scholar]
- 29.Xie X; Liu K; Liu F; Chen H; Wang X; Zu X; Ma X; Wang T; Wu Q; Zheng Y; Bode AM; Dong Z; Kim DJ, Gossypetin is a novel MKK3 and MKK6 inhibitor that suppresses esophageal cancer growth in vitro and in vivo. Cancer Lett. 2019, 442, 126. [DOI] [PubMed] [Google Scholar]
- 30.McGregor LM; Jain T; Liu DR, Identification of Ligand-Target Pairs from Combined Libraries of Small Molecules and Unpurified Protein Targets in Cell Lysates. J. Am. Chem. Soc 2014, 136, 3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan AI; McGregor LM; Jain T; Liu DR, Discovery of a Covalent Kinase Inhibitor from a DNA-Encoded Small-Molecule Library × Protein Library Selection. J. Am. Chem. Soc 2017, 139, 10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molnar J; Somberg JC, The Clinical Pharmacology of Ethacrynic Acid. Am. J. Ther 2009, 16. [DOI] [PubMed] [Google Scholar]
- 33.Dhillon AS; Hagan S; Rath O; Kolch W, MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279. [DOI] [PubMed] [Google Scholar]
- 34.Wang X; Destrument A; Tournier C, Physiological roles of MKK4 and MKK7: Insights from animal models. Biochim. Biophys. Acta 2007, 1773, 1349. [DOI] [PubMed] [Google Scholar]
- 35.Donna B-C; Michael SS; Robert LH, Targeting the JNK Pathway for Therapeutic Benefit in CNS Disease. CNS Neurol. Disord. Drug Targets 2002, 1, 31. [DOI] [PubMed] [Google Scholar]
- 36.Ham J; Eilers A; Whitfield J; Neame SJ; Shah B, c-Jun and the transcriptional control of neuronal apoptosis. Biochem. Pharmacol 2000, 60, 1015. [DOI] [PubMed] [Google Scholar]
- 37.Lai TW; Zhang S; Wang YT, Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol 2014, 115, 157. [DOI] [PubMed] [Google Scholar]
- 38.Chang T-S, Isolation, bioactivity, and production of ortho-hydroxydaidzein and ortho-hydroxygenistein. Int. J. Mol. Sci 2014, 15, 5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DE; Lee KW; Byun S; Jung SK; Song N; Lim SH; Heo Y-S; Kim JE; Kang NJ; Kim BY; Bowden GT; Bode AM; Lee HJ; Dong Z, 7,3″,4″ trihydroxyisoflavone, a metabolite of the soy isoflavone daidzein, suppresses ultraviolet B-induced skin cancer by targeting COT and MKK4. J. Biol. Chem 2011, 286, 14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rundhaug JE; Fischer SM, Cyclo-oxygenase-2 Plays a Critical Role in UV-induced Skin Carcinogenesis. Photochem. Photobiol 2008, 84, 322. [DOI] [PubMed] [Google Scholar]
- 41.Jung SK; Lee KW; Byun S; Kang NJ; Lim SH; Heo Y-S; Bode AM; Bowden GT; Lee HJ; Dong Z, Myricetin Suppresses UVB-Induced Skin Cancer by Targeting Fyn. Cancer Res. 2008, 68, 6021. [DOI] [PubMed] [Google Scholar]
- 42.Mantena SK; Katiyar SK, Grape seed proanthocyanidins inhibit UV-radiation-induced oxidative stress and activation of MAPK and NF-κB signaling in human epidermal keratinocytes. Free Radical Biol. Med 2006, 40, 1603. [DOI] [PubMed] [Google Scholar]
- 43.Wang L; Liu L; Shi Y; Cao H; Chaturvedi R; Calcutt MW; Hu T; Ren X; Wilson KT; Polk DB; Yan F, Berberine Induces Caspase-Independent Cell Death in Colon Tumor Cells through Activation of Apoptosis-Inducing Factor. PLOS ONE 2012, 7, e36418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goto H; Kariya R; Shimamoto M; Kudo E; Taura M; Katano H; Okada S, Antitumor effect of berberine against primary effusion lymphoma via inhibition of NF-κB pathway. Cancer Sci. 2012, 103, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim N; Park J; Gadhe CG; Cho SJ; Oh Y; Kim D; Song K, A Protoberberine Derivative HWY336 Selectively Inhibits MKK4 and MKK7 in Mammalian Cells: The Importance of Activation Loop on Selectivity. PLOS ONE 2014, 9, e91037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walter ED, Genistin (an Isoflavone Glucoside) and its Aglucone, Genistein, from Soybeans. J. Am. Chem. Soc 1941, 63, 3273. [Google Scholar]
- 47.Severson RK; Nomura AMY; Grove JS; Stemmermann GN, A Prospective Study of Demographics, Diet, and Prostate Cancer among Men of Japanese Ancestry in Hawaii. Cancer Res. 1989, 49, 1857. [PubMed] [Google Scholar]
- 48.Magee PJ; Rowland IR, Phyto-oestrogens, their mechanism of action: current evidence for a role in breast and prostate cancer. Br. J. Nutr 2004, 91, 513. [DOI] [PubMed] [Google Scholar]
- 49.Xu L; Bergan RC, Genistein Inhibits Matrix Metalloproteinase Type 2 Activation and Prostate Cancer Cell Invasion by Blocking the Transforming Growth Factor β-Mediated Activation of Mitogen-Activated Protein Kinase-Activated Protein Kinase 2–27-kDa Heat Shock Protein Pathway. Mol. Pharmacol 2006, 70, 869. [DOI] [PubMed] [Google Scholar]
- 50.Lakshman M; Xu L; Ananthanarayanan V; Cooper J; Takimoto CH; Helenowski I; Pelling JC; Bergan RC, Dietary Genistein Inhibits Metastasis of Human Prostate Cancer in Mice. Cancer Res. 2008, 68, 2024. [DOI] [PubMed] [Google Scholar]
- 51.Huang X; Chen S; Xu L; Liu Y; Deb DK; Platanias LC; Bergan RC, Genistein Inhibits p38 Map Kinase Activation, Matrix Metalloproteinase Type 2, and Cell Invasion in Human Prostate Epithelial Cells. Cancer Res. 2005, 65, 3470. [DOI] [PubMed] [Google Scholar]
- 52.Xu L; Ding Y; Catalona WJ; Yang XJ; Anderson WF; Jovanovic B; Wellman K; Killmer J; Huang X; Scheidt KA; Montgomery RB; Bergan RC, MEK4 function, genistein treatment, and invasion of human prostate cancer cells. J. Natl. Cancer Inst 2009, 101, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z; Li J; Mo B; Hu C; Liu H; Qi H; Wang X; Xu J, Genistein induces cell apoptosis in MDA-MB-231 breast cancer cells via the mitogen-activated protein kinase pathway. Toxicol. In Vitro 2008, 22, 1749. [DOI] [PubMed] [Google Scholar]
- 54.Zhang H; Gordon R; Li W; Yang X; Pattanayak A; Fowler G; Zhang L; Catalona WJ; Ding Y; Xu L; Huang X; Jovanovic B; Kelly DL; Jiang H; Bergan R, Genistein treatment duration effects biomarkers of cell motility in human prostate. PLOS ONE 2019, 14, e0214078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deibler KK; Schiltz GE; Clutter MR; Mishra RK; Vagadia PP; O’Connor M; George MD; Gordon R; Fowler G; Bergan R; Scheidt KA, Synthesis and Biological Evaluation of 3-Arylindazoles as Selective MEK4 Inhibitors. ChemMedChem 2019, 14, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deibler KK; Mishra RK; Clutter MR; Antanasijevic A; Bergan R; Caffrey M; Scheidt KA, A Chemical Probe Strategy for Interrogating Inhibitor Selectivity Across the MEK Kinase Family. ACS Chem. Bio 2017, 12, 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mishra RK; Deibler KK; Clutter MR; Vagadia PP; O’Connor M; Schiltz GE; Bergan R; Scheidt KA, Modeling MEK4 Kinase Inhibitors through Perturbed Electrostatic Potential Charges. J. Chem. Inf. Model 2019, 59, 4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki T; Kikuchi H; Ogura M; Homma MK; Oshima Y; Homma Y, Weight Loss by Ppc-1, a Novel Small Molecule Mitochondrial Uncoupler Derived from Slime Mold. PLOS ONE 2015, 10, e0117088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogura M; Kikuchi H; Shakespear N; Suzuki T; Yamaki J; Homma MK; Oshima Y; Homma Y, Prenylated quinolinecarboxylic acid derivative prevents neuronal cell death through inhibition of MKK4. Biochem. Pharmacol 2019, 162, 109. [DOI] [PubMed] [Google Scholar]
- 60.Yamasaki T; Kawasaki H; Nishina H, Diverse Roles of JNK and MKK Pathways in the Brain. J. Sig. Transduct 2012, 2012, 459265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mao Q-Q; Zhong X-M; Feng C-R; Pan A-J; Li Z-Y; Huang Z, Protective Effects of Paeoniflorin Against Glutamate-Induced Neurotoxicity in PC12 Cells via Antioxidant Mechanisms and Ca2+ Antagonism. Cell Mol. Neurobiol 2010, 30, 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cong C; Kluwe L; Li S; Liu X; Liu Y; Liu H; Gui W; Liu T; Xu L, Paeoniflorin inhibits tributyltin chloride-induced apoptosis in hypothalamic neurons via inhibition of MKK4-JNK signaling pathway. J. Ethnopharmacol 2019, 237, 1. [DOI] [PubMed] [Google Scholar]
- 63.Vercelli A; Biggi S; Sclip A; Repetto IE; Cimini S; Falleroni F; Tomasi S; Monti R; Tonna N; Morelli F; Grande V; Stravalaci M; Biasini E; Marin O; Bianco F; di Marino D; Borsello T, Exploring the role of MKK7 in excitotoxicity and cerebral ischemia: a novel pharmacological strategy against brain injury. Cell Death Dis. 2015, 6, e1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rega C; Russo R; Focà A; Sandomenico A; Iaccarino E; Raimondo D; Milanetti E; Tornatore L; Franzoso G; Pedone PV; Ruvo M; Chambery A, Probing the interaction interface of the GADD45β/MKK7 and MKK7/DTP3 complexes by chemical cross-linking mass spectrometry. Int. J. Biol. Macromol 2018, 114, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Centeno C; Repici M; Chatton JY; Riederer BM; Bonny C; Nicod P; Price M; Clarke PGH; Papa S; Franzoso G; Borsello T, Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 2007, 14, 240. [DOI] [PubMed] [Google Scholar]
- 66.Xu Q-H; Song B-J; Liu D; Chen Y-H; Zhou Y; Liu W-B; Li H; Long T-L; Zhang R; Liu W, The MKK7 inhibitor peptide GADD45β-I attenuates ER stress-induced mitochondrial dysfunction in HT22 cells: Involvement of JNK-Wnt pathway. Brain Res. 2018, 1691, 1. [DOI] [PubMed] [Google Scholar]
- 67.Sogabe Y; Matsumoto T; Hashimoto T; Kirii Y; Sawa M; Kinoshita T, 5Z-7-Oxozeaenol covalently binds to MAP2K7 at Cys218 in an unprecedented manner. Bioorg. Med. Chem. Lett 2015, 25, 593. [DOI] [PubMed] [Google Scholar]
- 68.London N; Miller RM; Krishnan S; Uchida K; Irwin JJ; Eidam O; Gibold L; Cimermančič P; Bonnet R; Shoichet BK; Taunton J, Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol 2014, 10, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shraga A; Olshvang E; Davidzohn N; Khoshkenar P; Germain N; Shurrush K; Carvalho S; Avram L; Albeck S; Unger T; Lefker B; Subramanyam C; Hudkins RL; Mitchell A; Shulman Z; Kinoshita T; London N, Covalent Docking Identifies a Potent and Selective MKK7 Inhibitor. Cell Chem. Biol 2019, 26, 98. [DOI] [PubMed] [Google Scholar]
- 70.Engel J; Becker C; Lategahn J; Keul M; Ketzer J; Mühlenberg T; Kollipara L; Schultz-Fademrecht C; Zahedi RP; Bauer S; Rauh D, Insight into the Inhibition of Drug-Resistant Mutants of the Receptor Tyrosine Kinase EGFR. Angew. Chem. Int. Ed 2016, 55, 10909. [DOI] [PubMed] [Google Scholar]
- 71.Wolle P; Hardick J; Cronin SJF; Engel J; Baumann M; Lategahn J; Penninger JM; Rauh D, Targeting the MKK7-JNK (Mitogen-Activated Protein Kinase Kinase 7-c-Jun N-Terminal Kinase) Pathway with Covalent Inhibitors. J. Med. Chem 2019, 62, 2843. [DOI] [PubMed] [Google Scholar]
- 72.Yang Y; Yang WS; Yu T; Yi Y-S; Park JG; Jeong D; Kim JH; Oh JS; Yoon K; Kim J-H; Cho JY, Novel anti-inflammatory function of NSC95397 by the suppression of multiple kinases. Biochem. Pharmacol 2014, 88, 201. [DOI] [PubMed] [Google Scholar]
- 73.Schepetkin IA; Karpenko AS; Khlebnikov AI; Shibinska MO; Levandovskiy IA; Kirpotina LN; Danilenko NV; Quinn MT, Synthesis, anticancer activity, and molecular modeling of 1,4-naphthoquinones that inhibit MKK7 and Cdc25. Eur. J. Med. Chem 2019, 183, 111719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Regan CP; Li W; Boucher DM; Spatz S; Su MS; Kuida K, Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci 2002, 99, 9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sohn SJ; Sarvis BK; Cado D; Winoto A, ERK5 MAPK Regulates Embryonic Angiogenesis and Acts as a Hypoxia-sensitive Repressor of Vascular Endothelial Growth Factor Expression. J. Biol. Chem 2002, 277, 43344. [DOI] [PubMed] [Google Scholar]
- 76.Watson FL; Heerssen HM; Bhattacharyya A; Klesse L; Lin MZ; Segal RA, Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci 2001, 4, 981. [DOI] [PubMed] [Google Scholar]
- 77.Cavanaugh JE; Ham J; Hetman M; Poser S; Yan C; Xia Z, Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J. Neurosci 2001, 21, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hayashi M; Fearns C; Eliceiri B; Yang Y; Lee J-D, Big Mitogen-Activated Protein Kinase 1/Extracellular Signal-Regulated Kinase 5 Signaling Pathway Is Essential for Tumor-Associated Angiogenesis. Cancer Res. 2005, 65, 7699. [DOI] [PubMed] [Google Scholar]
- 79.McCracken SRC; Ramsay A; Heer R; Mathers ME; Jenkins BL; Edwards J; Robson CN; Marquez R; Cohen P; Leung HY, Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 2008, 27, 2978. [DOI] [PubMed] [Google Scholar]
- 80.Drew BA; Burow ME; Beckman BS, MEK5/ERK5 pathway: the first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsieh F-C; Cheng G; Lin J, Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem. Biophys. Res. Commun 2005, 335, 292. [DOI] [PubMed] [Google Scholar]
- 82.Tatake RJ; O’Neill MM; Kennedy CA; Wayne AL; Jakes S; Wu D; Kugler SZ; Kashem MA; Kaplita P; Snow RJ, Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun 2008, 377, 120. [DOI] [PubMed] [Google Scholar]
- 83.Razumovskaya E; Sun J; Rönnstrand L, Inhibition of MEK5 by BIX02188 induces apoptosis in cells expressing the oncogenic mutant FLT3-ITD. Biochem. Biophys. Res. Commun 2011, 412, 307. [DOI] [PubMed] [Google Scholar]
- 84.Moy LY; Tsai L-H, Cyclin-dependent Kinase 5 Phosphorylates Serine 31 of Tyrosine Hydroxylase and Regulates Its Stability. J. Biol. Chem 2004, 279, 54487. [DOI] [PubMed] [Google Scholar]
- 85.Flaherty PT; Chopra I; Jain P; Yi S; Allen E; Cavanaugh J, Identification of benzimidazole-based inhibitors of the mitogen activated kinase-5 signaling pathway. Bioorg. Med. Chem. Lett 2010, 20, 2892. [DOI] [PubMed] [Google Scholar]
- 86.Flaherty PT; Chopra I; Jain P; Monlish D; Cavanaugh J, Structure- activity relationships of benzimidazole-based selective inhibitors of the mitogen activated kinase-5 signaling pathway. Biorg. Med. Chem 2010, 18, 8054. [DOI] [PubMed] [Google Scholar]
- 87.Chakrabarty S; Monlish DA; Gupta M; Wright TD; Hoang VT; Fedak M; Chopra I; Flaherty PT; Madura J; Mannepelli S; Burow ME; Cavanaugh JE, Structure activity relationships of anthranilic acid-based compounds on cellular and in vivo mitogen activated protein kinase-5 signaling pathways. Bioorg. Med. Chem. Lett 2018, 28, 2294. [DOI] [PubMed] [Google Scholar]
- 88.Fischmann TO; Smith CK; Mayhood TW; Myers JE; Reichert P; Mannarino A; Carr D; Zhu H; Wong J; Yang R-S; Le HV; Madison VS, Crystal Structures of MEK1 Binary and Ternary Complexes with Nucleotides and Inhibitors. Biochemistry 2009, 48, 2661. [DOI] [PubMed] [Google Scholar]
- 89.Xue Z; Vis DJ; Bruna A; Sustic T; van Wageningen S; Batra AS; Rueda OM; Bosdriesz E; Caldas C; Wessels LFA; Bernards R, MAP3K1 and MAP2K4 mutations are associated with sensitivity to MEK inhibitors in multiple cancer models. Cell Res. 2018, 28, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]