

Summary
Mycobacterium tuberculosis subverts host immunity to proliferate within host tissues. Non-selective transient receptor potential (TRP) ion channels are involved in host responses and altered upon bacterial infections. Altered expression and localization of TRPV4 in macrophages upon virulent M. tuberculosis infection together with differential distribution of TRPV4 in human tuberculosis (TB) granulomas indicate a role of TRPV4 in TB. Compared with wild-type mice, Trpv4-deficient littermates showed transiently higher mycobacterial burden and reduced proinflammatory responses. In the absence of TRPV4, activation failed to render macrophages capable of controlling mycobacteria. Surprisingly, Trpv4-deficient mice were superior to wild-type ones in controlling M. tuberculosis infection in the chronic phase. Thus, Trpv4 is important in host responses to mycobacteria, although with opposite functions early versus late in infection. Ameliorated chronic infection in the absence of Trpv4 and its expression in human TB lesions indicate TRPV4 as putative target for host-directed therapy.
Subject Areas: Molecular Biology, Immunology
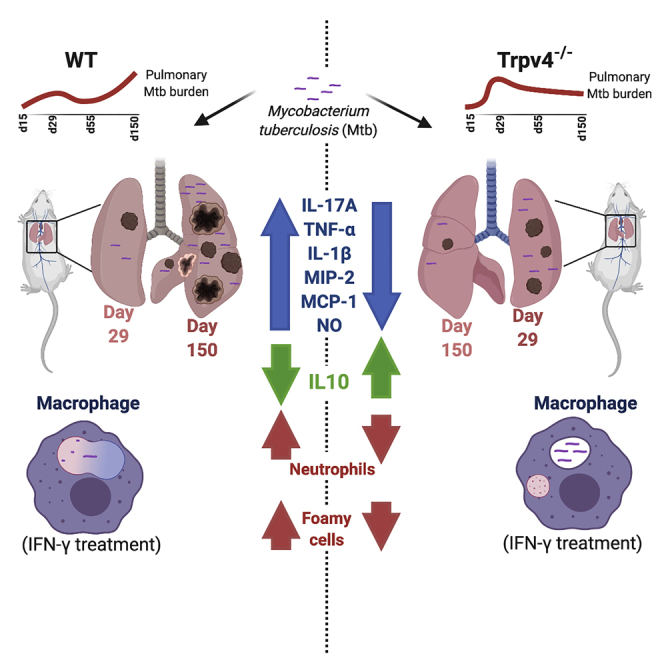
Graphical Abstract
Highlights
-
•
Mtb down-modulates TRPV4 expression in macrophages
-
•
Trpv4−/− macrophages cannot be activated to drive phagosome maturation and NO production
-
•
Trpv4-deficient mice are more resistant to Mtb
-
•
TRPV4-positive macrophages in the periphery of human granuloma but not at the center
Molecular Biology; Immunology
Introduction
Emerging data indicate a role for calcium ion channels in bacterial infections (Deretic and Fratti, 1999, King et al., 2020). Bacterial endotoxin can activate transient receptor potential (TRP) channels including TRPV4 in airway epithelial cells, which triggers proinflammatory responses (Alpizar et al., 2017). The host endeavors to eliminate a pathogen, whereas the pathogen strives to control or escape host defenses. Macrophages are in principle equipped to eliminate bacterial pathogens, but are exploited by virulent Mycobacterium tuberculosis as their primary resident host cells by circumventing the macrophage's host defense armamentarium. M. tuberculosis, the causative agent of human tuberculosis (TB), successfully escapes host defense to establish infection by modulating intracellular trafficking and intracellular calcium signaling as well as through induction of necrotic cell death. However, exposure of macrophages to extracellular ATP and interferon-gamma (IFN-γ) promotes phagosome acidification, generation of reactive nitrogen species (RNS), and apoptosis over necrotic cell death, and hence intracellular killing of M. tuberculosis. Mutants lacking the region of differentiation-1 (RD1) encompassing the esx1-encoded type 7 secretion system, beside others, are attenuated by failing both, inhibition of phagosome maturation and induction of necrotic cell death. The Esx1-associated small secreted proteins, early secretory antigen target-6 (ESAT-6) and culture filtrate protein 10 (CFP10), are involved in inhibition of phagosome acidification (de Jonge et al., 2007).
Among the protein family of calcium ion channels, TRP ion channels are involved in immune responses within the lung microenvironment (Venkatachalam and Montell, 2007). TRPA1 is expressed in foamy macrophages and regulates cholesterol metabolism and anti-inflammatory response. TRPM8 and TRPV1 are expressed in lung epithelial cells and are crucial for the synthesis of proinflammatory cytokines, e.g., interleukin (IL)-6, IL-8, tumor necrosis factor (TNF)-α, IL-1α, and IL-1β (Sabnis et al., 2008; Reilly et al., 2005). TRPV4 is highly expressed in alveolar epithelial cells, endothelial cells, neutrophils, and macrophages and important for pro-inflammatory responses, goblet cell recruitment, and airway wall thickening during inflammatory lung diseases (Jia et al., 2004, Moran et al., 2011, Suresh et al., 2015; Gombedza et al., 2017). Consequently, pharmacological inhibitors of TRPV4 have been considered as therapeutics of acute lung injury (Balakrishna et al., 2014). Inhibition of TRPV4 is associated with impaired in vitro neutrophil responses including chemotaxis (Yin et al., 2016). M. tuberculosis is known to inhibit calcium signaling in macrophages to reduce phago-lysosome fusion and secure intracellular survival (Malik et al., 2000). These studies strongly suggest that calcium regulation by TRPV4 may be crucially involved in inflammatory lung diseases. To date, there is no report on the role of TRPV4 in M. tuberculosis infection. We found that wild-type (WT) M. tuberculosis, but not an attenuated ΔRD1 mutant, can down-regulate Trpv4 expression, thereby inhibiting intracellular calcium mobilization. IFN-γ-activated Trpv4−/− macrophages failed to restrict M. tuberculosis growth due to limited phagosome maturation and nitrite (NO2-) production. Trpv4−/− mice showed higher M. tuberculosis lung burden associated with lower proinflammatory responses at early time points of infection. However, in the chronic phase of infection, Trpv4-deficient mice were superior to WT mice in controlling mycobacterial growth. Our data indicate that M. tuberculosis alters TRPV4 expression to facilitate the infection progress. At the late phase of infection, though, TRPV4 facilitates mycobacterial growth indicating TRPV4 as a host-directed therapeutic target for subsidiary treatment of antibiotic therapy of TB.
Results
Trpv4 Expression in Macrophages Is Altered by M. tuberculosis
To understand Trpv4 regulation during M. tuberculosis infection, we analyzed TRPV4 in M. tuberculosis-H37Rv-infected murine bone marrow-derived macrophages (BMDM) at protein level. Confocal microscopy revealed association of TRPV4 with the macrophage plasma membrane at 2 h (Figure 1A, upper panel) but dispersed localization at 24 h post-infection (p.i.) (Figure 1A, lower panel). Quantification of TRPV4 protein expression in infected BMDM lysates by western blot showed M. tuberculosis-H37Rv induced TRPV4 down-regulation at both 24 and 48 h p.i. (Figure 1B). In contrast, infection with M. tuberculosis ΔRD1 transiently down-regulated TRPV4 expression at 24 h but not 48 h p.i. Analysis of Trpv4 mRNA expression by qRT-PCR in human monocyte-derived macrophages revealed a multiplicity of infection (MOI)-dependent increase in transcript numbers between MOI 1 and 3, which became, however, reduced again at MOI 10 (Figure 1C). These results demonstrate that infection of macrophages with virulent M. tuberculosis-H37Rv redistributes TRPV4 and regulates its expression in an MOI-dependent manner and that loss of TRPV4 is probably not due to reduced, but rather partially compensated, by higher transcription rates.
Figure 1.

Trpv4 Expression and Mycobacterial Survival in Infected Macrophages
(A) Bone marrow-derived macrophages (BMDM) isolated from C57BL6/J WT mice were infected with M. tuberculosis-GFP (Mtb-GFP) at MOI 5, and the cells were fixed with 4% PFA 2 and 24 h p.i. TRPV4 was detected by immuno-cytochemistry using Cy3-conjugated TRPV4 antibody. Images were acquired using Leica TSC SP5 confocal microscope (scale bar = 5 μm). Mean fluorescence intensity (MFI) of TRPV4 staining was quantified by using Fiji ImageJ software. Statistical analysis was performed with two-way ANOVA. Mean ± SEM (n = 3), ∗p < 0.05.
(B) BMDM from C57BL6/J WT mice were infected with M. tuberculosis-H37Rv and M. tuberculosis ΔRD1 (MOI 5). Western blot analysis was performed to determine the TRPV4 expression using TRPV4-specific antibody at 2, 24 and 48 h p.i. Densitometry analysis of protein bands was performed using Fiji Image J software.
(C) Human monocyte-derived macrophages were infected with M. tuberculosis-H37Rv for 24 h at different MOI as indicated, and the mRNA level of TRPV4 was studied with respect to the reference gene HPRT (hypoxanthine-guanine phosphoribosyltransferase).
(D–F) BMDM (D), alveolar macrophages (E), and peritoneal macrophages (F) were isolated from WT and Trpv4−/− mice; pre-treated with IFN-γ (500 units/mL) overnight; and then infected with M. tuberculosis (MOI 1) for 2 h. Cells were lysed with 0.5% Triton X-100 at different time points, and the intracellular M. tuberculosis counts were assessed by CFU assay.
(G) WT and Trpv4−/− mice BMDM were infected with M. tuberculosis or M. tuberculosis ΔRD1, and the intracellular bacterial burden was assessed by CFU assay after cell lysis with 0.5% Triton X-100 at indicated time points. Western blot and confocal images are representative of three independent experiments. In case of CFU assay, “n” corresponds to number of independent experiments. Statistical analysis was performed with two-way ANOVA Bonferroni post-tests. For mRNA expression of TRPV4, statistical analysis was performed with one-way ANOVA. Mean ± SD, ∗p < 0.05,∗∗p < 0.01, ∗∗∗p < 0.001.
M. tuberculosis Survives in Activated Macrophages in the Absence of Trpv4
To assess the role of Trpv4 in intracellular survival and growth of M. tuberculosis, we compared mycobacterial burden in WT and Trpv4−/− BMDM, alveolar, as well as peritoneal, macrophages, which were either left at the resting state or were stimulated with IFN-γ (Figures 1D–1F). Compared with WT cells, we observed a slightly better growth of M. tuberculosis-H37Rv in resting Trpv4-deficient BMDM and alveolar, but not peritoneal, macrophages. However, IFN-γ-activated Trpv4−/− macrophages of all three types were significantly less capable to restrict the growth of M. tuberculosis counts at 48 and 72 h p.i. when compared with WT cells (Figures 1D–1F). Notably, resting WT but not Trpv4−/− BMDM were able to control intracellular growth of the M. tuberculosis ΔRD1 mutant 48 and 72 h p.i. (Figure 1G). Similarly, Mycobacterium smegmatis, otherwise controlled by WT BMDM were able to grow in Trpv4−/− cells (Figure S1A). These data were further corroborated by experiments wherein we pretreated the RAW264.7 macrophage cell line with the pharmacological TRPV4 inhibitor RN1734 (10 μM) followed by M. tuberculosis infection. Higher M. tuberculosis counts were found in RN1734-treated RAW cells when compared with mock-treated controls at 48 h p.i. (Figure S1B). To control for differential phagocytosis rates between resting and IFN-γ-activated WT and Trpv4−/− macrophages, which may influence subsequent growth, we counted M. tuberculosis numbers at 2 h p.i., which, however, were comparable between resting and activated WT and Trpv4−/− BMDMs (Figure S1C). Taken together, these findings pinpoint TRPV4 as an important host factor for the control of intracellular M. tuberculosis by macrophages, in particular when the anti-microbial potential of these cells is potentiated by IFN-γ activation.
Trpv4 Is Involved in Intracellular Trafficking of M. tuberculosis
Virulent mycobacteria are able to survive in resting macrophages by interfering with phagosome maturation by several mechanisms, including attenuation of intracellular calcium (Deretic and Fratti, 1999 ;Tejle et al., 2002), whereas IFN-γ-activated macrophages control mycobacteria, in part by promoting phagosome maturation (Ni Cheallaigh et al., 2016). We studied intracellular trafficking of mycobacteria in WT versus Trpv4−/− macrophages between 5 min and 24 to 48 h p.i. M. tuberculosis-GFP co-localized with the early endosomal GTPase, Rab5, in both, IFN-γ-activated WT and Trpv4−/− macrophages 5 min p.i. At 24 h p.i., mycobacteria in Trpv4−/− macrophages were still mostly co-localizing with Rab5, indicating retention in early phagosomes in the absence of Trpv4, whereas mycobacteria-containing phagosomes in WT macrophages were Rab5 negative (Figures 2A, 2B, and S2A). In line with this view, M. tuberculosis-H37Rv were found in LysoTracker-positive compartments in IFN-γ-activated WT macrophages 48 h p.i. but only to a small extent in Trpv4−/− BMDM (Figures 2C and S2B). These results indicate that in the absence of Trpv4, IFN-γ-activated macrophages failed to effectively deliver M. tuberculosis into acidic phago-lysosomal compartments.
Figure 2.

Localization of M. tuberculosis and Intracellular pH Measurement in BMDM Isolated from WT and Trpv4−/− Mice
(A) WT and Trpv4−/− BMDM were pre-treated with IFN-γ (500 units/mL) overnight. Cells were infected with M. tuberculosis-GFP (MOI 5), fixed with 4% PFA at indicated time points, and co-localization of M. tuberculosis with Rab5 (arrow marked) was studied using Rab5-specific antibody under Leica TSC SP5 confocal microscope. Images were processed with IMARIS software (Scale bar = 5 μm).
(B) At least 100 cells were analyzed to quantify the M. tuberculosis and Rab5 co-localization.
(C) IFN-γ-stimulated or unstimulated WT and Trpv4−/− mice BMDM were infected with M. tuberculosis-GFP (MOI 5). LysoTrackerRed (100nM) was added to the cells 48 h p.i., incubated for 1 h in dark at 37°C and 5% CO2, and fixed with 4% PFA. Colocalization of M. tuberculosis-GFP and lysosome was studied using Leica TCS SP5 confocal microscope, and images were processed with IMARIS software (Scale bar = 10 μm).
(D) WT and Trpv4−/− BMDM were incubated with PFA-fixed FITC-labeled Escherichia coli, and live-cell imaging was performed by confocal microscopy with 488- and 458-nm filter to track E. coli containing phagosome. Phagosomal pH was measured according to pH sensitivity of FITC at ratio 488/458 nm. Experiments were performed under live-cell imaging system with TSC SP5 confocal microscope. “n” corresponds to number of FITC-E. coli-containing phagosomes. Statistical analysis was performed with two-way ANOVA Bonferroni post-tests. Experiments were performed in triplicates. Mean ± SD, ∗p < 0.05, ∗∗<p < 0.01, ∗∗∗p < 0.001.
The influence of TRPV4 on phagosome maturation was further analyzed by determining the phagosomal pH in WT and Trpv4−/− macrophages upon uptake of non-viable model particles, i.e., fluorescein isothiocyanate (FITC)-labeled paraformaldehyde (PFA)-fixed (PF) E. coli. We employed the pH-sensitive property of FITC, which is quenched at acidic pH when excited at 490 nm, but not at 440 nm (Nunes et al., 2015). In TRPV4-deficient macrophages, acidification of PF E. coli phagosomes was significantly delayed when compared with WT cells (Figure 2D). These results demonstrate that Trpv4 plays a crucial role in phagosome acidification and maturation, which are important prerequisites to control intracellular pathogens.
Trpv4 Influences Generation of Reactive Nitrogen Species and Cell Death upon M. tuberculosis Infection
RNS production and apoptotic cell death represent anti-mycobacterial effector mechanisms of IFN-γ-activated macrophages controlling M. tuberculosis, at least in mice. It has been shown before that activation of TRPV4 induces RNS production in macrophages (Hamanaka et al., 2010). Therefore, we measured NO2- production of IFN-γ-activated WT and Trpv4−/− BMDM in response to either WT or ΔRD1 M. tuberculosis-H37Rv (MOI 5) 24 h p.i. When compared with WT macrophages, we found significantly less nitrite in Trpv4−/− ones upon infection with either WT or ΔRD1 M. tuberculosis (Figure 3A), which correlated with enhanced intracellular survival of M. tuberculosis in Trpv4−/− macrophages (Figure 1G).
Figure 3.

Determination of Nitrite Production and Cell Death in Mycobacteria-Infected WT and Trpv4−/− Mice BMDM
(A) WT and Trpv4−/− BMDM were incubated with or without IFN-γ overnight. Then cells were infected with M. tuberculosis or M. tuberculosis ΔRD1, and nitrite (NO2-) production was measured by Griess reagent after 24 h of infection using Biotek multiplate reader.
(B) BMDM were infected with M. tuberculosis or M. tuberculosis ΔRD1 (MOI 5), and cell death was studied by Sytox green (5 μM) assay with a fluorescence Ex/Em of 504/523 nm.
(C) WT and Trpv4−/− BMDM were incubated with or without IFN-γ overnight and infected with M. tuberculosis-DsRed. After 3 h of infection, extracellular bacteria were removed by washing, and 10 μM CellEvent caspase3/7 detection reagent in DMEM media was added. Live-cell imaging for 72 h was performed using the biostation imaging system. M. tuberculosis-DsRed (red), and caspase3/7 activity (green) was acquired in Cy3 and FITC channel, respectively. Arrows indicates M. tuberculosis (red) in the extracellular environment after cell death (Scale bar of magnified image = 10 μm). Each experiment was performed in triplicate. Statistical analysis was performed with two-way ANOVA Bonferroni post-tests. Mean ± SD, ∗p < 0.05,∗∗<p < 0.01, ∗∗∗p < 0.001.
Intracellular growth of M. tuberculosis drives host cell death through oxidative stress-mediated genomic instability (Mohanty et al., 2016). Therefore, we employed Sytox green to assess necrotic cell death in WT versus Trpv4−/− BMDM upon infection with WT or ΔRD1 M. tuberculosis. We observed significantly higher amounts of DNA released from the cells indicating necrotic cell death in both WT and ΔRD1 M. tuberculosis-infected Trpv4−/−macrophages when compared with WT cells. Of note, even the ΔRD1 M. tuberculosis mutant, which usually causes apoptotic rather than necrotic macrophage cell death, led to higher necrosis rates in Trpv4−/− than in WT macrophages (Figure 3B). To monitor cell death kinetics, we performed live-cell imaging of IFN-γ-activated M. tuberculosis-H37Rv-infected BMDM incubated with the fluorogenic caspase3/7 substrate as indicator for apoptosis between 3 and 72 h p.i. (Figure 3C, and Video S1). Trpv4−/−macrophages succumbed much earlier, i.e., between 24 and 48 h p.i., to M. tuberculosis infection likely due to higher intracellular mycobacterial burden. Removal of dead cell aggregates by bystander macrophages eventually resulted in higher rates of cell death in Trpv4−/− cell cultures when compared with WT ones. Notably, we also observed non-apoptotic cell death independent of caspase3/7-mediated apoptosis corroborating the necrotic cell death results shown in Figure 3B and Video S1. Taken together, these data indicate that intact TRPV4 protects host cells from M. tuberculosis-induced cell death.
Live-cell imaging video of caspase3/7 assay in Mtb-infected WT and Trpv4−/− macrophages. M. tuberculosis-DsRed (red) and caspase3/7 activity (green) was acquired in Cy3 and FITC channel, respectively. Related to Figure 3C.
Role of Trpv4 in the Restriction of M. tuberculosis Growth in Infected Mice
Next, we investigated the role of Trpv4 in vivo during M. tuberculosis infection. Upon aerosol infection with M. tuberculosis-H37Rv (100 colony-forming unit [CFU]), no significant differences in the survival rates of WT and Trpv4−/− mice were observed during an observation period of up to 150 days p.i. (data not shown). Body weight analysis showed no difference between infected and non-infected WT mice up to 90 days p.i. In contrast, reduced weight gain was observed in M. tuberculosis-infected Trpv4−/− mice of the same age starting at day 21 p.i. when compared with uninfected ones (Figure 4A). It should be noted that uninfected Trpv4−/− mice, when compared with WT ones, continued to gain weight over a 7-week observation period.
Figure 4.

Determination of M. tuberculosis-H37Rv Survival in WT and Trpv4−/−Mice
(A) Body weights of M. tuberculosis-H37Rv-infected and uninfected WT and Trpv4−/− mice were measured during the course of infection and presented as mean weight in g.
(B) WT and Trpv4−/− mice were exposed to M. tuberculosis aerosols (100 CFU), and bacterial burden in lungs was assessed at indicated time points by CFU assay. “n” indicates number of mice. Each point corresponds to one mouse. Statistical analysis was performed with two-way ANOVA Bonferroni post-tests. ∗∗p < 0.01.
(C) Acid fast staining was performed to determine the M. tuberculosis (pink and arrow marked) burden in infected WT and Trpv4−/− mice lungs at day 29 p.i. Images were taken from three different fields (scale bar= 20 μm).
(D) M. tuberculosis-infected lung sections from WT mice were prepared at indicated time points after infection, immune-stained with Trpv4 antibody (ACC034 Alomone lab 1:200), and then developed with oxidation of 3,3'-diaminobenzidin (DAB) staining (brown) to check Trpv4 expression. Uninfected Trpv4−/− and WT lung sections were used as negative and positive controls, respectively (scale bar of magnified image= 20 μm).
(E–G) (E) Lung lesions (indicated by arrow) in M. tuberculosis-infected WT and Trpv4−/− mice are depicted for the different time points p.i. H & E staining of (F) day 29 and (G) day 55 post M. tuberculosis-infected WT and Trpv4−/− mice lung section indicates immune cell infiltration. Images were acquired with Olympus BX41 light microscope with 10X magnification.
At 29 days p.i., both, CFU and Ziel-Nielssen's staining showed higher M. tuberculosis counts in Trpv4−/− lungs than in WT ones (Figures 4B and 4C). At day 55 and 90 p.i. both, WT and Trpv4−/− mice were similarly able to control the mycobacteria. To our surprise, significantly lower M. tuberculosis counts were found in lungs from Trpv4−/− mice at day 150 p.i. when compared with lungs from WT animals. In spleens of Trpv4−/− mice, we also found less M. tuberculosis counts at day 150 p.i. when compared with WT ones, despite almost similar counts at earlier time points p.i. (Figure S3A). No significant differences in mycobacterial counts between WT and Trpv4−/− mice were seen in the livers during the entire course of infection (Figure S3B). Immuno-histological analysis of WT lungs showed TRPV4-expressing cells at the periphery of granuloma, whereas cells present in the center showed reduced TRPV4 expression (arrow marked) (Figure 4D). Pulmonary inflammatory lesions (arrow marked) were larger in WT compared with Trpv4−/− mice at days 55, 90, and 150 p.i. (Figure 4E). H&E staining revealed larger areas of inflammatory infiltrates at the site of infection in M. tuberculosis-infected WT mouse lungs at day 29 (Figure 4F) and day 55 (Figure 4G) p.i. when compared with M. tuberculosis-infected Trpv4−/−mouse lungs. These data indicate that TRPV4 is involved in the immune responses, which control M. tuberculosis burden in mice, however, with opposite functions, i.e., protective versus pathological, in the early versus late stage of infection.
M. tuberculosis utilizes host lipid droplets as nutrient source for growth in granulomas (Daniel et al., 2011). Using oil red O staining of lung tissue sections, we already noticed more lipid droplets in lungs from uninfected WT mice when compared with Trpv4−/−ones (Figure S3D). More importantly, at day 55 p.i., M. tuberculosis-infected WT mice showed intense accumulation of lipid bodies in the lungs, which was more pronounced when compared with Trpv4−/−mice (Figures 5A and S3C).
Figure 5.

Analysis of Lipid Bodies, Cytokine, Chemokine, and Nitrite Production in M. tuberculosis-Infected WT and Trpv4−/− Mice
(A) Lipid staining of lung sections obtained from WT and Trpv4−/− mice was performed using oil red O at day 55 post infection (nuclei, blue; lipid bodies, red). Lung lysates from M. tuberculosis-infected WT and Trpv4−/− mice were prepared at different time points (Scale bar = 200 μm).
(B–H) Protein estimation was performed to ensure use of equal protein concentration in analysis. The production of (B) IFN-γ, (C) IL-17A, (D) TNF-α, (E) IL-1β, (F) MCP-1, (G) MIP-2, and (H) IL-10 were determined using MSD assay kit.
(I) Nitrite production was measured in lung lysates prepared from M. tuberculosis-infected WT and Trpv4−/− mice at 15 day p.i. using nitrate reductase and Griess reagent. The NO2- production was represented as the absorbance of Griess reagent at 543 nm. “n” corresponds to number of mice. Statistical analysis was performed with two-way ANOVA Bonferroni post-tests. Mean ± SEM, ∗p < 0.05, ∗∗<p < 0.01, ∗∗∗p < 0.001.
Reduced Pro-inflammatory Responses in Trpv4−/− Mice upon M. tuberculosis Infection
Infiltration of immune cells to the site of infection is crucial for the restriction of M. tuberculosis growth in vivo. Analysis of a panel of cytokines and chemokines in lung lysates of M. tuberculosis-infected mice showed higher production of the pro-inflammatory cytokines, IFN-γ, IL-17A, TNF-α, and IL-1β in WT than in Trpv4−/− mice. IFN-γ production was enhanced in both, WT and Trpv4−/− compared with uninfected mice, although WT mice showed moderately higher IFN-γ production than Trpv4−/− ones (Figure 5B). We also observed higher concentrations of IL-17A, TNF-α, and IL-1β in WT than in TRPV4−/− lungs at day 29 p.i. (Figures 5C–5E). Upon infection, the production of keratinocyte chemoattractant (KC) was increased in WT when compared with Trpv4−/− lungs, whereas IL-6 secretion was only moderately but not significantly enhanced (Figures S4A and S4B). Similarly, we observed higher concentrations of the chemokines, monocyte chemoattractant protein-1 (MCP-1), and macrophage inflammatory protein 2 (MIP-2 or CXCL2) in lungs from WT when compared with those from Trpv4−/− mice (Figures 5F and 5G). In contrast, the anti-inflammatory cytokine, IL-10, was significantly increased in Trpv4−/− lung tissue samples taken at day 15 and, again, at day 55 p.i. when compared with WT lungs. Comparable IL-10 concentrations were observed at day 29 p.i. (Figure 5H).
Determination of NO2- production at day 15 p.i. with M. tuberculosis showed enhanced NO2- concentrations in the lungs from WT but not Trpv4−/− mice. No differences in nitrite concentrations were observed in serum samples (Figures 5I and S4C).
Differential TRPV4 Distribution in Granulomas from Patients with TB
To investigate whether TRPV4 is also present in granulomas of human patients with TB, we analyzed TRPV4 expression in tissue section of lung granulomas obtained from three patients with TB, who had undergone lung tissue resection. We observed more TRPV4-positive cells at the periphery of the granuloma in all three patients, whereas cells in the center showed much weaker TRPV4 signals (Figure 6A). Dual staining with antibodies against the CD68 macrophage marker and TRPV4 in consecutive sections showed that TRPV4 expression did partially overlap with CD68-positive macrophages but was also associated with CD68-negative cells. CD68-positive cells present at the periphery of granulomas expressed more TRPV4, whereas those present at the center of the granuloma showed less TRPV4 (Figures 6B and 6C).
Figure 6.

Analysis of TRPV4 Expression in Lung Granulomas of a Patient with TB
Lung tissue sections were obtained from patients with TB, who underwent anti-TB therapy and lung tissue resection as part of the treatment.
(A) Immunohistochemistry was performed using an antibody to TRPV4, followed by a species-specific secondary antibody and DAB development (TRPV4, brown; nuclei, blue).
(B and C) Human lung tissue sections of patients with TB were stained for TRPV4 and CD68 and developed by DAB staining showing expression of TRPV4 and CD68 in TB granuloma (TRPV4 and CD68, brown; nuclei, blue; Scale bar = 100 μm).
Neutrophils represent the predominant cell population infected with M. tuberculosis in pulmonary samples from patients with active TB and are thought to fail controlling the infection and rather drive exacerbation of inflammation (Dallenga et al., 2017, Eum et al., 2010). Using immunofluorescence staining for neutrophil elastase (NE), we observed increasing numbers of NE-positive neutrophils in lungs from M. tuberculosis-infected WT mice between day 55 and 150 p.i., whereas only few NE signals were found in Trpv4−/− lungs (Figures 7A and 7B). Notably, we also observed TRPV4-positive neutrophil aggregates in blood vessels in close proximity to the granulomatous tissue from patients with active TB (Figure 7C). In summary, TRPV4 in human TB lesions was primarily associated with macrophages and neutrophils, and in mice, recruitment of neutrophils was reduced in the absence of TRPV4 at later time points of M. tuberculosis infection, indicating a link between TRPV4 expression and neutrophil-associated inflammation.
Figure 7.

Expression of Neutrophil Elastase in M. tuberculosis-Infected Mouse Lung
(A–C) (A and B) WT and Trpv4−/−mice were infected with M. tuberculosis (100 CFU) via aerosol, and 4-μm cryosections of lung tissue samples were prepared after days 55 and 150 p.i. Tissue sections were stained by fluorescence immunohistochemistry assay for neutrophil elastase (green). Images were acquired from three different zones of infection (field) from each mouse; Scale bar=20 μm). Mean fluorescence intensity (MFI) of neutrophil elastase staining was quantified by using Fiji ImageJ software. (C) Human lung tissue sections of patients with TB were stained for TRPV4 showing positive signals in polymorphonuclear neutrophils in a blood vessel at the site of infection (Scale bar of R1, R2 = 20 μm).
Discussion
In immune cells, different ion channels such as voltage-gated calcium channels, non-selective calcium channel, and transient receptor potential (TRP) ion channels are crucial for the regulation of calcium levels to facilitate cellular signaling pathways and effector functions (Song et al., 2015). Here, we report that virulent, but not attenuated M. tuberculosis, down-regulated Trpv4 expression in murine macrophages, indicating that M. tuberculosis can interfere with TRPV4 function. Importantly, we revealed that M. tuberculosis-regulated Trpv4 expression may critically be involved in modulating TB disease progression. In the absence of Trpv4, resting but even more prominently, IFN-γ activated Trpv4−/−macrophages failed to control growth of intracellular M. tuberculosis. This in vitro susceptibility phenotype corresponded to the failure of Trpv4−/− mice in restricting pulmonary mycobacterial numbers in the early stage of infection during the growth phase of M. tuberculosis. In contrast, the lack of Trpv4 was associated with better control of mycobacteria at the later chronic stage of infection. Thus, TRPV4 has a biphasic function associated with an early protective but later permissive phenotype. Association of functional TRPV4 with higher susceptibility to M. tuberculosis at later stages of infection in the murine TB model was paralleled by the presence of TRPV4-expressing macrophages and neutrophils in the periphery of granulomas from chronically infected WT mice and patients with active TB.
As a hallmark of M. tuberculosis virulence, the pathogen is able to inhibit phagosome acidification, a prerequisite for phagosome maturation and phago-lysosome fusion (Wong et al., 2011). We found that, at 24 h p.i., WT but not ΔRD1 M. tuberculosis infection moderately reduced intracellular calcium levels in WT macrophages, indicating that M. tuberculosis virulence genes encoded by the RD1 region are important for TRPV4 down-regulation and hence, reduction of calcium ion concentration, which otherwise is a prerequisite for phago-lysosome fusion (Figures S4D and S4E). Thereby, M. tuberculosis is able to maintain an immature phagosomal state as niche for intracellular growth, even in IFN-γ-activated macrophages (Trimble and Grinstein, 2007). This notion is supported by the retention of Rab-5 in M. tuberculosis phagosomes in TRPV4-deficient macrophages. This is not seen in mycobacterial phagosomes of WT macrophages, which efficiently delivered WT M. tuberculosis into phago-lysosomes. In fact, we also observed lower calcium uptake in resting Trpv4−/− compared with WT macrophages when incubated with ionomycin Ca2+ (Figure S4F). Increased cytosolic calcium levels enhance phagosome acidification. Accordingly, inhibition of calcium signaling by M. tuberculosis can interfere with phago-lysosome fusion, thereby promoting the pathogen's intracellular survival (Malik et al., 2000). Beyond mycobacteria, hampered phagosome maturation in the absence of TRPV4 seems a general phenotype as acidification of phagosomes containing fixed E. coli cells was also delayed. Taken together, our results suggest that virulent M. tuberculosis, in addition to other virulence mechanisms, target Trpv4, thereby impairing phagosome acidification and maturation to secure intracellular survival and growth. To see whether the in vitro susceptibility phenotype of Trpv4−/− macrophages is also relevant for bacterial infections other than mycobacterial ones, we infected WT versus Trpv4−/− BMDM with Salmonella typhimurium. In contrast to mycobacteria, the salmonellae grew similarly in WT and Trpv4−/− macrophages (data not shown), indicating that the observed phenotype is mycobacteria specific.
M. tuberculosis infection promotes apoptosis of human alveolar macrophages (Keane et al., 1997). Staining for active caspase3/7 revealed that Trpv4−/−macrophages infected with M. tuberculosis succumbed to apoptotic cell death earlier than WT cells, which was likely due to higher intracellular mycobacterial burden 36 h p.i. Using live-cell imaging, we observed that apoptotic cells were engulfed by bystander macrophages leading to removal of more than 90% Trpv4-/-macrophages by 72 h p.i. These results indicate a relevance of Trpv4 in M. tuberculosis-mediated host cell death.
Aerosol infection of Trpv4−/−mice and their WT littermates with M. tuberculosis revealed that the lack of Trpv4 led to higher bacterial burden at day 29 p.i., but equal ones at days 55 and 90, and significantly lower ones at days 150 p.i. Reduced control of M. tuberculosis in mice lacking Trpv4 in the early stage of infection is likely due to the failure of TRP4-deficient macrophages to promote phagosome maturation and RNS production. Moreover, inflammatory responses were reduced in Trpv4−/− lungs when compared with WT littermates including smaller inflammatory infiltrates and granuloma-like structures at the site of infection. This could be due to lower concentrations of MCP-1 and MIP-2, which are involved in the recruitment of immune cells to the infection sites, in M. tuberculosis-infected Trpv4−/− versus WT lungs (Figures 4F and 4G), which corroborates previous observations (Ye et al., 2012). IFN-γ and IL-17 production was increased in lungs of WT, but not of Trpv4−/−, mice at day 29 post M. tuberculosis infection and associated with reduced TNF-α production otherwise important for optimal macrophage activation. These data suggest that Trpv4−/− mice failed to develop sufficient innate immune responses to combat M. tuberculosis. In contrast, enlarged lung lesions and higher concentrations of pro-inflammatory cytokines and effectors produced in WT when compared with Trpv4−/− mice were likely instrumental in controlling M. tuberculosis growth in the presence of Trpv4. Increased secretion of the anti-inflammatory cytokine, IL-10, in Trpv4−/− versus WT mice infected with M. tuberculosis is likely responsible for this reduced Th1 response observed. Enhanced influx of neutrophils in WT compared with TRPV4−/− mice at the later stage of infection is likely contributing to enhanced inflammation in the presence of TRPV4. Inhibited TRPV4 has been shown to limit neutrophil functions including chemotaxis (Yin et al., 2016). Excessive secretion of neutrophil elastase by activated neutrophils is involved in the control of M. tuberculosis, but at the same time, can also cause tissue damage and pathology. Thus, TRPV4 has a protective role in M. tuberculosis infection during the early innate immune response phase before T cell immunity kicks in, but a detrimental one later in infection when exacerbated inflammatory responses including neutrophil influx maintain mycobacterial growth.
The appearance of lipid-storing macrophages as characterized by lipid droplets (LD) is a histopathological hallmark of the chronically M. tuberculosis-infected murine lung later in infection. LD formation in M. tuberculosis-infected macrophages has been suggested to be a programmed host response coordinated by IFN-γ (Knight et al., 2018). However, M. tuberculosis can utilize fatty acids stored in LDs as carbon source for intracellular growth. Thus, our observation that pulmonary lesions in Trpv4−/− mice contained less LDs than those in WT mice may also account for lower mycobacterial loads in Trpv4−/− compared with WT lungs at late stage of infection, i.e., day 150. This phenotype is likely caused by a reduced IFN-γ and neutrophil-driven inflammatory environment promoted by enhanced IL-10 production in the absence of Trpv4. Apart from providing a food source for mycobacteria, LDs are also involved in the production of pro-inflammatory eicosanoids such as prostaglandin E2, which promote neutrophil influx into the infected tissue. As such, lower numbers of LDs in lungs of M. tuberculosis-infected Trpv4−/− mice might be responsible for reduced inflammatory responses and neutrophil aggregations when compared with WT lungs (Kaul et al., 2012, Saka and Valdivia, 2012).
The relevance of TRPV4 in M. tuberculosis infection identified by our experimental studies in mice prompted us to analyze its presence in human TB lesions. Immuno-histopathological analyses of lung granuloma sections from three patients with TB showed TRPV4-expressing macrophages at the periphery of the necrotic granuloma, whereas those in the centers showed less TRPV4 expression. Exposure of macrophages to M. tuberculosis and its metabolites, which happens more likely in the center of TB granulomas, as well as subsequent necrotic cell death may account for the reduced TRPV4 expression observed. It should, however, be noted that the patients with TB, from which the tissue samples were derived, had a history of long-term anti-TB treatment. Nevertheless, surgery was needed to overcome prolonged culture positivity and imminent treatment failure. Consequently, bacterial loads through acid-fast staining could not be visualized despite the strong and obvious granulomatous lesions present. Notably, TRPV4 was also observed in neutrophils in the granuloma-adjacent blood vessels. This finding supports previous reports that TRPV4 deficiency impairs neutrophil response to pro-inflammatory stimuli, production of reactive oxygen species, adhesion, and chemotaxis (Yin et al., 2016).
Taken together, we report a novel bifunctional role of the TRPV4 ion channel in host responses to M. tuberculosis infection in mice, in which TRPV4 is protective during the early innate but exacerbating and proinflammatory during the late chronic stage of infection. To this end, the presence of TRPV4-expressing myeloid cells in lung granulomas indicates TRPV4 as a potential target to be explored for host-directed therapy to control TB disease progression and support antibiotic treatment.
Limitations of the Study
In this study, although we have observed down-regulation of TRPV4 by pathogenic M. tuberculosis H37Rv in macrophages, we believe that TRPV4 expression can also be regulated by other intracellular factors during in vitro culture in the absence of an infectious stimulus (Figure 1B uninfected condition). In our follow-up studies, we will compare TRPV4 expression in different innate and acquired immune cell populations from un-infected versus infected mice to delineated regulatory mechanisms, as well as the kinetics of immune cell frequencies during the infectious process to identify responses responsible for higher and lower susceptibility of Trpv4−/− mice early versus later in infection. Future availability of specific TRPV4 inhibitors employable in vivo, will allow to explore the host-directed therapy approach targeting TRPV4 in late stage of infection using a susceptible mouse model of TB. Ultimately, the cellular and molecular functions of TRPV4 and its putative interaction partners during macrophage responses to M. tuberculosis need to be approached using cell biology and biochemistry approaches. Our observation of a decreased neutrophil influx in lungs of M. tuberculosis-infected Trpv4−/− mice late in infection also requires further investigations to understand the underlaying molecular and immunological mechanisms of how TRPV4 is involved here and how this knowledge can be explored to improve treatment of tuberculosis.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Prof. Dr. Ulrich E. Schaible (uschaible@fz-borstel.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets/code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by grant (BT/PR23317/MED/29/1186/2017) from Department of Biotechnology, Government of India, and Alexander von Humboldt Fellowship (Ref 3.3-IND-1152176-HFST-E) to A.S. We would like to thank the members of the Sonawane as well as the Schaible labs for fruitful suggestions and discussions. S.K.N. would like to acknowledge the University Grant Commission, Government of India, for Rajiv Gandhi national fellowship (RGNF-2013-14-ST-ORI-44090) and German Academic Exchange Service for DAAD Scholarship. We are thankful to Dr. Chandan Goswami lab, NISER, Bhubaneswar, India for their help in providing reagents for intracellular calcium analysis. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author Contributions
S.K.N. designed, performed and analyzed the experiments and wrote the manuscript; K.P. performed the experiments and analyzed the data; J.E. and V.S. provided technical assistance and training; M.H. wrote the ethical approval for animal experiment; B.K. as clinician recruited patients with TB; B.K. and N.R. provided the lung samples from patients with TB; W.L. created the Trpv4−/− mice; W.M.K. provided the Trpv4−/− mice and revised the manuscript; U.E.S. designed experiments, analyzed the data, and contributed to writing the manuscript; A.S. designed experiments, analyzed the data, obtained funding, and wrote the manuscript.
Declaration of Interests
The authors have no conflict of interest.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101206.
Contributor Information
Ulrich E. Schaible, Email: uschaible@fz-borstel.de.
Avinash Sonawane, Email: asonawane@iiti.ac.in.
Supplemental Information
References
- Alpizar Y.A., Boonen B., Sanchez A., Jung C., Lopez-Requena A., Naert R., Steelant B., Luyts K., Plata C., De Vooght V. TRPV4 activation triggers protective responses to bacterial lipopolysaccharides in airway epithelial cells. Nat. Commun. 2017;8:1059. doi: 10.1038/s41467-017-01201-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishna S., Song W., Achanta S., Doran S.F., Liu B., Kaelberer M.M., Yu Z., Sui A., Cheung M., Leishman E. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;307:L158–L172. doi: 10.1152/ajplung.00065.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallenga T., Repnik U., Corleis B., Eich J., Reimer R., Griffiths G.W., Schaible U.E. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe. 2017;22:519–530.e3. doi: 10.1016/j.chom.2017.09.003. [DOI] [PubMed] [Google Scholar]
- Daniel J., Maamar H., Deb C., Sirakova T.D., Kolattukudy P.E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7:e1002093. doi: 10.1371/journal.ppat.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge M.I., Pehau-Arnaudet G., Fretz M.M., Romain F., Bottai D., Brodin P., Honore N., Marchal G., Jiskoot W., England P. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J. Bacteriol. 2007;189:6028–6034. doi: 10.1128/JB.00469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V., Fratti R.A. Mycobacterium tuberculosis phagosome. Mol. Microbiol. 1999;31:1603–1609. doi: 10.1046/j.1365-2958.1999.01279.x. [DOI] [PubMed] [Google Scholar]
- Eum S.Y., Kong J.H., Hong M.S., Lee Y.J., Kim J.H., Hwang S.H., Cho S.N., Via L.E., Barry C.E., 3rd Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137:122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombedza F., Kondeti V., Al-Azzam N., Koppes S., Duah E., Patil P., Hexter M., Phillips D., Thodeti C.K., Paruchuri S. Mechanosensitive transient receptor potential vanilloid 4 regulates Dermatophagoides farinae-induced airway remodeling via 2 distinct pathways modulating matrix synthesis and degradation. FASEB J. 2017;31:1556–1570. doi: 10.1096/fj.201601045R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka K., Jian M.Y., Townsley M.I., King J.A., Liedtke W., Weber D.S., Eyal F.G., Clapp M.M., Parker J.C. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;299:L353–L362. doi: 10.1152/ajplung.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y., Wang X., Varty L., Rizzo C.A., Yang R., Correll C.C., Phelps P.T., Egan R.W., Hey J.A. Functional TRPV4 channels are expressed in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;287:L272–L278. doi: 10.1152/ajplung.00393.2003. [DOI] [PubMed] [Google Scholar]
- Kaul V., Bhattacharya D., Singh Y., Van Kaer L., Peters-Golden M., Bishai W.R., Das G. An important role of prostanoid receptor EP2 in host resistance to Mycobacterium tuberculosis infection in mice. J. Infect. Dis. 2012;206:1816–1825. doi: 10.1093/infdis/jis609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J., Balcewicz-Sablinska M.K., Remold H.G., Chupp G.L., Meek B.B., Fenton M.J., Kornfeld H. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect. Immun. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M.M., Kayastha B.B., Franklin M.J., Patrauchan M.A. Calcium regulation of bacterial virulence. Adv. Exp. Med. Biol. 2020;1131:827–855. doi: 10.1007/978-3-030-12457-1_33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight M., Braverman J., Asfaha K., Gronert K. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-gamma/HIF-1alpha signaling and supports host defense. PLoS Pathog. 2018;14:e1006874. doi: 10.1371/journal.ppat.1006874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik Z.A., Denning G.M., Kusner D.J. Inhibition of Ca(2+) signaling by Mycobacterium tuberculosis is associated with reduced phagosome-lysosome fusion and increased survival within human macrophages. J. Exp. Med. 2000;191:287–302. doi: 10.1084/jem.191.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty S., Dal Molin M., Ganguli G., Padhi A., Jena P., Selchow P., Sengupta S., Meuli M., Sander P., Sonawane A. Mycobacterium tuberculosis EsxO (Rv2346c) promotes bacillary survival by inducing oxidative stress mediated genomic instability in macrophages. Tuberculosis (Edinb.) 2016;96:44–57. doi: 10.1016/j.tube.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Moran M.M., McAlexander M.A., Biro T., Szallasi A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 2011;10:601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Ni Cheallaigh C., Sheedy F.J., Harris J., Munoz-Wolf N., Lee J., West K., McDermott E.P., Smyth A., Gleeson L.E., Coleman M. A common variant in the adaptor mal regulates interferon gamma signaling. Immunity. 2016;44:368–379. doi: 10.1016/j.immuni.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes P., Guido D., Demaurex N. Measuring phagosome pH by ratiometric fluorescence microscopy. J. Vis. Exp. 2015:e53402. doi: 10.3791/53402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly C.A., Johansen M.E., Lanza D.L., Lee J., Lim J.O., Yost G.S. Calcium-dependent and independent mechanisms of capsaicin receptor (TRPV1)-mediated cytokine production and cell death in human bronchial epithelial cells. J. Biochem. Mol. Toxicol. 2005;19:266–275. doi: 10.1002/jbt.20084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabnis A.S., Reilly C.A., Veranth J.M., Yost G.S. Increased transcription of cytokine genes in human lung epithelial cells through activation of a TRPM8 variant by cold temperatures. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;295:L194–L200. doi: 10.1152/ajplung.00072.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka H.A., Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 2012;28:411–437. doi: 10.1146/annurev-cellbio-092910-153958. [DOI] [PubMed] [Google Scholar]
- Song L., Cui R., Yang Y., Wu X. Role of calcium channels in cellular antituberculosis effects: potential of voltage-gated calcium-channel blockers in tuberculosis therapy. J. Microbiol. Immunol. Infect. 2015;48:471–476. doi: 10.1016/j.jmii.2014.08.026. [DOI] [PubMed] [Google Scholar]
- Suresh K., Servinsky L., Reyes J., Baksh S., Undem C., Caterina M., Pearse D.B., Shimoda L.A. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015;309:L1467–L1477. doi: 10.1152/ajplung.00275.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejle K., Magnusson K.E., Rasmusson B. Phagocytosis and phagosome maturation are regulated by calcium in J774 macrophages interacting with unopsonized prey. Biosci. Rep. 2002;22:529–540. doi: 10.1023/a:1022025903688. [DOI] [PubMed] [Google Scholar]
- Trimble W.S., Grinstein S. TB or not TB: calcium regulation in mycobacterial survival. Cell. 2007;130:12–14. doi: 10.1016/j.cell.2007.06.039. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K., Montell C. TRP channels. Annu. Rev. Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D., Bach H., Sun J., Hmama Z., Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc. Natl. Acad. Sci. U S A. 2011;108:19371–19376. doi: 10.1073/pnas.1109201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., Kleiner S., Wu J., Sah R., Gupta R.K., Banks A.S., Cohen P., Khandekar M.J., Bostrom P., Mepani R.J. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell. 2012;151:96–110. doi: 10.1016/j.cell.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Michalick L., Tang C., Tabuchi A., Goldenberg N., Dan Q., Awwad K., Wang L., Erfinanda L., Nouailles G. Role of transient receptor potential vanilloid 4 in neutrophil activation and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2016;54:370–383. doi: 10.1165/rcmb.2014-0225OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live-cell imaging video of caspase3/7 assay in Mtb-infected WT and Trpv4−/− macrophages. M. tuberculosis-DsRed (red) and caspase3/7 activity (green) was acquired in Cy3 and FITC channel, respectively. Related to Figure 3C.
Data Availability Statement
This study did not generate datasets/code.