

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has caused the coronavirus disease 2019 (COVID‐19) pandemic. Accurate detection of SARS‐CoV‐2 using molecular assays is critical for patient management and the control of the COVID‐19 pandemic. However, there is an increasing number of SARS‐CoV‐2 viruses with mutations at the primer or probe binding sites, and these mutations may affect the sensitivity of currently available real‐time reverse transcription‐polymerase chain reaction (RT‐PCR) assays targeting the nucleocapsid (N), envelope (E), and open reading frame 1a or 1b genes. Using sequence‐independent single‐primer amplification and nanopore whole‐genome sequencing, we have found that the nonstructural protein 1 (nsp1) gene, located at the 5′ end of the SARS‐CoV‐2 genome, was highly expressed in the nasopharyngeal or saliva specimens of 9 COVID‐19 patients of different clinical severity. Based on this finding, we have developed a novel nsp1 real‐time RT‐PCR assay. The primers and probes are highly specific for SARS‐CoV‐2. Validation with 101 clinical specimens showed that our nsp1 RT‐PCR assay has a sensitivity of 93.1% (95% confidence interval [CI]: 86.2%‐97.2%), which was similar to those of N and E gene RT‐PCR assays. The diagnostic specificity was 100% (95% CI: 92.9%‐100%). The addition of nsp1 for multitarget detection of SARS‐CoV‐2 can avoid false‐negative results due to mutations at the primers/probes binding sites of currently available RT‐PCR assays.
Keywords: COVID‐19, diagnosis, nanopore sequencing, nsp1, RT‐PCR, SARS‐CoV‐2
Highlights
Nanopore sequencing is useful in identifying highly expressed gene region for diagnostic tests.
nsp1 is a suitable alternative gene target for SARS‐CoV‐2 RT‐PCR.
1. INTRODUCTION
In 2003, severe acute respiratory syndrome coronavirus (SARS‐CoV) cross species barrier and became the first coronavirus to be associated with a high fatality rate in humans. 1 In 2020, SARS‐CoV‐2, first reported from Hubei province of China in December 2019, has become the first coronavirus to cause a pandemic. 2 , 3 Most patients with coronavirus disease 2019 (COVID‐19) present with respiratory symptoms, while 18% exhibit gastrointestinal symptoms. 4 Radiologically, COVID‐19 is characterized by multifocal and peripheral ground‐glass opacities, but 2.9% of severe cases did not show any abnormalities in their lung computed tomography. 2 , 5 Complications include acute respiratory distress syndrome, arrhythmia, secondary bacterial infection, and multiorgan failure. 6 , 9 Pathologically, COVID‐19 was characterized by diffuse alveolar damage, endothelialitis interstitial lymphocyte infiltrates, and multinucleated syncytial cells in the lung, and microvascular steatosis in the liver. 10 , 11
The importance of diagnostic testing is well demonstrated with COVID‐19. Due to a shortage of diagnostic tests, a large number of undiagnosed patients with relatively mild symptoms were unknowingly spreading the virus in the community, and has led to the large outbreaks in some countries. 12 Undocumented infection has been estimated to be the source of 79% of laboratory‐confirmed infections. 13
Laboratory confirmation of SARS‐CoV‐2 relies on accurate molecular assays. Many groups have shared their in‐house real‐time reverse transcription‐polymerase chain reaction (RT‐PCR) protocol with the World Health Organization during the early period of COVID‐19, which have tremendously helped clinical microbiology laboratories around the world in the detection of SARS‐CoV‐2. 14 Currently, the gene targets of real‐time RT‐PCR for SARS‐CoV‐2 include the open reading frame 1a or 1b (ORF1a or 1b), RNA‐dependent RNA polymerase (RdRp)/helicase (Hel), spike (S), envelope (E), and nucleocapsid (N) genes. 2 , 14 , 15 The choice of these gene targets were based on previous experience with SARS‐CoV and Middle East respiratory syndrome coronavirus (MERS‐CoV). 16 , 17
Similar to other RNA respiratory viruses, SARS‐CoV‐2 can mutate quickly due to the intrinsic infidelity of viral RNA polymerase. The mean evolutionary rate of SARS‐CoV‐2 has been estimated to be 1.8 × 10−3 substitutions per site per year. 18 Mutations arising at the current gene targets can lower the sensitivity of the existing assays. Currently, three major phylogenetic types have been identified, including two subclusters of type A (ancestral type), type B (characterized by T8782C of nsp4 gene and C28144T of orf8 gene) and type C (characterized by G26144T of orf3a gene). 19 According to an analysis by GISAID, 0.02% to 0.53% of the genomes contain mutations in the last five nucleotides of the 3′ ends of the published RT‐PCR primer regions. 20 Therefore, there is an urgent need to expand the number of gene targets that can be used for RT‐PCR diagnosis.
The aim of this study is to identify novel targets for molecular detection of SARS‐CoV‐2. In our previous study, we have successfully identified highly‐expressed gene regions for influenza A virus using nanopore sequencing, and have developed highly sensitive and specific novel PB2 and NS RT‐PCR for detection of human and zoonotic influenza A viruses. 21 Using the same strategy, we first used nanopore sequencing to identify highly‐expressed gene regions of the SARS‐CoV‐2 genome in clinical specimens. After identifying nonstructural protein 1 (nsp1) gene as a highly‐expressed gene region, we have designed the nsp1 RT‐PCR which demonstrated high sensitivity and specificity using clinical specimens.
2. MATERIALS AND METHODS
2.1. Clinical specimens
For nanopore sequencing, we have retrieved 22 original clinical specimens of 14 patients with high viral load. For the determination of analytical specificity, we have retrieved the total nucleic acid (TNA) that were extracted from 13 nasopharyngeal aspirate specimens previously found to be positive for human coronaviruses (OC43, NL63, HKU1, 229E) by FilmArray Respiratory Panel 2 (BioFire; Biomerieux), which werecollected between November 2018 and December 2019 (Table S1).
For clinical validation, we retrieved specimens that tested positive for SARS‐CoV‐2 using our in‐house RdRp/Hel assay reported previously. 15 These specimens included 101 clinical specimens collected from COVID‐19 patients admitted to Queen Mary Hospital, or from COVID‐19 patients admitted to Princess Margaret Hospital for whom specimens were sent to Queen Mary Hospital for viral load testing. For the 87 patients of Queen Mary Hospital, the specimens were collected within 24 hours after hospital admission. For the 14 patients from Princess Margaret Hospital, we have retrieved their first specimens that were sent to our laboratory for viral load testing. We have also retrieved 50 nasopharyngeal specimens that tested negative by LightMix Modular SARS and Wuhan CoV E‐gene kit, which were collected between 11 April and 14 April 2020. The TNA of all specimens were extracted using NucliSENS easyMAG extraction system (BioMerieux, Marcy l'Étoile, France). Specimens with insufficient volume of TNA were excluded from analysis.
This study was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (HKU/HK HKW IRB) (UW 13‐372). This study is reported according to the Standards for Reporting of Diagnostic Accuracy Studies (STARD) guideline. 22
2.2. Nanopore sequencing library
Nanopore sequencing was performed as we described previously with modifications. 2 , 3 To deplete host cells, nasopharyngeal or saliva specimens were centrifuged at 16 000g for 2 minutes, and supernatant was used for subsequent RNA extraction. RNA was extracted from 140 μL of supernatant using QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) as we described previously. RNA was DNase treated, concentrated and cleaned using RNA Clean & Concentrator‐5 (Zymo Research, Irvine, CA).
Sequence‐independent single‐primer amplification (SISPA) was performed as described previously. 2 , 3 Briefly, DNase‐treated RNA was reverse transcribed to single strand complementary DNA (cDNA) using primer A (5′‐GTTTCCCACTGGAGGATA‐N9‐3′). Second strand cDNA synthesis was performed using Klenow Fragment (3′→5′ exo‐) (New England BioLabs, Ipswich, MA). PCR using primer B (5′‐GTTTCCCACTGGAGGATA‐3′) was used in generating the amplified cDNA libraries. Nanopore sequencing library preparation was performed according to manufacturer's instructions for Ligation Sequencing Kit (SQK‐LSK109, Oxford Nanopore Technologies). Briefly, amplified PCR products were purified by 1× AMPure XP bead (Beckman Coulter, California, CA). Equal molar of each amplified PCR products were then subjected to DNA repair, end preparation, and native barcode ligation (EXP‐NBD104, Oxford Nanopore Technologies). Barcoded samples were pooled and were ligated to sequencing adaptor. Sequencing was performed with Oxford Nanopore MinION device using R9.4.1 flow cell for 12 to 48 hours.
After sequencing, Guppy v3.4.5 was used in converting the raw signal data into FASTQ format, demultiplexing, removal of nanopore and SISPA adaptor sequences. Only reads with a minimum Q score of 7 were included for subsequent analysis. The sequencing run was quality‐checked using MinIONQC. 23 Human reads were depleted by mapping to reference human genome hg38, and unmapped reads were extracted using SAMTools. 24 The nonhuman reads were mapped to the reference genome SARS‐CoV‐2 isolate Wuhan‐Hu‐1 (NCBI GenBank: MN908947.3). BCFtools Mpileup was used in creating a variant file. 25 BCFtools call, 25 vcfutils.pl, 24 and Seqtk seq 26 were used in generating the FASTA consensus sequence. Finally, the coverage data were obtained using SAMtools. 24 Only specimens with a mean coverage of 250× were included for further analysis. The consensus sequences of five specimens from four patients (HKU‐902a, HKU‐903a, HKU‐903b, HKU‐907b, and HKU‐908a) were previously deposited into NCBI GenBank (accession number: MT114412, MT114414, MT114415, MT114417, MT114418). 3 The consensus sequences of HKU‐904a, HKU‐905a, HKU‐911a, HKU‐913a, and HKU‐915a have been deposited into NCBI GenBank (accession number: MT365028‐MT365032). Raw reads, after excluding human reads, have been deposited into BioProject (Bioproject ID: PRJNA627286).
2.3. Phylogenetic analysis
The phylogenetic tree of the whole SARS‐CoV‐2 genome was constructed using neighbour‐joining method using MEGA software package version 7.0. The bootstrap values from 1000 replicates were calculated to evaluate the reliability of the phylogenetic trees. Phylogenetic types A, B, and C were defined as described previously. 19
2.4. Selection of primers and probes
Primers and probes targeting the nsp1 region was designed by the multiple alignment of SARS‐CoV‐2, other human coronaviruses in the genus Betacoronavirus (including lineage A HCoV‐OC43 and HCoV‐HKU1, lineage B SARS‐CoV, and lineage C MERS‐CoV), and a bat‐like SARS‐CoV (Bat/Yunnan/RaTG13/2013) which was closely related to the human SARS‐CoV‐2 (Figure 1). 27
Figure 1.
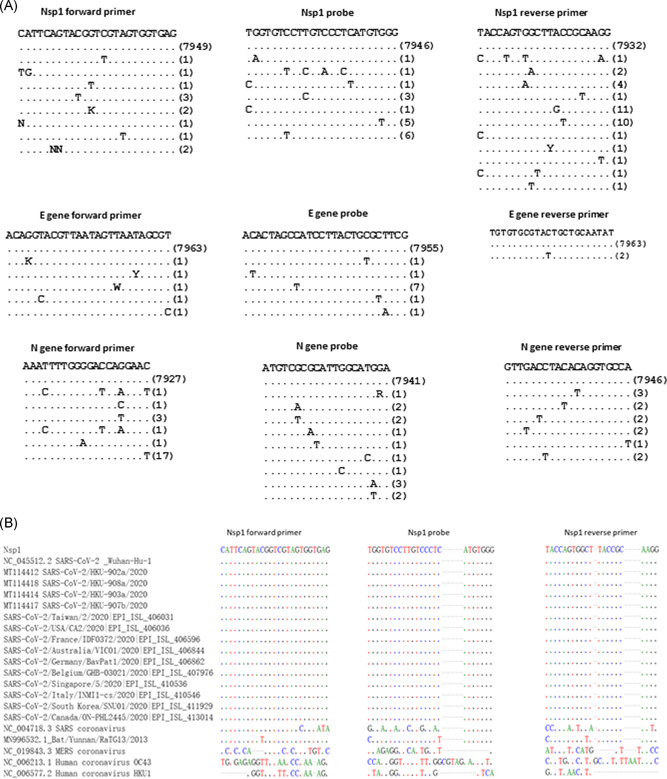
Alignment of primers and probes in this study. A, In silico analysis of nsp1, E and N gene primers/probe for SARS‐CoV‐2. Sequences from GISAID are shown in Table S2. B, Alignment of our novel nsp1 primers and probes with SARS‐CoV‐2 and other human coronaviruses in the genus Betacoronavirus, and the bat coronavirus Bat/Yunnan/RaTG13/2013. Sequences from GISAID are shown in Table S2. E, envelope; N, nucleocapsid; nsp1, nonstructural protein 1; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
2.5. Analytical sensitivity and specificity
The limit of detection (LOD) was determined using serially‐diluted SARS‐CoV‐2 virus culture isolates as described previously. 15 , 28 SARS‐CoV‐2 virus was cultured in VeroE6 cells. The concentration of the virus culture stock was 1.8 × 107 50% tissue culture infective doses (TCID50)/mL. Triplicates were performed for each dilution in two independent experiments.
Analytical specificity was determined using 13 clinical specimens positive for human coronaviruses 229E (n = 3), NL63 (n = 3), OC43 (n = 2), and HKU1 (n = 5), and from 17 virus culture isolates of SARS‐CoV, MERS‐CoV, HCoV‐229E, HCoV‐NL63, HCoV‐OC43, influenza viruses (A [H1N1], A [H3N2], B, C), respiratory syncytial virus, parainfluenza viruses 1 to 4, human metapneumovirus, rhinovirus/enterovirus and adenovirus as described previously. 28
2.6. Nsp1 gene real‐time RT‐PCR for nsp1 gene
Real‐time RT‐PCR targeting nsp1 gene was performed by QuantiNova Probe RT‐PCR Kit (Qiagen). A 20 μL reaction containing 4 μL of TNA, 10 μL 2× QuantiNova Probe RT‐PCR Master Mix, 0.2 μL QN Probe RT Mix, 1.6 μL of each 10 μM forward and reverse primer, 0.4 μL of 10 μM probe, and 2.2 μL nuclease‐free water. Thermal cycling was performed at 45°C for 10 minutes for reverse transcription, followed by 95°C for 5 minutes and then 45 cycles of 95°C for 5 seconds, 55°C for 30 seconds. The sequences of primers and probes are listed in Table 1.
Table 1.
Primers and probes used in this study
| Target (source) | Primer/probe | Sequence (5′‐3′) |
|---|---|---|
| nsp1 Gene (this study) | Forward | CATTCAGTACGGTCGTAGTGGTGAG |
| nsp1 Gene (this study) | Reverse | CCTTGCGGTAAGCCACTGGTA |
| nsp1 Gene (this study) | Probe | FAM‐CCCACATGAGGGACAAGGACACCA‐IABkFQ |
| E Gene (Corman et al 29 ) | Forward | ACAGGTACGTTAATAGTTAATAGCGT |
| E Gene (Corman et al 29 ) | Reverse | ATATTGCAGCAGTACGCACACA |
| E Gene (Corman et al 29 ) | Probe | FAM‐ACACTAGCCATCCTTACTGCGCTTCG‐BBQ |
| N Gene (Shirato et al 30 ) | Forward | AAATTTTGGGGACCAGGAAC |
| N Gene (Shirato et al 30 ) | Reverse | TGGCACCTGTGTAGGTCAAC |
| N Gene (Shirato et al 30 ) | Probe | FAM‐ATGTCGCGCATTGGCATGGA‐BHQ |
Abbreviations: E, envelope; N, nucleocapsid; nsp1, nonstructural protein 1.
2.7. E gene real‐time RT‐PCR
Real‐time RT‐PCR for E gene was performed according to a published protocol, except that the total reaction volume was reduced to 20 μL instead of 25 μL. 14 , 29 Briefly, superscript III one‐step RT‐PCR system with Platinum Taq Polymerase (Thermo Fisher Scientific, Waltham, MA) was used. A 20 μL reaction containing 4 μL of TNA, 10 μL 2× Reaction mix (containing 0.4 mM of each deoxyribonucleotide triphosphates (dNTP) and 3.2 mM magnesium sulfate), 0.8 μg of nonacetylated bovine albumin, 0.32 μL of a 50 mM magnesium sulfate solution, 0.8 μL of each 10 μM forward and reverse primer, 0.4 μL of 10 μM probe, 0.8 μL of Superscript III reverse transcriptase/Platinum Taq Mix, and 2.08 μL of nuclease‐free water. Thermal cycling was performed at 55°C for 10 minutes for reverse transcription, followed by 95°C for 3 minutes and then 45 cycles of 95°C for 15 seconds, 58°C for 30 seconds.
2.8. N gene real‐time RT‐PCR
Real‐time RT‐PCR for N gene was performed using primers and probes as described previously. 30 QuantiNova Probe RT‐PCR Kit (Qiagen) was used. A 20 μL reaction containing 4 μL of TNA, 10 μl 2× QuantiNova Probe RT‐PCR Master Mix, 0.2 μL QN Probe RT Mix, 1.0 and 1.4 μL of each 10 μM forward and reverse primer, respectively, 0.4 μL of 10 μM probe and 3.0 μL of nuclease‐free water. Thermal cycling was performed at 45°C for 10 minutes for reverse transcription, followed by 95°C for 5 minutes and then 45 cycles of 95°C for 5 seconds and 60°C for 30 seconds.
2.9. Statistical analysis
Statistical analysis was performed using PRISM 6.0 for Windows. The sensitivity of nsp1 and E gene real‐time RT‐PCT was compared using Fisher's exact test.
3. RESULTS
3.1. Nanopore sequencing of SISPA‐amplified genome
Nanopore sequencing of SISPA‐amplified genome was performed for a total of 22 specimens from 14 patients. Ten specimens from nine patients had a mean coverage of at least 250× and were included for further analysis. For one patient, both the nasopharyngeal and saliva specimen was included. For the other eight patients, there were three saliva and five nasopharyngeal specimens. All patients were hospitalized at Princess Margaret Hospital. The median age was 62 years. Four patients were female. Four patients required oxygen supplementation, two patients were admitted to the intensive care unit, and one patient died. Phylogenetic analysis using the whole viral genome showed that the virus strains from two patients belonged to 20905C subcluster of phylogenetic type A, and those from seven other patients belonged to type B (Figure 2).
Figure 2.
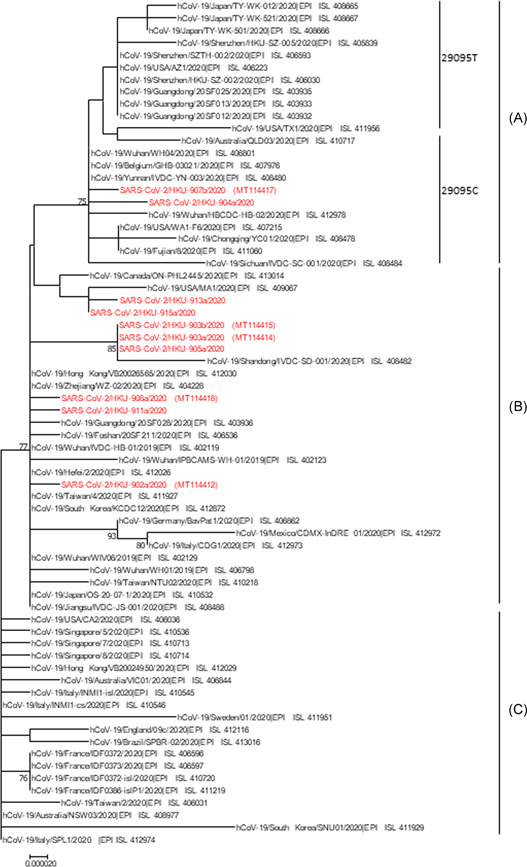
Phylogenetic tree of SARS‐CoV‐2 genomes. Nucleotide sequences were downloaded from NCBI Genbank and GISAID (Table S2). The phylogenetic tree was constructed by neighbour‐joining method with bootstrap values were calculated from 1000 trees. Sequences with coverage map showing in Figure 3A are highlighted in red. SARS‐CoV‐2, s evere acute respiratory syndrome coronavirus 2
To identify gene regions that were highly expressed, coverage map was visualized using integrative genomics viewer. The nsp1 gene region was highly expressed in all specimens (Figure 3A).
Figure 3.
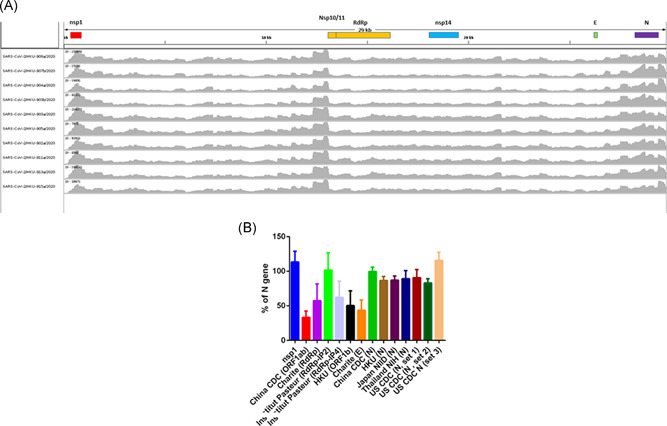
A, Coverage map of the nanopore sequencing of SISPA‐amplified viral genome. X‐axis shows the nucleotide position, while Y‐axis shows the number of reads. The coverage map was generated by integrative genomics viewer. B, Coverage information of nanopore sequencing for each real‐time RT‐PCR target region. The mean coverage of each RT‐PCR amplicon (nucleotide positions indicated in Table S3) is expressed as the percentage of the mean coverage of the entire N gene (nucleotide position 28274‐29533). Error bar indicates one standard deviation. RT‐PCR, reverse transcription‐polymerase chain reaction; SISPA, sequence‐independent single‐primer amplification
3.2. Nsp1 primers and probes
Specific primers and probes targeting SARS‐CoV‐2 were selected by aligning more than 7000 SARS‐CoV‐2 sequences downloaded from GISAID on 22 April 2020, other human Betacoronaviruses, and a bat SARS‐like coronavirus (Figure 1). The nsp1 primer and probe sequences are well‐matched with SARS‐CoV‐2 genome, but are distinct from 2003 SARS‐CoV, MERS‐CoV, OC43, and HKU1. Analysis showed that our nsp1 real‐time RT‐PCR target region was expressed more abundantly than the gene targets of other published real‐time RT‐PCR assay (Figure 3B).
The LOD of nsp1 gen was 18 TCID50/mL. Nsp1 RT‐PCR had tested negative for all 13 clinical specimens known to be positive for coronaviruses, five virus culture isolates of coronavirus (SARS‐CoV, MERS‐CoV, HCoV‐229E, HCoV‐NL63, HCoV‐OC43), and 12 virus culture isolates of other respiratory viruses (influenza virus A [H1N1] and A [H3N2], influenza B virus, influenza C virus, rhinovirus, adenovirus, respiratory syncytial virus, human metapneumovirus, and parainfluenza virus types 1‐4).
3.3. Diagnostic performance of nsp1 RT‐PCR
A total of 101 archived respiratory tract specimens collected between 29th January and 7th April 2020, and tested positive for SARS‐CoV‐2 by our‐house RdRp/Hel assay, were retrieved. Of these 101 COVID‐19 patients, SARS‐CoV‐2 was detected by at least one of nsp1, N or E gene RT‐PCR assays in 99 patients (98.0%), and 85 patients (84.2%) were detected by all three RT‐PCR assays (Table 2). Two patients (2%) were positive by nsp1 gene RT‐PCR only. The sensitivity was 93.1% (95% confidence interval [CI]: 86.2%‐97.2%) for nsp1 gene RT‐PCR, 95.1% (95% CI: 88.8‐98.4) for N gene RT‐PCR, and 89.1% (95% CI: 81.3‐94.4) for E gene RT‐PCR (Table 3).
Table 2.
Concordance of nsp1, N, and E gene RT‐PCR assays
| RT‐PCR | Number (%) (n = 101) | ||
|---|---|---|---|
| nsp1 | N | E | |
| + | + | + | 85 (84.2) |
| + | + | − | 6 (5.9) |
| + | − | + | 1 (1.0) |
| + | − | − | 2 (2.0) |
| − | + | + | 4 (4.0) |
| − | + | − | 1 (1.0) |
| − | − | + | 0 (0) |
| − | − | − | 2 (2.0) |
Note: +, positive; −, negative.
Abbreviations: E, envelope; N, nucleocapsid; nsp1, nonstructural protein 1; RT‐PCR, reverse transcription‐polymerase chain reaction.
Table 3.
Sensitivity and specificity of nsp1 gene RT‐PCR when compared with those of E gene and N gene RT‐PCR
| RT‐PCR target | Patients with COVID‐19 (n = 101) a | Patients without COVID‐19 (n = 50) | Sensitivity (95% CI) (%) |
|---|---|---|---|
| nsp1 | 94 | 0 | 93.1 (86.2‐97.2) |
| N | 96 | 0 | 95.1 (88.8‐98.4) |
| E | 90 | 0 | 89.1 (81.3‐94.4) |
Abbreviations: CI, confidence interval; COVID‐19, coronavirus disease 2019; E, envelope; N, nucleocapsid; nsp1, nonstructural protein 1; RT‐PCR, reverse transcription‐polymerase chain reaction.
Detected by RdRp‐Hel RT‐PCR.
To determine the diagnostic specificity of the assay, we tested nsp1 gene real‐time RT‐PCR on 50 nasopharyngeal specimens from non‐COVID‐19 patients. All 50 specimens tested negative by nsp1 RT‐PCR, and therefore, the specificity was 100% (95% CI: 92.9%‐100%)
4. DISCUSSION
In this study, we have developed a highly sensitive and specific nsp1 real‐time RT‐PCR for the detection of SARS‐CoV‐2. We first identified nsp1 to be a highly‐expressed gene target in clinical specimens using nanopore whole‐genome sequencing, and designed a real‐time RT‐PCR protocol based on nsp1 gene. This novel nsp1 real‐time RT‐PCR did not cross react with other human coronaviruses or other respiratory viruses, and has a sensitivity of 93.1%, which is similar to real‐time RT‐PCR targeting N and E genes. In silico analysis of the primers/probe regions showed that nsp1 is better than N gene which has fewer mutations in the 3′ end of the primers/probe region (Figure 1A). Together with the analysis done by GISAID, 20 which showed higher mutation rate in the published N gene primers/probe region, nsp1 can serve as an alternative target for the detection of SARS‐CoV‐2.
Identifying alternative targets for the detection of SARS‐CoV‐2 is important because genetic variations can affect the sensitivity of RT‐PCR. There are two major sources of genetic variations in coronaviruses. First, the mutation rates in CoV are moderate to high compared to other single‐stranded RNA viruses. It was estimated that the average substitution rate of SARS‐CoV and MERS‐CoV is about 10−3 substitutions per year per site. 31 Second, recombination is well known to occur for coronaviruses. These recombination events can occur between viruses in the same genus, same lineage, or even in different lineages. 31 The recombination occurs in both animal and human coronaviruses. 32 , 33 SARS‐CoV‐2 is also believed to arise due to recombination event that involves the receptor‐binding motif of a closely related BatcoV RaTG13. 34 Recombination is especially important because the current real‐time RT‐PCR targets are mainly located in the middle or 3′ end of the genome. In contrast, nsp1 gene is located in the 5′ end, and therefore, may not be affected by recombination events occurring between the nsp1 gene and RdRp gene. Unlike other targets commonly used for diagnosis which are located in the middle or 3' end of the viral genome, nsp1 is located in the 5′ end of the genome. 35 Since recombination can occur, it is important to have a target at the 5′ end of the genome.
Nsp1 is usually not considered to be highly expressed. However, our nanopore sequencing of clinical specimens has consistently shown that nsp1 gene region is highly expressed. Traditionally, it was thought that the subgenomic sequences arise from the leader sequence. However, recently, it was shown that the subgenomic sequences arise at the nsp1‐truncated nsp2 and/or truncated nsp3 region. 36
Shortly after the announcement of SARS‐CoV‐2 as the causative pathogen of COVID‐19, Shirato et al 30 has reported the use of a conventional nested RT‐PCR targeting nsp1 gene, but require Sanger sequencing for confirmation. In this early report, the sensitivity and specificity of their conventional nsp1 gene nested RT‐PCR was not reported.
The coverage map for SARS‐CoV‐2 from our study using of SISPA and nanopore sequencing suggests that nsp1 was highly expressed in clinical specimens. In contrast, in a previous study, nsp1 was not shown to be highly expressed in the coverage map from Illumina sequencing. 37 One possibility is the difference in library preparation. In the paper by Wu et al, ribosomal RNA depletion was performed to reduce the amount of human RNA in the specimen before reverse transcription. Ribosomal RNA depletion has been shown to introduce biased distribution of read coverage for influenza virus. 38 In contrast, our library preparation did not involve ribosomal RNA depletion, which may have avoided this bias.
Another potential technique to survey expression is direct RNA sequencing using nanopore technology. However, this technique requires a large amount of high quality and pure viral RNA as starting material, which is often unfeasible from clinical specimens. Moreover, the protocol utilizes oligodT primers to capture the polyA tail to allow the complex to be brought to the sequencing pore where the RNA is read from the 3′ to 5′ direction. This technology may not be useful in assessing abundance of particular regions as efficiency of detection is higher in early reads resulting in coverage bias towards the 3′ end of the genome.
Studies from SARS‐CoV indicate that NSP1 protein suppresses the antiviral response. 35 NSP1 can bind to 40S ribosome to block the assembly of ribosome and induce endonucleolytic cleavage and the degradation of host messenger RNAs. Furthermore, NSP1 can change the nuclear‐cytoplasmic distribution of nucleolin. 39 Since nsp1 gene is highly expressed in our SARS‐CoV‐2 patients, it remains to be determined whether the function of NSP1 is also enhanced in SARS‐CoV‐2.
There are several limitations of this study. First, although our nanopore SISPA‐based whole‐genome sequencing allowed us to identify highly‐expressed gene regions, there can still be bias during the library preparation which may affect the coverage of each position in the viral genome. Second, our nanopore sequencing did not encompass phylogenetic type C strains, which is mainly found in Europe and in America.
5. CONCLUSIONS
We have found that viral gene nsp1 is highly expressed in patients with COVID‐19. Our novel real‐time RT‐PCR assay targeting nsp1 was highly sensitive and specific in the detection of SARS‐CoV‐2 in clinical specimens. Our real‐time RT‐PCR is unique in that nsp1 gene is located at the 5′ end of the SARS‐CoV‐2 genome, while other currently published real‐time RT‐PCR protocols target genes located in the middle or 3′ end of the viral genome. The incorporation of the nsp1 gene in multiplex RT‐PCR assays will likely improve the detection of SARS‐CoV‐2.
AUTHOR CONTRIBUTIONS
WMC, JDI, AWHC, CCYY, LSL, RWSP performed the experiment and analyzed the data. KHC, ACKN, WKT, QTYT, WSL, MYWK, GTC collected samples and performed extraction. TWHC, IFNH, KHK, VCCC, JFWC discussed the results and commented the manuscript. KYY and KKWT designed the experiments and wrote the manuscript.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We gratefully acknowledge the originating and submitting laboratories who contributed sequences to GISAID (Supporting Information Data). This study was partly supported by the Theme‐Based Research Scheme (T11/707/15) of the Research Grants Council, HKSAR; and the donations of Richard Yu and Carol Yu, May Tam Mak Mei Yin, Michael Seak‐Kan Tong, Respiratory Viral Research Foundation Limited, Hui Ming, Hui Hoy and Chow Sin Lan Charity Fund Limited, Chan Yin Chuen Memorial Charitable Foundation, Marina Man‐Wai Lee, the Hong Kong Hainan Commercial Association South China Microbiology Research Fund, the Jessie & George Ho Charitable Foundation, Perfect Shape Medical Limited, and Kai Chong Tong.
Chan W‐M, Ip JD, Chu AW‐H, et al. Identification of nsp1 gene as the target of SARS‐CoV‐2 real‐time RT‐PCR using nanopore whole‐genome sequencing. J Med Virol. 2020;92:2725–2734. 10.1002/jmv.26140
Wan‐Mui Chan and Jonathan Daniel Ip contribute equally.
REFERENCES
- 1. Peiris JS, Lai ST, Poon LL, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319‐1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chan JFW, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person‐to‐person transmission: a study of a family cluster. Lancet. 2020;395:514‐523. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. To KK, Tsang OT, Leung WS, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS‐CoV‐2: an observational cohort study. Lancet Infect Dis. 2020;20:565‐574. 10.1016/S1473-3099(20)30196-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheung KS, Hung IF, Chan PP, et al. Gastrointestinal manifestations of SARS‐CoV‐2 infection and virus load in fecal samples from the Hong Kong cohort and systematic review and meta‐analysis. Gastroenterology. 2020;S0016‐5085(20):30448. 10.1053/j.gastro.2020.03.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi H, Han X, Jiang N, et al. Radiological findings from 81 patients with COVID‐19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. 2020;20:425‐434. 10.1016/S1473-3099(20)30086-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–1069. 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507‐513. 10.1016/S0140-6736(20)30211-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS‐CoV‐2 pneumonia in Wuhan, China: a single‐centered, retrospective, observational study. The lancet Respir Med. 2020;8:475‐481. 10.1016/S2213-2600(20)30079-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. The lancet Respir Med. 2020;8:420‐422. 10.1016/S2213-2600(20)30076-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ackermann M, Verleden SE, Kuehne M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020. 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharfstein JM, Becker SJ, Mello MM. Diagnostic testing for the novel coronavirus. JAMA. 2020;323:1437. 10.1001/jama.2020.3864 [DOI] [PubMed] [Google Scholar]
- 13. Li R, Pei S, Chen B, et al. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS‐CoV2). Science. 2020;368:489‐493. 10.1126/science.abb3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. World Health organization . Coronavirus disease (COVID‐19) technical guidance: Laboratory testing for 2019‐nCoV in humans. Available at https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf?sfvrsn=de3a76aa_2. Accessed on April 18, 2020.
- 15. Chan JF, Yip CC, To KK, et al. Improved molecular diagnosis of COVID‐19 by the novel, highly sensitive and specific COVID‐19‐RdRp/Hel real‐time reverse transcription‐polymerase chain reaction assay validated in vitro and with clinical specimens. J Clin Microbiol. 2020;58(5):e00310–e00320. 10.1128/JCM.00310-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chan JF, Lau SK, To KK, Cheng VC, Woo PC, Yuen KY. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS‐like disease. Clin Microbiol Rev. 2015;28(2):465‐522. 10.1128/CMR.00102-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng VC, Lau SK, Woo PC, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20(4):660‐694. 10.1128/CMR.00023-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Wang W, Zhao X, et al. Transmission dynamics and evolutionary history of 2019‐nCoV. J Med Virol. 2020;92(5):501‐511. 10.1002/jmv.25701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forster P, Forster L, Renfrew C, Forster M. Phylogenetic network analysis of SARS‐CoV‐2 genomes. Proc Natl Acad Sci U S A. 2020;117(17):9241‐9243. 10.1073/pnas.2004999117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. GISAID . Full genome nucleotide alignments for high quality genomes. Available at https://www.epicov.org/epi3/frontend#lightbox1588038909. Accessed on April 21, 2020.
- 21. Yip CC, Chan WM, Ip JD, et al. Nanopore sequencing reveals novel targets for the diagnosis and surveillance of human and avian influenza A virus. J Clin Microbiol. 2020;58:e02127‐19. 10.1128/JCM.02127-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bossuyt PM, Reitsma JB, Bruns DE, et al. STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies. Radiology. Dec. 2015;277(3):826‐832. 10.1148/radiol.2015151516 [DOI] [PubMed] [Google Scholar]
- 23. Lanfear R, Schalamun M, Kainer D, Wang W, Schwessinger B. MinIONQC: fast and simple quality control for MinION sequencing data. Bioinformatics. 2019;35(3):523‐525. 10.1093/bioinformatics/bty654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078‐2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Narasimhan V, Danecek P, Scally A, Xue Y, Tyler‐Smith C, Durbin R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next‐generation sequencing data. Bioinformatics. 2016;32(11):1749‐1751. 10.1093/bioinformatics/btw044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. https://github.com/lh3/seqtk
- 27. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yip CC, Ho CC, Chan JF, et al. Development of a novel, genome subtraction‐derived, SARS‐CoV‐2‐specific COVID‐19‐nsp2 real‐time RT‐PCR assay and its evaluation using clinical specimens. Int J Mol Sci. 2020;21:2574. 10.3390/ijms21072574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Corman VM, Landt O, Kaiser M. Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Euro Surveill. 2020;25(3):2000045. 10.2807/1560-7917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shirato K, Nao N, Katano H, et al. Development of genetic diagnostic methods for novel coronavirus 2019 (nCoV‐2019) in Japan. Jpn J Infect Dis. 2020. 10.7883/yoken.JJID.2020.061 [DOI] [PubMed] [Google Scholar]
- 31. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490‐502. 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lau SKP, Luk HKH, Wong ACP, et al. Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China. Viruses. 2019;11(11):980. 10.3390/v11110980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Woo PC, Lau SK, Yip CC, et al. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol. 2006;80(14):7136‐7145. 10.1128/JVI.00509-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cagliani R, Forni D, Clerici M, Sironi M. Computational inference of selection underlying the evolution of the novel coronavirus, SARS‐CoV‐2. J Virol. 2020;94(12):e00411–e00420. 10.1128/JVI.00411-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221‐236. 10.1080/22221751.2020.1719902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim D, Lee JY, Yang JS, Kim JW, Kim N, Chang H. The architecture of SARS‐CoV‐2 transcriptome. Cell. 2020;181:914‐921. 10.1016/jcell202004011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li D, Li Z, Zhou Z, et al. Direct next‐generation sequencing of virus‐human mixed samples without pretreatment is favorable to recover virus genome. Biol Direct. 2016;11(1):3. 10.1186/s13062-016-0105-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gomez GN, Abrar F, Dodhia MP, Gonzalez FG, Nag A. SARS coronavirus protein nsp1 disrupts localization of Nup93 from the nuclear pore complex. Biochem Cell Biol. 2019;97(6):758‐766. 10.1139/bcb-2018-0394 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information