

Abstract
Bats harbour diverse coronaviruses (CoVs), some of which are associated with zoonotic infections, as well as inter‐species transmission. In this study, a total of 512 bat faecal samples from the bat habitats at different geographical locations in South Korea were investigated between 2016 and 2019. Seventy‐eight samples were positive for coronaviruses (15.2%), comprising 68 alphacoronaviruses (13.3%) and 10 betacoronaviruses (2.0%). The positive rates tended to increase during the awakening (April) period. Notably, betacoronaviruses were only found in the site where Rhinolophus ferrumequinum was the major species of bats, and were related to SARS‐ and MERS‐related CoVs identified in China and South Korea, respectively. No betacoronaviruses were closely related to SARS‐CoV‐2 in this study. Alphacoronaviruses were detected in the sites where Hypsugo alaschanicus, Miniopterus fuliginosus, Miniopterus schreibersii, Rhinolophus ferrumequinum, Myotis bombinus, Myotis macrodactylus and Myotis petax were found to be the major bat species. Furthermore, alphacoronaviruses had higher genetic diversity than betacoronaviruses and had a wider distribution in Korea. Considering that different bat species are co‐roosting in crowded conditions in the same habitat, the diverse coronaviruses in Korean bats are likely to undergo cross‐species transmission events due to the richness in host species. Therefore, continuous monitoring should be performed, especially at the awakening time of the hibernating bats in the habitats where diverse bat species co‐roost, to better understand the evolution of coronaviruses in bats.
Keywords: alphacoronavirus, bats, coronavirus, diversity, host sharing, Korea
1. INTRODUCTION
Coronaviruses (CoVs) are the largest groups of positive‐sense, single‐stranded RNA viruses belonging to the Coronaviridae family. CoVs are divided into four genera, alpha‐, beta‐, gamma‐ and delta‐coronaviruses (de Groot et al., 2011). Many species of CoVs exist among mammals and birds. Alpha‐ and betacoronavirus have been reported only in mammals so far and can cause respiratory and gastrointestinal diseases with high mortality rates, for example transmissible gastroenteritis virus (TGEV) and porcine epidemic diarrhoea virus (PEDV) in young pigs, severe acute respiratory syndrome coronavirus (SARS‐CoV) in 2003 and Middle East Respiratory syndrome coronavirus (MERS‐CoV) in humans (Brian & Baric, 2005; Lee et al., 2003; Lin, Saif, Marthaler, & Wang, 2016; Zaki, Van Boheemen, Bestebroer, Osterhaus, & Fouchier, 2012).
Bats, a diverse group of mammals in the order Chiroptera, are not only mammals with the capability of powered flight but also the second‐largest order among mammals and distributed worldwide (Wang & Anderson, 2019). Bats are known as the primary reservoir for viruses, such as lyssaviruses, paramyxoviruses and filoviruses. Bats are also recognized as a natural reservoir host of the coronaviruses (Allocati et al., 2016). SARS‐CoV and MERS‐CoV are suggested to have originated from bats, as palm civets and dromedary camel are intermediate hosts, respectively (Azhar et al., 2014; Cui, Li, & Shi, 2019). Swine enteric alphacoronavirus (SeACoV), also known as swine acute diarrhoea syndrome coronavirus (SADS‐CoV), was genetically most closely related to HKU2‐CoV and was found in bats. These viruses have originated from a common ancestor (Pan et al., 2017; Zhou et al., 2018).
The newly emerging coronavirus (also referred to as SARS‐CoV‐2) causing acute respiratory illnesses, such as pneumonia (COVID‐19), was first identified in late December 2019 in Wuhan, China, and has emerged as a pandemic (https://www.who.int/emergencies/diseases/novel‐coronavirus‐2019). Bats were proposed as the most likely natural reservoir for SARS‐CoV‐2 even though the transmission from bats to human is still unknown (Zhou et al., 2020).
Since the emergence of SARS‐CoV in 2003 and MERS‐CoV in 2012, several studies have shown that bat CoVs are found in North and South America, Europe, Africa, Australia and Asia (Gouilh et al., 2018; August, Mathews, & Nunn, 2012; Carrington et al., 2008; De Sabato et al., 2019; Dominguez, O’Shea, Oko, & Holmes, 2007; Lazov et al., 2018; Liang et al., 2017; Lin et al., 2017; Rizzo et al., 2017; Smith et al., 2016; Tang et al., 2006; Tao et al., 2012; Wacharapluesadee et al., 2015; Watanabe et al., 2010; Woo et al., 2006). In South Korea, SARS‐like and MERS‐like coronaviruses were reported in bat faeces since 2015 (Kim et al., 2016). Another group also conducted surveillance of coronaviruses in bats in 2016 and showed that these were genetically similar to bat betacoronaviruses identified in China (Lee et al., 2018). However, long‐term coronavirus surveillance has not been previously described in South Korea and the surrounding islands. The aim of the present study was to identify genetic characteristics of coronaviruses circulating in bats in South Korea. Furthermore, we also focus on describing the distribution and seasonal pattern of bat coronaviruses in Korea between 2016 and 2019.
Here, we collected 512 samples of bat faeces from 2016 to 2019 in Korean bat species. Phylogenetic analyses were performed under a Bayesian statistical framework implemented in BEAST.
2. MATERIALS AND METHODS
2.1. Ethics statement
To avoid animal disturbance, the study was focused on the analysis of guano collected beneath bat roosting sites/colonies in Korea. This sampling design did not require ethical approval for the study. The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to.
2.2. Study sites and samples collection
Between 2016 and 2019, we obtained 512 faecal samples (n2016 = 135, n2017 = 108, n2018 = 108, n2019 = 161), which were collected from the bat habitats in 10 provinces and islands of South Korea (total of 111 sampling sites). The bat habitats were randomly chosen by the expert from the Korean Institute of Biospeleology among caves, abandoned mines, and under a bridge where bats inhabited. All the sampling locations were close to human settlements.
We sampled faecal pellets directly on the cave floor with sterile swabs. Fresh faecal samples were preferred. The faecal sample (0.3 g of faeces) was immediately placed into transport medium (1:10 wt/vol)(Universal Transport Medium, Noble Biosciences™). The samples were transported to the laboratory and ultimately stored at −80°C until analysis.
The accurate sampling locations were carefully recorded by noting the place name, latitude and longitude.
2.3. Coronavirus detection and sequencing
All samples were cleared by centrifugation at 3,000 × g for 15 min at 4°C. RNA was extracted by TRIzol LS (Invitrogen) from 250 µl of each sample supernatant and eluted in 40 µl DEPC water. RNA was extracted according to manufacturer's instructions. cDNA was synthesized with the M‐MLV reverse transcriptase kit (Invitrogen) using random hexamer following the manufacturer's protocol. Coronaviruses were detected by a pan‐coronavirus nested PCR specific for the RNA‐dependent RNA polymerase (RdRp) sequences, as described previously (Chu et al., 2011). Briefly, the initial PCR reaction was made with 2 µl of cDNA, 2× AccuPower® PCR Master Mix (Bioneer, USA), 1 µM of each primer and nuclease‐free water in a total volume of 20 µl. The first PCR used Chu‐RdRp‐N1‐F (5′‐GGKTGGGAYTAYCCKAARTG‐3′) and Chu‐RdRp‐N1‐R (5′‐TGYTGTSWRCARAAYTCRTG‐3′) primers. The PCR reaction mixture was made with 1 µl of the first PCR reaction, 2× AccuPower® PCR Master Mix (Bioneer), 1 µM of each primer and nuclease‐free water in a total volume of 20 µl. The second nested PCR used Chu‐RdRp‐N2‐F (5′‐GGTTGGGACTATCCTAAGTGTGA‐3′) and Chu‐RdRp‐N2‐R (5′‐CCATCATCAGATAGAATCATCAT‐3′) primers and amplified a 440 bp product within the first PCR. The same thermal conditions were applied for both PCR: 95°C for 10 min; then 40 cycles of 95°C for 30 s, 48°C for 30 s and 72°C for 40 s; followed by a final extension step of 72°C for 5 min. The PCR products were revealed by electrophoresis on 1.5% agarose gels and were gel purified using QIAquick Gel Extraction kit (Qiagen). The PCR products were submitted to the Cosmogenetech company in Seoul, Korea for sequencing.
2.4. Distribution and seasonal pattern analysis
The geocoding of each sampling site was plotted using the Google Maps API (Application Programming Interface) with the ‘ggmap’ R package based on obtained latitude and longitude for each sample. For seasonal pattern analysis, the percentage of positive coronavirus samples were recorded by the month in each year. A box plot for each month in the year within the surveillance time frame was plotted using the ‘boxplot’ function. Both analyses were conducted in R software (version 3.5.1).
2.5. Phylogenetic analysis
Nucleotide sequences were aligned using ClustalW software implemented in BioEdit (version 7.1.9). Each sequence was trimmed to where both forward and reverse reads were in 100% agreement with the consensus sequence. Preliminary phylogenetic analyses using the Maximum Likelihood method based on the Tamura‐Nei model were conducted. The main phylogenetic analyses were performed under a Bayesian statistical framework implemented in BEAST (version 1.10.4) (Drummond & Rambaut, 2007), using the best‐fit model according to the corrected Bayesian Information Criterion (BIC) obtained in jModelTest 2 (Darriba, Taboada, Doallo, & Posada, 2012). The general time‐reversible (GTR) model of substitution with a gamma distribution (GTR + G + I) and the coalescent model (constant size) was used in BEAST. Phylogenetic trees were displayed in FigTree (version 1.4.4).
2.6. Biomolecular species identification
As the faecal sample was collected from caves which harbours multiple bat species, total DNA extraction was performed only for PCR‐positive samples starting from the original suspensions using the QIAamp DNA Mini kit (Qiagen), and following the manufacturer protocol. To confirm species identification, PCR was performed using specific primers for the Cytochrome b gene (Cytb) as previously described (Schlegel et al., 2012). PCR products were visualized on 1% agarose gels, purified using the QIAquick Gel Extraction kit (Qiagen) and sequenced as above.
Species identification was conducted by comparing the obtained sequences to available reference sequences (BLAST alignment, NCBI web site).
Accession number(s). The sequence data have been submitted to the GenBank databases under accession numbers MK991901 to MK991951.
3. RESULTS
3.1. Virus detection
In total, 512 samples were tested for the presence of coronavirus by nested RT‐PCR detection of an RNA‐dependent RNA polymerase (RdRp) gene fragment (Chu et al., 2011). Coronaviruses were detected in 15.2% (78/512) of samples obtained from the bat habitats in 8 of 9 provinces and islands of Korea where the most prevalent bat species were found to be: Hypsugo alaschanicus (1), Minopterus fuliginosus (1), Miniopterus schreibersii (18), Myotis bombinus (1), Myotis marcodactylus (16), Myotis petax (5) and Rhinolophus ferrumequinum (36).
3.2. Distribution and seasonal pattern of bat coronaviruses
A map showing sampling sites is presented in Figure 1, based on the coronavirus‐positive sampling locations. From the total of 111 sampling sites, there are six sites belonging to four mainland provinces where betacoronaviruses were found, whereas alphacoronaviruses were detected at 25 sites in both mainland and island sites (Jeju Island and Island of Jeonam).
Figure 1.
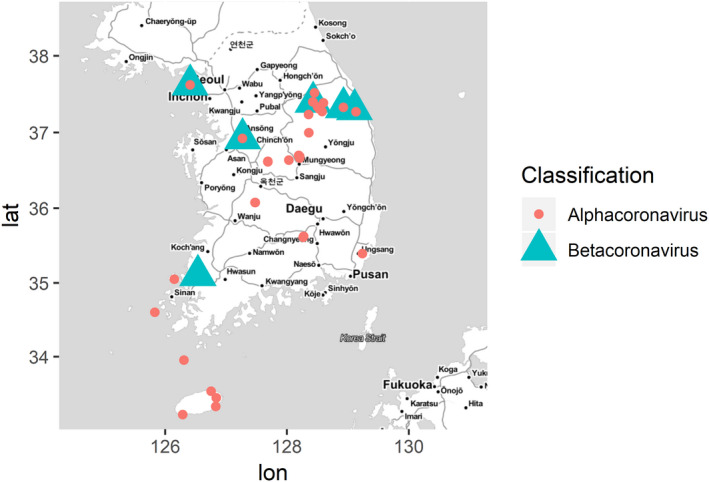
Map of geographical distribution of coronaviruses investigated in this study. Alphacoronavirus and betacoronavirus are depicted with red circle and green triangle, respectively. The map was built using ggmap and ggplot2 packages on R version 3.5.1
The seasonal pattern of bat coronavirus distribution during the study period between 2016 and 2019 is shown in Figure 2. Overall, the average and amplitude of the positivity rate fluctuated for each month over four years. Looking at the median, it seems clear that April exhibited relatively higher viral shedding rate, compared to winter (around January to March).
Figure 2.
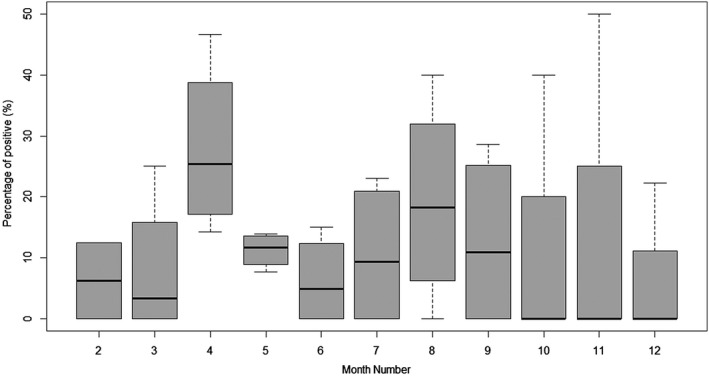
Box plot of the coronavirus positivity rate by month. Each box plot was pooled monthly for the percentage of coronavirus‐positive samples from 2016 to 2019. Data are presented as median and interquartile range; minimum and maximum values are shown by whiskers
3.3. Phylogenetic analysis
Phylogenetic analysis based on the nucleotide sequences of the partial RdRp gene (279 nt) in BEAST (v1.10.4) showed that the detected coronaviruses belonged to the genera Alphacoronavirus (13.3%: 68/512) and Betacoronavirus (2.0%: 10/512).
Alphacoronaviruses found at the site where seven bat species were predominant, namely Hypsugo alaschanicus, Minopterus fuliginosus, Miniopterus schreibersii, Myotis bombinus, Myotis marcodactylus, Myotis petax and Rhinolophus ferrumequinum. These 68 alphacoronaviruses were differently clustered. The sequences sharing ≥ 90% nucleotide identity are collapsed into the same clade with ≥ 95% of the posterior probability support. The alphacornaviruses found in this study were divided into eight clades: K1, K2, K3, K4, HKU6, HKU7, HKU8 and HKU10‐related (Figure 3). All sequences of K2 and K3 clades show high identity to BtCoV/Myotis emarginatus/LUX (90.4 to 91.5% nucleotide sequence identity) found in Luxembourg and BtCoV/A910/2005 (95.2 to 96.6% nucleotide sequence identity) found in Hong Kong, respectively. Only B19‐69 (K4 clade) clustered with PEDV, sharing 72.3% nucleotide sequence identity. Apparently, the K1 clade was most likely a new clade, clustered with the HKU6 clade. Each sequence of K1 clade has <89% pairwise nucleotide sequence identity to the closest reference sequence (Bat coronavirus Mm_CoV‐2 was found in Japan, accession number: LC469030.1) based on partial analysis of the RdRp. Furthermore, twenty‐four sequences of this clade were diversely identified in the sites and were found in different bat species, including Myotis petax, Miniopterus schreibersii, Myotis macrodactylus and Rhinolophus ferrumequinum, involving 14 sampling sites in Chungbuk, Gangwon and Jeju Island.
Figure 3.
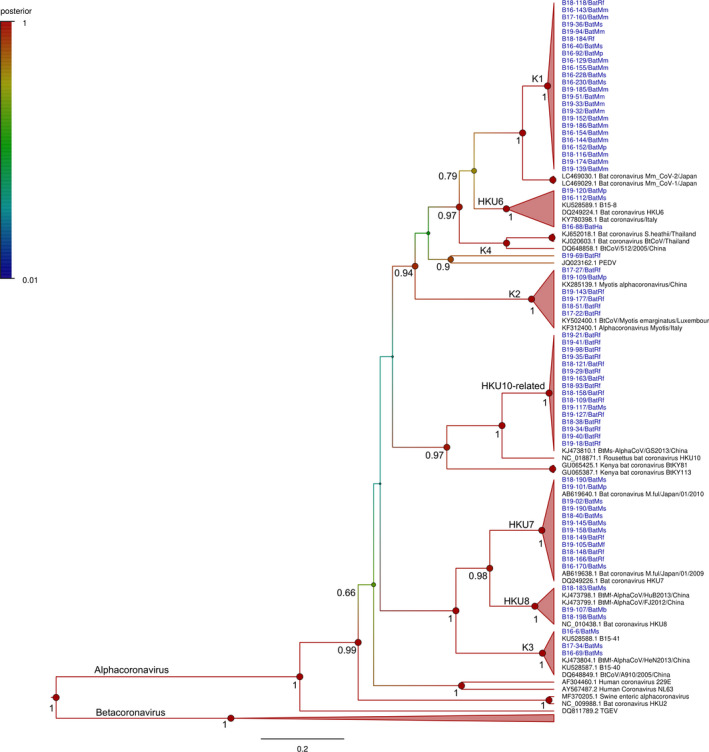
Bayesian phylogeny of 279 nucleotides from the alphacoronaviruses detected in this study with 30 reference sequences. Statistical support (posterior probability) of nodes is depicted using a gradual colour code of the tree, red indicating significant posterior probability values (> 0.95). For clarity of presentation, only posterior probability values above 0.65 are shown and values at crown positions were removed. Sequence names and main information is provided in taxa labels. Viruses detected in this study are coloured in blue
Ten betacoronaviruses were associated with Rhinolophus ferrumequinum only, which were divided into two groups (Figure 4). B17‐155 and B18‐50 were related to MERS‐CoV from South Korea and MERS‐related CoV from the United Arab Emirates identified in 2015, showing 85.1% sequence identity. The other eight sequences (B16‐75, B17‐60, B17‐62, B18‐82, B18‐83, B18‐92, B18‐168 and B18‐171) were grouped with the SARS‐like CoV clade, sharing 99.6% sequence identity (B16‐75, B17‐60, B17‐62, B18‐82, B18‐83, B18‐92, B18‐168 and B18‐171) were grouped. In addition, our SARS‐like CoVs only shared 86.7 to 87.0% nucleotide identity with SARS‐CoV‐2 and the bat coronavirus RaTG13 based on partial analysis of the RdRp.
Figure 4.
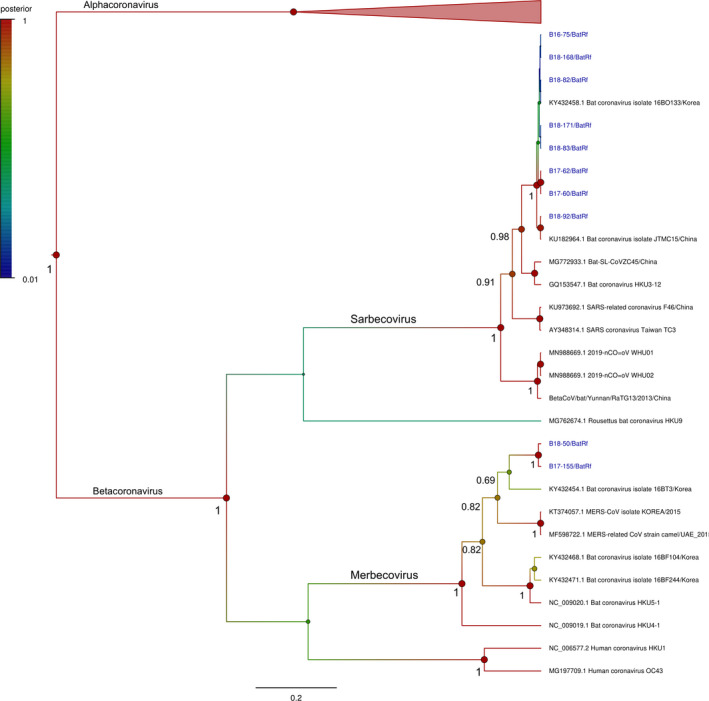
Bayesian phylogeny of 279 nucleotides from the betacoronaviruses detected in this study with 19 reference sequences. Statistical support (posterior probability) of nodes is depicted using a gradual colour code of the tree, red indicating significant posterior probability values (> 0.95). For clarity of presentation, only posterior probability values above 0.65 are shown and values at crown positions were removed. Sequence names and main information is provided in taxa labels. Viruses detected in this study are coloured in blue
4. DISCUSSION AND CONCLUSION
This research sought to comprehensively investigate coronaviruses in bats at different geographical locations in South Korea from 2016 to 2019, to improve our understanding of bat species and bat CoVs strains, and to find relationships with bat coronavirus identified in other countries.
4.1. Seasonal pattern of the bat coronaviruses in Korea
Previous studies indicated that the season affects the percentage of coronavirus positivity, with a higher coronavirus prevalence in the dry season in comparison with the wet season in Africa and Asia (Anthony et al., 2017). In addition, hibernating bats are a factor affecting the prevalence and diversity of coronavirus in the region during winter. Thus, it seems reasonable that during the awakening period (April), the average rate of coronavirus detection was higher than other months of the year, as observed in this study. By contrast, through the period of hibernation, the lower presence of coronavirus was observed. Especially in January, we did not find fresh faeces in the cave. Besides, the mean of positive rate was also high in August due to physical contact over the reproductive season. Thus, the physical contact and/or airborne transmission, and co‐roosting of several bat species pre‐ and post‐hibernation might lead to an increased viral transmission.
4.2. Distribution and diversity
From the prevalence of coronavirus in the studied sampling sites, it was assumed that coronaviruses are circulating in Korean bats throughout the country. Betacoronaviruses were only detected in the north (Gangwon and Incheon) and the southwest (Gyeonggi and Jeonnam), whereas alphacoronaviruses were more widely detected at both mainland and island sampling sites (Figure 1), and were detected at higher rates than betacoronaviruses. In our study, the prevalence of alphacoronaviruses was approximately seven times higher. It is likely that bats are more susceptible to—and wider hosts of—alphacoronavius than betacoronavirus (Anthony et al., 2017; Lazov et al., 2018; Lin et al., 2017; Wacharapluesadee et al., 2015). To date, seven of twenty‐four Korean bat species were found to be associated with alphacoronaviruses, and only four species were found to be associated with betacoronaviruses. Among these species, only Rhinolophus ferrumequinum was the major bat species from the site where both alphacoronaviruses and betacoronaviruses were found (Table 1).
Table 1.
Information of the positive sample
| No. | Clade/cluster | No. of positive | Bat species | Local detection |
|---|---|---|---|---|
| 1 | K1 | 24 | Miniopterus schreibersii | Chungbuk |
| Myotis macrodactylus | Gangwon | |||
| Myotis petax | Jeju Island | |||
| Rhinolophus ferrumequinum | ||||
| 2 | K2 | 6 | Myotis petax | Chungbuk |
| Rhinolophus ferrumequinum | Gangwon | |||
| Gyeonggi | ||||
| 3 | K3 | 3 | Miniopterus schreibersii | Gyeongbuk |
| Gyeongnam | ||||
| 4 | K4 | 1 | Rhinolophus ferrumequinum | Gyeongbuk |
| 5 | HKU6 | 3 | Hypsugo alaschanicus | Chungbuk |
| Miniopterus schreibersii | Gyeongbuk | |||
| Myotis petax | ||||
| 6 | HKU7 | 12 | Miniopterus fuliginosus | Gangwon |
| Miniopterus schreibersii | Gyeongbuk | |||
| Myotis petax | Gyeongnam | |||
| Rhinolophus ferrumequinum | Jeju Island | |||
| 7 | HKU8 | 3 | Miniopterus schreibersii | Gyeongbuk |
| Myotis bombinus | Jeju Island | |||
| 8 | HKU10‐related | 16 | Miniopterus schreibersii | Chungbuk |
| Rhinolophus ferrumequinum | Chungnam | |||
| Gangwon | ||||
| Gyeongbuk | ||||
| Incheon | ||||
| Jeju Island | ||||
| Jeonnam | ||||
| 9 | SARS‐like | 8 | Rhinolophus ferrumequinum | Gangwon |
| Gyeonggi | ||||
| Incheon | ||||
| Jeonnam | ||||
| 10 | MERS‐related | 2 | Rhinolophus ferrumequinum | Gangwon |
Globally, the diversity and distribution of bat coronaviruses were indicated in association with biogeography and diversity of their host (Leopardi et al., 2018). This hypothesis probably accounts for heterogeneous circulation of alphacoronavirus. It is especially important to explain the appearance and diversity of this putative new species of K1 clade (24 sequences) observed in four bat species (Table 2). Despite this, most of these alphacoronaviruses identified in Korean bats were close to bat coronavirus found in China in the same as well as different bat species.
Table 2.
Potential sharing of the certain bat coronaviruses among the major bat species of the sample collection sites. The bat species were confirmed by nucleotide sequence analysis of the Cytochrome b (Cytb) gene
| Cases | Name | Date of collection | Sampling position | Latitude | Longitude | Bat species | Clade |
|---|---|---|---|---|---|---|---|
| 1 | B19‐36/BatMs | 4/11/2019 | Gangwon | 37.310306 | 128.528028 | Miniopterus schreibersii | K1 |
| B19‐51/BatMm | 5/9/2019 | Gangwon | 37.310306 | 128.528028 | Myotis macrodactylus | ||
| B19‐94/BatMm | 7/1/2019 | Gangwon | 37.310306 | 128.528028 | Myotis macrodactylus | ||
| 2 | B16‐144/BatMm | 7/19/2016 | Gangwon | 37.235667 | 128.355278 | Myotis macrodactylus | K1 |
| B16‐152/BatMp | 7/19/2016 | Gangwon | 37.235667 | 128.355278 | Myotis petax | ||
| 3 | B19‐105/BatMf | 7/22/2019 | Gyeongbuk | 36.683083 | 128.20775 | Miniopterus fuliginosus | HKU7 |
| B19‐190/BatMs | 9/25/2019 | Gyeongbuk | 36.683083 | 128.20775 | Miniopterus schreibersii | ||
| 4 | B18‐166/BatRf | 9/29/2018 | Gangwon | 37.397861 | 128.426528 | Rhinolophus ferrumequinum | HKU7 |
| B19‐101/BatMp | 7/2/2019 | Gangwon | 37.397861 | 128.426528 | Myotis petax | ||
| B19‐145/BatMs | 8/20/2019 | Gangwon | 37.397861 | 128.426528 | Miniopterus schreibersii | ||
| B19‐158/BatMs | 8/24/2019 | Gangwon | 37.397861 | 128.426528 | Miniopterus schreibersii | ||
| 5 | B18‐198/BatMs | 12/25/2018 | Gyeongbuk | 36.683083 | 128.20775 | Miniopterus schreibersii | HKU8 |
| B19‐107/BatMb | 7/22/2019 | Gyeongbuk | 36.683083 | 128.20775 | Myotis bombinus |
4.3. The potential of cross‐species transmission and host switching
The mechanisms of cross‐species transmission or spillover and host switching are still unclear. However, these mechanisms are considered the dominant evolutionary events involving recombination between different viruses, associated with the expansion of viral host ranges or creation of a novel virus. It is widely accepted that cross‐species transmission events have contributed to the genetic diversity and evolution of coronaviruses (e.g. SARS‐CoV and HCoV) (Banerjee, Misra, Schountz, & Baker, 2018; Cui et al., 2019; Leopardi et al., 2018; Menachery, Graham, & Baric, 2017). Several different bat species crowding, co‐roosting and co‐hibernating may increase the potential for intraspecies and inter‐species transmission of viral infections.
For instance, in the putative new K1 clade, three alphacoronaviruses (B19‐36 in Miniopterus schreibersii, and B19‐51 and B19‐94 from Myotis macrodactylus) were detected in the same roost from different bat genera. In addition, B18‐166 from Rhinolophus ferrumequium, B19‐101 from Myotis petax, and B19‐145 and B19‐158 from Miniopterus schreibersii belonging to HKU7 clade were also found in the same cave. In HKU8 clade, B18‐198 and B19‐107 were identified in Miniopterus schreibersii and Myotis bombinus, respectively, found in the same cave from different sampling year. Furthermore, the potential of host switching in family species may explain the detection of these alphacoronaviruses belonging to new K1 clade in Myotis petax and Myotis macrodactylus from the same (Table 2, case 2) or neighbouring roosting sites in Gangwon.
In our study regions, co‐roosting behaviour was frequently observed in Myotis spp. and Miniopterus schreibersii with Rhinolophus ferrumeqium. Consequently, the frequency of interaction among these bat species provides conditions suitable for the spillover of a coronavirus. With respect to the frequent clustering of coronaviruses, the clusters associated with PEDV and BtCoV/512/2005 were most commonly contained Murina, Myotis, Schotophilus and Pipistrellus spp. (Leopardi et al., 2018). Thus, Rhinolophus ferrumeqium is likely to be a new host of these clusters as a result of cross‐species transmission events, which were achieved by jumping from the bat genera Myotis or Miniopterus to Rhinolophus.
Overall, this study provided recent molecular epidemiology of bat coronaviruses in Korea through long‐term and widespread surveillance. We also detected SARS‐related and MERS‐related CoVs that were closely related to bat coronaviruses identified in South Korea and China, and those betacoronaviruses detected in this study belonged to the major two clades, compared to the alphacoronaviruses (eight clades). The alphacoronaviruses were widely found in diverse bat species and were heterogeneous, which might be due to cross‐species transmission events. While Rhinolophus ferrumequinum was the only species harbouring betacoronaviruses, alphacoronaviruses were observed to have considerable richness in terms of host species in Korea. However, this study has several limitations because only eleven of the twenty‐four bat species in Korea were included in this study, and direct collection of faecal samples from bats was not possible. There should be follow‐up studies with direct sample collection from captured bats, and identification of the captured bat species based on genetic analysis.
These findings lead us to believe that further studies are needed to better understand the diversity, circulation and evolution of coronaviruses in bats. Certainly, the long‐term surveillance of bat CoVs should be maintained and expanded to include more geographical locations and bat species.
CONFLICT OF INTEREST
The authors declare no competing interests.
ACKNOWLEDGEMENTS
This study was supported by The Bio & Medical Technology Development Program of the National Research Foundation (NRF), funded by The Ministry of Science & ICT (NRF‐2018M3A9H4056347) and supported by The BioNano Health‐Guard Research Center, funded by The Ministry of Science and ICT(MSIT) of Korea as a Global Frontier Project (grant No. H‐GUARD_2013M3A6B2078954).
Lo VT, Yoon S‐W, Noh JY, et al. Long‐term surveillance of bat coronaviruses in Korea: Diversity and distribution pattern. Transbound Emerg Dis. 2020;67:2839–2848. 10.1111/tbed.13653
Van Thi Lo, Sun‐Woo Yoon and Ji Yeong Noh contributed equally.
Contributor Information
Dae Gwin Jeong, Email: dgjeong@kribb.re.kr.
Hye Kwon Kim, Email: khk1329@chungbuk.ac.kr.
DATA AVAILABILITY STATEMENT
The sequence data that support the findings of this study are openly available in Genbank (https://www.ncbi.nlm.nih.gov/genbank/) with the accession numbers MK991901 to MK991951.
REFERENCES
- Allocati, N. , Petrucci, A. , Di Giovanni, P. , Masulli, M. , Di Ilio, C. , & De Laurenzi, V. (2016). Bat–man disease transmission: Zoonotic pathogens from wildlife reservoirs to human populations. Cell Death Discovery, 2, 1–8. 10.1038/cddiscovery.2016.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony, S. J. , Johnson, C. K. , Greig, D. J. , Kramer, S. , Che, X. , Wells, H. , … Goldstein, T. (2017). Global patterns in coronavirus diversity. Virus Evolution, 3(1), 10.1093/ve/vex012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ar Gouilh, M. , Puechmaille, S. J. , Diancourt, L. , Vandenbogaert, M. , Serra‐Cobo, J. , Lopez Roïg, M. , … Manuguerra, J.‐C. (2018). SARS‐CoV related Betacoronavirus and diverse Alphacoronavirus members found in western old‐world. Virology, 517, 88–97. 10.1016/j.virol.2018.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- August, T. A. , Mathews, F. , & Nunn, M. A. (2012). Alphacoronavirus detected in bats in the United Kingdom. Vector‐Borne and Zoonotic Diseases, 12, 530–533. 10.1089/vbz.2011.0829 [DOI] [PubMed] [Google Scholar]
- Azhar, E. I. , El‐Kafrawy, S. A. , Farraj, S. A. , Hassan, A. M. , Al‐Saeed, M. S. , Hashem, A. M. , & Madani, T. A. (2014). Evidence for camel‐to‐human transmission of MERS coronavirus. New England Journal of Medicine, 370, 2499–2505. 10.1056/NEJMoa1401505 [DOI] [PubMed] [Google Scholar]
- Banerjee, A. , Misra, V. , Schountz, T. , & Baker, M. L. (2018). Tools to study pathogen‐host interactions in bats. Virus Research, 248, 5–12. 10.1016/j.virusres.2018.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian, D. , & Baric, R. (2005). Coronavirus genome structure and replication. In Enjuanes L. (Eds.), Coronavirus replication and reverse genetics (vol. 287, pp. 1–30), Current topics in microbiology and immunology. Berlin, Heidelberg: Springer. 10.1007/3-540-26765-4_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington, C. V. F. , Foster, J. E. , Zhu, H. C. , Zhang, J. X. , Smith, G. J. D. , Thompson, N. , … Guan, Y. I. (2008). Detection and phylogenetic analysis of group 1 coronaviruses in South American bats. Emerging Infectious Diseases, 14, 1890. 10.3201/eid1412.080642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, D. K. W. , Leung, C. Y. H. , Gilbert, M. , Joyner, P. H. , Ng, E. M. , Tse, T. M. , … Poon, L. L. M. (2011). Avian coronavirus in wild aquatic birds. Journal of Virology, 85, 12815–12820. 10.1128/JVI.05838-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, J. , Li, F. , & Shi, Z.‐L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17(3), 181–192. 10.1038/s41579-018-0118-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba, D. , Taboada, G. L. , Doallo, R. , & Posada, D. (2012). jModelTest 2: More models, new heuristics and parallel computing. Nature Methods, 9, 772. 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, R. J. , Baker, S. C. , Baric, R. , Enjuanes, L. , Gorbalenya, A. , Holmes, K. V. , … Ziebuhr, J. (2011). Coronaviridae. In: Virus Taxonomy, Classification and Nomenclature of Viruses. International Union of Microbiological Societies, Virology Division, Ninth Report of the International Committee on Taxonomy of Viruses, 806–828. [Google Scholar]
- De Sabato, L. , Lelli, D. , Faccin, F. , Canziani, S. , Di Bartolo, I. , Vaccari, G. , & Moreno, A. (2019). Full genome characterization of two novel Alpha‐coronavirus species from Italian bats. Virus Research, 260, 60–66. 10.1016/j.virusres.2018.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez, S. R. , O’Shea, T. J. , Oko, L. M. , & Holmes, K. V. (2007). Detection of group 1 coronaviruses in bats in North America. Emerging Infectious Diseases, 13, 1295. 10.3201/eid1309.070491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7, 214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. K. , Yoon, S.‐W. , Kim, D.‐J. , Koo, B.‐S. , Noh, J. Y. , Kim, J. H. , … Jeong, D. G. (2016). Detection of severe acute respiratory syndrome‐like, middle east respiratory syndrome‐like bat coronaviruses and group H rotavirus in faeces of Korean bats. Transboundary and Emerging Diseases, 63, 365–372. 10.1111/tbed.12515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazov, C. , Chriél, M. , Baagøe, H. , Fjederholt, E. , Deng, Y. U. , Kooi, E. , … Rasmussen, T. (2018). Detection and characterization of distinct alphacoronaviruses in five different bat species in Denmark. Viruses, 10, 486. 10.3390/v10090486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, N. , Hui, D. , Wu, A. , Chan, P. , Cameron, P. , Joynt, G. M. , … Sung, J. J. Y. (2003). A major outbreak of severe acute respiratory syndrome in Hong Kong. New England Journal of Medicine, 348, 1986–1994. 10.1056/NEJMoa030685 [DOI] [PubMed] [Google Scholar]
- Lee, S. , Jo, S.‐D. , Son, K. , An, I. , Jeong, J. , Wang, S.‐J. , … Oem, J.‐K. (2018). Genetic characteristics of coronaviruses from Korean bats in 2016. Microbial Ecology, 75, 174–182. 10.1007/s00248-017-1033-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopardi, S. , Holmes, E. C. , Gastaldelli, M. , Tassoni, L. , Priori, P. , Scaravelli, D. , … De Benedictis, P. (2018). Interplay between co‐divergence and cross‐species transmission in the evolutionary history of bat coronaviruses. Infection, Genetics and Evolution, 58, 279–289. 10.1016/j.meegid.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, J. , Yang, X.‐L. , Li, B. , Liu, Q. I. , Zhang, Q. , Liu, H. , … Zhang, L. (2017). Detection of diverse viruses in alimentary specimens of bats in Macau. Virologica Sinica, 32, 226–234. 10.1007/s12250-017-3976-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.‐M. , Saif, L. J. , Marthaler, D. , & Wang, Q. (2016). Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Research, 226, 20–39. 10.1016/j.virusres.2016.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X.‐D. , Wang, W. , Hao, Z.‐Y. , Wang, Z.‐X. , Guo, W.‐P. , Guan, X.‐Q. , … Zhang, Y.‐Z. (2017). Extensive diversity of coronaviruses in bats from China. Virology, 507, 1–10. 10.1016/j.virol.2017.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery, V. D. , Graham, R. L. , & Baric, R. S. (2017). Jumping species—a mechanism for coronavirus persistence and survival. Current Opinion in Virology, 23, 1–7. 10.1016/j.coviro.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Y. , Tian, X. , Qin, P. , Wang, B. , Zhao, P. , Yang, Y.‐L. , … Huang, Y.‐W. (2017). Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Veterinary Microbiology, 211, 15–21. 10.1016/j.vetmic.2017.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, F. , Edenborough, K. M. , Toffoli, R. , Culasso, P. , Zoppi, S. , Dondo, A. , … Mandola, M. L. (2017). Coronavirus and paramyxovirus in bats from Northwest Italy. BMC Veterinary Research, 13, 396. 10.1186/s12917-017-1307-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel, M. , Ali, H. S. , Stieger, N. , Groschup, M. H. , Wolf, R. , & Ulrich, R. G. (2012). Molecular identification of small mammal species using novel cytochrome B gene‐derived degenerated primers. Biochemical Genetics, 50, 440–447. 10.1007/s10528-011-9487-8 [DOI] [PubMed] [Google Scholar]
- Smith, C. , De Jong, C. , Meers, J. , Henning, J. , Wang, L.‐F. , & Field, H. (2016). Coronavirus infection and diversity in bats in the Australasian region. EcoHealth, 13, 72–82. 10.1007/s10393-016-1116-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. C. , Zhang, J. X. , Zhang, S. Y. , Wang, P. , Fan, X. H. , Li, L. F. , … Guan, Y. (2006). Prevalence and genetic diversity of coronaviruses in bats from China. Journal of Virology, 80, 7481–7490. 10.1128/JVI.00697-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, Y. , Tang, K. , Shi, M. , Conrardy, C. , Li, K. S. M. , Lau, S. K. P. , … Tong, S. (2012). Genomic characterization of seven distinct bat coronaviruses in Kenya. Virus Research, 167, 67–73. 10.1016/j.virusres.2012.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacharapluesadee, S. , Duengkae, P. , Rodpan, A. , Kaewpom, T. , Maneeorn, P. , Kanchanasaka, B. , … Hemachudha, T. (2015). Diversity of coronavirus in bats from Eastern Thailand. Virology Journal, 12, 57. 10.1186/s12985-015-0289-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L.‐F. , & Anderson, D. E. (2019). Viruses in bats and potential spillover to animals and humans. Current Opinion in Virology, 34, 79–89. 10.1016/j.coviro.2018.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, S. , Masangkay, J. S. , Nagata, N. , Morikawa, S. , Mizutani, T. , Fukushi, S. , … Akashi, H. (2010). Bat coronaviruses and experimental infection of bats, the Philippines. Emerging Infectious Diseases, 16, 1217. 10.3201/eid1608.100208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. Y. , Lau, S. K. P. , Li, K. S. M. , Poon, R. W. S. , Wong, B. H. L. , Tsoi, H.‐W. , … Yuen, K.‐Y. (2006). Molecular diversity of coronaviruses in bats. Virology, 351, 180–187. 10.1016/j.virol.2006.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki, A. M. , Van Boheemen, S. , Bestebroer, T. M. , Osterhaus, A. D. , & Fouchier, R. A. (2012). Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. New England Journal of Medicine, 367, 1814–1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- Zhou, P. , Fan, H. , Lan, T. , Yang, X.‐L. , Shi, W.‐F. , Zhang, W. , … Ma, J.‐Y. (2018). Fatal swine acute diarrhoea syndrome caused by an HKU2‐related coronavirus of bat origin. Nature, 556, 255–258. 10.1038/s41586-018-0010-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X.‐L. , Wang, X.‐G. , Hu, B. , Zhang, L. , Zhang, W. , … Shi, Z.‐L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequence data that support the findings of this study are openly available in Genbank (https://www.ncbi.nlm.nih.gov/genbank/) with the accession numbers MK991901 to MK991951.